GSK3 and Alzheimer’s disease: facts and fiction…
- Experimental Genetics Group – LEGTEGG, Department Human Genetics, KULeuven, Leuven, Belgium
The physiological functions and pathological roles of the Glycogen synthase kinase-type 3 (GSK3) kinases in peripheral and central systems are diverse and complex, and therefore hard to unravel in molecular detail in vivo. Our assignment to review and discuss available data to clarify the actual position of these kinases in the pathology of Alzheimer’s dementia (AD) was both ambitious and easy. On the one hand, numerous studies are available in isolated, recombinant, or cell-based systems, which have resulted in very diverse data-sets that are hardly informative for the brain in vivo. At the other extreme, reliable, and relevant models for the role of GSK3 in CNS are rare, if not lacking. Moreover, (too) many in vivo studies used Li+ as “specific” inhibitor of GSK3, which is factually not valid because lithium ions are neither specific nor potent inhibitors of GSK3 in vivo. More specific pharmacological inhibitors of GSK3 have met with considerable problems, and are reviewed by others in this issue or elsewhere. We concentrate here on AD-related aspects of GSK3 in brain in vivo, mainly studied in transgenic mice and highlight some of the more important issues, among many remaining: activation of GSK3 by amyloid, phosphorylation of protein tau, effects on or interference with synaptic activity, differentiation between both GSK3 isoforms. These relate directly to brain function, and brain dysfunction in AD, and are to be resolved if we want to understand the molecular pathology of this dreadful disease.
Alzheimer’s Disease
The term dementia stems from latin words meaning “without mind” or “de-humanized,” without any doubt the most horrifying condition a human being can experience. Dementia comprises the age-related deterioration of cognitive abilities and memory, associated with severe changes in personality, in character, in interests (or rather lack thereof), in social, and in general behavior. Progressive dementia closely connects to aging and is therefore rapidly becoming a prime problem for the aging populations of any nation where life expectancy exceeds 65 years. In parallel with improved health and medical care, the ever increasing life expectancy promotes age-related dementia as a major medical and socio-economic problem, next to vascular disease and cancer. It is estimated that already over 10 million people suffer dementia in Europe, and about 40 million world-wide (data 2010). These numbers will likely double every next 20 years (Alzheimer’s disease International; World Alzheimer Report, 2009). Because dementing patients are to be cared for around the clock, the number of care-takers needed in the future will surpass all other medical needs.
While dementia is the general medical term for various diseases that distort brain functioning, the most common types are Alzheimer’s dementia (AD), vascular dementia, various types of frontotemporal dementia (FTD) and dementia with Lewy bodies (DLB). AD is most conspicuously named after – not by – Alois Alzheimer, MD, who described the first case in 1907. It is by far the most frequent dementia, accounting for 50–70% of all cases, with rapidly increasing incidence and prevalence. The prevalence is only a few percent around ages 65–69 but increases to 25–35% in octogenarians (Ferri et al., 2005). Despite the small fraction of cases with early onset (before age 65), these are without doubt the most poignant, with most severe personal, familial, and social impact. Moreover, more women than men are becoming demented, and apparently not only because women live longer. According to some studies the increase is “exponential with age,” meaning that all humans will eventually become demented when old enough – a daunting prospect that is fortunately not yet supported by all facts.
Alzheimer’s dementia is a chronic, progressive disorder beginning with mild symptoms that are hard to recognize, mostly only in retrospect, and as yet impossible to diagnose objectively. The end-stage comprises very severe cognitive defects and deformed personality, inevitably evolving into complete dependence on external care. The associated physical weakening leads eventually to death from various causes, e.g., lung infections, stroke, coronary disease, and cancer. The entire disease process covers extended periods of 10–20 years or more, suspected to depend on physical, genetic, and epigenetic factors, all to be identified.
Even to date, more than a century after its first description, and despite massive fundamental and clinical research, AD remains hard to diagnose. The only conclusive diagnostic tool is identical to that used by professor Alzheimer: post-mortem silver-impregnation of brain sections, to reveal the two main pathological lesions, the extracellular amyloid plaques and the many neurons filled with tangles. Importantly, and often overlooked, Alzheimer also described the active glial processes, indicative of ongoing brain inflammation, which we believe is an essential element in the neurodegeneration process (Jaworski et al., 2009, 2010a,b, 2011).
In the absence of objective early diagnosis, AD is an exclusion diagnosis by clinical examination, with initial (un-)certainty around 50%, when the patients’ condition is labeled “probable AD.” Including biomarkers, i.e., biochemical in CSF or various types of brain imaging, diagnosis is becoming gradually more accurate, but remains statistical, yielding no more than about 80% certainty during life. Post-mortem analysis of the brain according to Alzheimer still remains the only certain diagnosis. Although this might not be the most important problem faced by clinicians in daily practice, the large margin of diagnostic uncertainty in early phases of AD, i.e., in mild cognitive impairment (MCI), poses a tremendous problem for clinical-phase therapeutic studies: stratification of the AD population at risk is needed, by genetic and diagnostic means.
The currently available pharmacological therapy for AD aims largely on increasing brain acetyl-choline levels, which is totally symptomatic. Given the rapidly rising incidence and prevalence, development of disease-modifying therapy is overdue and must go hand-in-hand with improved diagnosis, for reasons outlined above. Fundamental and applied research are to be fostered to understanding the mechanisms that cause and underlie, especially the sporadic forms of AD, to surpass the hurdle of most current studies and models that are largely restricted to familial, purely genetic types of AD.
Genetics: Amyloid First
The isolation and sequencing of amyloid peptides from brain of AD and Down’s patients (Glenner and Wong, 1984) was the most important biochemical finding in this field and marks the beginning of the molecular era of AD research. It allowed molecular cloning and sequencing of the cDNA coding for their precursor amyloid precursor protein (APP). In close parallel, but less conspicuous, phosphorylated protein Tau was detected as the major component of the intraneuronal tangled fibrils observed and described by Alzheimer (Brion, 1985; Grundke-Iqbal et al., 1986). Both findings boosted research into these enigmatic proteins, although less intense and less prominent for protein Tau than for amyloid.
The majority of AD cases are sporadic, with onset typically in persons over 65 years, and with “age” as the only known determining factor. An experiment of nature turned out “fortunately for science”: a small fraction, 1% or less of all AD cases are familial, purely genetic and inherited dominantly, causing early onset familial AD (EOFAD). The responsible mutations have been pin-pointed to the genes coding for the APP (12 families world-wide) and presenilins (PS1, 70+ mutations; PS2, 7 families; Goate et al., 1991; Sherrington et al.,1995; St George-Hyslop, 2000). These familial forms of AD are characterized by an age of onset between 35 and 55 years, and are most aggressive, progressing more rapid than sporadic AD over 5–10 years.
For sporadic AD, aging is a genetically or molecularly largely undefined condition. Only one, indirect genetic factor has been identified and stands out: the ε4 allele of the APOE gene, with an allele frequency of 15% in the general population, but associated to 40–50% of sporadic AD cases. The connection is again statistical and again not diagnostically useful, but the effect is marked: carriers’ risk is 5- to 10-fold higher, with an earlier age of onset (Corder et al., 1993).
More recently, three new genes, i.e., CLU, CR1, and PICALM, were found associated with sporadic AD in more global studies (Harold et al., 2009; Lambert et al., 2009). These genes do not, however, reach the statistical significance of the ApoE4 allele, and are yet to be studied as interesting research tools, hopefully helping to understand the overall disease process and their contribution to it. Nevertheless, even 17 years after the discovery of the AD–ApoE4 connection, we still do not understand to the fullest how this lipoprotein operates in brain, and how ApoE4 differs from the main ApoE3 isotype in brain functions. Whether scavenging and transport of ApoE-lipoproteins include removal of amyloid peptides from the interstitial spaces between neurons remains to be analyzed in detail (Castellano et al., 2011).
Genetic analysis of EOFAD cases provided knowledge of the genes that became the starting points for all biochemical, cell-biological and transgenic modeling of the mechanisms underlying familial AD – and by extrapolation sporadic AD, we all hope. These genes, i.e., APP and PS1/2, constitute the basis for the amyloid hypothesis posting that amyloid peptides are the prime cause of AD.
Genetics: A Strong Case for Tau in Neurodegeneration
Importantly, the known EOFAD cases are caused by mutations that affect amyloidogenic processing of APP, resulting either in more amyloid peptides, or in longer, less soluble amyloid peptides. Nevertheless, these EOFAD cases are very similar to sporadic AD, and always accompanied by severe tauopathy that is co-diagnostic for all AD cases – even those struck at young age!
Although protein Tau is thereby inherently involved, diagnostically as well as mechanistically, the recognized mutations in the MAPT gene on chromosome 17q21 coding for protein Tau, cause not AD but another type of dementia: FTD with parkinsonism, linked to chromosome 17 (FTDP-17). This sub-type of the FTD spectrum of dementias is clinically and diagnostically close to AD (Delacourte and Buée, 2000; Ingram and Spillantini, 2002). Tauopathies in general are a class of widely varying neuro-degenerative diseases, all characterized by pathological aggregation of protein Tau in specified brain regions and types of neurons, the most prominent of which are FTD, corticobasal degeneration and Pick’s disease (review Sergeant et al., 2005; Ludolph et al., 2009).
Importantly, the mutations in the MAPT gene that cause the FTDP-17 sub-type are situated not only in exons, but also in introns. The exonic mutations give rise to the expression of mutant protein Tau in the central neurons of patients in these FTD families. Physiologically, Tau is subject to alternative splicing resulting in the generation of six different isoforms, containing three or four microtubule (MT) binding domains (Tau3R or 4R respectively) and 0, 1, or 2 N-terminal repeats. Surprisingly, many mutations in introns surrounding exon 10 (encoding MT-binding domain 2) were located in, or close to the DNA signals that control mRNA splicing. Consequently, more or less – pending the mutation – species of wild-type protein Tau are produced containing MT-binding domain 2, encoded by exon 10, resulting in a distorted Tau3R/4R ratio. We can deduce from data in developing brain with mostly or even only Tau3R species expressed, that the Tau3R/4R ratio defines or allows more axonal morphological modulation and plasticity. The precise consequences of the Tau3R/4R ratio in adult and aging brain are to be defined, in FTD and eventually in AD. Despite the lack of a direct genetic link of the MAPT gene to AD, the link might be indirect by interaction with the GSK3β gene (Kwok et al., 2008).
In all primary tauopathies, the cognitive demise cannot but be due to the evident progressive Tau pathology – although molecularly it is not clear by what actions or mechanisms. In AD, the co-diagnostic tauopathy is generally described as “secondary” to amyloid pathology. The hypothesis that tauopathy is pathologically responsible in AD is supported by clinical and pathological observations, demonstrating that the spatial and temporal pattern, not of the amyloid but of the Tau pathology correlates most closely with the cognitive decline in AD (Braak and Braak, 1991; Reitz et al., 2009). MCI is the most likely prodrome of AD, but the underlying molecular changes and factors that mediate – or prevent – the conversion MCI to AD are not understood. Unexpectedly, recent pathological data demonstrate beginning tauopathy in CNS already at young age, i.e., decades before other defects, like amyloid deposition become apparent (Braak and Del Tredici, 2011a,b; Duyckaerts, 2011).
In this respect one wonders again why these dementias, even or in particular the familial types, take decades to develop? This can be related to, or even because of, the inherent resilience of neurons and their post-mitotic nature. Alternatively, or in addition, more than one or even more than two hits might be needed to functionally deteriorate, and eventually kill a neuron.
Tau, a Naturally Unfolded, Phosphorylated MAP
Protein Tau was first identified in early microtubule (MT) preparations and subsequently described to promote MT assembly, with its designated name (τ) referring to “tubule forming” (Weingarten et al., 1975). The only known function of Tau is as microtubule associated protein (MAP), with binding to MT regulated by a complex interplay of isoform expression and phosphorylation. Alternative mRNA-splicing yields Tau3R or Tau4R isoforms containing three or four MT-binding domains, as stated above (Mandelkow et al., 1996). Phosphorylation of serine and threonine residues is performed by kinases, with most prominent proline-directed kinases [e.g., Glycogen synthase kinase-type 3 (GSK3), cdk5, MAPK], and non-proline-dependent kinases (e.g., MARK, PKA, PKC; reviews Lau et al., 2002; Johnson and Stoothoff, 2004; Hanger et al., 2009).
While protein Tau contains a large number of serine/threonine phosphorylation sites, only about 15 have been demonstrated to be physiologically or pathologically important in vivo. Nevertheless, therein hides a major analytical problem: the potential 215 differently phosphorylated sub-forms of protein Tau are impossible to analyze individually. The balance between kinase and phosphatase activities determines the actual level of phosphorylation of protein Tau, like for all proteins in any biological system. In contrast to the relatively small set of kinases, the known phosphatases have been less well-studied with respect to the function of protein Tau in health and disease, although PP2B/calcineurin and PP2A are potentially important (Louis et al., 2011; and references therein).
The main action of phosphorylation of protein Tau as a MAP is in its interaction with MT: decreasing its affinity for, preventing its binding to and provoking its detachment from MT. In AD – and in all primary tauopathies – protein Tau becomes “hyper-phosphorylated”: rising from an average 2–5 phosphate groups per molecule, to up to 10 phosphate groups per Tau molecule in diseased and tangled neurons. The resulting pathological defects are mechanistically not well understood, but are presumed to comprise deregulation of MT-mediated transport, not only in axons but also in somata and dendrites, by the aggregated Tau-fibrils.
Amyloid and Tau in AD: A Troubled Relation…
Overall, the genetic findings briefly summarized in previous sections, boosted interest in protein Tau, because they made clear that defective Tau is sufficient to cause neurodegeneration and dementia in FTD, not unlike the disease process in AD. On the other hand, the findings also re-fueled the “amyloid-first cascade” hypothesis, albeit a re-phrased version: accumulation and aggregation of amyloid peptides causes “also” tauopathy – by unknown mechanisms – and thereby the clinical symptoms and pathological defects, from mild early cognitive defects to severe dementia – again by unknown mechanisms (Hardy and Allsop, 1991; Hardy and Selkoe, 2002; Duyckaerts et al., 2009; Jaworski et al., 2010).
In the last decade it gradually became clear that not the “cemented” protein-aggregates, but rather smaller, even soluble aggregates of amyloid and Tau are synapto- and neuro-toxic (Moechars et al., 1999; Lesné et al., 2006; Lasagna-Reeves et al., 2011; review Jaworski et al., 2010). Indeed, the earliest cognitive dysfunction in AD is likely caused by functional and/or structural weakening of synapses in affected limbic regions (Gonatas et al., 1967; Hamos et al., 1989; Dekosky and Scheff, 1990). These precede deposition and formation of solid amyloid plaques and tangles, and neuronal loss, thereby marking MCI, or a sub-type of MCI. We are challenged to understand this sub-type because it is the prodromal stage of AD, and we need to define its cause: amyloid or Tau or – most likely – both.
We now believe that synaptic dysfunction is caused by the accumulation of aggregated amyloid peptides and/or soluble phosphorylated Tau, but their exact relation remains enigmatic and burdened with questions. How do amyloid peptides in the secretory pathway or secreted affect the cytoplasmic protein Tau? Through a receptor for amyloid-aggregates and its downstream kinases? Is this part of a normal physiological mechanism, or merely a pathological problem?
The physiological relation between amyloid and Tau – if any – is not clear in normal brain, while their pathological relation is evident in AD brain, although complicated and still largely to be clarified. The amyloid cascade hypothesis initially posed aggregation of amyloid into plaques to cause synaptic dysfunction and neurodegeneration. Later the Tau pathology became more prominently included, still subordinate to amyloid pathology as outlined above, and defining AD as the combination of amyloid plaques and neurofibrillary tangles, conform the original description by A. Alzheimer.
Interestingly, recent findings in experimental models shed some light on the complex relation between amyloid and Tau, in as far as deficiency of protein Tau prevents amyloid-induced cognitive defects (Roberson et al., 2007, 2011; Ittner et al., 2010; Shipton et al., 2011). Moreover, protein Tau appears to be needed for amyloid to disrupt axonal transport in primary neurons (Vossel et al., 2010), although in vivo confirmation is needed. Additionally, specific phosphorylation of protein Tau could be involved in its mis- or re-localization from axons to the somatodendritic compartments, and perhaps even to dendritic spines (Hoover et al., 2010; Ittner et al., 2010; Kremer et al., submitted). These considerations and hypotheses, although still littered with large open spaces of unknowns, implicate kinases most deeply in the fundamental pathological mechanism that cause AD and related tauopathies.
For one, we showed that early in the amyloid pathology both GSK3 isozymes become activated, as demonstrated by increased tyrosine phosphorylation, in mouse brain in vivo (Terwel et al., 2008). Our finding then raises the important issue of the molecular mechanism by which amyloid peptides – and which species – augment the proposedly autocatalytic tyrosine phosphorylation of GSK3 (Cole et al., 2004; discussed below). These and related in vivo findings firmly establish GSK3 as essentially contributing to the pathogenesis of AD, linking amyloid to tau phosphorylation and confirming its original designation as Tau kinase I (Ishiguro et al., 1993; Spittaels et al., 2000; Muyllaert et al., 2006, 2008; Terwel et al., 2008; reviews Takashima, 2006; Jaworski et al., 2010). Most interestingly, GSK3 is also becoming intimately implicated in normal physiological mechanisms underlying synaptic plasticity, learning, and memory (Hooper et al., 2007; Peineau et al., 2007; Dewachter et al., 2009; Hur and Zhou, 2010; Smillie and Cousin, 2011). Consequently, we must besides amyloid and Tau consider direct contributions of (activated) GSK3 to synaptic defects in AD (Figure 1; Terwel et al., 2008; Jaworski et al., 2010a,b; and references therein).
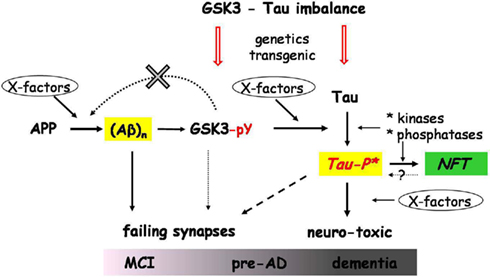
Figure 1. Schematic relations between amyloid, GSK3, protein Tau, and other factors. The scheme depicts the activation by amyloid peptides of GSK3α/β by increasing tyrosine phosphorylation, and leading to increased phosphorylation of protein Tau as the central event in AD pathogenesis. Condensed from in vivo observations in transgenic models, both proven (solid arrows) and proposed effects (broken arrows) are represented. The unknown molecular factors (X-factors) and mechanisms behind the relations and connections in this scheme are not yet fully understood as discussed in the text. Our most recent results did not confirm the proposed feedback effect of GSK3 on APP processing (data not shown). The amyloid and pTau species that cause synaptic defects, and eventually neurodegeneration, are not aggregates, but soluble oligomers (marked in yellow boxes). The phosphorylation of Tau by GSK3 and other kinases, produces a neurotoxic species, represented here as Tau-P*. This hypothetical intermediate is a soluble single, dimer, or small aggregate, in a transitional conformational state that can be directed either into aggregation (NFT; green box) or toward synaptic and neuronal toxicity. Tau-P* causes synaptic dysfunction, which in various combinations with amyloid peptides and aberrant activated GSK3 results in various synaptic defects, initiated in the earliest phases MCI or pre-AD, and evolving to dementia, as highlighted in the scheme. The genetic imbalance between GSK3 and Tau genes depicted in the scheme refers to the proposed interaction between the Tau (MAPT) and GSK3β genes in humans, discussed in the text. This interaction might impact on both GSK3 activation or availability and the Tau3R/4R ratio, thereby also contributing to the propensity of Tau phosphorylation. The imbalance is also generated in the various single and bigenic models, discussed in the text. The combination of all actors and factors and their interactions lead to a variety of clinical and pathological symptoms, observed in sporadic AD patients.
Glycogen Synthase Kinase-Type 3
Glycogen synthase kinase-type 3 was first described as the major regulator of glycogen metabolism, by phosphorylating and thereby inhibiting glycogen synthase (Embi et al., 1980; Woodgett, 1990). GSK3 denotes the proline-directed S/T kinases that exist as two isozymes, GSK3α and GSK3β encoded by different genes on chromosomes 19 and 3, respectively (Woodgett, 1990; Shaw et al., 1998). The GSK3 isozymes share overall 84% sequence identity, but 98% in the kinase domain indicating similar substrate specificities (Woodgett, 1990). Nevertheless, they are functionally not identical as demonstrated by in vitro, cellular, and in vivo data (Hoeflich et al., 2000; Kaidanovich-Beilin et al., 2010; Soutar et al., 2010). In addition to the overall similar structure, the α isozyme contains an extended glycine-rich N-terminal region that could define cellular localizations and interactions unique to this isozyme (Azoulay-Alfaguter et al., 2011).
Importantly, total absence of GSK3β is embryonically lethal in mice, implicating that GSK3α cannot compensate for the lack of its counterpart (Hoeflich et al., 2000). In contrast, GSK3α can be completely eliminated without obvious major adverse effects on viability or health, with the possible exception of male sterility (Kaidanovich-Beilin et al., 2010). The very different outcomes in mice lacking either GSK3 isozyme has been attributed to differences in their mediation or regulation of transcriptional activity by CREB, NF-κB, EGR-1, Smad3/4, or others (Liang and Chuang, 2007; Mines et al., 2011). For one, GSK3β deficient mouse pups most likely die because of the inability of NF-κB to regulate liver development in the absence of GSK3β (Hoeflich et al., 2000) and/or from a patterning malformation in the heart (Kerkela et al., 2008). In general, the GSK3 isozymes affect cell survival and death, which is of course essential, or detrimental to genesis and normal functioning of any organ (review Mines et al., 2011).
The molecular basis for the distinct functions of both GSK3 isozymes in normal physiology remains largely unknown, which is evidently not helping to define their similar or different roles in pathology. Differential expression of GSK3α and GSK3β mRNA and proteins indicate, not unexpectedly that they are subject to different regulation in different tissues to fulfill diverse biological roles. Recent advances in mice lacking one or the other GSK3 isoform, completely for GSK3α or in a cell-specific manner for either isoform are becoming known and confirm these general impressions. For instance, mice lacking GSK3α display improved glucose tolerance and increased glycogen synthesis as opposed to tissue-specific GSK3β knock-out mice (MacAulay et al., 2007; Patel et al., 2008).
Recently, progress is made in understanding the physiology of GSK3 isozymes in CNS. Both GSK3 isozymes differentially regulate neuronal survival, but both appear needed for axon formation, despite acting on different substrates in brain (Kim et al., 2006, 2009; Garrido et al., 2007; Liang and Chuang, 2007; Soutar et al., 2010). GSK3β is located throughout neuronal compartments, as well as in astrocytes, and associated with rough endoplasmatic reticulum, free ribosomes, mitochondria, and interestingly also with the post-synaptic density in dendritic spines (Perez-Costas et al., 2010). This fact links snugly to data that GSK3β mediates interaction between two major forms of synaptic plasticity: NMDA-dependent long-term potentiation (LTP) and long-term depression (LTD). Responsible signaling mechanisms involve the classic PI3K/AKT/GSK3 connection that regulates phosphorylation of S9 on GSK3β, and thereby its kinase activity. This link appears as crucial in the formation of LTD or LTP, and thereby for synaptic plasticity and cognition (Hooper et al., 2007; Peineau et al., 2007, 2008; Dewachter et al., 2009). GSK3 further contributes to NMDA receptor trafficking and functioning in cortical neurons, again important in cognition (Chen et al., 2007). Besides these post-synaptic actions, GSK3 also contributes to pre-synaptic functions in developing and mature nerve terminals (review Smillie and Cousin, 2011).
Several excellent reviews are concerned with various structural and physiological functions of the GSK3 isozymes in peripheral organs and in brain. We therefore concentrate on data – and even more on hypotheses – that eventually link either or both GSK3 isozymes to the pathology in AD. In any case it must be remembered that the GSK3α isozyme is by far the least well-studied isoform, largely because of the historically stronger interest in GSK3β in AD. We impose here an additional restriction by concentrating on data obtained in vivo, mainly in genetically modified mice. We believe that in vivo models are the only relevant for physiology and pathology alike, however, not without acknowledging that mouse brain will never equal the complexity of human brain.
GSK3 in AD Pathology
The overriding hypothesis in AD maintains that amyloid peptides indirectly affect the activity of phosphatases and even more so of kinases, in particular GSK3β but also others that all contribute – in concert – to the increased phosphorylation of protein Tau typical for all tauopathies. Moreover, it is highly likely that changes in kinase activity are an intrinsic aspect of the pathological problem, because they can and will negatively affect, or even disrupt, other synaptic signals essential for learning and memory. Despite considerable technical efforts, it is as yet impossible to separate and define the contributions of amyloid, of GSK3 and other kinases, and of phosphorylated protein Tau on synaptic functions (Figure 1).
The central role of tauopathy in AD and of the increased phosphorylation of protein Tau therein, was recognized early (Brion et al., 1986; Grundke-Iqbal et al., 1986). Hyper-phosphorylated protein Tau forms filamentous aggregates that become deposited mainly as neuropil threads in axons and dendrites, and as the better known neurofibrillary tangles in soma of neurons in cortex and hippocampus. The various intracellular deposits of aggregated Tau are proposed to cause problems with all sorts of transport systems, leading to impaired synaptic transmission, dendritic amputation, generalized neuronal dysfunction, and eventually neurodegeneration. Nevertheless, it is by no means certain that the large fibrillar aggregates are responsible, but rather the putative smaller aggregates we termed “Tau-P*” (Figure 1; Jaworski et al., 2009, 2010a,b, 2011).
In FTD the eventual hyper-phosphorylation is ascribed to the mutation in Tau or to the distorted Tau3R/4R ratio. Nevertheless, neither of these defects is a priori expected to evoke or solicit more phosphorylation than wild-type Tau or than a normal isoform ratio. Obviously, mutations in Tau or excess of one or the other isoform changes the molecular parameters and control mechanisms, in as far that over a time-period of decades tau becomes phosphorylated to a degree that cannot be reversed. From that point the sequence of events is rather straightforward, and well recapitulated in transgenic mice: increased phosphorylation causes conformational collapse, dimerization, and further aggregation into fibrils (Terwel et al., 2008; Patterson et al., 2011). In sporadic AD, protein tau is not mutated, and the isoform ratio is normal, leading to the conclusion that out-of-balance kinase activity must explain the hyper-phosphorylation of protein tau in AD brain (Figure 1).
GSK3β was identified in this respect as the major kinase, denoted Tau Kinase I, first in vitro and established firmly in vivo in transgenic mouse brain by us (Ishiguro et al., 1993; Spittaels et al., 2000; Takashima, 2006; Terwel et al., 2008). To date over 30 distinct phosphorylation sites are known that can potentially be phosphorylated by GSK3β on protein Tau (Hanger et al., 2009). Although in vivo, GSK3β appears to be the most dominant Tau kinase, it is by no means the only kinase, indirectly observed in vivo in our different transgenic mouse strains. Increasing GSK3 activity transgenically in itself did not cause major pathological problems related to AD or tauopathy (Spittaels et al., 2000, 2002). Remarkably, increased GSK3β activity alleviated the axonopathy of our Tau4R mice but did not cause tauopathy (Spittaels et al., 2000), in contrast to the dramatic tauopathy evoked by the same GSK3β transgene in combination with Tau.P301L mice, denominated biGT mice (Terwel et al., 2008).
The former demonstrated for the first time in vivo that GSK3 effectively phosphorylates protein Tau4R in brain, and thereby decreased or prevented binding of Tau4R to MT to restore normal transport. Nevertheless, phosphorylation of wild-type human Tau4R by GSK3 was not sufficient to promote aggregation into tangles (Spittaels et al., 2000). Conversely, the biGT mice demonstrated that also Tau.P301L was effectively phosphorylated by GSK3 in brain and to a degree sufficient to form abundant tangles in forebrain neurons (Terwel et al., 2008). Obviously, while these data proved that the P301L mutation contributed additionally and essentially to the aggregation of Tau in conjunction with GSK3β, the nature of the P301L contribution to the process of Tau aggregation is not yet revealed.
We cannot but conclude that GSK3β is a most efficient Tau kinase, needed but not sufficient to force tau to aggregate into fibrils.
De-Regulated GSK3 Activity in Transgenic Mouse Brain
We documented the increased tyrosine phosphorylation of both GSK isozymes in brain of relatively young APP.V717I mice, i.e., at age 4–8 months before their brain becomes littered with amyloid deposits (Moechars et al., 1999; Terwel et al., 2008). At that age most amyloid peptides are present as small aggregates, and inside or in close proximity of the neurons they are produced by (Figure 2).
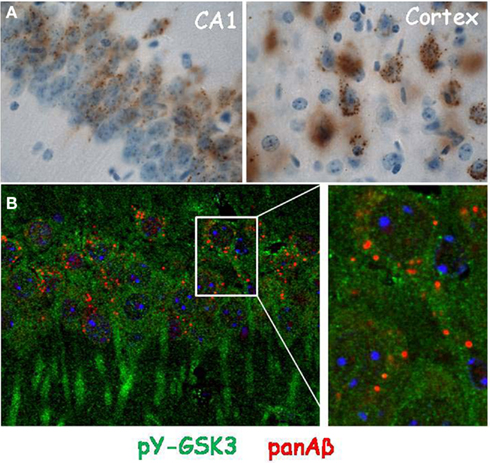
Figure 2. Localization of amyloid peptides and pY-GSK3 in transgenic mouse brain. (A) Intracellular amyloid (panAβ) in granules in pyramidal neurons of CA1 (left) and cortical layers III/IV (right) in brain of APP.V717I mice (age 6 months); (B) localization of activated pY-GSK3 (green) and panAβ (red) in the cytoplasm of CA1 neurons in the brain of APP.V717I mice (age 6 months), at right higher magnification of boxed area.
In brain, the regulation of the activity of GSK3α/β by phosphorylation of S21/S9 (inhibition) and/or Y279/216 (activation) is a matter of controversy – not only in AD but in normal physiology. In this respect a fundamental question refers to the structure-function relation of both GSK3 isozymes: they are known as S/T kinases that act on primed and non-primed substrates and modulated by tyrosine phosphorylation (Y279/Y216 in GSK3α/β, respectively; review Medina and Wandosell, 2011; references therein). Both kinases are inactivated by the well-known serine phosphorylation (S21/S9 in GSK3α/β, respectively) which evidently overrides activation by tyrosine phosphorylation. We cannot indulge here in extensive structural considerations on the tyrosine phosphorylation, i.e., whether it is autocatalytic or intra- or inter-molecular, which would promote GSK3 to the status of “dual activity kinases” (Cole et al., 2004; Medina and Wandosell, 2011; references therein). Nevertheless, the problem is most actual and of direct interest for AD, because of our observations in APP.V717I mice (Terwel et al., 2008) and observations in AD patients (Leroy et al., 2007; next section).
The implications are profound: amyloid peptides, which are located in the secretory pathway or are secreted, apparently are able to act on GSK3 to increase its tyrosine (auto-)phosphorylation, and thereby increase the kinase activity toward protein Tau (Figure 1). This hypothesis explains the observations in our transgenic and viral models for AD while raising important mechanistic questions (reviews Jaworski et al., 2010a,b).
GSK3 in Human Brain, not Enough Data…
Solid data on the physiological and pathological aspects of GSK3 in healthy human brain and in neurological diseases and dementia are rare and far apart.
In the frontal cortex of AD patients a non-significant trend of increased total levels of GSK3β was noted, with no difference in pS9-GSK3β. That contrasted to the significant increase in pY216-GSK3β (Leroy et al., 2007) recapitulated in our APP.V717I mice, discussed in the previous section (Terwel et al., 2008). In AD-frontal cortex pY216-GSK3 co-localized in somatodendritic compartments with highly phosphorylated protein Tau, a finding that is not too surprising, because enzymes and substrates must eventually meet. Neurons containing pY216-GSK3β were evident and widespread in brain of AD patients of all Braak-stages, from pre-tangle to stage VI, while biochemically no differences in pS9-GSK3 were noted (Leroy et al., 2007).
This implicates pY216-GSK3β as a pathological marker for tangled neurons, prevailing from early to late stages, and therefore a likely candidate to cause, or at least contribute to the tauopathy. Nevertheless, the mechanism behind the activating tyrosine phosphorylation of pY216-GSK3β in neurons destined or forced to form tangles still eludes us, as discussed in the previous section.
In contrast, co-localization of pS9-GSK3β and Tau was evident in tangles, dystrophic neurites and neuropil threads in AD brain, while biochemical fractionation revealed considerable pS9-GSK3 in the sarkosyl insoluble Tau fraction (Ferrer et al., 2002). Granulovascular degeneration, a neuronal lesion often observed in AD, is immunoreactive for GSK3α/β and for pY216-GSK3β. The co-localization with phosphorylated Tau (pS262, AT100) in these granules could imply that activated pY216-GSK3 becomes preferentially localized in special cellular compartments, e.g., autophagosomes, contributing to a potentially protective mechanism for neurons (Taelman et al., 2010; references therein).
To be complete, GSK3β can also become inactive after phosphorylation of S389 by p38MAPK, at least in mouse brain (Thornton et al., 2008) but no data are available for human brain. We conclude that we lack sufficient data in healthy and diseased human brain, to answer or counteract the questions that are raised by the studies in transgenic models.
GSK3, APP Processing, and Li+
A further problem in this sub-field is the eventual contribution of GSK3 to the pathogenesis of AD in terms of increased production of amyloid peptides. The question mainly originated from the finding that the cytoplasmic region of APP can become phosphorylated, with GSK3 one of the potential kinases (Aplin et al., 1996). Subsequently, and most amazingly, the less well-known GSK3α isozyme was proposed to regulate amyloidogenic processing of APP (Phiel et al., 2003). The reported evidence was largely based on cellular systems, using non-specific inhibitors and Li+, which evidently do not discriminate between the GSK3 isozymes. Additionally, Li+ produces diverse pleiotropic effects in biological systems, unrelated to GSK3, even involving NMDA-R and nitric oxide signaling as most recently demonstrated (reviews Agam et al., 2009; Ghasemi and Dehpour, 2011).
We have analyzed this issue more extensively in vivo in mouse brain, to help define physiological functions and pathological roles of GSK3α in normal brain and in AD-related neuronal processes in vivo. We generated and analyzed GSK3α deficient mice and observed normal processing of endogenous murine APP, as well as human APP produced by intracerebrally injected AAV–APP.SLA constructs (Jaworski et al., 2009; data not shown). Moreover, we analyzed bigenic GSK3α.KO × APP.V717I mice, yielding essentially the same negative outcome. We conclude that at least the GSK3α isozyme does not contribute significantly to the processing of APP in mouse brain in vivo. We are extending these studies to the GSK3β isozyme, by generating mice with neuron-specific deficiency of the GSK3β isozyme.
Finally, we advocate strongly against the use of lithium salts in cell-biological and in vivo studies and even more strongly against the generalized claim that Li+ is “a specific inhibitor” of GSK3 isozymes. Three points need to be brought to the attention of those engaging in this direction: Li+ ions are easy to use or administer, but by no means specific inhibitors of the GSK3 isozymes, nor does Li+ discriminate between isoforms, and lastly, Li+ is not even a potent kinase inhibitor. We consistently find that at least 5 mM Li+ is needed to inhibit GSK3 activity effectively in human retinoblastoma cell-based assays, while in vivo this concentration is highly toxic for mice on regimes that last for more than a day. Moreover, while LiCl incorporated in mouse food-pellets (2 g/kg) is not toxic for wild-type mice for up to 3 months, this diet effectively worsened the axonopathy in Tau4R mice and the tauopathy in Tau.P301L mice, without much effect on the phosphorylation status of protein Tau (Muyllaert et al., 2006, 2008; data not shown). Obviously, any effect noted using too low concentrations of lithium ions in any system, but particularly in vivo, cannot be attributed to inhibition of GSK3 but must be caused by other enzymes affected by Li+, of which more and more being discovered (reviews Agam et al., 2009; Ghasemi and Dehpour, 2011).
Concluding Remarks: GSK3 and Protein Tau
In contrast to APP processing, the relation of GSK3 to phosphorylation of Tau and tauopathy became stronger than ever. Most informative were GSK3β mice crossed with Tau4R and Tau.P301L mice expressing wild-type or mutant human Tau, respectively (Spittaels et al., 2000; Terwel et al., 2008), proving that GSK3β effectively phosphorylates protein Tau in brain in vivo.
Tau.P301L mice develop tauopathy first in their brainstem and progressively suffer severe motor defects, eventually dying from breathing defects and asphyxia around an average age of 9.4 months, with none surviving beyond 12 months of age (Dutschmann et al., 2010; data not shown). Interestingly, the combination with GSK3β alleviates tauopathy in the brainstem of Tau.P301L mice, rescuing their early mortality despite the dramatically increased tangle-load in forebrain (Terwel et al., 2008).
A comparable effect on Tau pathology, albeit less strong, was apparent in the second bigenic model that combines APP.V717I and Tau.P301L (biAT), capturing both AD pathologies in one model (Terwel et al., 2008). A considerable fraction of biAT mice survive longer than the parental Tau.P301L mice, although with a strong gender effect: survivors are mostly females, but again with more severe and aggravated Tau pathology than Tau.P301L mice. Remarkably, the amyloid pathology in female biAT mice is comparable to that in the parental single transgenic APP.V717I mice (Moechars et al., 1999; Terwel et al. 2008).
These mouse models convincingly proved that insoluble Tau is not the neurotoxic lethal factor – at least not in mouse brain. Because in the parental APP.V717I mice both GSK3 isozymes are activated by increased phosphorylation of Y279/216, the similarity in effects on tauopathy in both bigenic models is explained by this increased GSK3 activity, which posts GSK3 even more firmly as the major link between the two hallmark pathologies in AD (Figure 1).
Overall, our studies in transgenic mouse models add considerable weight to the hypothesis of direct involvement of GSK3 in AD pathogenesis, although the lack of any effect on APP processing in vivo focuses their involvement completely onto the increased phosphorylation of protein Tau. This outcome does, however, not change the general sequence of events, starting with aberrant production of amyloid peptides that activate GSK3 kinases to increase phosphorylation of protein Tau, as proposed by us (Muyllaert et al. 2008; Terwel et al. 2008; Jaworski et al. 2010a,b). Moreover, the bypass of amyloid via the activation of GSK3 to increased phosphorylation of Tau considerably strengthens the importance of tauopathy in the overall pathological process in AD. The obvious condition is that we define tauopathy no longer as the classical pathological fibrillar deposits in neuronal somata and processes, but as the phosphorylated molecular species, yet to be identified, which we denoted as Tau-P* (Figure 1; Jaworski et al. 2010a,b).
These toxic tau-species are likely dimers or small aggregates of protein Tau, and balance between further aggregation into the well-known large deposits that are obviously inert, or to remain soluble and be the toxic species that cause clinical dysfunctions known in AD patients, and in other tauopathies.
Remaining challenges are to define molecular identity and exact mechanisms, as well as find the “X-factors” (Figure 1), which are likely a combination of genetic, epigenetic, and environmental influences, besides disease and all sorts of stress. Besides the elusive Tau-species, also the amyloid peptides and aggregates, as well as the activated GSK3 isozymes can, or rather will affect synaptic functions. Eventually, the final neurodegeneration will turn out to be a combination of these different molecular players in different combinations, affected by different X-factors, which then explains the large variation in clinical picture that characterizes sporadic AD.
A final important consideration refers to the potential of GSK3, either or both isozymes, as therapeutic targets in dementia, AD, or related tauopathies. Judging from available data, originating from studies on physiology of CNS and peripheral systems, from studies on brain pathology, from pharmacological studies, including Li+, from epidemiological studies, from experimental mice (transgenic and knock-out) the prospect looks dim. GSK3 is involved in many essential pathways in many if not all physiological systems. Pharmacological inhibitors will have to hit the real target, i.e., GSK3 in neurons in entorhinal cortex, hippocampus, and cortex, while GSK3 in all other cells of the human body must not be affected. Potential alternatives, e.g., local application, or aiming at neuron-specific interactions of GSK3 with its controlling partners, or others that aim more specifically.
To make this happen, we need to collect more facts…
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The investigations were made possible by funding and support from the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (FWO-Vlaanderen), the Instituut Wetenschap and Techniek (IWT), EEC 6th and 7th Framework, the KULeuven Research-Fund, KULeuven-R&D. We sincerely thank many collaborators and scientists for technical assistance, advice, materials, scientific, and moral support. Anna Kremer and Tomasz Jaworski were recipients of EU-Marie Curie doctoral fellowships (MEST CT 2005-020013 NEURAD and MEST-CT-2005-020589 – EURON PhD School for Neurosciences, respectively).
References
Agam, G., Bersudsky, Y., Berry, G. T., Moechars, D., Lavi-Avnon, Y., and Belmaker, R. H. (2009). Knockout mice in understanding the mechanism of action of lithium. Biochem. Soc. Trans. 37(Pt 5), 1121–1125.
Aplin, A. E., Gibb, G. M., Jacobsen, J. S., Gallo, J. M., and Anderton, B. H. (1996). In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J. Neurochem. 67, 699–707.
Azoulay-Alfaguter, I., Yaffe, Y., Licht-Murava, A., Urbanska, M., Jaworski, J., Pietrokovski, S., Hirschberg, K., and Eldar-Finkelman, H. (2011). Distinct molecular regulation of glycogen synthase kinase-3alpha isozyme controlled by its N-terminal region: functional role in calcium/calpain signaling. J. Biol. Chem. 286, 13470–13480.
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259.
Braak, H., and Del Tredici, K. (2011a). The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 121, 171–181.
Braak, H., and Del Tredici, K. (2011b). Alzheimer’s pathogenesis: is there neuron-to-neuron propagation? Acta Neuropathol. 121, 589–595.
Brion, J. P. (1985). Immunological demonstration of tau protein in neurofibrillary tangles of Alzheimer’s disease. J. Alzheimers Dis. 9(3 Suppl.), 177–185.
Brion, J. P., Flament-Durand, J., and Dustin, P. (1986). Alzheimer’s disease and tau proteins. Lancet 2, 1098.
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., Demattos, R. B., Patterson, B. W., Fagan, A. M., Morris, J. C., Mawuenyega, K. G., Cruchaga, C., Goate, A. M., Bales, K. R., Paul, S. M., Bateman, R. J., and Holtzman, D. M. (2011). Human apoE isoforms differentially regulate rain amyloid-{beta} peptide clearance. Sci. Transl. Med. 3, 89ra57.b
Chen, P., Gu, Z., Liu, W., and Yan, Z. (2007). Glycogen synthase kinase 3 regulates NMDA receptor channel trafficking and function in cortical neurons. Mol. Pharmacol. 72, 40–51.
Cole, A., Frame, S., and Cohen, P. (2004). Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 377(Pt 1), 249–255.
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., Roses, A. D., Haines, J. L., and Pericak-Vance, M. A. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923.
Dekosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 30, 572–580.
Delacourte, A., and Buée, L. (2000). Tau pathology: a marker of neurodegenerative disorders. Curr. Opin. Neurol. 13, 371–376.
Dewachter, I., Ris, L., Jaworski, T., Seymour, C. M., Kremer, A., Borghgraef, P., De Vijver, H., Godaux, E., and Van Leuven, F. (2009). GSK3beta, a centre-staged kinase in neuropsychiatric disorders, modulates long term memory by inhibitory phosphorylation at serine-9. Neurobiol. Dis. 35, 193–200.
Dutschmann, M., Menuet, C., Stettner, G. M., Gestreau, C., Borghgraef, P., Devijver, H., Gielis, L., Hilaire, G., and Van Leuven, F. (2010). Upper airway dysfunction of Tau.P301L mice correlates with tauopathy in midbrain and ponto-medullary brainstem nuclei. J. Neurosci. 30, 1810–1821.
Duyckaerts, C. (2011). Tau pathology in children and young adults: can you still be unconditionally Baptist? Acta Neuropathol. 121, 145–147.
Duyckaerts, C., Delatour, B., and Potier, M. C. (2009). Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 118, 5–36.
Embi, N., Rylatt, D. B., and Cohen, P. (1980). Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 107, 519–527.
Ferrer, I., Barrachina, M., and Puig, B. (2002). Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 104, 583–591.
Ferri, C. P., Prince, M., Brayne, C., Brodaty, H., Fratiglioni, L., Ganguli, M., Hall, K., Hasegawa, K., Hendrie, H., Huang, Y., Jorm, A., Mathers, C., Menezes, P. R., Rimmer, E., Scazufca, M., and Alzheimer’s Disease International. (2005). Global prevalence of dementia: a Delphi consensus study. Lancet 366, 2112–2117.
Garrido, J. J., Simón, D., Varea, O., and Wandosell, F. (2007). GSK3 alpha and GSK3 beta are necessary for axon formation. FEBS Lett. 581, 1579–1586.
Ghasemi, M., and Dehpour, A. R. (2011). The NMDA receptor/nitric oxide pathway: a target for the therapeutic and toxic effects of lithium. Trends Pharmacol. Sci. 32, 420–434.
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890.
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving, N., James, L., Mant, R., Newton, P., Rooke, K., Roques, P., Talbot, C., Pericak-Vance, M., Roses, A., Williamson, R., Rossor, M., Owen, M., and Hardy, J. (1991). Segregation of a mis-sense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706.
Gonatas, N. K., Anderson, W., and Evangelista, I. (1967). The contribution of altered synapses in the senile plaque: an electron microscopic study in Alzheimer’s dementia. J. Neuropathol. Exp. Neurol. 26, 25–39.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917.
Hamos, J. E., DeGennaro, L. J., and Drachman, D. A. (1989). Synaptic loss in Alzheimer’s disease and other dementias. Neurology 39, 355–361.
Hanger, D. P., Anderton, B. H., and Noble, W. (2009). Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends. Mol. Med. 15, 112–119.
Hardy, J., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 12, 383–388.
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356.
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., Pahwa, J. S., Moskvina, V., Dowzell, K., Williams, A., Jones, N., Thomas, C., Stretton, A., Morgan, A. R., Lovestone, S., Powell, J., Proitsi, P., Lupton, M. K., Brayne, C., Rubinsztein, D. C., Gill, M., Lawlor, B., Lynch, A., Morgan, K., Brown, K. S., Passmore, P. A., Craig, D., McGuinness, B., Todd, S., Holmes, C., Mann, D., Smith, A. D., Love, S., Kehoe, P. G., Hardy, J., Mead, S., Fox, N., Rossor, M., Collinge, J., Maier, W., Jessen, F., Schürmann, B., van den Bussche, H., Heuser, I., Kornhuber, J., Wiltfang, J., Dichgans, M., Frölich, L., Hampel, H., Hüll, M., Rujescu, D., Goate, A. M., Kauwe, J. S., Cruchaga, C., Nowotny, P., Morris, J. C., Mayo, K., Sleegers, K., Bettens, K., Engelborghs, S., De Deyn, P. P., Van Broeckhoven, C., Livingston, G., Bass, N. J., Gurling, H., McQuillin, A., Gwilliam, R., Deloukas, P., Al-Chalabi, A., Shaw, C. E., Tsolaki, M., Singleton, A. B., Guerreiro, R., Mühleisen, T. W., Nöthen, M. M., Moebus, S., Jöckel, K. H., Klopp, N., Wichmann, H. E., Carrasquillo, M. M., Pankratz, V. S., Younkin, S. G., Holmans, P. A., O’Donovan, M., Owen, M. J., and Williams, J. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90.
Hooper, C., Markevich, V., Plattner, F., Killick, R., Schofield, E., Engel, T., Hernandez, F., Anderton, B., Rosenblum, K., Bliss, T., Cooke, S. F., Avila, J., Lucas, J. J., Giese, K. P., Stephenson, J., and Lovestone, S. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86.
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., Pitstick, R., Carlson, G. A., Lanier, L. M., Yuan, L. L., Ashe, K. H., and Liao, D. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081.
Hur, E. M., and Zhou, F. Q. (2010). GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551.
Ingram, E. M., and Spillantini, M. G. (2002). Tau gene mutations: dissecting the pathogenesis of FTDP-17. Trends Mol. Med. 8, 555–562.
Ishiguro, K., Shiratsuchi, A., Sato, S., Omori, A., Arioka, M., Kobayashi, S., Uchida, T., and Imahori, K. (1993). Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 325, 167–172.
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., Wölfing, H., Chieng, B. C., Christie, M. J., Napier, I. A., Eckert, A., Staufenbiel, M., Hardeman, E., and Götz, J. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397.
Jaworski, T., Dewachter, I., Lechat, B., Croes, S., Termont, A., Demedts, D., Borghgraef, P., Devijver, H., Filipkowski, R. K., Kaczmarek, L., Kügler, S., and Van Leuven, F. (2009). AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS ONE 4, e7280. doi: 10.1371/journal.pone.0007280
Jaworski, T., Kügler, S., and Van Leuven, F. (2010a). Modeling of tau-mediated synaptic and neuronal degeneration in Alzheimer’s disease. Int. J. Alzheimers Dis. 2010, pii: 573138.
Jaworski, T., Dewachter, I., Seymour, C. M., Borghgraef, P., Devijver, H., Kügler, S., and Van Leuven, F. (2010b). Alzheimer’s disease: old problem, new views from transgenic and viral models. Biochim. Biophys. Acta 1802, 808–818.
Jaworski, T., Lechat, B., Demedts, D., Gielis, L., Devijver, H., Borghgraef, P., Duimel, H., Verheyen, F. K., Kügler, S., and Van Leuven, F. (2011). Dendritic degeneration and neurovascular defects precede AAV-Tau-mediated neuronal loss. Am. J. Pathol. [Epub ahead of print].
Johnson, G. V., and Stoothoff, W. H. (2004). Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 117, 5721–5729.
Kaidanovich-Beilin, O., Lipina, T. V., Takao, K., van Eede, M., Hattori, S., Laliberté, C., Khan, M., Okamoto, K., Chambers, J. W., Fletcher, P. J., Macaulay, K., Doble, B. W., Henkelman, M., Miyakawa, T., Roder, J., and Woodgett, J. R. (2010). Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2, 35.
Kerkela, R., Kockeritz, L., Macaulay, K., Zhou, J., Doble, B. W., Beahm, C., Greytak, S., Woulfe, K., Trivedi, C. M., Woodgett, J. R., Epstein, J. A., Force, T., and Huggins, G. S. (2008). Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Invest. 118, 3609–3618.
Kim, W. Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., and Snider, W. D. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397.
Kim, W. Y., Zhou, F. Q., Zhou, J., Yokota, Y., Wang, Y. M., Yoshimura, T., Kaibuchi, K., Woodgett, J. R., Anton, E. S., and Snider, W. D. (2006). Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron 52, 981–996.
Kwok, J. B., Loy, C. T., Hamilton, G., Lau, E., Hallupp, M., Williams, J., Owen, M. J., Broe, G. A., Tang, N., Lam, L., Powell, J. F., Lovestone, S., and Schofield, P. R. (2008). Glycogen synthase kinase-3beta and tau genes interact in Alzheimer’s disease. Ann. Neurol. 64, 446–454.
Lambert, J. C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., Combarros, O., Zelenika, D., Bullido, M. J., Tavernier, B., Letenneur, L., Bettens, K., Berr, C., Pasquier, F., Fiévet, N., Barberger-Gateau, P., Engelborghs, S., De Deyn, P., Mateo, I., Franck, A., Helisalmi, S., Porcellini, E., Hanon, O., European Alzheimer’s Disease Initiative Investigators, de Pancorbo, M. M., Lendon, C., Dufouil, C., Jaillard, C., Leveillard, T., Alvarez, V., Bosco, P., Mancuso, M., Panza, F., Nacmias, B., Bossù, P., Piccardi, P., Annoni, G., Seripa, D., Galimberti, D., Hannequin, D., Licastro, F., Soininen, H., Ritchie, K., Blanché, H., Dartigues, J. F., Tzourio, C., Gut, I., Van Broeckhoven, C., Alpérovitch, A., Lathrop, M., and Amouyel, P. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099.
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Clos, A. L., Jackson, G. R., and Kayed, R. (2011). Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 6, 39.
Lau, L. F., Schachter, J. B., Seymour, P. A., and Sanner, M. A. (2002). Tau protein phosphorylation as a therapeutic target in Alzheimer’s disease. Curr. Top. Med. Chem. 2, 395–415.
Leroy, K., Yilmaz, Z., and Brion, J. P. (2007). Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 33, 43–55.
Lesné, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., Gallagher, M., and Ashe, K. H. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357.
Liang, M. H., and Chuang, D. M. (2007). Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J. Biol. Chem. 282, 3904–3917.
Louis, J. V., Martens, E., Borghgraef, P., Lambrecht, C., Sents, W., Longin, S., Zwaenepoel, K., Pijnenborg, R., Landrieu, I., Lippens, G., Ledermann, B., Götz, J., Van Leuven, F., Goris, J., and Janssens, V. (2011). Mice lacking phosphatase PP2A subunit PR61/B’delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3beta. Proc. Natl. Acad. Sci. U.S.A. 108, 6957–6962.
Ludolph, A. C., Kassubek, J., Landwehrmeyer, B. G., Mandelkow, E., Mandelkow, E. M., Burn, D. J., Caparros-Lefebvre, D., Frey, K. A., de Yebenes, J. G., Gasser, T., Heutink, P., Höglinger, G., Jamrozik, Z., Jellinger, K. A., Kazantsev, A., Kretzschmar, H., Lang, A. E., Litvan, I., Lucas, J. J., McGeer, P. L., Melquist, S., Oertel, W., Otto, M., Paviour, D., Reum, T., Saint-Raymond, A., Steele, J. C., Tolnay, M., Tumani, H., van Swieten, J. C., Vanier, M. T., Vonsattel, J. P., Wagner, S., Wszolek, Z. K., and Reisensburg Working Group for Tauopathies With Parkinsonism. (2009). Tauopathies with parkinsonism: clinical spectrum, neuropathologic basis, biological markers, and treatment options. Eur. J. Neurol. 16, 297–309.
MacAulay, K., Doble, B. W., Patel, S., Hansotia, T., Sinclair, E. M., Drucker, D. J., Nagy, A., and Woodgett, J. R. (2007). Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 6, 329–337.
Mandelkow, E. M., Schweers, O., Drewes, G., Biernat, J., Gustke, N., Trinczek, B., and Mandelkow, E. (1996). Structure, microtubule interactions, and phosphorylation of tau protein. Ann. N. Y. Acad. Sci. 777, 96–106.
Medina, M., and Wandosell, F. (2011). Deconstructing GSK-3: the fine regulation of its activity. Int. J. Alzheimers Dis. 2011, 479249.
Mines, M. A., Beurel, E., and Jope, R. S. (2011). Regulation of cell survival mechanisms in Alzheimer’s disease by glycogen synthase kinase-3. Int. J. Alzheimers Dis. 2011, 861072.
Moechars, D., Dewachter, I., Lorent, K., Reversé, D., Baekelandt, V., Naidu, A., Tesseur, I., Spittaels, K., Haute, C. V., Checler, F., Godaux, E., Cordell, B., and Van Leuven, F. (1999). Early phenotypic changes in transgenic mice that overexpres different mutants of amyloid precursor protein in brain. J. Biol. Chem. 274, 6483–6492
Muyllaert, D., Kremer, A., Jaworski, T., Borghgraef, P., Devijver, H., Croes, S., Dewachter, I., and Van Leuven, F. (2008). Glycogen synthase kinase-3beta, or a link between amyloid and tau pathology? Genes Brain Behav. 7(Suppl. 1), 57–66.
Muyllaert, D., Terwel, D., Borghgraef, P., Devijver, H., Dewachter, I., and Van Leuven, F. (2006). Transgenic mouse models for Alzheimer’s disease: the role of GSK-3B in combined amyloid and tau-pathology. Rev. Neurol. (Paris) 162, 903–907.
Patel, S., Doble, B. W., MacAulay, K., Sinclair, E. M., Drucker, D. J., and Woodgett, J. R. (2008). Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol. Cell. Biol. 28, 6314–6328.
Patterson, K. R., Remmers, C., Fu, Y., Brooker, S., Kanaan, N. M., Vana, L., Ward, S., Reyes, J. F., Philibert, K., Glucksman, M. J., and Binder, L. I. (2011). Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 286, 23063–23076.
Peineau, S., Bradley, C., Taghibiglou, C., Doherty, A., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2008). The role of GSK3 in synaptic plasticity. Br. J. Pharmacol. 153, S428–S437.
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., Lo, E., Wu, D., Saule, E., Bouschet, T., Matthews, P., Isaac, J. T., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Perez-Costas, E., Gandy, J. C., Melendez-Ferro, M., Roberts, R. C., and Bijur, G. N. (2010). Light and electron microscopy study of glycogen synthase kinase-3beta in the mouse brain. PLoS ONE 5, e8911. doi: 10.1371/journal.pone.0008911
Phiel, C. J., Wilson, C. A., Lee, V. M., and Klein, P. S. (2003). GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435–439.
Reitz, C., Honig, L., Vonsattel, J. P., Tang, M. X., and Mayeux, R. (2009). Memory performance is related to amyloid and tau patholgy in hippocampus. J. Neurol. Neurosurg. Psychiatr. 80, 715–721.
Roberson, E. D., Halabisky, B., Yoo, J. W., Yao, J., Chin, J., Yan, F., Wu, T., Hamto, P., Devidze, N., Yu, G. Q., Palop, J. J., Noebels, J. L., and Mucke, L. (2011). Amyloid-β-/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 31, 700–711.
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., Gerstein, H., Yu, G. Q., and Mucke, L. (2007). Reducing endogenous Tau ameliorates amyloidß–induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754.
Sergeant, N., Delacourte, A., and Buée, L. (2005). Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta 1739, 179–197.
Shaw, P. C., Davies, A. F., Lau, K. F., Garcia-Barcelo, M., Waye, M. M., Lovestone, S., Miller, C. C., and Anderton, B. H. (1998). Isolation and chromosomal mapping of human glycogen synthase kinase-3 alpha and -3 beta encoding genes. Genome 41, 720–727.
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., Tsuda, T., Mar, L., Foncin, J. F., Bruni, A. C., Montesi, M. P., Sorbi, S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D., Brookes, A., Sanseau, P., Polinsky, R. J., Wasco, W., Da Silva, H. A., Haines, J. L., Perkicak-Vance, M. A., Tanzi, R. E., Roses, A. D., Fraser, P. E., Rommens, J. M., and St George-Hyslop, P. H. (1995). Cloning of a gene bearing mis-sense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760.
Shipton, O. A., Leitz, J. R., Dworzak, J., Acton, C. E., Tunbridge, E. M., Denk, F., Dawson, H. N., Vitek, M. P., Wade-Martins, R., Paulsen, O., and Vargas-Caballero, M. (2011). Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J. Neurosci. 31, 1688–1692.
Smillie, K. J., and Cousin, M. A. (2011). The role of GS3 in presynaptic function. Int. J. Alzheimers Dis. 263673.
Soutar, M. P., Kim, W. Y., Williamson, R., Peggie, M., Hastie, C. J., McLauchlan, H., Snider, W. D., Gordon-Weeks, P. R., and Sutherland, C. (2010). Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 115, 974–983.
Spittaels, K., Van den Haute, C., Van Dorpe, J., Geerts, H., Mercken, M., Bruynseels, K., Lasrado, R., Vandezande, K., Laenen, I., Boon, T., Van Lint, J., Vandenheede, J., Moechars, D., Loos, R., and Van Leuven, F. (2000). Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J. Biol. Chem. 275, 41340–41349.
Spittaels, K., Van den Haute, C., Van Dorpe, J., Terwel, D., Vandezande, K., Lasrado, R., Bruynseels, K., Irizarry, M., Verhoye, M., Van Lint, J., Vandenheede, J. R., Ashton, D., Mercken, M., Loos, R., Hyman, B., Van der Linden, A., Geerts, H., and Van Leuven, F. (2002). Neonatal neuronal overexpression of glycogen synthase kinase-3 beta reduces brain size in transgenic mice. Neuroscience 113, 797–808.
St George-Hyslop, P. H. (2000). Genetic factors in the genesis of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 924, 1–7.
Taelman, V. F., Dobrowolski, R., Plouhinec, J. L., Fuentealba, L. C., Vorwald, P. P., Gumper, I., Sabatini, D. D., and De Robertis, E. M. (2010). Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148.
Takashima, A. (2006). GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 9, 309–317.
Terwel, D., Muyllaert, D., Dewachter, I., Borghgraef, P., Croes, S., Devijver, H., and Van Leuven, F. (2008). Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 172, 786–798.
Thornton, T. M., Pedraza-Alva, G., Deng, B., Wood, C. D., Aronshtam, A., Clements, J. L., Sabio, G., Davis, R. J., Matthews, D. E., Doble, B., and Rincon, M. (2008). Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 320, 667–670.
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., Cui, B., and Mucke, L. (2010). Tau reduction prevents Abeta-induced defects in axonal transport. Science 330, 198.
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862.
Keywords: GSK3, Alzheimer, tau
Citation: Kremer A, Louis JV, Jaworski T and Van Leuven F (2011) GSK3 and Alzheimer’s disease: facts and fiction… Front. Mol. Neurosci. 4:17. doi:10.3389/fnmol.2011.00017
Received: 08 July 2011;
Accepted: 08 August 2011;
Published online: 26 August 2011.
Edited by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaReviewed by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaSimon Lovestone, King’s College London, UK
Copyright: © 2011 Kremer, Louis, Jaworski and Van Leuven. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Fred Van Leuven, Experimental Genetics Group – LEGTEGG, Center for Human Genetics, KULeuven – Campus Gasthuisberg, ON1-06.602, Herestraat 49, B-3000 Leuven, Belgium. e-mail: fred.vanleuven@med. kuleuven.be