Role of GSK3 signaling in neuronal morphogenesis
- 1 Department of Orthopaedic Surgery, The Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 2 The Solomon H. Snyder Department of Neuroscience, The Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 3 Neuroscience Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Glycogen synthase kinase 3 (GSK3) is emerging as a key regulator of several aspects of neuronal morphogenesis including neuronal polarization, axon growth, and axon branching. Multiple signaling pathways have been identified that control neuronal polarization, including PI3K, Rho-GTPases, Par3/6, TSC–mTOR, and PKA–LKB1. However, how these pathways are coordinated is not clear. As GSK3 signaling exhibits crosstalk with each of these pathways it has the potential to integrate these polarity signals in the control neuronal polarization. After neurons establish polarity, GSK3 acts as an important signaling mediator in the regulation of axon extension and axon branching by transducing upstream signaling to reorganization of the axonal cytoskeleton, especially microtubules. Here we review the roles of GSK3 signaling in neuronal morphogenesis and discuss the underlying molecular mechanisms.
During neural development, differentiated neurons undergo multiple steps of morphogenesis, including directed neuronal migration, neuronal polarization, axon outgrowth, axon guidance, and axon branching, to form the architectural basis of neural circuits. A common cellular process underlying these morphogenetic events is reorganization of the neuronal cytoskeleton directed by extracellular cues. Therefore, understanding how extracellular signals are translated into cytoskeletal reorganization is a key question in study of neuronal morphogenesis. Glycogen synthase kinase 3s (GSK3s), including GSK3α and GSK3β, are key components of the canonical Wnt signaling pathway. Recent studies indicate that GSK3s are also key upstream regulators of neuronal microtubules, as a number of microtubule-binding proteins (MBPs) are GSK3 substrates. Further, in addition to canonical Wnt ligands GSK3 activity is modulated by many other extracellular factors and their associated downstream pathways. Therefore, GSK3 is emerging as a major signaling mediator that functions to orchestrate each step of neuronal morphogenesis. In this review, we summarize and evaluate studies that show the involvement of GSK3 signaling in neuronal polarization, axon growth, and axon branching. We also discuss the potential molecular and cellular mechanisms by which GSK3 signaling regulates these morphogenetic processes.
GSK3 Signaling in Neuronal Polarization
The development of axon/dendrite polarity forms the structural basis for directional transmission of neural activities between neurons. In the past decade, the cellular and molecular mechanisms by which neuronal polarity is regulated have been extensively studied (Barnes and Polleux, 2009). Based on observations in a seminal in vitro model (Craig and Banker, 1994), we know that neuronal polarization involves multiple cellular steps. First, an intracellular signal is locally activated in one of the unpolarized neurites that breaks the cell symmetry and initiates the polarization process. Additional polarity determinants, likely downstream of the initiation signal, are then asymmetrically accumulated in the same neurite, which later becomes the axon. Second, such asymmetric signaling then triggers reorganization of cytoskeletal elements (actin filaments and microtubules) in the future axon and leads to its preferential growth. Lastly, sustained activation of multiple cellular processes specifically in the axon, including polarized protein/membrane transport and local protein translation/degradation, act to maintain the established asymmetry and axon extension. At the same time, complementary signaling occurs in the dendrites to suppress neurite extension. During the past decade, many intracellular signaling pathways and molecules have been identified that control these polarization steps. These include the PI3K pathway, the Rho-GTPase pathways, the Par3/6 pathway, the TSC–mTOR pathway, and the PKA–LKB1 pathway (Shi et al., 2003; Nishimura et al., 2004, 2005; Schwamborn and Puschel, 2004; Barnes et al., 2007; Shelly et al., 2007; Choi et al., 2008; Li et al., 2008). However, how these pathways regulate neuronal polarization in a coordinated manner is not clear. GSK3 signaling has been shown to crosstalk with each of these pathways, suggesting that it may function to coordinate and integrate polarity signaling. Here, we review the roles of GSK3 signaling and its potential link to established polarity pathways during each step of neuronal polarization (Figure 1).
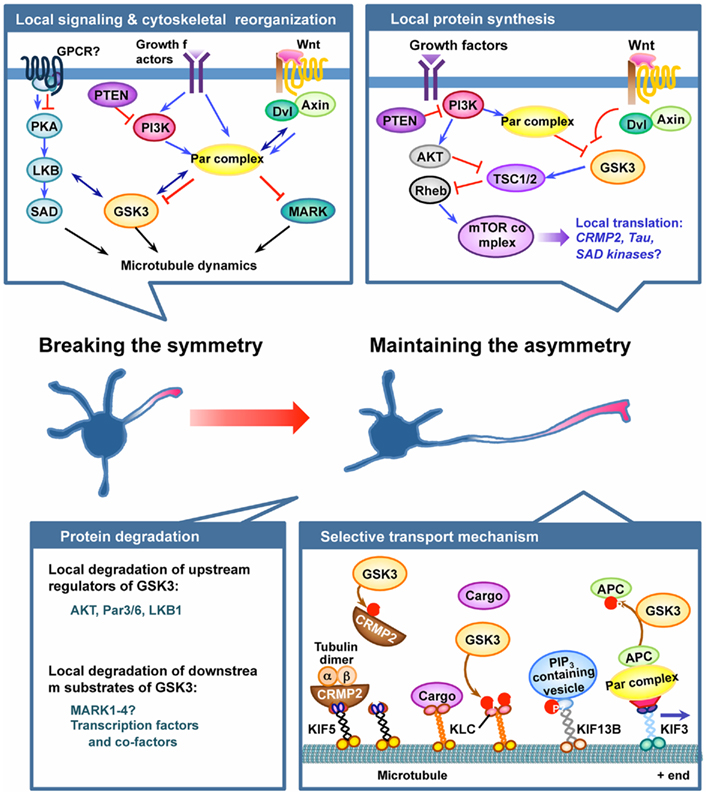
Figure 1. GSK3 plays pivotal roles in the establishment and maintenance of neuronal polarization. Neuronal polarization process begins by breaking cell symmetry, which is thought to be initiated by local activation of intracellular signaling in the nascent axon. Symmetry-breaking signals might involve confined activation of cell surface receptors, such as G protein-coupled receptors (GPCRs) or receptor tyrosine kinases, in the growth cone of the future axon. Several lines of evidence suggest GSK3 as a crucial node molecule of the initial symmetry-breaking pathways, such as the protein kinase A (PKA)–LKB1 pathway, the PI3K–partition defective protein (Par) pathway, and the Wnt–disheveled (Dvl) pathway. Local activation of symmetry-breaking signals leads to the reorganization of cytoskeletal elements, thereby establishing neuronal polarity. GSK3 might serve as a key molecule in the coordination of cytoskeletal elements by controlling microtubule dynamics and assembly via regulation of several microtubule-binding proteins (MBPs). Microtubule affinity-regulating kinase (MARK) has also been suggested to function downstream of the Par complex to control neuronal polarization by regulating MBPs. Once the symmetry is broken, maintaining the selective extension one neurite – the future axon – can be controlled by multiple mechanisms, including local protein synthesis and degradation, and selective transport mechanisms. GSK3 has been implicated in the local protein synthesis pathway by controlling the activity of the mammalian target of rapamycin (mTOR) complex via phosphorylating tuberous sclerosis 2 (TSC2), but the exact roles of the players involved and their regulation in the context of axon growth await to be determined. Local protein degradation through activation of the ubiquitin–proteasome system (UPS) is another possible mechanism to maintain asymmetrical accumulation of polarity proteins. Molecules that control GSK3 activity, such as Akt, PAR3/6, and LKB1, have been suggested as targets of the local degradation process. In addition, accumulating evidence suggests that protein levels of multiple downstream substrates of GSK3 are controlled by phosphorylation-dependent proteolytic degradation. Future studies are required to determine whether GSK3 activation in dendrites plays a part in maintaining the polarized growth of the axon and restricting the growth of the dendrites by controlling local protein degradation. Finally, selective delivery of membrane and cytoskeletal components and signaling proteins into defined compartments is an important mechanism to maintain neuronal polarity. GSK3 phosphorylates several motor proteins and polarity proteins to control their selective transport and localization. See text for further details for each of the mechanism depicted in the figure.
Breaking the Symmetry to Establish Neuronal Polarity
Before polarization, intracellular signaling molecules that control polarization are distributed evenly among unpolarized neurites. The breaking of cell symmetry is initiated by local activation of intracellular signals in one of the neurites, which later develops into the axon. One such symmetry-breaking signaling molecule is PI3K (Shi et al., 2003), which in neurons is a key downstream signaling mediator of receptor tyrosine kinases (RTKs; Reichardt, 2006). Activated PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to produce phosphatidylinositol-3,4,5-trisphosphate (PIP3) in the cell membrane, which then activates its downstream targets, such as AKT and Rho-GTPases (Cantley, 2002). Local activation of PI3K and the subsequent accumulation of PIP3 in the neurite that becomes the future axon have been shown to be early markers of neuronal polarization (Menager et al., 2004). Several lines of evidence suggest that GSK3 inactivation functions downstream of the PI3K–PIP3 pathway to control neuronal polarization. Thus, global inhibition of GSK3 with pharmacological inhibitors induces multiple axon formation, whereas overexpression of an active mutant of GSK3β (GSK3β-S9A) prevents axon formation (Jiang et al., 2005; Yoshimura et al., 2005). Importantly, overexpression of PTEN (a phosphatase that dephosphorylates PIP3) blocks neuronal polarization, which can be rescued by direct inhibition of GSK3 with pharmacological inhibitors (Jiang et al., 2005). Conversely, knocking down of PTEN induces multiple axon formation, which can be antagonized by overexpression of the active GSK3 mutant. However, how PI3K inactivates GSK3 to control neuronal polarization is still not clear.
One way that PI3K inactivates GSK3 is through AKT-mediated phosphorylation of GSK3 at an N-terminal domain serine residue (Cross et al., 1995). In support of the importance of Akt regulation, phosphorylated GSK3β co-localizes with phosphorylated AKT in the axon (Zhou et al., 2004; Jiang et al., 2005). Moreover, overexpression of a myristoylated active form of AKT leads to multiple axon formation which is antagonized by GSK3β-S9A expression (Jiang et al., 2005). However, neurons from the GSK3α-S21A/GSK3β-S9A double knockin mice, in which GSK3 cannot be phosphorylated by AKT, show no defect in neuronal polarization (Gartner et al., 2006), arguing against the idea that PI3K controls neuronal polarization via AKT-mediated GSK3 inactivation. PI3K has also been shown to regulate neuronal polarization through the Rho-GTPase Cdc42 and the subsequent activation of the Par3/6 cell polarity pathway (Shi et al., 2003). Par3/6 are mammalian homologs of the partition defective protein 3/6 of C. elegans. Interestingly, during directed cell migration locally activated Cdc42 at the cell leading edge can recruit the Par3/6 complex, which subsequently interacts with GSK3 and leads to GSK3 inactivation (Etienne-Manneville and Hall, 2003). Therefore, an alternative way that PI3K might inactivate GSK3 during neuronal polarization is via the Cdc42–Par3/6 pathway (Figure 1; Hur and Zhou, 2010). Further studies are required to test this hypothesis in neurons during polarization. How Par3/6 regulates GSK3 activity is unclear. A recent study showed that GSK3 activity could be regulated by DISC1 through protein–protein interactions independent of GSK3 serine phosphorylation (Mao et al., 2009). It is possible that Par3/6 also regulate GSK3 activity via the similar mechanism (Hur and Zhou, 2010). Interestingly, Par3 can directly bind to PI3K and enhance its activity (Itoh et al., 2010), suggesting that increased serine phosphorylation of GSK3 by Akt might be downstream of GSK3 inactivation by Par3. As a result, the levels of GSK3 serine phosphorylation can still be used as an indicator of GSK3 activity, even though GSK3 phosphorylation may not be a major mechanism by which GSK3 activity is inactivated (Hur and Zhou, 2010).
In addition to PI3K, another signaling molecule also activated downstream of RTKs or GPCRs that may act to break symmetry and initiate polarization is PKA. A recent study of neuronal polarization using live cell imaging of a cAMP biosensor suggests that local elevation of cAMP and activation of PKA in one undifferentiated neurite is an early signal to establish neuronal polarity (Shelly et al., 2010). The downstream mediator of PKA in regulation of neuronal polarization is the serine/threonine kinase LKB1 (Figure 1), which is the homolog of the partition defective protein 4 (Par4) of C. elegans (Barnes et al., 2007; Shelly et al., 2007). Activated PKA phosphorylates LKB1 and elevates its protein level. LKB1 then phosphorylates and activates SAD kinases, which have a similar kinase domain to that of partition defective protein 1 (Par1) and its mammalian homologs, MARK1–MARK4 (Kishi et al., 2005). Activated SAD kinases are believed to regulate neuronal polarization via the microtubule-binding protein Tau (Kishi et al., 2005). Interestingly, activation of PKA in neurons induces increased phosphorylation of both LKB1 and GSK3 (Shelly et al., 2010). Moreover, depletion of LKB1 abolishes GSK3 phosphorylation in all unpolarized neurites (Asada et al., 2007). These results suggest that GSK3 acts downstream of the PKA–LKB1 pathway to control neuronal polarization. Consistent with this idea, the Xenopus homolog of LKB1, XEEK1, is able to directly interact with GSK3β in vivo (Ossipova et al., 2003; Figure 1). In addition, a recent study showed that LKB1 inactivates GSK3 in mammalian neurons to control neuronal migration (Asada and Sanada, 2010). Taken together, these results suggest that GSK3 may act downstream of PI3K and PKA to establish neuronal polarity via direct interaction with Par3/6 and LKB1, respectively. While the cAMP–PKA pathway promotes axon formation during the symmetry-breaking process, reciprocal activation of the cGMP pathway acts to promote dendrite formation. A new study showed that semaphorin 3A acts upstream of cGMP to induce dendrite formation (Shelly et al., 2011). In contrast to the cAMP–PKA pathway that leads to GSK3 inactivation, semaphorin 3A has been shown to activate GSK3 (Eickholt et al., 2002). Consistent with this observation, semaphorin 3A treatment antagonizes PKA-induced GSK3 phosphorylation (Shelly et al., 2011). Together, these studies indicate that reciprocal regulation of cAMP/cGMP signaling controls axon/dendrite formation via opposite modulation of GSK3 activity.
Polarized Cytoskeletal Reorganization Preceding Asymmetrical Axon Extension
After polarity signals are accumulated in the future axon but before asymmetric axon extension occurs, the cytoskeletal elements in the future axon reorganize to complete the establishment of neuronal polarity (Witte and Bradke, 2008). Specifically, the actin filaments (F-actin) in the growth cone of the future axon become more dynamic than those in growth cones of other neurites (Bradke and Dotti, 1999). In addition, the microtubules in the future axon are preferentially stabilized (Witte et al., 2008). Importantly, these polarized reorganizations of F-actin and microtubules per se are sufficient to establish neuronal polarity, indicating that they play instructive roles in establishing neuronal polarization, rather than simply supporting axon extension (which will be discussed later in this review). However, how such polarized cytoskeletal rearrangement is regulated by upstream polarity signals remains unclear.
Among all the polarity pathways, GSK3 signaling is the best candidate for transducing the polarity signal into cytoskeletal reorganization, in particular microtubule stabilization (Zhou and Snider, 2005; Hur and Zhou, 2010). Many MBPs involved in neuronal polarization are GSK3 substrates, such as collapsin response-associated protein 2 (CRMP2; Cole et al., 2004), adenomatous polyposis coli (APC), Tau, and MAP1b. The function of these MBPs is to promote microtubule assembly and stability, and phosphorylation of them by GSK3 abolishes their ability to interact with microtubules. As a result, polarity signals can stabilize microtubules in the future axon by inactivation of GSK3. For instance, inhibition of GSK3 enhances the interactions between CRMP2 and tubulin dimers, which subsequently promotes microtubule assembly and stability. Overexpression of CRMP2 is sufficient to induce the formation of multiple axons (Yoshimura et al., 2005). Moreover, knocking down of CRMP2 antagonizes multiple axon formation induced by global GSK3 inactivation, indicating the CRMP2 acts downstream of GSK3 inactivation (Yoshimura et al., 2005). The microtubule plus end tracking protein (+TIP) APC localizes at the tips of all unpolarized neurites before neuronal polarization, and later becomes enriched in the growth cone of the future axon (Votin et al., 2005). Binding of APC to the microtubule plus ends functions to stabilize microtubules, a process negatively regulated by GSK3-mediated phosphorylation (Zumbrunn et al., 2001; Zhou et al., 2004). Therefore, local GSK3 inactivation in the future axon promotes the binding of APC to the microtubule plus ends, leading to asymmetrical microtubule stabilization in the future axon. However, neurons from APC knockout mice do not show polarization defects (Yokota et al., 2009), suggesting compensatory effect of its homolog APC-L (Nakagawa et al., 1998) or redundant function of other microtubule +TIPs.
How polarized cytoskeletal reorganization directs neuronal polarization is unknown. One potential mechanism is that it may affect post-translational modification of microtubules, which then facilitate the binding of motor proteins to the microtubules and the subsequent directed transport (see detailed discussion below). Indeed, activated kinesin-1 (or KIF5) is preferentially accumulated in the future axon before polarized axon extension, and global application of taxol or GSK3 inhibitors disrupts the polarized targeting of kinesin-1 (Hammond et al., 2010).
Maintaining Asymmetrical Axon Extension
After the establishment of initial polarity, neurons require sustained asymmetrical axon extension to complete the polarization process. Many cellular processes have been suggested to maintain such polarized axon extension, including polarized transport of proteins and membrane, local protein translation, and local protein degradation. GSK3 signaling has been shown to be involved in all of these processes.
Polarized transport of polarity determinants is important for their sustained accumulation in the axon to direct asymmetrical axon extension (Figure 1). Studies have shown that PI3K, PIP3, and Par3 are all specifically transported to the axon by kinesins, and that disruption of such transport blocks neuronal polarization (Shi et al., 2004; Horiguchi et al., 2006; Amato et al., 2011). GSK3 can directly phosphorylate several kinesin light chains and thus regulate the interactions between kinesins and their transported cargoes (Morfini et al., 2002, 2004; Szebenyi et al., 2002). In addition, many of the kinesin superfamily proteins have conserved GSK3 consensus sites (Taelman et al., 2010). Thus GSK3 may be a major regulatory factor of kinesin function. In support of this idea, Par3 is specifically delivered to the axon by forming a protein complex with APC and KIF3A, and such polarized transport requires local inactivation of GSK3 in the axon (Shi et al., 2004). Moreover, a recent study demonstrated that activation of the energy-sensing adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) suppressed neuronal polarization by phosphorylating the kinesin light chain (KCL2) of the KIF5 protein, which functions to target PI3K to the axon (Amato et al., 2011). Because GSK3 also phosphorylates KCL2 and negatively regulates its function (Morfini et al., 2002, 2004), it is possible that GSK3 inactivation promotes PI3K transport into the axon, thus forming a positive feedback loop to control neuronal polarization. Finally, PIP3 is specifically transported to the axon by KIF13B, which is also a potential GSK3 substrate (Horiguchi et al., 2006). In addition to regulating transport of proteins, GSK3 also regulates polarized axonal transport of vesicles containing membrane components (Morfini et al., 2004; Gartner et al., 2006). GSK3β is specifically accumulated in the Golgi region of hippocampal neurons in a membrane-bound form. Global inhibition of GSK3 with inhibitors disrupts preferential traffic of membrane containing vesicles to the axon, and directs them symmetrically toward all neurites (Gartner et al., 2006). However, how GSK3 regulates membrane transport is unknown. Together, these studies suggest that GSK3 functions to maintain asymmetrical axon extension via regulation of directed axonal transport.
Local and asymmetrical protein translation of polarity molecules has been reported to control neuronal polarization (Figure 1). One important pathway that regulates protein translation is the TSC–mTOR pathway (Ma and Blenis, 2009). Activation of AKT activates Rheb and the subsequent mammalian target of rapamycin (mTOR) kinase by inactivating their negative regulators TSC1/2. Activated mTOR then phosphorylates multiple translation regulators, such as ribosomal S6 kinase (S6K) and translation initiation factor 4E binding protein (4EBP), to initiate protein translation. During neuronal polarization phosphorylated S6K and 4EBP are enriched in the axon (Morita and Sobue, 2009), indicating local activation of mTOR signaling. Further, overexpression of constitutively activated Rheb and S6K induces multiple axon formation, whereas dominant negative Rheb and S6K block neuronal polarization (Morita and Sobue, 2009). Importantly, genetic knockout of TSC1 or TSC2 leads to the formation of multiple axons in vitro and in vivo (Choi et al., 2008). Evidence showing direct involvement of GSK3 signaling in the TSC–mTOR pathway during neuronal polarization is lacking. However, one study has shown that GSK3 directly phosphorylates TSC2 after AMPK-mediated priming, leading to inhibition of the mTOR pathway (Inoki et al., 2006). Conversely, inactivation of GSK3 is sufficient to activate mTOR-dependent protein translation. Therefore, it is possible that local inactivation of GSK3 in the axon plays a role in regulation of local translation through the mTOR pathway. Several polarity-related proteins are locally translated in the axon, including SAD kinase, CRMP2, Tau, and Par3 (Inoki et al., 2006; Choi et al., 2008; Hengst et al., 2009). Very interestingly, as mentioned earlier CRMP2 and Tau are GSK3 substrates, whereas Par3 acts upstream of GSK3, indicating additional examples of positive feedback loops involving GSK3 signaling through local protein translation.
Several studies have shown that local protein degradation via the ubiquitin–proteasome system (UPS) also controls neuronal polarization (Figure 1). Local degradation of polarity determinants in minor neurites is necessary to maintain their asymmetrical accumulation in the axon. In support, UPS has been shown to control the local degradation of AKT, Par3/6, and LKB1 in minor neurites during neuronal polarization, and global inhibition of UPS results in the formation of multiple axons (Yan et al., 2006; Cheng et al., 2011). Conversely, local degradation of RhoA (a suppressor of axon extension) is also required to maintain asymmetrical axon extension (Cheng et al., 2011). Although the GSK3 level in neurons is not regulated by UPS-mediated protein degradation, inhibition of UPS increases the level of phosphorylated GSK3β (Yan et al., 2006), suggesting that GSK3 inactivation might be involved in multiple axon formation induced by UPS inhibition. In support of this idea, the best studied function of GSK3 is regulation of the degradation of β-catenin in the canonical Wnt pathway via UPS (Hur and Zhou, 2010). A recent study demonstrates that GSK3 regulates UPS-mediated degradation of a much broader list of proteins (Kim et al., 2009a; Xu et al., 2009), raising the possibility that GSK3 may regulate the degradation of polarity determinants. For instance, Par3 and the aforementioned Par1 homologs, MARK1-4, contain three and six consecutive GSK3 phosphorylation sites (Taelman et al., 2010), respectively. It will be extremely interesting in the future to investigate whether these proteins can be regulated by GSK3-dependent, UPS-mediated protein degradation.
In summary, evidence is emerging that GSK3 is a key signal-integrating molecule that coordinates neuronal polarization by transducing early symmetry-breaking signaling, controlling cytoskeletal reorganization, and modulating polarized protein distribution. How GSK3 is asymmetrically regulated during neuronal polarization is not fully understood. One possible mechanism is that upstream regulators of GSK3 is locally distributed or activated. Another potential mechanism is local sequestration of GSK3 by scaffolding proteins. For instance, GSK3 has been shown to bind to the A-kinase anchoring proteins 220 (AKAP220), which mediates localized interaction between GSK3 and PKA (Tanji et al., 2002). In addition, a recent study showed that a GSK3 interacting protein could also function as an AKAP (Hundsrucker et al., 2010).
GSK3 Signaling in Axon Growth
After the neuron polarizes, the axon may extend a long distance to reach its targets and form synaptic connections. Axon growth is supported by extracellular factors, which transduce their signals into coordinated gene expression in the neuronal soma and local assembly of cytoskeleton and membrane in the growth cone (Zhou and Snider, 2006). GSK3 is a key mediator that translates extracellular signals into controlled axon assembly. Here we summarize how extracellular factors regulate GSK3 activity and discuss how GSK3 controls cytoskeletal reorganization in the growth cone during axon elongation.
GSK3 is Regulated by Multiple Extracellular Factors that Control Axon Growth
The most studied extracellular factors that support axon growth are growth factors, such as neurotrophins, IGF-1, and GDNF (Zhou and Snider, 2006). All these factors initiate their intracellular signals through RTKs. One of the major downstream mediators of RTKs is PI3K which plays a key role in controlling axon growth and is the best-known regulator of GSK3 signaling. Treatment of embryonic dorsal root ganglion (DRG) neurons with nerve growth factor (NGF) leads to PI3K activation and increased phosphorylation of GSK3β at its N-terminal serine-9 (Zhou et al., 2004). Functionally, overexpression of the active mutant of GSK3β (GSK3β-S9A) blocks NGF-induced axon growth. These results indicate that NGF inactivates GSK3 downstream of PI3K to promote axon growth. However, other studies have shown that NGF treatment leads to increased phosphorylation of MAP1b at its GSK3 sites, indicating that NGF also activates GSK3 (Goold and Gordon-Weeks, 2003; Trivedi et al., 2005). In contrast to GSK3β inactivation downstream of PI3K, GSK3 activation, and the subsequent phosphorylation of MAP1b is regulated by ERK1/2 (Goold and Gordon-Weeks, 2005), another major signaling mediator downstream of RTKs. Global application of netrin, a well-studied attractive axon guidance cue, also promotes axon growth through its receptor deleted in colon cancer (DCC) and the activation of the ERK1/2 pathway (Forcet et al., 2002). Similar to NGF, treatment of cultured neurons with netrin increased the level of phosphorylated MAP1b at its GSK3 site (Del Rio et al., 2004). Moreover, netrin treatment led to increased levels of GSK3β phosphorylation at both tyrosine-216, which is often associated with GSK3 activation, and serine-9, which indicates GSK3 inactivation (Del Rio et al., 2004). Thus, both NGF and netrin seem to inactivate and activate GSK3 in same cells downstream of PI3K and ERK1/2, respectively. One potential explanation is that regulation of GSK3 by NGF or netrin in neurons is segregated spatially or temporary allowing inactivation and activation at different times or places inside cell. Another possible explanation is that phosphorylation of GSK3β, either at serine-9 or at tyrosine-216, may not be a reliable indicator of GSK3 activity (Hur and Zhou, 2010). Future studies to directly examine GSK3 activity in live cells are required to test these hypotheses. Finally, one study has shown that Wnt3a can promote axon growth of mouse embryonic DRG neurons through its atypical receptor Ryk and the canonical Wnt pathway (Lu et al., 2004), suggesting the involvement of GSK3. However, the role of GSK3 in Wnt3a-induced axon growth has not been directly tested.
GSK3 Controls Axon Elongation Via Regulation of the Growth Cone Machinery
Axon elongation requires continuous addition of microtubules to the growing end, which is controlled by coordinated regulation of microtubule dynamics and actin reorganization in the growth cone (Zhou and Snider, 2006). Actin filaments are enriched in the peripheral domain (P-domain) of the growth cone, and are organized into actin bundles and actin meshwork. Microtubules project from the axon shaft to the central region (C-domain) of the growth cone in a bundled form. At the end of the microtubule bundles, individual microtubules splay apart and a subset of microtubules invade the actin-rich P-domain. Axon elongation is ultimately achieved through microtubule assembly at the nerve growth cones, in which a key step is microtubule extension toward the growth cone leading edge. Several factors have been shown to control microtubule extension in the growth cone. First, microtubules undergo cycles of polymerization and depolymerization, a process called dynamic instability (Burbank and Mitchison, 2006). There is an extensive list of microtubule or tubulin-binding proteins that act to regulate this process (Poulain and Sobel, 2010). For instance, some proteins can bind to the tubulin dimers and control tubulin polymerization (e.g., CRMP2) or depolymerization (e.g., stathmin). In addition, classic microtubule associated proteins (MAPs, e.g., Tau and MAP1b) bind along the microtubule lattices, whereas microtubule +TIPs (e.g., APC, EB1/3, and CLASP1/2) specifically interact with the plus ends, both of which function to stabilize polymerized microtubules. Lastly, there are proteins that function to disassemble microtubules, including microtubule severing proteins (e.g., katanin and spastin) and microtubule depolymerases (e.g., MCAK/KIF2c). Second, microtubule extension in the growth cone can be regulated by motor protein-mediated transport of microtubule polymers. The plus end directed motor kinesin-5, when anchored on the membrane, has been shown to transport microtubules away from the leading edge (Myers and Baas, 2007). Conversely, the minus end directed motor dynein, when immobilized at the growth cone leading edge, can pull the microtubules toward the leading edge (Myers et al., 2006). Third, dynamic interactions between actin filaments and microtubules in the growth cone are key processes that control the microtubule extension. Actin bundles function to guide microtubule extension toward the leading edge, whereas retrograde flow of actin meshwork powered by myosin II carries microtubules rearward, thus acting as a dynamic barrier for microtubule extension (Schaefer et al., 2002; Zhou et al., 2002; Medeiros et al., 2006).
Many of these cytoskeletal proteins are established GSK3 substrates, including CRMP2, APC, CLASP1/2, MAP1b, Tau, stathmin, and myosin II (Moreno and Avila, 1998; Kim et al., 2009a; Hur and Zhou, 2010), or contain conserved GSK3 sites, such as Katanin and KIF2c (Taelman et al., 2010). Thus, GSK3 is considered to be a major signaling pathway that regulates the growth cone machinery to drive axon elongation (Figure 2). However, because these molecules play different, and sometimes opposing roles in controlling cytoskeletal reorganization, the role of GSK3 signaling in regulation of axon elongation is complex. Expression of the active mutant of GSK3 (GSK3β-S9A) blocks axon growth (Zhou et al., 2004, 2006), indicating that inactivation of GSK3 is necessary for efficient axon elongation. Surprisingly, inhibition of GSK3 has been shown to either promote or impede axon growth depending on the extent of inhibition. Moderate GSK3 inhibition locally at the growth cone is required for efficient axon growth, whereas more complete inhibition of GSK3 impedes axon growth (Kim et al., 2006). One potential explanation for such pleiotropic effects is that different degrees of GSK3 inhibition differentially affect different GSK3 substrates. In support of this idea, phosphorylation of CRMP2 by GSK3 is more sensitive to GSK3 inhibition than phosphorylation of MAP1b (Kim et al., 2006). The identities of GSK3 substrates and the molecular mechanisms by which they specifically mediate axon growth promotion or axon growth inhibition upon GSK3 inactivation are being actively studied. Several molecules have been identified that promote axon growth downstream of GSK3 inactivation, including CRMP2 and APC. As mentioned earlier, inhibition of GSK3 facilitates the binding of CRMP2 to tubulin dimers and thus promotes axon growth by enhancing microtubule polymerization in the growth cone (Yoshimura et al., 2005). Similarly, dephosphorylated APC specifically bind to the microtubule plus ends and function to stabilize polymerizing microtubules, leading to increased microtubule extension in the growth cone (Zhou et al., 2004).
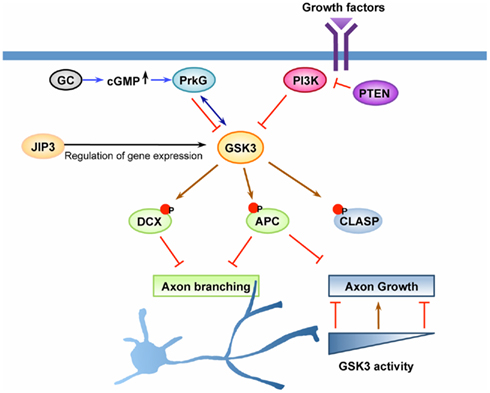
Figure 2. GSK3 is a key regulator of axon growth and branching. Axon growth is thought to be initiated by activation of cell surface receptors upon recognition of extrinsic cues, such as growth factors or other ligands. GSK3 has been suggested as a key molecule downstream of the PI3K pathway and the cyclic guanosine monophosphate (cGMP) pathway to control axon growth and/or branching. Upon activation of PI3K, GSK3 becomes inactivated, leading to dephosphorylation of GSK3 substrates that control the stability and dynamics of axonal microtubules. Dephosphorylation and subsequent binding of adenomatous polyposis coli (APC) on microtubule plus ends at certain locations along the axon enhances axonal branching, whereas its enrichment at the growth cone leads to the promotion of axon growth. CLASP, another substrate of GSK3 that binds to microtubules in response to GSK3 inactivation, plays a role to regulate axon growth downstream of GSK3. CLASP can both promote and inhibit axon growth depending on its microtubule-binding activity, a property which is tightly controlled by GSK3 activity. The cGMP pathway is another pathway that has been shown to control axonal branching. In response to elevation of cGMP by activation of guanylyl cyclase (GC), cGMP-dependent protein kinase G (PrkG) becomes activated and functions to increase axon branching by inactivating its substrate GSK3. In addition to the direct regulation of the kinase activity, GSK3 activity can be controlled by other mechanisms, such as regulation of its expression level. JNK-interacting protein (JIP) 3 has been suggested to control the expression level of GSK3 at the transcription level and/or by regulating mRNA stability. Down-regulation of JIP3, which decreases GSK3 level, increases axonal branching by controlling the phosphorylation status of doublecortin (DCX), a substrate of GSK3. Coordinated regulation of multiple GSK3 substrates likely defines the extent of axon growth and the locations at which axonal branching occurs.
Which substrates mediate impaired axon growth upon GSK3 inhibition to a greater extent? A new study has identified CLASP as a mediator to block axon growth upon greater GSK3 inhibition (Hur et al., 2011). Specifically, the study showed that in the absence of CLASP2, knocking down of GSK3 was unable to block axon growth. The study also revealed that in nerve growth cones CLASP could either track the microtubule plus ends or bind along the microtubule lattice depending on its phosphorylation states mediated by GSK3. Interestingly, these two microtubule-binding activities of CLASP regulate axon growth in an opposing manner: the microtubule lattice-binding activity mediates axon growth inhibition via preventing microtubule protrusion into the growth cone periphery, whereas the microtubule plus end-binding property supports axon extension via stabilizing the growing ends of axonal microtubules. As a result, when GSK3 activity becomes strongly suppressed the binding of CLASP along the microtubule lattice is increased, which subsequently induces microtubule looping in the growth cone and blocks axon growth. In addition to CLASP, other GSK3 substrates may also contribute to impaired axon growth upon greater GSK3 inhibition. For instance, the regulatory light chain of myosin II (MLC) was recently identified to be a GSK3 substrate, and phosphorylation of MLC by GSK3 promotes its degradation through UPS (Kim et al., 2009a). Moreover, both myosin IIA and IIB contain conserved GSK3 sites (Taelman et al., 2010), suggesting GSK3 as an important regulator of myosin II function. As a result, inhibition of GSK3 could block axon growth by enhancing myosin II activity in the growth cone. Indeed, similar to that of CLASP2, knocking down of GSK3 was unable to block axon growth when myosin II activity was inhibited (Hur et al., 2011). Further study is needed in the future to show directly that GSK3 phosphorylates myosin II and regulates its function in the growth cone. Another potential substrate is MAP1b, which plays a central role in maintaining microtubule dynamics in the growth cone when phosphorylated by GSK3 (Goold et al., 1999). Because microtubule dynamics are necessary for efficient axon growth, dephosphorylation of MAP1b upon greater inhibition of GSK3 may impede axon growth by reducing microtubule dynamics in the growth cone (Owen and Gordon-Weeks, 2003). In support of this idea, neurons from the MAP1b knockout mice show axon growth defects and exhibit markedly reduced microtubule dynamics in their growth cones (Gonzalez-Billault et al., 2002). Lastly, a recent study has provided some evidence that CRMP4 might act downstream of GSK3 inhibition to block axon growth (Alabed et al., 2010). Dephosphorylation of CRMP4 increases its binding to RhoA, a known negative regulator of axon growth.
In summary, abundant evidence indicates that an optimal level of GSK3 activity is required for efficient axon elongation (Figure 2). Under such conditions, some substrates are dephosphorylated (e.g., CRMP2, APC) to promote microtubule extension, whereas others are phosphorylated (e.g., CLASP, MAP1b) to maintain microtubule dynamics. Therefore, a delicate balance between activation and inactivation of GSK3 acts to support the most efficient axon elongation by coordinating the functions of multiple GSK3 substrates in the growth cone.
GSK3 Signaling in Axon Branching
When axons arrive at their destination, they usually form branches to innervate multiple targets. As with axon elongation, axon branching is tightly controlled by extracellular cues and their downstream intracellular signaling pathways that mediate reorganization of the axonal cytoskeleton to form axon branches. Based on morphology, axon branching can be grouped into arborization, bifurcation, and interstitial branching (Gibson and Ma, 2011). Time-lapse studies of live cells demonstrate that axon arborization and bifurcation usually occur at the distal axon through growth cone splitting, whereas interstitial branching forms de novo along the axon shaft (Zhou et al., 2002; Portera-Cailliau et al., 2005; Schmidt et al., 2007). Increasing evidence points to GSK3 as a converging signaling mediator that transduces branching signals into regulated cytoskeletal rearrangement at the growth cone or along the axon shaft. Here we review previous studies that show the involvement of GSK3 in axon branching and discuss the potential cellular mechanisms by which GSK3 signaling controls axon branching (Figure 2).
GSK3 functions have been carefully studied in sensory axons of DRG neurons that bifurcate at the dorsal root entry zone (DREZ) when they enter the spinal cord during development. Recent studies have identified C-type natriuretic peptide (CNP; also known as NPPC) as the extracellular factor that controls DRG axon bifurcation (Schmidt et al., 2009; Zhao and Ma, 2009). Downstream of CNP, cGMP, and cGMP-dependent protein kinase G1 (PrkG1) act to control axon branching via GSK3 inactivation (Zhao et al., 2009). Specifically, axon branching of cultured embryonic DRG neurons was substantially increased upon treatment of CNP or cGMP (8-Br-cGMP), and the effect was abolished in neurons from the PrkG1 knockout mice (Zhao and Ma, 2009). Moreover, PrkG1 inhibits GSK3β by binding and directly phosphorylating GSK3β at its serine-9 residue (Zhao et al., 2009). Importantly, cGMP-induced axon branching can be antagonized by overexpression of the active mutant of GSK3β (GSK3β-S9A), confirming that GSK3 inactivation is necessary for the cGMP–PrkG1 pathway to induce axon branching. As discussed earlier, serine phosphorylation may not be a major mechanism by which GSK3 is inactivated. Thus, it will be interested to examine if DRG axon bifurcation in the spinal cord during development is normal in the GSK3α-S21A/GSK3β-S9A double knockin mice, or to determine if PrkG1 is able to inactivate GSK3 via alternative mechanisms. After entering the spinal cord, axons of the NT3-responsive neurons in the DRG project ventrally and form terminal arborizations upon contact with the motor neurons, their synaptic targets. Wnt3 secreted by the motor neurons has been shown to cause such axon branching, suggesting that inhibition of GSK3 through the canonical Wnt pathway might be involved (Krylova et al., 2002). Indeed, inhibition of GSK3 with pharmacological inhibitors induced similar axonal arborization, mimicking the effect of Wnt3 (Krylova et al., 2002). In addition to the central axons in the spinal cord, the peripheral axons of sensory neurons also form terminal branches within their targeting areas (e.g., skin). The axon guidance cue Slit has been shown to play an important role in regulation of such peripheral axon branching of sensory neurons (Ma and Tessier-Lavigne, 2007). Whether GSK3 signaling is involved in Slit-induced axon branching is unknown. However, regulated GSK3 inactivation is involved in Slit-mediated neuronal migration (Higginbotham et al., 2006). In addition, the GSK3 substrate CLASP has been shown to act downstream of Slit to mediate axon repulsion (Lee et al., 2004). These results raise the possibility that Slit regulates sensory axon branching via GSK3 signaling.
In addition to extracellular cues, recent studies have provided evidence that intracellular molecules regulate axon branching via GSK3 signaling. One study (Bilimoria et al., 2010) showed that JNK-interacting protein 3 (JIP3) functioned to restrict axon branching of mouse cerebellar neurons via GSK3 and its substrate doublecortin (DCX). Knocking down of JIP3 resulted in significantly increased axon branching and a reduced level of GSK3β, protein suggesting that down-regulation of GSK3β mediated axon branching-induced by JIP3 depletion (Figure 2). Consistent with this idea, knocking down of GSK3β led to increased axon branching in a similar manner. Moreover, knocking down both JIP3 and GSK3β did not show an additive effect on axon branching, confirming that they share the same pathway. Lastly, DCX was identified as a substrate of GSK3β acting downstream of JIP3 and GSK3β to restrict axon branching. Finally, A few studies have demonstrated that PTEN deletion promotes axon branching (Kwon et al., 2006; Drinjakovic et al., 2010). As down-regulation of PTEN leads to increased activity of PI3K signaling, it is possible that axon branching-induced by loss of PTEN is through GSK3 inactivation. Together, these studies indicate that GSK3 inactivation is a common mechanism by which both extracellular cues and intracellular signals induce axon branching.
How GSK3 inactivation is translated into axon branching is not fully understood. High-resolution time-lapse studies of F-actin and microtubule dynamics indicate that coordinated regulation of F-actin reorganization and microtubule extension controls branch formation, similar to what occurs in the growth cone to drive axon elongation (Kalil et al., 2000; Dent and Kalil, 2001). At the growth cone, GSK3 inactivation is known to induce growth cone enlargement (Owen and Gordon-Weeks, 2003; Kim et al., 2006), presumably by promoting microtubule protrusion. Increased growth cone size with enhanced microtubule protrusion may therefore result in increased growth cone splitting to form axon branches. Along the axon shaft, axon branch formation requires coordinated cytoskeletal reorganization, including microtubule de-bundling and fragmentation, actin filopodia formation followed by microtubule extension (Dent and Kalil, 2001). Local inactivation of GSK3 along the axon shaft may promote these cytoskeletal changes through various GSK3-regulated cytoskeletal proteins. For instance, inactivation of GSK3 may modulate the function of DCX (Bilimoria et al., 2010), which is known to regulate microtubule organization along the axon shaft (Bielas et al., 2007; Tint et al., 2009; Figure 2). The microtubule severing protein Katanin is involved in microtubule fragmentation, a required process for initiating the branch formation (Qiang et al., 2010). The microtubule depolymerase KIF2a functions to impair microtubule polymerization and thus limiting the extension of axon branches (Homma et al., 2003). Interestingly, both Katanin and KIF2a are potential GSK3 substrates (Taelman et al., 2010), suggesting that inactivation of GSK3 may regulate their functions to control axon branching. Moreover, local inactivation of GSK3 along the axonal shaft has been shown to induce local accumulation of APC (Kim et al., 2006), which may stabilize polymerized microtubules to promote axon branching.
Conclusion and Future Directions
GSK3 is now considered to be a key signaling node molecule that acts to regulate multiple steps of neuronal morphogenesis during development by controlling a broad range of biological processes, such as cellular transport, protein translation and degradation, and cytoskeletal reorganization. Functions of GSK3 are achieved through its large number of substrates and multiple modes of regulation. Unlike most other kinases that are controlled by a simple activation/inactivation switch, it appears that regulation of GSK3 is more complex. Different levels of GSK3 activities regulated by upstream signals may affect different substrates, thus leading to distinct functions. Therefore, an important future direction is to identify and validate additional GSK3 substrates to understand how specific functions of GSK3 signaling are achieved via specific substrates. Moreover, the phosphorylation of GSK3 substrates is also regulated by various phosphatases, providing another level of regulation of GSK3 signaling.
As neuronal morphogenesis is a dynamic process, the ability to detect GSK3 activity at higher spatiotemporal resolution in live cells is necessary to fully understand the roles of GSK3 signaling. In addition to neuronal polarization, axon growth and axon branching, GSK3 signaling is also involved in other neuronal morphogenesis processes, such as directed neuronal migration, axon guidance, dendrite development, and synaptogenesis. Futures studies are necessary to establish the roles and the molecular mechanisms by which GSK3 signaling control these developmental processes. Finally, a number of neurological disorders, such as schizophrenia and autism, are associated with abnormal regulation of GSK3 signaling. It is therefore important in the future to identify the potential links between the functions of GSK3 signaling in neural development and the etiologies of these diseases.
The studies discussed in this review are mainly based on in vitro experiments. Most in vitro preparations are highly reduced and may not clearly reflect GSK3 functions in neurons in vivo, where a myriad of surface molecules and growth factors are present and a multiplicity of signaling pathways are active. Therefore, it is likely that the details of the developmental regulation by GSK3 signaling may not be entirely predictable from the in vitro studies that have appeared to date. Recently, decisive mouse genetic models (see other chapters) that enable cell-type specific GSK3 deletion have been generated. The first clear finding from these mice is that the two family members GSK3α and GSK3β can compensate for each other during brain development. Thus, when either GSK3α or GSK3β alone is deleted, gross morphological development proceeds. However, deletion of both family members results in dramatic neurodevelopmental abnormalities (Kim et al., 2006). The second clear finding is that GSK3 plays a major role in regulation of neural progenitors (Kim et al., 2009b). The neural progenitor cells of GSK3 deleted mice show striking disruption of apical–basal polarity, suggesting that GSK3 may also regulate cytoskeletal reorganization of neural progenitors. Work on GSK3 regulation of postmitotic neurons in vivo is just beginning. Experiments introducing constitutively activated GSK3 constructs via in utero electroporation strongly suggest a key role of GSK3 signaling in regulation of migration and cortical lamination (Asada and Sanada, 2010). It can be expected that further work with these mice will generate a decisive view of in vivo GSK3 functions in developmental processes discussed here, including neuronal polarization, axon extension and branching, as well as other key developmental processes, such as axon guidance, dendritic arborization, and synaptogenesis. However, it is important to keep in mind that because the regulation of GSK3 is complex, knockout studies, although very informative, may not fully reveal the molecular mechanisms by which GSK3 regulates neural development.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors were supported by the NIH grants R01NS064288 (to Feng-Quan Zhou), R01NS050968 (to William D. Snider), and the Christopher and Dana Reeve Foundation (to Eun-Mi Hur). We apologize for authors whose work we were unable to include due to space constraints.
References
Alabed, Y. Z., Pool, M., Ong Tone, S., Sutherland, C., and Fournier, A. E. (2010). GSK3 beta regulates myelin-dependent axon outgrowth inhibition through CRMP4. J. Neurosci. 30, 5635–5643.
Amato, S., Liu, X., Zheng, B., Cantley, L., Rakic, P., and Man, H. Y. (2011). AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science 332, 247–251.
Asada, N., and Sanada, K. (2010). LKB1-mediated spatial control of GSK3beta and adenomatous polyposis coli contributes to centrosomal forward movement and neuronal migration in the developing neocortex. J. Neurosci. 30, 8852–8865.
Asada, N., Sanada, K., and Fukada, Y. (2007). LKB1 regulates neuronal migration and neuronal differentiation in the developing neocortex through centrosomal positioning. J. Neurosci. 27, 11769–11775.
Barnes, A. P., Lilley, B. N., Pan, Y. A., Plummer, L. J., Powell, A. W., Raines, A. N., Sanes, J. R., and Polleux, F. (2007). LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 129, 549–563.
Barnes, A. P., and Polleux, F. (2009). Establishment of axon-dendrite polarity in developing neurons. Annu. Rev. Neurosci. 32, 347–381.
Bielas, S. L., Serneo, F. F., Chechlacz, M., Deerinck, T. J., Perkins, G. A., Allen, P. B., Ellisman, M. H., and Gleeson, J. G. (2007). Spinophilin facilitates dephosphorylation of doublecortin by PP1 to mediate microtubule bundling at the axonal wrist. Cell 129, 579–591.
Bilimoria, P. M., De La Torre-Ubieta, L., Ikeuchi, Y., Becker, E. B., Reiner, O., and Bonni, A. (2010). A JIP3-regulated GSK3beta/DCX signaling pathway restricts axon branching. J. Neurosci. 30, 16766–16776.
Bradke, F., and Dotti, C. G. (1999). The role of local actin instability in axon formation. Science 283, 1931–1934.
Burbank, K. S., and Mitchison, T. J. (2006). Microtubule dynamic instability. Curr. Biol. 16, R516–R517.
Cheng, P. L., Lu, H., Shelly, M., Gao, H., and Poo, M. M. (2011). Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron 69, 231–243.
Choi, Y. J., Di Nardo, A., Kramvis, I., Meikle, L., Kwiatkowski, D. J., Sahin, M., and He, X. (2008). Tuberous sclerosis complex proteins control axon formation. Genes Dev. 22, 2485–2495.
Cole, A. R., Knebel, A., Morrice, N. A., Robertson, L. A., Irving, A. J., Connolly, C. N., and Sutherland, C. (2004). GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 279, 50176–50180.
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789.
Del Rio, J. A., Gonzalez-Billault, C., Urena, J. M., Jimenez, E. M., Barallobre, M. J., Pascual, M., Pujadas, L., Simo, S., La Torre, A., Wandosell, F., Avila, J., and Soriano, E. (2004). MAP1B is required for Netrin 1 signaling in neuronal migration and axonal guidance. Curr. Biol. 14, 840–850.
Dent, E. W., and Kalil, K. (2001). Axon branching requires interactions between dynamic microtubules and actin filaments. J. Neurosci. 21, 9757–9769.
Drinjakovic, J., Jung, H., Campbell, D. S., Strochlic, L., Dwivedy, A., and Holt, C. E. (2010). E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 65, 341–357.
Eickholt, B. J., Walsh, F. S., and Doherty, P. (2002). An inactive pool of GSK-3 at the leading edge of growth cones is implicated in Semaphorin 3A signaling. J. Cell Biol. 157, 211–217.
Etienne-Manneville, S., and Hall, A. (2003). Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature 421, 753–756.
Forcet, C., Stein, E., Pays, L., Corset, V., Llambi, F., Tessier-Lavigne, M., and Mehlen, P. (2002). Netrin-1-mediated axon outgrowth requires deleted in colorectal cancer-dependent MAPK activation. Nature 417, 443–447.
Gartner, A., Huang, X., and Hall, A. (2006). Neuronal polarity is regulated by glycogen synthase kinase-3 (GSK-3beta) independently of Akt/PKB serine phosphorylation. J. Cell. Sci. 119, 3927–3934.
Gibson, D. A., and Ma, L. (2011). Developmental regulation of axon branching in the vertebrate nervous system. Development 138, 183–195.
Gonzalez-Billault, C., Owen, R., Gordon-Weeks, P. R., and Avila, J. (2002). Microtubule-associated protein 1B is involved in the initial stages of axonogenesis in peripheral nervous system cultured neurons. Brain Res. 943, 56–67.
Goold, R. G., and Gordon-Weeks, P. R. (2003). NGF activates the phosphorylation of MAP1B by GSK3beta through the TrkA receptor and not the p75(NTR) receptor. J. Neurochem. 87, 935–946.
Goold, R. G., and Gordon-Weeks, P. R. (2005). The MAP kinase pathway is upstream of the activation of GSK3beta that enables it to phosphorylate MAP1B and contributes to the stimulation of axon growth. Mol. Cell. Neurosci. 28, 524–534.
Goold, R. G., Owen, R., and Gordon-Weeks, P. R. (1999). Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J. Cell. Sci. 112(Pt 19), 3373–3384.
Hammond, J. W., Huang, C. F., Kaech, S., Jacobson, C., Banker, G., and Verhey, K. J. (2010). Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 21, 572–583.
Hengst, U., Deglincerti, A., Kim, H. J., Jeon, N. L., and Jaffrey, S. R. (2009). Axonal elongation triggered by stimulus-induced local translation of a polarity complex protein. Nat. Cell Biol. 11, 1024–1030.
Higginbotham, H., Tanaka, T., Brinkman, B. C., and Gleeson, J. G. (2006). GSK3beta and PKCzeta function in centrosome localization and process stabilization during Slit-mediated neuronal repolarization. Mol. Cell. Neurosci. 32, 118–132.
Homma, N., Takei, Y., Tanaka, Y., Nakata, T., Terada, S., Kikkawa, M., Noda, Y., and Hirokawa, N. (2003). Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell 114, 229–239.
Horiguchi, K., Hanada, T., Fukui, Y., and Chishti, A. H. (2006). Transport of PIP3 by GAKIN, a kinesin-3 family protein, regulates neuronal cell polarity. J. Cell Biol. 174, 425–436.
Hundsrucker, C., Skroblin, P., Christian, F., Zenn, H. M., Popara, V., Joshi, M., Eichhorst, J., Wiesner, B., Herberg, F. W., Reif, B., Rosenthal, W., and Klussmann, E. (2010). Glycogen synthase kinase 3beta interaction protein functions as an A-kinase anchoring protein. J. Biol. Chem. 285, 5507–5521.
Hur, E. M., Saijilafu, Lee, B. D., Kim, S. J., Xu, W. L., and Zhou, F. Q. (2011). GSK3 controls axon growth via CLASP-mediated regulation of growth cone microtubules. Genes Dev. 25, 1968–1981.
Hur, E. M., and Zhou, F. Q. (2010). GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551.
Inoki, K., Ouyang, H., Zhu, T., Lindvall, C., Wang, Y., Zhang, X., Yang, Q., Bennett, C., Harada, Y., Stankunas, K., Wang, C. Y., He, X., Macdougald, O. A., You, M., Williams, B. O., and Guan, K. L. (2006). TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126, 955–968.
Itoh, N., Nakayama, M., Nishimura, T., Fujisue, S., Nishioka, T., Watanabe, T., and Kaibuchi, K. (2010). Identification of focal adhesion kinase (FAK) and phosphatidylinositol 3-kinase (PI3-kinase) as Par3 partners by proteomic analysis. Cytoskeleton 67, 297–308.
Jiang, H., Guo, W., Liang, X., and Rao, Y. (2005). Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120, 123–135.
Kalil, K., Szebenyi, G., and Dent, E. W. (2000). Common mechanisms underlying growth cone guidance and axon branching. J. Neurobiol. 44, 145–158.
Kim, N. G., Xu, C., and Gumbiner, B. M. (2009a). Identification of targets of the Wnt pathway destruction complex in addition to beta-catenin. Proc. Natl. Acad. Sci. U.S.A. 106, 5165–5170.
Kim, W. Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., and Snider, W. D. (2009b). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397.
Kim, W. Y., Zhou, F. Q., Zhou, J., Yokota, Y., Wang, Y. M., Yoshimura, T., Kaibuchi, K., Woodgett, J. R., Anton, E. S., and Snider, W. D. (2006). Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron 52, 981–996.
Kishi, M., Pan, Y. A., Crump, J. G., and Sanes, J. R. (2005). Mammalian SAD kinases are required for neuronal polarization. Science 307, 929–932.
Krylova, O., Herreros, J., Cleverley, K. E., Ehler, E., Henriquez, J. P., Hughes, S. M., and Salinas, P. C. (2002). WNT-3, expressed by motoneurons, regulates terminal arborization of neurotrophin-3-responsive spinal sensory neurons. Neuron 35, 1043–1056.
Kwon, C. H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., Li, Y., Baker, S. J., and Parada, L. F. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388.
Lee, H., Engel, U., Rusch, J., Scherrer, S., Sheard, K., and Van Vactor, D. (2004). The microtubule plus end tracking protein Orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron 42, 913–926.
Li, Y. H., Werner, H., and Puschel, A. W. (2008). Rheb and mTOR regulate neuronal polarity through Rap1B. J. Biol. Chem. 283, 33784–33792.
Lu, W., Yamamoto, V., Ortega, B., and Baltimore, D. (2004). Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell 119, 97–108.
Ma, L., and Tessier-Lavigne, M. (2007). Dual branch-promoting and branch-repelling actions of Slit/Robo signaling on peripheral and central branches of developing sensory axons. J. Neurosci. 27, 6843–6851.
Ma, X. M., and Blenis, J. (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J., and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
Medeiros, N. A., Burnette, D. T., and Forscher, P. (2006). Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat. Cell Biol. 8, 215–226.
Menager, C., Arimura, N., Fukata, Y., and Kaibuchi, K. (2004). PIP3 is involved in neuronal polarization and axon formation. J. Neurochem. 89, 109–118.
Moreno, F. J., and Avila, J. (1998). Phosphorylation of stathmin modulates its function as a microtubule depolymerizing factor. Mol. Cell. Biochem. 183, 201–209.
Morfini, G., Szebenyi, G., Brown, H., Pant, H. C., Pigino, G., Deboer, S., Beffert, U., and Brady, S. T. (2004). A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 23, 2235–2245.
Morfini, G., Szebenyi, G., Elluru, R., Ratner, N., and Brady, S. T. (2002). Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 21, 281–293.
Morita, T., and Sobue, K. (2009). Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J. Biol. Chem. 284, 27734–27745.
Myers, K. A., and Baas, P. W. (2007). Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J. Cell Biol. 178, 1081–1091.
Myers, K. A., Tint, I., Nadar, C. V., He, Y., Black, M. M., and Baas, P. W. (2006). Antagonistic forces generated by cytoplasmic dynein and myosin-II during growth cone turning and axonal retraction. Traffic 7, 1333–1351.
Nakagawa, H., Murata, Y., Koyama, K., Fujiyama, A., Miyoshi, Y., Monden, M., Akiyama, T., and Nakamura, Y. (1998). Identification of a brain-specific APC homologue, APCL, and its interaction with beta-catenin. Cancer Res. 58, 5176–5181.
Nishimura, T., Kato, K., Yamaguchi, T., Fukata, Y., Ohno, S., and Kaibuchi, K. (2004). Role of the PAR-3-KIF3 complex in the establishment of neuronal polarity. Nat. Cell Biol. 6, 328–334.
Nishimura, T., Yamaguchi, T., Kato, K., Yoshizawa, M., Nabeshima, Y., Ohno, S., Hoshino, M., and Kaibuchi, K. (2005). PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat. Cell Biol. 7, 270–277.
Ossipova, O., Bardeesy, N., Depinho, R. A., and Green, J. B. (2003). LKB1 (XEEK1) regulates Wnt signalling in vertebrate development. Nat. Cell Biol. 5, 889–894.
Owen, R., and Gordon-Weeks, P. R. (2003). Inhibition of glycogen synthase kinase 3beta in sensory neurons in culture alters filopodia dynamics and microtubule distribution in growth cones. Mol. Cell. Neurosci. 23, 626–637.
Portera-Cailliau, C., Weimer, R. M., De Paola, V., Caroni, P., and Svoboda, K. (2005). Diverse modes of axon elaboration in the developing neocortex. PLoS Biol. 3, e272. doi:10.1371/journal.pbio.0030272
Poulain, F. E., and Sobel, A. (2010). The microtubule network and neuronal morphogenesis: dynamic and coordinated orchestration through multiple players. Mol. Cell. Neurosci. 43, 15–32.
Qiang, L., Yu, W., Liu, M., Solowska, J. M., and Baas, P. W. (2010). Basic fibroblast growth factor elicits formation of interstitial axonal branches via enhanced severing of microtubules. Mol. Biol. Cell 21, 334–344.
Reichardt, L. F. (2006). Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1545–1564.
Schaefer, A. W., Kabir, N., and Forscher, P. (2002). Filopodia and actin arcs guide the assembly and transport of two populations of microtubules with unique dynamic parameters in neuronal growth cones. J. Cell Biol. 158, 139–152.
Schmidt, H., Stonkute, A., Juttner, R., Koesling, D., Friebe, A., and Rathjen, F. G. (2009). C-type natriuretic peptide (CNP) is a bifurcation factor for sensory neurons. Proc. Natl. Acad. Sci. U.S.A. 106, 16847–16852.
Schmidt, H., Stonkute, A., Juttner, R., Schaffer, S., Buttgereit, J., Feil, R., Hofmann, F., and Rathjen, F. G. (2007). The receptor guanylyl cyclase Npr2 is essential for sensory axon bifurcation within the spinal cord. J. Cell Biol. 179, 331–340.
Schwamborn, J. C., and Puschel, A. W. (2004). The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat. Neurosci. 7, 923–929.
Shelly, M., Cancedda, L., Heilshorn, S., Sumbre, G., and Poo, M. M. (2007). LKB1/STRAD promotes axon initiation during neuronal polarization. Cell 129, 565–577.
Shelly, M., Cancedda, L., Lim, B. K., Popescu, A. T., Cheng, P. L., Gao, H., and Poo, M. M. (2011). Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron 71, 433–446.
Shelly, M., Lim, B. K., Cancedda, L., Heilshorn, S. C., Gao, H., and Poo, M. M. (2010). Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science 327, 547–552.
Shi, S. H., Cheng, T., Jan, L. Y., and Jan, Y. N. (2004). APC and GSK-3beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr. Biol. 14, 2025–2032.
Shi, S. H., Jan, L. Y., and Jan, Y. N. (2003). Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell 112, 63–75.
Szebenyi, G., Morfini, G., and Brady, S. T. (2002). Pictures in cell biology. GSK-3 and regulation of kinesin function. Trends Cell Biol. 12, 245.
Taelman, V. F., Dobrowolski, R., Plouhinec, J. L., Fuentealba, L. C., Vorwald, P. P., Gumper, I., Sabatini, D. D., and De Robertis, E. M. (2010). Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148.
Tanji, C., Yamamoto, H., Yorioka, N., Kohno, N., Kikuchi, K., and Kikuchi, A. (2002). A-kinase anchoring protein AKAP220 binds to glycogen synthase kinase-3beta (GSK-3beta) and mediates protein kinase A-dependent inhibition of GSK-3beta. J. Biol. Chem. 277, 36955–36961.
Tint, I., Jean, D., Baas, P. W., and Black, M. M. (2009). Doublecortin associates with microtubules preferentially in regions of the axon displaying actin-rich protrusive structures. J. Neurosci. 29, 10995–11010.
Trivedi, N., Marsh, P., Goold, R. G., Wood-Kaczmar, A., and Gordon-Weeks, P. R. (2005). Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J. Cell. Sci. 118, 993–1005.
Votin, V., Nelson, W. J., and Barth, A. I. (2005). Neurite outgrowth involves adenomatous polyposis coli protein and beta-catenin. J. Cell. Sci. 118, 5699–5708.
Witte, H., and Bradke, F. (2008). The role of the cytoskeleton during neuronal polarization. Curr. Opin. Neurobiol. 18, 479–487.
Witte, H., Neukirchen, D., and Bradke, F. (2008). Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol. 180, 619–632.
Xu, C., Kim, N. G., and Gumbiner, B. M. (2009). Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 8, 4032–4039.
Yan, D., Guo, L., and Wang, Y. (2006). Requirement of dendritic Akt degradation by the ubiquitin-proteasome system for neuronal polarity. J. Cell Biol. 174, 415–424.
Yokota, Y., Kim, W. Y., Chen, Y., Wang, X., Stanco, A., Komuro, Y., Snider, W., and Anton, E. S. (2009). The adenomatous polyposis coli protein is an essential regulator of radial glial polarity and construction of the cerebral cortex. Neuron 61, 42–56.
Yoshimura, T., Kawano, Y., Arimura, N., Kawabata, S., Kikuchi, A., and Kaibuchi, K. (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149.
Zhao, Z., and Ma, L. (2009). Regulation of axonal development by natriuretic peptide hormones. Proc. Natl. Acad. Sci. U.S.A. 106, 18016–18021.
Zhao, Z., Wang, Z., Gu, Y., Feil, R., Hofmann, F., and Ma, L. (2009). Regulate axon branching by the cyclic GMP pathway via inhibition of glycogen synthase kinase 3 in dorsal root ganglion sensory neurons. J. Neurosci. 29, 1350–1360.
Zhou, F. Q., and Snider, W. D. (2005). Cell biology. GSK-3beta and microtubule assembly in axons. Science 308, 211–214.
Zhou, F. Q., and Snider, W. D. (2006). Intracellular control of developmental and regenerative axon growth. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1575–1592.
Zhou, F. Q., Walzer, M., Wu, Y. H., Zhou, J., Dedhar, S., and Snider, W. D. (2006). Neurotrophins support regenerative axon assembly over CSPGs by an ECM-integrin-independent mechanism. J. Cell. Sci. 119, 2787–2796.
Zhou, F. Q., Waterman-Storer, C. M., and Cohan, C. S. (2002). Focal loss of actin bundles causes microtubule redistribution and growth cone turning. J. Cell Biol. 157, 839–849.
Zhou, F. Q., Zhou, J., Dedhar, S., Wu, Y. H., and Snider, W. D. (2004). NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron 42, 897–912.
Keywords: glycogen synthase kinase 3, neuronal polarity, axon growth, axon branching, microtubules, growth cone
Citation: Kim YT, Hur E-M, Snider WD and Zhou F-Q (2011) Role of GSK3 signaling in neuronal morphogenesis. Front. Mol. Neurosci. 4:48. doi: 10.3389/fnmol.2011.00048
Received: 30 August 2011;
Accepted: 06 November 2011;
Published online: 23 November 2011.
Edited by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaReviewed by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaCalum Sutherland, University of Dundee, UK
Copyright: © 2011 Kim, Hur, Snider and Zhou. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Feng-Quan Zhou, Department of Orthopaedic Surgery, Johns Hopkins University School of Medicine, 720 Rutland Avenue, 215 Ross Research Building, Baltimore, MD 21287, USA. e-mail: fzhou4@jhmi.edu