A Molecular toolbox for rapid generation of viral vectors to up- or down-regulate neuronal gene expression in vivo
- Centre for Integrative Physiology, University of Edinburgh, Edinburgh, Scotland, UK
We introduce a molecular toolbox for manipulation of neuronal gene expression in vivo. The toolbox includes promoters, ion channels, optogenetic tools, fluorescent proteins, and intronic artificial microRNAs. The components are easily assembled into adeno-associated virus (AAV) or lentivirus vectors using recombination cloning. We demonstrate assembly of toolbox components into lentivirus and AAV vectors and use these vectors for in vivo expression of inwardly rectifying potassium channels (Kir2.1, Kir3.1, and Kir3.2) and an artificial microRNA targeted against the ion channel HCN1 (HCN1 miRNA). We show that AAV assembled to express HCN1 miRNA produces efficacious and specific in vivo knockdown of HCN1 channels. Comparison of in vivo viral transduction using HCN1 miRNA with mice containing a germ line deletion of HCN1 reveals similar physiological phenotypes in cerebellar Purkinje cells. The easy assembly and re-usability of the toolbox components, together with the ability to up- or down-regulate neuronal gene expression in vivo, may be useful for applications in many areas of neuroscience.
Introduction
Tools for manipulation of gene expression in neurons in vivo are important for investigation of nervous system function and may be of therapeutic benefit. Using viruses to manipulate neuronal gene expression in vivo is increasingly popular due to the flexibility and economy of generating viruses compared with transgenic animals. Nevertheless, it can be time consuming to assemble new viral constructs containing different promoter, gene of interest, and reporter elements. Furthermore, many of these elements have not been widely validated for use in the central nervous system and their performance may vary between studies. A toolbox of standardized modular components that can be rapidly assembled into new viral constructs is therefore desirable. A critical component of such a toolbox will be methods to disrupt expression of targeted genes. One promising approach is gene knockdown by RNA interference (RNAi). However, current methods using RNAi in neurons are associated with off target effects and toxicity (Grimm et al., 2006; McBride et al., 2008; Ehlert et al., 2010; Martin et al., 2011). In non-neuronal cells intronic expression of miRNAs has been suggested to alleviate these problems, while simultaneously enabling stable, high level co-expression of reporter genes (Du et al., 2006). However, it is not clear if this approach will produce specific or selective gene knockdown when introduced into neurons in vivo using viruses.
To address the goal of establishing a molecular toolbox for in vivo up- and down-regulation of neuronal gene expression, we generated and validated a set of vectors containing cassettes encoding promoters, genes of interest, intronic miRNA constructs, and reporter proteins flanked by recombination sites. These cassettes can be assembled into various combinations and inserted into a viral backbone through a single recombination reaction. The system is extremely flexible, allowing rapid assembly of gene-reporter fusion constructs, bicistronic expression of genes, and expression of intronic interfering RNAs for gene knockdown. We show that viruses produced from these components give stable over-expression or efficient knockdown of genes in neurons in vivo, while simultaneously expressing reporter genes detectable by native fluorescence. By focusing on manipulation of the HCN1 gene, which encodes an ion channel that has been relatively well characterized using knockout mice (e.g., Nolan et al., 2003, 2004, 2007; Chen et al., 2009, 2010; Huang et al., 2009), we go on to provide evidence that intronic expression of artificial miRNAs causes effective knockdown of gene expression and does not appear to be associated with off-target effects on physiological properties of neurons. Because this molecular toolbox enables identification and investigation of transduced cells by a variety of techniques, it may be useful for addressing a broad range of questions within neuroscience.
Methods
Vector Construction
The principle behind the molecular toolbox is to allow rapid generation of new constructs through recombination cloning. To create the initial att-flanked PCR fragments for recombination, traditional digestion, and ligation methods were used for some constructs, whereas other were generated by PCR amplification or synthesized directly.
Restriction enzyme-based cloning
pEYFP-HCN1 was constructed by amplifying the HCN1 sequence from pEGFP-HCN1 (gift from Bina Santoro, Columbia University, New York) and cloning it into the EcoRI and BamHI sites of pEYFP-C1 (Clontech). pFCK-ChR2-mCherry was constructed by replacing the EGFP sequence in pFCK-ChR2-EGFP (from Ed Boyden, MIT, obtained via Addgene) with mCherry digested from pmCherry-N1 (Clontech) using AgeI and BsrGI. To enhance transgene expression we used a version of the woodchuck hepatitis virus post-transcriptional regulatory element optimized for safety (oPRE; Schambach et al., 2006). A lentivirus with oPRE cloned into the backbone (pLenti-oPRE) was generated by PCR amplifying the oPRE sequence from pMP71-GFP-opre (provided by Axel Schambach, Hannover Medical School, Hannover) and cloning it into the XhoI and XbaI sites of pLenti6/BLOCK-iT-DEST backbone (Invitrogen).
To create a Gateway-compatible adeno-associated virus (AAV) vector, pCAGW-ChR2-Venus (from Karel Svoboda, obtained via Addgene; Petreanu et al., 2009) was digested with AfeI and EcoRV to remove the CAGW promoter and the ChR2-Venus insert. Reading Frame B of the Gateway Vector Conversion System (Invitrogen) was ligated into the AAV backbone to create pAAV-Gateway.
Recombination cloning
While several technologies are available for recombination cloning (e.g., Ma et al., 1987; Bubeck et al., 1993; Oliner et al., 1993) here we use the Gateway system (Invitrogen). This recombination cloning system uses a three-step strategy to generate expression constructs (Hartley et al., 2000; Sasaki et al., 2004; Yahata et al., 2005). Cassettes encoding individual components of the constructs are amplified by PCR using primers containing att sites that allow recombination. The PCR cassettes are then recombined into a donor vector with matching att sites to generate an entry vector. By using alternative matching pairs of att sites different arrangements of entry vectors can be recombined into a destination vector to generate new expression constructs in a single reaction.
Generation of att-flanked cassettes. Cassettes encoding the toolbox components were generated by PCR amplification using primers containing the required att recombination site sequences in addition to 18–25 base pairs of template-specific sequence. Two hundred nanomolars of each primer was added to 1–2 μl of template DNA and 22.5 μl Platinum PCR SuperMix High Fidelity (Invitrogen). Thirty-five cycles of amplification were performed at 94°C for 30 s, 55°C for 30 s, and 68°C for 60 s.
Generation of entry vectors. To insert the att-flanked PCR cassettes into a vector, 50 fmol of purified att-flanked PCR products were added to 50 fmol of the appropriate pDONR vector (Invitrogen), 2 μl of BP Clonase II enzyme (Invitrogen), and TE Buffer pH 8.0 to a total of 10 μl. The recombination reactions were incubated at 25°C overnight generating entry vectors. One microliter of proteinase K solution (Invitrogen) was added and the reactions were incubated at 37°C for 10 min. Three microliters of each reaction was transformed into TOP10 cells (Invitrogen) and positive transformants were selected with kanamycin (Sigma-Aldrich).
Generation of viral destination vectors. To generate viral vectors, 10 fmol of each of the required entry vectors were added to 20 fmol of the destination vector (pLenti6/BLOCK-iT/DEST, pLenti-oPRE, or AAV-Gateway), 2 μl LR Clonase II Plus enzyme mix (Invitrogen), and TE Buffer pH 8.0 to a total volume of 10 μl. The reactions were incubated overnight at 25°C and treated with 1 μl proteinase K solution (Invitrogen) at 37°C for 10 min. The entire reaction was transformed into Stbl3 cells (Invitrogen) and positive transformants were selected using ampicillin (Invitrogen) and blasticidin (Autogen Bioclear).
General purpose cassettes. CAMKIIα short (0.4 kb) and long (1.3 kb) promoter variants (Dittgen et al., 2004) were amplified from mouse genomic DNA. Synapsin and enhanced synapsin promoters were amplified from pBSIISK-SYN-GFP-WPRE and pBSIISK-E/SYN-GFP-WPRE respectively (vectors provided by Hiroyuki Hioki, Kyoto University, Kyoto). The Netrin G1 putative promoter was amplified from mouse genomic DNA. A 1.1-kb sequence corresponding to the first 345 bp + 754 bp immediately upstream of the transcription start site of the Netrin G1 (Ntng1) transcript variant a, gene (NM_030699.2) was cloned. Fluorescent reporter cassettes were amplified from pEGFP-C1, pEYFP-C1, pECFP-C1, and pmCherry-C1 (Clontech). An IRES-EGFP cassette was amplified from pPRIG (provided by Patrick Martin, UNICE, Nice). A cassette containing oPRE was generated by flanking the oPRE sequence with the appropriate recombination sites to ensure it would always insert 3′ to any coding cassettes during recombination into a viral vector. EYFP-HCN1 cassettes were amplified from pEYFP-HCN1. Channelrhodopsin-mCherry was amplified from pFCK-ChR2-mCherry. CAMKII(0.4)-Halorhodopsin-EGFP was amplified from pFCK-Halo-EGFP (from Ed Boyden, MIT, obtained via Addgene; Han and Boyden, 2007). Following PCR, the amplicons were gel purified using a QIAquick gel extraction kit (Qiagen). To generate entry vectors, all cassettes were recombined into the appropriate donor vector.
Reporter constructs. To generate a reporter construct for visualization of neurons in vivo, pENTR-L1-CAMKII(0.4)-L4, pENTR-R4-EYFP-R3, and pENTR-L3-oPRE-L2 were recombined with pLenti6/BLOCK-iT-DEST to create pLenti6-CAMKII(0.4)-EYFP. To test the Netrin G1 promoter a reporter lentivirus was made by recombining pENTR-L1-NetrinG1-L4, pENTR-R4-EYFP-R3, and pENTR-L3-oPRE-L2 with pLenti6/BLOCK-iT-DEST to create pLenti6-NetrinG1-EYFP.
Ion channel constructs. The coding sequences of Kir2.1 (Mus musculus potassium inwardly rectifying channel, subfamily J, member 2 (Kcnj2) NM_008425) fused to a C-terminal mCherry sequence was synthesized by Mr Gene1. The sequence was flanked by L5-L2 att recombination sites to enable recombination cloning. pENTR-L1-CAMKII(0.4)-R5 and pENTR-L5-Kir2.1-mCherry-R2 were recombined with the pLenti-oPRE backbone to create the lentivector pLenti–CAMKII(0.4)-Kir2.1-mCherry. The coding sequences of Kir3.1 [M. musculus potassium inwardly rectifying channel, subfamily J, member 3 (Kcnj3) NM_008426.1] and Kir3.2 [M. musculus potassium inwardly rectifying channel, subfamily J, member 6 (Kcnj6), transcript variant Girk2A-1, NM_010606] fused to a C-terminal EGFP sequence and flanked by R4-R3 att sites were synthesized by Mr Gene. Entry vectors encoding the Kir3-EGFP constructs were recombined with pENTR-L1-CAMKII(0.4)-L4, pENTR-L3-oPRE-L2, and pLenti6/BLOCK-iT-DEST to create the lentivectors pLenti6–CAMKII(0.4)-Kir3.1-EGFP and pLenti6–CAMKII(0.4)-Kir3.2-EGFP.
miRNA constructs. BLOCK-iT miR RNAi sequences targeting mouse HCN1 (Mmi510951, Mmi510952, and Mmi510953; Invitrogen) were cloned into the synthetic intron of pSM155 (provided by Guangwei Du, University of Texas Health Science Center, Houston) using procedures described in detail elsewhere (Du et al., 2006). Briefly, RNAi sequences were designed so that once annealed, the duplexes had four nucleotide overhangs compatible with the cohesive ends produced by BsmBI digestion of pSM155. The RNAi duplexes were annealed and ligated into pSM155. A cassette containing the intronic miRNA upstream of EGFP was then amplified with B4r and B3r PCR primers and recombined to generate the entry vector pENTR-R4-HCN1miR-EGFP-R3. To create a negative control miRNA construct, a cassette containing a miRNA targeting luciferase upstream of an EGFP sequence was amplified from pSM155-Luc (also from Guangwei Du) with B4r and B3r primers and recombined to create the entry vector pENTR-R4-LucmiR-EGFP-R3. To generate a lentivirus expressing the HCN1 miRNA, pENTR-L1-ESYN-L4 was recombined with pENTR-R4-HCN1miR-EGFP-R3, pENTR-L3-oPRE-L2, and the pLenti6/BLOCK-iT-DEST backbone to create the lentivector pLenti6-ESYN-HCN1miR-EGFP. To generate AAV expressing miRNAs for experiments in Figures 5–7, pENTR-L1-ESYN-L4 and pENTR-L3-oPRE-L2 were recombined with pAAV-Gateway and pENTR-R4-HCN1miR-EGFP-R3 or pENTR-R4-LucmiR-EGFP-R3 to create pAAV-ESYN-HCN1miR-EGFP and pAAV-ESYN-LucmiR-EGFP.
PCR Primers
CAMKII(1.3) attB1: GGGGACAAGTTTGTACAAAAAAGCAGG CTCATTATGGCCTTAGGTCACTT
CAMKII(0.4) attB1: GGGGACAAGTTTGTACAAAAAAGCA GGCTACTTGTGGACTAAGTTTGTTC
CAMKII attB5r: GGGGACAACTTTTGTATACAAAGTTGTT GCCCCCAGAACTAGGGGCCACT
CAMKII attB4: GGGGACAACTTTGTATAGAAAAGTTGGG TGCCCCAGAACTAGGGGCCACT
SYN attB1: GGGGACAAGTTTGTACAAAAAAGCATACCTG CAGAGGGCCCTGCGTAT
SYN attB5r: GGGGACAACTTTTGTATACAAAGTTGTCGCC GCAGCGCAGATGGTC
SYN attB4:
GGGGACAACTTTGTATAGAAAAGTTGGGTGCGCCGCAG CGCAGATGGT
ESYN attB1: GGGGACAAGTTTGTACAAAAAAGCATACTA GTTATTAATAGTAATCAATT
NetrinG1 attB1:
GGGGACAAGTTTGTACAAAAAAGCAGGCTCTCCTACTT GGACTTCAAC
NetrinG1 attB4:
GGGGACAACTTTGTATAGAAAAGTTGGGTGCTTCCGAG GTCTCGGCAA
EXFP attB5: GGGGACAACTTTGTATACAAAAGTTGACCAT GGTGAGCAAGGGCGAGGA
EXFP attB2: GGGGACCACTTTGTACAAGAAAGCTGGGTAT TACTTGTACAGCTCGTCCA
EXFP attB4r: GGGGACAACTTTTCTATACAAAGTTGACCA TGGTGAGCAAGGGCGAGGA
EXFP attB3r: GGGGACAACTTTATTATACAAAGTTGTTTA CTTGTACAGCTCGTCCAT
IRES-EGFP attB3: GGGGACAACTTTGTATAATAAAGTTGG TTACTGGCCGAAGCCGCTT
oPRE attB3: GGGGACAACTTTGTATAATAAAGTTGGAGC ATCTTACCGCCATTTATACC
oPRE attB5: GGGGACAACTTTGTATACAAAAGTTGGAGC ATCTTACCGCCATTTATACC
oPRE attB2: GGGGACCACTTTGTACAAGAAAGCTGGGTA CGACAACACCACGGAATT
miR EXFP attB5: GGGGACAACTTTGTATACAAAAGTTGC TAAGGTAGCCTTGCAGAAGTTGG
miR EXFP attB4r: GGGGACAACTTTTCTATACAAAGTTGC TAAGGTAGCCTTGCAGAAGTTGG
ChR2 attB5: GGGGACAACTTTGTATACAAAAGTTGATGCC ACCATGGACTATGGCGGCG
ChR2 attB4r: GGGGACAACTTTTCTATACAAAGTTGCCAC CATGGACTATGGCGGCG
HCN1 attB5: GGGGACAACTTTGTATACAAAAGTTGATAT GGAAGGCGGCGGCAAACC
HCN1 attB4r: GGGGACAACTTTTCTATACAAAGTTGATAT GGAAGGCGGCGGCAAACC
HCN1 attB3r: GGGGACAACTTTATTATACAAAGTTGTTAA ATTCGAAGCAAAACGGGG
K2P9.1 attB4r: GGGGACAACTTTTCTATACAAAGTTGATA TGAAGCGGCAGAACGTGCGT
K2P9.1 attB3r: GGGGACAACTTTATTATACAAAGTTGTTT AGATGGACTTGCGACGGAGGTG
HCN1F_EcoRI: AATTCTAATGGAAGGCGGCG
HCN1R_BamHI: GATCTAAATTCGAAGCAAAACGG
Cell Culture and Transfections
HEK293FT cells (Invitrogen) were maintained at 37°C, 5% CO2 in DMEM with GlutaMAX I, 4,500 mg/l glucose and 110 mg/l sodium pyruvate (Gibco) supplemented with 50 U/ml each of penicillin and streptomycin (Sigma) and 10% v/v FCS (Invitrogen). DNA transfections of miRNA constructs were performed using FuGENE6 (Roche) according to manufacturer’s instructions.
Virus Production
Lentiviruses were generated by triple transfection of HEK293FT cells (Invitrogen) with the modified lentivector, psPAX2 (courtesy of Didier Trono, EPFL, Lausanne, obtained via Addgene) and the pLP/VSVG envelope plasmid (Invitrogen) using calcium phosphate transfection. The lentiviruses were harvested in serum-free medium after 3 days, filtered, and concentrated in primed Centricon Plus-80 filter devices (Millipore). Functional titer was determined by EGFP fluorescence after serial dilution on HEK293FT cells and was 0.5–2 × 108 TU/ml. Hybrid AAVs containing the ITR from AAV2 and Capsid from AAV1 were produced by Vectorbiolabs2. Titer was determined by measurement of vector genome copies and was 5 × 1013 GC/ml.
Stereotaxic Injection
All animal experiments were carried out according to the guidelines laid down by the University of Edinburgh’s Animal Welfare Committee and in accordance with the UK Animals (Scientific Procedures) Act 1986. C57BL6/JOlaHsd mice aged 4–6 weeks were anesthetized with isoflurane and mounted into a stereotaxic frame. 0.2–1 μl of virus was injected at 0.1 μl/min using pre-calibrated pulled glass pipettes with a tip diameter of 6–10 μm following a previously described protocol (Cetin et al., 2006). The pipette was left in place for at least 5 min following the injection before being slowly raised. For the hippocampal injections, co-ordinates measured in millimeter from bregma were: −1.8 A/P, ±1.6 M/L, −1.3 D/V for CA1, −1.8 A/P, ±1.4 M/L, −1.6 D/V for dentate gyrus. Cerebellum injections targeted the vermis and were measured relative to lambda. Co-ordinates were −2 A/P and 0–1.5 M/L. Injections of ∼200 nl of virus were made at Z = −3, −2.5, −2, −1.5, −1, and −0.5 either with the injecting arm angled back 10° and positioned over the midline, or angled back 30° and rotated 45° counter-clockwise to target more lobules. Following surgery, animals were returned to their home cages for at least 2 weeks before being used for analysis.
Imaging
Mice were terminally anesthetized with sodium pentobarbital and perfused with 4% PFA. The brains were post-fixed and immersed in 30% sucrose overnight before 30 μm coronal sections were cut on a freezing microtome. Native fluorescence of reporter genes was imaged using Leica TCS-NT and Zeiss LSM510 confocal microscopes. For imaging of spines, data were acquired using a 1,024 × 1,024 pixel image size and a Zeiss Plan NeoFLUAR 1.4 NA 63× oil immersion lens. Image data, acquired at Nyquist sampling rates, were deconvolved using Huygens software (Scientific Volume Imaging, Netherlands), and the resulting three-dimensional images were analyzed using NIH ImageJ software.
FACS Sorting
Lentivirus expressing Kir2.1-mCherry was stereotaxically targeted to the dentate gyrus of 4-week-old C57BL6/JOlaHsd mice. Two weeks after injections the mice were culled and the brains rapidly removed and placed in modified oxygenated artificial cerebrospinal fluid (ACSF) of the following composition (mM): NaCl 86, NaH2PO4 1.2, KCl 2.5, NaHCO3 25, CaCl2 0.5, MgCl2 7, glucose 25, sucrose 75, at 6°C for 10 min. Four hundred micrometers coronal slices were cut through the hippocampus on a Vibratome 3000 sectioning system. The slices were transferred to oxygenated ACSF + 10 mM HEPES at 37°C and incubated for 10 min. The dorsal dentate gyrus from each slice was microdissected out under a dissecting microscope. All samples from a single brain were pooled together in 1 well of a 12-well plate with 900 μl oxygenated ACSF + HEPES. One hundred microliters papain (200 U/ml, Sigma-Aldrich) was added to each well and the samples were incubated at 37°C in 5% CO2 for 45 min to dissociate the cells. The digestion reaction was stopped by the addition of 100 μl BSA (10 mg/ml; Sigma-Aldrich), 11 μl FCS (Invitrogen), 10 μl DNase (1,000 U/ml, Sigma-Aldrich), and 10 μl 5 mM Leupeptin (Sigma-Aldrich). The samples were triturated through a fire-polished Pasteur pipette until no clumps were visible. Two milliliters of cold ACSF + HEPES was added to each sample and they were passed through a 70-μm sieve (Becton Dickinson) to remove any remaining cell clumps. The cells were counted with a hemocytometer and resuspended in cold ACSF + HEPES to a density of 1 × 106 cells/ml. One microliter per milliliter of Live/Dead stain with a green fluorescent dye (Invitrogen) was added to each sample and they were incubated on ice for 30 min in the dark. The samples were centrifuged at 800 rpm and washed twice in 1 ml PBS + 1% FCS to remove unbound stain. Samples from an uninjected brain were prepared alongside the experimental brains to use as controls for the fluorescence-activated cell sorting (FACS) sorting and subsequent RT-PCR. A sample from the uninjected brain was heat-treated at 50°C to provide a positive control for calibration of the Live/Dead stain. The samples were sorted on a FACS Aria II (BD Biosciences, USA) with gating parameters set to collect live mCherry-fluorescent cells. The yield of positive cells isolated from the dentate gyrus of a single brain ranged from 5,249 to 14,624 cells.
Real Time PCR
Total mRNA was extracted using the Arcturus PicoPureAR RNA Isolation Kit (Applied Biosystems) in combination with the RNase-Free DNase Set (Qiagen) according to manufacturer’s instructions. Taqman Gene expression assays were used for amplification of target genes. Specific assays used were Mm00484848_m1 (Mcm6), Mm01326464_m1 (NeuroD6), Mm01253033_m1 (Gfap), and Mm00446968_m1 (Hprt1). RT-PCR reactions were set up using the TaqManAR RNA-to-CT 1-Step Kit (Applied Biosystems). One microliter of mRNA template was added to 1 μl of the gene-specific probe and primers mix, 10 μl 2× TaqMan RT-PCR mix, 0.5 μl 40× TaqMan RT Enzyme mix, and 7.5 μl H2O. Reverse transcription was performed at 48°C for 15 min followed by activation of AmpliTaq Gold DNA Polymerase at 95°C for 10 min and 40 cycles of amplification at 95°C for 15 s and 60°C for 1 min on an Mx3000P QPCR system (Stratagene). Quantification was performed using the comparative Ct method with normalization to Hprt1. Results were expressed as mean ± standard error of the mean and statistical analyses (ANOVA and Student’s t-test) were performed using Smith’s Statistical Package, version 2.80 or R (The R Foundation).
Electrophysiology
Cerebellar brain slices were prepared and whole-cell patch-recordings made from visually identified cerebellar Purkinje cells following previously described procedures (Nolan et al., 2003; Zonta et al., 2011). Briefly, sagittal cerebellar slices were cut in cold (4–6°C) modified oxygenated ACSF of the following composition (mM): NaCl 86, NaH2PO4 1.2, KCl 2.5, NaHCO3 25, CaCl2 0.5, MgCl2 7, glucose 25, and sucrose 75. Slices were then maintained in standard oxygenated ACSF of the following composition: NaCl 124, NaH2PO4 1.2, KCl 2.5, NaHCO3 25, CaCl2 2, MgCl2 1, and glucose 20. Slices were initially kept for 10–20 min at 37 ± 1°C and then at room temperature for up to 6 h before making recordings. Patch-clamp recordings were obtained in standard ACSF using electrodes with resistance 2–5 MΩ. Whole-cell voltage-clamp recordings of hyperpolarization-activated currents were made at room temperature (18–22°C) in the following ACSF modified to reduce activation of other voltage-gated conductances: (mM) NaCl 115, NaH2PO4 1.2, KCl 5, BaCl2 1, CdCl2 0.1, 4-AP 1, NiCl2 1, TEA 5, CaCl2 2, MgCl2 1, NBQX 0.2, D-AP5 0.2, picrotoxin 0.5, and tetrodotoxin 0.001. Virally transduced cells were identified by EGFP fluorescence. All recordings from non-transduced (EGFP negative) cells were in slices also containing transduced (EGFP positive) cells. Recordings of action potentials and voltage responses to current steps were made in cell-attached voltage-clamp and whole-cell current-clamp configurations in standard ACSF at 34–36°C. Electrophysiological data were acquired using an Instrutech ITC-18 AD boards (HEKA Elektronik) and Axograph software. Analysis used custom written routines in IGOR pro (Wavemetrics) and R (R Foundation). Comparison of group data used analysis of variance (ANOVA and Student’s t-test as appropriate). All data are expressed as mean ± standard error of the mean.
Results
A Molecular Toolbox for Rapid Generation of Viral Constructs
To enable rapid generation of new viral constructs we constructed a library of vectors containing cassettes compatible with the Gateway cloning system (Figure 1). The Gateway system uses site-specific recombination for directional cloning of PCR products and their subsequent subcloning into destination vectors. DNA fragments are amplified by PCR using primers that contain specific recombination sites (att sites). Entry vectors are generated using the BP Clonase enzyme to catalyze the insertion of the DNA fragment into a donor vector containing matching att sites. To generate the final construct the entry vector can then be recombined with a destination vector through the addition of LR Clonase. By including different att sites in each vector, att-flanked cassettes from up to four entry vectors can be directionally recombined into a destination vector in a single reaction (Figures 1A,C). Compared with conventional cloning approaches this reduces the number of steps and time required to generate new constructs and allows the same components to be easily rearranged to form new viral vectors. The att-flanked cassettes that we describe here encode neuronal promoters, genes of interest, reporter genes, or intronic miRNAs (Figure 1B). The promoter cassettes are flanked with the appropriate att sites to ensure they insert as the first cassette in the viral backbone and therefore drive expression of downstream cassettes. The reporter cassettes are flanked with att sites permitting their expression either alone or as N- or C-terminal fusion proteins. Because we wanted to transduce neurons with either lentivirus or AAV and as there are currently no AAV vectors available that are compatible with the Gateway cloning system, we converted pACAGW-ChR2-Venus-AAV (Petreanu et al., 2009) into a Gateway-compatible destination vector by removing the CAG promoter and the ChR2-Venus insert and replacing them with a recombination cassette. Functionality of all the modular components of the toolbox was confirmed in vitro before using a subset to generate viruses for further in vivo validation (Table 1).
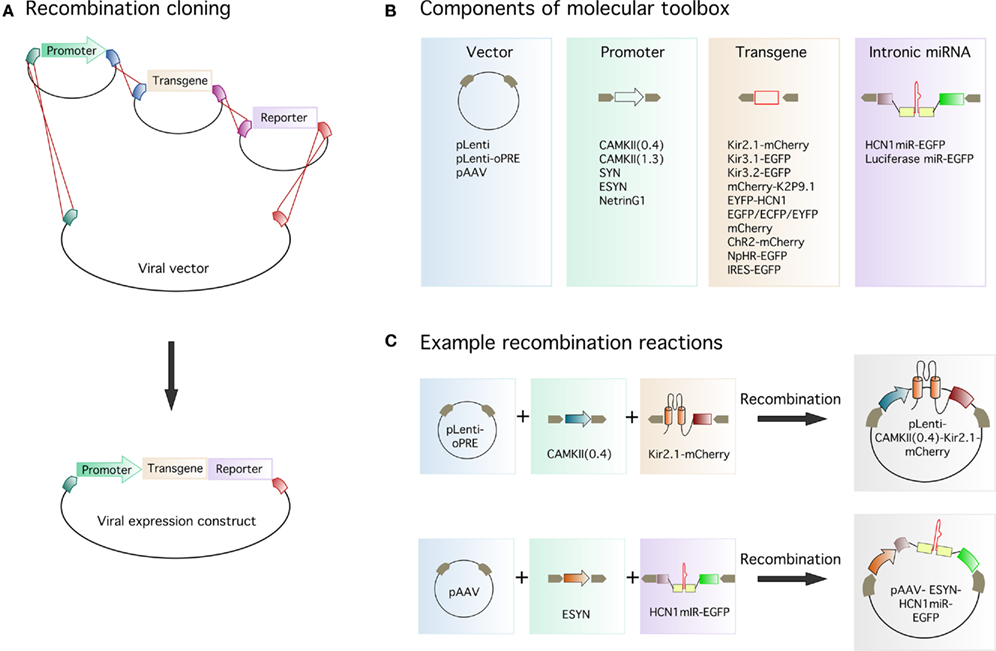
Figure 1. Schematic illustrating modular cloning system. (A) Schematic illustrating recombination of entry vectors carrying cassettes flanked by att sites with a viral destination vector. In a single reaction recombination between matching att sites inserts the cassettes into the vector in the correct order, generating a viral expression construct. (B) Components of the molecular toolbox flanked with att sites ready for recombination. For simplicity, only the att-flanked regions of the entry vectors are illustrated in (B,C). (C) Examples of recombination reactions to generate the lentiviral expression construct plenti-CAMKII(0.4)-Kir2.1-mCherry and the AAV expression construct pAAV-ESYN-HCN1miR-EGFP.
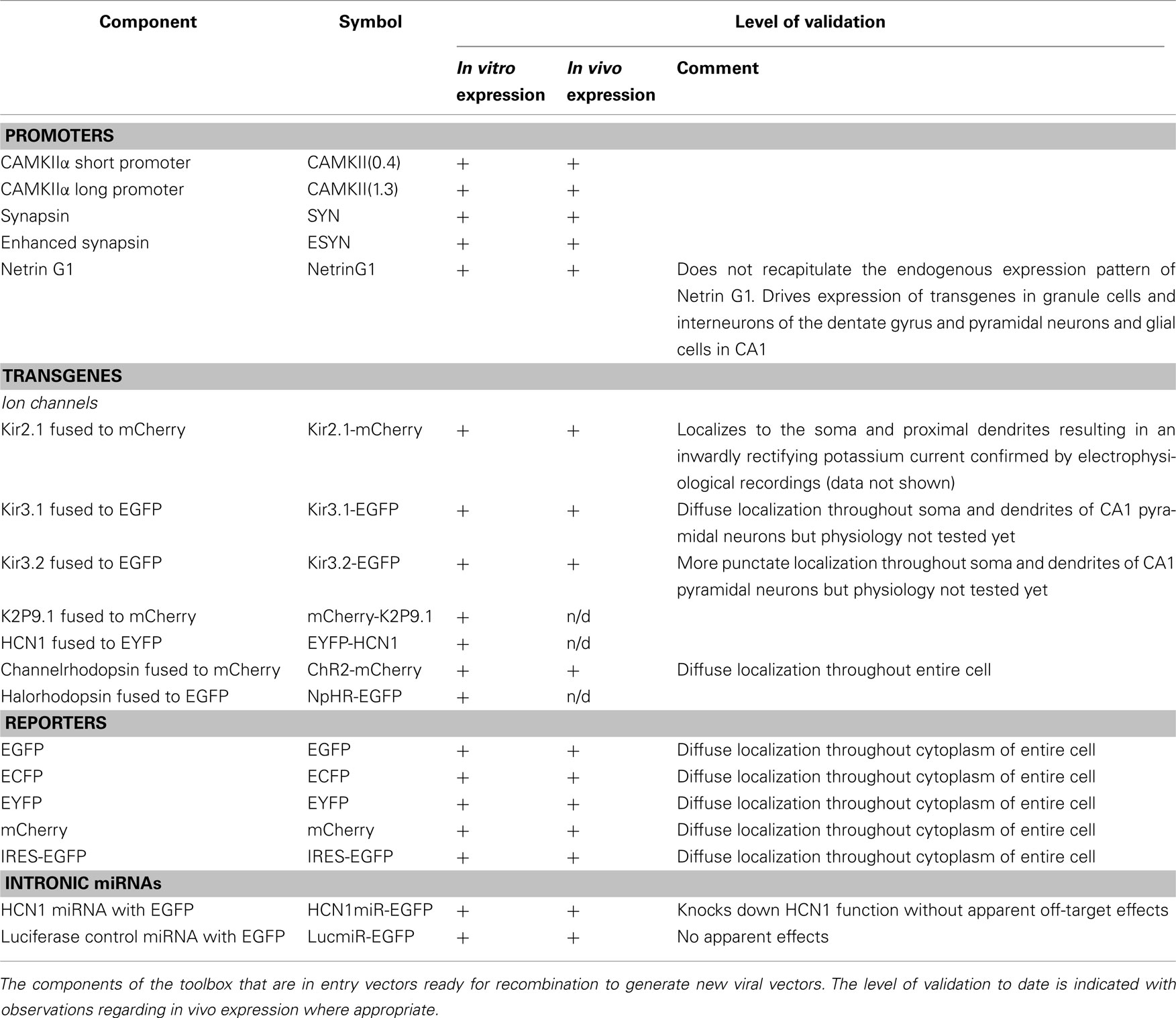
Table 1. Molecular toolbox components.
To evaluate expression of ion channels tagged with fluorescent reporter molecules, we first produced lentiviruses assembled from constructs encoding either an EYFP reporter alone, or Kir potassium channel-reporter fusion proteins, with expression driven by the short variant of the CAMKIIα promoter (CAMKII(0.4)-EYFP, CAMKII(0.4)-Kir2.1-mCherry, CAMKII(0.4)-Kir3.1-EGFP, and CAMKII(0.4)-Kir3.2-EGFP). To examine their expression in hippocampal neurons in vivo, we stereotaxically injected the viruses into CA1. Two weeks later, we sectioned the brains and imaged the native fluorescence in virally transduced cells. All four viruses expressed their transgenes at high levels within neurons. The subcellular localization of the reporter protein differed between the viruses, indicating differential trafficking of each ion channel (Figure 2A). The Kir2.1 channel was localized to the soma and proximal dendrites of CA1 pyramidal neurons, whereas Kir3.1 and Kir3.2 were present throughout the proximal and distal dendrites. Importantly, the differences in distribution of each ion channel reflect expression patterns for native channels in vivo (Miyashita and Kubo, 1997; Koyrakh et al., 2005) suggesting that this approach will be useful to study ion channel localization (cf. Piskorowski et al., 2011). Expression of Kir2.1-mCherry results in an inwardly rectifying potassium current confirmed by electrophysiological recordings from acute slices (data not shown) in agreement with previous findings using a similar construct in cultured neurons (Burrone et al., 2002). We are yet to assess the functional consequences of expression of Kir3.1-EGFP and Kir3.2-EGFP, but similar constructs yield functional channels in expression systems (Stevens et al., 1997).
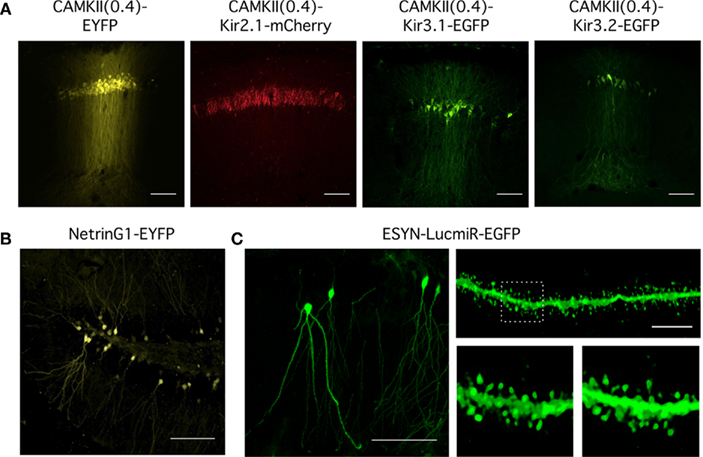
Figure 2. Lentiviral expression of transgenes in hippocampal neurons. Examples of gene expression from lentiviruses injected into the hippocampus. (A) Expression of EYFP or Kir-reporter fusion proteins in CA1 pyramidal neurons of the hippocampus driven by the CAMKIIα short promoter. (B) Expression of EYFP in dentate gyrus granule cells driven by the Netrin G1 promoter. (C) EGFP expression in dentate gyrus granule cells driven by the ESYN promoter. Reporter protein expression is bright enough to enable imaging of dendritic spines (upper and lower right panels). Higher magnification spine images (lower right panel) correspond to different depths from a Z-stack of the region indicated by the white box (upper right panel). Scale bars = 100 μm for all images except the spine image, where the scale bar = 5 μm.
Promoters other than CAMKIIα can also be successfully used to drive neuronal gene expression (Dittgen et al., 2004). To determine if a fragment of the putative Netrin G1 promoter can drive gene expression in vivo, we assembled the lentivirus NetrinG1-EYFP and injected it into the dentate gyrus. Consistent with in situ hybridization data from the Allen Brain Atlas which suggests the presence of Netrin G1 mRNA in the dentate gyrus, we found EYFP fluorescence in dentate gyrus granule cells transduced with NetrinG1-EYFP (Figure 2B). However, expression was not limited to granule cells, as we also observed occasional fluorescence in interneurons and glia (Table 1). The enhanced synapsin (ESYN) promoter also gave strong transgene expression in dentate gyrus granule cells transduced with the virus ESYN-LucmiR-EGFP (Figure 2C). The native fluorescence from this virus (and the other reporter viruses) was bright enough to allow imaging of dendritic spines (Figure 2C). Transgene expression was visible from 4 days post-injection, reached maximal intensity by 7–10 days post-injection and remained stable for at least 9–12 weeks (the longest time points we have analyzed for AAV and lentivirus respectively, data not shown).
Isolation of Neurons Transduced with Viruses in vivo for Downstream Analysis of Gene Expression
Expression of virally encoded fluorescent proteins in addition to interfering RNAs or transgenes enables identification of transduced neurons and in principle might allow their isolation from non-transduced neurons. We therefore assessed the amenability to FACS of neurons transduced using viruses generated from the toolbox (Figure 3).
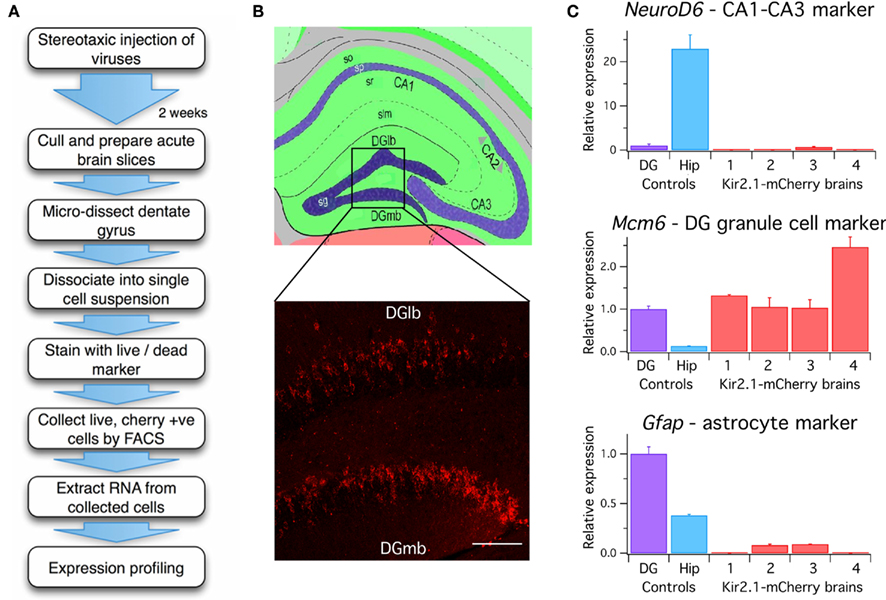
Figure 3. Analysis of mRNA expression from dentate gyrus granule cells transduced in vivo. (A) Flow diagram of procedure for ex vivo isolation of virally transduced cells from the dentate gyrus. (B) Upper panel: Schematic of the hippocampus adapted from the Allen Mouse Brain Reference Atlas. Virus was injected into the dentate gyrus. Lower panel: Expression of Kir2.1-mCherry in granule cells (sg) of the dentate gyrus lateral blade (DGlb) and medial blade (DGmb). Scale bar = 100 μm. (C) RT-PCR quantification of gene expression in Kir2.1-mCherry positive cell populations isolated from four lentivirally injected brains compared to control cells dissociated from the entire hippocampus (Hip) or microdissected dentate gyrus (DG) of pooled uninjected brains (n = 3). All reactions were performed in triplicate and normalized to the housekeeping gene Hprt1. Gene expression levels in the Kir2.1-mCherry positive cells are expressed relative to control cells isolated from the dentate gyrus of uninjected brains. A one-way ANOVA revealed a significant difference in gene expression between the brain samples for Neurod6 [F(5,9) = 50.713, p = 3 × 10−6], Mcm6 [F(5,9) = 8.204, p = 0.0036], and Gfap [F(5,9) = 154.442, p = 5 × 10−9], post hoc analysis showed the dissected dentate gyrus and the mCherry positive cells populations isolated from Kir2.1-mCherry injected brains had significantly lower Neurod6 expression than the entire hippocampus (p < 0.05 for DG, p < 0.005 for Kir2.1-mCherry brains, unpaired Student’s t-test). Expression of the granule cell marker Mcm6 was significantly higher in the Kir2.1-mCherry positive samples than in the entire hippocampus (p < 0.05, unpaired Student’s t-test). Gfap expression was significantly higher in the dentate gyrus than the entire hippocampus (p < 0.05, unpaired Student’s t-test), but the Kir2.1-mCherry positive samples expressed significantly less than both the dentate gyrus and the entire hippocampus (p < 0.005, unpaired Student’s t-test). so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; slm, stratum lacunosum-moleculare.
We stereotaxically injected CAMKII(0.4)-Kir2.1-mCherry into the dentate gyrus of mice aged 4–6 weeks (Figure 3B). Two weeks later the dentate gyrus was manually dissected out and dissociated (see Methods and Figure 3A). The cells were stained with a cell death marker and sorted by FACS to isolate a live Kir2.1-mCherry expressing population. We were able to harvest up to 14,624 positive cells from a single brain. RNA was extracted from these cells and screened by RT-PCR for markers of different hippocampal cell types to examine the purity of the sorted population. Samples of RNA from cells dissociated from either the entire dorsal hippocampus or just the dentate gyrus dissected from uninjected brains were used as controls.
To evaluate the accuracy of cell isolation and the specificity of neuronal transduction we used RT-PCR of mRNA isolated from sorted cells to compare expression of anatomical and cell type markers. Expression of Neurod6, which is a marker for the CA1 and CA3 regions of the hippocampus (Lein et al., 2004), was over 20-fold higher in the entire hippocampus compared with the dissected dentate gyrus. Neurod6 was undetectable in three out of four samples of sorted Kir2.1-mCherry positive cells and the remaining sample had extremely low expression of Neurod6 mRNA that was significantly less than the entire hippocampus (Figure 3C). This indicates minimal contamination of the samples with cells from CA1 or CA3. Expression of Mcm6, which is a marker for dentate gyrus granule cells (Lein et al., 2004), was over seven-fold higher in the dentate gyrus compared with the entire hippocampus. All four Kir2.1-mCherry positive samples expressed Mcm6 at a level equivalent to or higher than the dissected dentate gyrus and significantly higher than the entire hippocampus, suggesting they were enriched for dentate gyrus granule cells (Figure 3C). The dentate gyrus shows significantly increased mRNA expression of the glial marker Gfap compared to the entire hippocampus, possibly due to high levels of astrocytes in addition to Gfap-expressing neural progenitors in the subgranular zone (Liu et al., 2010). Gfap mRNA was only detectable in two of the four Kir2.1-mCherry positive samples and in both cases was at levels significantly less than both the dissected dentate gyrus and the entire hippocampus. The CAMKII(0.4) promoter should preclude expression of this construct in astrocytes but it may express in a small percentage of neural progenitors potentially explaining the low level of Gfap positivity (van Hooijdonk et al., 2009). Together, the RT-PCR results demonstrate that specific neuronal subpopulations can be transduced with viruses in vivo and then subsequently isolated for downstream analysis of gene expression.
Reduction of Neuronal Gene Expression by Viral Expression of Intronic MiRNAs
Whilst our previous constructs altered neuronal gene expression through over-expression of transgenes, it is also useful to be able to reduce expression of endogenous genes. This can be achieved through the use of RNAi. However, early approaches for RNAi that used RNA Pol-III promoters to express high levels of short hairpin RNAs (shRNA) can be toxic in vivo (McBride et al., 2008; Ehlert et al., 2010). More recently, Pol-II promoters have been used to express artificial miRNA sequences. This reduces the off-target effects (McBride et al., 2008) but can result in poor expression levels of cocistronic reporter genes (Du et al., 2006). To address this issue, vectors were developed in which the miRNA is contained within a synthetic intron upstream of the reporter gene (Du et al., 2006). This allows for efficient expression of both the reporter and the miRNA from a Pol-II promoter (Figure 4A). We speculated that viruses expressing artificial intronic miRNAs and reporter genes would have improved specificity of gene knockdown in neuronal populations in vivo.
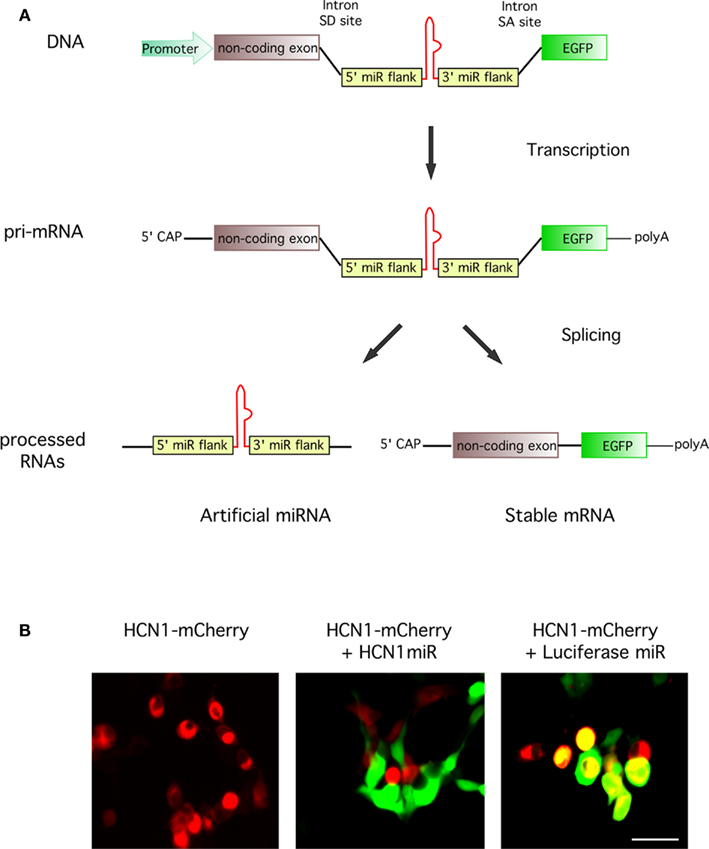
Figure 4. Knockdown of a target gene and high co-expression of reporter gene following expression of constructs containing intronic artificial miRNAs. (A) Schematic illustrating processing of an artificial intronic miRNA construct. DNA transcription produces a pri-mRNA with an intronic miRNA. Cellular splicing excises the miRNA component ready for downstream processing, leaving a stable mRNA with a 5′CAP and polyA tail for the expression of EGFP. (B) In vitro testing of an intronic miRNA construct targeting expression of HCN1. HEK293FT cells were transfected with plasmids expressing HCN1-mCherry (red) alone or as a cotransfection with a vector encoding a miRNA targeting either HCN1 (pSM155-HCN1miR) or luciferase (pSM155-LucmiR; green). Transfection with the HCN1 miRNA reduced expression of HCN1-mCherry whereas the luciferase miRNA did not. Scale bar = 50 μm.
To test this possibility, we constructed AAVs targeting expression of the HCN1 ion channel and evaluated their efficacy in cerebellar Purkinje neurons. Oligonucleotide sequences designed against HCN1 were cloned between the miRNA arms in the artificial intron of the pSM155 plasmid vector as previously described (Du et al., 2006). To check their effectiveness, the HCN1 miRNA vectors were cotransfected with an HCN1-mCherry plasmid into HEK293FT cells. The most effective HCN1 miRNA (Mmi510953) successfully reduced expression of HCN1-mCherry to almost undetectable levels whereas a negative control miRNA targeting luciferase (Luc miR) had no effect (Figure 4B). We therefore generated AAV vectors in which a cassette encoding the intronic HCN1 miRNA and EGFP reporter was inserted downstream of the ESYN promoter in a modified AAV backbone (AAV-Gateway). A negative control vector encoding Luc miR-EGFP was also constructed and viruses produced.
Injection of the AAVs into the cerebellum demonstrated widespread, high level expression of the EGFP reporter in Purkinje neurons within 2 weeks (Figure 5A). We evaluated the efficacy of the knockdown approach by measuring the hyperpolarization-activated current (Ih) recorded from cerebellar Purkinje cells in brain slices. Previous experiments with knockout mice suggest that HCN1 channels are the major ion channel type responsible for Ih recorded from cerebellar Purkinje cells (Nolan et al., 2003). Consistent with this idea, and with the efficacy of the artificial miRNA approach, Ih was abolished in Purkinje cells transduced with AAV expressing the HCN1 miRNA (Figure 5B-C). In contrast, Purkinje cells transduced with AAV expressing the miRNA targeted against luciferase had Ih with amplitude and kinetics comparable to non-transduced neurons (Figure 5C-D). Thus, efficacious suppression of HCN1 function by the viral expression of HCN1 miRNA is not due to non-specific effects of viral transduction or expression of miRNAs.
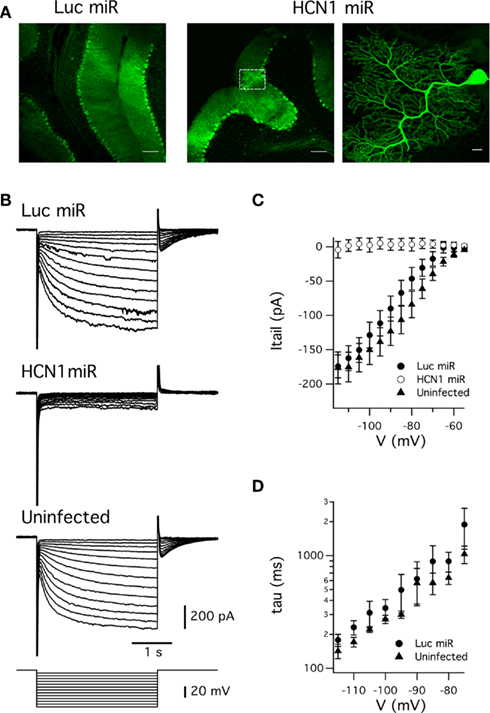
Figure 5. Expression of miRNAs targeted against HCN1 abolishes hyperpolarization-activated currents recorded from cerebellar Purkinje cells. (A) Native fluorescence of EGFP from cerebellar Purkinje cells transduced with AAVs expressing miRNAs targeting either luciferase (Luc miRNA) or HCN1 (HCN1 miR). The higher magnification image (right panel) corresponds to an HCN1miR expressing cell indicated by the white box (middle panel). Scale bars = 100 μm in left and middle panels, 10 μm in right panel. (B) Membrane current responses (upper three panels) to hyperpolarizing voltage steps (lower panel) recorded from a Purkinje cell infected with AAV expressing Luc miR (top), HCN1 miR (upper middle) or uninfected (lower middle). (C) Mean Ih tail current amplitude plotted as a function of test potential for each group of Purkinje cells. Tail current amplitudes differed between groups at membrane potentials ≤ −65 mV (p < 0.05, ANOVA). There was no significant difference between tail currents from Luc miR infected and uninfected neurons at any test potential (p > 0.05, unpaired Student’s t-test), whereas tail currents from cells infected with HCN1 miR differed from the Luc miR group at potentials ≤ −75 mV and from the uninfected group at all test potentials (p < 0.05, unpaired Student’s t-test). (D) Activation time constant plotted as a function of test potential for Ih recorded from uninfected Purkinje cells and from Purkinje cells infected with Luc miR. The time constant did not differ significantly between uninfected and Luc miR infected Purkinje cells at any test potential (p > 0.05, unpaired Student’s t-test). Group sizes for voltage-clamp experiments were as follows: Luc miR (n = 6 cells, 3 mice); HCN1 miR (n = 6 cells, 3 mice); uninfected (n = 6 cells, 4 mice).
To further address the specificity of the miRNA approach, we took advantage of the ability of electrophysiological techniques to quantify key physiological properties of neurons. We compared measures of spontaneous action potential firing, action potential waveform, and basic integrative properties, in control and HCN1 miRNA transduced neurons from mice with global deletion of HCN1 and wild-type littermate controls (Figures 6and 7). We reasoned that if knockdown with HCN1 miRNA is specific, then the physiological properties of Purkinje cells transduced with AAV expressing HCN1 miRNA should be similar to Purkinje cells from mice with global deletion of HCN1. Differences between physiological properties due to viral transduction or off-target effects of the HCN1 miRNA should be apparent by a similar effect of transduction with the HCN1 miRNA AAV on Purkinje cells from wild-type mice and mice with global deletion of HCN1.
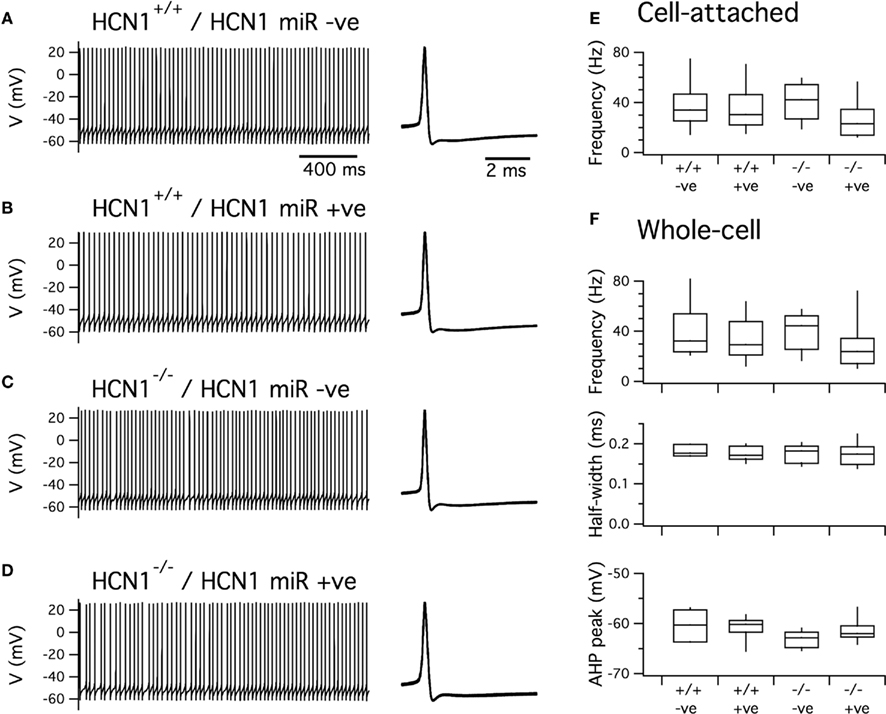
Figure 6. Expression of HCN1 miR does not affect spontaneous firing of cerebellar Purkinje cells. (A–D). Examples of spontaneous action potentials recorded from cerebellar Purkinje cells in brain slices obtained from control mice (A,B) or global HCN1 knockout mice (C,D). Purkinje cells were either positive for EGFP indicating expression of HCN1 miRNA (B,D) or were EGFP negative indicating they did not express HCN1 miRNA (A,C). (E) Frequency of spontaneous action potentials recorded from Purkinje cells in the cell-attached configuration. There was no significant effect of genotype (p = 0.59) or AAV infection (p = 0.14). (F) Frequency (upper panel), half-width (middle panel), and peak after-hyperpolarization (lower panel) for spontaneous action potentials recorded from Purkinje cells in the current-clamp configuration. There was no significant effect of AAV on any parameter (p > 0.14) and no significant effect of genotype on spike frequency or AHP duration. There was a significant effect of genotype on the AHP peak (p = 0.034). This effect is small and is inconsistent with previous comparisons suggesting it may simply reflect a chance outcome expected from repeated statistical testing. Statistical analysis uses ANOVA. Group sizes were as follows: HCN1+/+, HCN1 miR –ve (n = 7 cells, 3 mice); HCN1+/+, HCN1 miR+ve (n = 9 cells, 3 mice); HCN1+/+, HCN1 miR –ve (n = 9 cells, 3 mice); HCN1−/−, HCN1 miR+ve (n = 9 cells, 3 mice).
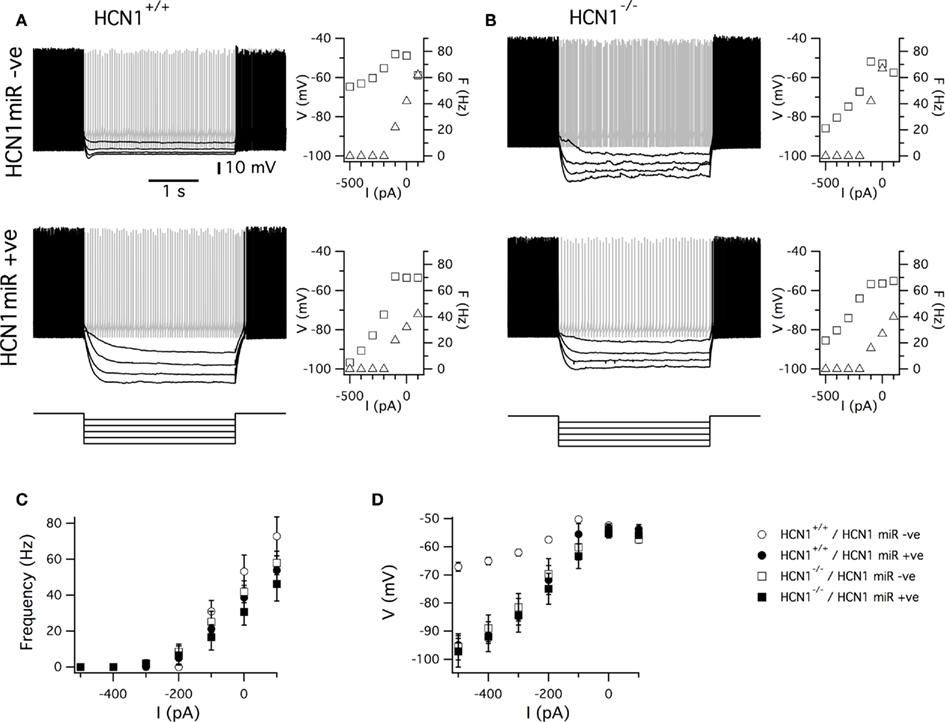
Figure 7. HCN1 miR expression and global deletion of HCN1 cause similar physiological phenotypes in cerebellar Purkinje cells. (A,B) Examples of responses of Purkinje cells from wild-type (A) and global HCN1 knockout mice (B) to series of current steps. Upper traces are recordings from uninfected Purkinje cells. Lower traces are recordings from Purkinje cells infected with AAV HCN1 miRNA EGFP. Graphs plot spike frequency and modal membrane potential as a function of the amplitude of the test current step. (C,D) Plot of the average across all neurons in each group of the spike frequency (C) and modal membrane potential (D) as a function of the amplitude of the test current step. There was no significant effect (p < 0.05) of genotype or AAV expression on the spike frequency at any amplitude of current step. In contrast, the amplitude of the membrane potential change depended on genotype and AAV expression (for −500 pA current steps, p = 0.0061 for genotype, p = 0.0044 for AAV infection, and p = 0.0050 for interaction between AAV infection and genotype). Numbers of neurons and mice are as in Figure 6.
We first examined spontaneous action potentials generated by cerebellar Purkinje cells. In agreement with previous studies of global HCN1 knockout mice (Nolan et al., 2003), we find that transduction with the HCN1 miRNA AAV does not affect the frequency of spontaneous action potentials recorded from Purkinje cells using either cell-attached or whole-cell configurations (Figures 6A–C). Properties of the action potential waveform were also similar in both uninfected and infected Purkinje cells from control mice and mice with global deletion of HCN1 (Figures 6A,D,E). These data are consistent with the notion that HCN1 channels do not play a pacemaker role in cerebellar Purkinje cells (Raman and Bean, 1999; Nolan et al., 2003; Carter and Bean, 2011) and also indicate that transduction with AAV expressing intronic microRNAs and EGFP does not have off target effects on ion channels important for generation of action potentials.
We next examined responses of Purkinje cells to injection of current steps though the recording electrode. In all experimental groups negative current steps switched off action potential firing. The relationship between action potential frequency and the amplitude of the current step was similar in Purkinje cells from wild-type and global HCN1 knockout mice that were uninfected or that were infected with the HCN1 miRNA AAV (Figures 7A–C). In contrast, the relationship between the amplitude of current steps and the modal membrane potential was significantly modified by deletion of HCN1 and also by knockdown of HCN1 following transduction with the HCN1 miRNA AAV (Figures 7A,B,D). Thus, whereas in Purkinje cells from wild-type mice the membrane potential remained close to spike threshold even during current steps that switched off spike firing, in Purkinje cells expressing HCN1 miRNA or from mice with deletion of HCN1, the membrane potential reached more hyperpolarized potentials. Importantly, there was no significant difference between any of the HCN1 knockdown or HCN1 deletion groups in the relationship between current and membrane potential. These results are consistent with the idea that HCN1 channels are primarily activated in Purkinje cells by stimuli that inhibit spontaneous action potential firing and that they act to stabilize the membrane potential close to spike threshold (Nolan et al., 2003). They also indicate that the effects of AAV mediated expression of HCN1 miRNA are specific to HCN1 channels and do not involve changes in the properties of other membrane ion channels, or general pathological effects.
Discussion
We have described a molecular toolbox of components to engineer lentiviral and AAV vectors for up- or down-regulation of neuronal gene expression in vivo. The system is extremely flexible and allows rapid generation of viral constructs. Viruses produced from these vectors yield stable expression of transgenes at levels high enough to detect native fluorescence in neurons. This allows easy identification of virally transduced cells for downstream analysis of the effects of gene manipulation.
A Modular Approach to Virus Assembly
Constructing viral vectors using traditional cloning methods is time consuming and inefficient as it requires restriction enzyme sites that must be compatible with insert sequences and the vector. Altering or rearranging elements within a vector often involves a new cloning strategy leading to unnecessary duplication of work. We have used a recombination cloning system to build a library of validated interchangeable components for the generation of viral vectors. New vectors can be quickly assembled using a single recombination reaction reducing both the time and expertise required. Entry vectors encoding the desired components must be generated initially, but they can then be utilized repeatedly in various combinations to create new expression constructs. By creating a Gateway-compatible AAV vector, the same entry vectors can now be used to produce both lentiviruses and AAV. Additional viral vectors, such as rabies or HSV could also be adapted to this system, further increasing the utility of the toolbox. In this way, the same components can be easily swapped between different viral systems, whilst minimizing the variability between them.
The toolbox could potentially be applied to diverse neuroscientific questions. Using different promoters we have expressed various reporter proteins, ion channels, and miRNA cassettes in neurons in vivo. The high level of native reporter fluorescence detectable from these viruses facilitates both imaging and electrophysiology-based techniques as well as ex vivo isolation of transduced cells. Using FACS to detect the reporter, we isolated relatively pure populations of transduced granule cells from the dentate gyrus. These cells could then be subjected to further ex vivo analysis of their DNA, RNA, or protein profiles to examine the consequences of the gene manipulation in more detail. This approach has the advantage of performing the manipulation in an in vivo context whilst allowing the use of a wide range of sensitive in vitro assays for the subsequent analysis of transduced cells. It is also possible to isolate many more transduced cells than is practical using other current techniques such as laser capture microscopy. It is important to note however, that the purity of the cell population isolated will depend on both the specificity of the transgene promoter and the accuracy of virus injection and tissue dissection.
In contrast to generation and maintenance of knockout and transgenic mice, which can be time consuming and expensive, the use of viruses to alter gene expression in vivo has several advantages. In particular, viruses can be rapidly produced and are easily targeted to well-defined brain regions at specific time points. However, there are potential disadvantages to consider when planning viral experiments. Transgene expression levels may vary unpredictably depending on how many viruses transduce each cell and where lentivirally expressed DNA integrates into the cell’s genome. In addition, while careful injection of lentiviruses and AAV directly into the brain parenchyma results in little or no immune response (Chamberlin et al., 1998; Mazarakis et al., 2001; Abordo-Adesida et al., 2005; Lowenstein et al., 2007) any surgical procedure still has the potential to induce local inflammatory responses. This can be counteracted through the use of careful surgical techniques, viral production and purification methods (Scherr et al., 2002; Baekelandt et al., 2003) and injection of lower viral titers (Chamberlin et al., 1998; Abordo-Adesida et al., 2005; Lowenstein et al., 2007). Nevertheless, where appropriate residual effects must still be excluded through the use of experimental controls.
Efficient and Specific Gene Knockdown Using Intronic Artificial microRNAs
Traditionally vectors for RNAi express shRNAs at high levels from strong Pol-III promoters. The shRNAs can result in toxicity by disrupting the biogenesis of natural miRNAs (Grimm et al., 2006), causing off-target silencing through accumulation of antisense RNAs (Birmingham et al., 2006) and stimulating innate immune responses to double stranded RNA (Samuel, 2001; Bridge et al., 2003; Fish and Kruithof, 2004). In vivo, these off-target effects can cause cell death and lethality (Grimm et al., 2006; Martin et al., 2011). Therefore, subsequent approaches have mimicked endogenous miRNAs by expressing artificial pri-miRNAs from Pol-II promoters. The pri-miRNAs are expressed at lower levels, but are more efficiently processed, minimizing their side effects (McBride et al., 2008). The use of Pol-II promoters also enables cocistronic expression of a reporter gene in a cell-specific and regulated manner. A weakness of this approach is that processing of the pri-miRNA can block translation of the cocistronic reporter gene as the resultant mRNA is unstable due to the lack of either a 5′ CAP or polyA tail (Du et al., 2006). Placing the miRNA within an artificial intron upstream of the reporter gene addresses this problem by allowing cellular splicing mechanisms to excise the miRNA whilst leaving an intact mRNA for co-expression of the reporter gene (Du et al., 2006). This approach has been used recently to generate a BAC transgenic mouse (Garbett et al., 2010), but viral delivery of artificial intronic miRNA to neurons has not been reported. Our results show that an AAV can be used to express an intronic miRNA targeting the HCN1 ion channel resulting in specific knockdown of Ih in cerebellar Purkinje neurons without apparent off-target effects.
The molecular toolbox we have developed allows for the rapid generation of lentiviral and AAV vectors. Nevertheless, it is still important to rigorously test each component in order to generate useful viruses. When expressed within a viral context, promoter sequences may not always yield the anticipated pattern of activity. Although our neuronal promoters gave the expected high levels of expression within principle neurons (Dittgen et al., 2004; Hioki et al., 2007; Nathanson et al., 2009a, b; van Hooijdonk et al., 2009), we also observed occasional expression in interneurons as has been reported previously (Nathanson et al., 2009a). Transgene expression driven by the Netrin G1 promoter sequence did not recapitulate endogenous Netrin G1 expression patterns and a Netrin G2 promoter sequence failed to express in vivo despite high activity in vitro (data not shown). Ensuring viral transgene expression is restricted to very specific subpopulations of neurons may require additional strategies, such as using viral constructs incorporating FLEX switches in combination with Cre-expressing transgenic animals (Schnütgen et al., 2003; Atasoy et al., 2008). As new technologies for the control of gene expression become available, they can also be incorporated into the molecular toolbox. We anticipate these tools will be of widespread use for investigation of nervous system function and pathology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Trudi Gillespi from the IMPACT Imaging facility at the University of Edinburgh for assistance with confocal microscopy and Bina Santoro, Hiroyuki Hioki, Patrick Martin, Axel Schambach, and Guangwei Du for generously providing plasmids. This work was funded by a Marie Curie Excellence Grant and grants from the BBSRC, the MRC, and NARSAD.
Footnotes
References
Abordo-Adesida, E., Follenzi, A., Barcia, C., Sciascia, S., Castro, M. G., Naldini, L., and Lowenstein, P. R. (2005). Stability of lentiviral vector-mediated transgene expression in the brain in the presence of systemic antivector immune responses. Hum. Gene Ther. 16, 741–751.
Atasoy, D., Aponte, Y., Su, H. H., and Sternson, S. M. (2008). A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J. Neurosci. 28, 7025–7030.
Baekelandt, V., Eggermont, K., Michiels, M., Nuttin, B., and Debyser, Z. (2003). Optimized lentiviral vector production and purification procedure prevents immune response after transduction of mouse brain. Gene Ther. 10, 1933–1940.
Birmingham, A., Anderson, E. M., Reynolds, A., Ilsley-Tyree, D., Leake, D., Fedorov, Y., Baskerville, S., Maksimova, E., Robinson, K., Karpilow, J., Marshall, W. S., and Khvorova, A. (2006). 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat. Methods 3, 199–204.
Bridge, A. J., Pebernard, S., Ducraux, A., Nicoulaz, A. L., and Iggo, R. (2003). Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 34, 263–264.
Bubeck, P., Winkler, M., and Bautsch, W. (1993). Rapid cloning by homologous recombination in vivo. Nucleic Acids Res. 21, 3601–3602.
Burrone, J., O’Byrne, M., and Murthy, V. N. (2002). Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420, 414–418.
Carter, B. C., and Bean, B. P. (2011). Incomplete inactivation and rapid recovery of voltage-dependent sodium channels during high-frequency firing in cerebellar Purkinje neurons. J. Neurophysiol. 105, 860–871.
Cetin, A., Komai, S., Eliava, M., Seeburg, P. H., and Osten, P. (2006). Stereotaxic gene delivery in the rodent brain. Nat. Protoc. 1, 3166–3173.
Chamberlin, N. L., Du, B., de Lacalle, S., and Saper, C. B. (1998). Recombinant adeno-associated virus vector: use for transgene expression and anterograde tract tracing in the CNS. Brain Res. 793, 169–175.
Chen, X., Shu, S., and Bayliss, D. A. (2009). HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. J. Neurosci. 29, 600–609.
Chen, X., Shu, S., Schwartz, L. C., Sun, C., Kapur, J., and Bayliss, D. A. (2010). Homeostatic regulation of synaptic excitability: tonic GABA(A) receptor currents replace I(h) in cortical pyramidal neurons of HCN1 knock-out mice. J. Neurosci. 30, 2611–2622.
Dittgen, T., Nimmerjahn, A., Komai, S., Licznerski, P., Waters, J., Margrie, T. W., Helmchen, F., Denk, W., Brecht, M., and Osten, P. (2004). Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc. Natl. Acad. Sci. U.S.A. 101, 18206–18211.
Du, G., Yonekubo, J., Zeng, Y., Osisami, M., and Frohman, M. A. (2006). Design of expression vectors for RNA interference based on miRNAs and RNA splicing. FEBS J. 273, 5421–5427.
Ehlert, E. M., Eggers, R., Niclou, S. P., and Verhaagen, J. (2010). Cellular toxicity following application of adeno-associated viral vector-mediated RNA interference in the nervous system. BMC Neurosci. 11, 20. doi:10.1186/1471-2202-11-20
Fish, R. J., and Kruithof, E. K. (2004). Short-term cytotoxic effects and long-term instability of RNAi delivered using lentiviral vectors. BMC Mol. Biol. 5, 9. doi: 10.1186/1471-2199-5-9
Garbett, K. A., Horváth, S., Ebert, P. J., Schmidt, M. J., Lwin, K., Mitchell, A., Levitt, P., and Mirnics, K. (2010). Novel animal models for studying complex brain disorders: BAC-driven miRNA-mediated in vivo silencing of gene expression. Mol. Psychiatry 15, 987–995.
Grimm, D., Streetz, K. L., Jopling, C. L., Storm, T. A., Pandey, K., Davis, C. R., Marion, P., Salazar, F., and Kay, M. A. (2006). Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441, 537–541.
Han, X., and Boyden, E. S. (2007). Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PLoS ONE 2, e299. doi:10.1371/journal.pone.0000299
Hartley, J. L., Temple, G. F., and Brasch, M. A. (2000). DNA cloning using in vitro site-specific recombination. Genome Res. 10, 1788–1795.
Hioki, H., Kameda, H., Nakamura, H., Okunomiya, T., Ohira, K., Nakamura, K., Kuroda, M., Furuta, T., and Kaneko, T. (2007). Efficient gene transduction of neurons by lentivirus with enhanced neuron-specific promoters. Gene Ther. 14, 872–882.
Huang, Z., Walker, M. C., and Shah, M. M. (2009). Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J. Neurosci. 29, 10979–10988.
Koyrakh, L., Luján, R., Colón, J., Karschin, C., Kurachi, Y., Karschin, A., and Wickman, K. (2005). Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J. Neurosci. 25, 11468–11478.
Lein, E. S., Zhao, X., and Gage, F. H. (2004). Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. J. Neurosci. 24, 3879–3889.
Liu, Y., Namba, T., Liu, J., Suzuki, R., Shioda, S., and Seki, T. (2010). Glial fibrillary acidic protein-expressing neural progenitors give rise to immature neurons via early intermediate progenitors expressing both glial fibrillary acidic protein and neuronal markers in the adult hippocampus. Neuroscience 166, 241–251.
Lowenstein, P. R., Mandel, R. J., Xiong, W. D., Kroeger, K., and Castro, M. G. (2007). Immune responses to adenovirus and adeno-associated vectors used for gene therapy of brain diseases: the role of immunological synapses in understanding the cell biology of neuroimmune interactions. Curr. Gene Ther. 7, 347–360.
Ma, H., Kunes, S., Schatz, P. J., and Botstein, D. (1987). Plasmid construction by homologous recombination in yeast. Gene 58, 201–216.
Martin, J. N., Wolken, N., Brown, T., Dauer, W. T., Ehrlich, M. E., and Gonzalez-Alegre, P. (2011). Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther. doi: 10.1038/gt.2011.10. [Epub ahead of print].
Mazarakis, N. D., Azzouz, M., Rohll, J. B., Ellard, F. M., Wilkes, F. J., Olsen, A. L., Carter, E. E., Barber, R. D., Baban, D. F., Kingsman, S. M., Kingsman, A. J., O’Malley, K., and Mitrophanous, K. A. (2001). Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum. Mol. Genet. 10, 2109–2121.
McBride, J. L., Boudreau, R. L., Harper, S. Q., Staber, P. D., Monteys, A. M., Martins, I., Gilmore, B. L., Burstein, H., Peluso, R. W., Polisky, B., Carter, B. J., and Davidson, B. L. (2008). Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. U.S.A. 105, 5868–5873.
Miyashita, T., and Kubo, Y. (1997). Localization and developmental changes of the expression of two inward rectifying K(+)-channel proteins in the rat brain. Brain Res. 750, 251–263.
Nathanson, J. L., Jappelli, R., Scheeff, E. D., Manning, G., Obata, K., Brenner, S., and Callaway, E. M. (2009a). Short promoters in viral vectors drive selective expression in mammalian inhibitory neurons, but do not restrict activity to specific inhibitory cell-types. Front. Neural Circuits 3:19. doi: 10.3389/neuro.04.019.2009
Nathanson, J. L., Yanagawa, Y., Obata, K., and Callaway, E. M. (2009b). Preferential labeling of inhibitory and excitatory cortical neurons by endogenous tropism of adeno-associated virus and lentivirus vectors. Neuroscience 161, 441–450.
Nolan, M. F., Dudman, J. T., Dodson, P. D., and Santoro, B. (2007). ‘HCN1 channels control resting and active integrative properties of stellate cells from layer II of the entorhinal cortex.’, J. Neurosci., 27(46), 12440–12451.
Nolan, M. F., Malleret, G., Dudman, J. T., Buhl, D. L., Santoro, B., Gibbs, E., Vronskaya, S., Buzsáki, G., Siegelbaum, S. A., Kandel, E. R., and Morozov, A. (2004). A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119, 719–732.
Nolan, M. F., Malleret, G., Lee, K. H., Gibbs, E., Dudman, J. T., Santoro, B., Yin, D., Thompson, R. F., Siegelbaum, S. A., Kandel, E. R., and Morozov, A. (2003). The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell 115, 551–564.
Oliner, J. D., Kinzler, K. W., and Vogelstein, B. (1993). In vivo cloning of PCR products in E. coli. Nucleic Acids Res.. 21, 5192–5197.
Petreanu, L., Mao, T., Sternson, S. M., and Svoboda, K. (2009). The subcellular organization of neocortical excitatory connections. Nature 457, 1142–1145.
Piskorowski, R., Santoro, B., and Siegelbaum, S. A. (2011). TRIP8b splice forms act in concert to regulate the localization and expression of HCN1 channels in CA1 pyramidal neurons. Neuron 70, 495–509.
Raman, I. M., and Bean, B. P. (1999). Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J. Neurosci. 19, 1663–1674.
Samuel, C. E. (2001). Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809; table of contents.
Sasaki, Y., Sone, T., Yoshida, S., Yahata, K., Hotta, J., Chesnut, J. D., Honda, T., and Imamoto, F. (2004). Evidence for high specificity and efficiency of multiple recombination signals in mixed DNA cloning by the Multisite Gateway system. J. Biotechnol. 107, 233–243.
Schambach, A., Bohne, J., Baum, C., Hermann, F. G., Egerer, L., von Laer, D., and Giroglou, T. (2006). Woodchuck hepatitis virus post-transcriptional regulatory element deleted from X protein and promoter sequences enhances retroviral vector titer and expression. Gene Ther. 13, 641–645.
Scherr, M., Battmer, K., Eder, M., Schüle, S., Hohenberg, H., Ganser, A., Grez, M., and Blömer, U. (2002). Efficient gene transfer into the CNS by lentiviral vectors purified by anion exchange chromatography. Gene Ther. 9, 1708–1714.
Schnütgen, F., Doerflinger, N., Calléja, C., Wendling, O., Chambon, P., and Ghyselinck, N. B. (2003). A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol. 21, 562–565.
Stevens, E. B., Woodward, R., Ho, I. H., and Murrell-Lagnado, R. (1997). Identification of regions that regulate the expression and activity of G protein-gated inward rectifier K+ channels in Xenopus oocytes. J. Physiol. 503(Pt 3), 547–562.
van Hooijdonk, L. W., Ichwan, M., Dijkmans, T. F., Schouten, T. G., de Backer, M. W., Adan, R. A., Verbeek, F. J., Vreugdenhil, E., and Fitzsimons, C. P. (2009). Lentivirus-mediated transgene delivery to the hippocampus reveals sub-field specific differences in expression. BMC Neurosci. 10, 2. doi: 10.1186/1471-2202-10-2
Yahata, K., Kishine, H., Sone, T., Sasaki, Y., Hotta, J., Chesnut, J. D., Okabe, M., and Imamoto, F. (2005). Multi-gene gateway clone design for expression of multiple heterologous genes in living cells: conditional gene expression at near physiological levels. J. Biotechnol. 118, 123–134.
Keywords: intronic miRNA, ion channel, lentivirus, AAV, RNAi, hippocampus, cerebellum, virus
Citation: White MD, Milne RVJ and Nolan MF (2011) A Molecular toolbox for rapid generation of viral vectors to up- or down-regulate neuronal gene expression in vivo. Front. Mol. Neurosci. 4:8. doi: 10.3389/fnmol.2011.00008
Received: 31 March 2011;
Paper pending published: 26 April 2011;
Accepted: 21 June 2011;
Published online: 04 July 2011.
Edited by:
Alistair N. Garratt, Max Delbrück Center for Molecular Medicine, GermanyCopyright: © 2011 White, Milne and Nolan. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Matthew F. Nolan, Centre for Integrative Physiology, University of Edinburgh, Edinburgh EH8 9XD, Scotland, UK. e-mail: mattnolan@ed.ac.uk