 Omid Kohannim1 Derrek P. Hibar1 Jason L. Stein1 Neda Jahanshad1 Xue Hua1 Priya Rajagopalan1 Arthur W. Toga1 Clifford R. Jack Jr.2 Michael W. Weiner3,4,5,6 Greig I. de Zubicaray7 Katie L. McMahon8 Narelle K. Hansell9 Nicholas G. Martin9 Margaret J. Wright9 Paul M. Thompson1* The Alzheimer’s Disease Neuroimaging Initiative†
Omid Kohannim1 Derrek P. Hibar1 Jason L. Stein1 Neda Jahanshad1 Xue Hua1 Priya Rajagopalan1 Arthur W. Toga1 Clifford R. Jack Jr.2 Michael W. Weiner3,4,5,6 Greig I. de Zubicaray7 Katie L. McMahon8 Narelle K. Hansell9 Nicholas G. Martin9 Margaret J. Wright9 Paul M. Thompson1* The Alzheimer’s Disease Neuroimaging Initiative†
- 1 Imaging Genetics Center at the Laboratory of Neuro Imaging, Department of Neurology, UCLA School of Medicine, Los Angeles, CA, USA
- 2 Mayo Clinic, Rochester, MN, USA
- 3 Department of Radiology, UC San Francisco, San Francisco, CA, USA
- 4 Department of Medicine, UC San Francisco, San Francisco, CA, USA
- 5 Department of Psychiatry, UC San Francisco, San Francisco, CA, USA
- 6 Department of Veterans Affairs Medical Center, San Francisco, CA, USA
- 7 School of Psychology, University of Queensland, Brisbane, QLD, Australia
- 8 Center for Advanced Imaging, University of Queensland, Brisbane, QLD, Australia
- 9 Queensland Institute of Medical Research, Brisbane, QLD, Australia
We implemented least absolute shrinkage and selection operator (LASSO) regression to evaluate gene effects in genome-wide association studies (GWAS) of brain images, using an MRI-derived temporal lobe volume measure from 729 subjects scanned as part of the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Sparse groups of SNPs in individual genes were selected by LASSO, which identifies efficient sets of variants influencing the data. These SNPs were considered jointly when assessing their association with neuroimaging measures. We discovered 22 genes that passed genome-wide significance for influencing temporal lobe volume. This was a substantially greater number of significant genes compared to those found with standard, univariate GWAS. These top genes are all expressed in the brain and include genes previously related to brain function or neuropsychiatric disorders such as MACROD2, SORCS2, GRIN2B, MAGI2, NPAS3, CLSTN2, GABRG3, NRXN3, PRKAG2, GAS7, RBFOX1, ADARB2, CHD4, and CDH13. The top genes we identified with this method also displayed significant and widespread post hoc effects on voxelwise, tensor-based morphometry (TBM) maps of the temporal lobes. The most significantly associated gene was an autism susceptibility gene known as MACROD2. We were able to successfully replicate the effect of the MACROD2 gene in an independent cohort of 564 young, Australian healthy adult twins and siblings scanned with MRI (mean age: 23.8 ± 2.2 SD years). Our approach powerfully complements univariate techniques in detecting influences of genes on the living brain.
Introduction
Genome-wide association studies (GWAS) offer a powerful approach to discover genes that affect individual risk for developing neurological and psychiatric disorders. Several high-density GWAS studies of Alzheimer’s disease (AD), in particular (e.g., Grupe et al., 2006), have identified genes that consistently affect AD risk across multiple populations and ethnic groups, with varying degrees of aggregated evidence. Among others, these include highly replicated risk genes such as ApoE,1CLU,2PICALM,3 and CR14 (Corder et al., 1993; Harold et al., 2009; Lambert et al., 2009), as well as others supported by moderate evidence GAB2,5GOLM1,6TRPC4AP,7LRAT,8ATXN1,9CD33,10 and FAM113B11 (Coon et al., 2007; Abraham et al., 2008; Bertram et al., 2008; Li et al., 2008; Beecham et al., 2009; Podulso et al., 2009). Most of these GWAS studies search for SNPs with alleles that are over-represented in very large samples of diseased versus matched control populations (often numbering tens of thousands of subjects). Diagnostic status, as defined in these case-control studies, is often ascertained based on a battery of cognitive tests, which are relatively remote from the molecular mechanisms of gene action in the brain. As such, studies of genetic variants that are over-represented in certain diagnostic groups often require samples of 30,000 subjects to find and replicate associations (Frayling et al., 2007).
Neuroimaging measures have been proposed as intermediate phenotypes (also known as endophenotypes) to overcome these limitations (Gottesman and Gould, 2003; Meyer-Lindenberg and Weinberger, 2006; Hall and Smoller, 2010). Several projects, such as the Alzheimer’s Disease Neuroimaging Initiative (ADNI12) have also sought biomarkers that are most highly associated with disease risk or with accelerated progression of the disease (Beckett et al., 2010). Imaging measures may offer several advantages for genetic analyses. As an entire 3D image of measurements is collected, rather than one single measure, the full arsenal of statistical methods developed for images – including voxel-based statistics and multivariate methods – may be used to identify spatial patterns of gene effects. Image-wide searches may also detect promising associations. Voxel-set selection may then be used to boost the power of replication attempts, by focusing on promising brain regions that may show larger effect sizes (Vounou et al., 2010, 2012). Such approaches boost statistical power by carrying only the most promising voxels into secondary analyses of independent datasets (Chen et al., 2010; Thompson et al., 2010).
Several neuroimaging measures are associated with AD (Frisoni et al., 2010; Jack et al., 2011), and some of these have already been analyzed using GWAS. Genome-wide studies of the ADNI dataset have identified new candidate AD risk genes including TOMM4013 (Potkin et al., 2009) and GRIN2B14 (Stein et al., 2010a). Traditionally, GWAS studies consider each genotype’s effect independently; they generally ignore the statistical interdependence between variants, such as their linkage disequilibrium (LD) structure, which makes certain variants more likely to be inherited together. More sophisticated GWAS approaches, using information on genes and even pathways, jointly consider groups of genetic variants that are correlated (Neale and Sham, 2004; Luo et al., 2010). Recently, neuroimaging studies have also begun to use these multi-SNP methods (Inkster et al., 2010; Hibar et al., 2011a,b; Kohannim et al., 2011; Wang et al., 2012). Here, we implement a gene-centric approach based on the least absolute shrinkage and selection operator (LASSO), to detect and model genetic influences on neuroimaging measures. LASSO is a form of regularized regression (Tibshirani, 1996), that assesses the combined effect of many correlated variables through a sparsity (or efficiency)-driven L1 penalty.
Here, we use the LASSO algorithm to select sparse groups of SNPs within genes. We then model the effects of the resulting SNPs jointly, to discover associations with neuroimaging measures in the ADNI dataset. Our aim was to detect more genes that influence brain structure with replication potential, as individual SNP variants have individual effect sizes that are usually hard to detect in neuroimaging datasets of the size available today, unless very large meta-analyses are performed (The ENIGMA Consortium, 2011; Stein et al., 2012). LASSO and similar penalized regression techniques have been successfully applied in the context of GWAS or candidate gene studies for selection of SNPs (Ayers and Cordell, 2010; Shi et al., 2011), detection of gene-gene interactions (D’Angelo et al., 2009; Li et al., 2011), and risk prediction from top GWAS hits (Kooperberg et al., 2010).
We hypothesized that using this approach, we would reduce the number of variants of interest in a gene to sparse sets of SNPs and thereby identify genes reliably associated with temporal lobe volume. (The method would work with other brain measures too; temporal lobe volume is just an example of specific interest, due to its role as a biomarker of neurodegenerative disease.) We tested the reliability and reproducibility of our top genetic association findings in an independent non-overlapping young adult cohort. Our goal was to see if the results could be replicated in a cohort scanned on a different continent, with a different scanner field strength, and with a roughly 50-year difference in mean age. Our motivation was to find gene effects that might persist across the human lifespan. Clearly, such a second sample presents challenges for replication, and we selected it to demonstrate the robustness of the results. We admit that this very stringent approach is only likely to find gene effects with an enduring influence throughout life, and would not serve to replicate biologically important effects present at only one part of the lifespan.
Materials and Methods
Subjects
Neuroimaging and genetic data were obtained from 818 subjects as part of ADNI, a 5-year study launched in 2004 by the NIH, private pharmaceutical companies, and non-profit organizations, as a public–private partnership. The goal of ADNI is to determine biological markers of Alzheimer’s disease through neuroimaging, genetics, neuropsychological tests, and other measures in order to develop and monitor new therapies, and reduce the time of clinical trials. Subjects were recruited from 58 sites across North America. The study was conducted according to the Good Clinical Practice guidelines, the Declaration of Helsinki, and U.S. 21 CFR Part 50 – Protection of Human Subjects, and Part 56 – Institutional Review Boards. Written informed consent was obtained from all participants before protocol-specific procedures were performed. All data acquired as part of this study are publicly available (see text footnote 12).
All ADNI subjects underwent thorough clinical and cognitive assessment at the time of scan acquisition to establish diagnosis. In this study, only the baseline 1.5-T scans were used (not the longitudinal follow-up data). The Mini-Mental State Exam (MMSE) was administered to provide a global measure of mental status (Cockrell and Folstein, 1988). The Clinical Dementia Rating (CDR) was used to assess dementia severity (Morris, 1993). Healthy volunteer status was determined if a subject had MMSE scores between 24 and 30 (inclusive), a CDR of 0, and was non-depressed, non-mild cognitive impairment (MCI), and non-demented. MCI diagnosis was determined if a subject had MMSE scores between 24 and 30 (inclusive), a memory complaint, objective memory loss measured by education adjusted scores on the Wechsler Memory Scale Logical Memory II, a CDR of 0.5, absence of significant levels of impairment in other cognitive domains, essentially preserved activities of daily living, and an absence of dementia. AD was diagnosed based on the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria for probable AD (McKhann et al., 1984), MMSE scores between 20 and 26 (inclusive), and CDR of 0.5 or 1.0. Definitive autopsy-based diagnosis of AD was not possible.
Our ADNI dataset consisted of 729 subjects (mean age: 75.5 ± 6.8 SD years; 173 AD, 358 MCI and 198 cognitively healthy controls) with available neuroimaging, genome-wide genetic data, and other relevant covariates (age, sex, and population structure parameters derivable from the GWAS). As our replication sample, we analyzed the Brisbane healthy young adult dataset, which consists of neuroimaging and genome-wide genetic data from 564 young adult healthy twins (a mixture of monozygotic and dizygotic) and siblings of European descent (mean age: 23.8 ± 2.2 SD years; Wright and Martin, 2004). None of the subjects had a history of significant head injury, neurological or psychiatric illness, substance abuse or dependence, or had a first-degree relative with a psychiatric disorder. All subjects were screened, using a detailed neurocognitive evaluation (de Zubicaray et al., 2008) to exclude cases of pathology known to affect brain structure. Handedness was assessed based on 12 items from Annett’s Handedness Questionnaire (Annett, 1970). This sample presents some challenges for replication, due to the wide age difference (of roughly 50 years) between it and the ADNI sample. As such we were aware that genes with age-dependent effects might be found in one cohort that might not replicate in the other, for biological reasons rather than limitations in power and sample sizes.
Neuroimaging
Structural brain MRI scans were acquired for ADNI subjects using 1.5 T MRI scanners. A subset of the ADNI subjects was also scanned at 3 T, but their scans were not used here to avoid any confounding effects of field strength. A sagittal 3D MP-RAGE sequence was used, that had been optimized for consistency across sites (Jack et al., 2008; TR/TE = 2,400/1,000 ms; flip angle = 8°; FOV = 24 cm; final reconstructed voxel resolution = 0.9375 × 0.9375 × 1.2 mm3). All scans were then linearly registered to the stereotaxic space defined by the International Consortium for Brain Mapping (ICBM-53; Mazziotta et al., 2001) using a 9-parameter transformation (three translations, three rotations, and three scales; Collins et al., 1994). Three-dimensional maps of regional brain volumes, computed relative to an average brain template, were generated with a method known as tensor-based morphometry (TBM), a well-established method for mapping volumetric differences in the brain (Hua et al., 2008a,b), using a minimal deformation template (MDT) from the healthy elderly group as reference. Temporal lobe volumes were obtained by integrating the Jacobian determinants of the deformation transform over the region defined as the temporal lobe on the mean anatomical template. Average temporal lobe volumes (average of lobar volumes in the left and right hemisphere) were considered as phenotypes for genome-wide association, and 3D maps were retained for use in post hoc analyses. The same temporal lobe volume phenotype was used in a prior study by Stein et al. (2010a).
In the Brisbane young adult cohort, all subjects were imaged on one scanner with structural whole-brain MRI at 4 T (Bruker Medspec). T1-weighted images were acquired with an inversion recovery rapid gradient echo sequence [TI/TR/TE = 700/1,500/3.35 ms; flip angle = 8° slice thickness = 0.9 mm, with a 2563 acquisition matrix]. All images were corrected for intensity non-uniformity (Sled et al., 1998) within an automatically delineated mask of the brain (Smith, 2002). Images were spatially normalized to the ICBM-152 template (Mazziotta et al., 2001) using a 9-parameter (global) transformation that rotated and scaled each image to minimize a normalized mutual information cost function (Jenkinson et al., 2002). Images were then resampled in the space of the template using sinc interpolation to yield 1 mm3 isotropic voxels. In this way, each brain was globally matched in size and mutually aligned, but local differences in shape and size remained intact. Similarly to ADNI, TBM was used to create maps of volumetric differences for each subject, using a reference MDT specially constructed for this young adult cohort.
Genotyping
The ADNI genotyping procedures are thoroughly described in Saykin et al. (2010). Briefly, genotypes were imputed, using Mach (version 1.0)15 as in Stein et al. (2010b). Imputation is commonly used in genetics to infer or impute unmeasured genotypes, based on available genotypes and known patterns of correlation among markers. We used imputation in ADNI to estimate haplotype phasing and remove missing genotype calls, yielding a set of SNPs without missing data. We used the Plink software16 to extract SNPs that had minor allele frequencies greater than 0.1 (10%), and Hardy–Weinberg equilibrium p-values less than 5.7 × 10−7. Hardy–Weinberg equilibrium refers to the principle that frequencies of alleles, and their corresponding genotypes, are in the expected proportions, under certain assumptions about the population. We only considered alleles with a minor allele frequency > 0.1 due to the very large sample sizes needed to detect effects of less common variants (Flint et al., 2010). As in Stein et al. (2010b), we also considered only unrelated Caucasian subjects identified by self-report and verified by multidimensional scaling (MDS) analysis (Stein et al., 2010a). This was done to decrease the effects of population stratification, although we additionally included MDS parameters as covariates in this study. As detailed in Hibar et al. (2011a), we extracted and grouped all intragenic SNPs, excluding those not located in any genes. Only SNPs within transcripts (i.e., introns and exons, including untranslated regions) were included; SNPs upstream or downstream from genes were not included to avoid arbitrary window sizes. Similarly, imputation of missing genotype calls based on haplotype phasing was performed in the Brisbane young adult cohort.
Gene-image Association
SNP selection with the LASSO algorithm
We incorporated all genotyped SNPs in a gene into LASSO regression models and obtained coefficients for each intragenic SNP. The LASSO (Tibshirani, 1996) is a form of regularized or “penalized” regression, where L1 regularization is introduced into the standard multiple linear regression procedure, using a compound cost function to optimize the regression coefficients:
Here, y represents the vector of our neuroimaging measure co-varied for age and sex (i.e., residuals of linear regression after adjustment for the covariates), X is the matrix of genotypes for a single gene coded additively for the number of minor alleles (i.e., 0, 1, or 2), and β* represents the vector of fitted regression coefficients for each SNP’s effect on the neuroimaging measure. λ is a positive, weighting parameter on the L1 penalty, which encourages sparsity in the resulting set of fitted regression coefficients. In other words, a regression model with a smaller number of coefficients is favored, but this desirable characteristic is traded-off with the need for a model that offers a good fit to the data (which is measured by the first term above). Leave-one-out cross-validation was performed to determine the optimal penalty parameter with the mean squared error criterion. The LASSO analysis was performed gene by gene, and relatively sparse subsets of SNPs within each gene were obtained with non-zero coefficients, by fitting each LASSO model using optimal parameters. For our analyses, we used the “glmnet” package (Friedman et al., 2010) implemented in R17, which optimizes model fitting parameters using a coordinate descent algorithm. We performed single leave-one-out cross-validation cycles with the coordinate descent algorithm for each fold and found regularization parameters that led to the smallest average mean squared errors across all folds.
Multi-SNP partial F-tests
Subsets of SNP genotypes within genes selected by the LASSO were incorporated into partial F-tests, and p-values were obtained for their joint effect on imaging phenotypes, after adjustment for age and sex. Partial F-tests were performed for each gene, where the full model included SNP genotypes selected by LASSO, along with covariates, and the reduced model contained only covariates. In ADNI, population structure parameters from MDS analysis were also included as covariates. For the Brisbane twin cohort, we used a version of the efficient mixed-modeling association (EMMA; Kang et al., 2008) software, modified to allow for subject relatedness when fitting multiple regressors in the regression model. The LASSO step used above serves as a filter to yield a less noisy and more sparse set of predictors, similarly to the data-adaptive collapsing method used in Chen et al. (2011) and the principal component regression method used in Hibar et al. (2011a). Although leave-one-out cross-validation is used to obtain the optimal parameters for LASSO, the SNPs that remain in the optimized LASSO model are incorporated into partial F-tests in the same dataset.
Correction for multiple comparisons
In genetics, there is a notion of genome-wide significance, based on the premise that credible results should surpass a very stringent statistical threshold that accounts for the very large number of separate statistical tests performed when imaging measure is associated with a large number of different variants on the genome. To compute an appropriate threshold to determine the genome-wide significance level for our gene-centric tests, we used a Bonferroni correction: p = 0.05/(number of genes; i.e., 18,284), which yields a p-value threshold of 2.73 × 10−6. To perform post hoc, exploratory tests on the top genes, we created voxelwise statistical maps using partial F-tests from a multiple linear regression fitted at each voxel. To correct for multiple spatial comparisons, we used a regional False Discovery Rate (FDR) method, which is now fairly standard in neuroimaging (Langers et al., 2007).
Results
Gene-Image Association Results
Sparse groups of SNPs within genes were selected by the LASSO algorithm and were subsequently incorporated into partial F-tests of association with the MRI-derived measure of temporal lobe volume (see Figure 1 for an example). We found a total of 22 genes to be genome-wide significant (after Bonferroni correction for the total number of genes assessed). Remarkably, the top gene (MACROD2), had a p-value of 7.94 × 10−12 (Table 1; also see later for independent replication of this top hit). We compared F-test p-values for significant genes with univariate p-values corresponding to single top SNPs in the same genes (similarly to Figure 2 in Hibar et al., 2011a). In the standard, univariate GWAS (Stein et al., 2010a), two genes passed genome-wide multiple comparison correction: GRIN2B and NRXN3. Here, we found the same two among our 22 significant genes, and the whole-gene p-values we obtained for both were “more significant” (i.e., had greater effect sizes) than their top SNP p-values reported in the GWAS (on the order of 10−9 and 10−8 instead of 10−7 and 10−6, respectively). The other 20 genes were undetectable with univariate GWAS as none of their SNPs attained an individual p-value that passed genome-wide significance. We confirmed that all top genes were expressed in the brain, using the Tissue-specific Gene Expression and Regulation (TIGER) database (Liu et al., 2008; Table 1). Although MACROD2 and GALNTL6 were not found in this database, their expression in the brain was evident from the GeneNote software (Shmueli et al., 2003).
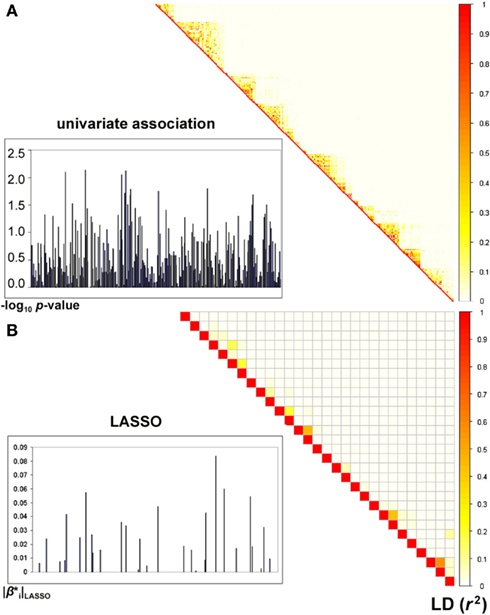
Figure 1. Comparison of univariate and LASSO association tests with an imaging-derived measure of temporal lobe volume. (A) p-values are shown, in the left panel, as −log10 p (i.e., after logarithmic transformation) for all 291 genotyped SNPs in the MACROD2 gene, using univariate association with temporal lobe volume. The most significant SNP in this gene has a p-value of 7.3 × 10−3. A matrix representing the pairwise correlation (i.e., r2), due to linkage disequilibrium (LD), between the SNPs is displayed in the right panel. (B) LASSO considers all SNPs in a gene jointly and assigns sparse coefficients to the SNPs. Here, the absolute values of the coefficients are displayed for all SNPs in MACROD2, 29 of which attained non-zero coefficients as part of the sparse regression model fitted by the LASSO method. When considered jointly in a multiple linear regression model, the selected SNPs yield a boosted p-value of 7.94 × 10−12 for MACROD2 (see Table 1). As in (A), the correlation structure between the selected SNPs is shown, demonstrating reduced multicollinearity.
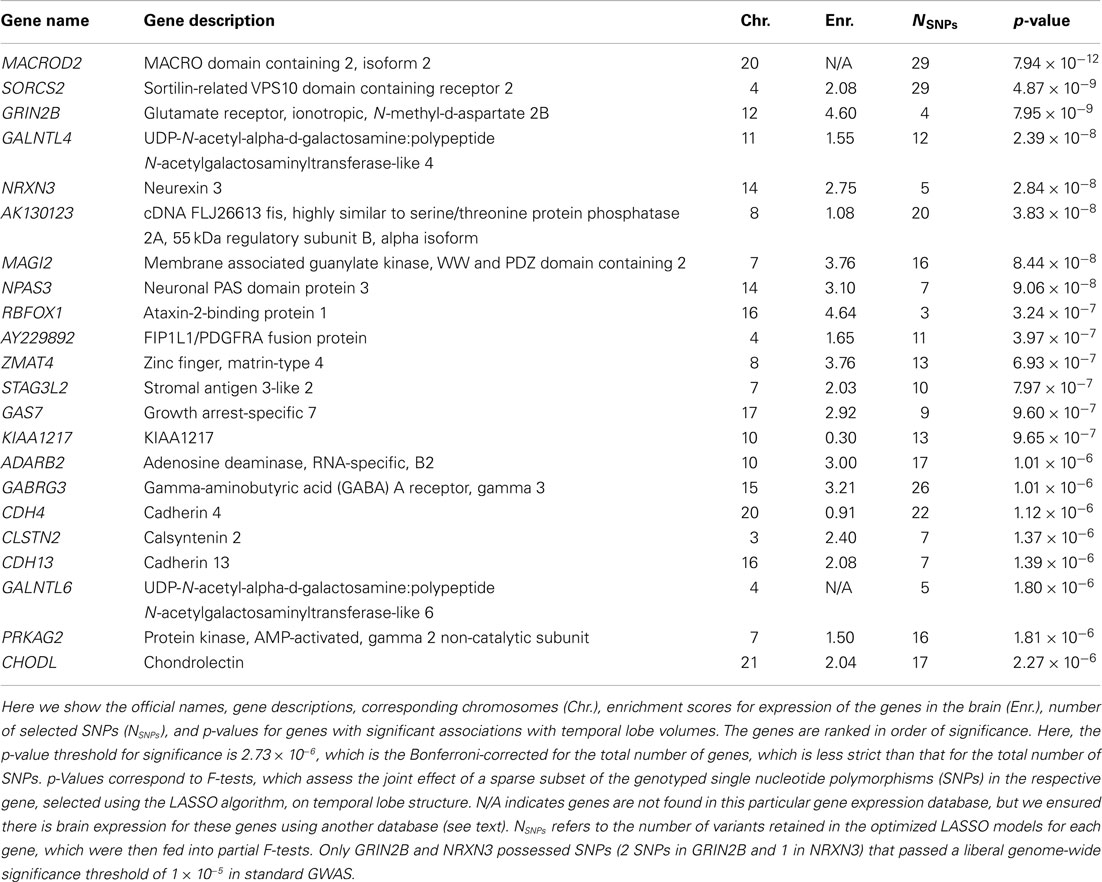
Table 1. Genes showing significant associations with an MRI-derived measure of temporal lobe volume.
Post hoc, Voxelwise Analyses
To study the effects of the genes we identified in more spatial detail, we conducted post hoc, voxelwise analyses, where the partial F-test analysis is repeated at each voxel, and p-value maps are obtained for the gene effects, after correction for multiple comparisons across all temporal lobe voxels. We note in advance that this type of follow-up analysis does not provide any additional statistical verification of the effects, above and beyond the evidence given to the gene for association with the overall temporal lobe volume, in the LASSO regression model. The purpose was mainly to explore whether the volumetric effect could be also detected diffusely at the voxel level, and if so how widespread the effect was in the brain.
The top genes we identified showed extensive, statistically significant effects on maps of temporal lobes’ volumetric differences (Figure 2). These are not to be considered as providing independent evidence of the effects, they simply pick up a pattern of regional effects that is likely to contribute to the aggregate effect of the gene on the temporal lobe volume.
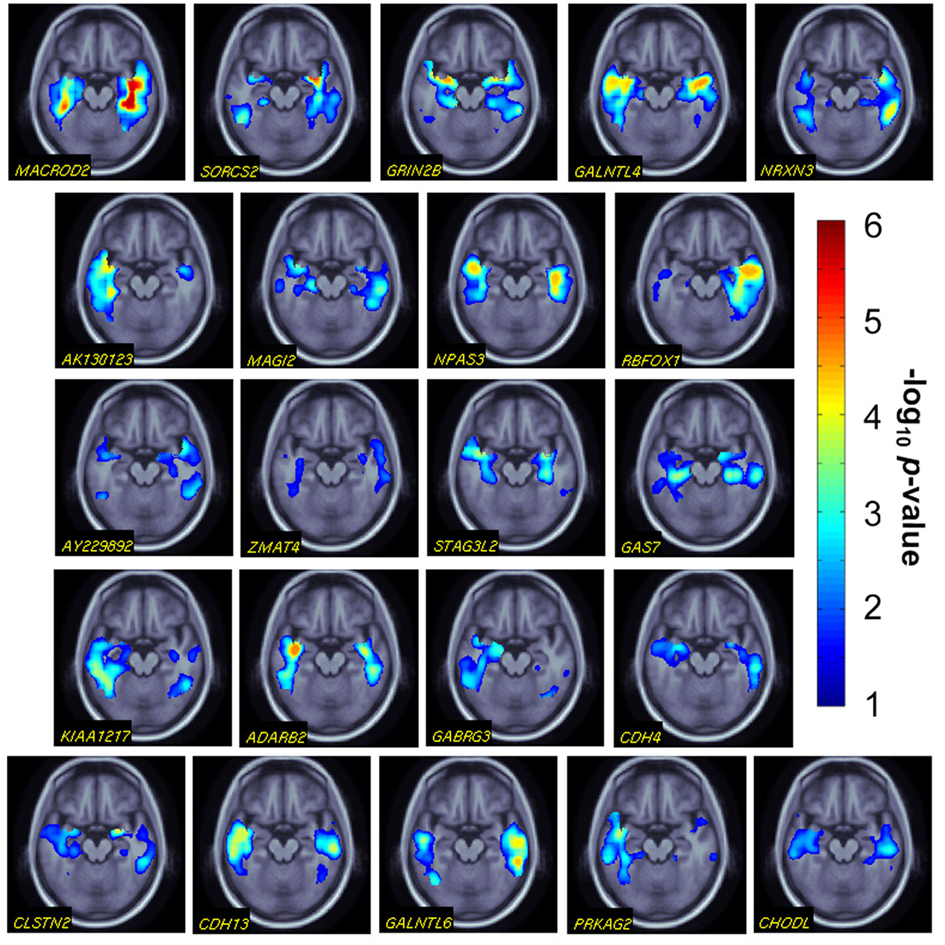
Figure 2. Post hoc gene effects on temporal lobe structure. We conducted voxelwise associations for the 22 significant genes we identified (gene names are shown at the bottom left of the images). A representative, axial slice is shown for all gene effects. Warmer colors represent more significant effects (i.e., higher effect sizes in the analysis of overall temporal lobe volume). p-values are corrected for multiple comparisons within the 3D image search region, using a regional false discovery rate method.
Replication of Gene-Image Associations
To replicate our most significant finding (i.e., MACROD2) from ADNI, we explored the voxelwise effects of this same gene on temporal lobe maps from N = 564 healthy young adult twins and siblings. We jointly considered the same group of SNPs in the gene, selected by the LASSO method in ADNI, and studied their effect on the younger subjects’ temporal lobe maps, adjusting for sex, age, and relatedness. After correcting for multiple comparisons, MACROD2’s association showed partial reproducibility, as it was statistically significant across ∼15% of the temporal lobe voxels (versus ∼52% of voxels in ADNI), much of which overlapped with those in ADNI (see Figure 3).
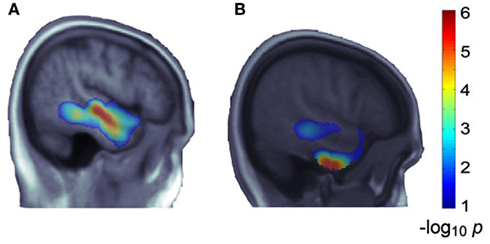
Figure 3. MACROD2’s effects in ADNI temporal lobes and partial replication in a younger cohort. The SNPs selected from MACROD2 in ADNI are strongly associated with mean temporal lobe volumes (p = 7.94 × 10−12), and reveal extensive and significant effects on TBM maps of temporal lobes (A). We studied the joint effect of the same group of SNPs in MACROD2 on temporal lobe maps of the Brisbane young adult dataset. The gene effect showed reproducibility, including effects at some specific locations overlapping with the ADNI findings, after correction for multiple comparisons across the temporal lobe voxels (B). A sagittal slice from the left hemisphere is shown; warmer colors represent more significant associations. Both (A,B) are taken from the same slice (x = 53 mm in Montreal Neurological Institute coordinates). The slices do not appear identical due to the age range differences between the populations [e.g., atrophy in (A)], but almost all of the significant voxels in the more central cluster in (B) overlap with the significant voxels in (A), implying replication.
Cross-Validation
As mentioned in the Section “Materials and Methods,” leave-one-out cross-validation was used to find optimal LASSO parameters, but no further loop of cross-validation was used for the F-tests. In separate analyses, we performed a nested cross-validation, where SNP selection with optimized LASSO was performed in a fifth of the dataset and the joint partial F-tests were then conducted in the rest of the data. This was repeated for fivefolds. The average p-values we obtained for our top genes with this approach did not reach genome-wide significance, mostly because the selection of SNPs with LASSO was unstable in the relatively small fractions (i.e., fifths) of the data.
We took another nested cross-validation approach, where a randomly selected half of the data was used to estimate LASSO coefficients, and the other half for partial F-tests. This, we presumed, might provide more balanced sample sizes for the SNP selection and for the F-test steps. After repeating this scheme across 10 trials, we obtained average p-values for our top genes. Only GRIN2B obtained gene-wide significance averaged across all trials (p = 1.47 × 10−6), and its association was not boosted when compared to that obtained from a univariate approach.
Discussion
We set out to discover and replicate gene effects on brain structure using a gene-centric LASSO regression approach. The goal of the method was to sift through the vast amount of genomic data and come up with a more efficient set of variants for association testing. LASSO allowed us to select sparse subsets of SNPs among all correlated SNPs within each gene and associate them jointly in partial F-tests with an MRI-derived temporal lobe volume measure. Using this approach, we identified over twenty genes with significant effects on temporal lobe structure in N = 729 elderly subjects from the ADNI cohort – a considerably larger number of genes, when compared to a univariate approach, which considers the association of single SNPs one-by-one. In all 22 genes identified, multi-SNP p-values (from partial F-tests), using SNPs selected by LASSO, were “more significant” (i.e., lower p-values, greater effect sizes) than the top genotyped SNP within each corresponding gene, as computed using standard univariate GWAS. GRIN2B and NRXN3, which were identified with univariate GWAS (Stein et al., 2010a), were boosted from p-values on the order of 10−7 and 10−6 to 10−9 and 10−8, respectively. In addition, new genes with more significant p-values were discovered, whose SNPs’ individual p-values were too weak to pass genome-wide significance in a more standard univariate GWAS experimental design. Furthermore, post hoc analysis revealed widespread and significant, voxelwise influences for the top genes on TBM maps of the temporal lobes. We also replicated, at least in part, the spatial effects of our most significant finding in the MACROD2 gene in an independent cohort of healthy, young adults.
Penalized regression techniques such as LASSO (Tibshirani, 1996), ridge regression (Hoerl, 1962), and the elastic net (Zou and Hastie, 2005) have recently been highly effective when used in GWAS. They all deal with (1) multicollinearity due to LD, (2) the large dimensionality of the genome, and (3) the problem of multiple comparisons (Malo et al., 2008; Cho et al., 2009, 2010; Lin et al., 2009; Shi et al., 2011). LASSO’s emphasis on sparsity is particularly useful in our study, as it helps point to a small set of independent variants in a given gene, which we can then incorporate into a multiple regression framework. This is similar to the approach taken by Chen et al. (2011), in the context of jointly considering rare and common variants. Here, we tested this algorithm in the context of finding genetic influences on an imaging-derived measure of temporal lobe volume. This allowed us to discover and replicate a great number of genes relative to our earlier imaging GWAS study (Stein et al., 2010a). We were also able to implicate several genes with previously identified relevance to brain disorders (see below).
In a recent study by our group, Hibar et al. (2011a) also considered gene-based associations with brain images with a new method based on principal component regression, which associates genes with images by capturing most of the variation among intragenic SNPs. Our approach complements this method, as it instead emphasizes sparsity of the model based on the available SNP data for each gene. In the case of principal components regression, a rather different line of analysis is taken in which the covariance in a set of N genotyped SNPs is analyzed to produce a reduced set of k (<N) predictors, that encode some of the genetic variance, but are more efficient than the original set. That method can also produce overall p-values for a specific gene to quantify their effects on brain structure. However, the results of principal components regression are less readily ascribed to any specific sets of SNPs on the genome.
Although the p-values we obtained for the two genes discovered in univariate GWAS were more significant than the univariate p-values of their top SNPs, this need not be the case for every gene. As also discussed in Hibar et al. (2011a), there are cases where a univariate test for the top SNP in a gene offers more power to detect an effect than a multivariate F-test for the whole gene. ADAMTS2, for instance, is a gene that contains a SNP with the lowest univariate p-value (rs12513486, p = 2.23 × 10−5) in our dataset just below the significance threshold considered in Stein et al. (2010a). With our F-test approach following LASSO regression, the gene actually obtained a weaker association (p = 4.83 × 10−5). Thus, our approach complements univariate GWAS, but does not always boost detection power by including multiple loci.
Several of the genes we identified have been well studied in the context of psychiatric and neurological disorders, including Alzheimer’s disease. Our most significant gene, MACROD2, which we also replicated in a new cohort, was recently discovered in the context of autism spectrum disorder (ASD), as the gene containing the top SNP (p < 5 × 10−8) in a GWAS of 1,558 families of whom some members had been diagnosed with ASD (Anney et al., 2010). The investigators of that study reported that although the precise function of this gene is mostly unknown, it is involved in several biological functions and the region comprising their top SNP may regulate PLD2, a gene coding for a member of a protein family with significant implications for ASD. MACROD2 has also been associated with schizophrenia, as the gene corresponding to a rare, copy number variant in a linkage analysis (Xu et al., 2009). The same gene has also been associated with MRI-defined brain infarcts, as the gene comprising the top SNP (p < 5 × 10−7) in a meta-analysis GWAS of >9,000 mostly white, European subjects from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium with an average age of 69.7 years (Debette et al., 2010). Interestingly, this study also found a suggestive association for a SNP in GALNTL4, another top gene in our list.
We additionally found boosted associations for another gene, GRIN2B, coding for a subunit of an N-methyl-D-aspartate (NMDA)-type glutamate receptor, and NRXN3, coding for a neurexin, important for synaptic function, both of which were previously identified in the same dataset with standard GWAS (Stein et al., 2010a).
We also found significant associations for SORCS2 and MAGI2, which are in the AlzGene18 database of genes that show promising associations with the risk for developing AD based on the literature (Rogaeva et al., 2007; Potkin et al., 2009). Additionally, NPAS3 has been linked to schizophrenia and bipolar disorder (Pickard et al., 2009), CLSTN2 has been associated with memory performance (Papassotiropoulos et al., 2006) and with Alzheimer’s disease (Liu et al., 2007), and RBFOX1 (A2BP1) has been very recently discovered as a splicing regulator of neuronal excitation and calcium homeostasis in the brain (Gehman et al., 2011). RBFOX1 has been associated with autism, among other brain disorders (Martin et al., 2007), and an RBFOX1 variant has interestingly been detected in another sparse regression study in ADNI (Vounou et al., 2012).
The discovery and replication populations in this study are quite different: the ADNI cohort consists of elderly subjects within the spectrum of Alzheimer’s disease and the Brisbane cohort consists of healthy young adults. Though the cohorts share a Caucasian background, which minimizes genetic heterogeneity, the age, and health differences between them implies that their brain structure may be influenced by genetic risks and mechanisms that partially overlap but also partially differ. By choosing cohorts that differ in age, we can find polymorphisms that are of enduring relevance over the lifespan, but may be less able to confirm gene effects that only matter in old age. Unfortunately, another elderly cohort with GWAS and MRI scans was not available to us at this time; we requested published GWAS data from other elderly cohorts whose MRI scans we have already analyzed, but our request was declined. However, as discussed in Stein et al. (2011) who discovered and replicated genetic variants on MRI-derived caudate volume in the same two cohorts, there may be genes with persistent effects over the human lifespan, so any such replication may be even stronger than one observed between more similar populations. In addition, though the genes we identified here may not all be AD genes, the effect of a well-known AD risk conferring polymorphism in the clusterin gene (CLU), and that of GAB2, another AD gene, have been replicated as showing associations with brain structure in the same young adult cohort (Braskie et al., 2011; Hibar et al., 2012). In other words, we knew in advance that genes associated with brain structure in the elderly may also exert detectable effects in scans from younger people. This was also the case for some SNP effects that were reliably replicated by two very large GWAS consortia analyzing brain scans from cohorts across the lifespan (Stein et al., 2012) or in elderly cohorts (Bis et al., 2012). In this study, we were similarly interested to see if our top gene’s effects on temporal lobe structure would replicate in the young, adult cohort, suggesting a more lasting influence on brain structure across a person’s lifetime.
In our study, SNPs were coded additively, i.e., using a value of 0, 1, or 2 for the number of minor alleles. This coding makes the assumption that all SNPs considered in the analysis exert their effects in an additive fashion, as opposed to alternative models such as recessive or dominant. This is certainly not the case for all SNPs, and the assumption likely affects the statistical power of our results, since greatest power is obtained when the true model of a causal allele is implemented (Lettre et al., 2007). This is a potential limitation of our study. We chose the additive model as previous genome-wide analyses of the same dataset relevant to our work implement the same allelic coding and were successful in finding genetic associations that were later replicated (Stein et al., 2010a; Hibar et al., 2011a). Furthermore, the additive model is the most commonly used association model. It is the model assumed in heritability calculations and has been argued to be closest to actual risk models for complex traits, such as our quantitative imaging-based measure (Balding, 2006).
A possible limitation of our results is that we do not implement a nested cross-validation approach, in which SNPs selected from LASSO regression would have been included in F-tests in non-overlapping subjects. Our implementation of LASSO here, however, fits into a filtering rather than predictive framework and similar data-adaptive filtering followed by F-tests in the same dataset has been done in previous work (Chen et al., 2011; Hibar et al., 2011a). This approach is potentially unfair, as fitting in LASSO is followed by another fitting with multiple linear regression (F-tests) in the same dataset, whereas fitting is only performed once in a univariate scheme. As GWAS are sensitive to sample size, a nested cross-validation scheme, though more robust, would most likely yield no significant results. We observed this power limitation, as we attempted nested approaches with varying numbers of folds, and were unable to obtain boosted gene-based associations. This may change in the future, as larger datasets become more widely available. Our use of a replication cohort, however, does add credibility to the top result, as the same set of SNPs selected in the discovery sample show significant, spatial effects on brain scans from a completely independent (non-overlapping) group of subjects scanned with a different scanner on a different continent. Another limitation of our approach is that we focus on genes, but exclude promoter and intergenic SNPs. This has the drawback of missing potentially important regulatory elements in the genome.
Our work has several possible future directions, biologically and methodologically. Further investigation is needed to clarify the roles of the genes we identified. We did create voxelwise maps for the top genes, but one could also use a more computationally demanding imaging GWAS approach by re-running the gene-centric, LASSO at each voxel in the brain (Stein et al., 2010b; Vounou et al., 2010; Hibar et al., 2011a), instead of running it on summary measures derived from the images. Sparse coding, used here to reduce the dimensionality of the genomic data, could also be used to zero in on the most promising voxels in the images, leading to a set of phenotypes in the images that show greatest association. Vounou et al. (2010, 2012), in particular, have proposed a general “reduced rank” method that distils a set of genes and brain measures from regions of interest into a more manageable set for assessing associations. Other approaches for dimension reduction, within both the image and the genome, involve variants of independent components analysis (Liu et al., 2009). In a recent advance, Chiang et al. (2011b) proposed to use genetic correlations to identify pairs of voxels in an image with common genetic determination, rather than simply phenotypic correlation. This could be more promising in principle than using phenotypic covariance, as it seems voxel sets are influenced by common (partially overlapping) sets of genes. By clustering these voxels into regions of interest, Chiang et al. (2011b) were able to boost power to detect genome-wide associations in a large DTI study. Clearly, the promise of multivariate methods for imaging genomics is high. Several variants of linear regression, penalized regression, and machine learning are now being adapted to handle images, with the main goal of boosting power and reducing the very large samples typically considered necessary for replicable findings in genetics (The ENIGMA Consortium, 2011).
In addition to GWAS, an alternative more hypothesis-driven approach is to use candidate gene studies to study the influence of genetic variants on brain structure. These have recently been successful in implicating genes as associated with brain white matter integrity measures derived from diffusion tensor imaging (e.g., CLU, Braskie et al., 2011; BDNF, Chiang et al., 2011a; HFE, Jahanshad et al., 2012). Furthermore, it will be interesting to study interactions between genes discovered through GWAS by considering the overall pathways or regulatory networks in which they act (Inkster et al., 2010; Potkin et al., 2010). Another type of genetic information, not considered here, is rare variants on the genome (Schork et al., 2009), or copy number variants, which may also be relevant in the determination of brain structure. Ongoing imaging studies are beginning to include proteomic and gene expression data, as well. Such studies may begin to integrate genetic information from different sources to probe the mechanisms of brain pathology and identify means to intervene and resist it.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Data collection and sharing for this project was funded by the ADNI (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., as well as non-profit partners the Alzheimer’s Association and Alzheimer’s Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California Los Angeles. This research was also supported by NIH grants P30 AG010129, K01 AG030514, and the Dana Foundation. We also thank the many contributors to ADNI-1 genotyping sample curation at NCRAD (Kelley Faber), performing BeadChip assays at TGen and initial quality control of genotypes (David Craig, April Allen, Jill Gerber, and Jason Corneveaux), and bioinformatics problem solving (Indiana U: Kwangsik Nho; Pfizer: Bryan DeChairo). The neuroimaging and genetic analysis of the twin dataset was supported by grant number RO1 HD050735 from the National Institute of Child Health and Human Development, USA, and Project Grant 496682 from the National Health and Medical Research Council, Australia. The collection of IQ data and zygosity typing was supported by the Australian Research Council (A7960034, A79906588, A79801419, and DP0212016). Greig I. de Zubicaray is supported by an Australian Research Council Future Fellowship (FT0991634). Additional support for algorithm development was provided by the NIA, NIBIB, and the National Center for Research Resources (AG016570, EB01651, and RR019771 to Paul M. Thompson). We are also grateful to the twins for their willingness to participate in our studies, and research nurses, Marlene Grace and Ann Eldridge, Queensland Institute of Medical Research, for twin recruitment. Derrek P. Hibar is partially supported by NSF GRFP grant DGE-0707424. Omid Kohannim is supported by NIH F30 AG041681 and the UCLA Graduate Division.
Footnotes
- ^Apolipoprotein E
- ^Clusterin
- ^Phosphatidylinositol binding clathrin assembly protein
- ^Complement component (3b/4b) receptor 1
- ^GRB2-associated binding protein 2
- ^Golgi membrane protein 1
- ^Transient receptor potential cation channel, subfamily C, member 4 associated protein
- ^Lecithin retinol acyltransferase
- ^Spinocerebellar ataxia type 1 protein
- ^Cluster of differentiation 33
- ^Family with sequence similarity 113, member B
- ^http://adni.loni.ucla.edu
- ^Translocase of outer mitochondrial membrane 40 homolog
- ^Glutamate receptor, ionotropic, N-methyl D-aspartate 2B
- ^http://www.sph.umich.edu/csg/abecasis/MaCH/index.html
- ^http://pngu.mgh.harvard.edu/∼purcell/plink/
- ^http://cran.r-project.org
- ^http://www.alzgene.org/
References
Abraham, R., Moskvina, V., Sims, R., Hollingworth, P., Morgan, A., Georgieva, L., Dowzell, K., Cichon, S., Hillmer, A. M., O’Donovan, M. C., Williams, J., Owen, M. J., and Kirov, G. (2008). A genome-wide association study for late-onset Alzheimer’s disease using DNA pooling. BMC Med. Genomics 1, 44. doi:10.1186/1755-8794-1-44
Annett, M. (1970). A classification of hand preference by association analysis. Br. J. Psychol. 61, 303–321.
Anney, R., Klei, L., Pinto, D., Regan, R., Conroy, J., Magalhaes, T. R., Correia, C., Abrahams, B. S., Sykes, N., Pagnamenta, A. T., Almeida, J., Bacchelli, E., Bailey, A. J., Baird, G., Battaglia, A., Berney, T., Bolshakova, N., Bölte, S., Bolton, P. F., Bourgeron, T., Brennan, S., Brian, J., Carson, A. R., Casallo, G., Casey, J., Chu, S. H., Cochrane, L., Corsello, C., Crawford, E. L., Crossett, A., Dawson, G., de Jonge, M., Delorme, R., Drmic, I., Duketis, E., Duque, F., Estes, A., Farrar, P., Fernandez, B. A., Folstein, S. E., Fombonne, E., Freitag, C. M., Gilbert, J., Gillberg, C., Glessner, J. T., Goldberg, J., Green, J., Guter, S. J., Hakonarson, H., Heron, E. A., Hill, M., Holt, R., Howe, J. L., Hughes, G., Hus, V., Igliozzi, R., Kim, C., Klauck, S. M., Kolevzon, A., Korvatska, O., Kustanovich, V., Lajonchere, C. M., Lamb, J. A., Laskawiec, M., Leboyer, M., Le Couteur, A., Leventhal, B. L., Lionel, A. C., Liu, X. Q., Lord, C., Lotspeich, L., Lund, S. C., Maestrini, E., Mahoney, W., Mantoulan, C., Marshall, C. R., McConachie, H., McDougle, C. J., McGrath, J., McMahon, W. M., Melhem, N. M., Merikangas, A., Migita, O., Minshew, N. J., Mirza, G. K., Munson, J., Nelson, S. F., Noakes, C., Noor, A., Nygren, G., Oliveira, G., Papanikolaou, K., Parr, J. R., Parrini, B., Paton, T., Pickles, A., Piven, J., Posey, D. J., Poustka, A., Poustka, F., Prasad, A., Ragoussis, J., Renshaw, K., Rickaby, J., Roberts, W., Roeder, K., Roge, B., Rutter, M. L., Bierut, L. J., Rice, J. P., Salt, J., Sansom, K., Sato, D., Segurado, R., Senman, L., Shah, N., Sheffield, V. C., Soorya, L., Sousa, I., Stoppioni, V., Strawbridge, C., Tancredi, R., Tansey, K., Thiruvahindrapduram, B., Thompson, A. P., Thomson, S., Tryfon, A., Tsiantis, J., Van Engeland, H., Vincent, J. B., Volkmar, F., Wallace, S., Wang, K., Wang, Z., Wassink, T. H., Wing, K., Wittemeyer, K., Wood, S., Yaspan, B. L., Zurawiecki, D., Zwaigenbaum, L., Betancur, C., Buxbaum, J. D., Cantor, R. M., Cook, E. H., Coon, H., Cuccaro, M. L., Gallagher, L., Geschwind, D. H., Gill, M., Haines, J. L., Miller, J., Monaco, A. P., Nurnberger, J. I. Jr., Paterson, A. D., Pericak-Vance, M. A., Schellenberg, G. D., Scherer, S. W., Sutcliffe, J. S., Szatmari, P., Vicente, A. M., Vieland, V. J., Wijsman, E. M., Devlin, B., Ennis, S., and Hallmayer, J. (2010). A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 19, 4072–4082.
Ayers, K. L., and Cordell, H. J. (2010). SNP selection in genome-wide and candidate gene studies via penalized logistic regression. Genet. Epidemiol. 34, 879–891.
Balding, D. J. (2006). A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 7, 781–791.
Beckett, L. A., Harvey, D. J., Gamst, A., Donohue, M., Kornak, J., Zhang, H., Kuo, J. H., and Alzheimer’s Disease Neuroimaging Initiative. (2010). The Alzheimer’s disease neuroimaging initiative: annual change in biomarkers and clinical outcomes. Alzheimers Dement. 6, 257–264.
Beecham, G. W., Martin, E. R., Li, Y.-J., Slifer, M. A., Gilbert, J. R., Haines, J. L., and Pericak-Vance, M. A. (2009). Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am. J. Hum. Genet. 84, 35–43.
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., Schjeide, B. M. M., Hooli, B., DiVito, J., Ionita, I., Jiang, H., Laird, N., Moscarillo, T., Ohlsen, K. L., Elliott, K., Wang, X., Hu-Lince, D., Ryder, M., Murphy, A., Wagner, S. L., Blacker, D., Becker, K. D., and Tanzi, R. E. (2008). Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83, 623–632.
Bis, J. C., Decarli, C., Smith, A. V., van der Lijn, F., Crivello, F., Fornage, M., Debette, S., Shulman, J. M., Schmidt, H., Srikanth, V., Schuur, M., Yu, L., Choi, S. H., Sigurdsson, S., Verhaaren, B. F., Destefano, A. L., Lambert, J. C., Jack, C. R. Jr., Struchalin, M., Stankovich, J., Ibrahim-Verbaas, C. A., Fleischman, D., Zijdenbos, A., den Heijer, T., Mazoyer, B., Coker, L. H., Enzinger, C., Danoy, P., Amin, N., Arfanakis, K., van Buchem, M. A., de Bruijn, R. F., Beiser, A., Dufouil, C., Huang, J., Cavalieri, M., Thomson, R., Niessen, W. J., Chibnik, L. B., Gislason, G. K., Hofman, A., Pikula, A., Amouyel, P., Freeman, K. B., Phan, T. G., Oostra, B. A., Stein, J. L., Medland, S. E., Vasquez, A. A., Hibar, D. P., Wright, M. J., Franke, B., Martin, N. G., Thompson, P. M., Enhancing Neuro Imaging Genetics through Meta-Analysis (ENIGMA) Consortium, Nalls, M. A., Uitterlinden, A. G., Au, R., Elbaz, A., Beare, R. J., van Swieten, J. C., Lopez, O. L., Harris, T. B., Chouraki, V., Breteler, M. M., De Jager, P. L., Becker, J. T., Vernooij, M. W., Knopman, D., Fazekas, F., Wolf, P. A., van der Lugt, A., Gudnason, V., Longstreth, W. T. Jr., Brown, M. A., Bennett, D. A., van Duijn, C. M., Mosley, T. H., Schmidt, R., Tzourio, C., Launer, L. J., Ikram, M. A., Seshadri, S., the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium. (2012). Common variants at 12q14 and 12q24 are associated with hippocampal volume. Nat. Genet. 44, 545–551.
Braskie, M. N., Jahanshad, N., Stein, J. L., Barysheva, M., McMahon, K. L., de Zubicaray, G. I., Martin, N. G., Wright, M. J., Ringman, J. M., Toga, A. W., and Thompson, P. M. (2011). Common Alzheimer’s disease risk variant within the CLU gene affects white matter microstructure in young adults. J. Neurosci. 31, 6764–6770.
Chen, H., Hendricks, A. E., Cheng, Y., Cupples, A. L., Dupuis, J., and Liu, C.-T. (2011). Comparison of statistical approaches to rare variant analysis of quantitative traits. BMC Proc. 5(Suppl. 9), S113. doi:10.1186/1753-6561-5-S9-S113
Chen, K., Langbaum, J. B., Fleisher, A. S., Ayutyanont, N., Reschke, C., Lee, W., Liu, X., Bandy, D., Alexander, G. E., Thompson, P. M., Foster, N. L., Harvey, D. J., de Leon, M. J., Koeppe, R. A., Jagust, W. J., Weiner, M. W., Reiman, E. M., Alzheimer’s Disease Neuroimaging Initiative. (2010). Twelve-month metabolic declines in probable Alzheimer’s disease and amnestic MCI assessed using an empirically pre-defined statistical region-of-interest: findings from the Alzheimer’s disease neuroimaging initiative. Neuroimage 51, 654–664.
Chiang, M.-C., Barysheva, M., Toga, A. W., Medland, S. E., Hansell, N. K., James, M. R., McMahon, K. L., de Zubicaray, G. I., Martin, N. G., Wright, M. J., and Thompson, P. M. (2011a). BDNF gene effects on brain circuitry replicated in 455 twins. Neuroimage 55, 448–454.
Chiang, M.-C., McMahon, K. L., de Zubicaray, G. I., Martin, N. G., Toga, A. W., Wright, M. J., and Thompson, P. M. (2011b). “Hierarchical clustering of the genetic connectivity matrix reveals the network topology of gene action on brain microstructure,” in International Symposium on Biomedical Imaging (ISBI), Chicago, 832–835.
Cho, S., Kim, H., Oh, S., Kim, K., and Park, T. (2009). Elastic-net regularization approaches for genome-wide association studies of rheumatoid arthritis. BMC Proc. 3(Suppl.), S7–S25. doi:10.1186/1753-6561-3-S7-S25
Cho, S., Kim, K., Kim, Y. J., Lee, J. K., Cho, Y. S., Lee, J. Y., Han, B. G., Kim, H., Ott, J., and Park, T. (2010). Joint identification of multiple genetic variants via elastic-net variable selection in a genome-wide association analysis. Ann. Hum. Genet. 74, 416–428.
Cockrell, J. R., and Folstein, M. F. (1988). Mini-Mental State Examination (MMSE). Psychopharmacol. Bull. 24, 689–692.
Collins, D. L., Neelin, P., Peters, T. M., and Evans, A. C. (1994). Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J. Comput. Assist. Tomogr. 18, 192–205.
Coon, K. D., Myers, A. J., Craig, D. W., Webster, J. A., Pearson, J. V., Hu Lince, D., Zismann, V. L., Beach, T. G., Leung, D., Bryden, L., Halperin, R. F., Marlowe, L., Kaleem, M., Walker, D. G., Ravid, R., Heward, C. B., Rogers, J., Papassotiropoulos, A., Reiman, E. M., Hardy, J., and Stephan, D. A. (2007). A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 68, 613–618.
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., Roses, A. D., Haines, J. L., and Pericak-Vance, M. A. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923.
D’Angelo, G. M., Rao, D., and Gu, C. C. (2009). Combining least absolute shrinkage and selection operator (LASSO) and principal-components analysis for detection of gene-gene interactions in genome-wide association studies. BMC Proc. 3(Suppl.), S7–S62. doi:10.1186/1753-6561-3-S7-S62
de Zubicaray, G. I., Chiang, M. C., McMahon, K. L., Shattuck, D. W., Toga, A. W., Martin, N. G., Wright, M. J., and Thompson, P. M. (2008). Meeting the challenges of neuroimaging genetics. Brain Imaging Behav. 2, 258–263.
Debette, S., Bis, J. C., Fornage, M., Schmidt, H., Ikram, M. A., Sigurdsson, S., Heiss, G., Struchalin, M., Smith, A. V., van der Lugt, A., DeCarli, C., Lumley, T., Knopman, D. S., Enzinger, C., Eiriksdottir, G., Koudstaal, P. J., DeStefano, A. L., Psaty, B. M., Dufouil, C., Catellier, D. J., Fazekas, F., Aspelund, T., Aulchenko, Y. S., Beiser, A., Rotter, J. I., Tzourio, C., Shibata, D. K., Tscherner, M., Harris, T. B., Rivadeneira, F., Atwood, L. D., Rice, K., Gottesman, R. F., van Buchem, M. A., Uitterlinden, A. G., Kelly-Hayes, M., Cushman, M., Zhu, Y., Boerwinkle, E., Gudnason, V., Hofman, A., Romero, J. R., Lopez, O., van Duijn, C. M., Au, R., Heckbert, S. R., Wolf, P. A., Mosley, T. H., Seshadri, S., Breteler, M. M., Schmidt, R., Launer, L. J., and Longstreth, W. T. Jr. (2010). Genome-wide association studies of MRI-defined brain infarcts: meta-analysis from the CHARGE Consortium. Stroke 41, 210–217.
Flint, J., Greenspan, R. J., and Kendler, K. S. (2010). How Genes Influence Behavior. Oxford: Oxford University Press.
Frayling, T. M., Timpson, N. J., Weedon, M. N., Zeggini, E., Freathy, R. M., Lindgren, C. M., Perry, J. R., Elliott, K. S., Lango, H., Rayner, N. W., Shields, B., Harries, L. W., Barrett, J. C., Ellard, S., Groves, C. J., Knight, B., Patch, A. M., Ness, A. R., Ebrahim, S., Lawlor, D. A., Ring, S. M., Ben-Shlomo, Y., Jarvelin, M. R., Sovio, U., Bennett, A. J., Melzer, D., Ferrucci, L., Loos, R. J., Barroso, I., Wareham, N. J., Karpe, F., Owen, K. R., Cardon, L. R., Walker, M., Hitman, G. A., Palmer, C. N., Doney, A. S., Morris, A. D., Smith, G. D., Hattersley, A. T., and McCarthy, M. I. (2007). A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894.
Friedman, J., Hastie, T., and Tibshirani, R. (2010). Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22.
Frisoni, G. B., Fox, N. C., Jack, C. R. Jr., Scheltens, P., and Thompson, P. M. (2010). The clinical use of structural MRI in Alzheimer’s disease. Nat. Rev. Neurol. 6, 67–77.
Gehman, L. T., Stoilov, P., Maguire, J., Damianov, A., Lin, C. H., Shiue, L., Ares, M. Jr., Mody, I., and Black, D. L. (2011). The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat. Genet. 43, 706–711.
Gottesman, I. I., and Gould, T. D. (2003). The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiatry 160, 636–645.
Grupe, A., Abraham, R., Li, Y., Rowland, C., Hollingworth, P., Morgan, A., Jehu, L., Segurado, R., Stone, D., Schadt, E., Karnoub, M., Nowotny, P., Tacey, K., Catanese, J., Sninsky, J., Brayne, C., Rubinsztein, D., Gill, M., Lawlor, B., Lovestone, S., Holmans, P., O’Donovan, M., Morris, J. C., Thal, L., Goate, A., Owen, M. J., and Williams, J. (2006). Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum. Mol. Genet. 16, 865–873.
Hall, M.-H., and Smoller, J. W. (2010). A new role for endophenotypes in the GWAS era: functional characterization of risk variants. Harv. Rev. Psychiatry 18, 67–74.
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., Pahwa, J. S., Moskvina, V., Dowzell, K., Williams, A., Jones, N., Thomas, C., Stretton, A., Morgan, A. R., Lovestone, S., Powell, J., Proitsi, P., Lupton, M. K., Brayne, C., Rubinsztein, D. C., Gill, M., Lawlor, B., Lynch, A., Morgan, K., Brown, K. S., Passmore, P. A., Craig, D., McGuinness, B., Todd, S., Holmes, C., Mann, D., Smith, A. D., Love, S., Kehoe, P. G., Hardy, J., Mead, S., Fox, N., Rossor, M., Collinge, J., Maier, W., Jessen, F., Schürmann, B., van den Bussche, H., Heuser, I., Kornhuber, J., Wiltfang, J., Dichgans, M., Frölich, L., Hampel, H., Hüll, M., Rujescu, D., Goate, A. M., Kauwe, J. S., Cruchaga, C., Nowotny, P., Morris, J. C., Mayo, K., Sleegers, K., Bettens, K., Engelborghs, S., De Deyn, P. P., Van Broeckhoven, C., Livingston, G., Bass, N. J., Gurling, H., McQuillin, A., Gwilliam, R., Deloukas, P., Al-Chalabi, A., Shaw, C. E., Tsolaki, M., Singleton, A. B., Guerreiro, R., Mühleisen, T. W., Nöthen, M. M., Moebus, S., Jöckel, K. H., Klopp, N., Wichmann, H. E., Carrasquillo, M. M., Pankratz, V. S., Younkin, S. G., Holmans, P. A., O’Donovan, M., Owen, M. J., and Williams, J. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093.
Hibar, D. P., Jahanshad, N., Stein, J. L., Kohannim, O., Toga, A. W., Medland, S. E., Hansell, N. K., McMahon, K. L., de Zubicaray, G. I., Montgomery, G. W., Martin, N. G., Wright, M. J., and Thompson, P. M. (2012). Alzheimer’s disease risk gene, GAB2, is associated with regional brain volume differences in N = 755 young, healthy twins. Twin Res. Hum. Genet. 15, 286–295.
Hibar, D. P., Stein, J. L., Kohannim, O., Jahanshad, N., Saykin, A. J., Shen, L., Kim, S., Pankratz, N., Foroud, T., Huentelman, M. J., Potkin, S. G., Jack, C. R. Jr., Weiner, M. W., Toga, A. W., Thompson, P. M., Alzheimer’s Disease Neuroimaging Initiative. (2011a). Voxelwise gene-wide association study (vGeneWAS). Multivariate gene-based association testing in 731 elderly subjects. Neuroimage 56, 1875–1891.
Hibar, D. P., Kohannim, O., Stein, J. L., Chiang, M. C., and Thompson, P. M. (2011b). Multilocus genetic analysis of brain images. Front. Genet. 2:73. doi:10.3389/fgene.2011.00073
Hoerl, A. E. (1962). Application of ridge analysis to regression problems. Chem. Eng. Prog. 58, 54–59.
Hua, X., Leow, A. D., Lee, S., Klunder, A. D., Toga, A. W., Lepore, N., Chou, Y.-Y., Brun, C., Chiang, M.-C., Barysheva, M., Jack, C. R. Jr., Bernstein, M. A., Britson, P. J., Ward, C. P., Whitwell, J. L., Borowski, B., Fleisher, A. S., Fox, N. C., Boyes, R. G., Barnes, J., Harvey, D., Kornak, J., Schuff, N., Boreta, L., Alexander, G. E., Weiner, M. W., and Thompson, P. M. (2008a). 3D characterization of brain atrophy in Alzheimer’s disease and mild cognitive impairment using tensor-based morphometry. Neuroimage 41, 19–34.
Hua, X., Leow, A. D., Parikshak, N., Lee, S., Chiang, M.-C., Toga, A. W., Jack, C. R. Jr., Weiner, M. W., and Thompson, P. M. (2008b). Tensor-based morphometry as a neuroimaging biomarker for Alzheimer’s disease: an MRI study of 676 AD MCI and normal subjects. Neuroimage 43, 458–469.
Inkster, B., Nichols, T. E., Saemann, P. G., Auer, D. P., Holsboer, F., Muglia, P., and Matthews, P. M. (2010). Pathway-based approaches to imaging genetics association studies: Wnt signaling, GSK3beta substrates and major depression. Neuroimage 53, 908–917.
Jack, C. R. Jr., Bernstein, M. A., Fox, N. C., Thompson, P., Alexander, G., Harvey, D., Borowski, B., Britson, P. J., L Whitwell, J., Ward, C., Dale, A. M., Felmlee, J. P., Gunter, J. L., Hill, D. L., Killiany, R., Schuff, N., Fox-Bosetti, S., Lin, C., Studholme, C., DeCarli, C. S., Krueger, G., Ward, H. A., Metzger, G. J., Scott, K. T., Mallozzi, R., Blezek, D., Levy, J., Debbins, J. P., Fleisher, A. S., Albert, M., Green, R., Bartzokis, G., Glover, G., Mugler, J., and Weiner, M. W. (2008). The Alzheimer’s Disease Neuroimaging Initiative (ADNI) MRI methods. J. Magn. Reson. Imaging 27, 685–691.
Jack, C. R. Jr., Cantillion, M., Cole, P., Dale, A., Fox, N. C., Foster, N., Frisoni, G. B., Johnson, K., Schwarz, A. J., Sperling, R., Suhy, J., Thompson, P. M., and Hill, D. L. G. (2011). Steps to standardization of hippocampal volumetry as a biomarker for clinical trials and for diagnostic criteria in Alzheimer’s disease. Alzheimers Dement. 7, 474–485.
Jahanshad, N., Kohannim, O., Hibar, D. P., Stein, J. L., McMahon, K. L., de Zubicaray, G. I., Medland, S. E., Montgomery, G. W., Whitfield, J. B., Martin, N. G., Wright, M. J., Toga, A. W., and Thompson, P. M. (2012). Brain structure in healthy adults is related to serum transferrin and the H63D polymorphism in the HFE gene. Proc. Natl. Acad. Sci. U.S.A. 109, E851–E859.
Jenkinson, M., Bannister, P., Brady, M., and Smith, S. (2002). Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage 17, 825–841.
Kang, H. M., Zaitlen, N. A., Wade, C. M., Kirby, A., Heckerman, D., Daly, M. J., and Eskin, E. (2008). Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723.
Kohannim, O., Hibar, D. P., Stein, J. L., Jahanshad, N., Jack, C. R. Jr., Weiner, M. W., Toga, A. W., and Thompson, P. M. (2011). “Boosting power to detect genetic associations in imaging using multi-locus, genome-wide scans and ridge regression,” in International Symposium on Biomedical Imaging (ISBI), Chicago, 1855–1859.
Kooperberg, C., LeBlanc, M., and Obenchain, V. (2010). Risk prediction using genome-wide association studies. Genet. Epidemiol. 34, 643–652.
Lambert, J. C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., Combarros, O., Zelenika, D., Bullido, M. J., Tavernier, B., Letenneur, L., Bettens, K., Berr, C., Pasquier, F., Fiévet, N., Barberger-Gateau, P., Engelborghs, S., De Deyn, P., Mateo, I., Franck, A., Helisalmi, S., Porcellini, E., Hanon, O., European Alzheimer’s Disease Initiative Investigatorsde Pancorbo, M. M., Lendon, C., Dufouil, C., Jaillard, C., Leveillard, T., Alvarez, V., Bosco, P., Mancuso, M., Panza, F., Nacmias, B., Bossù, P., Piccardi, P., Annoni, G., Seripa, D., Galimberti, D., Hannequin, D., Licastro, F., Soininen, H., Ritchie, K., Blanché, H., Dartigues, J. F., Tzourio, C., Gut, I., Van Broeckhoven, C., Alpérovitch, A., Lathrop, M., and Amouyel, P. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099.
Langers, D. R. M., Jansen, J. F. A., and Backes, W. H. (2007). Enhanced signal detection in neuroimaging by means of regional control of the global false discovery rate. Neuroimage 38, 43–56.
Lettre, G., Lange, C., and Hirschhorn, J. N. (2007). Genetic model testing and statistical power in population-based association studies of quantitative traits. Genet. Epidemiol. 31, 358–362.
Li, H., Wetten, S., Li, L., St Jean, P. L., Upmanyu, R., Surh, L., Hosford, D., Barnes, M. R., Briley, J. D., Borrie, M., Coletta, N., Delisle, R., Dhalla, D., Ehm, M. G., Feldman, H. H., Fornazzari, L., Gauthier, S., Goodgame, N., Guzman, D., Hammond, S., Hollingworth, P., Hsiung, G. Y., Johnson, J., Kelly, D. D., Keren, R., Kertesz, A., King, K. S., Lovestone, S., Loy-English, I., Matthews, P. M., Owen, M. J., Plumpton, M., Pryse-Phillips, W., Prinjha, R. K., Richardson, J. C., Saunders, A., Slater, A. J., St George-Hyslop, P. H., Stinnett, S. W., Swartz, J. E., Taylor, R. L., Wherrett, J., Williams, J., Yarnall, D. P., Gibson, R. A., Irizarry, M. C., Middleton, L. T., and Roses, A. D. (2008). Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 65, 45–53.
Li, J., Das, K., Fu, G., Li, R., and Wu, R. (2011). The Bayesian lasso for genome-wide association studies. Bioinformatics 27, 516–523.
Lin, Y., Zhang, M., Wang, L., Pungpapong, V., Fleet, J. C., and Zhang, D. (2009). Simultaneous genome-wide association studies of anti-cyclic citrullinated peptide in rheumatoid arthritis using penalized orthogonal-components regression. BMC Proc. 3(Suppl.), S17–S20. doi:10.1186/1753-6561-3-S7-S17
Liu, F., Arias-Vásquez, A., Sleegers, K., Aulchenko, Y. S., Kayser, M., Sanchez-Juan, P., Feng, B. J., Bertoli-Avella, A. M., van Swieten, J., Axenovich, T. I., Heutink, P., van Broeckhoven, C., Oostra, B. A., and van Duijn, C. M. (2007). A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am. J. Hum. Genet. 81, 17–31.
Liu, J., Pearlson, G., Windemuth, A., Ruano, G., Perrone-Bizzozero, N. I., and Calhoun, V. (2009). Combining fMRI and SNP data to investigate connections between brain function and genetics using parallel ICA. Hum. Brain Mapp. 30, 241–255.
Liu, X., Yu, X., Zack, D. J., Zhu, H., and Qian, J. (2008). TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinformatics 9, 271. doi:10.1186/1471-2105-9-271
Luo, L., Peng, G., Zhu, Y., Dong, H., Amos, C. I., and Xiong, M. (2010). Genome-wide gene and pathway analysis. Eur. J. Hum. Genet. 18, 1045–1053.
Malo, N., Libiger, O., and Schork, N. J. (2008). Accommodating linkage disequilibrium in genetic-association analysis via ridge regression. Am. J. Hum. Genet. 82, 375–385.
Martin, C. L., Duvall, J. A., Ilkin, Y., Simon, J. S., Arreaza, M. G., Wilkes, K., Alvarez-Retuerto, A., Whichello, A., Powell, C. M., Rao, K., Cook, E., and Geschwind, D. H. (2007). Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 869–876.
Mazziotta, J., Toga, A., Evans, A., Fox, P., Lancaster, J., Zilles, K., Woods, R., Paus, T., Simpson, G., Pike, B., Holmes, C., Collins, L., Thompson, P., MacDonald, D., Iacoboni, M., Schormann, T., Amunts, K., Palomero-Gallagher, N., Geyer, S., Parsons, L., Narr, K., Kabani, N., Le Goualher, G., Boomsma, D., Cannon, T., Kawashima, R., and Mazoyer, B. (2001). A probabilistic atlas and reference system for the human brain: International Consortium for Brain Mapping (ICBM). Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 1293–1322.
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944.
Meyer-Lindenberg, A., and Weinberger, D. R. (2006). Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat. Rev. Neurosci. 7, 818–827.
Morris, J. C. (1993). The Clinical Dementia Rating (CDR). current version and scoring rules. Neurology 43, 2412–2414.
Neale, B. M., and Sham, P. C. (2004). The future of association studies: gene-based analysis and replication. Am. J. Hum. Genet. 75, 353–362.
Papassotiropoulos, A., Stephan, D. A., Huentelman, M. J., Hoerndli, F. J., Craig, D. W., Pearson, J. V., Huynh, K. D., Brunner, F., Corneveaux, J., Osborne, D., Wollmer, M. A., Aerni, A., Coluccia, D., Hänggi, J., Mondadori, C. R., Buchmann, A., Reiman, E. M., Caselli, R. J., Henke, K., and de Quervain, D. J. (2006). Common Kibra alleles are associated with human memory performance. Science 314, 475–478.
Pickard, B. S., Christoforou, A., Thomson, P. A., Fawkes, A., Evans, K. L., Morris, S. W., Porteous, D. J., Blackwood, D. H., and Muir, W. J. (2009). Interacting haplotypes at the NPAS3 locus alter risk of schizophrenia and bipolar disorder. Mol. Psychiatry 14, 874–884.
Podulso, S. E., Huang, R., Huang, J., and Smith, S. (2009). Genome screen of late-onset Alzheimer’s extended pedigrees identifies TRPC4AP by haplotype analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 50–55.
Potkin, S. G., Guffanti, G., Lakatos, A., Turner, J. A., Kruggel, F., Fallon, J. H., Saykin, A. J., Orro, A., Lupoli, S., Salvi, E., Weiner, M., Macciardi, F., Alzheimer’s Disease Neuroimaging Initiative. (2009). Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s disease. PLoS ONE 4, e6501. doi:10.1371/journal.pone.0006501
Potkin, S. G., Macciardi, F., Guffanti, G., Wang, Q., Turner, J. A., Lakatos, A., Miles, M. F., Lander, A., Vawter, M. P., and Xie, X. (2010). Identifying gene regulatory networks in schizophrenia. Neuroimage 53, 839–847.
Rogaeva, E., Meng, Y., Lee, J. H., Gu, Y., Kawarai, T., Zou, F., Katayama, T., Baldwin, C. T., Cheng, R., Hasegawa, H., Chen, F., Shibata, N., Lunetta, K. L., Pardossi-Piquard, R., Bohm, C., Wakutani, Y., Cupples, L. A., Cuenco, K. T., Green, R. C., Pinessi, L., Rainero, I., Sorbi, S., Bruni, A., Duara, R., Friedland, R. P., Inzelberg, R., Hampe, W., Bujo, H., Song, Y. Q., Andersen, O. M., Willnow, T. E., Graff-Radford, N., Petersen, R. C., Dickson, D., Der, S. D., Fraser, P. E., Schmitt-Ulms, G., Younkin, S., Mayeux, R., Farrer, L. A., and St George-Hyslop, P. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39, 168–177.
Saykin, A. J., Shen, L., Foroud, T. M., Potkin, S. G., Swaminathan, S., Kim, S., Risacher, S. L., Nho, K., Huentelman, M. J., Craig, D. W., Thompson, P. M., Stein, J. L., Moore, J. H., Farrer, L. A., Green, R. C., Bertram, L., Jack, C. R. Jr., Weiner, M. W., Alzheimer’s Disease Neuroimaging Initiative. (2010). Alzheimer’s disease neuroimaging initiative biomarkers as quantitative phenotypes: genetics core aims progress and plans. Alzheimers Dement. 6, 265–273.
Schork, N. J., Murray, S. S., Frazer, K. A., and Topol, E. J. (2009). Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 19, 212–219.
Shi, G., Boerwinkle, E., Morrison, A. C., Gu, C. C., Chakravarti, A., and Rao, D. C. (2011). Mining gold dust under the genome wide significance level: a two-stage approach to analysis of GWAS. Genet. Epidemiol. 35, 111–118.
Shmueli, O., Horn-Saban, S., Chalifa-Caspi, V., Shmoish, M., Ophir, R., Benjamin-Rodrig, H., Safran, M., Domany, E., and Lancet, D. (2003). GeneNote: whole genome expression profiles in normal human tissues. C. R. Biol. 326, 1067–1072.
Sled, J. G., Zijdenbos, A. P., and Evans, A. C. (1998). A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans. Med. Imaging 17, 87–97.
Stein, J. L., Hibar, D. P., Madsen, S. K., Khamis, M., McMahon, K. L., de Zubicaray, G. I., Hansell, N. K., Montgomery, G. W., Martin, N. G., Wright, M. J., Saykin, A. J., Jack, C. R. Jr., Weiner, M. W., Toga, A. W., Thompson, P. M., Alzheimer’s Disease Neuroimaging Initiative. (2011). Discovery and replication of dopamine-related gene effects on caudate volume in young and elderly populations (N = 1198) using genome-wide search. Mol. Psychiatry 16, 927–937.
Stein, J. L., Hua, X., Morra, J. H., Lee, S., Hibar, D. P., Ho, A. J., Leow, A. D., Toga, A. W., Sul, J. H., Kang, H. M., Eskin, E., Saykin, A. J., Shen, L., Foroud, T., Pankratz, N., Huentelman, M. J., Craig, D. W., Gerber, J. D., Allen, A. N., Corneveaux, J. J., Stephan, D. A., Webster, J., DeChairo, B. M., Potkin, S. G., Jack, C. R. Jr., Weiner, M. W., Thompson, P. M., Alzheimer’s Disease Neuroimaging Initiative. (2010a). Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer’s disease. Neuroimage 51, 542–554.
Stein, J. L., Hua, X., Lee, S., Ho, A. J., Leow, A. D., Toga, A. W., Saykin, A. J., Shen, L., Foroud, T., Pankratz, N., Huentelman, M. J., Craig, D. W., Gerber, J. D., Allen, A. N., Corneveaux, J. J., DeChairo, B. M., Potkin, S. G., Weiner, M. W., Thompson, P. M., Alzheimer’s Disease Neuroimaging Initiative. (2010b). Voxelwise genome-wide association study (vGWAS). Neuroimage 53, 1160–1174.
Stein, J. L., Medland, S. E., Vasquez, A. A., Hibar, D. P., Senstad, R. E., Winkler, A. M., Toro, R., Appel, K., Bartecek, R., Bergmann, O., Bernard, M., Brown, A. A., Cannon, D. M., Chakravarty, M. M., Christoforou, A., Domin, M., Grimm, O., Hollinshead, M., Holmes, A. J., Homuth, G., Hottenga, J. J., Langan, C., Lopez, L. M., Hansell, N. K., Hwang, K. S., Kim, S., Laje, G., Lee, P. H., Liu, X., Loth, E., Lourdusamy, A., Mattingsdal, M., Mohnke, S., Maniega, S. M., Nho, K., Nugent, A. C., O’Brien, C., Papmeyer, M., Pütz, B., Ramasamy, A., Rasmussen, J., Rijpkema, M., Risacher, S. L., Roddey, J. C., Rose, E. J., Ryten, M., Shen, L., Sprooten, E., Strengman, E., Teumer, A., Trabzuni, D., Turner, J., Van Eijk, K., Van Erp, T. G., Van Tol, M. J., Wittfeld, K., Wolf, C., Woudstra, S., Aleman, A., Alhusaini, S., Almasy, L., Binder, E. B., Brohawn, D. G., Cantor, R. M., Carless, M. A., Corvin, A., Czisch, M., Curran, J. E., Davies, G., De Almeida, M. A., Delanty, N., Depondt, C., Duggirala, R., Dyer, T. D., Erk, S., Fagerness, J., Fox, P. T., Freimer, N. B., Gill, M., Göring, H. H., Hagler, D. J., Hoehn, D., Holsboer, F., Hoogman, M., Hosten, N., Jahanshad, N., Johnson, M. P., Kasperaviciute, D., Kent, J. W. Jr., Kochunov, P., Lancaster, J. L., Lawrie, S. M., Liewald, D. C., Mandl, R., Matarin, M., Mattheisen, M., Meisenzahl, E., Melle, I., Moses, E. K., Mühleisen, T. W., Nauck, M., Nöthen, M. M., Olvera, R. L., Pandolfo, M., Pike, G. B., Puls, R., Reinvang, I., Rentería, M. E., Rietschel, M., Roffman, J. L., Royle, N. A., Rujescu, D., Savitz, J., Schnack, H. G., Schnell, K., Seiferth, N., Smith, C., Steen, V. M., Valdés Hernández, M. C., Van Den Heuvel, M., Van Der Wee, N. J., Van Haren, N. E., Veltman, J. A., Völzke, H., Walker, R., Westlye, L. T., Whelan, C. D., Agartz, I., Boomsma, D. I., Cavalleri, G. L., Dale, A. M., Djurovic, S., Drevets, W. C., Hagoort, P., Hall, J., Heinz, A., Jack, C. R. Jr., Foroud, T. M., Le Hellard, S., Macciardi, F., Montgomery, G. W., Poline, J. B., Porteous, D. J., Sisodiya, S. M., Starr, J. M., Sussmann, J., Toga, A. W., Veltman, D. J., Walter, H., Weiner, M. W., The Alzheimer’s Disease Neuroimaging Initiative; (ADNI); EPIGEN Consortium; IMAGEN Consortium; Saguenay Youth Study Group (SYS)Bis, J. C., Ikram, M. A., Smith, A. V., Gudnason, V., Tzourio, C., Vernooij, M. W., Launer, L. J., Decarli, C., Seshadri, S., Cohorts For Heart And Aging Research In Genomic Epidemiology (CHARGE) Consortium; The Enhancing Neuro Imaging Genetics Through Meta-Analysis (ENIGMA) ConsortiumAndreassen, O. A., Apostolova, L. G., Bastin, M. E., Blangero, J., Brunner, H. G., Buckner, R. L., Cichon, S., Coppola, G., De Zubicaray, G. I., Deary, I. J., Donohoe, G., De Geus, E. J., Espeseth, T., Fernández, G., Glahn, D. C., Grabe, H. J., Hardy, J., Hulshoff Pol, H. E., Jenkinson, M., Kahn, R. S., McDonald, C., McIntosh, A. M., McMahon, F. J., McMahon, K. L., Meyer-Lindenberg, A., Morris, D. W., Müller-Myhsok, B., Nichols, T. E., Ophoff, R. A., Paus, T., Pausova, Z., Penninx, B. W., Potkin, S. G., Sämann, P. G., Saykin, A. J., Schumann, G., Smoller, J. W., Wardlaw, J. M., Weale, M. E., Martin, N. G., Franke, B., Wright, M. J., and Thompson, P. M. (2012). Identification of common variants associated with human hippocampal and intracranial volumes. Nat. Genet. 44, 552–561.
The ENIGMA Consortium. (2011). “Genome-wide association meta-analysis of hippocampal volume: results from the ENIGMA consortium,” in Organization for Human Brain Mapping Conference, Quebec City.
Thompson, P. M., Martin, N. G., and Wright, M. J. (2010). Imaging Genomics. Curr. Opin. Neurol. 23, 368–373.
Tibshirani, R. (1996). Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Series B Stat. Methodol. 58, 267–288.
Vounou, M., Janousova, E., Wolz, R., Stein, J. L., Thompson, P. M., Rueckert, D., Montana, G., Alzheimer’s Disease Neuroimaging Initiative. (2012). Sparse reduced-rank regression detects genetic associations with voxelwise longitudinal phenotypes in Alzheimer’s disease. Neuroimage 60, 700–716.
Vounou, M., Nichols, T. E., Montana, G., Alzheimer’s Disease Neuroimaging Initiative. (2010). Discovering genetic associations with high-dimensional neuroimaging phenotypes: a sparse reduced-rank regression approach. Neuroimage 53, 1147–1159.
Wang, H., Nie, F., Huang, H., Kim, S., Nho, K., Risacher, S. L., Saykin, A. J., Shen, L., Alzheimer’s Disease Neuroimaging Initiative. (2012). Identifying quantitative trait loci via group-sparse multitask regression and feature selection: an imaging genetics study of the ADNI cohort. Bioinformatics 28, 229–237.
Wright, M. J., and Martin, N. G. (2004). Brisbane Adolescent Twin Study: outline of study methods and research projects. Aust. J. Psychol. 56, 65–78.
Xu, B., Woodroffe, A., Rodriguez-Murillo, L., Roos, J. L., van Rensburg, E. J., Abecasis, G. R., Gogos, J. A., and Karayiorgou, M. (2009). Elucidating the genetic architecture of familial schizophrenia using rare copy number variant and linkage scans. Proc. Natl. Acad. Sci. U.S.A. 106, 16746–16751.
Keywords: neuroimaging, MRI, imaging genetics, GWAS, LASSO, MACROD2
Citation: Kohannim O, Hibar DP, Stein JL, Jahanshad N, Hua X, Rajagopalan P, Toga AW, Jack CR Jr., Weiner MW, de Zubicaray GI, McMahon KL, Hansell NK, Martin NG, Wright MJ, Thompson PM and The Alzheimer’s Disease Neuroimaging Initiative (2012) Discovery and replication of gene influences on brain structure using LASSO regression. Front. Neurosci. 6:115. doi: 10.3389/fnins.2012.00115
Received: 12 March 2012; Paper pending published: 10 April 2012;
Accepted: 12 July 2012; Published online: 06 August 2012.
Edited by:
Leonard C. Schalkwyk, King’s College London, UKCopyright: © 2012 Kohannim, Hibar, Stein, Jahanshad, Hua, Rajagopalan, Toga, Jack, Weiner, de Zubicaray, McMahon, Hansell, Martin, Wright, Thompson and The Alzheimer’s Disease Neuroimaging Initiative. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Paul M. Thompson, Imaging Genetics Center at the Laboratory of Neuro Imaging, Department of Neurology, University of California Los Angeles, School of Medicine, Neuroscience Research Building 225E, 635 Charles Young Drive, Los Angeles, CA 90095-1769, USA. e-mail: thompson@loni.ucla.edu
†Data used in preparing this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (www.loni.ucla.edu/ADNI). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. For a complete listing of ADNI investigators, please see: http://adni.loni.ucla.edu/wp-content/uploads/how_to_apply/ADNI_Manuscript_Citations.pdf