
- 1 Cellular and Molecular Biology Graduate Program, University of Wisconsin–Madison, Madison, WI, USA
- 2 Waisman Center, University of Wisconsin–Madison, Madison, WI, USA
- 3 Graduate Program in Neurobiology and Behavior, University of Washington, Seattle, WA, USA
- 4 Department of Neuroscience, University of Wisconsin–Madison, Madison, WI, USA
The process of neurogenesis includes neural stem cell proliferation, fate specification, young neuron migration, neuronal maturation, and functional integration into existing circuits. Although neurogenesis occurs largely during embryonic development, low levels but functionally important neurogenesis persists in restricted regions of the postnatal brain, including the subgranular zone of the dentate gyrus in the hippocampus and the subventricular zone of the lateral ventricles. This review will cover both embryonic and adult neurogenesis with an emphasis on the latter. Of the many endogenous mediators of postnatal neurogenesis, epigenetic pathways, such as mediators of DNA methylation, chromatin remodeling systems, and non-coding RNA modulators, appear to play an integral role. Mounting evidence shows that such epigenetic factors form regulatory networks, which govern each step of postnatal neurogenesis. In this review, we explore the emerging roles of epigenetic mechanisms particularly microRNAs, element-1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF), polycomb proteins, and methyl-CpG bindings proteins, in regulating the entire process of postnatal and adult neurogenesis. We further summarize recent data regarding how the crosstalk among these different epigenetic proteins forms the critical regulatory network that regulates neuronal development. We finally discuss how crosstalk between these pathways may serve to translate environmental cues into control of the neurogenic process.
Introduction
Neurogenesis is defined as the generation of new functional neurons, including the proliferation of neural stem/progenitor cells (NSCs), differentiation of these cells into new neurons, and maturation of new neurons that integrate into the neural circuitry. Currently, mammalian neurogenesis is divided into two phases: embryonic/developmental neurogenesis which forms the central nervous system (CNS) and adult neurogenesis which continues at low levels in postnatal and adult brains (Ming and Song, 2011). NSCs are the cellular basis of embryonic and adult neurogenesis and these cells share many regulatory factors and pathways. However, they have inherent differences and reside in distinct environments (Li and Zhao, 2008).
Epigenetic mechanisms, including DNA methylation, histone modifications, and non-coding RNAs (ncRNAs) have emerged as key regulators of gene expression that are required for NSC maintenance and fate specification. Recent literature has shown that maintaining the stemness of NSCs requires the epigenetic suppression of neuronal and glial genes, whereas NSC differentiation requires the removal of epigenetic suppression of genes necessary for neuronal or glial fate specification. In addition, many extrinsic signals, both under normal conditions and during disease and injury, can modulate this process (Hsieh and Eisch, 2010). In this review, we discuss the contribution of epigenetic mechanisms to NSC regulatory networks, with a particular emphasis on adult neurogenesis, and how epigenetic regulation mediates the environmental impact on neurogenesis. Understanding the regulatory mechanisms that govern NSCs, particularly when it comes to adult neurogenesis, is critical for regenerative medicine.
Neurogenesis
Neural stem cells are multipotent cells characterized by their abilities to self-renew and to generate differentiated cells in the nervous system. During development, radial glia in the neuroepithelium are the NSCs that generate the entire CNS. These radial glia produce cortical neurons either directly or indirectly through intermediate progenitor cells (Malatesta et al., 2000; Heins et al., 2002). At the end of neurogenesis, the neurogenic radial glia become translocating cells that are astrocytes (Kriegstein and Alvarez-Buylla, 2009). The three major cell types in the CNS arise from NSCs in a temporally defined sequence: neurons appear first, followed by astrocytes, and then oligodendrocytes (Kriegstein and Alvarez-Buylla, 2009).
In adult brains, neurogenesis has largely stopped, but multipotent NSCs have been found to exist in many brain regions. There are two areas of the adult CNS confirmed to have ongoing neurogenesis, a process defined by the production of new neurons. These neurogenic regions include the subgranular zone (SGZ) of the dentate gyrus (DG) in the hippocampus and the subventricular zone (SVZ) bordering the lateral ventricles (Li and Zhao, 2008). In these regions, radial glia-like stem cells (RGLs) give rise to intermediate or transit-amplifying progenitors that subsequently differentiate into immature and then mature neurons. Experimental evidence suggests that adult NSCs originate from the embryonic neuroepithelial cells, mostly radial glia, located in the ventricular zone (Li and Zhao, 2008).
The specific purpose of adult neurogenesis is not entirely clear; although still debated, mounting evidence points to important roles for it in adult learning and memory (Aimone et al., 2010; Ming and Song, 2011). For instance, ablating adult neuroprogenitors via mouse genetics, anti-proliferative drugs, or focal irradiation impairs hippocampus-dependent learning (Shors et al., 2002; Barkho et al., 2006, 2008; Saxe et al., 2006; Winocur et al., 2006; Dupret et al., 2008; Farioli-Vecchioli et al., 2008; Imayoshi et al., 2008; Clelland et al., 2009; Deng et al., 2009; Garthe et al., 2009; Jessberger et al., 2009); conversely, treatments that enhance neurogenesis also enhance hippocampus-dependent learning (Kitamura et al., 2009; Creer et al., 2010; Stone et al., 2011). Our recent publications, in which we enhanced or inhibited neurogenesis via mouse genetics, demonstrate a direct link between adult neurogenesis and hippocampus-dependent learning (Guo et al., 2011b, 2012). Moreover, both adult neurogenesis and learning are altered in several pathological conditions (Kempermann et al., 2008; Deng et al., 2010). As expected, the quiescence of RGLs, the cell-cycle progression and differentiation of IPCs, and the maturation of new neurons are all tightly controlled by intricate molecular networks that consist of intrinsic genetic and epigenetic programs modulated by extrinsic physiological and pathological conditions (Barkho et al., 2006; Smrt and Zhao, 2010; Barkho and Zhao, 2011). Therefore, adult NSCs (aNSCs) may play major roles in both normal brain functions, such as learning and memory, as well as the brain’s response to injury and disease.
Neural stem cells isolated from the rodent adult SVZ, DG, or forebrain can be maintained as multipotent progenitor cells in serum-free media with defined supplemental factors and the presence of the mitogens basic fibroblast growth factor (bFGF or FGF2) and epithelial growth factor (EGF). Clonal analyses have shown that these NSCs can be instructed to differentiate into all three major cell lineages of the brain (neurons, astrocytes, and oligodendrocytes). Therefore, the in vitro culture of NSCs makes not only a good system for studying neurogenesis, but also an excellent source of cells for potential cell-based therapies (Barkho et al., 2008; Barkho and Zhao, 2011). Understanding aNSCs and adult neurogenesis holds the key to therapeutic applications for not just aNSCs, but many other types of stem cells, as well. In addition, aNSCs make an excellent model system for studying neurodevelopment and related disorders that have a postnatal etiology, such as autism spectrum disorders. Extensive efforts have been invested in this goal. In this review, we will focus on how epigenetic mechanisms, and particularly how crosstalk among epigenetic mechanisms, regulate neurogenesis.
Epigenetic Mechanisms
The term “epigenetics” was crafted by Conrad H. Waddington in 1942, before the age of DNA, to indicate the “study of those processes by which genotype gives rise to phenotype.” Over the years, the meaning of “Epigenetics” has gone through a number of modifications as our knowledge of gene regulation grows. At present, “Epigenetics” is defined as “changes in the expression or function of genetic elements that are independent of changes to the DNA sequence.” This loose, modern definition includes three general mechanisms: histone modifications, DNA methylation and related modifications, and ncRNAs. This definition encompasses how a single fertilized egg can give rise to a vast array of cell types through the transmission of epigenetic programs to daughter cells (Hsieh and Eisch, 2010; Smrt and Zhao, 2010). Recent work has revealed that epigenetic mechanisms can also integrate external stimuli into regulation of the neurogenic process (Molfese, 2011). Epigenetic mechanisms are increasingly recognized as dynamic regulators of gene expression, especially in the field of neurogenesis. The distinct boundaries between epigenetic pathways are blurring as more interactions are uncovered. This crosstalk is increasingly recognized as an important component of NSC regulation.
DNA Methylation
DNA methylation is well known for its role in long-term gene silencing; it serves as the basis of imprinting, X chromosome inactivation, and the establishment of cell fate (Klose and Bird, 2006; Edwards and Ferguson-Smith, 2007; Reik, 2007). DNA methylation involves the covalent addition of a methyl group from the cofactor S-adenosyl-L-methionine (SAM) to C5 of cytosine in CpG dinucleotides and is catalyzed by a family of DNA methyltransferases (DNMTs). DNMT3a and DNMT3b establish de novo methylation, whereas DNMT1 maintains methylation patterns in daughter cells by recognizing hemi-methylated DNA and methylating the unmodified strand of newly synthesized DNA (Law and Jacobsen, 2010). DNA methylation is essential during development, as DNMT null mutations are embryonic lethal (Bestor, 2000). During the neural induction of embryonic stem cells (ESCs) to NSCs, many pluripotency genes are methylated and silenced, which shows the importance of DNA methylation during neurogenesis (Mohn et al., 2008). Interestingly, patterns of DNA methylation correlate more strongly with histone modification patterns than with the underlying genetic code, highlighting the interplay between these two systems (Meissner et al., 2008).
The role of dynamic DNA methylation in cell lineage specification is still murky. Until recently, methylation was thought to be a static DNA modification, with demethylation occurring only passively upon the reduction of DNMT levels, but a number of reports have suggested dynamic DNA methylation changes that involve active demethylation (Ooi et al., 2009; Chen and Riggs, 2011). The most convincing data include the neuronal activity-dependent demethylation mediated by a DNA excision repair protein, Gadd45b (Ma et al., 2009); however the existence and biological functions of active demethylation remain controversial. Hydroxymethylated cytosine (5hmC), which is particularly abundant in brain tissue and ESCs, is a critical intermediate in the demethylation pathway. Proteins from three families catalyze active DNA demethylation: ten-eleven translocation (TET) family proteins modify methylated cytosines by hydroxylation, and then by further oxidation to produce 5mC or 5hmC; enzymes of the AID/APOBEC family deaminate the base of 5mC or 5hmC; and finally, members of the UDG family of base excision repair (BER) glycosylases remove 5hmC via DNA repair mechanisms (Bhutani et al., 2011). 5hmC is hypothesized to play a role in maintaining the pluripotency of ESCs (Cimmino et al., 2011) and in activity-dependent gene expression regulation in the brain (Guo et al., 2011a); however, how 5hmC and DNA demethylation regulate neurogenesis remains a question.
Although DNA methylation may block gene repression by directly preventing the binding of certain transcription factors, DNA methylation-induced gene repression is primarily mediated by methyl-CpG-binding proteins (MBPs), which can recognize and bind to methylated DNA, and the recruitment of chromatin remodeling complexes (Defossez and Stancheva, 2011). The MBP family is divided into three branches: proteins containing methyl binding domain (MBD), including MBD1–5 and MeCP2; members of the Kaiso family of methyl-CpG binding zinc fingers; and UHRF1 and 2 proteins containing the SET and RING finger-associated domain (SRA; Defossez and Stancheva, 2011). Among MBPs, MBDs were discovered first and remain the best studied to date. Both MBD1 and MeCP2 are highly expressed in the brain and play important roles in neurodevelopment and plasticity (Fan and Hutnick, 2005), while many other MBDs are involved in cancer (Parry and Clarke, 2011). We have shown that members of the MBD family play significant roles in the regulation of adult neurogenesis, which we discuss further below. Two members of the Kaiso family of zinc finger DNA-binding proteins are expressed primarily in the brain and can bind methylated CpG and induce gene silencing (Filion et al., 2006). Although the function of UHRFs is more mysterious, UHRF1 (also called Np95) is essential for the maintenance of DNA methylation: UHRF1 has a high affinity for hemi-methylated DNA and recruits DNMT1 to newly synthesized strands of DNA to establish DNA methylation (Sharif et al., 2007). A critical step in understanding the function of MBPs is to identify their binding specificity and targets. Despite keen interest and some progress (Jorgensen et al., 2004; Klose et al., 2005; Clouaire et al., 2010), much remains unknown, although advances in high-throughput sequencing should help move this effort forward.
Histone Modifications
The basic component of chromatin is the nucleosome: hundred and forty-seven base pairs of DNA wrapped twice around an octamer of histone proteins, which contains two copies of each histone, H2A, H2B, H3, and H4. The integration of variant histones into nucleosomes can alter the properties of chromatin and introduces an additional level of regulatory control. The amino acid side chains of the N-terminus histone tail extend from the nucleosome and can be post-translationally modified by acetylation, methylation, ubiquitination, phosphorylation, ribosylation, and SUMOylation; collectively, these are known as the “histone code” (Bernstein et al., 2007). Approximately half of the mass of chromatin consists of non-histone proteins that are responsible for writing and reading the code, a complex process that involves the cooperative and dynamic engagement of several species (Ruthenburg et al., 2007).
Chromatin is present in two general states: highly condensed heterochromatin associated with gene silencing, and loosely packed euchromatin associated with gene expression. Alone, histone modifications are unlikely to be sufficient for gene activation or repression: interactions with activators or repressors are also necessary for efficient implementation. The enzymes that add and remove histone modifications are not specific to certain nucleosomes; rather, they are recruited by specific DNA-binding proteins. Thus, it is the interactions between the histone code, nucleosome-binding proteins, and DNA-binding proteins that regulate chromatin structure and gene expression (Bernstein et al., 2007).
The acetylation of lysine residues is catalyzed by histone acetyltransferases (HATs), and the reversal of this modification is catalyzed by histone deacetylase (HDAC). Acetylation of histone 4 (H4Ac) directly loosens chromatin by neutralizing the positive charge of lysine, eliminating its attraction to the negatively charged DNA backbone of the neighboring nucleosome. Acetyl transferases also regulate gene expression in trans by recruiting other effector chromatin remodeling complexes (Ruthenburg et al., 2007). Mammals have 18 HDACs that are grouped into four classes, based on homology with yeast counterparts (de Ruijter et al., 2003). Class I HDACs (HDAC1, 2, 3, and 8) are localized in the nucleus. Although Class I HDACs are believed to be ubiquitously expressed, HDAC2 is upregulated in aNSCs differentiated into neuronal lineages, whereas HDAC1 is enriched in glia in the adult brain (MacDonald and Roskams, 2008). We have found that HDAC1 and 2, but not the other HDACs, are expressed in NSCs isolated from the adult DG (Zhao, unpublished observation). Class II HDACs (HDAC4, 5, 6, 7, 9, and 10) are able to shuttle in and out of the nucleus in response to certain cellular signals, and these HDACs display interesting cell type-specific expression. In fact, the upregulation of HDAC4, 5, 7, and 9 has been found in differentiating NSCs (Ajamian et al., 2003), and HDAC5 regulates NSC neuronal differentiation (Schneider et al., 2008). HDAC4 and 5 are enriched in the brain and are involved in neuronal maturation and neuroprotection (Majdzadeh et al., 2008). However, HDAC 3 and 5 are also reported to be highly expressed in proliferating NSCs and required for their proliferation (Sun et al., 2007). Therefore, cell type-specific HDACs may serve as important regulators in aNSCs and neuronal development, though their roles are not completely defined.
Histone methylation of lysine or arginine residues is catalyzed by a variety of methyltransferases (HMT) and can be associated with either gene silencing or activation, depending on the number of methyl groups (me1, 2, or 3) and their specific location on the histone. So far, 20 unique methylation marks have been uncovered: repressive marks include, but are not limited to, H3K9me3/me2, H4K20me3, H3K27me, and H4K59; activating marks include H3K4me3, H3K36me3, and H3K79me3 (Mosammaparast and Shi, 2010). The levels of repressive trimethylation of lysine 27 on histone 3 (H3K27me3) and activating trimethylation of lysine 4 (H3K4me3) are controlled by antagonistic groups of proteins called Polycomb (PcG) and Trithorax (TrxG). The addition of PcG-catalyzed H3K27me3 marks leads to the formation of heterochromatin and gene repression, whereas TrxG-catalyzed H3K4me3 marks result in euchromatin formation and gene expression. When both repressive and activating modifications occupy the same genomic region, the result is a bivalent chromatin state that is poised for either activation or further repression (Ringrose and Paro, 2007). Many HMTs are associated with cancer (reviewed in Albert and Helin, 2010) and neurodevelopmental diseases, including Sotos syndrome, Wolf–Hirschhorn syndrome, and 9q syndrome (Nimura et al., 2010).
In recent years, histone methylation was recognized as a dynamic modification that plays a role in gene regulation (Mosammaparast and Shi, 2010). This realization began with the discovery of lysine-specific demethylase 1 (LSD1), also known as lysine (K)-specific demethylase 1A (KDM1A), which is part of the REST complex (Shi et al., 2004). KDMs are known to regulate neuronal differentiation and fate specification, while mutations of KDMs have been tied to X-linked mental retardation (XLMR) and many cancers (Pedersen and Helin, 2010).
Non-Coding RNAs
A large portion of the genome is transcribed into RNA but is never translated into protein. ncRNA is increasingly recognized as an important means of epigenetic regulation of cell fate determination and gene expression. The broad ncRNA category covers an array of small RNAs, including microRNAs (miRNAs), small nucleolar RNAs (snoRNAs), small interfering RNAs (siRNAs), and PIWI-interacting RNAs (piRNAs; Li and Zhao, 2008); of the small ncRNAs, this review will focus on miRNAs known to play a role in many facets of adult neurogenesis. miRNAs target mRNAs through base pairing between a short, 2- to 8-nucleotide seed sequence. Such a short complementation requirement means that a single miRNA may have dozens to hundreds of downstream targets (Bartel, 2009). The final processing steps of miRNA biosynthesis are mediated by Dicer and Drosha, members of the RNase III family, which cleave miRNA precursors into mature miRNA that is then loaded into silencing complexes (Krol et al., 2010). Although the mechanisms behind miRNA-mediated repression are not fully understood, the majority of miRNA-induced silencing effects result from the degradation of target mRNAs (Guo et al., 2010). Mechanistically, a miRNA binds to its target mRNA, frequently in the 3′ UTR, and recruits ribonucleoprotein complexes (miRNPs), also known as RNA-induced silencing complex (RISC), to deadenylate and degrade the mRNA. A number of miRNAs are known to play important roles in stem cells and development. For example, miR-124 is not only brain-enriched, but is also the most abundant miRNA in the embryonic and adult CNS. miR-124 levels increase during neuronal differentiation, and high levels of miR-124 promote neuronal differentiation; conversely, knockdown of endogenous miR-124 maintains proliferation and precursor status in several immature cell types, including adult NSCs. The downstream targets of miR-124 include splicing regulatory factor Ptbp1, pro-neuronal factors Ngn1 and NeuroD1, and REST (Makeyev et al., 2007; Visvanathan et al., 2007; Cheng et al., 2009; Yoo et al., 2009; Liu et al., 2011). The misregulation of miRNAs is involved in a number of neurodegenerative diseases, such as Alzheimer’s and Parkinson’s (Junn and Mouradian, 2012), and in many cancers (McDermott et al., 2011).
Another class of RNAs, long ncRNAs (lncRNAs), are gaining recognition as important mediators of cell fate specification, homeostasis, and plasticity in the CNS, though these roles have yet to be characterized fully (Wapinski and Chang, 2011). lncRNAs range in size from 200 bp to 10 kb, and though they do not code for protein, they undergo many of the same modifications as mRNA, including splicing and polyadenylation (Wang and Chang, 2011). Long intergenic ncRNAs (lincRNAs) are a type of lncRNAs found in gene introns; sometimes they are simply referred to as lncRNAs. There may be upward of 30,000 lncRNAs coded for in the human genome, and estimates are that over half of these are expressed in the CNS in a lineage-specific and developmentally regulated manner (Mercer et al., 2008). LncRNAs are involved in gene regulation at nearly every level, and the molecular mechanisms of their action can be characterized by four general archetypes: signal, decoy, guide, and scaffold (Wang and Chang, 2011). Many lncRNAs are involved in the regulation of chromatin structure; in fact, Khalil et al. (2009) discovered that approximately 20% of lincRNAs are bound to Polycomb repressive complex 2 (PRC2), either alone or with other chromatin-associated complexes, such as Cofactor of REST (CoREST). The expression and biological functions of lncRNAs in the CNS, as well as their role in a number of neurodevelopmental and neurodegenerative diseases, are reviewed by Qureshi et al. (2010b). These RNAs likely alter gene expression through crosstalk with other epigenetic mechanisms, including modulation of chromatin modification enzymes and MBPs. The many roles that ncRNAs and other epigenetic pathways play in embryonic and adult neurogenesis have sparked significant interest in unraveling the complex networks that govern these crucial processes. In the next sections, we will discuss the functional crosstalk among these mechanisms.
Crosstalk among Epigenetic Mechanisms Regulates Neurogenesis
Maintenance of multipotency, fate specification of NSCs, and phenotypic development of new neurons all require complex gene regulation. A host of experimental evidence has shown the critical roles of epigenetic regulation in this process (Li and Zhao, 2008; Liu and Zhao 2009). Not surprisingly, this complex regulation requires crosstalk among multiple epigenetic pathways. Here we present several examples of how interactions among epigenetic mechanisms control the neurogenic process.
Rest
Recent advances have shown that the element-1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) is part of an intricate and interconnected regulatory network of ncRNAs, chromatin remodeling complexes, and DNA-binding and -modifying complexes that ensures correct gene expression in the CNS during development and beyond (Qureshi et al., 2010a). Highlighting its importance, REST has been implicated in a number of disorders, ranging from Down syndrome, XLMR, and epilepsy syndromes to neurodegenerative disorders, such as Huntington’s disease, and cancers (Qureshi and Mehler, 2009). REST is a Krüppel-type zinc finger transcription factor that binds to 1/neuron-restrictive silencer element (RE1) sequences and other non-canonical genomic sites, where it acts as a scaffold for other multi-subunit complexes and DNA-binding proteins to activate, repress, or silence gene expression in a context-dependent manner (Qureshi et al., 2010a). RE1 is found in many neural-specific genes, including ion channels, neurotransmitter receptors, and neurosensory factors, and is highly expressed in non-neuronal lineages, suggesting that REST acts as a repressor of neuronal genes in non-neuronal lineages (Qureshi et al., 2010a). Since its initial discovery, REST has emerged as a central hub in a complex network of transcriptional and epigenetic mechanisms that precisely regulate neuronal development. REST recruits two main cofactors: mSin3 and CoREST. The N-terminus of REST recruits mSin3, which serves as a scaffold for HDAC1, 2, 4, and 5. The C-terminus of REST binds to CoREST and can recruit an astonishing array of proteins, including but not limited to HDAC1 and 2, MECP2, LSD1, G9a, Suc39h1, SCP1 (small C-terminal domain phosphatase), DNMTs, and other chromatin remodeling complexes (reviewed by Qureshi and Mehler, 2009). For example, MeCP2 targets the RE1 subset of neuronal genes and recruits HDACS, reflecting the possibility that both DNA methylation and histone remodeling are required to maintain these genes in a heterochromatin state. REST and CoREST are also found to complex with ATP-dependent chromatin remodeling mechanisms, such as BAF57, npBAF, and nBAF, in the suppression of neuronal gene function (Battaglioli et al., 2002). CoREST complexes can act independently of REST and may contribute to the dynamic regulation of different developmental stages (Ballas et al., 2005).
Deletion of REST is embryonic lethal (Chen et al., 1998), and the function of REST in stem cell lineage commitment is complex. In mouse ES cells, the transition from ESCs to NPCs is marked by a reduction in REST levels (Ballas et al., 2005); however, a knockdown of REST inhibits the formation of NSCs, NPCs, and neurons (Sun et al., 2008). Distinctly, the knockdown of CoREST impedes maintenance of NSCs and leads to altered neuronal differentiation (Abrajano et al., 2009). In adult neurogenic zones, the expression pattern of REST is biphasic; first, REST is expressed in quiescent, slowly dividing NSCs, then it diminishes in type 2b and type 3 transient amplifying cells, but reappears in immature and mature neurons (Gao et al., 2011). This study also shows that REST is required to maintain the adult NSC pool, as conditional knock down of REST in mice leads to a transient increase in neurogenesis that eventually depletes the NSC pool and leads to diminished neurogenesis (Gao et al., 2011). Further, beyond their roles in neurogenesis, REST and CoREST also modulate astrocyte and oligodendrocyte lineage specification and maintenance (Abrajano et al., 2009).
There is growing evidence to support close interactions between REST and small ncRNAs. A number of brain-enriched miRNAs, including miR-9, miR-124, and miR-132, as well as Dicer, are regulated by REST (Buckley et al., 2010). The relationship goes both ways: REST is itself a target of several of the miRNAs that it regulates. One of these, the bifunctional miR-9/miR-9*, targets both REST and CoREST and forms a double-negative feedback loop (Packer et al., 2008). miR-9 is also regulated by the cAMP response element-binding (CREB); in non-differentiated cells, REST represses transcription of the miR-9 gene miR-9-2, then, at the same time REST is dislodged, CREB is phosphorylated and activates miR-9 expression during in vitro neuronal differentiation (Laneve et al., 2010). miR-124, on the other hand, reduces the expression of the REST cofactors CoREST, MeCP2, and SCP1 (Visvanathan et al., 2007). REST has also been implicated in the regulation of ESC pluripotency via miR-21, though its role remains controversial (Singh et al., 2008).
Long ncRNAs may also play an important role in the REST regulatory network. Computational analysis comparing annotated lncRNAs and RE1 motifs suggests that REST could regulate 23% of lncRNAs (Johnson and Buckley, 2009). This study identified two candidate lncRNAs with brain-specific expression patterns, AK046052 and AK090153, which are silenced by REST. Other ncRNAs may act through other REST cofactors; in fact, 13% of the lncRNAs expressed in HeLa cells bind to CoREST (Khalil et al., 2009). In another example, the lncRNA AIR accumulates at promoters, where it recruits G9a, a REST cofactor and histone methyltransferase involved in H3K9 di- and trimethylation, to silence gene expression (Nagano et al., 2008).
Regulation of PcG and TrxG Proteins by DNA Methylation or MiRNAs
PcG and TrxG proteins are antagonistic, evolutionarily conserved members of a chromatin remodeling system that ensures proper expression of developmental programs and the maintenance of stem cell identities (Figure 1). During much of development, the target genes of PcG and TrxG complexes are marked by both activating H3K3me3 tags and repressing H2K27me3 tags. In this poised but repressed state, these genes can be quickly activated upon differentiation (Schuettengruber et al., 2007; Pietersen and van Lohuizen, 2008). In keeping with their role in maintaining stem cells, misregulation by PcG and TrxG proteins is frequently linked to the birth of cancer stem cells (Richly et al., 2011). PcG proteins are functional members of at least two multi-protein complexes that regulate chromatin structure: Polycomb repressive complexes 1 and 2 (PRC1 and PRC2; Li and Zhao, 2008; Pietersen and van Lohuizen, 2008). Each complex contains a distinct set of core proteins that were first identified and named in flies. In mammals, PRC2 is composed of four core components: enhancer of zeste 1 or 2 (EZH1 or EZH2), embryonic ectoderm development (EED), suppressor of zeste 12 (SUZ12), and RbAp46/48 (reviewed in Margueron and Reinberg, 2011). The PRC1 components are Chromobox (CBX) 2/4/6/7/8, PH1/2/3, RING1A/1B, and Bmi-1/Mel13/NSPC1 (Vidal, 2009). How the complexes are recruited to specific chromatin targets is not fully understood, but recent evidence indicates recruitment relies on not only DNA-binding proteins but ncRNAs (Khalil et al., 2009; Wang and Chang, 2011). In a simple model, PRC2 is recruited to target genes and catalyzes the trimethylation of H3K27, and PRC1 recognizes the H3K27me3 mark of PRC2 and mediates ubiquitination of H2AK119 to maintain gene repression; however, this step-wise model of recruitment is not always accurate, as the complexes can act independently of each other (Margueron and Reinberg, 2011). Interactions among PcG protein components and between PcG members and other regulatory proteins, including HATs, HDACs, REST, and TrxG proteins, are essential for PcG functions (Cao et al., 2005; Buchwald et al., 2006; Vidal, 2009).
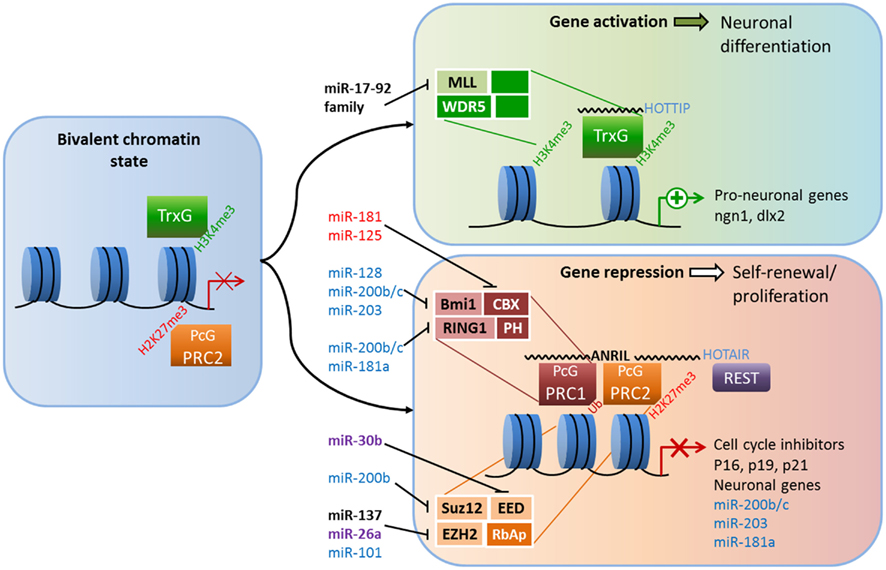
Figure 1. Crosstalk between epigenetic pathways and Polycomb group (PcG) and Trithorax group (TrxG) proteins during adult neurogenesis. The figure illustrates a possible model of crosstalk between epigenetic pathways and PcG and TrxG protein complexes during adult neurogenesis. In the bivalent chromatin state, genes are poised for either rapid induction or further repression; activating H3K4me3 marks and repressing H2K27me3 marks are both present, as are the antagonistic TrxG and PcG complexes (left panel). TrxG and other activating complexes displace PRC2 and loosen the chromatin which enables the expression of neuronal genes (top right panel). Conversely, the recruitment of PRC1 and other repressive complexes (possibly REST) condenses chromatin and suppresses cell-cycle inhibitors and neuronal genes, leading to neural stem cell (NSC) self-renewal (bottom right).The addition of activating H3K4me3 marks and the removal of repressive H2K27me3 marks is associated with chromatin loosening while further H2K27 trimethylation and the addition of repressive H2AUb marks condenses chromatin. Members of PcG and TrxG proteins that have been shown to regulate neurogenesis are shown in black (see text for references). Crosstalk among microRNAs, long non-coding RNAs and PcG and TrxG proteins are shown in black (adult neurogenesis), purple (development; Brett et al., 2011; Song et al., 2011a), red for ESC differentiation (O’Loghlen et al., 2012), and blue (cancer; Lau et al., 2008; Wong and Tellam, 2008; Friedman et al., 2009; Shimono et al., 2009; Wellner et al., 2009; Iliopoulos et al., 2010; Cao et al., 2011).
Evidence for the importance of PRC1 in neurogenesis comes from the neurodevelopmental phenotypes of mice lacking functional B-cell-specific Moloney murine leukemia virus integration site 1 (Bmi-1; Lobo et al., 2007). Mounting data point to an important role for Bmi-1 in controlling self-renewal and senescence of different types of stem cells, among them cancer stem cells and NSCs (Shi et al., 2008; Schuringa and Vellenga, 2010). Bmi-1 deficiency leads to reduced proliferation and self-renewal of SVZ neural progenitors, and these effects are mediated in part through the transcriptional repression of three cell-cycle inhibitors: p16Ink4a, p19Arf, and p21 (Molofsky et al., 2003; Fasano et al., 2007, 2009). Conversely, overexpression of Bmi-1 increases the self-renewal of SVZ-derived cells in vitro and increases proliferation in vivo (Yadirgi et al., 2011). However, one study found that transgenic Bmi-1 overexpression did not have significant effect on aNSC proliferation in either the SVZ or the DG (He et al., 2009); therefore demanding further analysis in future studies. The core component of PRC2, EZH2, is essential for stem cell maintenance and fate specification in many cell lineages, including neurons (reviewed in Yu et al., 2011). The role of EZH2 has not been explored directly in adult neurogenesis, but several lines of evidence indicate it might mirror the effects of Bmi-1 in postnatal neuronal development. In embryonic NSCs, Ezh2 is highly expressed in proliferating cells and its expression declines as cells differentiate into neurons (Sher et al., 2008). In embryonic epidural progenitors, the loss of Ezh2 does not change the fate of these cells; rather, it reduces their proliferation through the upregulation of p16Ink4a and p19Arf (Ezhkova et al., 2009). Moreover, in human embryonic fibroblast cells, the ability of Bmi-1 to repress p16 and p19 was dependent on EZH2 (Bracken et al., 2007). In cortical progenitor cells, the loss of EZH2 led to increased production of neurons at the expense of the progenitor population and improper developmental timing (Pereira et al., 2010). Other PcG members of PRC2 are also necessary to maintain neural progenitor cells. For example, ESCs from SUZ12−/− mice were unable to form neurons under normally permissive neuronal differentiation conditions in vitro (Pasini et al., 2007). Furthermore, knocking down EED enhanced neuronal differentiation in embryonic NSCs (Hirabayashi et al., 2009).
The question becomes how PcG proteins themselves are regulated. Evidence suggests that other epigenetic pathways contribute significantly to the regulation of PcG proteins in the context of neurogenesis. For example, Bmi-1 is targeted by several miRNAs, including miR-128 in glioblastoma cells (Cui et al., 2010; Dong et al., 2011), meaning such regulation might also exist in NSCs. Ezh2 is also regulated by miRNAs. In adult NSCs derived from the mouse forebrain, overexpression of miR-137 increases proliferation, whereas reduction of miR-137 enhances the neuronal differentiation of these cells (Szulwach et al., 2010). This modulation is believed to occur through miR-137’s post-transcriptional repression Ezh2, which leads to a global reduction in the repression of H3K27me3 marks. Moreover, miR-137 is regulated by MeCP2 (which will be discussed later), a fact that highlights the extensive crosstalk between epigenetic pathways (Szulwach et al., 2010). Recently, microdeletions of miR-137 were associated with intellectual disability (Willemsen et al., 2011), underscoring the significance of its interaction with Ezh2 and MeCP2.
LncRNAs may also be involved in the regulation of PcG. As discussed previously, components of PRC1 and PRC2, Bmi-1 and EZH2, respectively, are required to correctly regulate the switch between proliferation and differentiation through the expression of two cell-cycle inhibitors/tumor suppressors, p16Ink4a and p19Arf. The expression of the Ink4a/ARF/Ink4b locus, which codes for p16 and p19, as well as p15 and p14, is regulated by the lncRNA ANRIL (antisense ncRNA in the INK4 locus) via direct interaction with subunits of PRC1 and PRC2. ANRIL is a 3.8-kb ncRNA that is expressed in the opposite direction of and overlaps with the Ink4a locus. CBX7, an H3k27me3-recognizing component of PRC1, can bind directly to both ANRIL and H3K27me3, and both interactions are required for CBX7 to repress the INK4A and INK4B loci (Yap et al., 2010). ANRIL is also involved in the recruitment of PRC2, through SUZ12, to p15INK4B in the INK4A/ARF/INK4B region, and is necessary for its repression (Kotake et al., 2011). In this regulatory scheme, ANRIL acts as a scaffold that recruits PRC1, PRC2, and possibly other factors to regulate gene expression (Wang et al., 2011). LncRNAs are emerging as significant regulators of development and fate specification. Considering the intersection of regional and cell-specific epigenetic pathways and the diversity of chromatin remodeling complexes, the capacity for fine-tuned regulation of gene expression is extraordinary. HOTAIR is a 2.2-kb lincRNA encoded in the HOXC locus that represses transcription of HOXD genes in trans by recruiting PRC2 (Rinn et al., 2007). Specific phosphorylation of EZH2 is found to increase its affinity to HOTAIR, resulting in increased recruitment of PRC2 to Hox genes (Kaneko et al., 2010). Through its 3′ end, HOTAIR also recruits CoREST along with the lysine-specific histone methyltransferase, LSD1, which recognizes activating H3K4me3 marks (Tsai et al., 2010). Thus, lncRNA can recruit different chromatin remodeling complexes and resolve bivalent chromatin states via H3K4 demethylation and H3K27 methylation. Recently, the HOTAIRM1 was shown to be elevated in early differentiating neurons generated from human iPS (induced pluripotent stem) cells, as was JARID, a regulator of Polycomb repressive complexes (Lin et al., 2011). Both HOTAIR and ANRIL have been implicated in a number of cancers (Wapinski and Chang, 2011), but whether these ncRNAs play a role in adult neurogenesis remains to be seen.
The composition of TrxG complexes is heterogeneous; most contain MLL1–3, a SET domain factor, and its associated proteins, such as WDR5 and ASH2L, whereas others contain NURF complexes or ATP-dependent SWI/SNF complexes (Schuettengruber et al., 2007). Mixed-lineage leukemia 1 (Mll-1) is a TrxG member that contains a SET domain with H3K4 methyltransferase activity. MLL family members also interact with UTX, a di- and tri-methyl-H3K27 transferase that is part of the Jumonji-C family of proteins (Lee et al., 2007). Thus, Mll1 complexes may have the ability to identify genes for transcription and remove repressive marks, resolving bivalent chromatin situations. In 2009, Lim et al. revealed that Mll1 is specifically required for neuronal differentiation in the adult SVZ, but not for glial or oligodendrocyte differentiation; in the absence of Mll1, the level of the neuron-specific transcription factor dlx2 was reduced, but the levels of MASH1 or olig2, factors necessary for glial and oligodendrocyte generation, were not. Surprisingly, they found that while all three genes possessed activating H3K4me3 marks, only dlx2 were held in the bivalent chromatin state with repressing H3K27me3 marks (Lim et al., 2009). Thus, epigenetic control of the bivalent chromatin state is essential for proper adult neurogenesis. TrxG proteins are also regulated by miRNAs. For example, miR-17-92 cluster miRNAs are known regulators of MLL, and dysregulation of these clusters of miRNAs contributes to cellular transformation (Mi et al., 2010; Wong et al., 2010). Since multiple components of repressing PcG complexes are subject to regulation by ncRNAs, it is not surprising that lncRNAs are involved in the antagonistic, activating TrxG complexes. For example, the long intergenic RNA (lincRNA) HOXA transcript at the distal tip (HOTTIP) is found to be necessary for maintaining MLL complexes on HoxA genes (Wang et al., 2011).
MBD1 and MeCP2 Control the Expression of Other Epigenetic Factors
Both MBD1 and MeCP2 are highly expressed in the brain and play important roles in neurodevelopment and plasticity (Fan and Hutnick, 2005). As discussed below, we and others have found that both MBD1 and MeCP2 regulate NSCs and neurogenesis through transcriptional regulation of other epigenetic factors (summarized in Figure 2). Mutations in MeCP2 are the cause of the neurodevelopmental disorder Rett syndrome (RTT), but they are also associated with a number of other neurological disorders, including cases of Angelman syndrome, Prader–Willi syndrome, autism, and non-syndromic mental retardation (Chahrour and Zoghbi, 2007). Mouse models have replicated key phenotypes of RTT and revealed that MeCP2 plays a critical role in neuronal maturation and synaptogenesis, in part through the regulation of dendritic morphology, synaptic transmission, and long-term plasticity (Zhao et al., 2007; Smrt et al., 2011). Both clinically and in mouse models of Rett syndrome, loss of MeCP2 and increased dosage both result in similar pathology, highlighting the necessity for its precise control (Chahrour and Zoghbi, 2007). In adult NSCs from the DG, MeCP2 is not critical for early neurogenesis; rather, MeCP2 is important during the transition from immature to mature neurons (Smrt et al., 2007). Indeed, MeCP2 is highly expressed in post-mitotic neurons in many regions of the brain, and its loss results in global alterations in chromatin structure, including increased histone acetylation and increased H1 nucleosome occupancy (Skene et al., 2010). MeCP2 expression in astrocytes has recently been shown to have significant impact on neuronal function and RTT pathology (Ballas et al., 2009; Lioy et al., 2011). Since astrocyte-“specific” manipulations utilize the promoter of GFAP, a protein that is also expressed in NSCs, it is possible that some of these effects might be, at least in part, due to changes in NSCs.
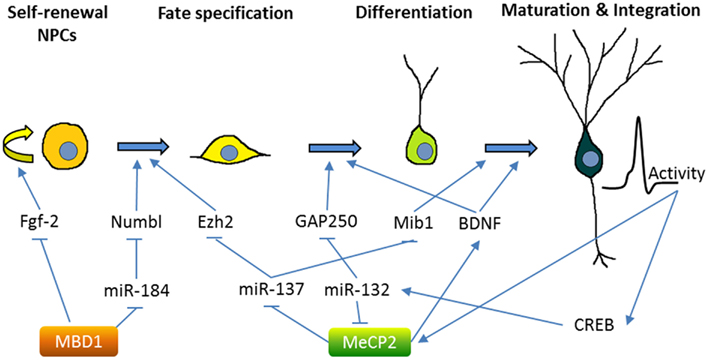
Figure 2. Methyl-CpG binding proteins MeCP2 and MBD1 regulate multiple stages of adult neurogenesis. The figure demonstrates the neurogenic process in the adult hippocampus, although some of the mechanisms were obtained by studying SVZ neurogenesis. MicroRNAs, molecular mediators, and other inputs that are known to regulate the neurogenesis process are summarized. MBD1 represses the self-renewal and promotes differentiation of NSCs through either direct regulation of critical stem cell genes (e.g., Fgf-2) or indirect crosstalk with miRNA pathways (e.g., miR-184). MeCP2 plays important roles at multiple stages of neurogenesis from NSC fate specification to maturation and integration of new neurons. Similar to MBD1, MeCP2 is known to both directly regulate the expression of developmental genes (e.g., BDNF) and indirectly modulate gene expression through miRNA pathways (e.g., miR-137 and miR-132). On the other hand, MeCP2 is regulated by neuronal activity and at least in part through CREB-regulated miRNA (see text for references).
MeCP2 may translate signaling cascades into epigenetic gene regulation. In neurons, neuronal activity leads to phosphorylation of MeCP2 at specific sites, which differentially affects its binding to gene promoters, such as brain-derived neurotrophic factor (BDNF), a neurotrophic factor critical for neuronal development and synaptic plasticity (Na and Monteggia, 2011). BDNF signaling via its receptor, TrkB, is essential for neurogenesis in hippocampal NSCs (Li et al., 2008b). miRNAs are emerging as important regulators at many points in the MeCP2 regulatory pathway. MeCP2 mRNA transcript is targeted by miRNA-132, a neuronal activity and CREB-regulated miRNA that regulates neuronal maturation and dendritic morphology in neonate rat hippocampal neurons (Klein et al., 2007; Wayman et al., 2008). The transgenic overexpression of miR-132 decreased MeCP2 levels and increased dendritic spine density in isolated primary hippocampal neurons (Hansen et al., 2010). Undoubtedly, neuronal maturation is regulated by an extensive, intertwined network of epigenetic pathways.
In addition to being a target of miRNAs, MeCP2 is also involved in miRNA regulation. miR-137 is enriched in the brain and miR-137 levels increase upon neuronal differentiation of adult forebrain-derived NSCs (Silber et al., 2008; Smrt et al., 2010b). miR-137 was found to modulate the proliferation and differentiation of forebrain aNSCs; the translation of miR-137 is repressed by MeCP2 and Sox2, a transcription factor that regulates stem cell self-renewal (Szulwach et al., 2010). One target of miR-137 in NSCs is the PRC2 component EZH2 (Szulwach et al., 2010). Interestingly, EZH2 has been shown to recruit DNMT to promoters of genes destined for DNA methylation and gene repression (Viré et al., 2006). Therefore, the crosstalk among these epigenetic mechanisms may coordinately regulate NSC fate. miR-137 also regulates later stages of neurogenesis: in immature neurons, overexpression of miR-137 inhibited hippocampal DG neuronal maturation by targeting Mind Bomb-1, a RING ubiquitin ligase (Smrt et al., 2010a). Significantly, in embryonic NSCs, miR-137 seems to play a role in the transition from proliferation to differentiation, as miR-137 positively regulates neuronal differentiation of embryonic NSCs by targeting LSD1, a component of a transcriptional complex containing HDACs and nuclear receptor TLX, an essential regulator in stem cell self-renewal (Sun et al., 2011).
Methyl binding domain proteins are essential in linking DNA methylation to the regulation of gene expression. In humans, mutations in MBD1 have been linked to a subset of individuals with autism (Li et al., 2005; Cukier et al., 2010), as well as to some cancers (Sansom et al., 2007). MBD1 null mice exhibit decreased neurogenesis in the DG, impaired spatial learning, and reduced long-term potentiation in the hippocampus (Zhao et al., 2003). These mice also show signs of depression, believed to be a result of decreased adult neurogenesis (Allan et al., 2008). MBD1 binds directly to the promoter of Fgf-2 (fibroblast growth factor 2), a molecule that promotes proliferation and is commonly used to expand NSC populations, to regulate its expression. Furthermore, a DNA methylation inhibitor blocked the effects of MBD1 in forebrain-derived aNSCs and increased Fgf-2 expression, underscoring the important role the methylation state of the promoter plays (Li et al., 2008a). In part, MBD1 regulates neurogenesis by silencing miR-184; an increased level of miR-184 promotes proliferation and hinders the differentiation of hippocampal GD adult NSCs in vivo and forebrain-derived NSCs in vitro. In turn, miR-184 binds the 3′ UTR of Numb like (Numbl), a signaling protein required for differentiation in adult neurogenesis, and targets it for degradation (Liu et al., 2010). Numbl is known to inhibit the Notch pathway (Petersen et al., 2006), which has a significant impact on neurogenesis. Interestingly, Notch signaling was recently shown to be required for FGF-2 receptor-dependent growth in embryonic cortical cells (Rash et al., 2011), bringing together a possible regulatory loop involving MBD1, FGF-2, miR-184, Notch, and Numbl. It is conceivable that regulation of miRNA expression by MBDs requires cooperation from other epigenetic machinery. In fact, HDAC1, 2, and 3 can facilitate the regulation of gene expression by MBD1 in cancer cells (Ng et al., 2000; Villa et al., 2006). Inhibition of both DNA methylation and HDAC is used for cancer treatment (Griffiths and Gore, 2008). Moreover, miRNA and HDAC-mediated pathways can cooperate to reprogram somatic cells to pluripotency (Anokye-Danso et al., 2011). It will be interesting to see whether coordinated manipulation of all three epigenetic pathways will be more effective at controlling cell fate.
Environmental Governance of Epigenetic Crosstalk
It is well known that environmental stimuli can modulate neurogenesis, both during early development and in adulthood (Li and Zhao, 2008; Barkho and Zhao, 2011). Numerous factors are found to promote adult neurogenesis, including exercise and environmental stimulation. Furthermore, depression, brain injury, and stress have all been linked to reduced adult neurogenesis. The relative impact of nature versus nurture on human neurodevelopment and brain function has been a topic of hot discussion and an active research area. The significant overlap among epigenetic pathways could explain how environmental factors regulate neurogenesis and NSC fates with a high degree of complexity. Here we give some examples as possible mechanisms whereby environmental stimulation can be translated into changes in gene expression.
Neuronal Activities Lead to Epigenetic Changes
A number of studies show that long-term potentiation and memory formation require histone acetylation, particularly at H3 and H4, and that extrinsic stimuli inhibiting histone acetylation, such as treatment with HDAC inhibitors and alcohol exposure, inhibit learning and memory (Crepaldi and Riccio, 2009). Interestingly, these treatments also impact NSCs and neurogenesis, with potential downstream effectors of these treatments including neurotrophins.
Neurotrophins, such as BDNF, seem to play a crucial role in the environmental influence on neurogenesis. BDNF is known to govern maturation, including the dendritic branching and synaptic development of new neurons (Smrt and Zhao, 2010). At a neurogenic level, BDNF promotes neuronal fate choice and terminal differentiation of NSCs both in vitro and in adult DG and SVZ (Li et al., 2008b). An enriched environment, physical exercise, and neuronal activity are all found to increase levels of BDNF (Smrt and Zhao, 2010). It is not surprising that BDNF, as a key point of regulation for neurogenesis, is itself regulated by multiple mechanisms, including epigenetic pathways. MeCP2 is known to work with REST/NRSF to recruit HDACS and suppress transcription of the BDNF promoter, and neuronal depolarization results in the release of MeCP2 from the BDNF promoter, thereby allowing for its transcription (Ballas et al., 2005; Zhou et al., 2006). In addition, miR-132 levels increase in response to both BDNF and neuronal activity in the hippocampus, olfactory bulb, and striatum, indicating that miR-132 may act in a feedback loop that increases neural plasticity (Nudelman et al., 2010; Remenyi et al., 2010). Such crosstalk may explain how depolarization is translated into neuronal termination differentiation and maturation, and why there is a reduced BDNF level in MeCP2-deficient mice.
It has long been known that electroconvulsive therapy, a treatment for depression, can promote hippocampal neurogenesis, which might be a causal link to better cognitive function; however, the mechanism underlying this is unclear (Ming and Song, 2011). One study shows that ECT leads to greater expression of the immediate early gene Gadd45b in hippocampal neurons, which catalyzes DNA demethylation of several important genes involved in adult neurogenesis, such as Bdnf, Fgf-1, and NR2 subunit of the NMDA receptor (Ma et al., 2009). In adult brains, neuronal activity-dependent DNA demethylation involves the activity of Tet1 through the 5hmC pathway (Guo et al., 2011a).
Neural Inflammation, Stress, and Disease Lead to Changes in Epigenetic States
Many negative factors can impact neurogenesis, but the mechanism remains unclear for most of these influences; epigenetic changes seen in some of these conditions may explain the missing link.
Drug and alcohol abuse is known to reduce NSC self-renewal, neuronal differentiation, neuronal maturation, and neurogenesis (Cho and Kim, 2010). The CREB protein and its cofactor, CREB-binding protein (CBP), are known to regulate NSCs, as well as neurogenesis and memory, and such function is dependent on the HAT function of CBP (Lee et al., 2009; Lopez-Atalaya et al., 2011). We and others have found that fetal alcohol exposure leads to a significant reduction in CBP expression, to levels similar to those seen in human Rubinstein–Taybi syndrome (heterozygotic loss of CBP gene; Constantinescu et al., 2004; Guo et al., 2011c). Alcohol exposure also leads to changes in DNA methylation in the promoter of key genes involved in development and NSCs (Zhou et al., 2011). Crosstalk among epigenetic pathways is a likely mechanism behind drug and alcohol abuse-induced cognitive deficits (Mandrekar, 2011).
Stroke is also found to cause DNA methylation changes, and altered DNA methylation may be a biomarker for cardiovascular disease and stroke. Inhibition of DNMTs and HDACs could be used as a potential treatment for reducing the neuronal death and tissue damage that result from stroke (for a recent review, see Kim et al., 2009; Baccarelli et al., 2010; Qureshi and Mehler, 2010). Stroke is known to result in enhanced neurogenesis in the SVZ and such enhanced endogenous neurogenesis has been considered as a potential endogenous cell therapy for over a decade. However, the mechanism behind this neurogenic response is unclear (Barkho and Zhao, 2011). Recently, focal cerebral ischemia in a rodent stroke model was found to result in reduced expression of miR-124, which targets Jagged-1 (JAG1), a ligand of Notch (Liu et al., 2011). The activation of the JAG-Notch signaling pathway leads to enhanced SVZ NSC proliferation and might prove to be at least one of the key mechanisms behind stroke-induced neurogenesis. Treatment with sodium butyrate, a HDAC inhibitor, stimulates SVZ and DG neurogenesis in the ischemic brain, possibly by promoting the expression of CREB and BDNF (Kim et al., 2009).
Aberrant DG neurogenesis as a signature of temporal lobe epilepsy has been appreciated for over a decade; however, whether abnormal neurons are the cause or consequence of seizure is still unclear (Schneider-Mizell et al., 2010). Alterations in histone modifications, DNA methylation, and ncRNA profiles have been seen in epileptic brains and animal models, therefore epigenetic mechanisms are proposed to be part of the etiology, as well as a possible treatment for, epilepsy (for a recent review, see Urdinguio et al., 2009). Valproic acid (VPA), a HDAC inhibitor and an antiepileptic drug, blocks seizure-induced neurogenesis and protects animals from seizure-induced deficits in a hippocampus-dependent learning task (Jessberger et al., 2007), although the specific targets of HDAC inhibitor treatment remain unclear. Quite a few miRNAs exhibit altered expression profiles in epileptic tissues (Aronica et al., 2010; Nudelman et al., 2010; Hu et al., 2011; Risbud et al., 2011; Song et al., 2011b), and some of them, such as miR-21, let-7e, miR-125, and miR-132, are known to regulate neurogenesis (Liu and Zhao, 2009). Future studies are needed to determine whether the expressions of these miRNAs are likely controlled by histone modification and DNA methylation, as well as whether these miRNAs can also be used as therapeutic targets, along with HDAC inhibitors, for the treatment of epilepsy.
The link between depression, neurogenesis, and epigenetic changes has been discussed extensively for years (Hsieh and Eisch, 2010). HDAC inhibitors are widely considered a treatment for depression (Grayson et al., 2010), and DNA methylation inhibitors, such as 5-aza cytidine, can also lead to dose-dependent increases in BDNF expression and anti-depression effects (Sales et al., 2011). Not surprisingly, changes in miRNA expression are seen in cases of human depression and in animal models, making the role of non-coding miRNAs in major depression apparent (for recent reviews, see Dwivedi, 2011; Ha, 2011; O’Connor et al., 2012). However, as is the case with epilepsy, how depression affects miRNA expression is still unclear. Epigenetic crosstalk in the etiology of depression is likely to become a fruitful area of future study. Of relevance here, the regulation of the stress pathway is also known be to under epigenetic control. Maternal care during the early postnatal period modulates the DNA methylation status of glucocorticoid receptors and glutamic acid decarboxylase 1 (GAD1) promoter (Zhang et al., 2010), and treatment with both HDAC inhibitor and DNA methylation inhibitor can alter the maternal effects (Weaver, 2009).
Conclusion
Neurogenesis is a complex process requiring coordinated control at multiple levels in a time- and stage-dependent manner, and the sensitivity of neurogenesis to environmental factors is what allows us to make necessary adjustments to fit better into our environment. Epigenetic mechanisms, with their flexibility and versatility, are uniquely suited to fulfill such a requirement. Although a large body of literature presents data on the epigenetic regulation of NSCs and neurogenesis, and its subsequent impact on cognitive functions, particularly learning and memory, few studies have dealt with the crosstalk among these individual mechanisms. At a conceptual level, crosstalk among these mechanisms is inevitable. As a flood of publications offer evidence of miRNA regulation of neurogenesis and related diseases, it is the downstream targets of these miRNAs that have been heavily focused on. The control of these miRNAs, particularly by DNA methylation and chromatin remodeling, as well as processes altered by these diseases, have not been given full attention. Future studies, with the goal of dissecting this crosstalk, both at the individual pathway level and global network level, will help us understand how neurogenesis is regulated and how the environment impacts these processes. Breakthroughs from such studies will likely yield new therapeutic targets for research to pursue.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank C. T. Strauss for editing the manuscript. This work was supported by grants from the NIH to Xinyu Zhao (RO1MH080434 and RO1MH078972), from International Rett Syndrome Foundation (IRSF) to Xinyu Zhao, a center grant from the NIH to the Waisman Center (P30HD03352). Emily M. Jobe is funded by a NIH Molecular Biosciences Training Grant (MBTG: T32 GM07215).
References
Abrajano, J. J., Qureshi, I. A., Gokhan, S., Zheng, D., Bergman, A., and Mehler, M. F. (2009). Differential deployment of REST and CoREST promotes glial subtype specification and oligodendrocyte lineage maturation. PLoS ONE 4, e7665. doi: 10.1371/journal.pone.0007665
Aimone, J. B., Deng, W., and Gage, F. H. (2010). Adult neurogenesis: integrating theories and separating functions. Trends Cogn. Sci. 14, 325–337.
Ajamian, F., Suuronen, T., Salminen, A., and Reeben, M. (2003). Upregulation of class II histone deacetylases mRNA during neural differentiation of cultured rat hippocampal progenitor cells. Neurosci. Lett. 346, 57–60.
Albert, M., and Helin, K. (2010). Histone methyltransferases in cancer. Semin. Cell Dev. Biol. 21, 209–220.
Allan, A. M., Liang, X., Luo, Y., Pak, C., Li, X., Szulwach, K. E., Chen, D., Jin, P., and Zhao, X. (2008). The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits. Hum. Mol. Genet. 17, 2047–2057.
Anokye-Danso, F., Trivedi, C. M., Juhr, D., Gupta, M., Cui, Z., Tian, Y., Zhang, Y., Yang, W., Gruber, P. J., Epstein, J. A., and Morrisey, E. E. (2011). Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 8, 376–388.
Aronica, E., Fluiter, K., Iyer, A., Zurolo, E., Vreijling, J., van Vliet, E. A., Baayen, J. C., and Gorter, J. A. (2010). Expression pattern of miR-146a, an inflammation-associated microRNA, in experimental and human temporal lobe epilepsy. Eur. J. Neurosci. 31, 1100–1107.
Baccarelli, A., Wright, R., Bollati, V., Litonjua, A., Zanobetti, A., Tarantini, L., Sparrow, D., Vokonas, P., and Schwartz, J. (2010). Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology 21, 819–828.
Ballas, N., Grunseich, C., Lu, D., Speh, J., and Mandel, G. (2005). REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657.
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317.
Barkho, B. Z., Munoz, A. E., Li, X., Li, L., Cunningham, L. A., and Zhao, X. (2008). Endogenous matrix metalloproteinase (MMP)-3 and MMP-9 promote the differentiation and migration of adult neural progenitor cells in response to chemokines. Stem Cells 26, 3139–3149.
Barkho, B. Z., Song, H., Aimone, J. B., Smrt, R. D., Kuwabara, T., Nakashima, K., Gage, F. H., and Zhao, X. (2006). Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Dev. 15, 407–421.
Barkho, B. Z., and Zhao, X. (2011). Adult neural stem cells: response to stroke injury and potential for therapeutic applications. Curr. Stem Cell Res. Ther. 6, 327–338.
Battaglioli, E., Andres, M. E., Rose, D. W., Chenoweth, J. G., Rosenfeld, M. G., Anderson, M. E., and Mandel, G. (2002). REST repression of neuronal genes requires components of the hSWI.SNF complex. J. Biol. Chem. 277, 41038–41045.
Bernstein, B. E., Meissner, A., and Lander, E. S. (2007). The mammalian epigenome. Cell 128, 669–681.
Bracken, A. P., Kleine-Kohlbrecher, D., Dietrich, N., Pasini, D., Gargiulo, G., Beekman, C., Theilgaard-Monch, K., Minucci, S., Porse, B. T., Marine, J. C., Hansen, K. H., and Helin, K. (2007). The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 21, 525–530.
Brett, J. O., Renault, V. M., Rafalski, V. A., Webb, A. E., and Brunet, A. (2011). The microRNA cluster miR-106b∼25 regulates adult neural stem/progenitor cell proliferation and neuronal differentiation. Aging 3, 108–124.
Buchwald, G., van der Stoop, P., Weichenrieder, O., Perrakis, A., van Lohuizen, M., and Sixma, T. K. (2006). Structure and E3-ligase activity of the Ring–Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. 25, 2465–2474.
Buckley, N. J., Johnson, R., Zuccato, C., Bithell, A., and Cattaneo, E. (2010). The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol. Dis. 39, 28–39.
Cao, Q., Mani, R. S., Ateeq, B., Dhanasekaran, S. M., Asangani, I. A., Prensner, J. R., Kim, J. H., Brenner, J. C., Jing, X., Cao, X., Wang, R., Li, Y., Dahiya, A., Wang, L., Pandhi, M., Lonigro, R. J., Wu, Y. M., Tomlins, S. A., Palanisamy, N., Qin, Z., Yu, J., Maher, C. A., Varambally, S., and Chinnaiyan, A. M. (2011). Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell 20, 187–199.
Cao, R., Tsukada, Y., and Zhang, Y. (2005). Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell 20, 845–854.
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437.
Chen, Z. F., Paquette, A. J., and Anderson, D. J. (1998). NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 20, 136–142.
Chen, Z. X., and Riggs, A. D. (2011). DNA methylation and demethylation in mammals. J. Biol. Chem. 286, 18347–18353.
Cheng, L., Pastrana, E., Tavazoie, M., and Doetsch, F. (2009). miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 12, 399–408.
Cho, K. O., and Kim, S. Y. (2010). Effects of brain insults and pharmacological manipulations on the adult hippocampal neurogenesis. Arch. Pharm. Res. 33, 1475–1488.
Cimmino, L., Abdel-Wahab, O., Levine, R. L., and Aifantis, I. (2011). TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell 9, 193–204.
Clelland, C. D., Choi, M., Romberg, C., Clemenson, G. D. Jr., Fragniere, A., Tyers, P., Jessberger, S., Saksida, L. M., Barker, R. A., Gage, F. H., and Bussey, T. J. (2009). A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 325, 210–213.
Clouaire, T., de Las Heras, J. I., Merusi, C., and Stancheva, I. (2010). Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA. Nucleic Acids Res. 38, 4620–4634.
Constantinescu, A., Wu, M., Asher, O., and Diamond, I. (2004). cAMP-dependent protein kinase type I regulates ethanol-induced cAMP response element-mediated gene expression via activation of CREB-binding protein and inhibition of MAPK. J. Biol. Chem. 279, 43321–43329.
Creer, D. J., Romberg, C., Saksida, L. M., van Praag, H., and Bussey, T. J. (2010). Running enhances spatial pattern separation in mice. Proc. Natl. Acad. Sci. U.S.A. 107, 2367–2372.
Cui, J. G., Zhao, Y., Sethi, P., Li, Y. Y., Mahta, A., Culicchia, F., and Lukiw, W. J. (2010). Micro-RNA-128 (miRNA-128) down-regulation in glioblastoma targets ARP5 (ANGPTL6), Bmi-1 and E2F-3a, key regulators of brain cell proliferation. J. Neurooncol. 98, 297–304.
Cukier, H. N., Rabionet, R., Konidari, I., Rayner-Evans, M. Y., Baltos, M. L., Wright, H. H., Abramson, R. K., Martin, E. R., Cuccaro, M. L., Pericak-Vance, M. A., and Gilbert, J. R. (2010). Novel variants identified in methyl-CpG-binding domain genes in autistic individuals. Neurogenetics 11, 291–303.
de Ruijter, A. J., van Gennip, A. H., Caron, H. N., Kemp, S., and van Kuilenburg, A. B. (2003). Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749.
Defossez, P. A., and Stancheva, I. (2011). Biological functions of methyl-CpG-binding proteins. Prog. Mol. Biol. Transl. Sci. 101, 377–398.
Deng, W., Aimone, J. B., and Gage, F. H. (2010). New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 11, 339–350.
Deng, W., Saxe, M. D., Gallina, I. S., and Gage, F. H. (2009). Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J. Neurosci. 29, 13532–13542.
Dong, P., Kaneuchi, M., Watari, H., Hamada, J., Sudo, S., Ju, J., and Sakuragi, N. (2011). MicroRNA-194 inhibits epithelial to mesenchymal transition of endometrial cancer cells by targeting oncogene BMI-1. Mol. Cancer 10, 99.
Dupret, D., Revest, J. M., Koehl, M., Ichas, F., De Giorgi, F., Costet, P., Abrous, D. N., and Piazza, P. V. (2008). Spatial relational memory requires hippocampal adult neurogenesis. PLoS ONE 3, e1959. doi: 10.1371/journal.pone.0001959
Dwivedi, Y. (2011). Evidence demonstrating role of microRNAs in the etiopathology of major depression. J. Chem. Neuroanat. 42, 142–156.
Edwards, C. A., and Ferguson-Smith, A. C. (2007). Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 19, 281–289.
Ezhkova, E., Pasolli, H. A., Parker, J. S., Stokes, N., Su, I. H., Hannon, G., Tarakhovsky, A., and Fuchs, E. (2009). Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 136, 1122–1135.
Fan, G., and Hutnick, L. (2005). Methyl-CpG binding proteins in the nervous system. Cell Res. 15, 255–261.
Farioli-Vecchioli, S., Saraulli, D., Costanzi, M., Pacioni, S., Cina, I., Aceti, M., Micheli, L., Bacci, A., Cestari, V., and Tirone, F. (2008). The timing of differentiation of adult hippocampal neurons is crucial for spatial memory. PLoS Biol. 6, e246. doi: 10.1371/journal.pbio.0060246
Fasano, C. A., Dimos, J. T., Ivanova, N. B., Lowry, N., Lemischka, I. R., and Temple, S. (2007). shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell 1, 87–99.
Fasano, C. A., Phoenix, T. N., Kokovay, E., Lowry, N., Elkabetz, Y., Dimos, J. T., Lemischka, I. R., Studer, L., and Temple, S. (2009). Bmi-1 cooperates with Foxg1 to maintain neural stem cell self-renewal in the forebrain. Genes Dev. 23, 561–574.
Filion, G. J., Zhenilo, S., Salozhin, S., Yamada, D., Prokhortchouk, E., and Defossez, P. A. (2006). A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell. Biol. 26, 169–181.
Friedman, J. M., Liang, G., Liu, C. C., Wolff, E. M., Tsai, Y. C., Ye, W., Zhou, X., and Jones, P. A. (2009). The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res. 69, 2623–2629.
Gao, Z., Ure, K., Ding, P., Nashaat, M., Yuan, L., Ma, J., Hammer, R. E., and Hsieh, J. (2011). The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J. Neurosci. 31, 9772–9786.
Garthe, A., Behr, J., and Kempermann, G. (2009). Adult-generated hippocampal neurons allow the flexible use of spatially precise learning strategies. PLoS ONE 4, e5464. doi: 10.1371/journal.pone.0005464
Grayson, D. R., Kundakovic, M., and Sharma, R. P. (2010). Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol. Pharmacol. 77, 126–135.
Griffiths, E. A., and Gore, S. D. (2008). DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin. Hematol. 45, 23–30.
Guo, H., Ingolia, N. T., Weissman, J. S., and Bartel, D. P. (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840.
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., and Song, H. (2011a). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434.
Guo, W., Allan, A. M., Zong, R., Zhang, L., Johnson, E. B., Schaller, E. G., Murthy, A. C., Goggin, S. L., Eisch, A. J., Oostra, B. A., Nelson, D. L., Jin, P., and Zhao, X. (2011b). Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat. Med. 17, 559–565.
Guo, W., Crossey, E. L., Zhang, L., Zucca, S., George, O. L., Valenzuela, C. F., and Zhao, X. (2011c). Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS ONE 6, e19351. doi: 10.1371/journal.pone.0019351
Guo, W., Murthy, A. C., Zhang, L., Johnson, E. B., Schaller, E. G., Allan, A. M., and Zhao, X. (2012). Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum. Mol. Genet. 21, 681–691.
Ha, T. Y. (2011). MicroRNAs in human diseases: from autoimmune diseases to skin, psychiatric and neurodegenerative diseases. Immune Netw. 11, 227–244.
Hansen, K. F., Sakamoto, K., Wayman, G. A., Impey, S., and Obrietan, K. (2010). Transgenic miR132 alters neuronal spine density and impairs novel object recognition memory. PLoS ONE 5, e15497. doi: 10.1371/journal.pone.0015497
He, S., Iwashita, T., Buchstaller, J., Molofsky, A. V., Thomas, D., and Morrison, S. J. (2009). Bmi-1 over-expression in neural stem/progenitor cells increases proliferation and neurogenesis in culture but has little effect on these functions in vivo. Dev. Biol. 328, 257–272.
Heins, N., Malatesta, P., Cecconi, F., Nakafuku, M., Tucker, K. L., Hack, M. A., Chapouton, P., Barde, Y. A., and Gotz, M. (2002). Glial cells generate neurons: the role of the transcription factor Pax6. Nat. Neurosci. 5, 308–315.
Hirabayashi, Y., Suzki, N., Tsuboi, M., Endo, T. A., Toyoda, T., Shinga, J., Koseki, H., Vidal, M., and Gotoh, Y. (2009). Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63, 600–613.
Hsieh, J., and Eisch, A. J. (2010). Epigenetics, hippocampal neurogenesis, and neuropsychiatric disorders: unraveling the genome to understand the mind. Neurobiol. Dis. 39, 73–84.
Hu, K., Zhang, C., Long, L., Long, X., Feng, L., Li, Y., and Xiao, B. (2011). Expression profile of microRNAs in rat hippocampus following lithium-pilocarpine-induced status epilepticus. Neurosci. Lett. 488, 252–257.
Iliopoulos, D., Lindahl-Allen, M., Polytarchou, C., Hirsch, H. A., Tsichlis, P. N., and Struhl, K. (2010). Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol. Cell 39, 761–772.
Imayoshi, I., Sakamoto, M., Ohtsuka, T., Takao, K., Miyakawa, T., Yamaguchi, M., Mori, K., Ikeda, T., Itohara, S., and Kageyama, R. (2008). Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 11, 1153–1161.
Jessberger, S., Clark, R. E., Broadbent, N. J., Clemenson, G. D., Consiglio, A., Lie, D. C., Squire, L. R., and Gage, F. H. (2009). Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learn. Mem. 16, 147–154.
Jessberger, S., Nakashima, K., Clemenson, G. D. Jr., Mejia, E., Mathews, E., Ure, K., Ogawa, S., Sinton, C. M., Gage, F. H., and Hsieh, J. (2007). Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci. 27, 5967–5975.
Johnson, R., and Buckley, N. J. (2009). Gene dysregulation in Huntington’s disease: REST, microRNAs and beyond. Neuromol. Med. 11, 183–199.
Jorgensen, H. F., Ben-Porath, I., and Bird, A. P. (2004). Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol. Cell. Biol. 24, 3387–3395.
Junn, E., and Mouradian, M. M. (2012). MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol. Ther. 133, 142–150.
Kaneko, S., Li, G., Son, J., Xu, C. F., Margueron, R., Neubert, T. A., and Reinberg, D. (2010). Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 24, 2615–2620.
Kempermann, G., Krebs, J., and Fabel, K. (2008). The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr. Opin. Psychiatry 21, 290–295.
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D., Thomas, K., Presser, A., Bernstein, B. E., van Oudenaarden, A., Regev, A., Lander, E. S., and Rinn, J. L. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672.
Kim, H. J., Leeds, P., and Chuang, D. M. (2009). The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J. Neurochem. 110, 1226–1240.
Kitamura, T., Saitoh, Y., Takashima, N., Murayama, A., Niibori, Y., Ageta, H., Sekiguchi, M., Sugiyama, H., and Inokuchi, K. (2009). Adult neurogenesis modulates the hippocampus-dependent period of associative fear memory. Cell 139, 814–827.
Klein, M., Lioy, D., Ma, L., Impey, S., Mandel, G., and Goodman, R. (2007). Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 10, 1513–1514.
Klose, R. J., and Bird, A. P. (2006). Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97.
Klose, R. J., Sarraf, S. A., Schmiedeberg, L., McDermott, S. M., Stancheva, I., and Bird, A. P. (2005). DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell 19, 667–678.
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M., and Xiong, Y. (2011). Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30, 1956–1962.
Kriegstein, A., and Alvarez-Buylla, A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184.
Krol, J., Loedige, I., and Filipowicz, W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610.
Laneve, P., Gioia, U., Andriotto, A., Moretti, F., Bozzoni, I., and Caffarelli, E. (2010). A minicircuitry involving REST and CREB controls miR-9-2 expression during human neuronal differentiation. Nucleic Acids Res. 38, 6895–6905.
Lau, P., Verrier, J. D., Nielsen, J. A., Johnson, K. R., Notterpek, L., and Hudson, L. D. (2008). Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J. Neurosci. 28, 11720–11730.
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220.
Lee, M. G., Villa, R., Trojer, P., Norman, J., Yan, K. P., Reinberg, D., Di Croce, L., and Shiekhattar, R. (2007). Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318, 447–450.
Lee, S., Lee, B., Lee, J. W., and Lee, S. K. (2009). Retinoid signaling and neurogenin2 function are coupled for the specification of spinal motor neurons through a chromatin modifier CBP. Neuron 62, 641–654.
Li, H., Yamagata, T., Mori, M., Yasuhara, A., and Momoi, M. Y. (2005). Mutation analysis of methyl-CpG binding protein family genes in autistic patients. Brain Dev. 27, 321–325.
Li, X., Barkho, B., Luo, Y., Smrt, R., Santistevan, N., Liu, C., Kuwabara, T., Gage, F., and Zhao, X. (2008a). Epigenetic regulation of the stem cell mitogen Fgf-2 by Mbd1 in adult neural stem/progenitor cells. J. Biol. Chem. 283, 27644–27652.
Li, Y., Luikart, B. W., Birnbaum, S., Chen, J., Kwon, C. H., Kernie, S. G., Bassel-Duby, R., and Parada, L. F. (2008b). TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59, 399–412.
Li, X., and Zhao, X. (2008). Epigenetic regulation of mammalian stem cells. Stem Cells Dev. 17, 1043–1052.
Lim, D., Huang, Y., Swigut, T., Mirick, A., Garcia-Verdugo, J., Wysocka, J., Ernst, P., and Alvarez-Buylla, A. (2009). Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 458, 529–533.
Lin, M., Pedrosa, E., Shah, A., Hrabovsky, A., Maqbool, S., Zheng, D., and Lachman, H. M. (2011). RNA-Seq of human neurons derived from iPS cells reveals candidate long non-coding RNAs involved in neurogenesis and neuropsychiatric disorders. PLoS ONE 6, e23356. doi: :10.1371/journal.pone.0023356
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., Hirrlinger, P. G., Kirchhoff, F., Bissonnette, J. M., Ballas, N., and Mandel, G. (2011). A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500.
Liu, C., Teng, Z. Q., Santistevan, N. J., Szulwach, K. E., Guo, W., Jin, P., and Zhao, X. (2010). Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell 6, 433–444.
Liu, C., and Zhao, X. (2009). MicroRNAs in adult and embryonic neurogenesis. Neuromol. Med. 11, 141–152.
Liu, X. S., Chopp, M., Zhang, R. L., Tao, T., Wang, X. L., Kassis, H., Hozeska-Solgot, A., Zhang, L., Chen, C., and Zhang, Z. G. (2011). MicroRNA profiling in subventricular zone after stroke: MiR-124a regulates proliferation of neural progenitor cells through Notch signaling pathway. PLoS ONE 6, e23461. doi: 10.1371/journal.pone.0023461
Lobo, N. A., Shimono, Y., Qian, D., and Clarke, M. F. (2007). The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 23, 675–699.
Lopez-Atalaya, J. P., Ciccarelli, A., Viosca, J., Valor, L. M., Jimenez-Minchan, M., Canals, S., Giustetto, M., and Barco, A. (2011). CBP is required for environmental enrichment-induced neurogenesis and cognitive enhancement. EMBO J. 30, 4287–4298.
Ma, D. K., Jang, M. H., Guo, J. U., Kitabatake, Y., Chang, M. L., Pow-Anpongkul, N., Flavell, R. A., Lu, B., Ming, G. L., and Song, H. (2009). Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323, 1074–1077.
MacDonald, J. L., and Roskams, A. J. (2008). Histone deacetylases 1 and 2 are expressed at distinct stages of neuro-glial development. Dev. Dyn. 237, 2256–2267.
Majdzadeh, N., Wang, L., Morrison, B. E., Bassel-Duby, R., Olson, E. N., and D’Mello, S. R. (2008). HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol 68, 1076–1092.
Makeyev, E., Zhang, J., Carrasco, M., and Maniatis, T. (2007). The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448.
Malatesta, P., Hartfuss, E., and Gotz, M. (2000). Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development 127, 5253–5263.
Mandrekar, P. (2011). Epigenetic regulation in alcoholic liver disease. World J. Gastroenterol. 17, 2456–2464.
Margueron, R., and Reinberg, D. (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349.
McDermott, A. M., Heneghan, H. M., Miller, N., and Kerin, M. J. (2011). The therapeutic potential of microRNAs: disease modulators and drug targets. Pharm. Res. 28, 3016–3029.
Meissner, A., Mikkelsen, T. S., Gu, H., Wernig, M., Hanna, J., Sivachenko, A., Zhang, X., Bernstein, B. E., Nusbaum, C., Jaffe, D. B., Gnirke, A., J, R., and Lander, E. S. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770.
Mercer, T. R., Dinger, M. E., Sunkin, S. M., Mehler, M. F., and Mattick, J. S. (2008). Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. U.S.A. 105, 716–721.
Mi, S., Li, Z., Chen, P., He, C., Cao, D., Elkahloun, A., Lu, J., Pelloso, L. A., Wunderlich, M., Huang, H., Luo, R. T., Sun, M., He, M., Neilly, M. B., Zeleznik-Le, N. J., Thirman, M. J., Mulloy, J. C., Liu, P. P., Rowley, J. D., and Chen, J. (2010). Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc. Natl. Acad. Sci. U.S.A. 107, 3710–3715.
Ming, G. L., and Song, H. (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702.
Mohn, F., Weber, M., Rebhan, M., Roloff, T. C., Richter, J., Stadler, M. B., Bibel, M., and Schubeler, D. (2008). Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 30, 755–766.
Molfese, D. L. (2011). Advancing neuroscience through epigenetics: molecular mechanisms of learning and memory. Dev. Neuropsychol. 36, 810–827.
Molofsky, A. V., Pardal, R., Iwashita, T., Park, I. K., Clarke, M. F., and Morrison, S. J. (2003). Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 425, 962–967.
Mosammaparast, N., and Shi, Y. (2010). Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 79, 155–179.
Na, E. S., and Monteggia, L. M. (2011). The role of MeCP2 in CNS development and function. Horm. Behav. 59, 364–368.
Nagano, T., Mitchell, J. A., Sanz, L. A., Pauler, F. M., Ferguson-Smith, A. C., Feil, R., and Fraser, P. (2008). The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720.
Ng, H. H., Jeppesen, P., and Bird, A. (2000). Active repression of methylated genes by the chromosomal protein MBD1. Mol. Cell. Biol. 20, 1394–1406.
Nimura, K., Ura, K., and Kaneda, Y. (2010). Histone methyltransferases: regulation of transcription and contribution to human disease. J. Mol. Med. (Berl.) 88, 1213–1220.
Nudelman, A. S., DiRocco, D. P., Lambert, T. J., Garelick, M. G., Le, J., Nathanson, N. M., and Storm, D. R. (2010). Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus 20, 492–498.
O’Connor, R. M., Dinan, T. G., and Cryan, J. F. (2012). Little things on which happiness depends: microRNAs as novel therapeutic targets for the treatment of anxiety and depression. Mol. Psychiatry 17, 359–376.
O’Loghlen, A., Muñoz-Cabello, A. M., Gaspar-Maia, A., Wu, H. A., Banito, A., Kunowska, N., Racek, T., Pemberton, H. N., Beolchi, P., Lavial, F., Masui, O., Vermeulen, M., Carroll, T., Graumann, J., Heard, E., Dillon, N., Azuara, V., Snijders, A. P., Peters, G., Bernstein, E., and Gil, J. (2012). MicroRNA regulation of Cbx7 mediates a switch of Polycomb orthologs during ESC differentiation. Cell Stem Cell 10, 33–46.
Ooi, S. K., O’Donnell, A. H., and Bestor, T. H. (2009). Mammalian cytosine methylation at a glance. J. Cell. Sci. 122, 2787–2791.
Packer, A. N., Xing, Y., Harper, S. Q., Jones, L., and Davidson, B. L. (2008). The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 28, 14341–14346.
Parry, L., and Clarke, A. R. (2011). The roles of the methyl-CpG binding proteins in cancer. Genes Cancer 2, 618–630.
Pasini, D., Bracken, A. P., Hansen, J. B., Capillo, M., and Helin, K. (2007). The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 27, 3769–3779.
Pedersen, M. T., and Helin, K. (2010). Histone demethylases in development and disease. Trends Cell Biol. 20, 662–671.
Pereira, J. D., Sansom, S. N., Smith, J., Dobenecker, M. W., Tarakhovsky, A., and Livesey, F. J. (2010). Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 107, 15957–15962.
Petersen, P. H., Tang, H., Zou, K., and Zhong, W. (2006). The enigma of the numb-Notch relationship during mammalian embryogenesis. Dev. Neurosci. 28, 156–168.
Pietersen, A. M., and van Lohuizen, M. (2008). Stem cell regulation by polycomb repressors: postponing commitment. Curr. Opin. Cell Biol. 20, 201–207.
Qureshi, I. A., Gokhan, S., and Mehler, M. F. (2010a). REST and CoREST are transcriptional and epigenetic regulators of seminal neural fate decisions. Cell Cycle 9, 4477–4486.
Qureshi, I. A., Mattick, J. S., and Mehler, M. F. (2010b). Long non-coding RNAs in nervous system function and disease. Brain Res. 1338, 20–35.
Qureshi, I. A., and Mehler, M. F. (2009). Regulation of non-coding RNA networks in the nervous system – what’s the REST of the story? Neurosci. Lett. 466, 73–80.
Qureshi, I. A., and Mehler, M. F. (2010). Epigenetic mechanisms underlying human epileptic disorders and the process of epileptogenesis. Neurobiol. Dis. 39, 53–60.
Rash, B. G., Lim, H. D., Breunig, J. J., and Vaccarino, F. M. (2011). FGF signaling expands embryonic cortical surface area by regulating Notch-dependent neurogenesis. J. Neurosci. 31, 15604–15617.
Reik, W. (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432.
Remenyi, J., Hunter, C., Cole, C., Ando, H., Impey, S., Monk, C., Martin, K., Barton, G., Hutvagner, G., and Arthur, J. (2010). Regulation of the miR-212/132 locus by MSK1 and CREB in response to neurotrophins. Biochem. J. 428, 281–291.
Richly, H., Aloia, L., and Di Croce, L. (2011). Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis. 2, e204.
Ringrose, L., and Paro, R. (2007). Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 134, 223–232.
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A., Goodnough, L. H., Helms, J. A., Farnham, P. J., Segal, E., and Chang, H. Y. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323.
Risbud, R. M., Lee, C., and Porter, B. E. (2011). Neurotrophin-3 mRNA a putative target of miR21 following status epilepticus. Brain Res. 1424, 53–59.
Ruthenburg, A. J., Li, H., Patel, D. J., and Allis, C. D. (2007). Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 8, 983–994.
Sales, A. J., Biojone, C., Terceti, M. S., Guimaraes, F. S., Gomes, M. V., and Joca, S. R. (2011). Antidepressant-like effect induced by systemic and intra-hippocampal administration of DNA methylation inhibitors. Br. J. Pharmacol. 164, 1711–1721.
Sansom, O. J., Maddison, K., and Clarke, A. R. (2007). Mechanisms of disease: methyl-binding domain proteins as potential therapeutic targets in cancer. Nat. Clin. Pract. Oncol. 4, 305–315.
Saxe, M. D., Battaglia, F., Wang, J. W., Malleret, G., David, D. J., Monckton, J. E., Garcia, A. D., Sofroniew, M. V., Kandel, E. R., Santarelli, L., Hen, R., and Drew, M. R. (2006). Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc. Natl. Acad. Sci. U.S.A. 103, 17501–17506.
Schneider, J. W., Gao, Z., Li, S., Farooqi, M., Tang, T. S., Bezprozvanny, I., Frantz, D. E., and Hsieh, J. (2008). Small-molecule activation of neuronal cell fate. Nat. Chem. Biol. 4, 408–410.
Schneider-Mizell, C. M., Parent, J. M., Ben-Jacob, E., Zochowski, M. R., and Sander, L. M. (2010). From network structure to network reorganization: implications for adult neurogenesis. Phys. Biol. 7, 046008.
Schuettengruber, B., Chourrout, D., Vervoort, M., Leblanc, B., and Cavalli, G. (2007). Genome regulation by polycomb and trithorax proteins. Cell 128, 735–745.
Schuringa, J. J., and Vellenga, E. (2010). Role of the polycomb group gene BMI1 in normal and leukemic hematopoietic stem and progenitor cells. Curr. Opin. Hematol. 17, 294–299.
Sharif, J., Muto, M., Takebayashi, S., Suetake, I., Iwamatsu, A., Endo, T. A., Shinga, J., Mizutani-Koseki, Y., Toyoda, T., Okamura, K., Mitsuya, K., Okano, M., and Koseki, H. (2007). The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450, 908–912.
Sher, F., Rossler, R., Brouwer, N., Balasubramaniyan, V., Boddeke, E., and Copray, S. (2008). Differentiation of neural stem cells into oligodendrocytes: involvement of the polycomb group protein Ezh2. Stem Cells 26, 2875–2883.
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., and Casero, R. A. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953.
Shi, Y., Sun, G., Zhao, C., and Stewart, R. (2008). Neural stem cell self-renewal. Crit. Rev. Oncol. Hematol. 65, 43–53.
Shimono, Y., Zabala, M., Cho, R. W., Lobo, N., Dalerba, P., Qian, D., Diehn, M., Liu, H., Panula, S. P., Chiao, E., Dirbas, F. M., Somlo, G., Pera, R. A., Lao, K., and Clarke, M. F. (2009). Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138, 592–603.
Shors, T. J., Townsend, D. A., Zhao, M., Kozorovitskiy, Y., and Gould, E. (2002). Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus 12, 578–584.
Silber, J., Lim, D., Petritsch, C., Persson, A. I., Maunakea, A. K., Yu, M., Vandenberg, S. R., Ginzinger, D. G., James, C. D., Costello, J. F., Bergers, G., Weiss, W. A., Alvarez-Buylla, A., and Hodgson, J. G. (2008). miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 6, 14. doi: 10.1186/1741-7015-6-14
Singh, S. K., Kagalwala, M. N., Parker-Thornburg, J., Adams, H., and Majumder, S. (2008). REST maintains self-renewal and pluripotency of embryonic stem cells. Nature 453, 223–227.
Skene, P. J., Illingworth, R. S., Webb, S., Kerr, A. R., James, K. D., Turner, D. J., Andrews, R., and Bird, A. P. (2010). Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 37, 457–468.
Smrt, R., Eaves-Egenes, J., Barkho, B., Santistevan, N., Zhao, C., Aimone, J., Gage, F., and Zhao, X. (2007). Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol. Dis. 27, 77–89.
Smrt, R. D., Pfeiffer, R. L., and Zhao, X. (2011). Age-dependent expression of MeCP2 in a heterozygous mosaic mouse model. Hum. Mol. Genet. 20, 1834–1843.
Smrt, R. D., Szulwach, K. E., Pfeiffer, R. L., Li, X., Guo, W., Pathania, M., Teng, Z. Q., Luo, Y., Peng, J., Bordey, A., and Zhao, X. (2010a). MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells 28, 1060–1070.
Smrt, R. D., Szulwach, K. E., Pfeiffer, R. L., Li, X., Guo, W., Pathania, M., Teng, Z. Q., Luo, Y., Peng, J., and Bordey, A. et al. (2010b). MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells 28, 1060–1070.
Smrt, R. D., and Zhao, X. (2010). Epigenetic regulation of neuronal dendrite and dendritic spine development. Front. Biol. 5, 304–323.
Song, P. P., Hu, Y., Liu, C. M., Yan, M. J., Song, G., Cui, Y., Xia, H. F., and Ma, X. (2011a). Embryonic ectoderm development protein is regulated by microRNAs in human neural tube defects. Am. J. Obstet. Gynecol. 204, 544.e9–544.e17.
Song, Y. J., Tian, X. B., Zhang, S., Zhang, Y. X., Li, X., Li, D., Cheng, Y., Zhang, J. N., Kang, C. S., and Zhao, W. (2011b). Temporal lobe epilepsy induces differential expression of hippocampal miRNAs including let-7e and miR-23a/b. Brain Res. 1387, 134–140.
Stone, S. S., Teixeira, C. M., Devito, L. M., Zaslavsky, K., Josselyn, S. A., Lozano, A. M., and Frankland, P. W. (2011). Stimulation of entorhinal cortex promotes adult neurogenesis and facilitates spatial memory. J. Neurosci. 31, 13469–13484.
Sun, G., Ye, P., Murai, K., Lang, M. F., Li, S., Zhang, H., Li, W., Fu, C., Yin, J., Wang, A., Ma, X., and Shi, Y. (2011). miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat. Commun. 2, 529.
Sun, G., Yu, R. T., Evans, R. M., and Shi, Y. (2007). Orphan nuclear receptor TLX recruits histone deacetylases to repress transcription and regulate neural stem cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 104, 15282–15287.
Sun, Y. M., Cooper, M., Finch, S., Lin, H. H., Chen, Z. F., Williams, B. P., and Buckley, N. J. (2008). Rest-mediated regulation of extracellular matrix is crucial for neural development. PLoS ONE 3, e3656. doi: 10.1371/journal.pone.0003656
Szulwach, K., Li, X., Smrt, R., Li, Y., Luo, Y., Lin, L., Santistevan, N., Li, W., Zhao, X., and Jin, P. (2010). Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell Biol. 189, 127–141.
Tsai, M. C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., Shi, Y., Segal, E., and Chang, H. Y. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693.
Urdinguio, R. G., Sanchez-Mut, J. V., and Esteller, M. (2009). Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 8, 1056–1072.
Vidal, M. (2009). Role of polycomb proteins Ring1A and Ring1B in the epigenetic regulation of gene expression. Int. J. Dev. Biol. 53, 355–370.
Villa, R., Morey, L., Raker, V. A., Buschbeck, M., Gutierrez, A., De Santis, F., Corsaro, M., Varas, F., Bossi, D., Minucci, S., Pelicci, P. G., and Di Croce, L. (2006). The methyl-CpG binding protein MBD1 is required for PML-RARalpha function. Proc. Natl. Acad. Sci. U.S.A. 103, 1400–1405.
Viré, E., Brenner, C., Deplus, R., Blanchon, L., Fraga, M., Didelot, C., Morey, L., Van Eynde, A., Bernard, D., Vanderwinden, J. M., Bollen, M., Esteller, M., Di Croce, L., de Launoit, Y., and Fuks, F. (2006). The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874.
Visvanathan, J., Lee, S., Lee, B., Lee, J. W., and Lee, S.-K. (2007). The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev. 21, 744–749.
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914.
Wang, K. C., Yang, Y. W., Liu, B., Sanyal, A., Corces-Zimmerman, R., Chen, Y., Lajoie, B. R., Protacio, A., Flynn, R. A., Gupta, R. A., Wysocka, J., Lei, M., Dekker, J., Helms, J. A., and Chang, H. Y. (2011). A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124.
Wapinski, O., and Chang, H. Y. (2011). Long noncoding RNAs and human disease. Trends Cell Biol. 21, 354–361.
Wayman, G. A., Davare, M., Ando, H., Fortin, D., Varlamova, O., Cheng, H. Y., Marks, D., Obrietan, K., Soderling, T. R., Goodman, R. H., and Impey, S. (2008). An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc. Natl. Acad. Sci. U.S.A. 105, 9093–9098.
Weaver, I. C. (2009). Epigenetic effects of glucocorticoids. Semin. Fetal Neonatal Med. 14, 143–150.
Wellner, U., Schubert, J., Burk, U. C., Schmalhofer, O., Zhu, F., Sonntag, A., Waldvogel, B., Vannier, C., Darling, D., zur Hausen, A., Brunton, V. G., Morton, J., Sansom, O., Schüler, J., Stemmler, M. P., Herzberger, C., Hopt, U., Keck, T., Brabletz, S., and Brabletz, T. (2009). The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, 1487–1495.
Willemsen, M. H., Vallès, A., Kirkels, L. A., Mastebroek, M., Olde Loohuis, N., Kos, A., Wissink-Lindhout, W. M., de Brouwer, A. P., Nillesen, W. M., Pfundt, R., Holder-Espinasse, M., Vallée, L., Andrieux, J., Coppens-Hofman, M. C., Rensen, H., Hamel, B. C., van Bokhoven, H., Aschrafi, A., and Kleefstra, T. (2011). Chromosome 1p21.3 microdeletions comprising DPYD and MIR137 are associated with intellectual disability. J. Med. Genet. 48, 810–818.
Winocur, G., Wojtowicz, J. M., Sekeres, M., Snyder, J. S., and Wang, S. (2006). Inhibition of neurogenesis interferes with hippocampus-dependent memory function. Hippocampus 16, 296–304.
Wong, C. F., and Tellam, R. L. (2008). MicroRNA-26a targets the histone methyltransferase enhancer of zeste homolog 2 during myogenesis. J. Biol. Chem. 283, 9836–9843.
Wong, P., Iwasaki, M., Somervaille, T. C., Ficara, F., Carico, C., Arnold, C., Chen, C. Z., and Cleary, M. L. (2010). The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res. 70, 3833–3842.
Yadirgi, G., Leinster, V., Acquati, S., Bhagat, H., Shakhova, O., and Marino, S. (2011). Conditional activation of Bmi1 expression regulates self-renewal, apoptosis, and differentiation of neural stem/progenitor cells in vitro and in vivo. Stem Cells 29, 700–712.
Yap, K. L., Li, S., Munoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., Gil, J., Walsh, M. J., and Zhou, M. M. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674.
Yoo, A. S., Staahl, B. T., Chen, L., and Crabtree, G. R. (2009). MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature 460, 642–646.
Yu, Y. L., Chou, R. H., Chen, L. T., Shyu, W. C., Hsieh, S. C., Wu, C. S., Zeng, H. J., Yeh, S. P., Yang, D. M., Hung, S. C., and Hung, M. C. (2011). EZH2 regulates neuronal differentiation of mesenchymal stem cells through PIP5K1C-dependent calcium signaling. J. Biol. Chem. 286, 9657–9667.
Zhang, T. Y., Hellstrom, I. C., Bagot, R. C., Wen, X., Diorio, J., and Meaney, M. J. (2010). Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J. Neurosci. 30, 13130–13137.
Zhao, X., Pak, C., Smrt, R. D., and Jin, P. (2007). Epigenetics and neural developmental disorders: Washington DC, September 18 and 19, 2006. Epigenetics 2, 126–134.
Zhao, X., Ueba, T., Christie, B. R., Barkho, B., McConnell, M. J., Nakashima, K., Lein, E. S., Eadie, B. D., Willhoite, A. R., Muotri, A., Summers, R. G., Chun, J., Lee, K. F., and Gage, F. H. (2003). Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc. Natl. Acad. Sci. U.S.A. 100, 6777–6782.
Zhou, F. C., Balaraman, Y., Teng, M., Liu, Y., Singh, R. P., and Nephew, K. P. (2011). Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol. Clin. Exp. Res. 35, 735–746.
Zhou, Z., Hong, E. J., Cohen, S., Zhao, W. N., Ho, H. Y., Schmidt, L., Chen, W. G., Lin, Y., Savner, E., Griffith, E. C., Hu, L., Steen, J. A., Weitz, C. J., and Greenberg, M. E. (2006). Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255–269.
Keywords: neurogenesis, neural stem cells, epigenetic regulation, DNA methylation, chromatin, non-coding RNA, microRNA
Citation: Jobe EM, McQuate AL and Zhao X (2012) Crosstalk among epigenetic pathways regulates neurogenesis. Front. Neurosci. 6:59. doi: 10.3389/fnins.2012.00059
Received: 30 January 2012; Accepted: 04 April 2012;
Published online: 08 May 2012.
Edited by:
Yanhong Shi, City of Hope, USAReviewed by:
Jenny Hsieh, University of Texas Southwestern Medical Center, USAAshok K. Shetty, Institute for Regenerative Medicine, Texas A&M Health Science Center College of Medicine at Scott & White, USA
Gonzalo Alvarez-Bolado, University of Heidelberg, Germany
Copyright: © 2012 Jobe, McQuate and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Xinyu Zhao, Department of Neuroscience and Waisman Center, University of Wisconsin–Madison School of Medicine and Public Health, Madison, WI 53705, USA. e-mail: xzhao@waisman.wisc.edu