Transgenic strategy for identifying synaptic connections in mice by fluorescence complementation (GRASP)
- Department of Molecular and Cellular Biology and Center for Brain Science, Harvard University, Cambridge, MA, USA
In the “GFP reconstitution across synaptic partners” (GRASP) method, non-fluorescent fragments of GFP are expressed in two different neurons; the fragments self-assemble at synapses between the two to form a fluorophore. GRASP has proven useful for light microscopic identification of synapses in two invertebrate species, Caenorhabditis elegans and Drosophila melanogaster, but has not yet been applied to vertebrates. Here, we describe GRASP constructs that function in mammalian cells and implement a transgenic strategy in which a Cre-dependent gene switch leads to expression of the two fragments in mutually exclusive neuronal subsets in mice. Using a transgenic line that expresses Cre selectively in rod photoreceptors, we demonstrate labeling of synapses in the outer plexiform layer of the retina. Labeling is specific, in that synapses made by rods remain labeled for at least 6 months whereas nearby synapses made by intercalated cone photoreceptors on many of the same interneurons remain unlabeled. We also generated antisera that label reconstituted GFP but neither fragment in order to amplify the GRASP signal and thereby increase the sensitivity of the method.
Introduction
Analysis of neural circuits requires identification of neurons that are synaptically connected to each other. A major impediment to mapping these connections is that many neurites in the central nervous system are too small to be resolved by conventional light microscopy: synaptic sites can be visualized but the neurites from which they arise cannot be identified unambiguously. At present, therefore, connectivity is generally assessed by electron microscopic and electrophysiological methods. Both of these techniques are extremely laborious, however, and both are difficult to apply when the pre- and postsynaptic somata are separated by long-distances. Accordingly, many groups have sought improved light microscopic methods for circuit analysis, and three have shown considerable promise. The first is super-resolution microscopy, sometimes called nanoscopy, in which even the most slender neurites can be resolved (Huang et al., 2010). The second is transsynaptic tracing, in which genetically encoded labels are secreted at synaptic sites and selectively internalized by the apposed pre- or postsynaptic membrane (Horowitz et al., 1999; Yoshihara et al., 1999; Wickersham et al., 2007; Lo and Anderson, 2011). The third is “GFP Reconstitution Across Synaptic Partners” or GRASP (Feinberg et al., 2008).
GRASP is based on the use of non-fluorescent fragments of GFP that can be fused to other proteins, yet retain the ability to self-assemble and form a fluorophore. This complementation strategy was initially used to detect intracellular protein-protein interactions (Hu and Kerppola, 2003; Zhang et al., 2004; Cabantous et al., 2005). To adapt the method for detection of synapses, Feinberg et al. (2008) used a GFP derivative that had been engineered for efficient complementation (“superfolder GFP”; Pedelacq et al., 2006), split it into two fragments (sGFP1–10 and sGFP11), fused them to the ectodomains of cell surface proteins, and expressed one fusion protein in each of two neurons in C. elegans that were known to be synaptically connected. They showed that the fragments reconstituted fluorescent GFP specifically at sites of synapses between the two neurons. Subsequently, Gordon and Scott (2009) and Gong et al. (2010) used the method to mark synapses in Drosophila.
GRASP is particularly useful because it is the only light microscopic method to date capable of unequivocally marking synaptic contacts between two genetically specified neurons. We, therefore, asked whether it can be applied to mammals. We report here generation of knock-in mice in which sGFP1–10 and sGFP11 are fused to components of synaptic membrane proteins, neuroligin (NLG), and neurexin (NXN) (Südhof, 2008), and expressed in mutually exclusive neuronal subsets. We demonstrate that GRASP specifically marks synapses between members of the two subsets in retina, without marking synapses made by other axons on the same postsynaptic cells.
Materials and Methods
GRASP Constructs
We modified the DNA sequence of superfolder GFP (Pedelacq et al., 2006) by optimizing codon usage for mice and removing potential splicing donor and acceptor motifs without altering the encoded amino acid sequence (GenBank accession number JQ341914). The modified gene was synthesized (DNA2.0, Menlo Park, CA) and cloned into a backbone derived from CMV-EGFP-N1 (Clontech, Mountain View, CA). The GFP1–10 and GFP11 fragments were retrieved from this vector by PCR and fused to rat neuroligin-1 or neurexin-1β (obtained from Alice Ting, MIT), or human CD4 (obtained from Evan Feinberg and Cori Bargmann, Rockefeller University).
Cell Lines
Human embryonic kidney 293 cells (HEK293; ATCC, Manassas, VA) were subcloned and a derivative was selected that could be cloned with high efficiency; we call this line 293PL. 293PL cells in 24-well tissue culture dishes were transfected using DMRIE-C (Life Technologies, Grand Island, NY), then trypsinized 3 days later, plated on 10 cm tissue culture dishes, and selected in 1 mg/ml G418 (Life Technologies). Colonies were split and tested by immunostaining for expression of the gene product. More than 70% of the cells in each clone showed high expression of the introduced gene.
Each clone was cultured to confluency, trypsinized, and plated onto glass coverslips, either alone or mixed with cells of a second clone. After 2–3 days, cultures were fixed with 100% methanol or 4% paraformaldehyde/PBS for 15 min at room temperature, then rinsed with PBS. Methanol-fixed cultures had lower background than paraformaldehyde-fixed cultures, so this treatment was usually used for detecting native GRASP signals. For immunostaining, paraformaldehyde cultures were processed as described previously (Yamagata and Sanes, 2010). Finally, cultures were mounted with FluoroGel (Electron Microscopy Sciences, Hatfield, PA) and imaged with a Pascal LSM-510 confocal microscope (Zeiss).
Use of stably transfected lines was required for these assays because co-expression of two complementary split GFP fragments in the same cell leads to efficient cis-reconstitution that hampers detection of the much lower levels of trans-reconstitution between apposed cells. Surprisingly, we observed such co-expression when we mixed populations that had been transiently transfected with complementary fragments. We suspect that low levels of DNA adhere to cells even when the cells were trypsinized and rinsed multiple times prior to co-culture.
Antibodies to GFP
Full length superfolder GFP was cloned into pEcoli-6xHN (Clontech) and introduced into BL21(DE3) pLysS cells. The cells were grown in Magic Media (Life Technologies) and processed with Bugbuster (EMD Chemicals, Philadelphia, PA), and the GFP was purified with Talon columns (Clontech). Hens were immunized with the recombinant GFP (Covance), eggs from immunized hens were harvested, and the IgY fraction from egg yolk was purified with IgY purification kit (Pierce, Rockford, IL).
To obtain antibody that reacted selectively with holo-GFP, the sGFP1–10 fragment was expressed in E. coli using pEcoli-6xHN. This fragment was insoluble, so inclusion bodies were purified, dissolved with 4M urea (in 50 mM TrisHCl, pH 7.4), purified using Talon columns, and coupled to NHS-Activated Agarose (Pierce). The anti-GFP IgY was absorbed to this resin and the non-binding fraction was further absorbed with acetone powder prepared from mouse brain. This purified antibody is called anti-rGFP.
A hybridoma producing a mouse monoclonal antibody GFP-G1 (subclass, IgG1) was obtained by immunizing a female Balb/c mouse. Its splenocytes were fused to FOX-NY myeloma line, and selected by a standard procedure for generating hybridomas. Hybridomas were screened by staining tissue from GFP-expressing mice to obtain an antibody suitable for immunostaining. One antibody selected, GFP-G1, recognizes all the GFP derivatives tested (EYFP, Venus, superfolder GFP, and cerulean). IgG was purified from cultured supernatants using Protein-G affinity gels (GenScript, Piscataway, NJ).
Knock-in Mouse Line
To generate a GRASP knock-in targeting vector, pRosa26-PAS (Srinivas et al., 2001) was modified in the following series of steps. (1) A phosphorylated i-SceI linker (AGTTACGCTAGGGATAACAGGGTAATATAG) was ligated into the SwaI site, producing a recognition site for linearization of the targeting vector. (2) A PacI-FRT-neo-FRT-AscI selection cassette was cloned into pROSA26PAS-i-SceI. (3) A PacI-CAG-RfA-WPRE-PacI cassette containing the chicken β-actin promoter and CMV immediate-early enhancer (together called CAG), a Gateway RfA destination cassette (Gateway Vector Conversion system; Life Technologies), and a WPRE fragment (woodchuck hepatitis virus post-transcriptional element) was assembled in a modified pBluescriptKS+ that had been generated by inserting GAGCTCAGTTACGCTAGGGATAACAGGGTAATATAGAATTCTT AATTAAGCGGCCGCGATCGCCCGGGCATTTAAATGGCCTG CAGGGCCGTTTAAACGGCCGGCCGTCGACTCGAGCGTAA CTATAACGGTCCTAAGGTAGCGAAGGTACC into the Kpn I/SacI sites. (4) This cassette was cloned into a PacI site of pROSA26PAS-FNF-iSceI. (5) sGFP11::NXN was cloned to pCR8-Topo (Life Technologies) with an appended I-CeuI sequence at its N-terminus. (6) sGFP1–10::NLG and a polyadenylation signal were cloned into a modified pBluescript+ that had generated by inserting CCGGGAGCTCCGTAACTATAACGGTCCTAAGGTA GCGAATTCTTAATTAAGCGGCCGCGATCGCCCGGGCATTT AAATGGCCTGCAGGGCCGTTTAAACGGCCGGCCGTCGAC TCGAGCGTAACTATAACGGTCCTAAGGT AGCGAAGGTACCGCGC and 2 loxP sites into the KpnI/SacI sites. (7) This sGFP1–10::NLG with a polyadenylation signal was excised with I-CeuI and cloned into an I-CeuI site of 11::NXN in pCR8-Topo. (8) This sGFP-10::NLG-sGFP11-NXN cassette was transferred to the product of step 4 above using LR clonase (Life Technologies).
The i-SceI linearized vector was electroporated into a 129/B6 F1 hybrid ES cell line, V6.5. G418-resistant, targeted ES clones were identified by PCR. Correct targeting efficiency was >50%. ES cell transfections and blastocyst injections were performed by the Genome Modification Facility, Harvard University. After germ-line transmission, the FRT-neo-FRT sequence was removed by crossing to mice that express Flp recombinase ubiquitously (Farley et al., 2000).
Rhodopsin-Cre transgenic mice (Li et al., 2005) were obtained from Shiming Chen (Washington University). Animal procedures were in compliance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal and Care and Use Program at Harvard University.
Immunostaining
Immunostaining was performed as described by Yamagata and Sanes (2010). Briefly, tissues were dissected, fixed in 4% paraformaldehyde/PBS overnight at 4°C, sunk in 15% (w/v) and 30% (w/v) sucrose/PBS, mounted in OCT compound, and cryosectioned. The sections were treated with 0.1% Triton X-100/PBS followed by Image-iT FX signal enhancer (Life Technologies), blocked with 5% skim milk/PBS, incubated with primary antibodies overnight at 4°C, rinsed, incubated with secondary antibodies with Neurotrace 435 (Life Technologies) overnight at 4°C, rinsed, and mounted with FluoroGel.
Primary antibodies used were: rabbit anti-Cre (Abcam, Cambridge, MA); mouse anti-neuroligin-1 and anti-neurexin-1β (NeuroMab, Davis, CA); mouse anti-bassoon (GeneTex, Irvine, CA); mouse anti-SV2 (Developmental Studies Hybridoma Bank, Iowa City, IA), mouse anti-β-dystroglycan (Leica Microsystems, Buffalo Grove, IL), mouse anti-PSD95 (Affinity Bioreagents, IgG1); monoclonal anti-GFP (GFP-Mab20, Sigma) and the two anti-GFP antibodies described above. Rhodamine-conjugated Peanut agglutinin was from Vector Lab (Burlingame, CA) and secondary antibodies were from Jackson Immunoresearch Laboratories (West Grove, PA).
Results
GRASP Between Cultured Mammalian Cells
Superfolder GFP (Pedelacq et al., 2006) was designed for use in E. coli. To improve expression in mammalian cells, we optimized its codon usage for mice (GenBank, JQ341914), and fused the sGFP1–10 and sGFP11 fragments to mammalian transmembrane cell surface proteins: human CD4 glycoprotein with a cytoplasmically fused monomeric cherry, rat neurexin-1β (NXN) and rat neuroligin-1 (NLG; Figure 1A). The CD4-cherry fusion has been used for GRASP in C. elegans (Feinberg et al., 2008) and Drosophila (Gordon and Scott, 2009), and NXN and NLG are localized to the pre- and postsynaptic membranes, respectively, of most mammalian synapses (Südhof, 2008; Shen and Scheiffele, 2010).
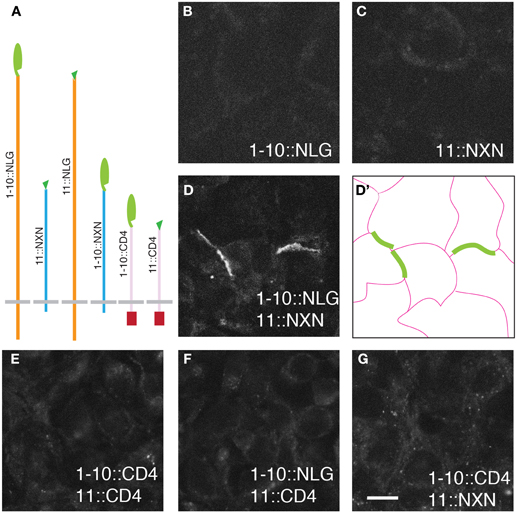
Figure 1. GRASP signals in cultured mammalian cells. (A) Superfolder GFP fragments sGFP1–10 and sGFP11 were fused to mammalian transmembrane proteins neurexin (NXN), neuroligin (NLG), and CD4 for display on the cell surface. Assembly of sGFP1–10 with sGFP11 reconstitutes fluorescent GFP. (B–D) 293 cells stably transfected with either 1–10::NLG (B) or 11::NXN (C) show no GRASP fluorescence. However, GRASP signal was observed at contacts between sGFP1–10::NLG- and sGFP11::NXN-expressing cells (D). (D') shows the outline of cells in (D). (E–G) No complementation was detected with CD4 fusions. Bar: 10 μm.
The six fusions were introduced into HEK293PL cells and stably transfected populations were selected, passaged and cultured either alone or in pairwise combinations. We observed no reconstituted GFP fluorescence when any one of the six populations was cultured alone or in most combinations (Figure 1 and Table 1). In only two cases were contacts between cells fluorescent, indicating reconstitution of GFP in mixtures of sGFP1–10::NLG cells with sGFP11::NXN cells and in mixtures of sGFP1–10::NXN cells with sGFP11::NLG cells (Figure 1D and Table 1). GRASP signal was readily detectable in live cells viewed with an inverted microscope, supporting the possibility that the technique could be used to image synapses in live tissue. Immunolabeling with antibodies to NXN and NLN confirmed that in each case, signal was present only at contacts between NXN-expressing and NLN-expressing cells (data not shown). The failure to observe complementation between sGFP1–10::CD4 and sGFP11::CD4, was surprising in that these fusions generate GRASP signals in C. elegans and Drosophila (Feinberg et al., 2008; Gordon and Scott, 2009).

Table 1. GRASP signal at cell-cell contacts of various configurations.
Generation of GRASP Knock-in Mice
To mark synapses in vivo, we generated mice in which sGFP1–10::NLG and sGFP11::NXN are expressed in complementary sets of neurons. Tests in HEK293PL cells had shown that the two fragments complement efficiently when expressed in the same cell, resulting in intense signals that prevent detection of the relatively weak signals resulting from complementation at opposed membranes between cells (see Methods). To prevent such co-expression, we designed a vector that contained both fragments, with sGFP1–10::NLG expressed by default and sGFP11-NXN expressed only following Cre-dependent excision of sGFP1–10::NLG (Figures 2A,B). The cassette (CAG-LoxP-sGFP1–10::NLG-pA-LoxP-11:: sGFP11::NXN-pA) was introduced into HEK293PL cells using a piggyBac-transposon (Yamagata and Sanes, 2008, 2010) to generate a cell line harboring a single copy. The cells expressed sGFP1–10::NLG but not sGFP11::NXN. Following transfection with a plasmid encoding Cre, the cells expressed sGFP11::NXN but not sGFP1–10::NLG (data not shown).
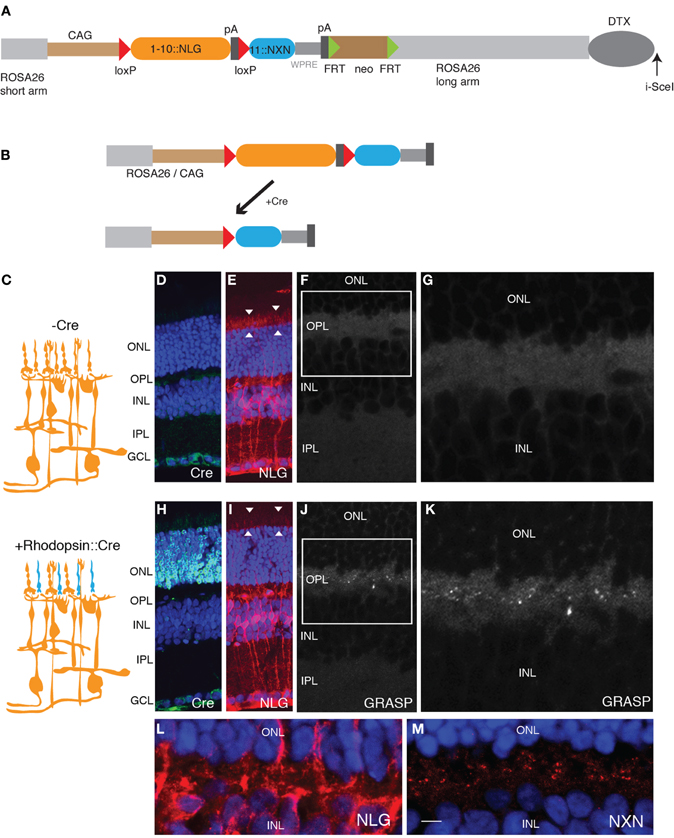
Figure 2. Generation of GRASP knock-in mice. (A) Targeting construct for generation of mGRASP1 mice. sGFP1–10::NLG was placed between LoxP sites, followed by sGFP11::NXN. WPRE, Woodchuck post-transcriptional regulatory element; pA, polyadenylation signal. FRT-SV40-neo-FRT and DTX denote selection cassettes used for positive and negative selection of correctly targeted ES cells, respectively; these cassettes were absent from the final mGRASP1 line. (B) Cre recombinase deletes sGFP1–10::NLG, thereby activating expression of sGFP11::NXN. (C) Diagram of gene switching in retina. sGFP1–10::NLG is expressed in all cells of mGRASP1 mice (top). Following cross to rhodopisin-Cre, mice, expression of sGFP1–10::NLG is extinguished in rod photoreceptors but persists in other cells; concurrently, expression of sGFP11::NXN is activated specifically in rod photoreceptors. (D–G) Sections of retina from P30 mGRASP mice. (D,E) Sections stained with anti-Cre (D), and anti-NLG (E). Neurotrace 435 counterstain is blue. No Cre is detectable, but NLG immunoreactivity is present throughout the retina. Because NLG immunoreactivity is barely detectable in wild-type retina (not shown). signal in (E) is due to sGFP1–10::NLG. Note strong signal associated with photoreceptor outer segments (arrowheads in E). (F,G) No GRASP signals are detectable in retina. (G) shows high-power view of OPL from area boxed in (F). (H–K) Sections of retina from P30 mGRASP; rhodopsin-Cre mice. (H,I) Sections stained with anti-Cre (H), anti-NLG (I), and Neurotrace 435 (blue). Cre is abundant in photoreceptor nuclei in the ONL (H) and NLG immunoreactivity is lost from photoreceptor outer segments (arrowheads in I). (J,K) GRASP signals are detectable in OPL. (K) shows high-power view of area boxed in (J). (L,M) Sections of retina from P30 mGRASP; rhodopsin-Cre mice were stained with anti-NLG (L) and anti-NXN (M). NXN is concentrated in nerve terminals in the OPL. Because NXN immunoreactivity is barely detectable in wild-type retina (not shown), signal in (M) is due to sGFP11::NXN. Bar, 10 μm for D,E,H,I; 5 μm for F,J; 2.5 μm for G,K–M. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
We then generated mice using this gene switch vector. We used a knock-in strategy to ensure that the genome contained only a single copy of the cassette. Thus, any individual cell can express either sGFP1–10::NLG or sGFP11::NXN, but can never express both. In contrast, transgenes introduced by oocyte injection often comprise multiple copies in tandem, so recombination of only a subset of the copies would lead to cis-complementation. To maximize levels of expression, we used the strong composite CAG (CMV + chicken β-actin) promoter-enhancer, added a WPRE to stabilize mRNA, and inserted the vector into the ROSA26 locus, which is suitable for ubiquitous expression of transgenes (Soriano, 1999; Zong et al., 2005; Madisen et al., 2010). Homologous recombinants were selected and used to generate germ-line chimeras by standard methods. We call the resulting line mGRASP1.
For initial tests, we used a transgenic line that expresses Cre recombinase specifically in rod photoreceptors under control of regulatory elements from the rhodopsin gene (Li et al., 2005). Immunostaining for Cre recombinase confirmed the specificity of the transgene (Figures 2D,H). We expected that sGFP1–10::NLG would be broadly expressed and sGFP11-::NXN would be undetectable in mGRASP1 mice, whereas sGFP11-NXN would be expressed only in rods, and sGFP1–10::NLG would be expressed in all other retinal cells in double transgenic mGRASP1; rhodopsin-Cre mice (Figure 2C). Indeed, in mGRASP1 mice, NLG immunoreactivity was present throughout the retina, including in photoreceptor outer segments (Figure 2E). NLG immunoreactivity was barely detectable in wild-type retina (data not shown), indicating that the signal in mGRASP1 mice reflected expression of sGFP1–10::NLG. Little NXN reactivity was detected in retinas of wild-type or mGRASP1 mice. In mGRASP1; rhodopsin-Cre mice, NLG immunoreactivity was lost from outer retina, confirming loss of sGFP1–10::NLG (Figures 2E,I). Likewise, immunostaining with NXN confirmed accumulation of sGFP11::NXN in the outer plexiform layer of mGRASP1; rhodopsin-Cre mice (Figures 2L,M). Most important, complementation led to appearance of GRASP signal in the outer plexiform layer of mGRASP1 (Figures 2F,G,J,K); rhodopsin-Cre mice; this is layer in which axon terminals of photoreceptors form synapses on dendrites of bipolar and horizontal cells (Rao-Mirotznik et al., 1995; Sterling and Matthews, 2005).
Immunoamplification of GRASP Signals
Although GRASP signals in mGRASP1; rhodopsin-Cre mice were clear and specific, they were weak. We attempted to enhance the signal using a commercially available monoclonal antibody, GFP-Mab20, which binds to an epitope present in holo-GFP but not in either the sGFP1–10 or the sGFP11 fragment (Gordon and Scott, 2009). This antibody was useful but inadequate, because it recognizes only a single epitope and therefore, provides limited amplification, and also because anti-mouse secondary antibodies lead to non-specific staining of mouse tissues. We, therefore, generated polyclonal antisera to native GFP, and purified from the sera those antibodies that recognized native GFP but not sGFP1–10 (see Methods and Figures 3A–D). The purified antibodies specifically recognized reconstituted GFP at borders between cells transfected with sGFP1–10::NLG and sGFP11::NXN cells, whereas a monoclonal antibody to GFP, which we also generated, recognized both the reconstituted GFP and the sGFP1–10::NLG fragment (Figures 3E–G). We used the holo-GFP-specific antibody, which we call anti-rGFP, for further characterization of mGRASP1 mice.
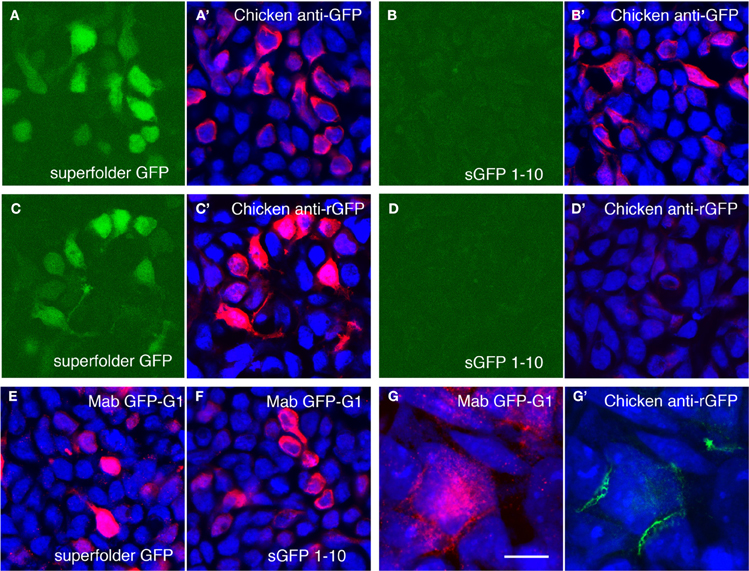
Figure 3. Amplification of GRASP signal using antibodies recognizing reconstituted GFP. (A,B) Superfolder GFP is fluorescent when expressed in 293 cells (A) whereas split GFP1–10 is not (B). Chicken antibodies to bacterially expressed superfolder GFP recognize both proteins (A',B'). Blue channel shows counter-staining with Neurotrace 435. (C,D) Following absorption to sGFP1–10 and tissue, the same antibodies react with fluorescent superfolder GFP (C,C'), but not sGFP1–10 (D,D'). (E,F) Mouse monoclonal antibody Mab GFP-G1 recognizes both superfolder GFP (E) and split GFP1–10 (F). (G,G') Cells transfected with 1–10::NLG or 11::NXN were co-cultured and doubly stained with Mab GFP-G1 and chicken anti-rGFP. Mab GFP-G1 stained the sGFP1–10::NLG expressing cell. In contrast, anti-rGFP stains only contacts between sGFP1–10::NLG- and sGFP11::NXN-expressing cells (mab GFP-G1-positive and -negative, respectively). Bar, 20 μm for A–F; 5 μm for (G).
Characterization and Specificity of GRASP Signals in Photoreceptor Synapses
To test whether the GRASP signal in the outer plexiform layer of mGRASP1; rhodopsin-Cre mice is localized to rod synapses, we double-labeled sections with anti-rGFP plus antibodies to synaptic components. Bassoon marks active zones in photoreceptor nerve terminals (Brandstätter et al., 1999). GRASP signals abutted Bassoon-positive puncta (Figures 4A,B). In one set of sections from P60 retina, we found that 73 ± 4% of Bassoon-positive puncta in the OPL were anti-rGFP-positive. This is an underestimate, because Bassoon marks cone as well as rod synapses and the latter are unlabeled (see below). In many cases, a pair of GRASP-positive puncta were localized directly adjacent to a larger Bassoon-positive punctum. In these triplets, the Bassoon-positive structure was generally closer to the outer nuclear layer, in which photoreceptors reside, while the rGFP-positive puncta were closer to the inner nuclear layer, which contains horizontal and bipolar cells. It is known that each rod synapse comprises a presynaptic rod terminal, invaginating processes from two horizontal cells, and a central bipolar process; the horizontal cell processes directly about the photoreceptor membrane whereas the bipolar process does not (Figure 4I; also see Rao-Mirotznik et al., 1995; Sterling and Matthews, 2005). Thus, the pattern we observed suggests that GRASP signals mark direct contacts between the rod terminal and the horizontal processes. Lack of GRASP signal associated with the bipolar process is consistent with the idea that directly abutting membranes are required for assembly of sGFP1–10 with sGFP11.
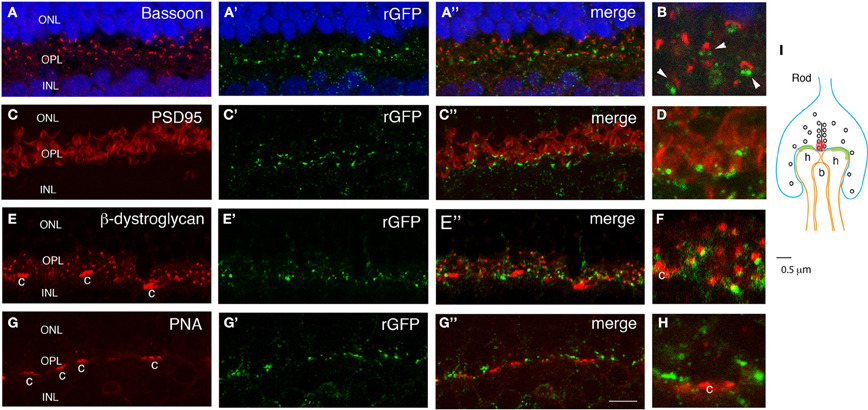
Figure 4. GRASP signal in OPL is confined to rod synapses. Sections from retina of double transgenic mGRASP1; rhodopsin-Cre mice were double-stained with anti-rGFP plus anti-bassoon (A,B), anti-PSD95 (C,D), anti-β-dystroglycan (E,F) or peanut agglutinin (PNA, G,H). (B,D,F,H) show high-power images from fields shown in (A,C,E,G), respectively. (I) is a sketch of a rod photoreceptor synapse. (A,B) Pairs of GFP-positive puncta are apposed to sites labeled with anti-Bassoon, which marks active zones in rod terminals (red spot in I). This arrangement suggests that GRASP signals are present at synaptic contacts made by rod terminals onto two invaginating horizontal cell processes (I). Blue color shows Neurotrace 435. (C,D) GFP-positive puncta are apposed to sites labeled with anti-PSD95, which marks perisynaptic regions widely within rod terminals. (E,F) GFP-positive puncta are apposed to sites labeled with anti-β-dystroglycan, which marks perisynaptic regions within rod terminals. β-dystroglycan is also present in cone terminals (c). (G,H) GFP-positive puncta are absent from sites labeled with Peanut agglutinin-positive, which marks cone photoreceptors terminals (c). This arrangement suggests that GRASP signals are present at synapses of rod but not cone photoreceptors on horizontal and bipolar cell processes. (I) A sketch of a rod spherule. Two horizontal cell dendrites (h) directly contact a rod terminal, and a bipolar dendrite (b) is distant from a synaptic ribbon. Bar, 3 μm for A,C,E,G; 1 μm for B,D,F,H.
We also stained sections with antibodies to PSD95 and β-dystroglycan. In many central synapses, PSD95 is associated with the postsynaptic membrane, but in photoreceptor synapses it is localized in perisynaptic areas within photoreceptor nerve terminals, adjacent to active zones (Koulen et al., 1998; Yamagata and Sanes, 2010). Likewise, β-dystroglycan is present in expanded portions of photoreceptor terminals, but not at active zones (Blank et al., 1999). Double labeling revealed that rGFP-positive puncta were near PSD95- and β-dystroglycan-positive puncta (Figures 4C–F). These patterns support the idea that GRASP signals line rod nerve terminals.
Finally, we asked whether GRASP signals are confined to synapses made by rod photoreceptors onto horizontal cells, or whether they are also present at closely apposed synapses made by cone photoreceptors on the same horizontal cells. In mice, 95–97% of photoreceptors are rods, and only 3–5% are cones, so if GRASP signals were not confined to appropriate synapses on horizontal cells we would expected them to be present in nearby cone synapses. To mark cone pedicle synapses, we incubated sections with peanuts agglutinin (PNA), which labels cone but not rod terminals in mouse retina (Blanks and Johnson, 1983). GRASP signals were excluded from PNA-positive structures (Figures 4G,H), indicating that the method can distinguish synapses made by two different presynaptic cells onto the same postsynaptic cell.
Long-Term Expression of Mammalian GRASP System
The GRASP method generates a transsynaptic link that is not present endogenously. It is, therefore, a concern that the long-term presence of this link could affect synaptic structure. Moreover, our implementation of the GRASP method requires expression of NXN and NLG both of what can affect synapse number when overexpressed (Chubykin et al., 2007; Dahlhaus et al., 2010). We, therefore, analyzed the number and molecular architecture of rod photoreceptor synapses in 7 month-old mGRASP1; rhodopsin-Cre mice. The presence of GRASP links over this prolonged period had no detectable effect on the levels or localization of any synaptic marker tested, including SV2, bassoon, PSD95, and β-dystroglycan (Figure 5). In addition, the number of photoreceptor terminals (bassoon-positive puncta) in the OPL of 7 month-old retinas did not differ significantly between mice expressing both mGRASP and Rhodopsin-Cre, and controls expressing only Rhodopsin-Cre (8.9 ± 2.3 and 9.1 ± 2 puncta/100 μm2, respectively; p > 0.1 by t-test). Thus, long-term expression of GRASP linkages at synapses has no detectable effect on the persistence or molecular architecture of these synapses.
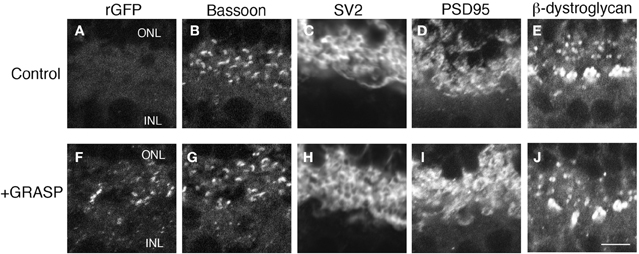
Figure 5. Prolonged expression of GRASP has no obvious effect on the molecular architecture of photoreceptor synapses. (A–E) OPL of 7 month-old control mice were stained with anti-rGFP (A) and antibodies to synaptic markers: bassoon at active zones (B), SV2 in synaptic vesicles (C), and PSD95 and β-dystroglycan in regions of nerve terminal membrane abutting active zones (D,E). (F–J) OPL of 7 month-old mGRASP1; rhodopsin-Cre mouse, stained as in (A–E). Bar, 2 μm.
Discussion
We have shown that the GRASP method, which was originally developed to detect synaptic connections in C. elegans (Feinberg et al., 2008), can also be used to detect synapses in mice. As a test, we labeled synapses between rod photoreceptors and their postsynaptic partners, horizontal cells; we showed that labeling is specific in that synapses made by cone receptors on the same postsynaptic partners were unlabeled.
While we were preparing this work for publication, Kim et al. (2012) described an alternative method for implement GRASP in mice. They, like us, found that fusing fragments to NXN and NLG was effective whereas fusions to CD4 were ineffective. In our cases, GRASP signals from CD4 fusions were unworkable faint, whereas for Kim et al. (2012), signals were detectable but not synaptically localized. Whereas we used a transgenic strategy to express the split GFP fragments, they used a viral strategy. A major advantage of the transgenic strategy is that labeling is completely non-invasive whereas a major advantage of the viral strategy is that post- as well as presynaptic cells can be selected. Another difference between the methods is that whereas we used unmodified NXN and NLG, they mutated NLG to prevent strong interactions with NXN, which, as noted above, might affect synaptic size or strength (Chubykin et al., 2007; Dahlhaus et al., 2010). Although we have noted no ill effects of overexpression, it may be more prudent to adopt the strategy of Kim et al. (2012) in the future.
Together the two sets of results make a strong case that GRASP will be generally useful for circuit analysis in mice. First, both transgenic (this paper) and viral methods (Kim et al., 2012) can be used to deliver GRASP components. Second, both light (this paper) and electron microscopic (Kim et al., 2012) methods demonstrate the specificity of GRASP labeling. Third, marking of synapses in retina (this paper) and forebrain (Kim et al., 2012) indicate that the method can be applied to multiple synaptic types.
The major drawback to our instantiation of the GRASP method is that it is insufficiently sensitive. Although signals in photoreceptor synapses are robust, these are especially large synapses, and we have not been able to consistently detect signals at smaller synapses in the inner plexiform layer of the retina, the spinal or the cortex using other Cre transgenic lines. We suspect that this limitation reflects the relatively low expression of the postsynaptic partner, sGFP1–10::NLG in mGRASP1 mice. This, in turn, may result from fact that the WPRE in the construct, which greatly enhances expression (Madisen et al., 2010) would be expected to stabilize the mRNA encoding sGFP11::NXN but not that encoding sGFP1–10::NLG. In addition, our experience with gene transfer methods in vivo suggests that the viral and electroporation methods used by Kim et al. (2012) lead to considerably higher levels of expression than does the knock-in method we used. To circumvent this limitation, and thereby expand the range of applications for the method, we are currently engineering transgenes in which both components are expressed at higher levels.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Genome Modification at Harvard University for generating germ-line chimeras, Dr. Shiming Chen for providing rhodopsin-Cre mice on behalf of Dr. Jason Chen, and Dr. Evan Feinberg for valuable comments. This work was supported by the Gatsby Foundation, NIH (U24NS063931), and Collaborative Innovation Award #43667 from HHMI.
References
Blank, M., Koulen, P., Blake, D. J., and Kröger, S. (1999). Dystrophin and beta dystroglycan in photoreceptor terminals from normal and mdx3Cv mouse retinae. Eur. J. Neurosci. 11, 2121–2133.
Blanks, J. C., and Johnson, L. V. (1983). Selective lectin binding of the developing mouse retina. J. Comp. Neurol. 221, 31–41.
Brandstätter, J. H., Fletcher, E. L., Garner, C. C., Gundelfinger, E. D., and Wässle, H. (1999). Differential expression of the presynaptic cytomatrix protein bassoon among ribbon synapses in the mammalian retina. Eur. J. Neurosci. 11, 3683–3693.
Cabantous, S., Terwilliger, T. C., and Waldo, G. S. (2005). Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23, 102–107.
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R., and Südhof, T. C. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931.
Dahlhaus, R., Hines, R. M., Eadie, B. D., Kannangara, T. S., Hines, D. J., Brown, C. E., Christie, B. R., and El-Husseini, A. (2010). Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus 20, 305–322.
Farley, F. W., Soriano, P., Steffen, L. S., and Dymecki, S. M. (2000). Widespread recombinase expression using FLPeR (flipper) mice. Genesis 28, 106–110.
Feinberg, E. H., Vanhoven, M. K., Bendesky, A., Wang, G., Fetter, R. D., Shen, K., and Bargmann, C. I. (2008). GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 57, 353–363.
Gong, Z. F., Liu, J. Q., Guo, C., Zhou, Y. Q., Teng, Y., and Liu, L. (2010). Two pairs of neurons in the central brain control Drosophila innate light preference. Science 330, 499–502.
Gordon, M. D., and Scott, K. (2009). Motor control in a Drosophila taste circuit. Neuron 61, 373–384.
Horowitz, L. F., Montmayeur, J. P., Echelard, Y., and Buck, L. B. (1999). A genetic approach to trace neural circuits. Proc. Natl. Acad. Sci. U.S.A. 96, 3194–3199.
Hu, C. D., and Kerppola, T. K. (2003). Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21, 539–545.
Huang, B., Babcock, H., and Zhuang, X. (2010). Breaking the diffraction barrier: super-resolution imaging of cells. Cell 143, 1047–1058.
Kim, J., Zhao, T., Petralia, R. S., Yu, Y., Peng, H., Myers, E., and Magee, J. C. (2012). mGRASP enables mapping mammalian synaptic connectivity with light microscopy. Nat. Methods 9, 96–102.
Koulen, P., Fletcher, E. L., Craven, S. E., Bredt, D. S., and Wassle, H. (1998). Immunocytochemical localization of the postsynaptic density protein PSD-95 in the mammalian retina. J. Neurosci. 18, 10136–10149.
Li, S., Chen, D., Sauvé, Y., McCandless, J., Chen, Y. J., and Chen, C. K. (2005). Rhodopsin-iCre transgenic mouse line for cre-mediated rod-specific gene targeting. Genesis 41, 73–80.
Lo, L., and Anderson, D. J. (2011). A cre-dependent, anterograde transsynaptic viral tracer for mapping output pathways of genetically marked neurons. Neuron 72, 938–950.
Madisen, L., Zwingman, T. A., Sunkin, S. M., Oh, S. W., Zariwala, H. A., Gu, H., Ng, L. L., Palmiter, R. D., Hawrylycz, M. J., Jones, A. R., Lein, E. S., and Zeng, H. (2010). A robust and high-throughput cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140.
Pedelacq, J. D., Cabantous, S., Tran, T., Terwilliger, T. C., and Waldo, G. S. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79–88.
Rao-Mirotznik, R., Harkins, A. B., Buchsbaum, G., and Sterling, P. (1995). Mammalian rod terminal: architecture of a binary synapse. Neuron 14, 561–569.
Shen, K., and Scheiffele, P. (2010). Genetics and cell biology of building specific synaptic connectivity. Annu. Rev. Neurosci. 33, 473–507.
Soriano, P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71.
Srinivas, S., Watanabe, T., Lin, C. S., William, C. M., Tanabe, Y., Jessell, T. M., and Costantini, F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4.
Sterling, P., and Matthews, G. (2005). Structure and function of ribbon synapses. Trends Neurosci. 28, 20–29.
Südhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911.
Wickersham, I. R., Lyon, D. C., Barnard, R. J., Mori, T., Finke, S., Conzelmann, K. K., Young, J. A., and Callaway, E. M. (2007). Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 53, 639–647.
Yamagata, M., and Sanes, J. R. (2008). Dscam and Sidekick proteins direct lamina-specific synaptic connections in vertebrate retina. Nature 451, 465–469.
Yamagata, M., and Sanes, J. R. (2010). Synaptic localization and function of Sidekick recognition molecules require MAGI scaffolding proteins. J. Neurosci. 30, 3579–3588.
Yoshihara, Y., Mizuno, T., Nakahira, M., Kawasaki, M., Watanabe, Y., Kagamiyama, H., Jishage, K., Ueda, O., Suzuki, H., Tabuchi, K., Sawamoto, K., Okano, H., Noda, T., and Mori, K. (1999). A genetic approach to visualization of multisynaptic neural pathways using plant lectin transgene. Neuron 22, 33–41.
Zhang, S., Ma, C., and Chalfie, M. (2004). Combinatorial marking of cells and organelles with reconstituted fluorescent proteins. Cell 119, 137–144.
Keywords: GRASP, GFP, synapse, retina, photoreceptor, rod, neuroligin, neurexin
Citation: Yamagata M and Sanes JR (2012) Transgenic strategy for identifying synaptic connections in mice by fluorescence complementation (GRASP). Front. Mol. Neurosci. 5:18. doi: 10.3389/fnmol.2012.00018
Received: 14 January 2012; Accepted: 03 February 2012;
Published online: 16 February 2012.
Edited by:
Joshua A. Weiner, The University of Iowa, USAReviewed by:
Christian Lohmann, Netherlands Institute for Neuroscience, NetherlandsJames Jontes, Ohio State University, USA
Copyright: © 2012 Yamagata and Sanes. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Joshua R. Sanes, Department of Molecular and Cellular Biology and Center for Brain Science, Harvard University, 52 Oxford Street, Cambridge, MA 02138, USA. e-mail: sanesj@mcb.harvard.edu