 Jesus Avila
Jesus Avila George Perry
George Perry Bryan A. Strange
Bryan A. Strange Felix Hernandez
Felix Hernandez- 1Neurobiology, Centro de Biologia Molecular Severo Ochoa (CSIC-UAM), Madrid, Spain
- 2Centro de Investigacion Biomedica en Red de Enfermedades Neurodegenerativas, Madrid, Spain
- 3College of Sciences, The University of Texas at San Antonio, San Antonio, TX, USA
- 4Department of Neuroimaging, Reina Sofia Foundation, Center for Alzheimer Research, FCIEN, Madrid, Spain
- 5Laboratory for Clinical Neuroscience, CTB, Universidad Politecnica de Madrid, Madrid, Spain
It is well established that some individuals with normal cognitive capacity have abundant senile plaques in their brains. It has been proposed that those individuals are resilient or have compensation factors to prevent cognitive decline. In this comment, we explore an alternative mechanism through which cognitive capacity is maintained. This mechanism could involve the impairment of alternative neural circuitry. Also, the proportion of molecules such as Aβ or tau protein present in different areas of the brain could be important.
Introduction
Loss of episodic memory is the most well known feature of Alzheimer disease (AD). Braak and Braak (1996), suggested that damage of the connections between the entorhinal cortex (EC) and hippocampal area could play an important role in the memory impairment of AD (Gomez-Isla et al., 1996). Within the hippocampus, other studies have suggested that the CA1 hippocampal subregion could be the minimal region that is required for acquisition of episodic memory (Zola-Morgan et al., 1986; Volpe et al., 1992; Tsien et al., 1996; Shimizu et al., 2000; Bendel et al., 2005; Buenz et al., 2006; Bueters et al., 2008). There are several works indicating the possible (and different) pathways that connect the EC with CA1 (for a review see Moser et al., 2014 and Figure 1). Some of these pathways go through the dentate gyrus, in which adult neurogenesis could be involved in the formation of new memories (Zola-Morgan et al., 1986; Deng et al., 2010).
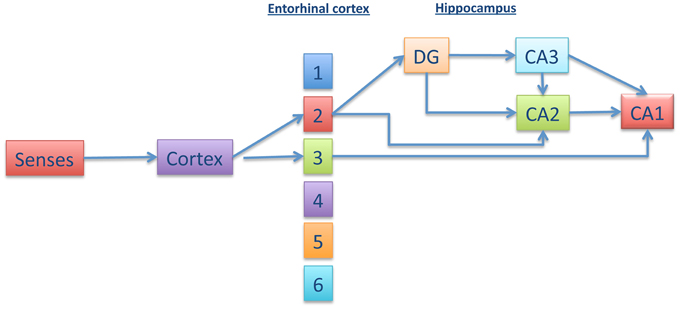
Figure 1. Different pathways connecting entorhinal cortex with CA1. Information from the senses is transmitted to the cortex and, afterwards, to the enthorhinal cortex (EC) (upper layers). From EC, the information could go directly to CA1 or, indirectly, through dentate gyrus (DG), CA3 (or CA2), and CA1. This trisynaptic pathway involves adult neurogenesis.
Damage to the EC and hippocampal region in AD is associated with the appearance of senile plaques and neurofibrillary tangles, aberrant structures first described by Alzheimer (1907). Indeed, it is typically considered that a proper diagnosis of AD is only really complete when, at autopsy, the appearance of plaques and tangles are observed in the brain of the patient (Khachaturian, 1985).
However, the causal nature of the correlation between appearance of plaques and loss of memory (or cognitive impairment) is not clear. Thus, it has been proposed that the disease should be treated, not the lesions (Zhu et al., 2007), because the pathology may be a protective, possibly antioxidant response to the primary pathogenesis. Furthermore, studies on familial AD (FAD) cases reveal an asymptomatic phase in which there are plaques without cognitive impairment (Bateman et al., 2012), although it may also suggest that the presence of plaques is a first step that could favor the subsequent onset of the disease and that their presence is not immediately related to cognitive impairment (Bateman et al., 2012).
Plaques and Cognitive Impairment
There is controversy surrounding a total correlation between the presence of plaques and the presence of cognitive impairment. Some time ago, Katzman et al described cases of cognitively normal people bearing plaques (Katzman et al., 1989). In a more recent study, analyzing those people without cognitive impairment, it was found that some people met the criteria for high likelihood AD, based on the presence of plaques (Bennett et al., 2006), indicating that AD pathology can be found in the brain of those without cognitive impairment (Aizenstein et al., 2008). Some functional magnetic resonance analysis have also reported the presence of Aβ aggregates in people without cognitive impairment (Dickerson et al., 2004; Sperling et al., 2009; Mormino et al., 2012).
More recently, Elman et al reported that some older people may maintain normal cognition despite the presence of plaques observed by positron emission tomography (PET) analyses (Elman et al., 2014). There are at least two additional explanations from those discussed above, a possible resilience, based on personal characteristics such as having a higher cognitive reserve (Xu et al., 2014), or compensation of a degenerated pathway by using an alternative functional pathway (Elman et al., 2014). In addition, it can be suggested that failure of more than one neuronal circuit can be needed for cognitive impairment.
This led us to consider another alternative to that involving the connections from EC to CA1, indicated in Figure 1, an alternative that might prevent cognitive decline.
In the work of Elman et al. (2014), there are explanations that involve different types of connections, based on fMRI studies, suggesting that CA1 can connect with the cortex without going through the EC. In those studies, Elman et al. (2014) took advantage of the system by analyzing two networks that were previously identified. One of them links those areas that respond to a specific cognitive activity, like visualization of a photograph (Fox et al., 2005; Xu et al., 2014). This is the task-positive network (TPN) (Fox et al., 2005). The other one indicates the regions that are activated in the resting state (Buckner et al., 2008). This is the default mode network (DMN). The DMN can be disrupted in some neurodegenerative disorders (Buckner et al., 2008).
In human subjects, with normal cognition and no plaques, engaging in a task activates the TPN while the DMN shuts down. However, Elman et al. (2014) found that in the same conditions, people with plaques and without cognitive impairment also displayed higher TPN activity but—critically—DMN was less deactivated. The authors focused on this decrease of DMN shut down to try to explain the maintenance of cognition. Indeed, there are several works, using fMRI analysis, or other image techniques, indicating the connection between dysfunction of DMN and cognitive decline. We include some examples of those works (Petrella et al., 2011; Wang et al., 2013; Garces et al., 2014; Gardini et al., 2015).
Alternative Pathways to CA1
The activated areas in DMN include prefrontal cortex and posterior cingulate cortex (Buckner et al., 2008). Cingulate cortex is connected to other structures involved in memory, including the mammillary bodies (MB) (Vann and Aggleton, 2004; Shah et al., 2012). A connection between MB and CA2, which in turn is connected to CA1, has also been found (Haglund et al., 1984; Kohara et al., 2014). Thus, we propose this MB-CA2-CA1 circuit as an alternative pathway through which information from the cerebral cortex can reach CA1 and avoid the EC (Figure 2).
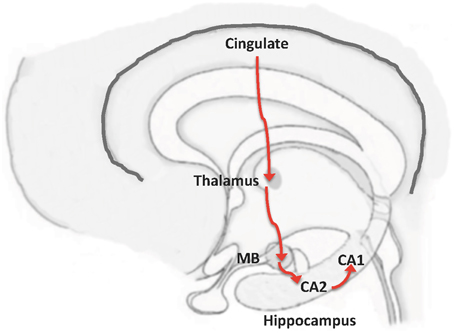
Figure 2. From neocortex to hippocampus, by-passing entorhinal cortex: the MB-CA2-CA1 pathway. A possible pathway to connect the cortex with CA1, without transmission through EC, could be based on a proposed connection between mammillary bodies (MB) to CA2, and from CA2 to CA1 (Haglund et al., 1984; Kohara et al., 2014). In this way, the previously known connection shown in the Figure could be complemented by the following pathway: Cortex  cingulum mammillary bodies CA2 CA1.
cingulum mammillary bodies CA2 CA1.
We suggest that, for severe cognitive impairment to occur, disruption of both circuits, that involving EC-CA1 and that involving cingulate cortex-CA1, is needed. Indeed, abnormal connectivity between the posterior cingulate and hippocampus has been described in patients with cognitive impairment (Zhou et al., 2008). In addition, it has been also indicated that a decrease in the functional connectivity at the posterior cingulate cortex can be critical for the conversion from mild cognitive impairment to AD (Bozzali et al., 2014). Also, in the pioneering works of Stern (2002, 2006) on resilience (cognitive reserve) or compensation, the loss of connectivity at the posterior cingulate cortex was already proposed. In this comment, our main focus is not on resilience (see on that subject the recent work of Pereira et al., 2014), but on compensation.
If, indeed, there is first a loss of connectivity at the hippocampal area (location of the dentate gyrus), followed by a loss of connectivity at the posterior cingulate cortex (Zhou et al., 2008), risk factors, such as aging, could also play a role in this process. It has been recently reported that there is a brain network that links development, aging, and vulnerability to AD (Douaud et al., 2014). This link is based on the so-called Ribot's law indicating that the destruction of memories progresses in reverse order to that of their formation (Douaud et al., 2014). Brain structures, like dentate gyrus, that play a role in recent memory, are assembled very late in development but can lose their functionality very early in the neurodegeneration process.
We suggest that, it is possible that both mechanisms, that involving EC-CA1 and that involving posterior cingulate cortex-CA1, could be damaged and are needed to develop a severe cognitive impairment and AD. Thus, both mechanisms should be needed for disease progression.
Distribution of Aβ and Tau in Cingulate Gyrus and Enthorinal Cortex
As previously indicated, the brain of AD patients contain senile plaques, composed of amyloid-beta peptide (Aβ) and neurofibrillary tangles, containing tau protein polymers. By looking at the causes of FAD, genetic analyses have indicated that mutations in APP (amyloid precursor protein), PSEN-1 (presenilin 1) or PSEN-2 (presenilin 2) genes are the cause of the different types of familial disease. The main consequence of these mutations is an increased production of Aβ. This observation resulted in the proposed amyloid cascade hypothesis of AD. This hypothesis indicates that Aβ accumulation in brain is the primary factor driving AD pathology (Selkoe, 1991; Hardy and Higgins, 1992).
Afterwards, it was shown that the presence of tau protein is essential to the amyloid-beta induced neurotoxicity, occurring in AD (Rapoport et al., 2002; Roberson et al., 2007; Ittner et al., 2010). Based on these and other reports, it was suggested that Aβ could initiate the pathological process but the presence of tau is needed for the progression of the process. In this way, the cell co-localization of Aβ and tau could increase the possibility of neuronal damage.
At the molecular level, a double dissociation in regional distribution of tau and amyloid-beta has been reported when comparing cingulate gyrus and enthorhinal cortex in post-mortem Alzheimer's brains (Shukla and Bridges, 1999). It was described that tau load was almost twice as great in the enthorinal cortex than elsewhere in the brain, whereas Aβ levels were much higher in the cingulate gyrus compared to enthorinal cortex (Shukla and Bridges, 1999). It should be known if those differences may play a role in the development of the disease, mainly within the cingulate cortex, although we know that EC and posterior cingulate cortex could be interconnected and that modified tau could spread from EC to the cingulate cortex (Yassa, 2014). That transport time of modified tau from EC to posterior cingulate cortex may determine the progression time of the disease during the transition from MCI to AD.
In many transgenic animal models, there are no differences in the expression of tau or Aβ at specific locations of the brain. This is because, sometimes, the expression of tau or APP mRNAs is under strong promoters and it may facilitate the expression of a protein throughout the whole brain in a non-physiological way. Thus, in these animal models the progression of degeneration may take place in a different fashion than that occurring in the human disease.
If both, Aβ and tau aggregates, play a common, important role in the pathogenesis of AD, it will be of interest to determine the localization and overlap of those aggregates in different brain regions, given that the presence of plaques in the absence of tau, in some regions, might not be sufficient for cognitive decline. In this way, decreasing the amount of tau at specific brain regions could have a therapeutic function if, as indicated, the common presence of Aβ and tau aggregates is needed for the progression of the disease. Determining Aβ and tau overlap may, however, be complex as clear differences in the amount of tau can be found even between subregions of memory-related brain areas, e.g., hippocampus CA1 (high tau amount), vs. CA3 and CA2 (low tau amount) (our preliminary results). In addition, the presence (or absence) of other molecules related to Aβ or tau pathology could play a role in the development of neurodegeneration. A recent example of this is the telomerase protein TERT that apart from being protective against oxidative damage has a protective role against tau pathology (Spilsbury et al., 2015). Previously it was reported that a coordinated expression of tau and heme oxygenase 1 may play a pivotal role in the cytoprotection of neuronal cells (Takeda et al., 2004). The oxidative imbalance in AD has been extensively reviewed (Zhu et al., 2005; Mondragon-Rodriguez et al., 2013).
Funding
This study was funded by grants from the Spanish Ministry of Health (SAF 2011-24841, BFU-2008-03980, BFU-2010-21507), the Comunidad de Madrid (SA2010/BMD2331), the Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED, ISCIII) (CB401), and the Fundación R. Areces.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aizenstein, H. J., Nebes, R. D., Saxton, J. A., Price, J. C., Mathis, C. A., Tsopelas, N. D., et al. (2008). Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 65, 1509–1517. doi: 10.1001/archneur.65.11.1509
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bateman, R. J., Xiong, C., Benzinger, T. L., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bendel, O., Bueters, T., von Euler, M., Ove Ogren, S., Sandin, J., and von Euler, G. (2005). Reappearance of hippocampal CA1 neurons after ischemia is associated with recovery of learning and memory. J. Cereb. Blood Flow Metab. 25, 1586–1595. doi: 10.1038/sj.jcbfm.9600153
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bennett, D. A., Schneider, J. A., Arvanitakis, Z., Kelly, J. F., Aggarwal, N. T., Shah, R. C., et al. (2006). Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bozzali, M., Dowling, C., Serra, L., Spano, B., Torso, M., Marra, C., et al. (2014). The impact of cognitive reserve on brain functional connectivity in Alzheimer's disease. J. Alzheimers Dis. 44, 243–250. doi: 10.3233/JAD-141824
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braak, H., and Braak, E. (1996). Evolution of the neuropathology of Alzheimer's disease. Acta Neurol. Scand. Suppl. 165, 3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buckner, R. L., Andrews-Hanna, J. R., and Schacter, D. L. (2008). The brain's default network: anatomy, function, and relevance to disease. Ann. N.Y. Acad. Sci. 1124, 1–38. doi: 10.1196/annals.1440.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buenz, E. J., Rodriguez, M., and Howe, C. L. (2006). Disrupted spatial memory is a consequence of picornavirus infection. Neurobiol. Dis. 24, 266–273. doi: 10.1016/j.nbd.2006.07.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bueters, T., von Euler, M., Bendel, O., and von Euler, G. (2008). Degeneration of newly formed CA1 neurons following global ischemia in the rat. Exp. Neurol. 209, 114–124. doi: 10.1016/j.expneurol.2007.09.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deng, W., Aimone, J. B., and Gage, F. H. (2010). New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 11, 339–350. doi: 10.1038/nrn2822
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dickerson, B. C., Salat, D. H., Bates, J. F., Atiya, M., Killiany, R. J., Greve, D. N., et al. (2004). Medial temporal lobe function and structure in mild cognitive impairment. Ann. Neurol. 56, 27–35. doi: 10.1002/ana.20163
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Douaud, G., Groves, A. R., Tamnes, C. K., Westlye, L. T., Duff, E. P., Engvig, A., et al. (2014). A common brain network links development, aging, and vulnerability to disease. Proc. Natl. Acad. Sci. U.S.A. 111, 17648–17653. doi: 10.1073/pnas.1410378111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Elman, J. A., Oh, H., Madison, C. M., Baker, S. L., Vogel, J. W., Marks, S. M., et al. (2014). Neural compensation in older people with brain amyloid-beta deposition. Nat. Neurosci. 17, 1316–1318. doi: 10.1038/nn.3806
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fox, M. D., Snyder, A. Z., Vincent, J. L., Corbetta, M., Van Essen, D. C., and Raichle, M. E. (2005). The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc. Natl. Acad. Sci. U.S.A. 102, 9673–9678. doi: 10.1073/pnas.0504136102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garces, P., Angel Pineda-Pardo, J., Canuet, L., Aurtenetxe, S., Lopez, M. E., Marcos, A., et al. (2014). The default mode network is functionally and structurally disrupted in amnestic mild cognitive impairment - a bimodal MEG-DTI study. Neuroimage Clin. 6, 214–221. doi: 10.1016/j.nicl.2014.09.004
Gardini, S., Venneri, A., Sambataro, F., Cuetos, F., Fasano, F., Marchi, M., et al. (2015). Increased functional connectivity in the default mode network in mild cognitive impairment: a maladaptive compensatory mechanism associated with poor semantic memory performance. J. Alzheimers Dis. 45, 457–470. doi: 10.3233/JAD-142547
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gomez-Isla, T., Price, J. L., McKeel, D. W. Jr., Morris, J. C., Growdon, J. H., and Hyman, B. T. (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J. Neurosci. 16, 4491–4500.
Haglund, L., Swanson, L. W., and Kohler, C. (1984). The projection of the supramammillary nucleus to the hippocampal formation: an immunohistochemical and anterograde transport study with the lectin PHA-L in the rat. J. Comp. Neurol. 229, 171–185. doi: 10.1002/cne.902290204
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Katzman, R., Aronson, M., Fuld, P., Kawas, C., Brown, T., Morgenstern, H., et al. (1989). Development of dementing illnesses in an 80-year-old volunteer cohort. Ann. Neurol. 25, 317–324. doi: 10.1002/ana.410250402
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khachaturian, Z. S. (1985). Diagnosis of Alzheimer's disease. Arch. Neurol. 42, 1097–1105. doi: 10.1001/archneur.1985.04060100083029
Kohara, K., Pignatelli, M., Rivest, A. J., Jung, H. Y., Kitamura, T., Suh, J., et al. (2014). Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nat. Neurosci. 17, 269–279. doi: 10.1038/nn.3614
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mondragon-Rodriguez, S., Perry, G., Zhu, X., Moreira, P. I., Acevedo-Aquino, M. C., and Williams, S. (2013). Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: implications for Alzheimer's disease. Oxid. Med. Cell. Longev. 2013:940603. doi: 10.1155/2013/940603
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mormino, E. C., Brandel, M. G., Madison, C. M., Marks, S., Baker, S. L., and Jagust, W. J. (2012). Abeta Deposition in aging is associated with increases in brain activation during successful memory encoding. Cereb. Cortex 22, 1813–1823. doi: 10.1093/cercor/bhr255
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moser, E. I., Roudi, Y., Witter, M. P., Kentros, C., Bonhoeffer, T., and Moser, M. B. (2014). Grid cells and cortical representation. Nat. Rev. Neurosci. 15, 466–481. doi: 10.1038/nrn3766
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pereira, A. C., Lambert, H. K., Grossman, Y. S., Dumitriu, D., Waldman, R., Jannetty, S. K., et al. (2014). Glutamatergic regulation prevents hippocampal-dependent age-related cognitive decline through dendritic spine clustering. Proc. Natl. Acad. Sci. U.S.A. 111, 18733–18738. doi: 10.1073/pnas.1421285111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petrella, J. R., Sheldon, F. C., Prince, S. E., Calhoun, V. D., and Doraiswamy, P. M. (2011). Default mode network connectivity in stable vs progressive mild cognitive impairment. Neurology 76, 511–517. doi: 10.1212/WNL.0b013e31820af94e
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rapoport, M., Dawson, H. N., Binder, L. I., Vitek, M. P., and Ferreira, A. (2002). Tau is essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 99, 6364–6369. doi: 10.1073/pnas.092136199
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Selkoe, D. J. (1991). The molecular pathology of Alzheimer's disease. Neuron 6, 487–498. doi: 10.1016/0896-6273(91)90052-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shah, A., Jhawar, S. S., and Goel, A. (2012). Analysis of the anatomy of the Papez circuit and adjoining limbic system by fiber dissection techniques. J. Clin. Neurosci. 19, 289–298. doi: 10.1016/j.jocn.2011.04.039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shimizu, E., Tang, Y. P., Rampon, C., and Tsien, J. Z. (2000). NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science 290, 1170–1174. doi: 10.1126/science.290.5494.1170
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shukla, C., and Bridges, L. R. (1999). Regional distribution of tau, beta-amyloid and beta-amyloid precursor protein in the Alzheimer's brain: a quantitative immunolabelling study. Neuroreport 10, 3785–3789. doi: 10.1097/00001756-199912160-00012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sperling, R. A., Laviolette, P. S., O'Keefe, K., O'Brien, J., Rentz, D. M., Pihlajamaki, M., et al. (2009). Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63, 178–188. doi: 10.1016/j.neuron.2009.07.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spilsbury, A., Miwa, S., Attems, J., and Saretzki, G. (2015). The role of telomerase protein TERT in Alzheimer's Disease and in Tau-related pathology in vitro. J. Neurosci. 35, 1659–1674. doi: 10.1523/JNEUROSCI.2925-14.2015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stern, Y. (2002). What is cognitive reserve? Theory and research application of the reserve concept. J. Int. Neuropsychol. Soc. 8, 448–460. doi: 10.1017/S1355617702813248
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stern, Y. (2006). Cognitive reserve and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 20, 112–117. doi: 10.1097/01.wad.0000213815.20177.19
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takeda, A., Itoyama, Y., Kimpara, T., Zhu, X., Avila, J., Dwyer, B. E., et al. (2004). Heme catabolism and heme oxygenase in neurodegenerative disease. Antioxid. Redox Signal. 6, 888–894. doi: 10.1089/ars.2004.6.888
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsien, J. Z., Huerta, P. T., and Tonegawa, S. (1996). The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87, 1327–1338. doi: 10.1016/S0092-8674(00)81827-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vann, S. D., and Aggleton, J. P. (2004). The mammillary bodies: two memory systems in one? Nat. Rev. Neurosci. 5, 35–44. doi: 10.1038/nrn1299
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Volpe, B. T., Davis, H. P., Towle, A., and Dunlap, W. P. (1992). Loss of hippocampal CA1 pyramidal neurons correlates with memory impairment in rats with ischemic or neurotoxin lesions. Behav. Neurosci. 106, 457–464. doi: 10.1037/0735-7044.106.3.457
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Risacher, S. L., West, J. D., McDonald, B. C., Magee, T. R., Farlow, M. R., et al. (2013). Altered default mode network connectivity in older adults with cognitive complaints and amnestic mild cognitive impairment. J. Alzheimers Dis. 35, 751–760. doi: 10.3233/JAD-130080
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, W., Yu, J. T., Tan, M. S., and Tan, L. (2014). Cognitive reserve and Alzheimer's disease. Mol. Neurobiol. 51, 187–208. doi: 10.1007/s12035-014-8720-y
Yassa, M. A. (2014). Ground zero in Alzheimer's disease. Nat. Neurosci. 17, 146–147. doi: 10.1038/nn.3631
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, Y., Dougherty, J. H. Jr., Hubner, K. F., Bai, B., Cannon, R. L., and Hutson, R. K. (2008). Abnormal connectivity in the posterior cingulate and hippocampus in early Alzheimer's disease and mild cognitive impairment. Alzheimers Dement. 4, 265–270. doi: 10.1016/j.jalz.2008.04.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhu, X., Avila, J., Perry, G., and Smith, M. A. (2007). Treating the lesions, not the disease. Am. J. Pathol. 170, 1457–1459. doi: 10.2353/ajpath.2007.070193
Zhu, X., Lee, H. G., Casadesus, G., Avila, J., Drew, K., Perry, G., et al. (2005). Oxidative imbalance in Alzheimer's disease. Mol. Neurobiol. 31, 205–217. doi: 10.1385/MN:31:1-3:205
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Alzheimer disease (AD), cognitive decline, tau proteins, abeta, tauopathies
Citation: Avila J, Perry G, Strange BA and Hernandez F (2015) Alternative neural circuitry that might be impaired in the development of Alzheimer disease. Front. Neurosci. 9:145. doi: 10.3389/fnins.2015.00145
Received: 18 February 2015; Paper pending published: 29 March 2015;
Accepted: 08 April 2015; Published: 23 April 2015.
Edited by:
Irving E. Vega, Michigan State University, USAReviewed by:
Cheng-Xin Gong, New York State Institute for Basic Research in Developmental Disabilities, USAMarcelo Febo, University of Florida, USA
Copyright © 2015 Avila, Perry, Strange and Hernandez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesus Avila, Neurobiology, Centro de Biología Molecular Severo Ochoa CSIC-UAM, 208, Madrid 28049, Spain javila@cbm.uam.es