 Elena A. Kosenko1*
Elena A. Kosenko1* Lyudmila A. Tikhonova1
Lyudmila A. Tikhonova1 Carmina Montoliu2
Carmina Montoliu2 George E. Barreto3,4
George E. Barreto3,4 Gjumrakch Aliev5*
Gjumrakch Aliev5* Yury G. Kaminsky1
Yury G. Kaminsky1- 1Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Pushchino, Russia
- 2Fundación Investigación Hospital Clínico, INCLIVA Instituto Investigación Sanitaria, Valencia, Spain
- 3Departamento de Nutrición y Bioquímica, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá, Colombia
- 4Instituto de Ciencias Biomédicas, Universidad Autónoma de Chile, Santiago, Chile
- 5GALLY International Biomedical Research Institute Inc., San Antonio, TX, United States
Alzheimer's disease (AD) is a slowly progressive, neurodegenerative disorder of uncertain etiology. According to the amyloid cascade hypothesis, accumulation of non-soluble amyloid β peptides (Aβ) in the Central Nervous System (CNS) is the primary cause initiating a pathogenic cascade leading to the complex multilayered pathology and clinical manifestation of the disease. It is, therefore, not surprising that the search for mechanisms underlying cognitive changes observed in AD has focused exclusively on the brain and Aβ-inducing synaptic and dendritic loss, oxidative stress, and neuronal death. However, since Aβ depositions were found in normal non-demented elderly people and in many other pathological conditions, the amyloid cascade hypothesis was modified to claim that intraneuronal accumulation of soluble Aβ oligomers, rather than monomer or insoluble amyloid fibrils, is the first step of a fatal cascade in AD. Since a characteristic reduction of cerebral perfusion and energy metabolism occurs in patients with AD it is suggested that capillary distortions commonly found in AD brain elicit hemodynamic changes that alter the delivery and transport of essential nutrients, particularly glucose and oxygen to neuronal and glial cells. Another important factor in tissue oxygenation is the ability of erythrocytes (red blood cells, RBC) to transport and deliver oxygen to tissues, which are first of all dependent on the RBC antioxidant and energy metabolism, which finally regulates the oxygen affinity of hemoglobin. In the present review, we consider the possibility that metabolic and antioxidant defense alterations in the circulating erythrocyte population can influence oxygen delivery to the brain, and that these changes might be a primary mechanism triggering the glucose metabolism disturbance resulting in neurobiological changes observed in the AD brain, possibly related to impaired cognitive function. We also discuss the possibility of using erythrocyte biochemical aberrations as potential tools that will help identify a risk factor for AD.
Introduction
Alzheimer's disease (AD) is a slowly progressing, systemic neurodegenerative disorder of uncertain etiology. Clinical manifestation of this disorder usually consists of cognitive deficits in memory in the elderly. Some estimates suggest that 50% of the population over the age of 80 years suffers from this type of dementia. With increases in life expectancy of our population, AD is already approaching epidemic proportions with no cure or preventative therapy yet available. Now, AD affects ~24 million people worldwide with 4.6 million new cases of dementia every year (one new case every 7 s), and if existing trends continue, 115 million individuals worldwide will have Alzheimer's disease (AD) by 2050 (Wimo and Prince, 2010; Fita et al., 2011).
AD develops sporadically in 95–98% of the AD population (Bird, 2005; Reddy, 2006; Kaminsky et al., 2010). However, the genetic-linked cases have provided a great deal of biochemical insights in the disease process. The research field has been focused on the role of Aβ in the brain stemming from the fact that accumulation of these peptides results in aggregation and formation of insoluble plaques, which trigger a cascade of deleterious changes, leading to neuronal death and thus causing AD. This train of events has been called the amyloid-cascade hypothesis of AD (Hardy and Higgins, 1992). It is significant that accumulation of aggregated Aβ is the primary abnormality in AD and that its deposition is required for postmortem diagnosis. Now, however, a large body of evidence exists, and new data continues to accumulate indicating that the number of Aβ deposits in the brain does not correlate well with the degree of cognitive impairment (Braak and Braak, 1991; Terry et al., 1991; Giannakopoulos et al., 2003; Guillozet et al., 2003). Indeed, Aβ deposition may occur in normal non-demented elderly people (Joachim et al., 1989; Mann et al., 1992; Lue et al., 1999; Schmitt et al., 2000; Pike et al., 2007), that is in agree with the fact that virtually all humans start to accumulate Aβ in the brain upon aging (Funato et al., 1998; Wang et al., 1999; Morishima-Kawashima et al., 2000). Besides, amyloid plaques are not specific for Alzheimer's disease and have been found in many other pathological conditions, including transmissible spongiform encephalopathies (Liberski, 2004), Down's syndrome (Glenner and Wong, 1984), Lewy body in Parkinson's disease (Arai et al., 1992), acute traumatic brain injury with diffuse axonal damage (Smith et al., 2003) and chronic traumatic brain injury associated with boxing (Roberts et al., 1990; Jordan, 2000) and football (Omalu et al., 2005). What is clear from these studies is that the presence of brain plaques alone is insufficient to produce cognitive decline in AD (Jack et al., 2009) and that such studies support the basis for the formation of a new hypothesis.
Recently, a modified Aβ-cascade hypothesis has been formulated that predicts intraneuronal accumulation of soluble Aβ oligomers, but not monomer or insoluble amyloid fibrils, as the first step of a fatal cascade in AD (McLean et al., 1999; Wirths et al., 2004; Selkoe, 2007). The amyloid oligomerization is observed to occur intracellularly (Connolly and Volpe, 1990) and Aβ1−42 oligomers turn out to be potent neurotoxins in animal brain and neuronal cultures where they are able to disrupt glutamatergic synaptic function (Lambert et al., 1998; Hsia et al., 1999; Klein et al., 2001; Hardy and Selkoe, 2002; Kamenetz et al., 2003; Walsh and Selkoe, 2004; Roselli et al., 2005) and neuronal calcium homeostasis (Bapat et al., 1983; Mattson et al., 1992; Demuro et al., 2005), promote abnormal release of glutamate in hippocampal neurons (Brito-Moreira et al., 2011), induce oxidative stress (De Felice et al., 2007), incite tau hyperphosphorylation (De Felice et al., 2008), and synapse loss (Lue et al., 1999; Selkoe, 2008), inhibit long-term potentiation in the hippocampus (Walsh et al., 2002), which is required for memory formation, and in turn leads to the cognitive deficits in the animal. Using oligomer-sensitive immunoassay, the soluble Aβ oligomers have been found in brains of AD patients (Kuo et al., 1996; Lue et al., 1999; Gong et al., 2003). This confirms the prediction that soluble oligomeric Aβ-forms are characteristic of AD pathology. However, the soluble Aβ burden displayed considerable individual variation in the brain of AD patients. Thus, the mean level of soluble Aβ can increase 3-fold (McLean et al., 1999), 6-fold, 12-fold (Kuo et al., 1996), and 70-fold (Gong et al., 2003) in brain of AD patients compared to age-matched control, at that, the majority of soluble peptides was Aβ1−42 (Kuo et al., 1996). On the other hand, it was found that the levels of soluble Aβ1−42 were smallest in the AD brain (0.7%) and that the soluble pools of Aβ1−40 and Aβ1−42 were the largest fractions of total Aβ in the normal brain (50 and 23% respectively, Wang et al., 1999). Other authors also showed that the Aβ1−42 levels were found in the brains of normal elderly subjects (Tabaton and Piccini, 2005) and that in subjects with AD these concentrations increased slightly compared with the age-matched control (Lue et al., 1999). These studies suggest that within individual AD subjects, the areas with greater numbers of soluble Aβ oligomers did not, as a rule, and whether the levels of these “concentration-jumping” oligomers correlate with the memory decline in AD remains to be determined. Indeed, previous studies have shown that these Aβ forms were observed in the brains of patients with Down's syndrome (DS) (Teller et al., 1996; Gyure et al., 2001; Tabaton and Gambetti, 2006) indicating that the accumulation of soluble oligomers are not specific for AD. Moreover, in brains of patients with DS, increased levels of oxidative damage occur prior to the onset of Aβ deposition (Nunomura et al., 2000). Hence, the formation of diffuse amyloid plaques may be considered as the message talking about the disruption of brain homeostasis or as a compensatory response to remove reactive oxygen species (Atwood et al., 2003). Thus, these facts provide the opportunity to investigate the pathological conditions that precede the formation of the Aβ deposits in the human brain.
It is well known that a characteristic reduction of cerebral perfusion and metabolism occurs in patients with AD (de la Torre, 2000b; Aliev et al., 2003a). It was suggested that capillary distortions commonly found in the AD brain elicit hemorheological changes that altered the delivery and transport of essential nutrients, particularly glucose, and oxygen required for its aerobic oxidation in brain cells (de la Torre and Mussivand, 1993; de la Torre, 2002a; Chang et al., 2007; Aliev, 2011) resulting in an energy metabolic breakdown of the biosynthetic and synaptic pathways, subsequently leading to the death of neurons as a consequence of cognitive deterioration. In fact, it was proposed that AD may originate as a vascular disorder with the resultant impairment of oxygen delivery to the brain with the plaques and tangles found in the brain secondary to the effects of the vascular pathology (de la Torre, 2002a). Another important factor in tissue oxygenation is the ability of red blood cells (RBC) to the binding, transport and delivery of oxygen to tissues that depends, first of all, on RBC energy metabolism and antioxidant status (Brewer et al., 1974) that is extremely important for the functioning and regulation of oxygen affinity to hemoglobin (van Wijk and van Solinge, 2005).
Surprisingly, despite the main role of RBC metabolism in the delivery of oxygen to the tissues, no systematic programs of research have examined the relationship between the breach of the energy metabolism of these cells in destabilization of glucose metabolism in the brain pathology and this relationship is still not sufficiently discussed in the literature. Therefore, our current hypothesis is that RBC metabolism plays a key role in AD brain disorders. We propose that the long-term lack of sufficient energy, disturbance of glycolytic, antioxidant RBSs pathways, and sodium potassium pump in oldest subjects [caused by different reasons and also in contact with Aβ, which is located on the luminal surfaces of cerebral microvessels (Grammas et al., 2002; Michaud et al., 2013)] can cause a decrease in the ability of RBC to transfer oxygen to tissue, leading to inadequate oxygenation and can result in abnormal glucose/energy metabolism, oxidative stress and, thereby, increase the susceptibility of neurons to damage, and reduce mental capacity as a consequence thereof. We have called this chain of events as “the erythrocytic hypothesis of Alzheimer disease” (Kosenko et al., 2016). In support of this hypothesis we also believe that erythrocyte biochemical aberrations might be used as potential tools in the early detection of the brain pathology development. This hypothesis provides ideas for the development of innovative personalized medical technologies allowing recovering the energy metabolism and the system of antioxidant defense in erythrocytes.
Brain Glucose Metabolism, Glutamate Toxicity, and Aβ Accumulation: Cause or Effect?
The brain is normally dependent on glucose for oxidative metabolism and function, therefore it is extremely sensitive to fluctuation in the blood glucose concentration, and since no satisfactory brain endogenous substitute exists. In spite of the fact that under certain conditions such as starvation or diabetes the ketone bodies can supply up to 50% of the brain's energy needs, the rest of the energy anyway must come from glucose. Therefore, within even just a few minutes glucose and oxygen deprivation induces significant dysfunction, and a longer time period can ultimately result in cell death (Blass, 2002). In addition to ATP production, the oxidation of glucose can produce other important intermediate such as lactate, which does not enter necessarily in the tricarboxylic acid cycle, but rather can be released and transported by the circulation into the liver for glucose synthesis de novo. Glucose also can be incorporated into lipids, proteins, and glycogen, and it is also the precursor of certain neurotransmitters such as γ-aminobutyric acid (GABA) (Plum and Posner, 1972), glutamate (Hamberger et al., 1979), and acetylcholine (Gibson et al., 1975). Thus, circulating glucose regulates many brain functions, including brain vitality, activity, learning, and memory (Korol and Gold, 1998).
Whereas the cerebral energy status is only slightly decreased during the normal aging process, glucose metabolism, and cellular ATP production are severely reduced in sporadic AD (Kyles et al., 1993; Hoyer, 1996). Certain neuronal populations are especially vulnerable to cut glucose oxidation, specifically neurons in the CA1, subiculum, and dentate gyrus of the hippocampus, and neurons in the outer layers of the cortex (Auer and Siesjö, 1993). A substantial proportion of neurons in these regions is glutamatergic and evidence suggests that hypoglycemic injury in these neurons is initiated almost entirely by hyperactivation of glutamate receptor (Auer et al., 1985), followed by the glutamate cascade and oxidative stress. The numerous studies have provided conclusive proof that glutamate becomes neurotoxic via the NMDA receptor when intracellular energy levels are reduced (Novelli et al., 1988; Beal et al., 1991; Albin and Greenamyre, 1992; Beal, 1992; Storey et al., 1992; Kosenko et al., 1994; Gonzalez et al., 2015). On the other hand, there is a direct relationship between disturbances in energy metabolism and mental disorder. For example, in 1932 Quastel J. first put forward a general suggestion that disturbances in energy metabolism would impair the neurological function, including particularly cognition (Quastel, 1932). During the past decades, a lot of work has proved Quastel's theory to be prescient and showed that the cause-effect relation is nonspecific as impairing cerebral energy metabolism can induce mental disorders to varying degrees (confusion, mental fatigue, agnosia, or dementia) in different pathological situations. Thus, impaired mental function has been reported in association with hypoglycemia (Bruce et al., 2009), inadequate transportation of glucose across the blood-brain barrier (Klepper and Voit, 2002; Pascual et al., 2004), defective astroglial glutamate transportation (Rönnbäck and Hansson, 2004), hypoxia (Gibson et al., 1981), diabetes (Richardson, 1990), heart failure (Riegel et al., 2002), reduced glucose tolerance (Vanhanen et al., 1997), bradycardia, hypotension (Ackerman, 1974), high intracranial pressure (Yoshida et al., 1996), stroke (van der Zwaluw et al., 2011), hypothermia, alcohol intoxication, thiamine and vitamin C deficiency, sedative-hypnotic drugs, opioids consumption (Martindale et al., 2010), general anesthesia (Parikh and Chung, 1995; Xie et al., 2006b), hypocapnia (Dodds and Allison, 1998), chronic stress (Conrad et al., 1996; Conrad, 2006), chronic noise stress (Arnsten and Goldman-Rakic, 1998; Manikandan et al., 2006), mixed brain pathologies (Schneider et al., 2007), hepatic encephalopathy (Butterworth, 2003), hyperammonemia (Llansola et al., 2007), trauma (Brooks et al., 2000), and so forth. Interestingly, after trauma, a large number of Aβ positive neurons appeared in human (Chen et al., 2004; Uryu et al., 2007) and animal brain (Kamal et al., 2001; Kasa et al., 2001; Papp et al., 2002; Hamberger et al., 2003). APP (amyloid precursor protein) accumulation is also observed following rat (Li et al., 1995) and human spinal cord injury (Ahlgren et al., 1996; Cornish et al., 2000). Long-term presence of APP and accumulation of Aβ in the rat thalamus were observed after middle cerebral artery occlusion (van Groen et al., 2005) and in cultured cells that had been treated with spirochetes or bacterial lipopolysaccharide (LPS) (Miklossy et al., 2006) and other infectious agents (Balin and Appelt, 2001).
A number of studies have also demonstrated that abnormal activation of β-adrenergic receptors (β-ARs), which mediate the effect of stress, might contribute to Aβ peptides production resulting in accelerating amyloid plaque formation in vitro and in vivo by enhancing γ-secretase activity (Ni et al., 2006) and that blocking β-ARs attenuates acute stress-induced Aβ peptide production (Yu et al., 2010). Indeed, the common inhalation anesthetic isoflurane has been reported to increase brain Aβ protein levels in vitro (Xie et al., 2006a) and in vivo (Xie et al., 2008; Zhang et al., 2008; Dong et al., 2009). Hypocapnia can also increase Aβ production in H4 human neuroglioma cells (Xie et al., 2004). Nanoscale particulates, a major component airborne pollution, inducing the blood-brain barrier disruption and neuroinflammation (Murr et al., 2006), result in AD-associated Aβ1−42, accumulation in the brains of children living in the high-pollution area (Calderón-Garcidueñas et al., 2008a,b). Upon careful analysis of these pathologies, one can see that there is a steady disruption of brain aerobic metabolism and the subsequent increase in APP processing and the formation of amyloids (Gabuzda et al., 1994; Webster et al., 1998; Velliquette et al., 2005). Thus, according to positron emission tomography (PET), isoflurane anesthesia can cause a 50% decrease in the rate of glucose uptake by the brain (Alkire et al., 1995, 1997), which leads to a sharp inhibition of aerobic oxidation in the cells and development of severe hypoxia, decreased neuronal activity (Hodes et al., 1985), and the appearance of amyloid in the brain 6-24 h after application of the anesthetic (Xie et al., 2006a, 2008). In ischemia-reperfusion, in addition to increasing oxidative stress, there is a decrease in the rate of blood flow, since migration of neutrophils to the site damaged by hypoxia can cause blockage of capillaries (Simpson et al., 1988), which impairs the entry of glucose and oxygen into the brain and promotes the formation of amyloids in damaged brain structures (van Groen et al., 2005; Tesco et al., 2007). In hypoglycemia, the limited supply of glucose from the blood to the brain also contributes to the accumulation of amyloids in the brain (Shi et al., 1997).
Altogether, these findings suggest that a transient insult, e.g., trauma, ischemia, neuroinflammation, anesthesia, or infectious agents could lead to secondary and persistent brain injuries and that the initial production of Aβ and its precursor, perhaps, are associated with physiological compensatory mechanisms for repair or protection of neurons exposed to significant disturbances in homeostasis (Smith et al., 2000; Lee et al., 2004). These facts are consistent with the numerous data showing that amyloid exhibits trophic and neuroprotective (Whitson et al., 1989; Koo et al., 1993; Singh et al., 1994; Luo et al., 1996), antioxidant (Smith et al., 1998, 2002a; Kontush et al., 2001; Atwood et al., 2002) properties and accumulates in the tissue after impairment of the energy metabolism with non-specific stimulus (Gabuzda et al., 1994; Webster et al., 1998; Velliquette et al., 2005), while under physiological conditions the diurnal fluctuation of brain Aβ levels is strictly regulated (Kang et al., 2009). Additionally, scores obtained on mini-mental state examination in AD subjects correlate highly with reductions of glucose metabolism (Blass, 2003), suggesting that the metabolic lesion precedes the development of neuropsychological abnormalities (Gibson and Huang, 2002) and support the conclusion that sporadic AD is a hypometabolic disorder which is provoked by a dysfunctional cerebral energy metabolism (Hoyer et al., 1988; Blass and Gibson, 1991; Meier-Ruge and Bertoni-Freddari, 1996; Perry et al., 1998; Smith et al., 2002b; Aliev et al., 2003b, 2004; Zhu et al., 2007). Obviously, the detection of mechanisms of disturbance of aerobic glucose metabolism in the brain is one of the most pressing tasks which will facilitate further progress on to determine not only to the midlife AD risk factor, but also on the lifespan of the older persons. Therefore, any pharmacological intervention, directed at correcting the chronic hypoperfusion state would possibly change the natural course of development of dementing neurodegeneration (Aliev et al., 2003a).
The Possible Role of RBC in Pathogenesis of AD
The pathologic causes of brain glucose metabolism disorders in AD may vary in signs and symptoms, which are as follows: desensitization of the neuronal insulin receptor (Hoyer, 2000), a decrease in the enzymes of the tricarbonic acid cycle activities (Meier-Ruge et al., 1984; Marcus et al., 1989; Marcus and Freedman, 1997; Bubber et al., 2005), impaired glucose transporter at the blood-brain barrier (Kalaria and Harik, 1989), depressed glucose transport into neurons (Simpson and Davies, 1994; Simpson et al., 1994), hippocampal region atrophy (Jobst et al., 1992; Villain et al., 2008) neuronal loss in the affected areas (McGeer et al., 1990), NO-dependent endothelial dysfunction and degeneration (De Jong et al., 1999; de la Torre, 2000a, 2002b) in brain capillaries that affect the capillary blood flow and optimal delivery of glucose and oxygen to neuronal cells (de la Torre, 2000b; Aliev et al., 2003a).
Another important factor in tissue oxygenation is the ability of RBC to bind, transport and release oxygen to tissues. For this, the RBC requires several essential metabolic pathways such as (i) anaerobic glycolysis, which is the only source of energy (ATP production) for sustaining cell structure and function; (ii) maintenance of the electrolyte gradient between plasma and red cell cytoplasm through the activity of adenosine triphosphate (ATP)-driven membrane pumps; (iii) pentose phosphate shunt (PPS) that controls the anti-oxidant pathways by produced NADPH, which plays an important role in maintaining glutathione in the reduced state; (iv) antioxidant pathways necessary for the protection of RBC proteins and hemoglobin against oxidation; and (v) nucleotide metabolism for the maintenance of the purine and pyrimidine homeostasis. Moreover, erythrocytes possess a unique glycolytic bypass, Rapoport-Luebering shunt to produce 2,3-diphosphoglycerate (2,3-DPG), a crucial metabolite in the regulation of hemoglobin affinity for oxygen (Cho et al., 2008). Thus, the mature erythrocyte retains a strictly regulated system of soluble enzymes, structural proteins, carbohydrates, lipids, anions, cations, cofactors, metabolites, antioxidants all of which are required in balance for effective metabolism and functioning of the cell. A change of at least one component of this system will lead to an imbalance and loss of RBC functional capacity. Indeed, a significant loss in ATP (Rabini et al., 1997), Mg2+, Na+, and ATP-ase activity (Ajmani and Rifkind, 1998) all of which may decrease erythrocyte deformability (Sakuta, 1981; Kucukatay et al., 2009), changes morphology (Gov and Safran, 2005) and increases RBC volume (Kowluru et al., 1989; Kucukatay et al., 2009). Extensive diminution of intracellular antioxidant GSH promotes oxidative damage of protein and lipids and compromises structural integrity of the RBC (Morris et al., 2008). Decreased 2,3-DPG, operating as a regulator of the oxygen affinity of RBC (Duhm, 1971) reduces the ability of RBC to release oxygen, resulting in tissue hypoxia (MacDonald, 1977; Nakamura et al., 1995; Figure 1). Considering the cause-effect relationship between various intracellular metabolic pathways and RBC function, it may be inferred that intact biochemical intracellular pathways are a major factor controlling the paramount RBC function associated with the ability to bind, transport, and release oxygen to tissues.
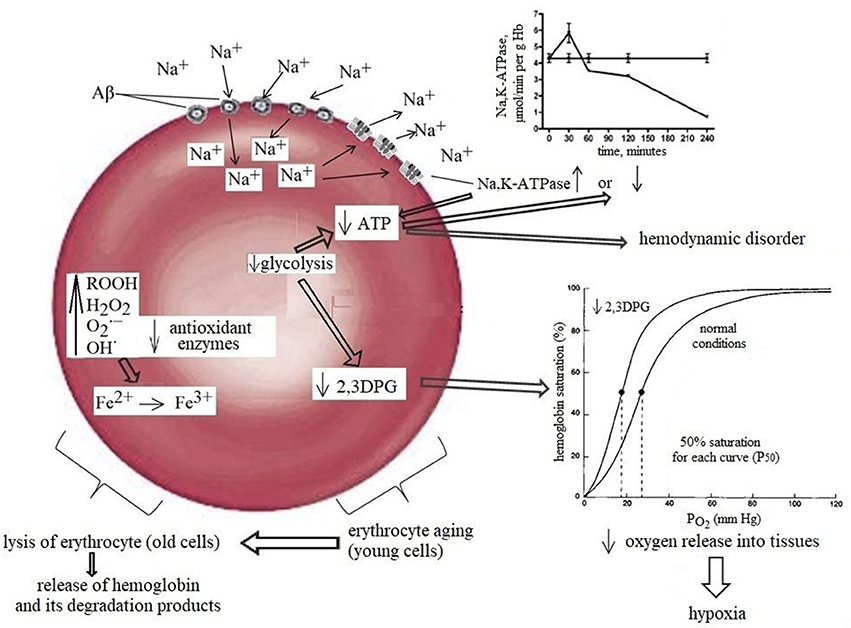
Figure 1. Normal aging diminishes RBC functions, including a detectable decrease in the activity of glycolytic and antioxidant enzymes. The combined effects of these damage together with a slight but significant decrease in 2,3-DPG are most likely contributor to the morphological changes in oldster subject which may result in decreased erythrocyte deformability, alter rheology, loss of adequate oxygen delivery and reduce the threshold for the development of neuropathology. The left part of the scheme: Amyloids possess gramicidin D-like action and upon contact with erythrocytes rapidly increase the concentration of sodium in the cells causing rapid activation of the Na+, K+-ATPase leading to the increase in ATP and 2,3DPG hydrolysis and can increase in Hb affinity to oxygen, that may be one of the factors contributing to brain hypoxia which lead to glucose hypometabolism and memory dysfunction in AD. The right part of the scheme: Prolonged contact with erythrocytes depletes ATP stores, causing Na+, K+-ATPase pumps and Na+- dependent ion channels to stop working and, consequently, the erythrocytes to swell and lyse. RBCs release hemoglobin, which is a source of iron. In turn, this metal catalyses the formation of toxic reactive oxygen species that mediate neuronal injury.
Recently, we measured some parameters of adenine nucleotide metabolism, glycolysis, pentose phosphate pathway, 2,3-DPG shunt (Kaminsky et al., 2013), oxidant and antioxidant enzymes and metabolites (Kosenko et al., 2012) in RBCs samples from Alzheimer's subjects (AD) and non-Alzheimer's dementia (NA) patients. We found that activities of all glycolytic, pentose phosphate pathway and 2,3-DPG shunt enzymes, Na+, K+-ATPase, as well as NAD/NADH ratio, pyruvate and lactate levels evidently decreased in aging and increased equally in AD and NA to levels or above levels of the YC (young controls) group indicating an increase in RBC glycolysis and ion fluxes. Elevated Na+, K+-ATPase activity and decreased ATP levels imply that ATP loss was mostly based on energy-expending redistribution of Na+ and K+ across the plasma membrane in erythrocytes from AD patients. These results confirm the fact that in AD, as in certain other diseases the balance between ATP formation and ion pumping may be disordered resulting in a decrease in intercellular energy charge, and an increase in lactate formation and catabolism of adenylates (Ronquist and Waldenström, 2003). These defects were accompanied by a significant decrease [relatively to both age-matched controls (AMC) and young adult controls (YC)] in the 2,3-DPG concentration that was accompanied by increases in the activity of diphosphoglycerate phosphatase (DPGP-ase), an enzyme that converts 2,3-DPG to 3PG (Kosenko et al., 2016). Of course, other factors besides of 2,3-DPG may affect the affinity of oxygen to hemoglobin (Samaja et al., 2003), but the relationship between the 2,3-DPG concentration in RBC as a biological indicator of tissue hypoxia in diabetic neuropathy (Nakamura et al., 1995), as well as in preterm infants with perinatal problems (Tsirka et al., 1990; Cholevas et al., 2008), in patients with the nondeletion genotype of hemoglobinopathy (Papassotiriou et al., 1998), with hypertension (Resnick et al., 1994), in experimental endotoxin shock (Matsumoto, 1995), severe hypophosphatemia (Larsen et al., 1996), and some types of glycolytic enzymes disturbances (McCully et al., 1999) was well established. Thus, the results generated the hypothesis that chronic enhancement in the rate of active transport in AD (Ronquist and Waldenström, 2003) leading to the increase in ATP and 2,3-DPG hydrolysis and can increase in Hb affinity to oxygen, loss of adequate oxygen delivery to tissues that may be one of the factors contributing to brain hypoxia (Aliev et al., 2004), glucose hypometabolism, and memory dysfunction in AD. It should be noted, however, that RBC of even cognitively stable aging persons (AMC) was characterized by a slight but significant decrease in 2,3-DPG when compared with the young adult control group. The tendency for the ATP production, adenylate energy charge, adenine nucleotide pool size, and ATP/ADP ratio (Kosenko et al., 2016) was a decrease in aging with no notable changes in dementia. There were no differences between AMC, AD, and NA groups in GSH levels, as well as in GSSG levels and the GSH/GSSG ratio in RBCs (Kosenko et al., 2012). Activities of calpain and caspase-3 in RBCs from aged subjects, on the contrary, were three times higher than those in young controls and were equally high in both dementia types (Kaminsky et al., 2012). The trend for the hydroperoxide generation was an increase in aging with no dramatic changes in dementia. There were no significant differences between AC, AD, and NA subjects in H2O2, organic hydroperoxide and the sum of H2O2 plus organic hydroperoxides content of RBC (Kaminsky et al., 2013). The results suggest that oxidative stress to some extent is already present in the RBC of the AMC subjects (Kosenko et al., 2012) and that together with the disturbances of glycolytic and transport processes and proteolysis increasing are a general feature of aging and not a feature of dementia. This view is supported by data comparing AD with normal aging, where was documented the same profile of damage (Smith et al., 1991; Moreira et al., 2006) suggesting that RBC oxidative damage is no longer an end stage but rather a signal of underlying changes of state (Moreira et al., 2006).
Although endogenous oxidative stress may damage the RBC itself the mass effect of large quantities of free radicals leaving the red cell has a prodigious potential to damage other components of the circulation (Johnson et al., 2005) including endothelial cells resulting in the microvascular pathology (Kiefmann et al., 2008). The combined effects of these damages most likely contribute to the morphological changes in oldster subjects (Richards et al., 2007), which may result in decreased erythrocyte deformability (Kuypers et al., 1990) and alter rheology and reduce the threshold for the development of neuropathology (Ajmani et al., 2003). We propose that the long-term lack of sufficient energy, disturbances of glycolytic pathway and sodium/potassium pump in aged subjects can decrease the ability of RBC to transfer oxygen, leading to inadequate tissue oxygenation and abnormal glucose metabolism in the brain and thereby reducing mental capacity and cognition. Thus, the reduced mental capacity may be, to a large extent, due to the imbalance in the metabolic processes in RBC. Obviously, other factors may be operative, but the role of RBC biochemical alterations as possible preclinical indicator of mental disorders must be critically examined. During the last 10 years, numerous biochemical abnormalities in RBC of subjects suffering from various mental disturbances have been detected (Danon et al., 1992; Rifkind et al., 1999; Ponizovsky et al., 2003; Pankowska et al., 2005; Lang et al., 2015; Pretorius et al., 2016). We believe that obligatory measurement of RBC biochemical parameters in peoples older than 50 years in the dynamics will help identify the risk factor for AD.
The problem is clear, but a number of questions arise in connection with the above-mentioned. If oxidative stress is more or less present in the erythrocytes of all elderly people and is a risk factor for dementia, why does this risk factor “work” for some people, while others, with the same risk factor, live to a very old age, maintaining “bright mind” and working capacity? The same question arises with regard to the concentration of 2,3-DPG reduction and the energy metabolism rate in the erythrocytes in general. It is obvious that the answers to these questions can only be obtained after identification of the reasons causing a global energy metabolism disorder, an increase of oxidative stress that are the basis of quick aging, affection of erythrocytes and that lead to a disruption of their functional capacity and early death. In other words, it is necessary to find out, under what influence factors (endogenous and exogenous) the reserve capacity of erythrocytes to withstand the stress that they are constantly exposed to, which circulate from the lungs to the tissues, decreases too soon.
Another problem is the lack of absolute knowledge of the hemopoiesis status in older people and especially in stressful situations that require intensification of the formation of blood cells. Numerous data indicate that the functions of basal hemopoiesis, which maintains the number of blood cells within the norm, changes insignificantly with age (Sansoni et al., 1993; Bagnara et al., 2000), whereas the reserve capacity of the bone marrow to resist stressful situations requiring its activation, even in healthy elderly people, reduces significantly with age (Williams et al., 1986; Globerson, 1999). For example, during bacterial infection or other periods of high hematopoietic demand, the formation of blood becomes “flaccid” and badly regulated, paradoxically (Rothstein, 1993), which makes it possible to assume that there is a hidden defect in the achievement of hematopoietic equilibrium in older people. Hence, the main question arises. Is such a hidden bone marrow defect typical for people with dementia? And does this defect lead only to the disruption of cellular equilibrium, or does it also cause the appearance of defective cells in the circulation that have not received adequate “strength reserve” in the bone marrow, and therefore they quickly age and get damaged in the bloodstream? It is clear that without the answers to these questions, it is impossible to evaluate the contribution of the impaired functional capacity of erythrocytes to the clinical symptoms of AD and other types of dementia. However, when dementia is exclusively referred to brain diseases, the attention of scientists is concentrated only on neurological symptoms, whereas all possible “defects” of erythrocytes, which cause pathological consequences for the brain, remain unexplored. We found only a few literature sources discussing the role of morphological changes that characterize the violation of the architecture of the erythrocyte membranes in the development of neurological symptoms characteristic of dementia (Mohanty et al., 2008). In particular, it has been shown that the appearance of atypical cells with altered morphological features, that is giant elongated erythrocytes with a nonhomogeneous membrane (acanthocytes or erythrocytes with numerous random spur-like cytoplasmic outgrowths) (Brecher and Bessis, 1972; Lan et al., 2015), occurs in advance (for several years) before the onset of memory disorders (Goodall et al., 1994). The mechanism of the acanthocytes emergence in the bloodstream is unknown, but since atypical cells are only a small part of the general population of normal erythrocytes, it has been suggested that the cause of their formation is related to a disruption in the synthesis of membrane structural proteins that occurs in the stage of erythrocyte formation in the bone marrow, although the possibility of the cell damage under the influence of unknown factors immediately after they come out from the bone marrow into the blood is not ruled out. Soluble amyloid peptides that are found in various cerebral vessels of the patients with AD [cerebral amyloid angiopathy (CAA)] and which, on the one hand, are capable to contact the cellular elements of blood, on the other—to damage the walls of blood vessels and cause a hemorrhage in the brain (Thanvi and Robinson, 2006), have recently been recognized as one of these factors. However, it is possible that the appearance of erythrocytes of the atypical form is associated with amyloids that circulate in the bloodstream and bind to the cell membrane (Kuo et al., 2000; Kiko et al., 2012) leading to its damage.
The Role of Amyloid Angiopathy in Erythrocyte Damage
The fact that AD is a systemic disease has been known for a long time, dating back to the last century, when amyloid peptides were first detected in small vessels of the brains of patients with AD (Scholz, 1938). For the sake of justice, it is worth mentioning that the existence of amyloid peptides was known back in 1878 when Atkins discovered amyloids in the brain and blood vessels of the brain in a young patient with dementia caused by a head injury (Atkins, 1878). At that time, it has been also identified that amyloids accumulate in the brain vessels in patients with syphilis (Atkins, 1875), and epilepsy (Blocq and Marinesco, 1892) indicating that cerebral amyloidosis and CAA are accompanying a number of diseases.
As it is now well known, in patients with DA, amyloids (mainly Aβ1−40) are found in the capillaries (Attems and Jellinger, 2004), arteries, arterioles, veins, and venules (Thal et al., 2002), which penetrate the leptomeningeal, cortical and subcortical areas of the brain (Weller et al., 2009), as well as in blood vessels supplying the hippocampus (Masuda et al., 1988). Localized in various structures of blood vessels (Wisniewski and Wegiel, 1994) and in contact with numerous cells (myocytes, pericytes), amyloids cause their damage (Vonsattel et al., 1991; Dalkara et al., 2011), as a result of which the membrane of the vessels seems to loosen, becoming unstable, which can eventually lead to the formation of an aneurysms, its rupture and cerebral hemorrhage (Thanvi and Robinson, 2006), that is, a condition that usually occurs with strokes and which irrespective of AD causes the formation of hematoma, lysis of erythrocytes, brain edema (Xi et al., 2006), local cell death, and memory damage (Pfeifer et al., 2002). Interestingly, the multiple microvascular pathology, mediated by the presence of amyloids in different structures of the blood vessels, was confirmed not only at postmortem examination, but also in life in virtually all patients with AD (Kalaria and Hedera, 1995; Farkas and Luiten, 2001; Bailey et al., 2004; Smith and Greenberg, 2009) regardless of the presence of atherosclerotic changes in the vessels. It should be noted, however, that CAA in AD patients is observed in 90–100% of cases, while brain zones with hemorrhage are detected only in 20–25% of patients with AD (Urbach, 2011). This means that brain damage in AD can occur for reasons not associated with CAA-induced hemorrhage. Indeed, a significant accumulation of amyloids in the brain vessels can cause their occlusion, thereby blocking blood flow, supplying the brain with oxygen and glucose (de la Torre and Stefano, 2000), and causing neurodegeneration of neurons and memory impairment (Thal et al., 2008, 2009). In addition, localized in endothelial cells lining the lumen of blood vessels (Michaud et al., 2013), amyloids are constantly in contact with erythrocytes circulating in the bloodstream, causing, on the one hand, their adhesion to endothelial cells, thereby violating the blood flow (Ravi et al., 2004), on the other—interacting with the membrane of erythrocytes, cause its modification and damage (Nicolay et al., 2007).
It was true that some studies have demonstrated that AD patients have increased RBC membrane injury suggesting the increased capability for erythrocyte lysis in vivo (Bosman et al., 1991; Goodall et al., 1994; Mattson et al., 1997; Solerte et al., 2000; Kosenko et al., 2009), as evidenced by the accumulation of free hemoglobin and iron in the brain of AD patients (Wu et al., 2004; Perry et al., 2008). The consequences of RBC lysis for the brain are well known (Xi et al., 1998). It has been shown that the appearance of free hemoglobin in the brain leads to rapid destruction of the hemato-encephalic barrier, DNA fragmentation, increased lipid peroxidation and global oxidative stress, development of the inflammatory process, vasoconstriction, hypoperfusion, brain atrophy (Alexander and LoVerme, 1980), memory impairment and death (Hackett and Anderson, 2000).
The lytic effect of amyloids was confirmed on the general population of erythrocytes. In vitro Aβ induces rapid lysis of human and rat erythrocytes that can be either attenuated by antioxidants (Mattson et al., 1997) or amplified in the presence of inhibitors of glycolytic and antioxidant enzymes or Na+, K+-ATPase (Kosenko et al., 2008a), and suggested the role of RBC glycolysis, ion pumping capacity and antioxidant status in the bioactivity and erythrotoxicity of amyloids. Given the above, we assumed that the constant contact of erythrocytes with amyloids can cause not only the change and damage on the membrane structures, but also the metabolic/energy metabolism in the erythrocytes underlying the aging, integrity, and functional ability of the cells. This assumption does not contradict the known pathological consequence of chronic brain hypoperfusion, leading to reducing oxygen delivery to the brain (de la Torre, 2017). On the contrary, it clearly points to the possible existence of additional unspecified mechanisms, restricting the oxygen supply to the brain and, therefore, participating in the development of hypoxia and neurodegenerative processes specific to AD (Thal et al., 2009). However, the population of erythrocytes is not homogeneous, and the facts that the least resistance of old erythrocytes to endogenous and exogenous pathological factors caused by a reduced rate of energy metabolism, antioxidant defense, and strengthening of catabolic processes (Bonsignore et al., 1964; Shinozuka et al., 1994) are well established.
Data on the effect of amyloid peptides on erythrocytes of different ages at present are currently not available and are of special interest, since patients with AD are characterized by accelerated aging of erythrocytes in the bloodstream (Bosman et al., 1991). We have recently showed that sensibility of RBC to βA-induced hemolysis was in proportion to both cell age and βA concentration (Tikhonova et al., 2014). The inhibition of glucose consumption and lactate production by βA was found to occur in both cells type. However, greater demand for ATP of the Na+, K+ -ATPase, in combination with a more reduced capacity of the glycolytic pathway and 2,3-DPG levels in old cells lead to more pronounced imbalance between ATP and 2,3-DPG formation, total nucleotide changes and ion pumping in aging erythrocytes exposed to the amyloid. Interestingly, the decline in the levels of antioxidative, glycolytic enzymes, 2,3DPG, ATP, adenine nucleotide pool and the adenylate energy charge in young cells treated with amyloid were similar to that we found as occurring during in vivo red cell aging (control old erythrocytes). Thus, our data obtained show that even a limited contact of amyloid with erythrocytes is sufficient to transform young erythrocytes into old ones and that similar biochemical mechanisms can underlie the accelerated aging of cells in the bloodstream of patients with AD (Bosman et al., 1991) (Figure 2), that may be one of the main reasons of both inadequate supply of oxygen to the brain, and lysis of cells in circulation.
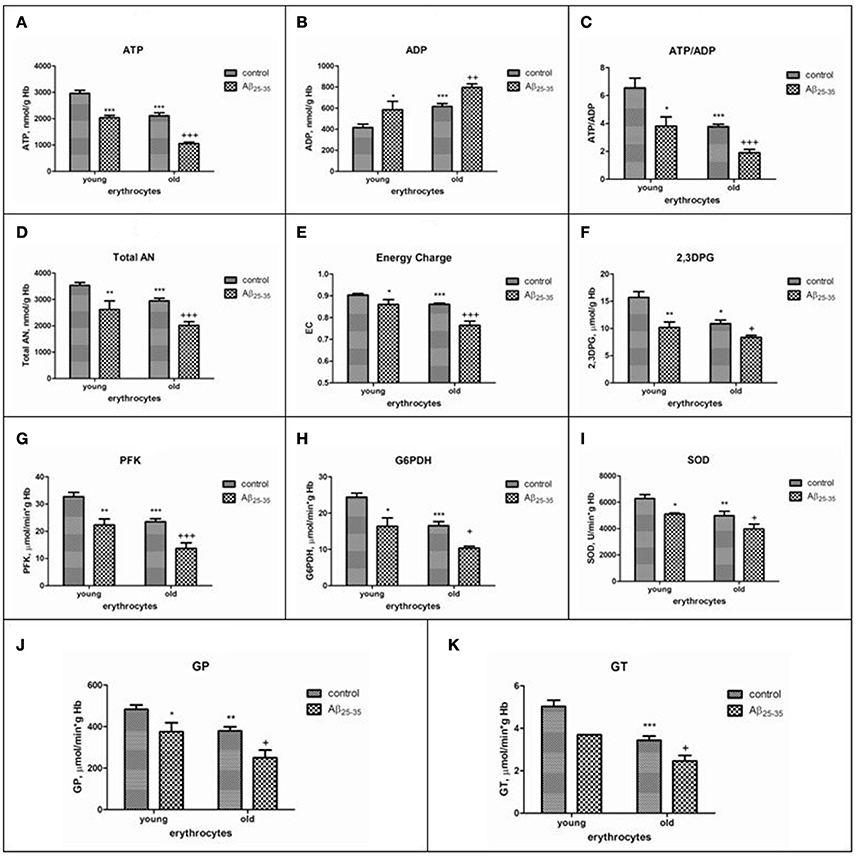
Figure 2. The effects of Aβ25-35 on the parameters of the adenylate system, concentration of 2,3–DPG and activities of some glycolytic and antioxidant enzymes activities in young and old erythrocytes (RBCs). (A) ATP, (B) ADP, (C) ratio ATP/ADP, (D) total adenine nucleotide pool size, (E) energy charge, (F) 2,3DPG, (G–K) activities of phosphofructokinase, glucose-6-phosphate dehydrogenase, superoxide dismutase, glutathione peroxidase, glutathione transferase, respectively. ATP and AN are expressed as micromol/g Hb, ADP as nmol/g Hb. AN, total adenine nucleotide pool size; EC, adenylate energy charge [EC = (ATP + 1/2ADP)/AN]; phosphofructokinase (PFK), glucose-6-phosphate dehydrogenase (G-6PDH), glutathione peroxidase (GP), glutathione transferase (GT) activities are expressed as IU/g hemoglobin (Hb); superoxide dismutase (SOD) is expressed as units/min per g Hb. One unit of SOD activity is defined as the amount of enzyme required to produce a 50% inhibition of the rate of p-nitrotetrazolium blue reduction. The results are the mean±SEM of 16 rats. Cells were incubated at 25°C for 30 min in 10 mmol/L potassium phosphate buffer, pH 7.4, containing 0.9% NaCl, 5 mmol/L KCl, and 10 μmol/L Aβ25-35. Control was incubated with nontoxic Aβ35-25. Significant differences are indicated: *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to young cells; +P < 0.05, ++P < 0.01, and +++P < 0.001 as compared to the old control (one-way analysis of variance [ANOVA] with Bonferroni's multiple comparison test). Aβ indicates amyloid β.
Novel Therapeutic Strategy: Problems and Possible Solutions
At present there are no medical drugs which are able to increase and improve perfusion of the brain of AD patients, since due to the absence of early diagnostics the use of any drug therapy, when the brain tissue is irreparably damaged, is late and inefficient (Hachinski and Munoz, 1997). Thus, it is very problematic “to repair” chronically damaged blood vessels of the brain and to restore their functional state with the preparations available, as well as it is hard “to cure diseased erythrocytes.” This is connected, first of all, with the fact that real causes of “chronic disease” of erythrocytes during natural aging of the organism are unknown. One of the important problems, as noted above, is the absence of total knowledge on the hematopoietic status of the elderly, especially during long bed rest, accompanied by undernourishment, leads to a decrease in metabolism rate.
It is interesting to note that the problems in relation to the “disease” of erythrocytes arise during transfusion of the whole donor blood or packed RBC to the patients with different diseases in order to restore oxygen transport to the tissues and release carbonic acid from them. The main challenge is that all intracellular indices of erythrocytes change very quickly during the storage period of the donated blood leading to rapid cell aging (Lang et al., 2016). And if these indices are not corrected before blood transfusion this may result in irreparable consequences (Beutler et al., 1969). It has been shown, for example, that if during red blood cell transfusion intracellular ATP concentration of erythrocytes was lower by 40% compared to the normal cells, these erythrocytes were lysed to an excessive degree in blood flow of the recipient (Hamasaki et al., 1981). Transfusion of erythrocytes with low intracellular content of 2,3-DPG did not allow for quick restoration of adequate delivery of oxygen to the tissues. Taken together, these observations require development of the ways to increase the concentrations of ATP and 2,3-DPG in erythrocytes immediately before RBC transfusion to the patient (Beutler et al., 1969). Further studies in this field are actively undertaken, and multiple developments directed toward restoration of energy exchange in the stored erythrocytes are successively utilized by the physicians to save the patients life (Valeri and Hirsch, 1969). The main components restoring energy exchange in erythrocytes are glucose, adenine, ascorbate-2-phosphate, phosphoenolpyruvat or the cations and activators of glycolysis, which can penetrate into erythrocytes (Moore et al., 1981). It has been shown that different activators of enzymes introduced into the medium, where erythrocytes are stored, maintain normal concentration of ATP and 2,3-DPG for 1,5 months (Vora, 1987). However, although the scientists have made a considerable progress in solving the problems with regard to restoration of energy exchange disturbed during storage of erythrocytes, all the developments use modulators and activators, which are able to quickly and easily pass through the cell membrane of erythrocytes. This is a limitation for the use of the wider class of active compounds that are unable to be transported into the cells. We tried to circumvent this problem and developed a technology of the encapsulation of substrates and high molecular enzymes in erythrocytes under hypotonic conditions leading to the formation of pores in the membranes of erythrocytes (Seeman et al., 1973), enabling the enzymes with great molecular mass (Baker, 1967; Kosenko et al., 2008b; Godfrin et al., 2012; Kaminsky and Kosenko, 2012; Alexandrovich et al., 2017) to pass through the cells. For instance, we developed an approach on how to introduce regulatory glycolytic enzymes into erythrocytes, where the activity of these enzymes in erythrocytes of old animals and in the elderly decreased by 30–50% (Kaminsky et al., 2013). The data obtained showed that the encapsulation of even one regulatory enzyme in erythrocytes stimulated glycolysis to considerable extent. The signs of it were the increased rate of glucose consumption and the formation of lactate. Moreover, the erythrocytes obtained circulated in the animal's blood flow within many days, sustaining the activity of encapsulated enzymes, the normal level of ATP, 2,3-DPG and other metabolites of energy exchange and antioxidant defense (data not shown). These results obtained are important by two reasons. First, such technology can be applied to restore ATP, 2,3-DPG and other metabolites of energy exchange, the concentration of which is decreased sharply in erythrocytes in aging of the organism. Transfusion of own erythrocytes, possessing encapsulated enzymes, should theoretically reduce the risk of the onset of inadequate oxygen supply to the brain both in the elderly and in patients with AD. Secondly, investigations make the basis for further development of innovative personalized therapeutic strategy.
Conclusions
Presently, non-genetic Alzheimer's disease is classified as a neurodegenerative disorder. However, there is an impressive body of evidence indicating that AD is a systemic metabolic disease (Perry et al., 2003), and it has originated as a vascular disorder with the resultant impairment of the delivery and transport of essential nutrients, particularly glucose and oxygen resulting in an energy metabolic breakdown with the plaques and tangles found in the brain secondary to the effects of the vascular pathology (de la Torre, 2002a). Since erythrocyte serve as the only oxygen carrier and their ability to the binding, transport, and delivery of oxygen to tissues depends, first of all, on the energy metabolism and antioxidant status there is therefore a strong possibility that the disturbance of energy metabolism and oxidative enhancement in these cells may have a dramatic impact on destabilization of aerobic glucose metabolism in the brain and AD development. With regard to RBC-controlled brain vital activity there is incontrovertible evidence that even just a few minutes of oxygen deprivation (together with glucose) initiate significant brain dysfunction and chronic effect can ultimately result in the irreversible brain damage and permanent impairment of cognition. This implies that the cerebrometabolic abnormalities are the most common form of dementia (Chibber et al., 2016; Gonzalez-Reyes et al., 2016). However, although major mechanisms involved in brain damage due to metabolic abnormalities resulting from the oxygen deprivation including alterations in neurotransmission, defect of mitochondrial oxidative phosphorylation, disturbance of Ca2+ homeostasis, oxidative stress and eventually apoptotic or necrotic cell death are profound and obvious (Barreto et al., 2011; Cabezas et al., 2012, 2015; Avila Rodriguez et al., 2014; Toro-Urrego et al., 2016; Baez et al., 2017; Baez-Jurado et al., 2017a,b; Martin-Jiménez et al., 2017a,b; Shevtsova et al., 2017), no systematic programs of research have examined the relationship between the breach of the energy metabolism of erythrocytes in the causing of leading to cerebrometabolic abnormalities and dementia. One of the reasons of this paradox is the large number of reports stating that brain atrophy and degeneration of nerve cells, observed with dementia, can occur without cerebrovascular pathology, but only through the amyloid fault, leading to the struggle with amyloids, and not with the causes that “gave birth to them,” and made the AD a permanently incurable disease with unknown etiology. In our view, a careful examination and reversing age-related metabolic/energetic changes in erythrocytes is an achievable goal and will provide these cells as a marker of a risk of inadequate brain oxygen supply, resulting the irreversible brain damage and permanent impairment of cognition. We also strongly believe that biochemical erythrocyte indicators (ATP, 2,3DPG, glucose, lactate and others), as well as the enzymes of glycolysis, pentose phosphate and Rapoport-Luebering shunt, antioxidant pathways all of which are responsible for interrelated metabolism and functional capacity of RBC should be studied (especially in people over 50 years, and in the dynamics) not only in research laboratories, but also in clinical settings that may provide a basis for innovative personalized therapeutic strategies.
The development of technologies to assist in restoration of erythrocyte energy metabolism must form an integral part of new therapeutic strategies in the treatment of a great variety of disorders accompanied by inadequate oxygen delivery. Similar studies just are gathering pace but have already marked a turning-point in our knowledge regarding AD and amyloid peptides that cannot be the only pharmacological target in the struggle against this devastating illness of human beings.
Author Contributions
All of authors (EK, LT, CM, GB, GA, and YK) write manuscript, created figures, and proof final version of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The reported study was funded by RFBR and Moscow region according to the research project No 17-44-500561.
References
Ackerman, W. J. (1974). Stupor, bradycardia, hypotension and hypothermia. A presentation of Wernicke's encephalopathy with rapid response to thiamine. West. J. Med. 121, 428–429.
Ahlgren, S., Li, G. L., and Olsson, Y. (1996). Accumulation of beta-amyloid precursor protein and ubiquitin in axons after spinal cord trauma in humans: immunohistochemical observations on autopsy material. Acta Neuropathol. 92, 49–55. doi: 10.1007/s004010050488
Ajmani, R. S., and Rifkind, J. M. (1998). Hemorheological changes during human aging. Gerontology 44, 111–120. doi: 10.1159/000021993
Ajmani, R. S., Fleg, J. L., Demehin, A. A., Wright, J. G., O'Connor, F., Heim, J. M., et al. (2003). Oxidative stress and hemorheological changes induced by acute treadmill exercise. Clin. Hemorheol. Microcirc. 28, 29–40.
Albin, R. L., and Greenamyre, J. T. (1992). Alternative excitotoxic hypotheses. Neurology 42, 733–738. doi: 10.1212/WNL.42.4.733
Alexander, M. P., and LoVerme, S. R. (1980). Aphasia after left hemispheric intracerebral hemorrhage. Neurology 30, 1193–1202. doi: 10.1212/WNL.30.11.1193
Alexandrovich, Y. G., Kosenko, E. A., Sinauridze, E. I., Obydennyi, S. I., Kireev, I. I., Ataullakhanov, F. I., et al. (2017). Rapid elimination of blood alcohol using erythrocytes: mathematical modeling and in vitro study. BioMed Res. Int. 2017:5849593. doi: 10.1155/2017/5849593
Aliev, G. (2011). Oxidative stress induced-metabolic imbalance, mitochondrial failure, and cellular hypoperfusion as primary pathogenetic factors for the development of Alzheimer disease which can be used as a alternate and successful drug treatment strategy: past, present and future. CNS Neurol. Disord. Drug Targets 10, 147–148. doi: 10.2174/187152711794480492
Aliev, G., Obrenovich, M. E., Smith, M. A., and Perry, G. (2003a). Hypoperfusion, mitochondria failure, oxidative stress, and Alzheimer disease. J. Biomed. Biotechnol. 2003, 162–163. doi: 10.1155/S1110724303305029
Aliev, G., Smith, M. A., de la Torre, J. C., and Perry, G. (2004). Mitochondria as a primary target for vascular hypoperfusion and oxidative stress in Alzheimer's disease. Mitochondrion 4, 649–663. doi: 10.1016/j.mito.2004.07.018
Aliev, G., Smith, M. A., Obrenovich, M. E., de la Torre, J. C., and Perry, G. (2003b). Role of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Azheimer disease. Neurotox. Res. 5, 491–504. doi: 10.1007/BF03033159
Alkire, M. T., Haier, R. J., Barker, S. J., Shah, N. K., Wu, J. C., and Kao, Y. J. (1995). Cerebral metabolism during propofol anesthesia in humans studied with positron emission tomography. Anesthesiology 82, 393–403. discussion: 27A. doi: 10.1097/00000542-199502000-00010
Alkire, M. T., Haier, R. J., Shah, N. K., and Anderson, C. T. (1997). Positron emission tomography study of regional cerebral metabolism in humans during isoflurane anesthesia. Anesthesiology 86, 549–557. doi: 10.1097/00000542-199703000-00006
Arai, H., Lee, V. M., Hill, W. D., Greenberg, B. D., and Trojanowski, J. Q. (1992). Lewy bodies contain beta-amyloid precursor proteins of Alzheimer's disease. Brain Res. 585, 386–390. doi: 10.1016/0006-8993(92)91242-7
Arnsten, A. F., and Goldman-Rakic, P. S. (1998). Noise stress impairs prefrontal cortical cognitive function in monkeys: evidence for a hyperdopaminergic mechanism. Arch. Gen. Psychiatry 55, 362–368. doi: 10.1001/archpsyc.55.4.362
Atkins, R. (1875). Half-yearly report on mental disease. Dublin J. Med. Sci. 314–331. doi: 10.1007/BF02975691
Atkins, R. (1878). On the morbid histology of the spinal cord in five cases of insanity. Br. Med. J. 2, 96–99. doi: 10.1136/bmj.2.916.96
Attems, J., and Jellinger, K. A. (2004). Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology–a pilot study. Acta Neuropathol. 107, 83–90. doi: 10.1007/s00401-003-0796-9
Atwood, C. S., Obrenovich, M. E., Liu, T., Chan, H., Perry, G., Smith, M. A., et al. (2003). Amyloid-beta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Brain Res. Brain Res. Rev. 43, 1–16. doi: 10.1016/S0165-0173(03)00174-7
Atwood, C. S., Robinson, S. R., and Smith, M. A. (2002). Amyloid-β: redox-metal chelator and antioxidant. J. Alzheimers Dis. 4, 203–214. doi: 10.3233/JAD-2002-4310
Auer, R. N., and Siesjö, B. K. (1993). Hypoglycaemia: brain neurochemistry and neuropathology. Baillieres Clin. Endocrinol. Metab. 7, 611–625. doi: 10.1016/S0950-351X(05)80210-1
Auer, R., Kalimo, H., Olsson, Y., and Wieloch, T. (1985). The dentate gyrus in hypoglycemia: pathology implicating excitotoxin-mediated neuronal necrosis. Acta Neuropathol. 67, 279–288. doi: 10.1007/BF00687813
Avila Rodriguez, M., Garcia-Segura, L. M., Cabezas, R., Torrente, D., Capani, F., Gonzalez, J., et al. (2014). Tibolone protects T98G cells from glucose deprivation. J. Steroid Biochem. Mol. Biol. 144(Pt B), 294–303. doi: 10.1016/j.jsbmb.2014.07.009
Baez, E., Guio-Vega, G. P., Echeverria, V., Sandoval-Rueda, D. A., and Barreto, G. E. (2017). 4'-Chlorodiazepam protects mitochondria in T98G astrocyte cell line from glucose deprivation. Neurotox. Res. 32, 163–171. doi: 10.1007/s12640-017-9733-x
Baez-Jurado, E., Hidalgo-Lanussa, O., Guio-Vega, G., Ashraf, G. M., Echeverria, V., Aliev, G., et al. (2017a). Conditioned medium of human adipose mesenchymal stem cells increases wound closure and protects human astrocytes following scratch assay in vitro. Mol. Neurobiol. doi: 10.1007/s12035-017-0771-4. [Epub ahead of print].
Baez-Jurado, E., Vega, G. G., Aliev, G., Tarasov, V. V., Esquinas, P., Echeverria, V., et al. (2017b). Blockade of neuroglobin reduces protection of conditioned medium from human mesenchymal stem cells in human astrocyte model (T98G) under a scratch assay. Mol. Neurobiol. doi: 10.1007/s12035-017-0481-y. [Epub ahead of print].
Bagnara, G. P., Bonsi, L., Strippoli, P., Bonifazi, F., Tonelli, R., D'Addato, S., et al. (2000). Hemopoiesis in healthy old people and centenarians: well-maintained responsiveness of CD34+ cells to hemopoietic growth factors and remodeling of cytokine network. J. Gerontol. A Biol. Sci. Med. Sci. 55, B61–B66. discussion: B67–B70. doi: 10.1093/gerona/55.2.B61
Bailey, T. L., Rivara, C. B., Rocher, A. B., and Hof, P. R. (2004). The nature and effects of cortical microvascular pathology in aging and Alzheimer's disease. Neurol. Res. 26, 573–578. doi: 10.1179/016164104225016272
Baker, R. F. (1967). Entry of ferritin into human red cells during hypotonic haemolysis. Nature 215, 424–425. doi: 10.1038/215424a0
Balin, B. J., and Appelt, D. M. (2001). Role of infection in Alzheimer's disease. J. Am. Osteopath. Assoc. 101, S1–S6.
Bapat, R. D., Supe, A. N., and Sathe, M. J. (1983). Management of small bowel perforation with intra- and post-operative lavages with povidone iodine. (A prospective study). J. Postgrad. Med. 29, 29–33.
Barreto, G., White, R. E., Ouyang, Y., Xu, L., and Giffard, R. G. (2011). Astrocytes: targets for neuroprotection in stroke. Cent. Nerv. Syst. Agents Med. Chem. 11, 164–173. doi: 10.2174/187152411796011303
Beal, M. F. (1992). Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann. Neurol. 31, 119–130. doi: 10.1002/ana.410310202
Beal, M. F., Swartz, K. J., Hyman, B. T., Storey, E., Finn, S. F., and Koroshetz, W. (1991). Aminooxyacetic acid results in excitotoxin lesions by a novel indirect mechanism. J. Neurochem. 57, 1068–1073. doi: 10.1111/j.1471-4159.1991.tb08258.x
Beutler, E., Meul, A., and Wood, L. A. (1969). Depletion and regeneration of 2,3-diphosphoglyceric acid in stored red blood cells. Transfusion 9, 109–115. doi: 10.1111/j.1537-2995.1969.tb05527.x
Bird, T. D. (2005). Genetic factors in Alzheimer's disease. N. Engl. J. Med. 352, 862–864. doi: 10.1056/NEJMp058027
Blass, J. P. (2002). Glucose/mitochondria in neurological conditions. Int. Rev. Neurobiol. 51, 325–376. doi: 10.1016/S0074-7742(02)51010-2
Blass, J. P. (2003). Cerebrometabolic abnormalities in Alzheimer's disease. Neurol. Res. 25, 556–566. doi: 10.1179/016164103101201995
Blass, J. P., and Gibson, G. E. (1991). The role of oxidative abnormalities in the pathophysiology of Alzheimer's disease. Rev. Neurol. 147, 513–525.
Blocq, P., and Marinesco, G. (1892). Sur les le'sions et la pathoge'nie del 'epilepsie dite essentielle. Sem. Me'dical 12, 445–446.
Bonsignore, A., Fornaini, G., Fantoni, A., Leoncini, G., and Segni, P. (1964). Relationship between age and enzymatic activities in human erythrocytes from normal and fava bean-sensitive subjects. J. Clin. Invest. 43, 834–842. doi: 10.1172/JCI104969
Bosman, G. J., Bartholomeus, I. G., de Man, A. J., van Kalmthout, P. J., and de Grip, W. J. (1991). Erythrocyte membrane characteristics indicate abnormal cellular aging in patients with Alzheimer's disease. Neurobiol. Aging 12, 13–18. doi: 10.1016/0197-4580(91)90033-G
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Brecher, G., and Bessis, M. (1972). Present status of spiculed red cells and their relationship to the discocyte-echinocyte transformation: a critical review. Blood 40, 333–344.
Brewer, G. J., Oelshlegel, F. J., Moore, L. G., and Noble, N. A. (1974). In vivo red cell glycolytic control and DPG-ATP levels. Ann. N. Y. Acad. Sci. 241, 513–523. doi: 10.1111/j.1749-6632.1974.tb21907.x
Brito-Moreira, J., Paula-Lima, A. C., Bomfim, T. R., Oliveira, F. B., Sepúlveda, F. J., De Mello, F. G., et al. (2011). Aβ oligomers induce glutamate release from hippocampal neurons. Curr. Alzheimer Res. 8, 552–562. doi: 10.2174/156720511796391917
Brooks, W. M., Stidley, C. A., Petropoulos, H., Jung, R. E., Weers, D. C., Friedman, S. D., et al. (2000). Metabolic and cognitive response to human traumatic brain injury: a quantitative proton magnetic resonance study. J. Neurotrauma 17, 629–640. doi: 10.1089/089771500415382
Bruce, D. G., Davis, W. A., Casey, G. P., Clarnette, R. M., Brown, S. G. A., Jacobs, I. G., et al. (2009). Severe hypoglycaemia and cognitive impairment in older patients with diabetes: the Fremantle diabetes study. Diabetologia 52, 1808–1815. doi: 10.1007/s00125-009-1437-1
Bubber, P., Haroutunian, V., Fisch, G., Blass, J. P., and Gibson, G. E. (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol. 57, 695–703. doi: 10.1002/ana.20474
Butterworth, R. F. (2003). Hepatic encephalopathy–a serious complication of alcoholic liver disease. Alcohol Res. Health 27, 143–145.
Cabezas, R., Avila, M. F., Gonzalez, J., El-Bacha, R. S., and Barreto, G. E. (2015). PDGF-BB protects mitochondria from rotenone in T98G cells. Neurotox. Res. 27, 355–367. doi: 10.1007/s12640-014-9509-5
Cabezas, R., El-Bacha, R. S., Gonzalez, J., and Barreto, G. E. (2012). Mitochondrial functions in astrocytes: neuroprotective implications from oxidative damage by rotenone. Neurosci. Res. 74, 80–90 doi: 10.1016/j.neures.2012.07.008
Calderón-Garcidueñas, L., Mora-Tiscare-o, A., Ontiveros, E., Gómez-Garza, G., Barragán-Mejía, G., Broadway, J., et al. (2008a). Air pollution, cognitive deficits and brain abnormalities: a pilot study with children and dogs. Brain Cogn. 68, 117–127. doi: 10.1016/j.bandc.2008.04.008
Calderón-Garcidueñas, L., Solt, A. C., Henríquez-Roldán, C., Torres-Jardón, R., Nuse, B., Herritt, L., et al. (2008b). Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol. Pathol. 36, 289–310. doi: 10.1177/0192623307313011
Chang, C.-Y., Liang, H.-J., Chow, S.-Y., Chen, S.-M., and Liu, D.-Z. (2007). Hemorheological mechanisms in Alzheimer's disease. Microcirculation 14, 627–634. doi: 10.1080/10739680701411056
Chen, X.-H., Siman, R., Iwata, A., Meaney, D. F., Trojanowski, J. Q., and Smith, D. H. (2004). Long-term accumulation of amyloid-β, β-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 165, 357–371. doi: 10.1016/S0002-9440(10)63303-2
Chibber, S., Alexiou, A., Alama, M. N., Barreto, G. E., Aliev, G., and Ashraf, G. M. (2016). A synopsis on the linkage between age-related dementias and vascular disorders. CNS Neurol. Disord. Drug Targets 15, 250–258. doi: 10.2174/1871527315666160202121809
Cho, J., King, J. S., Qian, X., Harwood, A. J., and Shears, S. B. (2008). Dephosphorylation of 2,3-bisphosphoglycerate by MIPP expands the regulatory capacity of the Rapoport-Luebering glycolytic shunt. Proc. Natl. Acad. Sci. U.S.A. 105, 5998–6003. doi: 10.1073/pnas.0710980105
Cholevas, V., Challa, A., Lapatsanis, P. D., and Andronikou, S. (2008). Changes in red cell phosphate metabolism of preterm and fullterm infants with perinatal problems during their first month of life. Eur. J. Pediatr. 167, 211–218. doi: 10.1007/s00431-007-0464-5
Connolly, A. M., and Volpe, J. J. (1990). Clinical features of bilirubin encephalopathy. Clin. Perinatol. 17, 371–379.
Conrad, C. D. (2006). What is the functional significance of chronic stress-induced CA3 dendritic retraction within the hippocampus? Behav. Cogn. Neurosci. Rev. 5, 41–60. doi: 10.1177/1534582306289043
Conrad, C. D., Galea, L. A., Kuroda, Y., and McEwen, B. S. (1996). Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav. Neurosci. 110, 1321–1334. doi: 10.1037/0735-7044.110.6.1321
Cornish, R., Blumbergs, P. C., Manavis, J., Scott, G., Jones, N. R., and Reilly, P. L. (2000). Topography and severity of axonal injury in human spinal cord trauma using amyloid precursor protein as a marker of axonal injury. Spine 25, 1227–1233. doi: 10.1097/00007632-200005150-00005
Dalkara, T., Gursoy-Ozdemir, Y., and Yemisci, M. (2011). Brain microvascular pericytes in health and disease. Acta Neuropathol. 122, 1–9. doi: 10.1007/s00401-011-0847-6
Danon, D., Bologna, N. B., and Gavendo, S. (1992). Memory performance of young and old subjects related to their erythrocyte characteristics. Exp. Gerontol. 27, 275–285. doi: 10.1016/0531-5565(92)90055-5
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K., Fernandez, S. J., Ferreira, S. T., et al. (2007). Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. doi: 10.1074/jbc.M607483200
De Felice, F. G., Wu, D., Lambert, M. P., Fernandez, S. J., Velasco, P. T., Lacor, P. N., et al. (2008). Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol. Aging 29, 1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029
De Jong, G. I., Farkas, E., Stienstra, C. M., Plass, J. R., Keijser, J. N., de la Torre, J. C., et al. (1999). Cerebral hypoperfusion yields capillary damage in the hippocampal CA1 area that correlates with spatial memory impairment. Neuroscience 91, 203–210. doi: 10.1016/S0306-4522(98)00659-9
de la Torre, J. C. (2000a). Cerebral hypoperfusion, capillary degeneration, and development of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 14(Suppl. 1), S72–S81. doi: 10.1097/00002093-200000001-00012
de la Torre, J. C. (2000b). Impaired cerebromicrovascular perfusion. Summary of evidence in support of its causality in Alzheimer's disease. Ann. N. Y. Acad. Sci. 924, 136–152. doi: 10.1111/j.1749-6632.2000.tb05572.x
de la Torre, J. C. (2002a). Alzheimer disease as a vascular disorder: nosological evidence. Stroke 33, 1152–1162. doi: 10.1161/01.STR.0000014421.15948.67
de la Torre, J. C. (2002b). Alzheimer's disease: how does it start? J. Alzheimers Dis. 4, 497–512. doi: 10.3233/JAD-2002-4606
de la Torre, J. C. (2017). Are major dementias triggered by poor blood flow to the brain? Theoretical considerations. J. Alzheimers Dis. 57, 353–371. doi: 10.3233/JAD-161266
de la Torre, J. C., and Mussivand, T. (1993). Can disturbed brain microcirculation cause Alzheimer's disease? Neurol. Res. 15, 146–153. doi: 10.1080/01616412.1993.11740127
de la Torre, J. C., and Stefano, G. B. (2000). Evidence that Alzheimer's disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res. Brain Res. Rev. 34, 119–136. doi: 10.1016/S0165-0173(00)00043-6
Demuro, A., Mina, E., Kayed, R., Milton, S. C., Parker, I., and Glabe, C. G. (2005). Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280, 17294–17300. doi: 10.1074/jbc.M500997200
Dodds, C., and Allison, J. (1998). Postoperative cognitive deficit in the elderly surgical patient. Br. J. Anaesth. 81, 449–462.
Dong, Y., Zhang, G., Zhang, B., Moir, R. D., Xia, W., Marcantonio, E. R., et al. (2009). The common inhalational anesthetic sevoflurane induces apoptosis and increases beta-amyloid protein levels. Arch. Neurol. 66, 620–631. doi: 10.1001/archneurol.2009.48
Duhm, J. (1971). Effects of 2,3-diphosphoglycerate and other organic phosphate compounds on oxygen affinity and intracellular pH of human erythrocytes. Pflugers Arch. 326, 341–356. doi: 10.1007/BF00586998
Farkas, E., and Luiten, P. G. (2001). Cerebral microvascular pathology in aging and Alzheimer's disease. Prog. Neurobiol. 64, 575–611. doi: 10.1016/S0301-0082(00)00068-X
Fita, I. G., Enciu, A. M., and Stanoiu, B. P. (2011). New insights on Alzheimer's disease diagnostic. Rom. J. Morphol. Embryol. 52, 975–979.
Funato, H., Yoshimura, M., Kusui, K., Tamaoka, A., Ishikawa, K., Ohkoshi, N., et al. (1998). Quantitation of amyloid beta-protein (A beta) in the cortex during aging and in Alzheimer's disease. Am. J. Pathol. 152, 1633–1640.
Gabuzda, D., Busciglio, J., Chen, L. B., Matsudaira, P., and Yankner, B. A. (1994). Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 269, 13623–13628.
Giannakopoulos, P., Herrmann, F. R., Bussière, T., Bouras, C., Kövari, E., Perl, D. P., et al. (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology 60, 1495–1500. doi: 10.1212/01.WNL.0000063311.58879.01
Gibson, G. E., and Huang, H.-M. (2002). Oxidative processes in the brain and non-neuronal tissues as biomarkers of Alzheimer's disease. Front. Biosci. J. Virtual Libr. 7, d1007–d1015. doi: 10.2741/gibson
Gibson, G. E., Jope, R., and Blass, J. P. (1975). Decreased synthesis of acetylcholine accompanying impaired oxidation of pyruvic acid in rat brain minces. Biochem. J. 148, 17–23. doi: 10.1042/bj1480017
Gibson, G. E., Pulsinelli, W., Blass, J. P., and Duffy, T. E. (1981). Brain dysfunction in mild to moderate hypoxia. Am. J. Med. 70, 1247–1254. doi: 10.1016/0002-9343(81)90834-2
Glenner, G. G., and Wong, C. W. (1984). Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135. doi: 10.1016/0006-291X(84)91209-9
Globerson, A. (1999). Hematopoietic stem cells and aging. Exp. Gerontol. 34, 137–146. doi: 10.1016/S0531-5565(98)00069-2
Godfrin, Y., Horand, F., Franco, R., Dufour, E., Kosenko, E., Bax, B. E., et al. (2012). International seminar on the red blood cells as vehicles for drugs. Expert Opin. Biol. Ther. 12, 127–133. doi: 10.1517/14712598.2012.631909
Gong, Y., Chang, L., Viola, K. L., Lacor, P. N., Lambert, M. P., Finch, C. E., et al. (2003). Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422. doi: 10.1073/pnas.1834302100
Gonzalez, J., Jurado-Coronel, J. C., Avila, M. F., Sabogal, A., Capani, F., and Barreto, G. E. (2015). NMDARs in neurological diseases: a potential therapeutic target. Int. J. Neurosci. 125, 315–327. doi: 10.3109/00207454.2014.940941
Gonzalez-Reyes, R. E., Aliev, G., Avila-Rodrigues, M., and Barreto, G. E. (2016). Alterations in glucose metabolism on cognition: a possible link between diabetes and dementia. Curr. Pharm. Des. 22, 812–818. doi: 10.2174/1381612822666151209152013
Goodall, H. B., Reid, A. H., Findlay, D. J., Hind, C., Kay, J., and Coghill, G. (1994). Irregular distortion of the erythrocytes (acanthocytes, spur cells) in senile dementia. Dis. Markers 12, 23–41. doi: 10.1155/1994/493810
Gov, N. S., and Safran, S. A. (2005). Red blood cell membrane fluctuations and shape controlled by ATP-induced cytoskeletal defects. Biophys. J. 88, 1859–1874. doi: 10.1529/biophysj.104.045328
Grammas, P., Yamada, M., and Zlokovic, B. (2002). The cerebromicrovasculature: a key player in the pathogenesis of Alzheimer's disease. J. Alzheimers Dis. 4, 217–223. doi: 10.3233/JAD-2002-4311
Guillozet, A. L., Weintraub, S., Mash, D. C., and Mesulam, M. M. (2003). Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch. Neurol. 60, 729–736. doi: 10.1001/archneur.60.5.729
Gyure, K. A., Durham, R., Stewart, W. F., Smialek, J. E., and Troncoso, J. C. (2001). Intraneuronal abeta-amyloid precedes development of amyloid plaques in down syndrome. Arch. Pathol. Lab. Med. 125, 489–492. doi: 10.1043/0003-9985(2001)125<0489:IAAPDO>2.0.CO;2
Hachinski, V., and Munoz, D. G. (1997). Cerebrovascular pathology in Alzheimer's disease: cause, effect or epiphenomenon? Ann. N. Y. Acad. Sci. 826, 1–6. doi: 10.1111/j.1749-6632.1997.tb48456.x
Hackett, M. L., and Anderson, C. S. (2000). Health outcomes 1 year after subarachnoid hemorrhage: an international population-based study. The Australian cooperative research on subarachnoid hemorrhage study Group. Neurology 55, 658–662. doi: 10.1212/WNL.55.5.658
Hamasaki, N., Ideguchi, H., and Ikehara, Y. (1981). Regeneration of 2,3-bisphosphoglycerate and ATP in stored erythrocytes by phosphoenolpyruvate: a new preservative for blood storage. Transfusion 21, 391–396. doi: 10.1046/j.1537-2995.1981.21481275994.x
Hamberger, A. C., Chiang, G. H., Nylén, E. S., Scheff, S. W., and Cotman, C. W. (1979). Glutamate as a CNS transmitter. I. Evaluation of glucose and glutamine as precursors for the synthesis of preferentially released glutamate. Brain Res. 168, 513–530. doi: 10.1016/0006-8993(79)90306-8
Hamberger, A., Huang, Y.-L., Zhu, H., Bao, F., Ding, M., Blennow, K., et al. (2003). Redistribution of neurofilaments and accumulation of β-amyloid protein after brain injury by rotational acceleration of the head. J. Neurotrauma 20, 169–178. doi: 10.1089/08977150360547080
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hodes, J. E., Soncrant, T. T., Larson, D. M., Carlson, S. G., and Rapoport, S. I. (1985). Selective changes in local cerebral glucose utilization induced by phenobarbital in the rat. Anesthesiology 63, 633–639. doi: 10.1097/00000542-198512000-00013
Hoyer, S. (1996). Oxidative metabolism deficiencies in brains of patients with Alzheimer's disease. Acta Neurol. Scand. Suppl. 165, 18–24. doi: 10.1111/j.1600-0404.1996.tb05868.x
Hoyer, S. (2000). Brain glucose and energy metabolism abnormalities in sporadic Alzheimer disease. Causes and consequences: an update. Exp. Gerontol. 35, 1363–1372. doi: 10.1016/S0531-5565(00)00156-X
Hoyer, S., Oesterreich, K., and Wagner, O. (1988). Glucose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type? J. Neurol. 235, 143–148. doi: 10.1007/BF00314304
Hsia, A. Y., Masliah, E., McConlogue, L., Yu, G. Q., Tatsuno, G., Hu, K., et al. (1999). Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc. Natl. Acad. Sci. U.S.A. 96, 3228–3233. doi: 10.1073/pnas.96.6.3228
Jack, C. R., Lowe, V. J., Weigand, S. D., Wiste, H. J., Senjem, M. L., Knopman, D. S., et al. (2009). Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain J. Neurol. 132, 1355–1365. doi: 10.1093/brain/awp062
Joachim, C. L., Mori, H., and Selkoe, D. J. (1989). Amyloid beta-protein deposition in tissues other than brain in Alzheimer's disease. Nature 341, 226–230. doi: 10.1038/341226a0
Jobst, K. A., Smith, A. D., Barker, C. S., Wear, A., King, E. M., Smith, A., et al. (1992). Association of atrophy of the medial temporal lobe with reduced blood flow in the posterior parietotemporal cortex in patients with a clinical and pathological diagnosis of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 55, 190–194. doi: 10.1136/jnnp.55.3.190
Johnson, R. M., Goyette, G., Ravindranath, Y., and Ho, Y.-S. (2005). Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radic. Biol. Med. 39, 1407–1417. doi: 10.1016/j.freeradbiomed.2005.07.002
Jordan, B. D. (2000). Chronic traumatic brain injury associated with boxing. Semin. Neurol. 20, 179–185. doi: 10.1055/s-2000-9826
Kalaria, R. N., and Harik, S. I. (1989). Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J. Neurochem. 53, 1083–1088. doi: 10.1111/j.1471-4159.1989.tb07399.x
Kalaria, R. N., and Hedera, P. (1995). Differential degeneration of the cerebral microvasculature in Alzheimer's disease. Neuroreport 6, 477–480. doi: 10.1097/00001756-199502000-00018
Kamal, A., Almenar-Queralt, A., LeBlanc, J. F., Roberts, E. A., and Goldstein, L. S. (2001). Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414, 643–648. doi: 10.1038/414643a
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/S0896-6273(03)00124-7
Kaminsky, Y., Poghosyan, A., Tikhonova, L., Palacios, H. H., Kamal, M. A., Kosenko, E., et al. (2012). Glycolytic and proteolytic metabolism in erythrocytes from elderly and demented patients. Am. J. Neuroprotect. Neuroregeneration 4, 1–5. doi: 10.1166/ajnn.2012.1039
Kaminsky, Y. G., and Kosenko, E. A. (2012). Argocytes containing enzyme nanoparticles reduce toxic concentrations of arginine in the blood. Bull. Exp. Biol. Med. 153, 406–408. doi: 10.1007/s10517-012-1727-3
Kaminsky, Y. G., Marlatt, M. W., Smith, M. A., and Kosenko, E. A. (2010). Subcellular and metabolic examination of amyloid-β peptides in Alzheimer disease pathogenesis: evidence for Aβ (25-35). Exp. Neurol. 221, 26–37. doi: 10.1016/j.expneurol.2009.09.005
Kaminsky, Y. G., Reddy, V. P., Ashraf, G. M., Ahmad, A., Benberin, V. V., Kosenko, E. A., et al. (2013). Age-related defects in erythrocyte 2,3-diphosphoglycerate metabolism in dementia. Aging Dis. 4, 244–255. doi: 10.14336/AD.2013.0400244
Kang, J.-E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Kasa, P., Papp, H., and Pakaski, M. (2001). Presenilin-1 and its N-terminal and C-terminal fragments are transported in the sciatic nerve of rat. Brain Res. 909, 159–169. doi: 10.1016/S0006-8993(01)02679-8
Kiefmann, R., Rifkind, J. M., Nagababu, E., and Bhattacharya, J. (2008). Red blood cells induce hypoxic lung inflammation. Blood 111, 5205–5214. doi: 10.1182/blood-2007-09-113902
Kiko, T., Nakagawa, K., Satoh, A., Tsuduki, T., Furukawa, K., Arai, H., et al. (2012). Amyloid β levels in human red blood cells. PLoS ONE 7:e49620. doi: 10.1371/journal.pone.0049620
Klein, W. L., Krafft, G. A., and Finch, C. E. (2001). Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 24, 219–224. doi: 10.1016/S0166-2236(00)01749-5
Klepper, J., and Voit, T. (2002). Facilitated glucose transporter protein type 1 (GLUT1) deficiency syndrome: impaired glucose transport into brain– a review. Eur. J. Pediatr. 161, 295–304. doi: 10.1007/s00431-002-0939-3
Kontush, A., Berndt, C., Weber, W., Akopyan, V., Arlt, S., Schippling, S., et al. (2001). Amyloid-β is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic. Biol. Med. 30, 119–128. doi: 10.1016/S0891-5849(00)00458-5
Koo, E. H., Park, L., and Selkoe, D. J. (1993). Amyloid beta-protein as a substrate interacts with extracellular matrix to promote neurite outgrowth. Proc. Natl. Acad. Sci. U.S.A. 90, 4748–4752. doi: 10.1073/pnas.90.10.4748
Kosenko, E., Kaminsky, Y., Grau, E., Miñana„, M. D., Marcaida, G., Grisolía, S., et al. (1994). Brain ATP depletion induced by acute ammonia intoxication in rats is mediated by activation of the NMDA receptor and Na+,K+-ATPase. J. Neurochem. 63, 2172–2178. doi: 10.1046/j.1471-4159.1994.63062172.x
Kosenko, E. A., Aliev, G., and Kaminsky, Y. G. (2016). Relationship between chronic disturbance of 2,3-diphosphoglycerate metabolism in erythrocytes and Alzheimer disease. CNS Neurol. Disord. Drug Targets 15, 113–123. doi: 10.2174/1871527314666150821103444
Kosenko, E. A., Aliev, G., Tikhonova, L. A., Li, Y., Poghosyan, A. C., and Kaminsky, Y. G. (2012). Antioxidant status and energy state of erythrocytes in Alzheimer dementia: probing for markers. CNS Neurol. Disord. Drug Targets 11, 926–932. doi: 10.2174/1871527311201070926
Kosenko, E. A., Solomadin, I. N., and Kaminskii, I. G. (2009). Effect of the beta-amyloid peptide Abeta25-35 and fullerene C60 on the activity of enzymes in erythrocytes. Bioorg. Khim. 35, 172–177.
Kosenko, E. A., Solomadin, I. N., Marov, N. V., Venediktova, N. I., Pogosian, A. S., and Kaminskĭ, I. G. (2008a). [Role of glycolysis and antioxidant enzymes in the toxicity of amyloid beta peptide Aβ25-35 to erythrocytes]. Bioorg. Khim. 34, 654–660.
Kosenko, E. A., Venediktova, N. I., Kudryavtsev, A. A., Ataullakhanov, F. I., Kaminsky, Y. G., Felipo, V., et al. (2008b). Encapsulation of glutamine synthetase in mouse erythrocytes: a new procedure for ammonia detoxification. Biochem. Cell Biol. Biochim. Biol. Cell. 86, 469–476. doi: 10.1139/O08-134
Kowluru, R., Bitensky, M. W., Kowluru, A., Dembo, M., Keaton, P. A., and Buican, T. (1989). Reversible sodium pump defect and swelling in the diabetic rat erythrocyte: effects on filterability and implications for microangiopathy. Proc. Natl. Acad. Sci. U.S.A. 86, 3327–3331. doi: 10.1073/pnas.86.9.3327
Kucukatay, V., Erken, G., Bor-Kucukatay, M., and Kocamaz, E. (2009). Effect of sulfite treatment on erythrocyte deformability in young and aged rats. Toxicol. Mech. Methods 19, 19–23. doi: 10.1080/15376510802175788
Kuo, Y. M., Emmerling, M. R., Vigo-Pelfrey, C., Kasunic, T. C., Kirkpatrick, J. B., Murdoch, G. H., et al. (1996). Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 271, 4077–4081. doi: 10.1074/jbc.271.8.4077
Kuo, Y. M., Kokjohn, T. A., Kalback, W., Luehrs, D., Galasko, D. R., Chevallier, N., et al. (2000). Amyloid-β peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem. Biophys. Res. Commun. 268, 750–756. doi: 10.1006/bbrc.2000.2222
Kuypers, F. A., Scott, M. D., Schott, M. A., Lubin, B., and Chiu, D. T. (1990). Use of ektacytometry to determine red cell susceptibility to oxidative stress. J. Lab. Clin. Med. 116, 535–545.
Kyles, A. E., Waterman, A. E., Livingston, A., and Vetmed, B. (1993). Antinociceptive effects of combining low doses of neuroleptic drugs and fentanyl in sheep. Am. J. Vet. Res. 54, 1483–1488.
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lan, J., Liu, J., Zhao, Z., Xue, R., Zhang, N., Zhang, P., et al. (2015). The peripheral blood of Aβ binding RBC as a biomarker for diagnosis of Alzheimer's disease. Age Ageing 44, 458–464. doi: 10.1093/ageing/afv009
Lang, E., Pozdeev, V. I., Xu, H. C., Shinde, P. V., Behnke, K., Hamdam, J. M., et al. (2016). Storage of erythrocytes induces suicidal erythrocyte death. Cell. Physiol. Biochem. 39, 668–676. doi: 10.1159/000445657
Lang, F., Jilani, K., and Lang, E. (2015). Therapeutic potential of manipulating suicidal erythrocyte death. Expert Opin. Ther. Targets 19, 1219–1227. doi: 10.1517/14728222.2015.1051306
Larsen, V. H., Waldau, T., Gravesen, H., and Siggaard-Andersen, O. (1996). Erythrocyte 2,3-diphosphoglycerate depletion associated with hypophosphatemia detected by routine arterial blood gas analysis. Scand. J. Clin. Lab. Investig. Suppl. 224, 83–87. doi: 10.3109/00365519609088626
Lee, H.-G., Casadesus, G., Zhu, X., Takeda, A., Perry, G., and Smith, M. A. (2004). Challenging the amyloid cascade hypothesis: senile plaques and amyloid-beta as protective adaptations to Alzheimer disease. Ann. N.Y. Acad. Sci. 1019, 1–4. doi: 10.1196/annals.1297.001
Li, G. L., Farooque, M., Holtz, A., and Olsson, Y. (1995). Changes of β-amyloid precursor protein after compression trauma to the spinal cord: an experimental study in the rat using immunohistochemistry. J. Neurotrauma 12, 269–277. doi: 10.1089/neu.1995.12.269
Liberski, P. P. (2004). Amyloid plaques in transmissible spongiform encephalopathies (prion diseases). Folia Neuropathol. 42(Suppl. B), 109–119.
Llansola, M., Rodrigo, R., Monfort, P., Montoliu, C., Kosenko, E., Cauli, O., et al. (2007). NMDA receptors in hyperammonemia and hepatic encephalopathy. Metab. Brain Dis. 22, 321–335. doi: 10.1007/s11011-007-9067-0
Lue, L. F., Kuo, Y. M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., et al. (1999). Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–862. doi: 10.1016/S0002-9440(10)65184-X
Luo, Y., Sunderland, T., Roth, G. S., and Wolozin, B. (1996). Physiological levels of β-amyloid peptide promote PC12 cell proliferation. Neurosci. Lett. 217, 125–128. doi: 10.1016/0304-3940(96)13087-1
MacDonald, R. (1977). Red cell 2,3-diphosphoglycerate and oxygen affinity. Anaesthesia 32, 544–553. doi: 10.1111/j.1365-2044.1977.tb10002.x
Manikandan, S., Padma, M. K., Srikumar, R., Jeya Parthasarathy, N., Muthuvel, A., and Sheela Devi, R. (2006). Effects of chronic noise stress on spatial memory of rats in relation to neuronal dendritic alteration and free radical-imbalance in hippocampus and medial prefrontal cortex. Neurosci. Lett. 399, 17–22. doi: 10.1016/j.neulet.2006.01.037
Mann, D. M., Jones, D., South, P. W., Snowden, J. S., and Neary, D. (1992). Deposition of amyloid beta protein in non-Alzheimer dementias: evidence for a neuronal origin of parenchymal deposits of β protein in neurodegenerative disease. Acta Neuropathol. 83, 415–419. doi: 10.1007/BF00713534
Marcus, D. L., and Freedman, M. L. (1997). Decreased brain glucose metabolism in microvessels from patients with Alzheimer's disease. Ann. N. Y. Acad. Sci. 826, 248–253. doi: 10.1111/j.1749-6632.1997.tb48476.x
Marcus, D. L., de Leon, M. J., Goldman, J., Logan, J., Christman, D. R., Wolf, A. P., et al. (1989). Altered glucose metabolism in microvessels from patients with Alzheimer's disease. Ann. Neurol. 26, 91–94. doi: 10.1002/ana.410260114
Martindale, J. L., Senecal, E. L., Obermeyer, Z., Nadel, E. S., and Brown, D. F. M. (2010). Altered mental status and hypothermia. J. Emerg. Med. 39, 491–496. doi: 10.1016/j.jemermed.2010.03.021
Martin-Jiménez, C. A., Gáitan-Vaca, D. M., Echeverria, V., González, J., and Barreto, G. E. (2017a). Relationship between obesity, alzheimer's disease, and Parkinson's disease: an astrocentric view. Mol. Neurobiol. 54, 7096–7115. doi: 10.1007/s12035-016-0193-8
Martin-Jiménez, C. A., García-Vega, Á., Cabezas, R., Aliev, G., Echeverria, V., González, J., et al. (2017b). Astrocytes and endoplasmic reticulum stress: a bridge between obesity and neurodegenerative diseases. Prog. Neurobiol. 158, 45–68. doi: 10.1016/j.pneurobio.2017.08.001
Masuda, J., Tanaka, K., Ueda, K., and Omae, T. (1988). Autopsy study of incidence and distribution of cerebral amyloid angiopathy in Hisayama, Japan. Stroke 19, 205–210. doi: 10.1161/01.STR.19.2.205
Matsumoto, Y. (1995). [The function of red blood cells during experimental hemorrhagic and endotoxic shock]. Masui 44, 342–348.
Mattson, M. P., Begley, J. G., Mark, R. J., and Furukawa, K. (1997). Aβ25-35 induces rapid lysis of red blood cells: contrast with Abeta1-42 and examination of underlying mechanisms. Brain Res. 771, 147–153. doi: 10.1016/S0006-8993(97)00824-X
Mattson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., and Rydel, R. E. (1992). beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. Off. J. Soc. Neurosci. 12, 376–389.
McCully, K., Chance, B., and Giger, U. (1999). In vivo determination of altered hemoglobin saturation in dogs with M-type phosphofructokinase deficiency. Muscle Nerve 22, 621–627. doi: 10.1002/(SICI)1097-4598(199905)22:5<621::AID-MUS11>3.0.CO;2-D
McGeer, E. G., McGeer, P. L., Harrop, R., Akiyama, H., and Kamo, H. (1990). Correlations of regional postmortem enzyme activities with premortem local glucose metabolic rates in Alzheimer's disease. J. Neurosci. Res. 27, 612–619. doi: 10.1002/jnr.490270422
McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller, S. J., Smith, M. J., Vbeyreuther, K., et al. (1999). Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866. doi: 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M
Meier-Ruge, W., and Bertoni-Freddari, C. (1996). The significance of glucose turnover in the brain in the pathogenetic mechanisms of Alzheimer's disease. Rev. Neurosci. 7, 1–19. doi: 10.1515/REVNEURO.1996.7.1.1
Meier-Ruge, W., Iwangoff, P., and Reichlmeier, K. (1984). Neurochemical enzyme changes in Alzheimer's and Pick's disease. Arch. Gerontol. Geriatr. 3, 161–165. doi: 10.1016/0167-4943(84)90007-4
Michaud, J.-P., Bellavance, M.-A., Préfontaine, P., and Rivest, S. (2013). Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid β. Cell Rep. 5, 646–653. doi: 10.1016/j.celrep.2013.10.010
Miklossy, J., Kis, A., Radenovic, A., Miller, L., Forro, L., Martins, R., et al. (2006). Beta-amyloid deposition and Alzheimer's type changes induced by Borrelia spirochetes. Neurobiol. Aging 27, 228–236. doi: 10.1016/j.neurobiolaging.2005.01.018
Mohanty, J. G., Eckley, D. M., Williamson, J. D., Launer, L. J., and Rifkind, J. M. (2008). Do red blood cell-beta-amyloid interactions alter oxygen delivery in Alzheimer's disease? Adv. Exp. Med. Biol. 614, 29–35. doi: 10.1007/978-0-387-74911-2_4
Moore, G. L., Ledford, M. E., and Brummell, M. R. (1981). Improved red blood cell storage using optional additive systems (OAS) containing adenine, glucose and ascorbate-2-phosphate. Transfusion 21, 723–731. doi: 10.1046/j.1537-2995.1981.21682085764.x
Moreira, P. I., Zhu, X., Liu, Q., Honda, K., Siedlak, S. L., Harris, P. L., et al. (2006). Compensatory responses induced by oxidative stress in Alzheimer disease. Biol. Res. 39, 7–13. doi: 10.4067/S0716-97602006000100002
Morishima-Kawashima, M., Oshima, N., Ogata, H., Yamaguchi, H., Yoshimura, M., Sugihara, S., et al. (2000). Effect of apolipoprotein E allele epsilon4 on the initial phase of amyloid β-protein accumulation in the human brain. Am. J. Pathol. 157, 2093–2099. doi: 10.1016/S0002-9440(10)64847-X
Morris, C. R., Suh, J. H., Hagar, W., Larkin, S., Bland, D. A., Steinberg, M. H., et al. (2008). Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood 111, 402–410. doi: 10.1182/blood-2007-04-081703
Murr, L. E., Soto, K. F., Garza, K. M., Guerrero, P. A., Martinez, F., Esquivel, E. V., et al. (2006). Combustion-generated nanoparticulates in the El Paso, TX, USA/Juarez, Mexico Metroplex: their comparative characterization and potential for adverse health effects. Int. J. Environ. Res. Public. Health 3, 48–66. doi: 10.3390/ijerph2006030007
Nakamura, J., Koh, N., Sakakibara, F., Hamada, Y., Wakao, T., Hara, T., et al. (1995). Polyol pathway, 2,3-diphosphoglycerate in erythrocytes and diabetic neuropathy in rats. Eur. J. Pharmacol. 294, 207–214. doi: 10.1016/0014-2999(95)00531-5
Ni, Y., Zhao, X., Bao, G., Zou, L., Teng, L., Wang, Z., et al. (2006). Activation of β2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat. Med. 12, 1390–1396. doi: 10.1038/nm1485
Nicolay, J. P., Gatz, S., Liebig, G., Gulbins, E., and Lang, F. (2007). Amyloid induced suicidal erythrocyte death. Cell. Physiol. Biochem. 19, 175–184. doi: 10.1159/000099205
Novelli, A., Reilly, J. A., Lysko, P. G., and Henneberry, R. C. (1988). Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 451, 205–212. doi: 10.1016/0006-8993(88)90765-2
Nunomura, A., Perry, G., Pappolla, M. A., Friedland, R. P., Hirai, K., Chiba, S., et al. (2000). Neuronal oxidative stress precedes amyloid-β deposition in down syndrome. J. Neuropathol. Exp. Neurol. 59, 1011–1017. doi: 10.1093/jnen/59.11.1011
Omalu, B. I., DeKosky, S. T., Minster, R. L., Kamboh, M. I., Hamilton, R. L., and Wecht, C. H. (2005). Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57, 128–134. discussion: 128–134. doi: 10.1227/01.NEU.0000163407.92769.ED
Pankowska, E., Szypowska, A., Wysocka, M., and Lipka, M. (2005). [The role of 2,3-DPG in nerve conduction of children with type 1 diabetes]. Endokrynol. Diabetol. Chor. Przemiany Materii Wieku Rozw. 11, 207–210.
Papassotiriou, I., Kister, J., Griffon, N., Abraham, D. J., Kanavakis, E., Traeger-Synodinos, J., et al. (1998). Synthesized allosteric effectors of the hemoglobin molecule: a possible mechanism for improved erythrocyte oxygen release capability in hemoglobinopathy H disease. Exp. Hematol. 26, 922–926.
Papp, H., Pakaski, M., and Kasa, P. (2002). Presenilin-1 and the amyloid precursor protein are transported bidirectionally in the sciatic nerve of adult rat. Neurochem. Int. 41, 429–435. doi: 10.1016/S0197-0186(02)00014-1
Parikh, S. S., and Chung, F. (1995). Postoperative delirium in the elderly. Anesth. Analg. 80, 1223–1232.
Pascual, J. M., Wang, D., Lecumberri, B., Yang, H., Mao, X., Yang, R., et al. (2004). GLUT1 deficiency and other glucose transporter diseases. Eur. J. Endocrinol. 150, 627–633. doi: 10.1530/eje.0.1500627
Perry, G., Nunomura, A., Raina, A. K., Aliev, G., Siedlak, S. L., Harris, P. L. R., et al. (2003). A metabolic basis for Alzheimer disease. Neurochem. Res. 28, 1549–1552. doi: 10.1023/A:1025678510480
Perry, G., Smith, M. A., McCann, C. E., Siedlak, S. L., Jones, P. K., and Friedland, R. P. (1998). Cerebrovascular muscle atrophy is a feature of Alzheimer's disease. Brain Res. 791, 63–66. doi: 10.1016/S0006-8993(98)00006-7
Perry, R. T., Gearhart, D. A., Wiener, H. W., Harrell, L. E., Barton, J. C., Kutlar, A., et al. (2008). Hemoglobin binding to A β and HBG2 SNP association suggest a role in Alzheimer's disease. Neurobiol. Aging 29, 185–193. doi: 10.1016/j.neurobiolaging.2006.10.017
Pfeifer, L. A., White, L. R., Ross, G. W., Petrovitch, H., and Launer, L. J. (2002). Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58, 1629–1634. doi: 10.1212/WNL.58.11.1629
Pike, K. E., Savage, G., Villemagne, V. L., Ng, S., Moss, S. A., Maruff, P., et al. (2007). β-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain J. Neurol. 130, 2837–2844. doi: 10.1093/brain/awm238
Plum, F., and Posner, J. B. (1972). The diagnosis of stupor and coma. Contemp. Neurol. Ser. 10, 1–286.
Ponizovsky, A. M., Barshtein, G., and Bergelson, L. D. (2003). Biochemical alterations of erythrocytes as an indicator of mental disorders: an overview. Harv. Rev. Psychiatry 11, 317–332. doi: 10.1080/10673220390264258
Pretorius, E., Olumuyiwa-Akeredolu, O.-O. O., Mbotwe, S., and Bester, J. (2016). Erythrocytes and their role as health indicator: using structure in a patient-orientated precision medicine approach. Blood Rev. 30, 263–274. doi: 10.1016/j.blre.2016.01.001
Quastel, J. H. (1932). Biochemistry and mental disorder. Lancet 220, 1417–1419. doi: 10.1016/S0140-6736(00)97278-7
Rabini, R. A., Petruzzi, E., Staffolani, R., Tesei, M., Fumelli, P., Pazzagli, M., et al. (1997). Diabetes mellitus and subjects' ageing: a study on the ATP content and ATP-related enzyme activities in human erythrocytes. Eur. J. Clin. Invest. 27, 327–332. doi: 10.1046/j.1365-2362.1997.1130652.x
Ravi, L. B., Mohanty, J. G., Chrest, F. J., Jayakumar, R., Nagababu, E., Usatyuk, P. V., et al. (2004). Influence of beta-amyloid fibrils on the interactions between red blood cells and endothelial cells. Neurol. Res. 26, 579–585. doi: 10.1179/016164104225016227
Reddy, P. H. (2006). Amyloid precursor protein-mediated free radicals and oxidative damage: implications for the development and progression of Alzheimer's disease. J. Neurochem. 96, 1–13. doi: 10.1111/j.1471-4159.2005.03530.x
Resnick, L. M., Gupta, R. K., Barbagallo, M., and Laragh, J. H. (1994). Is the higher incidence of ischemic disease in patients with hypertension and diabetes related to intracellular depletion of high energy metabolites? Am. J. Med. Sci. 307(Suppl. 1), S66–S69.
Richards, R. S., Wang, L., and Jelinek, H. (2007). Erythrocyte oxidative damage in chronic fatigue syndrome. Arch. Med. Res. 38, 94–98. doi: 10.1016/j.arcmed.2006.06.008
Richardson, J. T. (1990). Cognitive function in diabetes mellitus. Neurosci. Biobehav. Rev. 14, 385–388. doi: 10.1016/S0149-7634(05)80060-0
Riegel, B., Bennett, J. A., Davis, A., Carlson, B., Montague, J., Robin, H., et al. (2002). Cognitive impairment in heart failure: issues of measurement and etiology. Am. J. Crit. Care Off. Publ. Am. Assoc. Crit. Care Nurses 11, 520–528.
Rifkind, J. M., Abugo, O. O., Peddada, R. R., Patel, N., Speer, D., Balagopalakrishna, C., et al. (1999). Maze learning impairment is associated with stress hemopoiesis induced by chronic treatment of aged rats with human recombinant erythropoietin. Life Sci. 64, 237–247. doi: 10.1016/S0024-3205(98)00559-1
Roberts, G. W., Allsop, D., and Bruton, C. (1990). The occult aftermath of boxing. J. Neurol. Neurosurg. Psychiatry 53, 373–378. doi: 10.1136/jnnp.53.5.373
Rönnbäck, L., and Hansson, E. (2004). On the potential role of glutamate transport in mental fatigue. J. Neuroinflammation 1:22. doi: 10.1186/1742-2094-1-22
Ronquist, G., and Waldenström, A. (2003). Imbalance of plasma membrane ion leak and pump relationship as a new aetiological basis of certain disease states. J. Intern. Med. 254, 517–526. doi: 10.1111/j.1365-2796.2003.01235.x
Roselli, F., Tirard, M., Lu, J., Hutzler, P., Lamberti, P., Livrea, P., et al. (2005). Soluble β-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J. Neurosci. Off. J. Soc. Neurosci. 25, 11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005
Rothstein, G. (1993). Hematopoiesis in the aged: a model of hematopoietic dysregulation? Blood 82, 2601–2604.
Sakuta, S. (1981). Blood filtrability in cerebrovascular disorders, with special reference to erythrocyte deformability and ATP content. Stroke 12, 824–828. doi: 10.1161/01.STR.12.6.824
Samaja, M., Crespi, T., Guazzi, M., and Vandegriff, K. D. (2003). Oxygen transport in blood at high altitude: role of the hemoglobin-oxygen affinity and impact of the phenomena related to hemoglobin allosterism and red cell function. Eur. J. Appl. Physiol. 90, 351–359. doi: 10.1007/s00421-003-0954-8
Sansoni, P., Cossarizza, A., Brianti, V., Fagnoni, F., Snelli, G., Monti, D., et al. (1993). Lymphocyte subsets and natural killer cell activity in healthy old people and centenarians. Blood 82, 2767–2773.
Schmitt, F. A., Davis, D. G., Wekstein, D. R., Smith, C. D., Ashford, J. W., and Markesbery, W. R. (2000). “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology 55, 370–376. doi: 10.1212/WNL.55.3.370
Schneider, J. A., Arvanitakis, Z., Bang, W., and Bennett, D. A. (2007). Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24
Scholz, W. (1938). Studien zur Pathologie der Hirngefasse II. Die drusige Entartung der Hirnarterien und capillarien. Z Ges Neurol. Psychiat. 162, 694–715. doi: 10.1007/BF02890989
Seeman, P., Cheng, D., and Iles, G. H. (1973). Structure of membrane holes in osmotic and saponin hemolysis. J. Cell Biol. 56, 519–527. doi: 10.1083/jcb.56.2.519
Selkoe, D. J. (2007). Developing preventive therapies for chronic diseases: lessons learned from Alzheimer's disease. Nutr. Rev. 65, S239–S243. doi: 10.1301/nr.2007.dec.S239-S243
Selkoe, D. J. (2008). Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113. doi: 10.1016/j.bbr.2008.02.016
Shevtsova, E. F., Vinogradova, D. V., Neganova, M. E., Avila-Rodriguez, M., Ashraf, G. M., Barreto, G. E., et al. (2017). Mitochondrial permeability transition pore as a suitable target for neuroprotective agents against Alzheimer's Disease. CNS Neurol. Disord. Drug Targets 16, 677–685. doi: 10.2174/1871527316666170424114444
Shi, J., Xiang, Y., and Simpkins, J. W. (1997). Hypoglycemia enhances the expression of mRNA encoding β-amyloid precursor protein in rat primary cortical astroglial cells. Brain Res. 772, 247–251. doi: 10.1016/S0006-8993(97)00827-5
Shinozuka, T., Miyamoto, T., Hirazono, K., Ebisawa, K., Murakami, M., Kuroshima, Y., et al. (1994). Follow-up laparoscopy in patients with ovarian cancer. Tokai J. Exp. Clin. Med. 19, 53–59.
Simpson, I. A., Chundu, K. R., Davies-Hill, T., Honer, W. G., and Davies, P. (1994). Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer's disease. Ann. Neurol. 35, 546–551. doi: 10.1002/ana.410350507
Simpson, I. A., and Davies, P. (1994). Reduced glucose transporter concentrations in brains of patients with Alzheimer's disease. Ann. Neurol. 36, 800–801. doi: 10.1002/ana.410360522
Simpson, P. J., Mickelson, J., Fantone, J. C., Gallagher, K. P., and Lucchesi, B. R. (1988). Reduction of experimental canine myocardial infarct size with prostaglandin E1: inhibition of neutrophil migration and activation. J. Pharmacol. Exp. Ther. 244, 619–624.
Singh, V. K., Cheng, J. F., and Leu, S. J. (1994). Effect of substance P and protein kinase inhibitors on beta-amyloid peptide-induced proliferation of cultured brain cells. Brain Res. 660, 353–356. doi: 10.1016/0006-8993(94)91313-7
Smith, C. D., Carney, J. M., Starke-Reed, P. E., Oliver, C. N., Stadtman, E. R., Floyd, R. A., et al. (1991). Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 88, 10540–10543. doi: 10.1073/pnas.88.23.10540
Smith, D. H., Chen, X.-H., Iwata, A., and Graham, D. I. (2003). Amyloid β accumulation in axons after traumatic brain injury in humans. J. Neurosurg. 98, 1072–1077. doi: 10.3171/jns.2003.98.5.1072
Smith, E. E., and Greenberg, S. M. (2009). β-amyloid, blood vessels, and brain function. Stroke 40, 2601–2606. doi: 10.1161/STROKEAHA.108.536839
Smith, M. A., Casadesus, G., Joseph, J. A., and Perry, G. (2002a). Amyloid-β and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radic. Biol. Med. 33, 1194–1199. doi: 10.1016/S0891-5849(02)01021-3
Smith, M. A., Drew, K. L., Nunomura, A., Takeda, A., Hirai, K., Zhu, X., et al. (2002b). Amyloid-β, tau alterations and mitochondrial dysfunction in Alzheimer disease: the chickens or the eggs? Neurochem. Int. 40, 527–531. doi: 10.1016/S0197-0186(01)00123-1
Smith, M. A., Hirai, K., Hsiao, K., Pappolla, M. A., Harris, P. L., Siedlak, S. L., et al. (1998). Amyloid-β deposition in Alzheimer transgenic mice is associated with oxidative stress. J. Neurochem. 70, 2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x
Smith, M. A., Joseph, J. A., and Perry, G. (2000). Arson. Tracking the culprit in Alzheimer's disease. Ann. N. Y. Acad. Sci. 924, 35–38. doi: 10.1111/j.1749-6632.2000.tb05557.x
Solerte, S. B., Ceresini, G., Ferrari, E., and Fioravanti, M. (2000). Hemorheological changes and overproduction of cytokines from immune cells in mild to moderate dementia of the Alzheimer's type: adverse effects on cerebromicrovascular system. Neurobiol. Aging 21, 271–281. doi: 10.1016/S0197-4580(00)00105-6
Storey, E., Hyman, B. T., Jenkins, B., Brouillet, E., Miller, J. M., Rosen, B. R., et al. (1992). 1-Methyl-4-phenylpyridinium produces excitotoxic lesions in rat striatum as a result of impairment of oxidative metabolism. J. Neurochem. 58, 1975–1978. doi: 10.1111/j.1471-4159.1992.tb10080.x
Tabaton, M., and Gambetti, P. (2006). Soluble amyloid-β in the brain: the scarlet pimpernel. J. Alzheimers Dis. 9, 127–132. doi: 10.3233/JAD-2006-9S315
Tabaton, M., and Piccini, A. (2005). Role of water-soluble amyloid-β in the pathogenesis of Alzheimer's disease. Int. J. Exp. Pathol. 86, 139–145. doi: 10.1111/j.0959-9673.2005.00428.x
Teller, J. K., Russo, C., DeBusk, L. M., Angelini, G., Zaccheo, D., Dagna-Bricarelli, F., et al. (1996). Presence of soluble amyloid beta-peptide precedes amyloid plaque formation in Down's syndrome. Nat. Med. 2, 93–95. doi: 10.1038/nm0196-93
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Tesco, G., Koh, Y. H., Kang, E. L., Cameron, A. N., Das, S., Sena-Esteves, M., et al. (2007). Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron 54, 721–737. doi: 10.1016/j.neuron.2007.05.012
Thal, D. R., Capetillo-Zarate, E., Larionov, S., Staufenbiel, M., Zurbruegg, S., and Beckmann, N. (2009). Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol. Aging 30, 1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017
Thal, D. R., Ghebremedhin, E., Rüb, U., Yamaguchi, H., Del Tredici, K., and Braak, H. (2002). Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61, 282–293. doi: 10.1093/jnen/61.3.282
Thal, D. R., Griffin, W. S. T., and Braak, H. (2008). Parenchymal and vascular Aβ-deposition and its effects on the degeneration of neurons and cognition in Alzheimer's disease. J. Cell. Mol. Med. 12, 1848–1862. doi: 10.1111/j.1582-4934.2008.00411.x
Thanvi, B., and Robinson, T. (2006). Sporadic cerebral amyloid angiopathy–an important cause of cerebral haemorrhage in older people. Age Ageing 35, 565–571. doi: 10.1093/ageing/afl108
Tikhonova, L. A., Kaminsky, Y. G., Reddy, V. P., Li, Y., Solomadin, I. N., Kosenko, E. A., et al. (2014). Impact of amyloid β25-35 on membrane stability, energy metabolism, and antioxidant enzymes in erythrocytes. Am. J. Alzheimers Dis. Other Demen. 29, 685–695. doi: 10.1177/1533317514534757
Toro-Urrego, N., Garcia-Segura, L. M., Echeverria, V., and Barreto, G. E. (2016). Testosterone protects mitochondrial function and regulates neuroglobin expression in astrocytic cells exposed to glucose deprivation. Front. Aging Neurosci. 8:152. doi: 10.3389/fnagi.2016.00152
Tsirka, A., Challa, A., and Lapatsanis, P. D. (1990). Red cell phosphate metabolism in preterm infants with idiopathic respiratory distress syndrome. Acta Paediatr. Scand. 79, 763–768. doi: 10.1111/j.1651-2227.1990.tb11552.x
Urbach, H. (2011). Comment on: Brain Microbleeds and Alzheimer's disease: innocent observation or key player?: Cordonnier C, van der Flier WM. Brain. 2011;134:335-44. Clin. Neuroradiol. 21, 43–44. doi: 10.1007/s00062-011-0063-8
Uryu, K., Chen, X.-H., Martinez, D., Browne, K. D., Johnson, V. E., Graham, D. I., et al. (2007). Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 208, 185–192. doi: 10.1016/j.expneurol.2007.06.018
Valeri, C. R., and Hirsch, N. M. (1969). Restoration in vivo of erythrocyte adenosine triphosphate, 2,3-diphosphoglycerate, potassium ion, and sodium ion concentrations following the transfusion of acid-citrate-dextrose-stored human red blood cells. J. Lab. Clin. Med. 73, 722–733.
van der Zwaluw, C. S., Valentijn, S. A. M., Nieuwenhuis-Mark, R., Rasquin, S. M. C., and van Heugten, C. M. (2011). Cognitive functioning in the acute phase poststroke: a predictor of discharge destination? J. Stroke Cerebrovasc. Dis. 20, 549–555. doi: 10.1016/j.jstrokecerebrovasdis.2010.03.009
van Groen, T., Puurunen, K., Mäki, H.-M., Sivenius, J., and Jolkkonen, J. (2005). Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 36, 1551–1556. doi: 10.1161/01.STR.0000169933.88903.cf
Vanhanen, M., Koivisto, K., Karjalainen, L., Helkala, E. L., Laakso, M., Soininen, H., et al. (1997). Risk for non-insulin-dependent diabetes in the normoglycaemic elderly is associated with impaired cognitive function. Neuroreport 8, 1527–1530. doi: 10.1097/00001756-199704140-00041
van Wijk, R., and van Solinge, W. W. (2005). The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood 106, 4034–4042. doi: 10.1182/blood-2005-04-1622
Velliquette, R. A., O'Connor, T., and Vassar, R. (2005). Energy inhibition elevates β-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J. Neurosci. 25, 10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005
Villain, N., Desgranges, B., Viader, F., de la Sayette, V., Mézenge, F., Landeau, B., et al. (2008). Relationships between hippocampal atrophy, white matter disruption, and gray matter hypometabolism in Alzheimer's disease. J. Neurosci. 28, 6174–6181. doi: 10.1523/JNEUROSCI.1392-08.2008
Vonsattel, J. P., Myers, R. H., Hedley-Whyte, E. T., Ropper, A. H., Bird, E. D., and Richardson, E. P. (1991). Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann. Neurol. 30, 637–649. doi: 10.1002/ana.410300503
Vora, S. (1987). Metabolic manipulation of key glycolytic enzymes: a novel proposal for the maintenance of red cell 2,3-DPG and ATP levels during storage. Biomed. Biochim. Acta 46, S285–S289.
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Walsh, D. M., and Selkoe, D. J. (2004). Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 44, 181–193. doi: 10.1016/j.neuron.2004.09.010
Wang, J., Dickson, D. W., Trojanowski, J. Q., and Lee, V. M. (1999). The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp. Neurol. 158, 328–337. doi: 10.1006/exnr.1999.7085
Webster, M. T., Pearce, B. R., Bowen, D. M., and Francis, P. T. (1998). The effects of perturbed energy metabolism on the processing of amyloid precursor protein in PC12 cells. J. Neural Transm. 105, 839–853. doi: 10.1007/s007020050098
Weller, R. O., Boche, D., and Nicoll, J. A. R. (2009). Microvasculature changes and cerebral amyloid angiopathy in Alzheimer's disease and their potential impact on therapy. Acta Neuropathol. 118, 87–102. doi: 10.1007/s00401-009-0498-z
Whitson, J. S., Selkoe, D. J., and Cotman, C. W. (1989). Amyloid β protein enhances the survival of hippocampal neurons in vitro. Science 243, 1488–1490. doi: 10.1126/science.2928783
Williams, L. H., Udupa, K. B., and Lipshitz, D. A. (1986). Evaluation of the effect of age on hematopoiesis in the C57BL/6 mouse. Exp. Hematol. 14, 827–832.
Wimo, A., and Prince, M. (2010). World Alzheimer Report 2010: The Global Economic Impact of Dementia. Alzheimer's Disease International, London.
Wirths, O., Multhaup, G., and Bayer, T. A. (2004). A modified β-amyloid hypothesis: intraneuronal accumulation of the β-amyloid peptide–the first step of a fatal cascade. J. Neurochem. 91, 513–520. doi: 10.1111/j.1471-4159.2004.02737.x
Wisniewski, H. M., and Wegiel, J. (1994). Beta-amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol. 87, 233–241. doi: 10.1007/BF00296738
Wu, C.-W., Liao, P.-C., Yu, L., Wang, S.-T., Chen, S.-T., Wu, C.-M., et al. (2004). Hemoglobin promotes Aβ oligomer formation and localizes in neurons and amyloid deposits. Neurobiol. Dis. 17, 367–377. doi: 10.1016/j.nbd.2004.08.014
Xi, G., Keep, R. F., and Hoff, J. T. (1998). Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J. Neurosurg. 89, 991–996. doi: 10.3171/jns.1998.89.6.0991
Xi, G., Keep, R. F., and Hoff, J. T. (2006). Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 5, 53–63. doi: 10.1016/S1474-4422(05)70283-0
Xie, Z., Culley, D. J., Dong, Y., Zhang, G., Zhang, B., Moir, R. D., et al. (2008). The common inhalation anesthetic isoflurane induces caspase activation and increases amyloid β-protein level in vivo. Ann. Neurol. 64, 618–627. doi: 10.1002/ana.21548
Xie, Z., Dong, Y., Maeda, U., Alfille, P., Culley, D. J., Crosby, G., et al. (2006a). The common inhalation anesthetic isoflurane induces apoptosis and increases amyloid β protein levels. Anesthesiology 104, 988–994. doi: 10.1097/00000542-200605000-00015
Xie, Z., Dong, Y., Maeda, U., Moir, R., Inouye, S. K., Culley, D. J., et al. (2006b). Isoflurane-induced apoptosis: a potential pathogenic link between delirium and dementia. J. Gerontol. A. Biol. Sci. Med. Sci. 61, 1300–1306. doi: 10.1093/gerona/61.12.1300
Xie, Z., Moir, R. D., Romano, D. M., Tesco, G., Kovacs, D. M., and Tanzi, R. E. (2004). Hypocapnia induces caspase-3 activation and increases Aβ production. Neurodegener. Dis. 1, 29–37. doi: 10.1159/000076667
Yoshida, K., Furuse, M., Izawa, A., Iizima, N., Kuchiwaki, H., and Inao, S. (1996). Dynamics of cerebral blood flow and metabolism in patients with cranioplasty as evaluated by 133Xe CT and 31P magnetic resonance spectroscopy. J. Neurol. Neurosurg. Psychiatry 61, 166–171. doi: 10.1136/jnnp.61.2.166
Yu, N.-N., Wang, X.-X., Yu, J.-T., Wang, N.-D., Lu, R.-C., Miao, D., et al. (2010). Blocking β2-adrenergic receptor attenuates acute stress-induced amyloid β peptides production. Brain Res. 1317, 305–310. doi: 10.1016/j.brainres.2009.12.087
Zhang, B., Dong, Y., Zhang, G., Moir, R. D., Xia, W., Yue, Y., et al. (2008). The inhalation anesthetic desflurane induces caspase activation and increases amyloid β-protein levels under hypoxic conditions. J. Biol. Chem. 283, 11866–11875. doi: 10.1074/jbc.M800199200
Keywords: Alzheimer's disease, amyloid β, erythrocytes, metabolic dysfunction, multilayered pathology, clinical manifestation
Citation: Kosenko EA, Tikhonova LA, Montoliu C, Barreto GE, Aliev G and Kaminsky YG (2018) Metabolic Abnormalities of Erythrocytes as a Risk Factor for Alzheimer's Disease. Front. Neurosci. 11:728. doi: 10.3389/fnins.2017.00728
Received: 24 October 2017; Accepted: 13 December 2017;
Published: 05 January 2018.
Edited by:
Ghulam Md Ashraf, King Abdulaziz University, Saudi ArabiaReviewed by:
Etheresia Pretorius, Stellenbosch University, South AfricaZemin Wang, Harvard Medical School, United States
Copyright © 2018 Kosenko, Tikhonova, Montoliu, Barreto, Aliev and Kaminsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena A. Kosenko, eakos@rambler.ru
Gjumrakch Aliev, aliev03@gmail.com