Optimization of adult sensory neuron electroporation to study mechanisms of neurite growth
- 1Spinal Cord Injury Center, Heidelberg University Hospital, Heidelberg, Germany
- 2Department of Neurosciences, University of California, San Diego, La Jolla, CA, USA
The development of eukaryotic transfection technologies has been rapid in recent years, providing the opportunity to better analyze cell-autonomous mechanisms influencing various cellular processes, including cell-intrinsic regulators of regenerative neurite growth and survival. Electroporation is one of the more effective methodologies for transfection of post-mitotic neurons demonstrating sufficient neuronal survival and transfection efficiency. To further maximize the number of transfected neurons especially with large plasmids, to limit the cellular exposure to serum, and to minimize the number of animals required for cell isolation per experiment, we compared two state-of-the-art electroporation devices for in vitro transfection of adult rat dorsal root ganglion (DRG) neuron cultures. By refining different parameters, transfection efficiencies of 39–42% could be achieved using the Lonza 4D-Nucleofector X-unit system, 1.5–2-fold higher rates than those that have been previously published for adult DRG neurons using smaller plasmid sizes. Our protocol further limits the number of cells required to 3 × 105 cells per 20 μl reaction using only 2 μg DNA/reaction and allows for the complete omission of serum post-transfection. Application of this optimized protocol will contribute to furthering the study of neuron-intrinsic mechanisms responsible for growth and survival under physiological and pathophysiological conditions.
Introduction
In vitro assays have been used extensively to study neuronal survival, neuron-glia interactions, neurophysiology, signal transduction, development, and neurite outgrowth. Cultivation of primary neurons facilitates the analysis of factors within a controlled environment, decreases the amount of time required for in vivo studies, and offers the opportunity to precisely manipulate single parameters. Over the last few decades, tools have been developed to manipulate transcriptional mechanisms, providing the ability to assess the role of gene expression in cell-intrinsic processes under various conditions.
In general, viral or non-viral gene delivery techniques can be used to manipulate gene expression. While viral delivery systems can be highly efficient, they are more expensive, time-consuming, and require additional safety measures. Non-viral methods developed in recent years offer an alternative with low cell-toxicity and high transfection rates previously only possible with viral vectors. At present, the most common non-viral methods include direct injection of naked DNA, biolistic, or gene gun technology (Dib-Hajj et al., 2009), sonoporation (Lin et al., 2010), lipid-based transfer, chemical vectors, cationic polymers, and electroporation (Inoue and Krumlauf, 2001; Washbourne and McAllister, 2002; Al-Dosari and Gao, 2009). Electroporation, in particular, has proven to be one of the most versatile non-viral techniques for studying adult neurons. The transfer of genetic material into cells via electroporation has been described in vitro as early as 1982 (Neumann et al., 1982) and in vivo initially in 1991 (Titomirov et al., 1991). Especially in the last decade, dramatic progress has been achieved in transfection of post-mitotic neurons, with examples in most major cultured cell types. In vitro cultivation of primary neurons is mostly restricted to cells isolated from embryonic and early postnatal animals. Neurons isolated from dorsal root ganglia (DRGs) are one of the few mammalian neurons that can be isolated from adult animals and cultivated for extended periods in vitro. These neurons provide an exceptional model for studying many aspects of neuronal functions, regulated by components of the extracellular environment and intrinsic mechanisms. For developmental studies, DRG neurons can be isolated as early as E10 (Forbes and Welt, 1981; Rhoades et al., 1991), and adult neurons survive even without neurotrophic factor support (Malin et al., 2007). Numerous studies have confirmed that DRG neurons cultivated in vitro retain many of their immunocytochemical and physiological characteristics (Baccaglini and Hogan, 1983; Wood et al., 1988; Gold et al., 1996; McCarter et al., 1999; Reid and Flonta, 2001).
DRG neurons also provide an excellent model to study the cell-intrinsic mechanisms of axonal regeneration. After injury to the peripheral branch of these sensory neurons, axons readily regenerate, whereas injury to the central branch does not result in regeneration. However, if a central spinal cord lesion is preceded by a lesion in the peripheral branch of sensory axons (a so-called conditioning lesion), central axon regeneration is stimulated (Richardson and Issa, 1984; Neumann and Woolf, 1999; Neumann et al., 2005). The mechanisms underlying the conditioning effect remain incompletely understood, but transcriptional programs are necessary for enhanced axonal growth after conditioning lesions (Smith and Skene, 1997). Microarray analyses have identified many potential candidate genes that can be tested for growth-promoting effects (Costigan et al., 2002; Stam et al., 2007; Michaelevski et al., 2010). While an in vivo screen would require much time and pose a challenge to isolating neuron-specific effects, in vitro transfection of DRG neurons in combination with a brief culture duration and reliable, immediate growth assessment would be most appropriate to narrow the list of candidate genes for further testing in vivo.
One of the limitations of electroporation is the transient nature of gene expression and the limited transfection efficiency with large plasmid constructs (Bloquel et al., 2004). For short-term in vitro studies, gene expression for several days after electroporation is usually sufficient. However, in vivo, expression for several weeks, months, or years might be required, something that is difficult to accomplish with plasmid electroporation. In addition, if several genes of interest are to be co-expressed or if a reporter gene is expressed from the same construct under a separate promoter, plasmid size increases, leading to a decline in electroporation rates (Ghosh et al., 2000). We, therefore, aimed to optimize transfection efficiency using a large plasmid suitable for co-expression of several genes and for the production of viral vectors for subsequent in vivo studies. Using the same expression construct for both delivery systems allows for viral production without subcloning and limits the possibility of differences between in vitro and in vivo results due to changes in promoters or other genetic elements. While comparing different transfection protocols, we also aimed to reduce the number of undefined factors in the post-transfection culture medium that may influence neuronal survival or neurite outgrowth.
Methods
Plasmid Production
For in vitro electroporation, a lentiviral vector plasmid derived from pCDH1-CMV-MCS EF-1α-copGFP (System Biosciences) was used. This plasmid was modified by replacing the CMV promoter with the CAG composite promoter (human cytomegalovirus immediate-early enhancer with a modified chicken beta-actin promoter and beta globin intron) for the expression of EGFP (Niwa et al., 1991). The EGFP cDNA in this plasmid can be exchanged for any other transgene of interest using the Gateway (Invitrogen) recombination system (Low et al., 2010). Expression of a second reporter gene, copGFP, is driven by the EF-1α promoter. The total size of this plasmid is approximately 10 kb. The combination of CAG and EF-1α promoters has previously been shown to allow for the simultaneous, long-term expression of two transgenes in adult CNS neurons after lentiviral vector delivery (Low et al., 2010). A lentiviral plasmid was used to allow for the rapid transition from in vitro electroporation to in vivo viral gene transfer. Plasmids were purified using EndoFree plasmid Mega preps (Invitrogen).
Isolation of Dorsal Root Ganglia from Rats
Adult Fischer 344 rats (10–14 weeks) were killed by an overdose of anesthesia mixture (125 mg/kg ketamine, 6.35 mg/kg xylazine, 1.25 mg/kg acepromazine). State and Institutional Animal Care and Use Committee and Society for Neuroscience guidelines on animal care were strictly followed. Following decapitation, the spinal column, including the muscles surrounding the vertebrae, was removed. The caudal end was cut so as to barely expose the opening of the vertebral canal. A 10 ml syringe filled with cold Hank's Balanced Salt Solution (HBSS) with an 18-gauge needle was inserted into the caudal opening of the vertebral canal, and the cord was quickly extruded through the rostral opening. The column was cut longitudinally in half along the dorsal and ventral axes. When necessary, the remaining dura mater was removed. At this point, all DRGs are exposed, lying in the intervertebral foramina along the vertebral canal. For each DRG, the dorsal root was grasped with microforceps to pull the ganglion gently away from the foramen, and the nerve distal to the DRG was cut with microscissors. The DRG was then carefully trimmed of nerve roots, cut half way into the center of the ganglion, and placed in Hibernate A medium (Invitrogen) on ice. DRGs from C1 to L6 from each animal were pooled.
Dissociation of Dorsal Root Ganglia
The DRGs were washed once in cold Ca/Mg-free HBSS and incubated in a 1:1 mixture of Collagenase XI (Sigma; 2.5 mg/ml final concentration; = 1200 collagenase digestive units/mg) and Dispase I (Worthington; 5 mg/ml final concentration; = 4 units/mg) at 37°C for 30 min, shifting every 10 min to equally distribute the DRGs in the tube. The enzyme solution was removed, and the cells were briefly washed once with warm complete medium [DMEM/F12 (Gibco), 1 × B27 (Invitrogen), 2 mM L-glutamine, and 1% penicillin and streptomycin (1000 U per 10 mg/ml final concentration)] with 10% fetal bovine serum (FBS) to stop the digestion and once with serum-free complete medium to remove any remaining serum. Mechanical trituration was carried out using a sterile fire-polished glass Pasteur pipette as follows: the ganglia were placed in 1 ml serum-free complete medium and triturated approximately 15 times, avoiding air bubbles. Cells were transferred to a 15 ml conical tube and triturated 15 times more, if necessary. Large remaining pieces of tissue debris were allowed to sink to the bottom of the tube (approximately 1–2 min), and the supernatant containing the single-cell suspension was transferred to a new 15 ml conical tube. Cells were counted under bright-field light microscopy using a hemocytometer. After cell counting, the appropriate amount of cells for the subsequent electroporation reactions was centrifuged at room temperature for 5 min at 800 rpm. From one animal, between 1.25 and 1.75 × 106 cells including neurons and glia were typically obtained after dissociation. Naïve, untransfected cells were plated at a density of 18,000 cells per well in 2 ml complete medium. All DRG neurons were cultured at 37°C in 5% CO2 for 48 h total.
Transfection
Unless otherwise stated in the Results, the amounts, volumes, and solutions of transfection reactions were those described here. The cell pellet was re-suspended in P3 buffer (as recommended by Lonza for neurons) at a concentration of 3 × 105 cells per 20 μl. 20 μl of cell suspension was transferred to a new tube and combined with 2.2 μg plasmid DNA per reaction, composing up to 10% of the reaction mixture volume (2.2 μl of 1 mg/ml DNA per 20 μl cell suspension). The DNA/cell mixture was then transferred to the 20 μl reaction strip. To ensure that the remaining wells were kept sterile, adhesive covers (MidSci) were applied and cut to fit to the top surface of the unused wells. Electroporation was carried out using the Lonza 4D-Nucleofector X-unit system, and the cells were allowed to sit at room temperature for 10 min following the reaction. Warm serum-free complete medium was added, and the cells were gently distributed equally into three 35 mm wells with slight shifting after each transfer, with a cell density of 105 cells/well in 2 ml medium. Three hours after electroporation, the medium was replaced with 2 ml warm serum-free complete medium per well.
Substrate for Plating
Six-well polystyrene plates were first coated with 1.5 ml per 35 mm well poly-D-lysine (Sigma; 16.67 μg/ml in sterile H2O) for 1 h at room temperature. Plates were washed twice with equal volumes of water. Diluted laminin (Sigma; 0.5 μg/ml in D'PBS) was placed in the wells at a volume of 1.5 ml/well and allowed to sit for 3 h at room temperature. After two wash steps with equal volumes of D'PBS, the wells were filled with 2 ml of complete culture medium and placed at 37°C until cell plating.
Immunocytochemistry and Imaging
After 48 h in culture, cells were fixed with 4% ice-cold paraformaldehyde in 0.1 M phosphate buffer and washed 4 × 5 min with 0.1 M tris-buffered saline (TBS). Cells were blocked for 1 h at room temperature in TBS, 0.1% Triton X, and 5% donkey serum and incubated with primary antibodies in TBS with 0.1% Triton X and 1% donkey serum overnight at 4°C. Cells were washed 4 × 5 min with TBS and incubated with secondary antibodies diluted in TBS for 2.5 h at room temperature in the dark. Cells were washed 4 × 5 min with TBS. In the third wash, 4′-6-Diamidino-2-phenylindole (DAPI; Sigma) was added at a concentration of 1:1000 (stock concentrations: 0.1 mg/ml). Antibodies used for fluorescent immunocytochemistry were the following: rabbit anti-copGFP (1:5000; Evrogen), mouse anti-beta-III-tubulin (1:1000; Promega), donkey anti-rabbit Alexa 488 (1:1000; Molecular Probes), and donkey anti-mouse Alexa 594 (1:1000; Molecular Probes).
Imaging of plates was automated using an Olympus IX81 inverted microscope equipped with a motorized stage and controlled by Olympus Cell-P Software using a 4× objective. Twelve imaged and automatically stitched non-overlapping montages of 4 × 5 frames covering more than 90% of the 35 mm well surface were used for quantification. Olympus imaging files were converted in NIH ImageJ to 8-bit RGB format for analysis using the NeuronJ plug-in.
Analysis and Statistics
Neurons were identified by beta-III-tubulin labeling and transfected neurons by copGFP double labeling. Neurite outgrowth assessments were done by manually tracing the longest neurite of each neuron double labeled for copGFP and beta-III-tubulin, starting at the base of the neurite along the edge of the cell soma. For neurons without neurites, a small line was traced along the edge of the soma, amounting to no more than 10 pixels (16 μm). Cell counts and neurite lengths were averaged for each well. Data are presented as means and standard errors of the mean. Group means were compared by unpaired two-tailed Student's t-test and two-way or one-way ANOVA followed by Fisher's post-hoc analysis to assess significant differences. A significance criterion of p < 0.05 was used in all statistical tests.
Results
The goal of the current experiments was to develop a transfection protocol for adult DRG neurons that (1) is suitable for a medium-throughput screen for neurite outgrowth and neuronal survival assessments, (2) limits the number of primary cells and animals necessary to achieve sufficient statistical power, (3) provides a consistently high percentage of transfected neurons, and (4) minimizes the use of undefined media components such as FBS in the culture medium, due to their potential influence on cell survival, glial proliferation, and neurite extension.
Transfection Device Comparison
We first compared two recently developed transfection/electroporation devices (Neon Transfection System, Invitrogen, Carlsbad, CA and 4D-Nucleofector System, Lonza, Basel, Switzerland) to determine which system would yield the highest number of transfected adult DRG neurons. DRGs were isolated, dissociated, and transfected with both devices using two different reaction size kits, and the cultures were assessed for the number of cells surviving and the number of copGFP-expressing neurons after cultivation for 48 h. The optimization guidelines for primary neurons provided by each manufacturer were generally followed, including the electroporation programs, use of serum in the 3 h post-electroporation incubation, and the reaction buffers (Lonza, P3 Solution; Neon, Buffer R). The suggested optimization protocols vary with regards to the cell number (Lonza recommends 4–5 × 106 cells; Neon recommends 5 × 105–2 × 106 cells) and DNA amount per transfection (Lonza recommends 0.5–3 μg DNA; Neon recommends 5–30 μg DNA). Using a 100 μl reaction volume in both systems, 106 cells per reaction, and 10 μg of plasmid DNA (10 kb; 10% of the reaction volume), the transfection efficiency with the Lonza 4D system (using electroporation code DR114, one of the codes recommended in the optimization guidelines for primary neurons) was more than two-fold higher than the rates observed with the best six of a total of 29 tested Neon programs, including 23 programs suggested in Invitrogen's optimization guidelines (Figure 1; best Neon program: 1500 V, 10 ms pulse width, three pulses; no transfected cells were observed in the negative control without current). Neuronal survival rates were slightly higher with the Neon, but the absolute number of copGFP-positive DRG neurons obtained with the best 4D X-unit program exceeded three-fold the number of transfected neurons using the Neon device. Due to the high cell number needed for the 100 μl reactions and the consistent results obtained in the assays, these experiments were only conducted in duplicate. The Neon system also provides the option of 10 μl pipettes, and the Lonza 4D offers 20 μl well strips. The best programs from the Neon were also assessed using the 10 μl pipettes with lower cell and DNA amounts (1.5 × 105 cells; 1.5 μg DNA) but resulted in similar low efficiency rates (data not shown). Therefore, further optimization was carried out only with the Lonza 4D-Nucleofector.
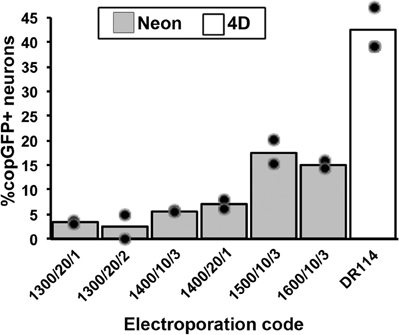
Figure 1. Comparison of transfection efficiency between electroporation devices and programs. Electroporation reactions were conducted in 100 μl vessels using an equal number of DRG cells and a 10 kb plasmid expressing copGFP. Neurons were identified by beta-III-tubulin labeling upon fixation after 48 h in culture. The Lonza 4D-Nucleofector system (white bar) resulted in significantly higher transfection rates compared to all programs used with the Invitrogen Neon device [gray bars; parameters listed are electric potential (V)/pulse width (ms)/number of pulses]. Only the six best electroporation codes chosen from previous pilot screens of 29 Neon programs are shown (symbols indicate individual data points for each condition).
Selection of Electroporation Vessel Size and Code for Transfecting Adult DRG Neurons
Since at least 106 cells are needed for the 100 μl cuvettes, the 20 μl strips were tested to lower the amount of cells required per reaction. As we and others have previously used up to 1.5–2.0 × 106 DRG cells and 10 μg DNA (10 kb) per 100 μl reaction using the previous Lonza electroporation system (manufacturer's protocol for DRG neurons; Amaxa Nucleofector II), that number was scaled down to 3 × 105 cells and 2 μg DNA, not exceeding 10% of the total reaction volume per 20 μl reaction. As a result, averaged across two experiments each, the efficiency rates were comparable between the 20 μl and 100 μl sizes, with 43% of neurons transfected using the 100 μl cuvette and 38% of neurons transfected using the 20 μl strip (n = 4–5 wells across two experiments; Figure 2A). It should be noted that FBS was not included in the incubation following the transfections with the 20 μl strips, potentially reducing the number of surviving cells (see below).
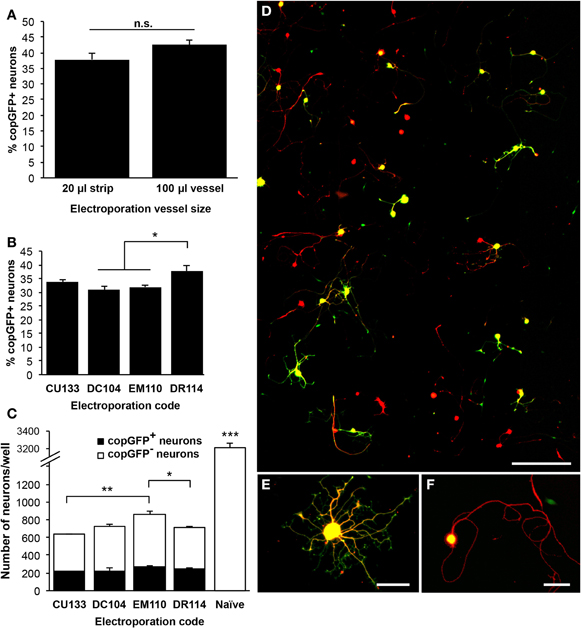
Figure 2. Optimization of the 4D-Nucleofector electroporation code for transfection of adult dorsal root ganglion neurons. (A) Comparison of the 100 μl cuvette and the 20 μl well strip using code DR114 indicated a slightly higher transfection rate with the 100 μl cuvette, but this difference did not reach statistical significance (n = 4–5 wells across two independent experiments). (B) Five 4D-Nucleofector codes were compared using the 20 μl well strips. DR114 consistently resulted in the highest transfection rate, measured 48 h after electroporation (n = 3–5 wells per condition; ANOVA p < 0.001). CU110 did not result in any transfected neurons (not shown). (C) For the four codes that resulted in successful transfection, total neuronal survival rates were examined. CU133 exhibited the most neuronal death, and EM110 produced the highest total neuronal survival rate. Compared to naïve, untransfected cell cultures, electroporated cultures demonstrated a neuronal survival rate of 18–27% (ANOVA p < 0.001). (D) A representative composite image showing approximately 0.75% of the total imaged surface of a 35 mm well and 9% of one 20-frame montage. Beta-III-tubulin labeling (red) indicates neurons, and copGFP labeling (green) identifies transfected cells. Neurons are sufficiently and consistently distributed throughout each well, and the low plating density allows for straightforward neurite tracing and quantification. (E and F) Higher magnification shows neurons with (E) high transgene expression, with copGFP detected in the cell soma and neurites, and (F) moderate expression, with copGFP present only in the cell soma. Scale bar = 300 μm in (D), 50 μm in (E and F). *p < 0.05; **p < 0.01; ***p < 0.001.
As smaller reaction volumes yielded similar transfection efficiencies, five Lonza 4D-Nucleofector reaction codes were tested under the 20 μl strip protocol for optimal performance, the first four of which are suggested in Lonza's optimization guidelines: CU110, DC104, DR114, EM110, and CU133. With the exception of code CU110, which did not result in any transfected neurons, the transfection efficiency rates ranged from 30% to 38% (n = 3–5 wells per condition; ANOVA p < 0.001; Figure 2B). All protocols resulted in significant neuronal loss compared to non-transfected control neurons, which received no electrical current (ANOVA p < 0.001; Figure 2C). Neuronal survival rates were between 18% and 27% of naïve, untransfected controls. In addition to demonstrating the highest neuronal transfection efficiency, qualitatively, expression levels were greatest in neurons transfected with the DR114 code: 65–87% of transfected neurons displayed copGFP-immunoreactivity in cell soma and all neuritic processes (Figure 2E). In the remaining transfected cells, only the cell soma showed copGFP-immunoreactivity (Figure 2F). Neurite outgrowth did not differ between high- and moderate-expression transfected neurons (data not shown). Therefore, due to the high neuronal transfection efficiency and the pattern of high expression level, DR114 was chosen as the program to use for further examination of other parameters.
Optimization of the Transfection Protocol
To determine whether the protocol described above produced the highest possible transfection efficiency and greatest survival of transfected DRG neurons, the amount of DNA and the percent volume of the reaction solution were compared between 1 μg and 2 μg (making up 10% of the total reaction volume), 3 μg (15% volume), and 6 μg plasmid DNA (30% volume) using the DR114 protocol in a 20 μl volume. Normalized across three experiments to the previously used condition of 2 μg DNA and 3 × 105 cells, reactions with 1 μg DNA resulted in significantly lower transfection efficiencies than those with 2 or 3 μg DNA, with the highest efficiency rates achieved with 2 μg DNA (37–43%; n = 3–9 wells per condition; ANOVA p < 0.01; Figure 3A, black bars). As expected, the lower transfection efficiency with 1 μg of DNA also resulted in 30% fewer transfected neurons compared to reactions with 2 μg DNA (p < 0.01; Figure 3A, white bars), despite comprising equivalent proportional volumes of DNA in the reaction mixture (10% of the cell suspension). Reactions with 6 μg DNA generated about 50% fewer transfected neurons than reactions with 2 or 3 μg DNA, likely due to DNA-mediated toxicity (Fisher's post-hoc analysis, p < 0.001 and p < 0.05, respectively).
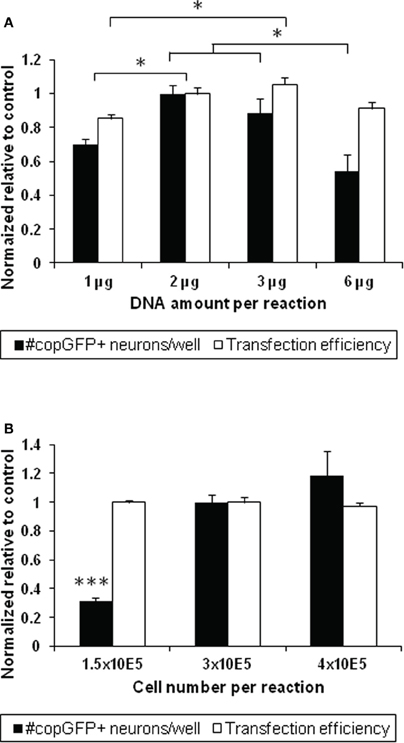
Figure 3. Optimization of DNA amount and cell number for transfecting adult dorsal root ganglion neurons. (A) Altering the amount of DNA and the percent volume of plasmid in the reaction mixture influences the number of transfected neurons at 48 h (n = 3–9 wells across three experiments; ANOVA p < 0.01; black bars). A low amount of DNA (1 μg) resulted in a significantly lower number of copGFP-positive neurons compared to 2 μg DNA comprising the same percent reaction volume (10% of the 20 μl vessel; p < 0.01). Increasing the DNA amount (6 μg comprising 30% of the reaction volume) also resulted in 39%–46% fewer transfected cells compared to reactions with 2 and 3 μg DNA, respectively (p < 0.05). Using 1 μg DNA also lowered transfection efficiencies from values achieved with 2 and 3 μg DNA (p < 0.05; white bars). The condition with 2 μg DNA was set as 100% to normalize across different experiments. (B) Three cell concentrations were tested to determine whether cell numbers would influence the transfection output using 2 μg of DNA. While no significant difference was seen in transfection efficiency at 48 h, lowering the cell number to 1.5 × 105 cells significantly diminished the absolute number of transfected neurons per well disproportionally by more than three-fold (ANOVA p < 0.001). All values are relative to the 2 μg DNA and 3 × 105 cells condition within the same experiment. *p < 0.05; ***p < 0.001.
Next, to determine whether altering the cell number per electroporation reaction would increase the transfection output, we compared 1.5 × 105, 3 × 105, and 4 × 105 DRG cells in the 20 μl vessel (manufacturer's recommendation: 2.5 × 105). While the percentage of transfected neurons was unaffected across all conditions, the number of copGFP-positive neurons per well dropped disproportionally by nearly 70% from 297 ± 47 copGFP-positive DRG neurons when half the number of cells was used and increased by less that 20% when cell numbers were increased by one-third (n = 3–9 wells across three experiments; ANOVA p < 0.0001; Figure 3B). Neurite lengths did not significantly differ between the conditions (data not shown).
Influence of Serum
As the process of electroporation can affect cellular integrity via mechanical damage to the membrane and toxicity from the infiltration of media-derived factors, steps to maximize cell survival while minimizing influences on cell physiological parameters have to be taken. As reported in Lonza's optimization guidelines and primary neuron manual, serum is highly recommended to support cell survival following electroporation. To explore whether serum is necessary for neuronal survival post-electroporation, DRG cells were transfected according to the above described optimized protocol (Lonza 4D, DR114 code, 20 μl volume, 2 μg DNA, 3 × 105 cells) and cultured with and without serum for 3 h or 15 h post-electroporation. Media were replaced completely with serum-free complete culture medium, and cells were cultivated for a total of 48 h.
Overall, transfection efficiencies were unaffected by the omission of FBS in the post-electroporation incubation period (Figure 4A). A Two-Way ANOVA with Fisher's post-hoc analysis indicated a significant interaction between serum and incubation time post-electroporation (p < 0.05) in influencing neuronal survival: if the post-electroporation incubation lasted overnight (15–16 h), survival dropped by nearly 40% when cells were incubated without FBS (Figure 4B). In contrast, cell survival was not affected by omission of serum if the medium was changed 3 h following electroporation with serum-free culture medium.
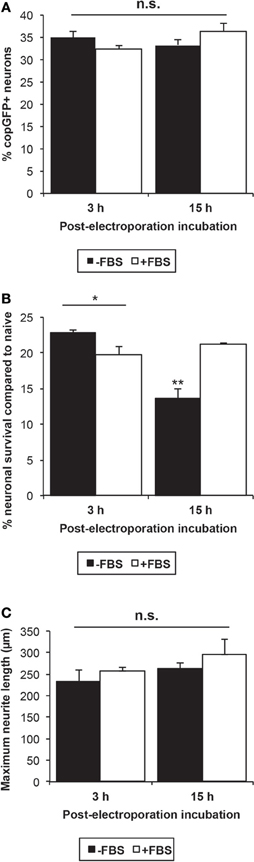
Figure 4. Influence of serum following neuronal transfection. (A) The omission of FBS and a shorter post-electroporation incubation time did not significantly influence the neuronal transfection efficiency at 48 h. (B) Neuronal survival was significantly affected by both the presence of serum and the incubation time between electroporation and medium change. Omission of FBS from the medium resulted in significant cell death if medium was not changed until 15 h post-electroporation (ANOVA p < 0.001, Fisher's post-hoc analysis; *p < 0.05; **p < 0.01). However, when the medium was changed 3 h after the electroporation, the omission of serum did not decrease cell survival. (C) Though no significant difference was detected, neurite length was increased on average by 12% after incubation in serum-containing medium for 3 h or 15 h, suggesting that even short-term serum exposure stimulates growth-related mechanisms in adult DRG neurons.
Previous studies have shown that neurite length is reduced by 25–60% at 24- and 72 h time points post-transfection in postnatal DRG neurons and adult retinal ganglion cells, respectively (Leclere et al., 2005; Seggio et al., 2008). Under our protocol, electroporation indeed diminished neurite extension by approximately 44% at 48 h (Student's t-test p < 0.05), compared to untreated naïve cultures, but not the percentage of neurons extending neurites (data not shown). Overnight (15–16 h) and even short-term incubation with FBS (3 h) increased neurite outgrowth, on average, by 12%, but these differences did not reach statistical significance (Figure 4C).
Discussion
The electroporation and neurite outgrowth assay method described here provides transfection efficiencies that are sufficiently high to analyze five individual transfections with five different large plasmid constructs using adult DRG neurons isolated from a single animal. On average, more than 3000 strongly transfected neurons dispersed across fifteen 35 mm wells total with a neuronal transfection efficiency between 39 and 42% can be achieved. These values are at least 1.5–2-fold higher than previously published methods using adult DRG neurons and smaller plasmids (Leclere et al., 2005; Hudmon et al., 2008; Dib-Hajj et al., 2009).
This optimization was conducted to develop a specific protocol for transfecting adult rat DRG neurons with large plasmids that can also be used for the production of viral vectors. In addition, we aimed to minimize exogenous factors in cell culture medium, maximize the transfection efficiency, and minimize the amount of tissue and animals needed per experiment. Compared to the manufacturer's suggested protocol for primary neurons, this method has refined the DNA amount, cell number, and post-electroporation culture media.
In contrast to the manufacturer's suggestion of 0.1–0.6 μg DNA, it was established that a higher amount of DNA (2 μg) per 20 μl reaction, composing 10% of the cell/DNA/buffer mixture, yields the highest transfection efficiency and the greatest number of transfected neurons per well. A further increase to 6 μg resulted in a 46% drop in the number of transfected neurons per well upon electroporation. Since we used the same plasmid DNA solution for all experiments, the volume of the reaction mixture allocated for DNA increased from 10% to 30% to test the 6 μg DNA condition. The manufacturer's protocol recommends that no less than 85–90% of the reaction volume consist of the provided P3 buffer. It is, therefore, possible that the decline in transfected neurons is due, at least in part, to the disproportionate mixture of P3 buffer and DNA-containing TE in the reaction vessel.
Serum is a well-known stimulant of cell growth and survival (Dulbecco, 1970; Lau and Nathans, 1985; Bruckstein and Higgins, 1988; Delree et al., 1993). Both manufacturers' protocols (Invitrogen and Lonza) indicate that FBS is an essential component of the post-transfection culture medium, at least for the approximate 3–15 h period (overnight incubation) immediately following the electroporation, to prevent neuronal death. For studies that focus on regulators of survival and neurite growth and transcriptional responses to gene expression, addition of serum is highly likely to influence outcome measures and should be avoided, if possible. Because incubation with FBS did not improve cell survival when medium was changed 3 h post-transfection, FBS can be omitted.
Electroporation of adult DRGs reduced neurite outgrowth at 48 h compared to naïve neurons. It is not surprising that neurite outgrowth is negatively influenced by the process of electroporation, whereby pores are transiently formed across the neuronal membrane (Xie et al., 1990). This inhibition is likely to be transient, as previously demonstrated with retinal ganglion neurons (Leclere et al., 2005). Further experiments are needed to confirm whether the effect is short- or long-term for adult DRG neurons. In summary, our optimized transfection and culture protocol employed the Lonza 4D-Nucleofector X-unit system with electroporation code DR114 and Lonza P3 buffer, 3 × 105 cells and 2 μg DNA (10% reaction volume) per 20 μl reaction well, no FBS in the medium following electroporation, and the change of all media at 3 h post-transfection to serum-free complete medium. Even higher transfection efficiencies can likely be achieved with smaller plasmid constructs. DRG neurons isolated from one adult rat, therefore, provide sufficient neurons for at least five transfections with three well replicates, supplying 150–250 strongly transfected neurons per well for quantification.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from NIH/NINDS (NS054883), International Foundation for Research in Paraplegia (IRP/IFP), and the EU (IRG268282) and a fellowship to Julianne McCall (Landesgraduiertenförderung).
References
Al-Dosari, M. S., and Gao, X. (2009). Nonviral gene delivery: principle, limitations, and recent progress. AAPS J. 11, 671–681.
Baccaglini, P. I., and Hogan, P. G. (1983). Some rat sensory neurons in culture express characteristics of differentiated pain sensory cells. Proc. Natl. Acad. Sci. U.S.A. 80, 594–598.
Bloquel, C., Fabre, E., Bureau, M. F., and Scherman, D. (2004). Plasmid DNA electrotransfer for intracellular and secreted proteins expression: new methodological developments and applications. J. Gene Med. 6(Suppl. 1), S11–S23.
Bruckstein, D. A., and Higgins, D. (1988). Morphological differentiation of embryonic rat sympathetic neurons in tissue culture: conditions under which neurons form axons but not dendrites. Dev. Biol. 128, 324–336.
Costigan, M., Befort, K., Karchewski, L., Griffin, R. S., D'Urso, D., Allchorne, A., Sitarski, J., Mannion, J. W., Pratt, R. E., and Woolf, C. J. (2002). Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 3, 16.
Delree, P., Ribbens, C., Martin, D., Rogister, B., Lefebvre, P. P., Rigo, J. M., Leprince, P., Schoenen, J., and Moonen, G. (1993). Plasticity of developing and adult dorsal root ganglion neurons as revealed in vitro. Brain Res. Bull. 30, 231–237.
Dib-Hajj, S. D., Choi, J. S., Macala, L. J., Tyrrell, L., Black, J. A., Cummins, T. R., and Waxman, S. G. (2009). Transfection of rat or mouse neurons by biolistics or electroporation. Nat. Protoc. 4, 1118–1126.
Dulbecco, R. (1970). Topoinhibition and serum requirement of transformed and untransformed cells. Nature 227, 802–806.
Forbes, D. J., and Welt, C. (1981). Neurogenesis in the trigeminal ganglion of the albino rat: a quantitative autoradiographic study. J. Comp. Neurol. 199, 133–147.
Ghosh, C., Song, W., and Lahiri, D. K. (2000). Efficient DNA transfection in neuronal and astrocytic cell lines. Mol. Biol. Rep. 27, 113–121.
Gold, M. S., Dastmalchi, S., and Levine, J. D. (1996). Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience 71, 265–275.
Hudmon, A., Choi, J. S., Tyrrell, L., Black, J. A., Rush, A. M., Waxman, S. G., and Dib-Hajj, S. D. (2008). Phosphorylation of sodium channel Na(v)1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J. Neurosci. 28, 3190–3201.
Inoue, T., and Krumlauf, R. (2001). An impulse to the brain–using in vivo electroporation. Nat. Neurosci. 4, 1156–1158.
Lau, L. F., and Nathans, D. (1985). Identification of a set of genes expressed during the G0/G1 transition of cultured mouse cells. EMBO J. 4, 3145–3151.
Leclere, P. G., Panjwani, A., Docherty, R., Berry, M., Pizzey, J., and Tonge, D. A. (2005). Effective gene delivery to adult neurons by a modified form of electroporation. J. Neurosci. Methods 142, 137–143.
Lin, C. R., Chen, K. H., Yang, C. H., Cheng, J. T., Sheen-Chen, S. M., Wu, C. H., Sy, W. D., and Chen, Y. S. (2010). Sonoporation-mediated gene transfer into adult rat dorsal root ganglion cells. J. Biomed. Sci. 17, 44.
Low, K., Blesch, A., Herrmann, J., and Tuszynski, M. H. (2010). A dual promoter lentiviral vector for the in vivo evaluation of gene therapeutic approaches to axon regeneration after spinal cord injury. Gene Ther. 17, 577–591.
Malin, S. A., Davis, B. M., and Molliver, D. C. (2007). Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat. Protoc. 2, 152–160.
McCarter, G. C., Reichling, D. B., and Levine, J. D. (1999). Mechanical transduction by rat dorsal root ganglion neurons in vitro. Neurosci. Lett. 273, 179–182.
Michaelevski, I., Segal-Ruder, Y., Rozenbaum, M., Medzihradszky, K. F., Shalem, O., Coppola, G., Horn-Saban, S., Ben-Yaakov, K., Dagan, S. Y., Rishal, I., Geschwind, D. H., Pilpel, Y., Burlingame, A. L., and Fainzilber, M. (2010). Signaling to transcription networks in the neuronal retrograde injury response. Sci. Signal. 3, ra53.
Neumann, E., Schaefer-Ridder, M., Wang, Y., and Hofschneider, P. H. (1982). Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1, 841–845.
Neumann, S., Skinner, K., and Basbaum, A. I. (2005). Sustaining intrinsic growth capacity of adult neurons promotes spinal cord regeneration. Proc. Natl. Acad. Sci. U.S.A. 102, 16848–16852.
Neumann, S., and Woolf, C. J. (1999). Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23, 83–91.
Niwa, H., Yamamura, K., and Miyazaki, J. (1991). Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199.
Reid, G., and Flonta, M. L. (2001). Physiology – Cold current in thermoreceptive neurons. Nature 413, 480.
Rhoades, R. W., Enfiejian, H. L., Chiaia, N. L., Macdonald, G. J., Miller, M. W., Mccann, P., and Goddard, C. M. (1991). Birthdates of trigeminal ganglion cells contributing axons to the infraorbital nerve and specific vibrissal follicles in the rat. J. Comp. Neurol. 307, 163–175.
Richardson, P. M., and Issa, V. M. (1984). Peripheral injury enhances central regeneration of primary sensory neurones. Nature 309, 791–793.
Seggio, A. M., Ellison, K. S., Hynd, M. R., Shain, W., and Thompson, D. M. (2008). Cryopreservation of transfected primary dorsal root ganglia neurons. J. Neurosci. Methods 173, 67–73.
Smith, D. S., and Skene, J. H. (1997). A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 17, 646–658.
Stam, F. J., Macgillavry, H. D., Armstrong, N. J., De Gunst, M. C., Zhang, Y., van Kesteren, R. E., Smit, A. B., and Verhaagen, J. (2007). Identification of candidate transcriptional modulators involved in successful regeneration after nerve injury. Eur. J. Neurosci. 25, 3629–3637.
Titomirov, A. V., Sukharev, S., and Kistanova, E. (1991). In vivo electroporation and stable transformation of skin cells of newborn mice by plasmid DNA. Biochim. Biophys. Acta 1088, 131–134.
Washbourne, P., and McAllister, A. K. (2002). Techniques for gene transfer into neurons. Curr. Opin. Neurobiol. 12, 566–573.
Wood, J. N., Winter, J., James, I. F., Rang, H. P., Yeats, J., and Bevan, S. (1988). Capsaicin-induced ion fluxes in dorsal-root ganglion-cells in culture. J. Neurosci. 8, 3208–3220.
Keywords: dorsal root ganglion, transfection, electroporation, neurite outgrowth, regeneration
Citation: McCall J, Nicholson L, Weidner N and Blesch A (2012) Optimization of adult sensory neuron electroporation to study mechanisms of neurite growth. Front. Mol. Neurosci. 5:11. doi: 10.3389/fnmol.2012.00011
Received: 08 November 2011; Accepted: 27 January 2012;
Published online: 08 February 2012.
Edited by:
Simone Di Giovanni, Universität Tübingen, GermanyReviewed by:
Hansjürgen Volkmer, Universität Tübingen, Germany;Frédérique Scamps, Institut National de la Santé et Recherche Médicale, France
Copyright: © 2012 McCall, Nicholson, Weidner and Blesch. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Armin Blesch, Spinal Cord Injury Center, Heidelberg University Hospital, Schlierbacher Landstrasse 200 a, 69118 Heidelberg, Germany. e-mail: armin.blesch@med.uni-heidelberg.de