
- Department of Neurology, University of Massachusetts Medical School, Worcester, MA, USA
During normal aging or neurodegenerative diseases, neuronal survival and function depend on protein homeostasis, which is regulated by multiple mechanisms, including the microRNA (miRNA) pathway. In different cells types, the absence of Dicer, a key miRNA processing enzyme, leads to neurodegeneration through cell-autonomous and non-cell-autonomous mechanisms. Loss of certain miRNAs also causes neurodegeneration in some model organisms. On the other hand, miRNA expression is misregulated in patients with different neurodegenerative diseases. Thus, the miRNA pathway appears to be essential in the pathogenesis of several age-dependent neurodegenerative conditions; however, our understanding of the underlying mechanism remains rudimentary. The precise causal relationships between specific miRNAs and neurodegeneration in humans need to be further investigated.
Introduction
Alzheimer’s disease (AD), frontotemporal dementia (FTD), Parkinson’s disease (PD), and other neurodegenerative disorders are a major health problem in both developed and developing countries (Hampel et al., 2011; Reitz et al., 2011; Wittchen et al., 2011). Since no effective treatments are available, it is unlikely that the adverse societal effects of these disorders will be substantially alleviated in the near future. These disorders are characterized by progressive neuronal dysfunction that initially affects selected groups of neurons in specialized neuronal circuits. A number of cellular and molecular mechanisms lead to neuronal demise. Among them, neurotoxicity induced by misfolding, mislocalization, or abnormally elevated concentrations of particular protein species seems to be a common theme (Jucker and Walker, 2011; Lee et al., 2012; Selkoe et al., 2012). Mutations in several genes that are apparently functionally unrelated can cause the same neurodegenerative disease. Multiple environmental factors (e.g., viral infections and exposure to certain toxins) might also contribute to the development of a neurodegenerative disease (Ahmed and Wicklund, 2011; Gao and Hong, 2011). Much remained to be learned about how the complex interactions of environmental and genetic factors initially lead to the misregulation of protein homeostasis and subsequently to neuronal dysfunction.
MicroRNAs (miRNAs) are small non-coding RNAs that post-transcriptionally regulate gene expression by degrading their target mRNAs or repressing their translation (Ambros et al., 2003; Bartel, 2004; Ghildiyal and Zamore, 2009). Initially discovered in Caenorhabditis elegans (Lee et al., 1993), miRNAs have been found in plants, invertebrates, mammals, and humans (Bartel, 2009). Two major features of miRNAs indicate their potential contributions to neurodegenerative diseases. First, miRNAs can simultaneously regulate many target transcripts, and up to 50% of all coding genes may be regulated by miRNAs (Krol et al., 2010). Thus, miRNAs are central regulators of genetic networks. Second, miRNAs ensure stable protein levels under variable conditions and are therefore essential for the robustness of biological processes (Herranz and Cohen, 2010). Given the importance of protein homeostasis and the diversity of cellular pathways potentially leading to neurodegeneration, it has been hypothesized that miRNAs might contribute to neurodegenerative diseases (Eacker et al., 2009; Lau and de Strooper, 2010; Sonntag, 2010).
In this review, we briefly describe the biogenesis of miRNAs and their potential involvement in the evolution of the human brain. Then we will discuss accumulating evidence that miRNAs are important contributors to neurodegenerative diseases. Indeed, some observations suggest that miRNA alterations can disrupt protein homeostasis and may be at the root of neurodegenerative processes (Figure 2). Conversely, other data strongly suggest that altered miRNA networks are a consequence of abnormal neuronal physiology (Figure 3). Examples in specific neurodegenerative diseases will be presented.
miRNA Biogenesis
Although several alternative mechanisms also exist (Yang and Lai, 2011), the canonical pathway for miRNA biogenesis involves a primary transcript generated by RNA polymerase II (Lee et al., 2004). The primary miRNA is cleaved by a nuclear complex containing Drosha and DGCR8, giving rise to a hairpin precursor molecule (pre-miRNA) of 70–100 nt (Lee et al., 2003; Han et al., 2004). This pre-miRNA is exported to the cytoplasm, where Dicer, a RNA-III nuclease, catalyzes the final cleavage in the maturation process, resulting in an imperfect RNA duplex (Hutvagner et al., 2001). One strand (guide strand) is loaded into an RNA-induced silencing complex (RISC) to bind the target mRNA; the other strand (passenger strand) is usually destroyed (Chendrimada et al., 2005; Bartel, 2009). miRNAs control gene expression at the post-transcriptional level through imperfect base pairing with specific sequences, located mostly in the 3′UTRs of mRNAs. After recognition, miRNA–target interactions often result in mRNA degradation or inhibition of mRNA translation (Krol et al., 2010; Figure 1).
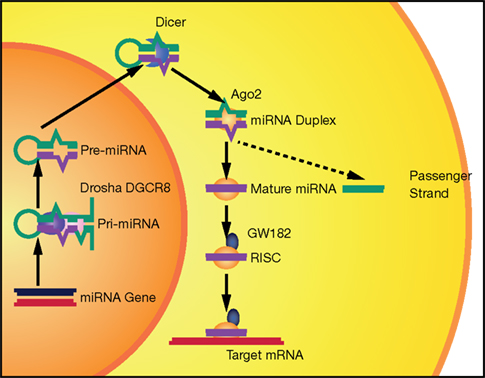
Figure 1. The canonical miRNA biogenesis pathway. miRNAs are produced from long Pol II transcripts (pri-miRNA). A nuclear complex containing Drosha (purple oval) and DGCR8 (pink oval) cleaves the primary transcript and generates a precursor miRNA (pre-miRNA). After nuclear export, pre-miRNA is further processed by Dicer (blue croissant). Then, Ago2 (yellow oval) binds to the complex formed by miRNA duplex and Dicer. Ago2 induces Dicer dissociation and the release of the passenger strand from the complex. Finally, other proteins, such as GW182 (dark blue), associate with Ago2 and form a RISC complex that recognizes and then silences (by mRNA degradation and/or translation inhibition) a target mRNA.
Role of miRNA in Brain Evolution
Neurodegenerative diseases are considered devastating disorders because they often impair cognitive and executive functions. Understanding how the human brain acquired such functions is a challenging task that might also provide important insights into the mechanisms of neurodegeneration. It was noted four decades ago that genetic differences among species do not account for brain divergence and that brain evolution could have been driven by changes in gene expression levels (King and Wilson, 1975). Early comparative transcriptome analyses confirmed this hypothesis and revealed more human-specific than chimpanzee-specific expression changes in the prefrontal cortex (PFC); no such changes were observed in blood, liver, or other tissues (Enard et al., 2002; Caceres et al., 2003). These findings supported the idea that brain-specific changes in gene expression levels shaped the evolution of the nervous system.
To determine whether miRNAs participated in this process, miRNA expression profiles in human and chimpanzee brains were compared (Berezikov et al., 2006). Many of the newly identified brain miRNAs were expressed only in humans, and many were restricted to primates. Another study reported that 10–35% of miRNAs were expressed in the human brain but not in chimpanzee or macaque brains (Hu et al., 2011). More importantly, developmental profiles of brain miRNAs and their target genes showed the fastest rates of human-specific evolutionary change (Somel et al., 2011). Although experimental evidence is still lacking, it is an attractive hypothesis that rapidly evolving miRNAs in the human brain are essential for neuronal function and maintenance.
Global Loss of miRNAs Causes Neurodegeneration
Genetic disruption of miRNAs biogenesis pathways has been used to probe the potential link between miRNAs and neurodegeneration. In mouse cerebellar Purkinje cells, conditional knockout of Dicer leads to age-dependent cerebellar degeneration and ataxia (Schaefer et al., 2007). Cell-type specific deletion of Dicer in striatal, retinal, spinal, and cortical neurons produced similar results (Cuellar et al., 2008; Damiani et al., 2008; Davis et al., 2008; Haramati et al., 2010). Dicer deletion also altered the phosphorylation pattern of tau before neuronal cell loss (Hebert et al., 2010), indicating that some mechanisms of neurodegeneration might be controlled through miRNAs. Of note, neurodegeneration in the absence of Dicer could also result from the toxic accumulation of pre-miRNAs or from the loss of other Dicer functions unrelated to miRNAs biogenesis. However, reduced production of a small proportion of miRNAs as a result of DGCR8 haploinsufficiency also leads to neuronal dysfunction (Stark et al., 2008; Fenelon et al., 2011; Schofield et al., 2011), supporting the notion that neurodegeneration could indeed arise from loss of miRNAs.
Glial cell defects may also profoundly influence neuronal survival (Ilieva et al., 2009; Prinz et al., 2011). Indeed, neurodegeneration ensues after targeted deletion of Dicer in astrocytes (Tao et al., 2011), oligodendrocytes (Shin et al., 2009), and Schwann cells (Pereira et al., 2010; Verrier et al., 2010; Wu et al., 2012). Thus, alteration of miRNA networks has the potential to disrupt neuronal function in a cell-autonomous or non-cell-autonomous manner and lead to neurodegenerative phenotypes.
Loss of Individual miRNAs Leads to Neurodegeneration
Genetic analyses have revealed essential roles for specific miRNAs in long-term neuronal survival, for example in mice lacking miR-124-1 (Sanuki et al., 2011), one of the most well studied miRNAs in neuronal development (Gao, 2010). The mouse genome has three miR-124 loci (miR-124-1, -2, and -3). Deletion of miR-124-1, the dominant source of this miRNA, increases apoptosis in the hippocampus and retina, causing a significant decrease in brain size. Furthermore, in the tail-suspension test, adult mutant mice exhibit a front and hind limb clasping response, a common phenotype in mouse models of neurodegenerative disorders. This effect seems to be mediated by regulation of the transcription factor Lhx2 (Sanuki et al., 2011). It is unclear whether the neurodegeneration also reflects the absence of miR-124’s well-documented developmental functions (Cao et al., 2007; Visvanathan et al., 2007; De Pietri Tonelli et al., 2008; Cheng et al., 2009; Maiorano and Mallamaci, 2009). This question could be answered by using a conditional knockout approach.
Another miRNA that might be involved in neuronal survival is miR-8 (Karres et al., 2007), which is not brain-specific and has a complex pattern of expression. Mutant flies lacking miR-8 have limb and wing defects and increased apoptosis in the central nervous system. Moreover, concomitant decrease of the transcriptional repressor atrophin in miR-8 mutant flies partially rescued the phenotypes (Karres et al., 2007). It remains to be determined whether miR-8 contributes to neurodegeneration in mammals.
miRNAs in Neurodegenerative Diseases
Microarray studies have shown that the brain expresses a wide range of miRNAs, suggesting that these small RNA molecules participate in nervous system physiology (Lagos-Quintana et al., 2002; Miska et al., 2004; Lim et al., 2005; Manakov et al., 2009). More importantly, profiling studies revealed profound changes in several miRNAs (i.e., miR-9, miR-29 cluster, miR-107, miR-125b, and miR-128) in patient brains such as that of AD patients (Kim et al., 2007; Lukiw, 2007; Hebert et al., 2008; Johnson et al., 2008; Wang et al., 2008, 2011; Nunez-Iglesias et al., 2010). Although these global changes in miRNA expression under pathological conditions should be interpreted cautiously, they support the notion that dysregulation of miRNAs networks is a common theme in neurodegenerative diseases.
Alzheimer’s Disease
A pathological hallmark of AD, the most prevalent neurodegenerative disease, is the accumulation of plaques formed by short β-amyloid (Aβ) peptides, commencing in the hippocampus, and spreading progressively throughout the brain (Ballard et al., 2011; Selkoe et al., 2012). It is likely the accumulation is caused by both increased production and impaired clearance of Aβ. Aβ peptides exert toxic effects and elicit an inflammatory response. Both may contribute to disrupt neuronal homeostasis and alter the integrity of neuronal networks involved in learning, memory, and other cognitive functions. Aβ peptides are generated through proteolytic cleavage of the amyloid precursor protein (APP) by γ-secretase and β-site APP-cleaving enzyme 1 (BACE1; O’Brien and Wong, 2011).
Amyloid precursor protein and BACE1 each contain several miRNA target sites in their 3′UTRs. Several miRNAs have been reported to repress APP expression, including miR-16, miR-101, miR-106a, and miR-520c (Patel et al., 2008; Liu et al., 2010; Long and Lahiri, 2011). miR-137 and miR-181c regulate serine palmitoyltransferase, which modulates the Aβ level (Geekiyanage and Chan, 2011). Interestingly, polymorphisms in miRNA binding sites in the 3′UTR of APP gene could influence the binding efficiency of these miRNAs. Thus, miRNAs might fine tune APP expression, which may enhance or limit the risk of AD (Delay et al., 2011).
A significant decrease in neuronal miR-107 expression and a parallel increase in BACE1 have been observed in AD patients (even those at the earliest stages of AD; Wang et al., 2008, 2011; Nelson and Wang, 2010). The 3′UTR of BACE1 mRNA has functional binding sites for miR-29 (Hebert et al., 2008), miR-107 (Wang et al., 2008), and miR-124 (Fang et al., 2012). Interestingly, miR-107 also controls the expression of other proteins relevant to AD pathology, such as cofilin (Yao et al., 2010), an actin-binding protein that accumulates in cytoplasmic inclusions known as Hirano bodies (Hirano, 1994). Thus, a single miRNA deregulation could activate multiple potentially pathogenic cascades upstream of Aβ accumulation.
Another possibility is that alterations in miRNAs in AD brain are a consequence of amyloid deposits. For instance, miR-106b is aberrantly expressed in APPswe/PSE9 mice (Wang et al., 2010a), and miR-146a levels are increased in AD brains and in several mouse models of AD (Li et al., 2011). In vitro exposure of hippocampal neurons to Aβ peptides preferentially decreases the levels of mature miRNAs (only a small fraction of miRNAs were upregulated; Schonrock et al., 2010). miRNAs were similarly deregulated in the hippocampus of APP23 mice at the onset of plaque formation. Overall, these studies suggest that miRNA deregulation is an essential pathogenic mechanism that is induced by Aβ aggregation and contributes to the progression and severity of AD.
Polyglutamine Diseases
Polyglutamine (polyQ) diseases are a group of nine neurodegenerative disorders caused by an unstable CAG expansion in the coding region of their respective associated genes (Orr and Zoghbi, 2007). Apart from this common feature, polyQ diseases have distinct clinical presentations, and the proteins involved in these diseases have no structural/functional homology (Table 1). Huntington disease (HD), the most frequent polyQ disease, is characterized by the progressive loss of striatal neurons and motor impairment (typically resulting in the involuntary writhing movements called chorea) and is often associated with cognitive and behavioral deficits (Shoulson and Young, 2011).
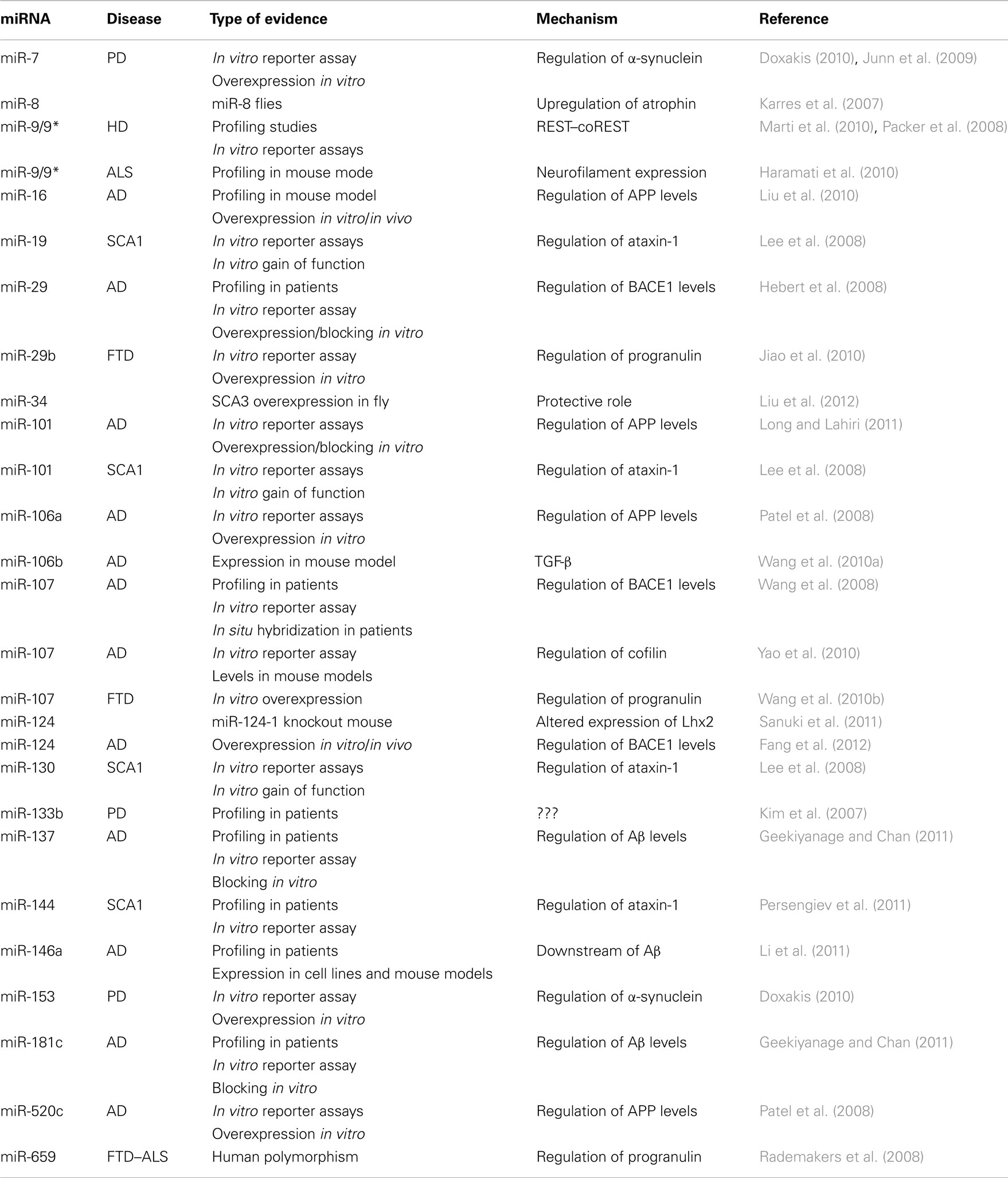
Table 1. List of miRNAs and the neurodegenerative diseases to which they have been associated.
The causal mutation in HD is an expanded repetition of the CAG trinucleotide in the first exon of the gene encoding huntingtin (HTT; Gilliam et al., 1987), a large protein (3300 amino acids) whose functions remains mostly unknown. HTT associates with Ago2 in P-bodies, and HTT depletion impairs miRNA-mediated gene silencing (Savas et al., 2008). Expanded HTT may sequester RNA processing factors in the cytoplasm (Jiang et al., 2011).
miRNAs were implicated in HD pathogenesis by two lines of evidence. First, the levels of repressor element 1 silencing transcription (REST) factor, a major pathogenic pathway in HD (Buckley et al., 2010), is elevated in HD neurons, resulting in repression of hundreds of key neuronal genes (Zuccato et al., 2003, 2007; Johnson et al., 2010b). Canonical and non-canonical REST binding motifs have been mapped in close proximity to 22 miRNA sites in the human genome, including several miRNAs that are abundant in neurons (Bruce et al., 2004; Jothi et al., 2008; Yu et al., 2011). Could abnormal REST deregulate miRNAs network in HD patients? REST and its cofactor coREST possess functional target sites for miR-9 and miR-9*, respectively (Packer et al., 2008), and miR-9 and miR-9* (together with miR-7, miR-124, miR-132, and other miRNAs) are downregulated in HD patients (Johnson et al., 2008; Packer et al., 2008; Marti et al., 2010). miRNA deregulation in HD was confirmed by profiling studies in animal models, although there was a high degree of variability among the models (Lee et al., 2011). These observations strongly suggest that altered miRNA transcription is a major event in HD pathogenesis.
Besides HD, there are eight other polyQ diseases: dentatorubral–pallidoluysian atrophy, spinal and bulbar muscular atrophy, and spinocerebellar ataxia (SCA) 1, 2, 3, 6, 7, and 17 (Gatchel and Zoghbi, 2005). In fly models of SCA3, reduction of miRNA processing after knockout of Dicer1 markedly enhances the toxicity induced by mutant ataxin-3 (Bilen et al., 2006). In parallel genetic screens, a single miRNA, bantam, was identified as a potent downstream modulator of both polyQ and tau toxicity in flies (Bilen et al., 2006). In a recent paper, miR-34 was shown to be protective against expanded SCA3 (Liu et al., 2012). Subsequent work suggested that miR-19, miR-101, and miR-130 are important for the post-translational regulation of ataxin-1 (Lee et al., 2008). Inhibition of those miRNAs enhanced the cytotoxicity of polyQ-expanded ataxin-1 in human cells. Moreover, miR-144, a highly conserved miRNA, also regulated ataxin-1 expression and appeared to be associated with aging. Ataxin-1 levels are higher in the cerebellum and cortex of SCA1 patients than in healthy aged brains (Persengiev et al., 2011). On the other hand, ataxin-2 might be required for miRNA function (McCann et al., 2011), further supporting the intimate association between miRNAs and polyQ diseases.
Parkinson’s Disease
Parkinson’s disease is a neurodegenerative disorder that primarily affects movement. Clinical symptoms include bradykinesia (decreased ability to start and continue movements), resting tremor, and rigidity. These symptoms are due to the relatively selective loss of dopaminergic neurons in the substantia nigra (Dauer and Przedborski, 2003) and reflect the impairment of neuronal networks important for regulating motor function. The past two decades have witnessed significant advances in the identification of distinct genetic loci at which pathogenic mutations are associated with parkinsonism (for review, see Lesage and Brice, 2009; Zimprich, 2011). Most research is focused on genes that have been conclusively linked to PD pathogenesis, including those encoding α-synuclein, leucine-rich repeat kinase 2, PTEN-induced putative kinase 1, parkin, and DJ-1.
Profiling studies of PD brains have revealed abnormalities in miRNA content (Kim et al., 2007; Minones-Moyano et al., 2011). One of the miRNAs found to be downregulated in these studies, miR-133b, plays a major role in the development of midbrain dopaminergic neurons by regulating the transcription factor Pitx3 (Kim et al., 2007). miR-7 and miR-153 control the expression of α-synuclein (Junn et al., 2009; Doxakis, 2010). Since intracellular levels of this protein appear to be critical in mediating its toxicity, deregulation of those miRNAs might lead to increased toxic levels of α-synuclein.
Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
Amyotrophic lateral sclerosis (ALS) is a progressive, lethal, degenerative disorder characterized by the selective death of motor neurons in the brain and spinal cord (Pasinelli and Brown, 2006; Ferraiuolo et al., 2011). ALS shares many clinical, pathological, and molecular features with FTD, the second most common early-onset dementia (Ferrari et al., 2011). Clinically, FTD progresses from an insidious onset of behavioral changes, impaired frontal executive functions, and language deficits to more severe cognitive defects and, finally, to generalized dementia (Boxer and Miller, 2005). In familial FTD cases, the mutated locus has been identified in the genes encoding tau (Hong et al., 1998; Hutton et al., 1998), VCP (Watts et al., 2004), CHMP2B (Skibinski et al., 2005), progranulin (Baker et al., 2006; Cruts et al., 2006), and C9ORF72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Among them, CHMP2B (Parkinson et al., 2006; Cox et al., 2010), VCP (Johnson et al., 2010a), and C9ORF72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Stewart et al., 2012) have also been implicated in ALS. Moreover, the RNA-binding proteins TDP-43 and FUS have been strongly implicated in both FTD and ALS (Arai et al., 2006; Neumann et al., 2006, 2009; Sreedharan et al., 2008; Vance et al., 2009).
As described above, Dicer deletion in spinal motor neurons mimics most of the clinical (e.g., progressive paralysis) and pathological (e.g., astrocytosis and signs of axonopathy) features of ALS (Haramati et al., 2010). Whether the miRNA pathway is involved in the molecular pathogenesis of FTD/ALS caused by C9ORF72 repeat expansion is unknown. However, a potential involvement for the miRNA pathway in other forms of FTD or ALS comes from limited studies on TDP-43. TDP-43 is mutated in a subset of ALS patients (Sreedharan et al., 2008), and the expression levels of some miRNAs are affected in TDP-43 mutant flies (Buratti et al., 2010). Biochemical interactions between TDP-43 and Drosha, a key miRNA processing enzyme (Han et al., 2004), have been observed (Gregory et al., 2004; Ling et al., 2010). These findings raise the possibility that TDP-43 may play a role in miRNA processing; however, the mechanism remains to be elucidated and whether endogenous Drosha and TDP-43 physically interact needs to be demonstrated.
Some miRNAs are emerging as important contributors to ALS pathogenesis. The muscle-specific miR-206 is upregulated upon nerve injury and is required for regeneration of neuromuscular synapses. Moreover, miR-206 deficiency accelerates disease progression in a mouse model of ALS (Williams et al., 2009). miR-9/9*, an evolutionarily conserved and multifunctional miRNA (Yuva-Aydemir et al., 2011), is also potentially involved in ALS. Profiling of miRNA expression in motor neurons harboring an SMN1mut allele found in pediatric spinal motor atrophy revealed decreases of more than 90% in miR-9 and miR-9* levels (Haramati et al., 2010). More importantly, changes in the expression levels of the neurofilament subunits likely contribute to the disease, and miR-9 is an upstream regulator of the neurofilament mRNAs.
Several other miRNAs might be linked to FTD–ALS through different mechanisms. For example, miR-29b and miR-107 regulate progranulin levels (Jiao et al., 2010; Wang et al., 2010b). Since progranulin haploinsufficiency can cause FTD114,115, excessive levels of those miRNAs might decrease progranulin levels and be a risk factor for the disease. Consistent with these observations, a genetic polymorphism in the 3′UTR of the progranulin gene is associated with a higher risk of FTD–ALS, and multiple miRNAs are misregulated in FTD with TDP-43 pathology (Rademakers et al., 2008; Kocerha et al., 2011). This genetic variant (rs5848) affects the miR-659 binding site, resulting in more efficient binding and, consequently, decreased progranulin levels. It is not known whether translational regulation by miRNAs is a common mechanism in FTD caused by progranulin deficiency or in other neurodegenerative diseases (Rollinson et al., 2011).
Summary
Although much progress has been made in our understanding of how miRNAs control gene expression at the post-transcriptional level during development, their contributions to neurodegenerative disease remain poorly understood. Many fundamental questions need to be addressed. Is miRNA pathway disruption a downstream consequence or a cause of neurodegeneration (Figure 2 vs. Figure 3)? Are miRNAs essential for the proper regulation of aggregation-prone proteins or do they control additional pathogenic pathways? Are individual miRNAs especially important in particular neurodegenerative diseases? Which miRNA target or targets are relevant for the disease? To address these questions, it will be essential to generate novel experimental models, such as conditional knockouts in which developmental defects can be circumvented, allowing assessment of the functions of specific miRNAs in the adult brain. We expect many exciting findings will be made in years to come.
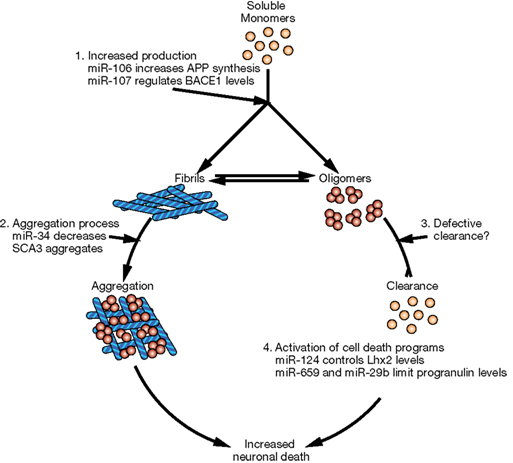
Figure 2. Potential mechanisms of neurodegeneration induced by altered miRNA networks. Schematic representation of cellular pathways that could be affected downstream of miRNAs. (1) Defects in miRNAs could increase the levels of aggregation-prone proteins either directly (i.e., miR-106a and APP in AD) or indirectly (i.e., miR-107 acting through BACE1 or miR-137 acting through serine palmitoyltransferase in AD). (2) miRNAs could control the expression of proteins involved in proper folding or quality control, increasing the risk of protein aggregation. (3) miRNAs could impair the removal of aggregated proteins and therefore increase their levels and toxicity. (4) Finally, altered miRNAs might result in neuronal cell death due to increased levels of certain transcription factors (i.e., miR-124 controls neuronal survival by limiting the expression of Lhx2) or the imbalance between pro-survival and pro-apoptotic signals (i.e., in FTD, polymorphism rs5848 results in more efficient binding of miR-659 and decreased levels of the pro-survival factor progranulin).
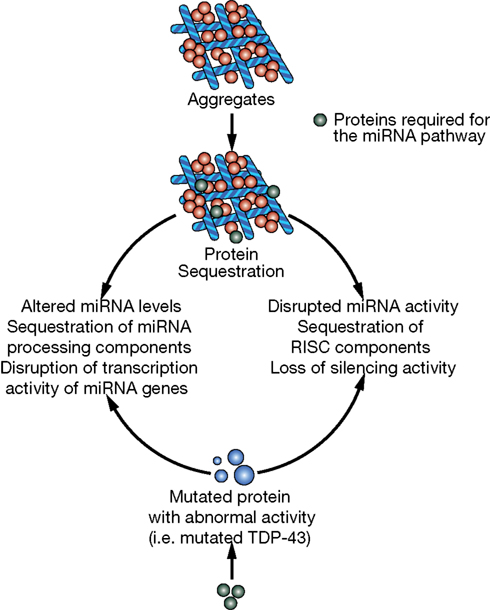
Figure 3. Potential mechanisms of miRNA deregulation downstream of protein aggregation/dysfunction. Two basic mechanisms could affect miRNA regulatory networks: (1) alteration of miRNA levels through deregulation of transcription (i.e., miR-9 downstream of HTT and REST) or processing (i.e., TDP-43 altering Drosha function) and (2) interference with RISC activity (i.e., ataxin-2 seems to be required for optimal miRNA silencing).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank S. Ordway for editorial assistance, A. Wilson for help with the figures, and Gao lab members for discussions. This work is supported by the National Institutes of Health (RO1NS066586 and RO1NS057553 to Fen-Biao Gao).
References
Ahmed, A., and Wicklund, M. P. (2011). Amyotrophic lateral sclerosis: what role does environment play? Neurol. Clin. 29, 689–711.
Ambros, V., Lee, R. C., Lavanway, A., Williams, P. T., and Jewell, D. (2003). MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr. Biol. 13, 807–818.
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., Mann, D., Tsuchiya, K., Yoshida, M., Hashizume, Y., and Oda, T. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611.
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., Snowden, J., Adamson, J., Sadovnick, A. D., Rollinson, S., Cannon, A., Dwosh, E., Neary, D., Melquist, S., Richardson, A., Dickson, D., Berger, Z., Eriksen, J., Robinson, T., Zehr, C., Dickey, C. A., Crook, R., McGowan, E., Mann, D., Boeve, B., Feldman, H., and Hutton, M. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919.
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer’s disease. Lancet 377, 1019–1031.
Berezikov, E., Thuemmler, F., van Laake, L. W., Kondova, I., Bontrop, R., Cuppen, E., and Plasterk, R. H. (2006). Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 38, 1375–1377.
Bilen, J., Liu, N., Burnett, B. G., Pittman, R. N., and Bonini, N. M. (2006). MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol. Cell 24, 157–163.
Boxer, A. L., and Miller, B. L. (2005). Clinical features of frontotemporal dementia. Alzheimer Dis. Assoc. Disord. 19(Suppl. 1), S3–S6.
Bruce, A. W., Donaldson, I. J., Wood, I. C., Yerbury, S. A., Sadowski, M. I., Chapman, M., Gottgens, B., and Buckley, N. J. (2004). Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc. Natl. Acad. Sci. U.S.A. 101, 10458–10463.
Buckley, N. J., Johnson, R., Zuccato, C., Bithell, A., and Cattaneo, E. (2010). The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol. Dis. 39, 28–39.
Buratti, E., De Conti, L., Stuani, C., Romano, M., Baralle, M., and Baralle, F. (2010). Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J. 277, 2268–2281.
Caceres, M., Lachuer, J., Zapala, M. A., Redmond, J. C., Kudo, L., Geschwind, D. H., Lockhart, D. J., Preuss, T. M., and Barlow, C. (2003). Elevated gene expression levels distinguish human from non-human primate brains. Proc. Natl. Acad. Sci. U.S.A. 100, 13030–13035.
Cao, X., Pfaff, S. L., and Gage, F. H. (2007). A functional study of miR-124 in the developing neural tube. Genes Dev. 21, 531–536.
Chendrimada, T. P., Gregory, R. I., Kumaraswamy, E., Norman, J., Cooch, N., Nishikura, K., and Shiekhattar, R. (2005). TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436, 740–744.
Cheng, L. C., Pastrana, E., Tavazoie, M., and Doetsch, F. (2009). miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 12, 399–408.
Cox, L. E., Ferraiuolo, L., Goodall, E. F., Heath, P. R., Higginbottom, A., Mortiboys, H., Hollinger, H. C., Hartley, J. A., Brockington, A., Burness, C. E., Morrison, K. E., Wharton, S. B., Grierson, A. J., Ince, P. G., Kirby, J., and Shaw, P. J. (2010). Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 5, e9872. doi:10.1371/journal.pone.0009872
Cruts, M., Gijselinck, I., van der Zee, J., Engelborghs, S., Wils, H., Pirici, D., Rademakers, R., Vandenberghe, R., Dermaut, B., Martin, J. J., van Duijn, C., Peeters, K., Sciot, R., Santens, P., De Pooter, T., Mattheijssens, M., Van den Broeck, M., Cuijt, I., Vennekens, K., De Deyn, P. P., Kumar-Singh, S., and Van Broeckhoven, C. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924.
Cuellar, T. L., Davis, T. H., Nelson, P. T., Loeb, G. B., Harfe, B. D., Ullian, E., and McManus, M. T. (2008). Dicer loss in striatal neurons produces behavioral and neuroanatomical phenotypes in the absence of neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 105, 5614–5619.
Damiani, D., Alexander, J. J., O’Rourke, J. R., McManus, M., Jadhav, A. P., Cepko, C. L., Hauswirth, W. W., Harfe, B. D., and Strettoi, E. (2008). Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J. Neurosci. 28, 4878–4887.
Dauer, W., and Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron 39, 889–909.
Davis, T. H., Cuellar, T. L., Koch, S. M., Barker, A. J., Harfe, B. D., McManus, M. T., and Ullian, E. M. (2008). Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J. Neurosci. 28, 4322–4330.
De Pietri Tonelli, D., Pulvers, J. N., Haffner, C., Murchison, E. P., Hannon, G. J., and Huttner, W. B. (2008). miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 135, 3911–3921.
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., Nicholson, A. M., Finch, N. A., Flynn, H., Adamson, J., Kouri, N., Wojtas, A., Sengdy, P., Hsiung, G. Y., Karydas, A., Seeley, W. W., Josephs, K. A., Coppola, G., Geschwind, D. H., Wszolek, Z. K., Feldman, H., Knopman, D. S., Petersen, R. C., Miller, B. L., Dickson, D. W., Boylan, K. B., Graff-Radford, N. R., and Rademakers, R. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256.
Delay, C., Calon, F., Mathews, P., and Hebert, S. S. (2011). Alzheimer-specific variants in the 3′UTR of amyloid precursor protein affect microRNA function. Mol. Neurodegener. 6, 70.
Doxakis, E. (2010). Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 285, 12726–12734.
Eacker, S. M., Dawson, T. M., and Dawson, V. L. (2009). Understanding microRNAs in neurodegeneration. Nat. Rev. Neurosci. 10, 837–841.
Enard, W., Khaitovich, P., Klose, J., Zollner, S., Heissig, F., Giavalisco, P., Nieselt-Struwe, K., Muchmore, E., Varki, A., Ravid, R., Doxiadis, G. M., Bontrop, R. E., and Paabo, S. (2002). Intra- and interspecific variation in primate gene expression patterns. Science 296, 340–343.
Fang, M., Wang, J., Zhang, X., Geng, Y., Hu, Z., Rudd, J. A., Ling, S., Chen, W., and Han, S. (2012). The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 209, 94–105.
Fenelon, K., Mukai, J., Xu, B., Hsu, P. K., Drew, L. J., Karayiorgou, M., Fischbach, G. D., Macdermott, A. B., and Gogos, J. A. (2011). Deficiency of Dgcr8, a gene disrupted by the 22q11.2 microdeletion, results in altered short-term plasticity in the prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 108, 4447–4452.
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630.
Ferrari, R., Kapogiannis, D., Huey, E. D., and Momeni, P. (2011). FTD and ALS: a tale of two diseases. Curr. Alzheimer Res. 8, 273–294.
Gao, F. B. (2010). Context-dependent functions of specific microRNAs in neuronal development. Neural Dev. 1, 5::25. doi: 10.1186/1749-8104-5-25
Gao, H. M., and Hong, J. S. (2011). Gene-environment interactions: key to unraveling the mystery of Parkinson’s disease. Prog. Neurobiol. 94, 1–19.
Gatchel, J. R., and Zoghbi, H. Y. (2005). Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 6, 743–755.
Geekiyanage, H., and Chan, C. (2011). MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid beta, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 31, 14820–14830.
Ghildiyal, M., and Zamore, P. D. (2009). Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 10, 94–108.
Gilliam, T. C., Tanzi, R. E., Haines, J. L., Bonner, T. I., Faryniarz, A. G., Hobbs, W. J., MacDonald, M. E., Cheng, S. V., Folstein, S. E., Conneally, P. M., Wexler, N. S., and Gusella, J. F. (1987). Localization of the Huntington’s disease gene to a small segment of chromosome 4 flanked by D4S10 and the telomere. Cell 50, 565–571.
Gregory, R. I., Yan, K. P., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., and Shiekhattar, R. (2004). The Microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240.
Hampel, H., Prvulovic, D., Teipel, S., Jessen, F., Luckhaus, C., Frolich, L., Riepe, M. W., Dodel, R., Leyhe, T., Bertram, L., Hoffmann, W., and Faltraco, F. (2011). The future of Alzheimer’s disease: the next 10 years. Prog. Neurobiol. 95, 718–728.
Han, J., Lee, Y., Yeom, K. H., Kim, Y. K., Jin, H., and Kim, V. N. (2004). The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 18, 3016–3027.
Haramati, S., Chapnik, E., Sztainberg, Y., Eilam, R., Zwang, R., Gershoni, N., McGlinn, E., Heiser, P. W., Wills, A. M., Wirguin, I., Rubin, L. L., Misawa, H., Tabin, C. J., Brown, R. J., Chen, A., and Hornstein, E. (2010). miRNA malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. U.S.A. 107, 13111–13116.
Hebert, S. S., Horre, K., Nicolai, L., Papadopoulou, A. S., Mandemakers, W., Silahtaroglu, A. N., Kauppinen, S., Delacourte, A., and De Strooper, B. (2008). Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U.S.A. 105, 6415–6420.
Hebert, S. S., Papadopoulou, A. S., Smith, P., Galas, M. C., Planel, E., Silahtaroglu, A. N., Sergeant, N., Buee, L., and De Strooper, B. (2010). Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 19, 3959–3969.
Herranz, H., and Cohen, S. M. (2010). MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev. 24, 1339–1344.
Hirano, A. (1994). Hirano bodies and related neuronal inclusions. Neuropathol. Appl. Neurobiol. 20, 3–11.
Hong, M., Zhukareva, V., Vogelsberg-Ragaglia, V., Wszolek, Z., Reed, L., Miller, B. I., Geschwind, D. H., Bird, T. D., McKeel, D., Goate, A., Morris, J. C., Wilhelmsen, K. C., Schellenberg, G. D., Trojanowski, J. Q., and Lee, V. M. (1998). Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282, 1914–1917.
Hu, H. Y., Guo, S., Xi, J., Yan, Z., Fu, N., Zhang, X., Menzel, C., Liang, H., Yang, H., Zhao, M., Zeng, R., Chen, W., Paabo, S., and Khaitovich, P. (2011). MicroRNA expression and regulation in human, chimpanzee, and macaque brains. PLoS Genet. 7, e1002327. doi:10.1371/journal.pgen.1002327
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., Pickering-Brown, S., Chakraverty, S., Isaacs, A., Grover, A., Hackett, J., Adamson, J., Lincoln, S., Dickson, D., Davies, P., Petersen, R. C., Stevens, M., de Graaff, E., Wauters, E., van Baren, J., Hillebrand, M., Joosse, M., Kwon, J. M., Nowotny, P., Che, L. K., Norton, J., Morris, J. C., Reed, L. A., Trojanowski, J., Basun, H., Lannfelt, L., Neystat, M., Fahn, S., Dark, F., Tannenberg, T., Dodd, P. R., Hayward, N., Kwok, J. B., Schofield, P. R., Andreadis, A., Snowden, J., Craufurd, D., Neary, D., Owen, F., Oostra, B. A., Hardy, J., Goate, A., van Swieten, J., Mann, D., Lynch, T., and Heutink, P. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705.
Hutvagner, G., McLachlan, J., Pasquinelli, A. E., Balint, E., Tuschl, T., and Zamore, P. D. (2001). A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293, 834–838.
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772.
Jiang, Y. J., Che, M. X., Yuan, J. Q., Xie, Y. Y., Yan, X. Z., and Hu, H. Y. (2011). Interaction with polyglutamine-expanded huntingtin alters cellular distribution and RNA processing of huntingtin yeast two-hybrid protein A (HYPA). J. Biol. Chem. 286, 25236–25245.
Jiao, J., Herl, L. D., Farese, R. V., and Gao, F. B. (2010). MicroRNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS ONE 5, e10551. doi:10.1371/journal.pone.0010551
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., Gibbs, J. R., Brunetti, M., Gronka, S., Wuu, J., Ding, J., McCluskey, L., Martinez-Lage, M., Falcone, D., Hernandez, D. G., Arepalli, S., Chong, S., Schymick, J. C., Rothstein, J., Landi, F., Wang, Y. D., Calvo, A., Mora, G., Sabatelli, M., Monsurro, M. R., Battistini, S., Salvi, F., Spataro, R., Sola, P., Borghero, G., Galassi, G., Scholz, S. W., Taylor, J. P., Restagno, G., Chio, A., and Traynor, B. J. (2010a). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864.
Johnson, R., Richter, N., Jauch, R., Gaughwin, P. M., Zuccato, C., Cattaneo, E., and Stanton, L. W. (2010b). The human accelerated region 1 noncoding RNA is repressed by REST in Huntington’s disease. Physiol. Genomics 41, 269–274.
Johnson, R., Zuccato, C., Belyaev, N. D., Guest, D. J., Cattaneo, E., and Buckley, N. J. (2008). A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 29, 438–445.
Jothi, R., Cuddapah, S., Barski, A., Cui, K., and Zhao, K. (2008). Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 36, 5221–5231.
Jucker, M., and Walker, L. C. (2011). Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 70, 532–540.
Junn, E., Lee, K. W., Jeong, B. S., Chan, T. W., Im, J. Y., and Mouradian, M. M. (2009). Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. U.S.A. 106, 13052–13057.
Karres, J. S., Hilgers, V., Carrera, I., Treisman, J., and Cohen, S. M. (2007). The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell 131, 136–145.
Kim, J., Inoue, K., Ishii, J., Vanti, W. B., Voronov, S. V., Murchison, E., Hannon, G., and Abeliovich, A. (2007). A MicroRNA feedback circuit in midbrain dopamine neurons. Science 317, 1220–1224.
King, M. C., and Wilson, A. C. (1975). Evolution at two levels in humans and chimpanzees. Science 188, 107–116.
Kocerha, J., Kouri, N., Baker, M., Finch, N., DeJesus-Hernandez, M., Gonzalez, J., Chidamparam, K., Josephs, K. A., Boeve, B. F., Graff-Radford, N. R., Crook, J., Dickson, D. W., and Rademakers, R. (2011). Altered microRNA expression in frontotemporal lobar degeneration with TDP-43 pathology caused by progranulin mutations. BMC Genomics 12, 527. doi:10.1186/1471-2164-12-527
Krol, J., Loedige, I., and Filipowicz, W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610.
Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., and Tuschl, T. (2002). Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739.
Lau, P., and de Strooper, B. (2010). Dysregulated microRNAs in neurodegenerative disorders. Semin. Cell Dev. Biol. 21, 768–773.
Lee, E. B., Lee, V. M., and Trojanowski, J. Q. (2012). Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50.
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854.
Lee, S. T., Chu, K., Im, W. S., Yoon, H. J., Im, J. Y., Park, J. E., Park, K. H., Jung, K. H., Lee, S. K., Kim, M., and Roh, J. K. (2011). Altered microRNA regulation in Huntington’s disease models. Exp. Neurol. 227, 172–179.
Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., Lee, J., Provost, P., Radmark, O., Kim, S., and Kim, V. N. (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415–419. doi:10.1186/1471-2164-10-419
Lee, Y., Kim, M., Han, J., Yeom, K. H., Lee, S., Baek, S. H., and Kim, V. N. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060.
Lee, Y., Samaco, R. C., Gatchel, J. R., Thaller, C., Orr, H. T., and Zoghbi, H. Y. (2008). miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat. Neurosci. 11, 1137–1139.
Lesage, S., and Brice, A. (2009). Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 18, R48–R59.
Li, Y. Y., Cui, J. G., Hill, J. M., Bhattacharjee, S., Zhao, Y., and Lukiw, W. J. (2011). Increased expression of miRNA-146a in Alzheimer’s disease transgenic mouse models. Neurosci. Lett. 487, 94–98.
Lim, L. P., Lau, N. C., Garrett-Engele, P., Grimson, A., Schelter, J. M., Castle, J., Bartel, D. P., Linsley, P. S., and Johnson, J. M. (2005). Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433, 769–773.
Ling, S. C., Albuquerque, C. P., Han, J. S., Lagier-Tourenne, C., Tokunaga, S., Zhou, H., and Cleveland, D. W. (2010). ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. U.S.A. 107, 13318–13323.
Liu, N., Landreh, M., Cao, K., Abe, M., Hendriks, G. J., Kennerdell, J. R., Zhu, Y., Wang, L. S., and Bonini, N. M. (2012). The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482, 519–523.
Liu, W., Liu, C., Zhu, J., Shu, P., Yin, B., Gong, Y., Qiang, B., Yuan, J., and Peng, X. (2010). MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 33, 522–534.
Long, J. M., and Lahiri, D. K. (2011). MicroRNA-101 downregulates Alzheimer’s amyloid-beta precursor protein levels in human cell cultures and is differentially expressed. Biochem. Biophys. Res. Commun. 404, 889–895.
Lukiw, W. J. (2007). Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 18, 297–300.
Maiorano, N. A., and Mallamaci, A. (2009). Promotion of embryonic cortico-cerebral neuronogenesis by miR-124. Neural Dev. 4, 40.
Manakov, S. A., Grant, S. G., and Enright, A. J. (2009). Reciprocal regulation of microRNA and mRNA profiles in neuronal development and synapse formation. BMC Genomics 10, 419.
Marti, E., Pantano, L., Banez-Coronel, M., Llorens, F., Minones-Moyano, E., Porta, S., Sumoy, L., Ferrer, I., and Estivill, X. (2010). A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 38, 7219–7235.
McCann, C., Holohan, E. E., Das, S., Dervan, A., Larkin, A., Lee, J. A., Rodrigues, V., Parker, R., and Ramaswami, M. (2011). The Ataxin-2 protein is required for microRNA function and synapse-specific long-term olfactory habituation. Proc. Natl. Acad. Sci. U.S.A. 108, E655–E662.
Minones-Moyano, E., Porta, S., Escaramis, G., Rabionet, R., Iraola, S., Kagerbauer, B., Espinosa-Parrilla, Y., Ferrer, I., Estivill, X., and Marti, E. (2011). MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 20, 3067–3078.
Miska, E. A., Alvarez-Saavedra, E., Townsend, M., Yoshii, A., Sestan, N., Rakic, P., Constantine-Paton, M., and Horvitz, H. R. (2004). Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 5, R68.
Nelson, P. T., and Wang, W. X. (2010). MiR-107 is reduced in Alzheimer’s disease brain neocortex: validation study. J. Alzheimers Dis. 21, 75–79.
Neumann, M., Rademakers, R., Roeber, S., Baker, M., Kretzschmar, H. A., and Mackenzie, I. R. (2009). A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–2931.
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., Bruce, J., Schuck, T., Grossman, M., Clark, C. M., McCluskey, L. F., Miller, B. L., Masliah, E., Mackenzie, I. R., Feldman, H., Feiden, W., Kretzschmar, H. A., Trojanowski, J. Q., and Lee, V. M. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133.
Nunez-Iglesias, J., Liu, C. C., Morgan, T. E., Finch, C. E., and Zhou, X. J. (2010). Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS ONE 5, e8898. doi:10.1371/journal.pone.0008898
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204.
Orr, H. T., and Zoghbi, H. Y. (2007). Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621.
Packer, A. N., Xing, Y., Harper, S. Q., Jones, L., and Davidson, B. L. (2008). The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 28, 14341–14346.
Parkinson, N., Ince, P. G., Smith, M. O., Highley, R., Skibinski, G., Andersen, P. M., Morrison, K. E., Pall, H. S., Hardiman, O., Collinge, J., Shaw, P. J., and Fisher, E. M. (2006). ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67, 1074–1077.
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723.
Patel, N., Hoang, D., Miller, N., Ansaloni, S., Huang, Q., Rogers, J. T., Lee, J. C., and Saunders, A. J. (2008). MicroRNAs can regulate human APP levels. Mol. Neurodegener. 3, 10.
Pereira, J. A., Baumann, R., Norrmen, C., Somandin, C., Miehe, M., Jacob, C., Luhmann, T., Hall-Bozic, H., Mantei, N., Meijer, D., and Suter, U. (2010). Dicer in Schwann cells is required for myelination and axonal integrity. J. Neurosci. 30, 6763–6775.
Persengiev, S., Kondova, I., Otting, N., Koeppen, A. H., and Bontrop, R. E. (2011). Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis. Neurobiol. Aging 32, 2316.e17–2316.e27.
Prinz, M., Priller, J., Sisodia, S. S., and Ransohoff, R. M. (2011). Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 14, 1227–1235.
Rademakers, R., Eriksen, J. L., Baker, M., Robinson, T., Ahmed, Z., Lincoln, S. J., Finch, N., Rutherford, N. J., Crook, R. J., Josephs, K. A., Boeve, B. F., Knopman, D. S., Petersen, R. C., Parisi, J. E., Caselli, R. J., Wszolek, Z. K., Uitti, R. J., Feldman, H., Hutton, M. L., Mackenzie, I. R., Graff-Radford, N. R., and Dickson, D. W. (2008). Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum. Mol. Genet. 17, 3631–3642.
Reitz, C., Brayne, C., and Mayeux, R. (2011). Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 7, 137–152.
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., Schymick, J. C., Laaksovirta, H., van Swieten, J. C., Myllykangas, L., Kalimo, H., Paetau, A., Abramzon, Y., Remes, A. M., Kaganovich, A., Scholz, S. W., Duckworth, J., Ding, J., Harmer, D. W., Hernandez, D. G., Johnson, J. O., Mok, K., Ryten, M., Trabzuni, D., Guerreiro, R. J., Orrell, R. W., Neal, J., Murray, A., Pearson, J., Jansen, I. E., Sondervan, D., Seelaar, H., Blake, D., Young, K., Halliwell, N., Callister, J. B., Toulson, G., Richardson, A., Gerhard, A., Snowden, J., Mann, D., Neary, D., Nalls, M. A., Peuralinna, T., Jansson, L., Isoviita, V. M., Kaivorinne, A. L., Holtta-Vuori, M., Ikonen, E., Sulkava, R., Benatar, M., Wuu, J., Chio, A., Restagno, G., Borghero, G., Sabatelli, M., Heckerman, D., Rogaeva, E., Zinman, L., Rothstein, J. D., Sendtner, M., Drepper, C., Eichler, E. E., Alkan, C., Abdullaev, Z., Pack, S. D., Dutra, A., Pak, E., Hardy, J., Singleton, A., Williams, N. M., Heutink, P., Pickering-Brown, S., Morris, H. R., Tienari, P. J., and Traynor, B. J. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268.
Rollinson, S., Rohrer, J. D., van der Zee, J., Sleegers, K., Mead, S., Engelborghs, S., Collinge, J., De Deyn, P. P., Mann, D. M., Van Broeckhoven, C., and Pickering-Brown, S. M. (2011). No association of PGRN 3’UTR rs5848 in frontotemporal lobar degeneration. Neurobiol. Aging 32, 754–755.
Sanuki, R., Onishi, A., Koike, C., Muramatsu, R., Watanabe, S., Muranishi, Y., Irie, S., Uneo, S., Koyasu, T., Matsui, R., Cherasse, Y., Urade, Y., Watanabe, D., Kondo, M., Yamashita, T., and Furukawa, T. (2011). miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat. Neurosci. 14, 1125–1134.
Savas, J. N., Makusky, A., Ottosen, S., Baillat, D., Then, F., Krainc, D., Shiekhattar, R., Markey, S. P., and Tanese, N. (2008). Huntington’s disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc. Natl. Acad. Sci. U.S.A. 105, 10820–10825.
Schaefer, A., O’Carroll, D., Tan, C. L., Hillman, D., Sugimori, M., Llinas, R., and Greengard, P. (2007). Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 204, 1553–1558.
Schofield, C. M., Hsu, R., Barker, A. J., Gertz, C. C., Blelloch, R., and Ullian, E. M. (2011). Monoallelic deletion of the microRNA biogenesis gene Dgcr8 produces deficits in the development of excitatory synaptic transmission in the prefrontal cortex. Neural Dev. 6, 11.
Schonrock, N., Ke, Y. D., Humphreys, D., Staufenbiel, M., Ittner, L. M., Preiss, T., and Gotz, J. (2010). Neuronal microRNA deregulation in response to Alzheimer’s disease amyloid-beta. PLoS ONE 5, e11070. doi:10.1371/journal.pone.0011070
Selkoe, D., Mandelkow, E., and Holtzman, D. (2012). Deciphering Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a011460.
Shin, D., Shin, J. Y., McManus, M. T., Ptacek, L. J., and Fu, Y. H. (2009). Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann. Neurol. 66, 843–857.
Shoulson, I., and Young, A. B. (2011). Milestones in Huntington disease. Mov. Disord. 26, 1127–1133.
Skibinski, G., Parkinson, N. J., Brown, J. M., Chakrabarti, L., Lloyd, S. L., Hummerich, H., Nielsen, J. E., Hodges, J. R., Spillantini, M. G., Thusgaard, T., Brandner, S., Brun, A., Rossor, M. N., Gade, A., Johannsen, P., Sorensen, S. A., Gydesen, S., Fisher, E. M., and Collinge, J. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808.
Somel, M., Liu, X., Tang, L., Yan, Z., Hu, H., Guo, S., Jiang, X., Zhang, X., Xu, G., Xie, G., Li, N., Hu, Y., Chen, W., Paabo, S., and Khaitovich, P. (2011). MicroRNA-driven developmental remodeling in the brain distinguishes humans from other primates. PLoS Biol. 9, e1001214. doi:10.1371/journal.pbio.1001214
Sonntag, K. C. (2010). MicroRNAs and deregulated gene expression networks in neurodegeneration. Brain Res. 1338, 48–57.
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., Ackerley, S., Durnall, J. C., Williams, K. L., Buratti, E., Baralle, F., de Belleroche, J., Mitchell, J. D., Leigh, P. N., Al-Chalabi, A., Miller, C. C., Nicholson, G., and Shaw, C. E. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672.
Stark, K. L., Xu, B., Bagchi, A., Lai, W. S., Liu, H., Hsu, R., Wan, X., Pavlidis, P., Mills, A. A., Karayiorgou, M., and Gogos, J. A. (2008). Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 40, 751–760.
Stewart, H., Rutherford, N. J., Briemberg, H., Krieger, C., Cashman, N., Fabros, M., Baker, M., Fok, A., DeJesus-Hernandez, M., Eisen, A., Rademakers, R., and Mackenzie, I. R. (2012). Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 123, 409–417.
Tao, J., Wu, H., Lin, Q., Wei, W., Lu, X. H., Cantle, J. P., Ao, Y., Olsen, R. W., Yang, X. W., Mody, I., Sofroniew, M. V., and Sun, Y. E. (2011). Deletion of astroglial Dicer causes non-cell-autonomous neuronal dysfunction and degeneration. J. Neurosci. 31, 8306–8319.
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., Hu, X., Smith, B., Ruddy, D., Wright, P., Ganesalingam, J., Williams, K. L., Tripathi, V., Al-Saraj, S., Al-Chalabi, A., Leigh, P. N., Blair, I. P., Nicholson, G., de Belleroche, J., Gallo, J. M., Miller, C. C., and Shaw, C. E. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211.
Verrier, J. D., Semple-Rowland, S., Madorsky, I., Papin, J. E., and Notterpek, L. (2010). Reduction of Dicer impairs Schwann cell differentiation and myelination. J. Neurosci. Res. 88, 2558–2568.
Visvanathan, J., Lee, S., Lee, B., Lee, J. W., and Lee, S. K. (2007). The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev. 21, 744–749.
Wang, H., Liu, J., Zong, Y., Xu, Y., Deng, W., Zhu, H., Liu, Y., Ma, C., Huang, L., Zhang, L., and Qin, C. (2010a). miR-106b aberrantly expressed in a double transgenic mouse model for Alzheimer’s disease targets TGF-beta type II receptor. Brain Res. 1357, 166–174.
Wang, W. X., Wilfred, B. R., Madathil, S. K., Tang, G., Hu, Y., Dimayuga, J., Stromberg, A. J., Huang, Q., Saatman, K. E., and Nelson, P. T. (2010b). miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am. J. Pathol. 177, 334–345.
Wang, W. X., Huang, Q., Hu, Y., Stromberg, A. J., and Nelson, P. T. (2011). Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: white matter versus gray matter. Acta Neuropathol. 121, 193–205.
Wang, W. X., Rajeev, B. W., Stromberg, A. J., Ren, N., Tang, G., Huang, Q., Rigoutsos, I., and Nelson, P. T. (2008). The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 28, 1213–1223.
Watts, G. D., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., Pestronk, A., Whyte, M. P., and Kimonis, V. E. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381.
Williams, A. H., Valdez, G., Moresi, V., Qi, X., McAnally, J., Elliott, J. L., Bassel-Duby, R., Sanes, J. R., and Olson, E. N. (2009). MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 326, 1549–1554.
Wittchen, H. U., Jacobi, F., Rehm, J., Gustavsson, A., Svensson, M., Jonsson, B., Olesen, J., Allgulander, C., Alonso, J., Faravelli, C., Fratiglioni, L., Jennum, P., Lieb, R., Maercker, A., van Os, J., Preisig, M., Salvador-Carulla, L., Simon, R., and Steinhausen, H. C. (2011). The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 21, 655–679.
Wu, D., Raafat, A., Pak, E., Clemens, S., and Murashov, A. K. (2012). Dicer-microRNA pathway is critical for peripheral nerve regeneration and functional recovery in vivo and regenerative axonogenesis in vitro. Exp. Neurol. 233, 555–565.
Yang, J. S., and Lai, E. C. (2011). Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol. Cell 43, 892–903.
Yao, J., Hennessey, T., Flynt, A., Lai, E., Beal, M. F., and Lin, M. T. (2010). MicroRNA-related cofilin abnormality in Alzheimer’s disease. PLoS ONE 5, e15546. doi:10.1371/journal.pone.0015546
Yu, H. B., Johnson, R., Kunarso, G., and Stanton, L. W. (2011). Coassembly of REST and its cofactors at sites of gene repression in embryonic stem cells. Genome Res. 21, 1284–1293.
Yuva-Aydemir, Y., Simkin, A., Gascon, E., and Gao, F. B. (2011). MicroRNA-9: functional evolution of a conserved small regulatory RNA. RNA Biol. 8, 557–564.
Zimprich, A. (2011). Genetics of Parkinson’s disease and essential tremor. Curr. Opin. Neurol. 24, 318–323.
Zuccato, C., Belyaev, N., Conforti, P., Ooi, L., Tartari, M., Papadimou, E., MacDonald, M., Fossale, E., Zeitlin, S., Buckley, N., and Cattaneo, E. (2007). Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci. 27, 6972–6983.
Keywords: Alzheimer’s disease, ALS, FTD, microRNAs, neurodegeneration, C9ORF72, CHMP2B, TDP-43
Citation: Gascon E and Gao F-B (2012) Cause or effect: misregulation of microRNA pathways in neurodegeneration. Front. Neurosci. 6:48. doi: 10.3389/fnins.2012.00048
Received: 13 February 2012; Paper pending published: 29 February 2012;
Accepted: 23 March 2012; Published online: 09 April 2012.
Edited by:
Jernej Ule, Medical Research Council Laboratory of Molecular Biology, UKReviewed by:
Tao Sun, Cornell University Weill Medical College, USAPhilip Wong, Johns Hopkins University School of Medicine, USA
Copyright: © 2012 Gascon and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Eduardo Gascon and Fen-Biao Gao, Department of Neurology, University of Massachusetts Medical School, Worcester, MA 01605, USA. e-mail: eduardo.gascon@umassmed.edu; fen-biao.gao@umassmed.edu