 John Alexander1†
John Alexander1† Hera Potamianou1†Jinchuan Xing2,3Li Deng2,3
Hera Potamianou1†Jinchuan Xing2,3Li Deng2,3 Iordanis Karagiannidis1
Iordanis Karagiannidis1 Fotis Tsetsos1Petros Drineas4
Fotis Tsetsos1Petros Drineas4 Zsanett Tarnok5
Zsanett Tarnok5 Renata Rizzo6Tomasz Wolanczyk7Luca Farkas5Peter Nagy5Urszula Szymanska7
Renata Rizzo6Tomasz Wolanczyk7Luca Farkas5Peter Nagy5Urszula Szymanska7 Christos Androutsos8Vaia Tsironi8Anastasia Koumoula8
Christos Androutsos8Vaia Tsironi8Anastasia Koumoula8 Csaba Barta9 TSGeneSEE10
Csaba Barta9 TSGeneSEE10 Paul Sandor11Cathy L. Barr12,13
Paul Sandor11Cathy L. Barr12,13 Jay Tischfield2,3
Jay Tischfield2,3 Peristera Paschou1
Peristera Paschou1 Gary A. Heiman2,3*
Gary A. Heiman2,3* Marianthi Georgitsi1,14*
Marianthi Georgitsi1,14*- 1Department of Molecular Biology and Genetics, Democritus University of Thrace, Alexandroupoli, Greece
- 2Department of Genetics, Rutgers, The State University of New Jersey, Piscataway, NJ, USA
- 3Human Genetics Institute of New Jersey, Rutgers, The State University of New Jersey, Piscataway, NJ, USA
- 4Computer Science Department, Purdue University, West Lafayette, USA
- 5Vadaskert Clinic for Child and Adolescent Psychiatry, Budapest, Hungary
- 6Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
- 7Department of Child Psychiatry, Medical University of Warsaw, Warsaw, Poland
- 8Child and Adolescent Psychiatry Clinic, Sismanoglio General Hospital of Attica, Athens, Greece
- 9Molecular Biology and Pathobiochemistry, Institute of Medical Chemistry, Semmelweis University, Budapest, Hungary
- 10Tourette Syndrome Genetics — Southern and Eastern Europe Initiative (TSGeneSEE Consortium)
- 11Department of Psychiatry, University of Toronto, Toronto, ON, Canada
- 12Genetics and Development Division, Krembil Research Institute, University Health Network, Toronto, ON, Canada
- 13Program in Neurosciences and Mental Health, The Hospital for Sick Children, Toronto, ON, Canada
- 14Laboratory of General Biology, Department of Medicine, Aristotle University of Thessaloniki, Thessaloniki, Greece
Although the genetic basis of Tourette Syndrome (TS) remains unclear, several candidate genes have been implicated. Using a set of 382 TS individuals of European ancestry we investigated four candidate genes for TS (HDC, SLITRK1, BTBD9, and SLC6A4) in an effort to identify possibly causal variants using a targeted re-sequencing approach by next generation sequencing technology. Identification of possible disease causing variants under different modes of inheritance was performed using the algorithms implemented in VAAST. We prioritized variants using Variant ranker and validated five rare variants via Sanger sequencing in HDC and SLITRK1, all of which are predicted to be deleterious. Intriguingly, one of the identified variants is in linkage disequilibrium with a variant that is included among the top hits of a genome-wide association study for response to citalopram treatment, an antidepressant drug with off-label use also in obsessive compulsive disorder. Our findings provide additional evidence for the implication of these two genes in TS susceptibility and the possible role of these proteins in the pathobiology of TS should be revisited.
Introduction
Tourette Syndrome (TS; OMIM #137580) is a complex neurodevelopmental disorder of childhood onset, characterized by motor and vocal tics. It often presents with co-morbidities such as attention deficit hyperactivity disorder (ADHD) and/or obsessive-compulsive disorder (OCD), as well as mood disorders, anxiety and sleep disorders. Apart from various environmental factors that contribute to TS complexity (Hoekstra et al., 2013; Mathews et al., 2014), twin and family studies have long established that TS bears a strong genetic component (Price et al., 1985; Pauls et al., 1991, 2014). However, despite extensive on-going genetic research, concrete evidence supporting specific pathogenetic mechanisms is still largely lacking. Several genes (such as SLITRK1, HDC, NLGN4, CNTNAP2, IMMP2L, SLC6A4 also known as SERT, and NTN4) and chromosomal loci have been implicated to date although, associations between genetic variation and TS are often limited to specific population-based cohorts or may be restricted to extremely rare mutations identified solely in unique multigenerational pedigrees.
A very promising candidate TS susceptibility region was implicated for the first time in 2005 by Abelson et al. (Abelson et al., 2005). The study reported a de novo inversion in a TS patient, occurring in the vicinity of the Slit and Trk-like family member 1 (SLITRK1) gene. The authors subsequently sequenced SLITRK1 in 174 unrelated TS probands and identified rare mutations. This spurred intense interest in TS genetics with multiple studies reporting rather mixed results (Abelson et al., 2005; Deng et al., 2006; Keen-Kim et al., 2006; Wendland et al., 2006; Chou et al., 2007; Scharf et al., 2008; Zimprich et al., 2008; O'Roak et al., 2010; Karagiannidis et al., 2012). Recently, our group and others have provided evidence for association of the disorder with common alleles and haplotypes being over-transmitted in TS cases; these findings support the hypothesis of the existence of an as of yet unidentified risk factor in linkage disequilibrium (LD) with the associated markers and possibly lying in gene regulatory regions (Miranda et al., 2009; Karagiannidis et al., 2012).
Abnormalities of cortico-striatal-thalamic-cortical pathways and dysfunction of both dopamine and serotonin neurotransmitter systems have long been considered in association to TS (Peterson et al., 2003; Kalanithi et al., 2005; Muller-Vahl et al., 2005). The recent implication of the L-histidine decarboxylase (HDC) gene in TS etiology has also raised the intriguing hypothesis of the involvement of histaminergic neural pathways in the onset of the disorder (Ercan-Sencicek et al., 2010; Lei et al., 2012). Moreover, a genome-wide scan for de novo or transmitted rare copy number variants in TS had found enrichment of genes within the histamine signaling pathways (Fernandez et al., 2012). Subsequently, our group found evidence for over-transmission of HDC alleles and significantly associated haplotypes in trios of European origin (Karagiannidis et al., 2013).
The serotonin transporter gene (SERT, 5-HTT), coding for a solute carrier family 6 member 4 protein (SLC6A4) was reported to carry both common and rare variants as well as a high-expressing haplotype associated with increased gene expression (5-HTTLPR/rs25531/rs25532) and protein activity (p.I425V), thus potentially contributing to serotonergic abnormalities in TS with or even more so, without OCD (Kilic et al., 2003; Moya et al., 2013). These high-expressing alleles have been previously significantly associated with OCD (Hu et al., 2006; Wendland et al., 2008), which is believed to share a degree of common genetic background with TS (Mathews and Grados, 2011; Yu et al., 2015).
Variants in the BTB/POZ domain-containing protein 9 gene (BTBD9) have been previously associated with restless legs syndrome (RLS) (Allen et al., 2005; Winkelmann et al., 2007). High RLS prevalence has also been reported in TS and, similar to TS, it has been linked to frontostriatal circuits dysfunction and is responsive to dopamine neurotransmission modification (Turjanski et al., 1999). BTBD9 variants were found to be associated with TS in French Canadian and Chinese patient cohorts (Riviere et al., 2009; Guo et al., 2012). Yet, all analysed variants were intronic and thus do not provide direct functional relevance; however, they could be in LD with potentially functional yet unknown variants, such as variants in gene regulatory regions or, tissue and neuroanatomical region-specific, naturally-occurring somatic mutations (Lodato et al., 2015).
The abovementioned TS susceptibility genes have emerged from candidate gene association studies (SLC6A4, BTBD9), chromosomal aberration studies (SLITRK1), and linkage analysis studies (HDC). Despite decades of efforts in the field of TS genetics, the overall number of identified variants likely predisposing to TS remains extremely small. With the advent of next-generation sequencing technology, there is a resurging interest toward the identification of rare variants of potentially intermediate-to-high penetrance effects that might aid in explaining part of the missing heritability of the TS phenotype.
In the present study we report results on a targeted re-sequencing approach in a cohort of 382 TS cases of European and Canadian origin, aiming to explore the existence of rare genetic variation across four genes that have attracted considerable interest in the past years, namely SLITRK1, HDC, BTBD9, and SLC6A4. Moreover, given the findings from our previous studies on association of SLITRK1 and HDC haplotypes with TS (Karagiannidis et al., 2012, 2013), we wanted to explore whether previously associated risk haplotypes are in linkage with rare coding, and thus directly functional variants in these two genes.
Materials and Methods
Study Subjects
A total of 382 individuals with TS were included in the study. The subjects represent affected cases from family trios who had been previously recruited within The Tourette Syndrome Genetics—Southern and Eastern Europe Initiative (TSGeneSEE) and originated from Hungary (n = 117), Italy (n = 95), Poland (n = 68), Greece (n = 17), and Albania (n = 8). In addition, 77 TS cases originating from Canada were also available. The majority of these individuals have been described previously (Karagiannidis et al., 2013). Assessment was performed by on-site clinicians using the tools provided by the TS Association International Consortium for Genetics (TSAICG, 2007). TS was ascertained according to DSM-IV-TR criteria for Italy, Hungary, Albania, and Greece, and DSM-IV for Poland. For Canadian samples, TS was ascertained using DSM-III-R criteria. Differences between DSM-III-R, DSM-IV, and DSM-IV-TR are minimal; the upper age limit of onset is 18 in DSM-IV (and DSM-IV-TR) and 21 in DSM-III-R, and the “marked distress” criterion, possibly pointing to more severe cases, only appears in DSM-IV (applied only to the Polish cases; Cath et al., 2011). For all samples, collection was approved by local Ethics Boards and written informed consent was taken from all participating individuals or their parents.
Genomic DNA Extraction
Peripheral blood samples were collected in EDTA-containing tubes and genomic DNA was purified using the Qiagen Gentra Puregene kit with minor protocol modifications (Qiagen GmbH, Hilden, Germany).
Exome Capture and Targeted Re-Sequencing
Using the Fluidigm custom panel design pipeline, the four candidate genes (HDC, SLITRK1, SLC6A4, BTBD9) were targeted for sequencing and custom primers were designed. For HDC and SLITRK1, the whole locus including 1 kb upstream and downstream were targeted. For SLC6A4 and BTBD9, exonic regions of the genes were targeted (Table S1).
DNA samples were tagged and amplified using Fluidigm Access Array System (Fluidigm Corporation, South San Francisco, USA). The 48.48 Access Array integrated fluidics circuit (IFC) protocol was followed. The sample mix solution was prepared using 50 ng/uL of genomic DNA. The gDNA was then combined with sample pre-mix and loaded into the 48.48 Access Array IFC. Upon amplification completion, the IFC was placed on the Post-PCR IFC Controller AX and the Harvest (151x) Script was run. After the completion of the script 10 uL of the product was removed and placed into a 96-well PCR plate. Barcoding PCR was then set up using the manufacturer's instructions on a conventional PCR thermal cycler. With the incorporation of a unique identifier or barcode for each sample and the necessary sequencing adaptors, it is possible to process all samples simultaneously on the sequencing platform.
Quality and quantity of the library were evaluated on the Agilent BioAnalyzer (Agilent Technologies, Santa Clara, CA, US) and quantified using KAPA quantification. The sequencing was then completed following the protocol for the Illumina (Illumina, San Diego, CA) MiSeq V3 chemistry 2 × 100-bp paired-end reads. Once the sequencing was completed, de-multiplexing was performed using Illumina's bcl2fastq2 v2.15 to generate sample specific fastq files.
Sequence Alignment and Genotype Calling
Sequence alignment was performed using Burroughs Wheeler alignment (BWA; Li and Durbin, 2009) using the reference human genome build hg19 from UCSC. Samtools (Li et al., 2009) was used to convert BAM alignment files to SAM format and then sort and index them. INDEL realignment was performed using IndelRealigner and base quality score recalibration was performed using BaseRecalibrator from GATK (McKenna et al., 2010). PCR and optical duplicates were removed from samples using Picard (http://picard.sourceforge.net). Variant calling was done on the targeted regions (Table S1) by combining all the samples using the GATK pipeline and following their best practices protocol. Further filtering was applied to keep only the variants that passed the quality control filters and 95% genotype call rate. Sequencing data (BAM files) can be accessed via http://paschou-lab.mbg.duth.gr/share_data.html.
Variant Ranker Analysis
Variants were then annotated using ANNOVAR (Wang et al., 2010) and ranked using Variant Ranker tool (http://paschou-lab.mbg.duth.gr/Software.html). Variant ranker aggregates annotation information based on allelic frequency scores, conservation scores, prediction algorithm scores, and clinical information. Using Variant Ranker, the variants are prioritized on the basis of their effect, novelty, and annotation information. A variant was designated as novel if not present in database of human variation dbSNP build 138 and had a minor allele frequency (MAF) ≤ 0.01 in 1000 Genomes, Exome Aggregation Consortium (ExAC) and National Heart, Lung, and Blood Institute Exome Sequencing Project (NHLBI ESP6500) database.
VAAST Analysis
Scoring and prioritization analysis of deleterious alleles was performed using VAAST 2.0 (Hu et al., 2013). We applied the likelihood ratio test in VAAST using both dominant and recessive disease models of inheritance, with the help of a background file comprising of 189 genomes from 1000 Genomes project (Abecasis et al., 2012), 184 Danish exomes (Li et al., 2010), 10Gen genome dataset (Reese et al., 2010), and 40 whole genomes from the Complete Genomics Diversity Panel.
Identification of Linkage Disequilibrium (LD) SNPs
We used the web-based application LDproxy (Machiela and Chanock, 2015) to identify proxy LD SNPs and LdOOKUP (http://purces04.u.hpc.mssm.edu/ldookup/ldookup.cgi) to browse for variants in LD with other GWAS studies from the Psychiatric Genomics Consortium, brain eQTL data and the NHGRI GWAS catalog (Welter et al., 2014).
Sanger Sequencing
PCR primers were designed using PRIMER3 (http://primer3plus.com) and confirmed to have unique genomic product by UCSC in-silico PCR (http://genome.ucsc.edu/cgi-bin/hgPcr), targeting amplicons of sizes between 300 and 400 bp. PCR products were purified using QIAquick PCR purification kit from Qiagen (QIAGEN, CA), followed by Sanger sequencing on an ABI 3730xl sequencer in Genscript (NJ, USA), using the BigDye Terminator v.3.1 chemistry.
Results
Identification of Variants
We explored whether coding variations in four genes, namely, HDC, SLITRK1, BTBD9 and SLC6A4 play a role in TS susceptibility. We utilized next-generation sequencing technology to perform targeted sequencing on a total of 60 kilobases, including the entire HDC and SLITRK1 genes, and spanning 15 exons of SLC6A4 and 11 exons of BTBD9 genes, across 382 TS cases of Caucasian origin (Table S1). Regarding data quality, more than 95% of bases were sequenced with >99.9% accuracy, covering most amplicons at >100X.
We obtained 336 single nucleotide polymorphisms (SNPs) with 31 exonic SNPs (see summary statistics on Table 1). We focused only on functionally important non-synonymous exonic variants and pursued validation via Sanger sequencing. We found a novel rare (MAF = 0.13%) variant in HDC residing in exon 12 (HDC:NM_002112:exon12:c.A1564C:p.I522L), two rare (MAF = 0.13%) novel SLITRK1 variants, at positions chr13:g.84454582 (SLITRK1:NM_052910 :exon1:c.G1061C:p.G354A). and chr13:g.84454485 (SLITRK1:NM_052910:exon1:c.G115 8T:p.K386N), residing in exon 1 of the gene, and two known rare variants, rs146746846 (MAF = 0.13%) (SLITRK1:NM_052910:exon1:c.C892T:p.H298Y) and rs150504822 (MAF = 0.26%; SLITRK1:NM_052910:exon1:c.A1252T:p.T418S), also in exon 1 (Table 2). The five confirmed variants were ranked within the top six potentially etiologically relevant variants by our Variant Ranker algorithm and were all predicted to be deleterious by MutationTaster functional prediction algorithm (Schwarz et al., 2010; Table S2). One confirmed SLITRK1 variant (rs146746846) was also identified by VAAST under a recessive model of inheritance (Table S3). Proxy LD SNPs available for rs150504822 on SLITRK1 were input into LdOOKUP to identify significant variants in other GWAS studies. A variant in LD with rs150504822, namely rs6563353, was identified (Tables S4, S5); rs6563353 has been positively associated (p = 2.06e-6) with increased citalopram-induced general side effect burden, in patients with depression (Adkins et al., 2012). Citalopram is an antidepressant drug of the selective serotonin reuptake inhibitor class, with an off-label use for OCD treatment.
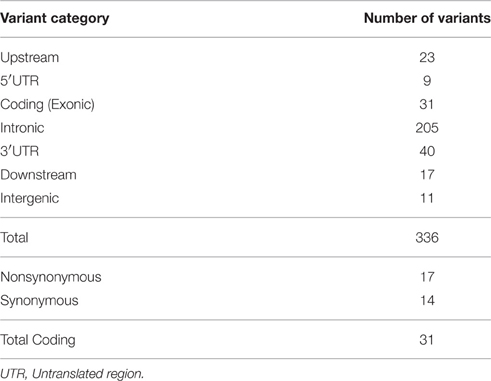
Table 1. Summary of functional annotation of identified variants.
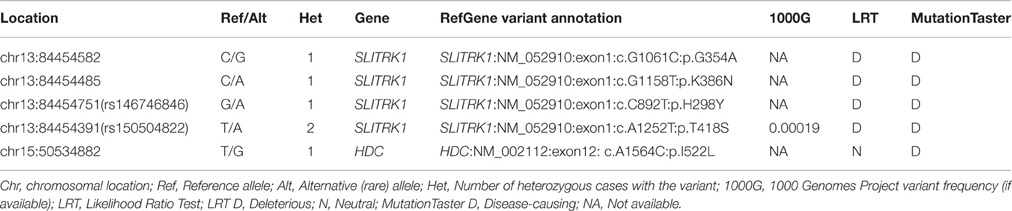
Table 2. Nonsynonymous variants confirmed by Sanger sequencing.
Discussion
To our knowledge, this is the first study applying next generation sequencing technology in quest for genetic susceptibility variants shaping the genomic landscape of TS. It is expected that at least part of the missing heritability of complex disorders, such as TS, will be attributable to low-frequency variants with intermediate penetrance effects (Manolio et al., 2009) and, if truly causative, they could account for a small proportion of TS cases.
In the present study, we identified and confirmed a rare novel HDC coding variant as well as two rare novel and two rare known SLITRK1 coding variants, all confirmed by Sanger sequencing (Table 2). We did not extensively cover the regulatory regions of the genes; thus, regulatory variants may have been missed resulting in under-estimation of the contribution of these genes to TS risk.
The recent implication of the L-histidine decarboxylase (HDC) gene in TS etiology has raised the intriguing hypothesis of the involvement of histaminergic neural pathways in the onset of the disorder. Ercan-Sencicek et al. studied a family with a father and all eight of his children affected with TS, and found a rare premature termination codon mutation (p.W317X) in HDC, also absent in 3,000 control individuals. Due to the demonstrated dominant-negative mode of function of the mutant HDC enzyme, the authors concluded that histaminergic neurotransmission is most likely diminished in their patients (Ercan-Sencicek et al., 2010). However, HDC coding variants seem to be extremely rare since apart from the mutation identified in the original pedigree, HDC has not been found altered in other TS cases of Caucasian or Asian origin (Ercan-Sencicek et al., 2010; Lei et al., 2012). Interestingly, a preceding genome-wide study of 95 French Canadian trios with familial history of TS (Riviere et al., 2010) had also shown association to a microsatellite marker on chromosome 15 (D15S1016), lying within the same interval found to be linked with TS in the original study that implicated HDC in TS etiology. Finally, a recent genome-wide scan for de novo or transmitted rare copy number variants in TS found enrichment of genes within the histamine receptor signaling pathways (Fernandez et al., 2012). Even though in the present study we did not detect the rare variants previously reported in the literature, we did find a novel exonic nonsynonymous variant, chr15:g.50534882 (c.A1564C:p.I522L), in one TS case.
Histamine plays a central role in gastric acid secretion, innate, and acquired immunity and immunomodulation, bronchoconstriction, vasodilation and neurotransmission. The neuronal histaminergic system is involved in a number of basic physiological functions such as circadian rhythmicity, energy metabolism, neuro-endocrine homeostasis, stress, sensory, and motor functions, cognition, attention, learning, and memory. Histidine decarboxylase (HDC) is the key enzyme for histamine production from histidine and in the brain its mRNA is expressed exclusively in the posterior hypothalamus (Haas et al., 2008). So far, the histaminergic system has not received as much attention as other monoaminergic systems of the brain. Classically established as a “peripherally” important mediator of inflammation, the importance of histamine in neurotransmission, and its role in neuropsychiatric disorders is only recently starting to become appreciated (Tiligada et al., 2011) and deserves further investigation also in the context of TS. Interestingly, Hdc−∕− deficient mice have several traits relevant to features of TS and have shown decreased brain histamine and increased sensitivity to stereotypic behaviors (i.e., hyperlocomotion) upon administration of dopamine agonists (Kubota et al., 2002) and repetitive grooming after induced fear (Xu et al., 2015). Such stimulant-induced movements, including rearing, sniffing, and biting have previously been proposed as a model of human tics (Saka and Graybiel, 2003; Castellan Baldan et al., 2014).
SLITRK1 has been extensively studied since its identification as a much promising TS candidate gene. However, with the exception of the variants reported by Abelson et al. (2005), additional novel non-synonymous mutations have not been identified in a large number of subsequent studies interrogating patient cohorts (Deng et al., 2006; Chou et al., 2007; Scharf et al., 2008; Zimprich et al., 2008) or single families (Robertson and Orth, 2006; Verkerk et al., 2006; Fabbrini et al., 2007; Pasquini et al., 2008). On the other hand, genetic association studies and haplotype analyses could not rule out the implication of SLITRK1 in TS etiology either via the existence of risk factors in LD with SLITRK1 (Miranda et al., 2009) or as of yet unidentified SLITRK1 regulatory variants (Karagiannidis et al., 2012). In our study, we did not detect the SLITRK1 variants that had been previously reported in association to TS but we report on the identification of two novel variants, chr13:g.84454582 and chr13:g.84454485, each found in one TS patient. We also identified two rare non-synonymous SLITRK1 variants (rs150504822 and rs146746846, identified in two TS cases and one TS case, respectively). Interestingly, rs150504822 was found in LD with rs6563353, a variant that has been previously associated with citalopram-induced general side-effect burden in patients with depression (Adkins et al., 2012). Citalopram, is a well-tolerated acute treatment drug conferring positive response in children and adolescents with OCD (Thomsen et al., 2001; Mukaddes et al., 2003).
Interestingly, SLITRK1 encodes a transmembrane protein necessary for dendritic growth, axon guidance and branching of neuronal cells. It is expressed in both the embryonic and postnatal brain (cortex, thalamus, and basal ganglia), reflecting neuroanatomical regions most commonly affected in TS (Proenca et al., 2011), as well as in mature neurons with large axons (Aruga and Mikoshiba, 2003). Slitrk1−∕− mice exhibited elevated anxiety and depression-like behaviour, consistent with TS symptoms, in which the anxiety-like behaviour was attenuated upon clonidine administration, a drug often used in TS treatment (Katayama et al., 2010).
Our previous and present studies suggest that SLITRK1 and HDC may have an under-appreciated role in TS aetiology. Unarguably, the functional role of the identified variants warrants further investigation by a wide array of experiments. Next-generation sequencing technology is rapidly becoming a routine, albeit still costly approach to target patient genomes that is expected to significantly expedite the quest for rare variants with reduced penetrance, not only in TS susceptibility but also other complex neurodevelopmental and neuropsychiatric disorders.
Author Contributions
JA, HP, JX, LD, and IK performed experimental procedures, data analysis and interpretation, and participated in manuscript writing. FT and PD participated in data analysis and interpretation, and in manuscript writing. ZT, RR, TW, LF, PN, US, CA, VT, AK, CB, PS, and CLB performed patient sample recruitment and ascertainment, and participated in data analysis and interpretation. JT, PP, GH, and MG designed the study, supervised experimental procedures, performed data analysis and interpretation, and wrote the manuscript. All authors read and approved the final version to be published and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to the TS individuals and their families for accepting to participate in the studies of the genetic basis of TS. This study was made possible thanks to the collaborative efforts of Tourette Syndrome Genetics—Southern and Eastern Europe Initiative (TSGeneSEE) and COST Action BM905: European Network for the Study of GTS (EUNETGTS). This project was financed by FP7-PEOPLE-2012-ITN, project: TS-EUROTRAIN, grant number 316978, and grants from the National Institute of Mental Health [R01MH092293; U24MH068457] and the Human Genetics Institute of New Jersey. The collection of Canadian families for this study was supported by grants from The Tourette Syndrome Association of America, N.I.H. grant MS40024-01, the Ontario Mental Health Foundation, and The Tourette Syndrome Foundation of Canada. CB was supported by the Merit-prize scholarship of Semmelweis University and the János Bolyai Research Scholarship of the Hungarian Academy of Sciences BO/00987/16/5.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2016.00428
References
Abecasis, G. R., Auton, A., Brooks, L. D., DePristo, M. A., Durbin, R. M., Handsaker, R. E., et al. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. doi: 10.1038/nature11632
Abelson, J. F., Kwan, K. Y., O'Roak, B. J., Baek, D. Y., Stillman, A. A., Morgan, T. M., et al. (2005). Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science 310, 317–320. doi: 10.1126/science.1116502
Adkins, D. E., Clark, S. L., Åberg, K., Hettema, J. M., Bukszár, J., McClay, J. L., et al. (2012). Genome-wide pharmacogenomic study of citalopram-induced side effects in STAR+D. Transl. Psychiatry 2, e129. doi: 10.1038/tp.2012.57
Allen, R. P., Walters, A. S., Montplaisir, J., Hening, W., Myers, A., Bell, T. J., et al. (2005). Restless legs syndrome prevalence and impact: REST general population study. Arch. Intern. Med. 165, 1286–1292. doi: 10.1001/archinte.165.11.1286
Aruga, J., and Mikoshiba, K. (2003). Identification and characterization of Slitrk, a novel neuronal transmembrane protein family controlling neurite outgrowth. Mol. Cell. Neurosci. 24, 117–129. doi: 10.1016/S1044-7431(03)00129-5
Castellan Baldan, L., Williams, K. A., Gallezot, J.-D., Pogorelov, V., Rapanelli, M., Crowley, M., et al. (2014). Histidine decarboxylase deficiency causes tourette syndrome: parallel findings in humans and mice. Neuron 81, 77–90. doi: 10.1016/j.neuron.2013.10.052
Cath, D. C., Hedderly, T., Ludolph, A. G., Stern, J. S., Murphy, T., Hartmann, A., et al. (2011). European clinical guidelines for Tourette syndrome and other tic disorders. Eur. Child Adolesc. Psychiatry 20, 155–171. doi: 10.1007/s00787-011-0164-6
Chou, I.-C., Wan, L., Liu, S.-C., Tsai, C.-H., and Tsai, F.-J. (2007). Association of the Slit and Trk-like 1 gene in Taiwanese patients with Tourette syndrome. Pediatr. Neurol. 37, 404–406. doi: 10.1016/j.pediatrneurol.2007.06.017
Deng, H., Le, W. D., Xie, W. J., and Jankovic, J. (2006). Examination of the SLITRK1 gene in Caucasian patients with Tourette syndrome. Acta Neurol. Scand. 114, 400–402. doi: 10.1111/j.1600-0404.2006.00706.x
Ercan-Sencicek, A. G., Stillman, A. A., Ghosh, A. K., Bilguvar, K., O'Roak, B. J., Mason, C. E., et al. (2010). L-histidine decarboxylase and Tourette's syndrome. N. Engl. J. Med. 362, 1901–1908. doi: 10.1056/NEJMoa0907006
Fabbrini, G., Pasquini, M., Aurilia, C., Berardelli, I., Breedveld, G., Oostra, B. A., et al. (2007). A large Italian family with Gilles de la Tourette syndrome: clinical study and analysis of the SLITRK1 gene. Mov. Disord. 22, 2229–2234. doi: 10.1002/mds.21697
Fernandez, T. V., Sanders, S. J., Yurkiewicz, I. R., Ercan-Sencicek, A. G., Kim, Y.-S., Fishman, D. O., et al. (2012). Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol. Psychiatry 71, 392–402. doi: 10.1016/j.biopsych.2011.09.034
Guo, Y., Su, L., Zhang, J., Lei, J., Deng, X., Xu, H., et al. (2012). Analysis of the BTBD9 and HTR2C variants in Chinese Han patients with Tourette syndrome. Psychiatr. Genet. 22, 300–303. doi: 10.1097/YPG.0b013e32835862b1
Haas, H. L., Sergeeva, O. A., and Selbach, O. (2008). Histamine in the nervous system. Physiol Rev. 88, 1183–1241. doi: 10.1152/physrev.00043.2007
Hoekstra, P. J., Dietrich, A., Edwards, M. J., Elamin, I., and Martino, D. (2013). Environmental factors in Tourette syndrome. Neurosci. Biobehav. Rev. 37, 1040–1049. doi: 10.1016/j.neubiorev.2012.10.010
Hu, H., Huff, C. D., Moore, B., Flygare, S., Reese, M. G., and Yandell, M. (2013). VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet. Epidemiol. 37, 622–634. doi: 10.1002/gepi.21743
Hu, X.-Z., Lipsky, R. H., Zhu, G., Akhtar, L. A., Taubman, J., Greenberg, B. D., et al. (2006). Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am. J. Hum. Genet. 78, 815–826. doi: 10.1086/503850
Kalanithi, P. S., Zheng, W., Kataoka, Y., DiFiglia, M., Grantz, H., Saper, C. B., et al. (2005). Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 13307–13312. doi: 10.1073/pnas.0502624102
Karagiannidis, I., Dehning, S., Sandor, P., Tarnok, Z., Rizzo, R., Wolanczyk, T., et al. (2013). Support of the histaminergic hypothesis in Tourette syndrome: association of the histamine decarboxylase gene in a large sample of families. J. Med. Genet. 50, 760–764. doi: 10.1136/jmedgenet-2013-101637
Karagiannidis, I., Rizzo, R., Tarnok, Z., Wolanczyk, T., Hebebrand, J., Nöthen, M. M., et al. (2012). Replication of association between a SLITRK1 haplotype and Tourette Syndrome in a large sample of families. Mol. Psychiatry 17, 665–668. doi: 10.1038/mp.2011.151
Katayama, K., Yamada, K., Ornthanalai, V. G., Inoue, T., Ota, M., Murphy, N. P., et al. (2010). Slitrk1-deficient mice display elevated anxiety-like behavior and noradrenergic abnormalities. Mol. Psychiatry 15, 177–184. doi: 10.1038/mp.2008.97
Keen-Kim, D., Mathews, C. A., Reus, V. I., Lowe, T. L., Herrera, L. D., Budman, C. L., et al. (2006). Overrepresentation of rare variants in a specific ethnic group may confuse interpretation of association analyses. Hum. Mol. Genet. 15, 3324–3328. doi: 10.1093/hmg/ddl408
Kilic, F., Murphy, D. L., and Rudnick, G. (2003). A human serotonin transporter mutation causes constitutive activation of transport activity. Mol. Pharmacol. 64, 440–446. doi: 10.1124/mol.64.2.440
Kubota, Y., Ito, C., Sakurai, E., Sakurai, E., Watanabe, T., and Ohtsu, H. (2002). Increased methamphetamine-induced locomotor activity and behavioral sensitization in histamine-deficient mice. J. Neurochem. 83, 837–845. doi: 10.1046/j.1471-4159.2002.01189.x
Lei, J., Deng, X., Zhang, J., Su, L., Xu, H., Liang, H., et al. (2012). Mutation screening of the HDC gene in Chinese Han patients with Tourette syndrome. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 159B, 72–76. doi: 10.1002/ajmg.b.32003
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, Y., Vinckenbosch, N., Tian, G., Huerta-Sanchez, E., Jiang, T., Jiang, H., et al. (2010). Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 42, 969–972. doi: 10.1038/ng.680
Lodato, M. A., Woodworth, M. B., Lee, S., Evrony, G. D., Mehta, B. K., Karger, A., et al. (2015). Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98. doi: 10.1126/science.aab1785
Machiela, M. J., and Chanock, S. J. (2015). LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557. doi: 10.1093/bioinformatics/btv402
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Mathews, C. A., and Grados, M. A. (2011). Familiality of Tourette syndrome, obsessive-compulsive disorder, and attention-deficit/hyperactivity disorder: heritability analysis in a large sib-pair sample. J. Am. Acad. Child Adolesc. Psychiatry 50, 46–54. doi: 10.1016/j.jaac.2010.10.004
Mathews, C. A., Scharf, J. M., Miller, L. L., MacDonald-Wallis, C., Lawlor, D. A., and Ben-Shlomo, Y. (2014). Association between pre- and perinatal exposures and Tourette syndrome or chronic tic disorder in the ALSPAC cohort. Br. J. Psychiatry 204, 40–45. doi: 10.1192/bjp.bp.112.125468
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Miranda, D. M., Wigg, K., Kabia, E. M., Feng, Y., Sandor, P., and Barr, C. L. (2009). Association of SLITRK1 to Gilles de la Tourette Syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 483–486. doi: 10.1002/ajmg.b.30840
Moya, P. R., Wendland, J. R., Rubenstein, L. M., Timpano, K. R., Heiman, G. A., Tischfield, J. A., et al. (2013). Common and rare alleles of the serotonin transporter gene, SLC6A4, associated with Tourette's disorder. Mov. Disord. 28, 1263–1270. doi: 10.1002/mds.25460
Mukaddes, N. M., Abali, O., and Kaynak, N. (2003). Citalopram treatment of children and adolescents with obsessive-compulsive disorder: a preliminary report. Psychiatry Clin. Neurosci. 57, 405–408. doi: 10.1046/j.1440-1819.2003.01139.x
Muller-Vahl, K. R., Meyer, G. J., Knapp, W. H., Emrich, H. M., Gielow, P., Brucke, T., et al. (2005). Serotonin transporter binding in Tourette Syndrome. Neurosci. Lett. 385, 120–125. doi: 10.1016/j.neulet.2005.05.031
O'Roak, B. J., Morgan, T. M., Fishman, D. O., Saus, E., Alonso, P., Gratacos, M., et al. (2010). Additional support for the association of SLITRK1 var321 and Tourette syndrome. Mol. Psychiatry 15, 447–450. doi: 10.1038/mp.2009.105
Pasquini, M., Fabbrini, G., Berardelli, I., Bonifati, V., Biondi, M., and Berardelli, A. (2008). Psychopathological features of obsessive-compulsive disorder in an Italian family with Gilles de la Tourette syndrome not linked to the SLITRK1 gene. Psychiatry Res. 161, 109–111. doi: 10.1016/j.psychres.2008.02.012
Pauls, D. L., Fernandez, T. V., Mathews, C. A., State, M. W., and Scharf, J. M. (2014). The Inheritance of Tourette Disorder: a review. J. Obsessive Compuls. Relat. Disord. 3, 380–385. doi: 10.1016/j.jocrd.2014.06.003
Pauls, D. L., Raymond, C. L., Stevenson, J. M., and Leckman, J. F. (1991). A family study of Gilles de la Tourette syndrome. Am. J. Hum. Genet. 48, 154–163.
Peterson, B. S., Thomas, P., Kane, M. J., Scahill, L., Zhang, H., Bronen, R., et al. (2003). Basal Ganglia volumes in patients with Gilles de la Tourette syndrome. Arch. Gen. Psychiatry 60, 415–424. doi: 10.1001/archpsyc.60.4.415
Price, R. A., Kidd, K. K., Cohen, D. J., Pauls, D. L., and Leckman, J. F. (1985). A twin study of Tourette syndrome. Arch. Gen. Psychiatry 42, 815–820. doi: 10.1001/archpsyc.1985.01790310077011
Proenca, C. C., Gao, K. P., Shmelkov, S. V., Rafii, S., and Lee, F. S. (2011). Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends Neurosci. 34, 143–153. doi: 10.1016/j.tins.2011.01.001
Reese, M. G., Moore, B., Batchelor, C., Salas, F., Cunningham, F., Marth, G. T., et al. (2010). A standard variation file format for human genome sequences. Genome Biol. 11:R88. doi: 10.1186/gb-2010-11-8-r88
Riviere, J.-B., St-Onge, J., Gaspar, C., Diab, S., Dion, Y., Lesperance, P., et al. (2010). Genome-wide TDT analysis in French-Canadian families with Tourette syndrome. Can. J. Neurol. Sci. 37, 110–112. doi: 10.1017/S0317167100009744
Riviere, J.-B., Xiong, L., Levchenko, A., St-Onge, J., Gaspar, C., Dion, Y., et al. (2009). Association of intronic variants of the BTBD9 gene with Tourette syndrome. Arch. Neurol. 66, 1267–1272. doi: 10.1001/archneurol.2009.213
Robertson, M. M., and Orth, M. (2006). Behavioral and affective disorders in Tourette syndrome. Adv. Neurol. 99, 39–60.
Saka, E., and Graybiel, A. M. (2003). Pathophysiology of Tourette's syndrome: striatal pathways revisited. Brain Dev. 25(Suppl. 1), S15–S19. doi: 10.1016/S0387-7604(03)90002-7
Scharf, J. M., Moorjani, P., Fagerness, J., Platko, J. V., Illmann, C., Galloway, B., et al. (2008). Lack of association between SLITRK1var321 and Tourette syndrome in a large family-based sample. Neurology 70, 1495–1496. doi: 10.1212/01.wnl.0000296833.25484.bb
Schwarz, J. M., Rodelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
Thomsen, P. H., Ebbesen, C., and Persson, C. (2001). Long-term experience with citalopram in the treatment of adolescent OCD. J. Am. Acad. Child Adolesc. Psychiatry 40, 895–902. doi: 10.1097/00004583-200108000-00010
Tiligada, E., Kyriakidis, K., Chazot, P. L., and Passani, M. B. (2011). Histamine pharmacology and new CNS drug targets. CNS Neurosci. Ther. 17, 620–628. doi: 10.1111/j.1755-5949.2010.00212.x
TSAICG (2007). Genome scan for Tourette disorder in affected-sibling-pair and multigenerational families. Am. J. Hum. Genet. 80, 265–272. doi: 10.1086/511052
Turjanski, N., Lees, A. J., and Brooks, D. J. (1999). Striatal dopaminergic function in restless legs syndrome: 18F-dopa and 11C-raclopride PET studies. Neurology 52, 932–937. doi: 10.1212/WNL.52.5.932
Verkerk, A. J., Cath, D. C., van der Linde, H. C., Both, J., Heutink, P., Breedveld, G., et al. (2006). Genetic and clinical analysis of a large Dutch Gilles de la Tourette family. Mol. Psychiatry 11, 954–964. doi: 10.1038/sj.mp.4001877
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi: 10.1093/nar/gkq603
Welter, D., MacArthur, J., Morales, J., Burdett, T., Hall, P., Junkins, H., et al. (2014). The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006. doi: 10.1093/nar/gkt1229
Wendland, J. R., DeGuzman, T. B., McMahon, F., Rudnick, G., Detera-Wadleigh, S. D., and Murphy, D. L. (2008). SERT Ileu425Val in autism, Asperger syndrome and obsessive-compulsive disorder. Psychiatr. Genet. 18, 31–39. doi: 10.1097/YPG.0b013e3282f08a06
Wendland, J. R., Kruse, M. R., and Murphy, D. L. (2006). Functional SLITRK1 var321, varCDfs and SLC6A4 G56A variants and susceptibility to obsessive-compulsive disorder. Mol. Psychiatry 11, 802–804. doi: 10.1038/sj.mp.4001848
Winkelmann, J., Schormair, B., Lichtner, P., Ripke, S., Xiong, L., Jalilzadeh, S., et al. (2007). Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat. Genet. 39, 1000–1006. doi: 10.1038/ng2099
Xu, M., Li, L., Ohtsu, H., and Pittenger, C. (2015). Histidine decarboxylase knockout mice, a genetic model of Tourette syndrome, show repetitive grooming after induced fear. Neurosci. Lett. 595, 50–53. doi: 10.1016/j.neulet.2015.03.067
Yu, D., Mathews, C. A., Scharf, J. M., Neale, B. M., Davis, L. K., Gamazon, E. R., et al. (2015). Cross-disorder genome-wide analyses suggest a complex genetic relationship between Tourette's syndrome and OCD. Am. J. Psychiatry 172, 82–93. doi: 10.1176/appi.ajp.2014.13101306
Keywords: next generation sequencing, targeted re-sequencing, genetic susceptibility, rare variants, TS candidate genes, HDC, SLITRK1
Citation: Alexander J, Potamianou H, Xing J, Deng L, Karagiannidis I, Tsetsos F, Drineas P, Tarnok Z, Rizzo R, Wolanczyk T, Farkas L, Nagy P, Szymanska U, Androutsos C, Tsironi V, Koumoula A, Barta C, TSGeneSEE, Sandor P, Barr CL, Tischfield J, Paschou P, Heiman GA and Georgitsi M (2016) Targeted Re-Sequencing Approach of Candidate Genes Implicates Rare Potentially Functional Variants in Tourette Syndrome Etiology. Front. Neurosci. 10:428. doi: 10.3389/fnins.2016.00428
Received: 31 March 2016; Accepted: 02 September 2016;
Published: 21 September 2016.
Edited by:
Ahmet O. Caglayan, Yale University, USAReviewed by:
Munis Dundar, Erciyes University, TurkeyRomina D'Aurizio, National Research Council (CNR), Italy
Copyright © 2016 Alexander, Potamianou, Xing, Deng, Karagiannidis, Tsetsos, Drineas, Tarnok, Rizzo, Wolanczyk, Farkas, Nagy, Szymanska, Androutsos, Tsironi, Koumoula, Barta, TSGeneSEE, Sandor, Barr, Tischfield, Paschou, Heiman and Georgitsi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary A. Heiman, heiman@dls.rutgers.edu
Marianthi Georgitsi, margeorgitsi@auth.gr
†These authors have contributed equally to this work.