Protein degradation and protein synthesis in long-term memory formation
 Timothy J. Jarome
Timothy J. Jarome Fred J. Helmstetter
Fred J. Helmstetter- 1Department of Neurobiology, University of Alabama at Birmingham, Birmingham, AL, USA
- 2Department of Psychology, University of Wisconsin-Milwaukee, Milwaukee, WI, USA
Long-term memory (LTM) formation requires transient changes in the activity of intracellular signaling cascades that are thought to regulate new gene transcription and de novo protein synthesis in the brain. Consistent with this, protein synthesis inhibitors impair LTM for a variety of behavioral tasks when infused into the brain around the time of training or following memory retrieval, suggesting that protein synthesis is a critical step in LTM storage in the brain. However, evidence suggests that protein degradation mediated by the ubiquitin-proteasome system (UPS) may also be a critical regulator of LTM formation and stability following retrieval. This requirement for increased protein degradation has been shown in the same brain regions in which protein synthesis is required for LTM storage. Additionally, increases in the phosphorylation of proteins involved in translational control parallel increases in protein polyubiquitination and the increased demand for protein degradation is regulated by intracellular signaling molecules thought to regulate protein synthesis during LTM formation. In some cases inhibiting proteasome activity can rescue memory impairments that result from pharmacological blockade of protein synthesis, suggesting that protein degradation may control the requirement for protein synthesis during the memory storage process. Results such as these suggest that protein degradation and synthesis are both critical for LTM formation and may interact to properly “consolidate” and store memories in the brain. Here, we review the evidence implicating protein synthesis and degradation in LTM storage and highlight the areas of overlap between these two opposing processes. We also discuss evidence suggesting these two processes may interact to properly form and store memories. LTM storage likely requires a coordinated regulation between protein degradation and synthesis at multiple sites in the mammalian brain.
Introduction
The ubiquitin-proteasome system (UPS) is a complex network of different ubiquitin ligases and interconnected protein structures involved in the regulation of protein degradation in neurons. This system has been reviewed extensively by others (Hegde, 2010; Mabb and Ehlers, 2010; Bingol and Sheng, 2011), but in general proteins become targeted for degradation through a series of steps in which the small protein modifier ubiquitin is covalently bound to a target substrate. The substrate can acquire anywhere from 1 to 7 ubiquitin modifiers, which link together at specific lysine residues forming polyubiquitin chains. In general, longer ubiquitin chains and lysine-48 linkage provide the maximal signal for degradation (Fioravante and Byrne, 2011) while lysine-63 and M1 linkage (linear ubiquitination) often target substrates for other non-proteolytic functions (Rieser et al., 2013). The ubiquitination process determines what proteins will be targeted for degradation by the proteasome, but the proteasome ultimately controls which of these polyubiquitinated substrates will be degraded.
The catalytic structure in the UPS is the 26S proteasome, which consists of a core (20S) and two regulatory particles (19S) (Bedford et al., 2010). The 20S proteasome is the catalytic core of the proteasome structure and it possesses three types of proteolytic activity controlled by the β-subunits. The activity of the 20S core is regulated by the 19S regulatory particles, which contain the only six ATP-sensitive subunits of the proteasome known as the Rpt subunits. The Rpt6 subunit has received the most attention as it has been shown that increases in its phosphorylation regulates increases in proteasome activity in vitro (Bingol et al., 2010; Djakovic et al., 2012) and correlates with increased proteasome activity in vivo (Jarome et al., 2013), suggesting that phosphorylation of Rpt6 (at Serine-120) may be the primary regulator of activity-dependent changes in proteasome activity in the brain. Additionally, the 19S proteasome contains numerous deubiquitinating enzymes which generally facilitate the degradation process by removing ubiquitin moieties as the substrate enters the proteasome, thus maintaining the ubiquitin pool (Kowalski and Juo, 2012). However, some deubiquitinating enzymes, such as the ubiquitin-specific protease 14 (USP14), actually seem to inhibit the degradation of certain substrates (Lee et al., 2010; Jin et al., 2012). This suggests that not only does the proteasome degrade polyubiquitinated substrates, but it can actually determine which of these substrates will ultimately be degraded.
In recent years numerous studies have suggested a role for the proteolytic activity of the UPS in activity-dependent synaptic plasticity. For example, bidirectional activity-dependent homeostatic scaling requires UPS-mediated protein degradation (Ehlers, 2003). Interestingly, this proteasome-dependent homeostatic scaling is largely regulated by phosphorylation of the Rpt6 subunit at Serine-120 (Rpt6-S120) (Djakovic et al., 2012) which enhances proteasome activity (Djakovic et al., 2009), suggesting that Rpt6-mediated increases in proteasome activity are critical for activity-dependent synaptic plasticity. Consistent with this, protein degradation is involved in new dendritic spine growth that is regulated by phosphorylation of Rpt6-S120 (Hamilton et al., 2012; Hamilton and Zito, 2013). Additionally, proteasome inhibitors alter long-term potentiation (LTP) in the hippocampus (Fonseca et al., 2006; Dong et al., 2008) and long-term facilitation (LTF) in Aplysia (Chain et al., 1999; Lee et al., 2012), suggesting that protein degradation is critical for various forms of synaptic plasticity.
Recently, attention has turned to the potential role of protein degradation in learning-dependent synaptic plasticity. Indeed, there is now convincing evidence that UPS-mediated protein degradation is likely involved in various different stages of memory storage. However, while some studies have suggested potential roles for protein degradation in long-term memory (LTM) formation and storage (Kaang and Choi, 2012), one intriguing question is whether protein degradation is linked to the well-known transcriptional and translational alterations thought to be critical for memory storage in the brain (Johansen et al., 2011). Here, we discuss evidence demonstrating a role for protein degradation and synthesis in the long-term storage of memories in the mammalian brain, highlighting instances in which a requirement for protein degradation correlates with a requirement for protein synthesis. Additionally, we discuss evidence suggesting that both protein degradation and synthesis may be regulated by CaMKII signaling during LTM formation. Collectively, we propose that LTM storage requires coordinated changes in protein degradation and synthesis in the brain, which may be primarily controlled through a CaMKII-dependent mechanism.
Memory Paradigms
A variety of different rodent behavioral paradigms have been used to study the molecular neurobiology of LTM formation, and these behavioral tasks often result in alterations in synaptic plasticity in many different brain regions. This review will focus mostly on fear-related conditioning paradigms, but will also discuss results from the Morris water maze (MWM) and the object recognition paradigm. One of the most common rodent behavioral procedures is Pavlovian fear conditioning, in which a neutral conditional stimulus (CS) becomes associated with a noxious or aversive unconditional stimulus (UCS). As a result of this association, the CS can elicit an emotional response based on the memory of the UCS. A simple form of Pavlovian fear conditioning is contextual fear conditioning, in which rodents learn to fear the specific context or training environment in which the UCS occurred. Memories in this paradigm require an intact and active amygdala and hippocampus for their formation and long-term storage (Kim and Fanselow, 1992; Phillips and Ledoux, 1992). Auditory delay fear conditioning involves a discrete auditory cue (CS) that coterminates with the UCS. Memories formed in this paradigm require an intact and active amygdala, but unlike contextual fear conditioning, do not require the hippocampus (Helmstetter, 1992a,b; Phillips and Ledoux, 1992; Ledoux, 2000). A more complex form of fear conditioning is auditory trace fear conditioning in which the auditory CS predicts the UCS but the two stimuli are separated in time. The introduction of this “trace interval” recruits the prefrontal cortex (Gilmartin and McEchron, 2005b; Gilmartin et al., 2013b). Trace fear conditioning also requires the hippocampus and amygdala (Gilmartin and McEchron, 2005a; Kwapis et al., 2011; Gilmartin et al., 2012). Fear conditioning can be measured in several ways including behavioral observation of freezing (e.g., Helmstetter, 1992a,b) or CS modulation of reflex responses (Hitchcock and Davis, 1991; Rosen et al., 1991). Inhibitory avoidance is another popular aversive learning procedure. In this task, rodents are placed into the lit (white) compartment of a black-white shuttle box. Once the partition is opened, the animal will go to the dark (black) side of the box where it will receive a shock. Animals will then learn to avoid the dark compartment to prevent receiving the shock again. Memories formed using this paradigm require the amygdala and hippocampus for their long-term storage (Taubenfeld et al., 2001; Milekic et al., 2007). Finally, another form of aversive classical conditioning is conditioned taste aversion, in which rodents will acquire an aversion to a specific food due to experiencing illness associated with it. Memories formed using this paradigm require the amygdala and insular cortex for their long-term storage (Rodriguez-Ortiz et al., 2011). In general, memories formed through aversive conditioning require the amygdala but the contribution of the hippocampus, prefrontal cortex and insular cortex depend on the specific behavioral paradigm used.
Two of the most common non-shock based spatial paradigms used to study LTM formation are the MWM and object recognition procedures. In the MWM, a spatial paradigm that can also be stressful and aversive (D'Hooge and De Deyn, 2001), rodents are typically placed into a pool where a hidden platform is positioned in one of four quadrants, each of which has specific spatial cues surrounding it. The animal is given several trials to learn where the platform is, and its latency to swim to the platform will decrease as it learns the task. On the probe day, the platform is removed and the amount of time the animal spends searching each quadrant is measured. If the animal learned the task, it will spend a majority of its time searching the target quadrant where the platform had been during training. Due to the spatial nature of the task, memories acquired using this paradigm require the hippocampus (Artinian et al., 2008). In the objection recognition paradigm, a non-aversive form of spatial learning, rodents are allowed to explore two objects for a period of time. At later test one of the objects is replaced with a new object and the amount of time the animal spends exploring the objects is recorded Object memory is indicated by more time spent exploring the novel object during the test phase. As in the MWM, memories formed in this paradigm require the hippocampus for their formation and long-term storage (Rossato et al., 2007).
The Role of Protein Synthesis in Memory Storage
For several decades there has been a general consensus that de novo protein synthesis is critical for the formation and stability of LTM (Davis and Squire, 1984; Helmstetter et al., 2008; Johansen et al., 2011). Consistent with this, numerous studies using pharmacological, molecular, and genetic approaches have implicated a role for increased transcriptional and translational regulation in various brain regions during LTM formation for a variety of different behavioral tasks. Table 1 summarizes some the findings for inhibitors of gene transcription and protein synthesis on LTM formation and stability following retrieval. Some of the first evidence suggesting that new protein synthesis may be necessary for LTM after fear conditioning came from a study showing that infusions of a broad spectrum mRNA synthesis inhibitor in the amygdala impaired memory for auditory and contextual fear conditioning (Bailey et al., 1999). Consistent with this, infusions of broad spectrum inhibitors of gene transcription or protein synthesis into the amygdala can impair LTM for auditory delay fear conditioning, auditory trace fear conditioning, contextual fear conditioning, fear potentiated startle, inhibitory avoidance, and conditioned taste aversion memories (Schafe and Ledoux, 2000; Bahar et al., 2003; Lin et al., 2003; Yeh et al., 2006; Milekic et al., 2007; Jarome et al., 2011; Kwapis et al., 2011), suggesting that increased protein synthesis is critical for the storage of fear memories in the amygdala (Hoeffer et al., 2011). Inhibiting protein synthesis in the hippocampus impairs LTM for contextual fear conditioning, MWM spatial memories, inhibitory avoidance memories, and object recognition memories (Bourtchouladze et al., 1998; Taubenfeld et al., 2001; Rossato et al., 2007; Artinian et al., 2008), while inhibiting protein synthesis impairs LTM for conditioned taste aversion memories in the insular cortex and trace fear memories in the medial prefrontal cortex (Blum et al., 2006; Moguel-Gonzalez et al., 2008; Reis et al., 2013). Additionally, infusions of more selective inhibitors of protein synthesis which block mTOR-mediated translation impair fear memory and object recognition memory formation in the amygdala and hippocampus, supporting that de novo translation is critical for LTM formation in these regions (Parsons et al., 2006b; Gafford et al., 2011; Jobim et al., 2012a,b). Collectively, these results suggest new protein synthesis is required for the processes of memory consolidation.
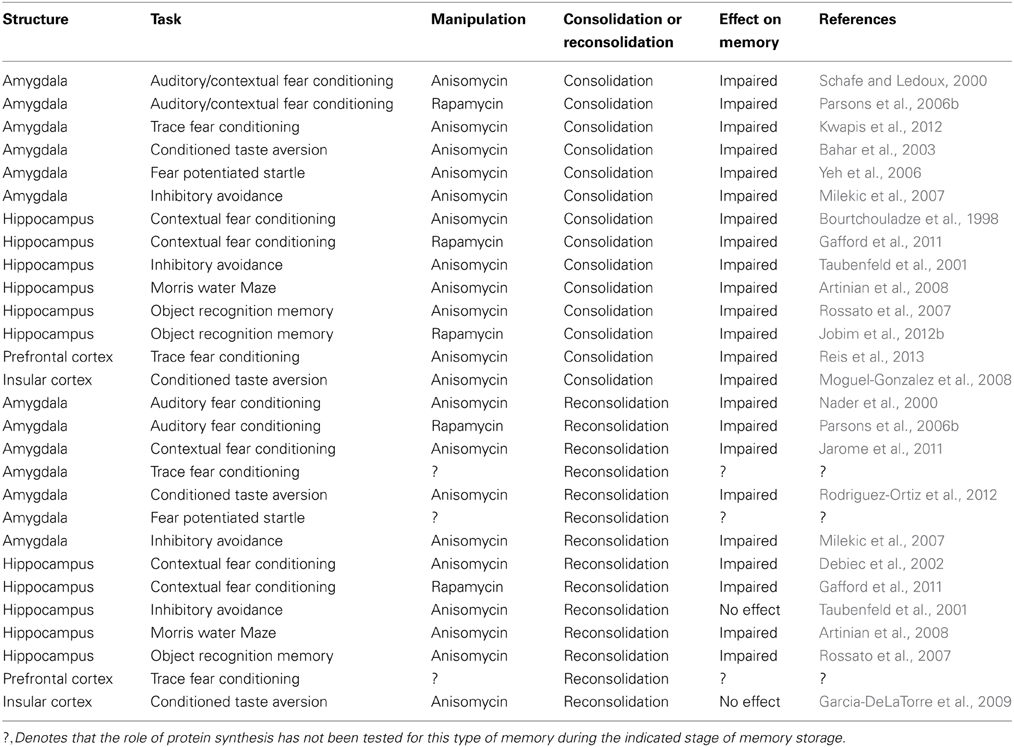
Table 1. The role of protein synthesis in memory consolidation and reconsolidation.
In addition to a role for new protein synthesis in LTM consolidation, numerous studies have shown that the use or retrieval of an established memory results in a second phase of increased protein synthesis, a process referred to as memory reconsolidation. Protein synthesis inhibitors infused into the amygdala can impair the reconsolidation of auditory delay fear memories, contextual fear memories, inhibitory avoidance memories, and conditioned taste aversion memories (Nader et al., 2000; Parsons et al., 2006a; Milekic et al., 2007; Jarome et al., 2011, 2012; Rodriguez-Ortiz et al., 2012). Additionally, inhibiting protein synthesis in the hippocampus impairs the reconsolidation of contextual fear memories (Debiec et al., 2002; Lee et al., 2008; Gafford et al., 2011), MWM spatial memories (Artinian et al., 2008), and object recognition memories (Rossato et al., 2007), though it has no effect on inhibitory avoidance memories in the hippocampus (Taubenfeld et al., 2001) or conditioned taste aversion memories in the insular cortex (Garcia-DeLaTorre et al., 2009). These results indicate that new protein synthesis is a necessary step in the transfer of a retrieved memory back to long-term storage, suggesting that both the consolidation and reconsolidation of memories requires de novo protein translation in several brain regions.
Consistent with the evidence from the broad spectrum inhibitors, several studies have implicated a role for specific intracellular signaling molecules thought to be “upstream” of protein synthesis in LTM formation and storage (Johansen et al., 2011). For example, inhibition of NMDA receptor (NMDAR) function impairs the consolidation of auditory delay fear and contextual fear memories, fear potentiated startle, and conditioned taste aversion memories in the amygdala (Walker and Davis, 2000; Yasoshima et al., 2000; Rodrigues et al., 2001), auditory trace and contextual fear memories in the prefrontal cortex (Gilmartin and Helmstetter, 2010; Gilmartin et al., 2013a), contextual fear memories, MWM and objection recognition spatial memories in the hippocampus (Liang et al., 1994; Izquierdo et al., 1999; Czerniawski et al., 2012; Da Silva et al., 2013; Warburton et al., 2013), and conditioned taste aversion memories in the insular cortex (Escobar et al., 1998). Inhibition of signaling molecules thought to be downstream of NMDAR activity but upstream of protein synthesis such as Protein Kinase A, ERK-MAPK, and CaMKII impairs memory consolidation for auditory fear memories, contextual fear memories, inhibitory avoidance memories, MWM spatial memories, and conditioned taste aversion memories (Schafe and Ledoux, 2000; Schafe et al., 2000; Sacchetti et al., 2001; Koh et al., 2002; Quevedo et al., 2004; Rodrigues et al., 2004; Leon et al., 2010; Ota et al., 2010; Chen et al., 2012; Halt et al., 2012). Many of these signaling molecules are thought to regulate the transient changes in gene expression hypothesized to be critical for LTM formation in the brain. Consistent with this, epigenetic modifications such as acetylation, phosphorylation, and methylation of histones and DNA methylation are critical for LTM formation in the amygdala and hippocampus (Levenson et al., 2004; Lubin et al., 2008; Gupta et al., 2010; Maddox and Schafe, 2011; Jarome and Lubin, 2013), suggesting that LTM formation requires dynamic changes in gene transcription and protein translation in multiple regions of the brain.
The Role of Protein Degradation in Memory Storage
The idea that protein degradation could contribute to activity-dependent synaptic plasticity was first identified more than a decade ago by a study demonstrating that the induction of LTF in Aplysia resulted in increased expression of Ap-uch, which encodes a ubiquitin C-terminal hydrolase, and that a loss of this gene impaired LTF (Hegde et al., 1997). Additionally, application of the proteasome inhibitor lactacystin impairs LTF (Chain et al., 1999), suggesting that functional proteasome activity is critical for synaptic plasticity. Consistent with this identified role of ubiquitin-proteasome mediated protein degradation in activity-dependent synaptic plasticity, homeostatic changes in synaptic strength that result from chronic stimulation or inhibition of cultured hippocampal neurons requires activity of the UPS (Ehlers, 2003). This activity-dependent homeostatic scaling requires phosphorylation of the proteasome subunit Rpt6 at Serine-120, a CaMKII target site (Djakovic et al., 2009; Bingol et al., 2010). Remarkably, enhancements in Rpt6 phosphorylation is sufficient to drive long-term changes in synaptic strength (Djakovic et al., 2012) and new dendritic spine growth in vitro (Hamilton et al., 2012), suggesting that protein degradation is a critical regulator of synaptic plasticity. Furthermore, numerous studies have indicated a role for protein degradation in LTP, the proposed cellular analog of memory (for review, see Hegde, 2010). For example, inhibiting protein degradation or protein synthesis individually impairs late-LTP (Fonseca et al., 2006), suggesting that protein degradation is critical for maintaining LTP following its induction. Interestingly, proteasome inhibitors can also enhance the induction of LTP (Dong et al., 2008), an effect that is largely due to the proteasomes targeting of translational activators during LTP induction (Dong et al., 2014). Thus, it is well-established that protein degradation is a critical regulator of synaptic plasticity in vitro.
While not considered in traditional memory models, the theory that protein degradation accompanies changes in protein synthesis during LTM formation has become increasingly popular. Indeed, numerous studies now clearly demonstrate a role for UPS-mediated protein degradation in LTM formation and storage in neurons (Felsenberg et al., 2012; Kaang and Choi, 2012; Jarome and Helmstetter, 2013). Interestingly, strong evidence suggests that changes in protein degradation are correlated with changes in protein synthesis in specific brain regions during both memory consolidation and reconsolidation. Table 2 summarizes the findings for genetic and pharmacological manipulations of ubiquitin-proteasome activity on LTM formation and stability following retrieval. Proteasome inhibitors infused in the amygdala alter LTM for contextual and auditory fear conditioning (Jarome et al., 2011), fear potentiated startle (Yeh et al., 2006) but not conditioned taste aversion memories (Rodriguez-Ortiz et al., 2011). Additionally, proteasome inhibitors impair MWM and inhibitory avoidance memories in the hippocampus (Lopez-Salon et al., 2001; Artinian et al., 2008), trace fear memories in the prefrontal cortex (Reis et al., 2013), and conditioned taste aversion memories in the insular cortex (Rodriguez-Ortiz et al., 2011). These results suggest a strong overlap between the protein degradation and synthesis processes during LTM formation. Consistent with this, fear conditioning simultaneously increases protein polyubiquitination and mTOR phosphorylation in the amygdala 1 h after behavioral training (Jarome et al., 2011), a time when protein synthesis is increased in the amygdala (Hoeffer et al., 2011), suggesting a potential overlap between protein degradation and synthesis processes. However, inhibiting proteasome activity in the hippocampus does not alter LTM for contextual fear conditioning. Consistent with this finding, a genetic loss of a ubiquitin E3 ligase alters memory for amygdala but not hippocampus dependent fear memories (Pick et al., 2013a,b), suggesting that there is not a perfect overlap between the protein degradation and synthesis processes during LTM formation. In general, these results do suggest though that protein degradation and protein synthesis are both critical for LTM formation in the same brain regions and they likely overlap in time following behavioral training.
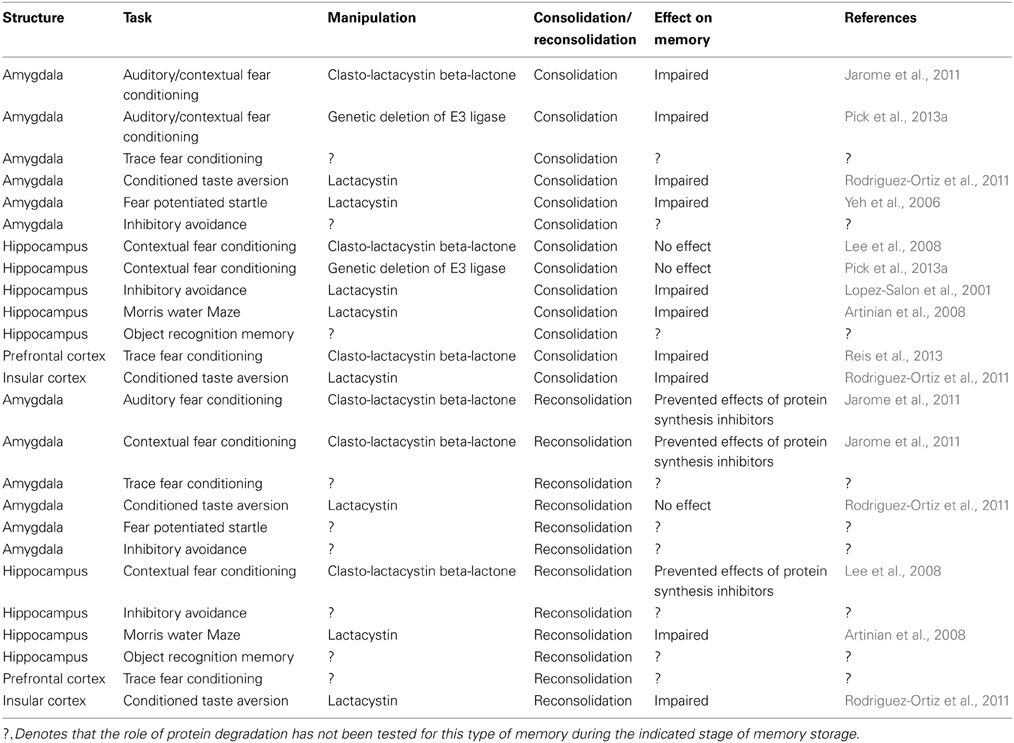
Table 2. The role of protein degradation in memory consolidation and reconsolidation.
Protein degradation is also critical for the reconsolidation of fear memories, though much less is known about the role of the UPS in this stage of memory storage. For example, proteasome inhibitors alter the reconsolidation of auditory and contextual fear memories in the amygdala (Jarome et al., 2011), contextual fear memories and MWM memories in the hippocampus (Artinian et al., 2008; Lee, 2008, 2010; Lee et al., 2008) and conditioned taste aversion memories in the insular cortex (Rodriguez-Ortiz et al., 2011), though it is currently unknown if protein degradation regulates the reconsolidation of trace fear memories, inhibitory avoidance memories, and object recognition memories in the brain. Despite this, there does appear to be a clear overlap between the protein degradation and protein synthesis processes during memory reconsolidation as manipulation of protein degradation in the amygdala and hippocampus prevent the effectiveness of protein synthesis inhibitors at disrupting LTM for fear conditioning tasks. Collectively, these results suggest a strong correlation between protein degradation and protein synthesis during the reconsolidation process.
In addition to the protein degradation function, non-proteolytic functions of the UPS are also critical for LTM and correlate with the demand for increased protein synthesis. For example, non-proteolytic monoubiquitination of the cytoplasmic polyadenylation element binding protein 3 (CPEB3) by the E3 ligase Neuralized1 is critical for the consolidation of hippocampus dependent memories (Pavlopoulos et al., 2011). Interestingly, monoubiquitination of CPEB3 by Neuralized1 was critical for activity dependent increases in GluR1 and GluR2, suggesting that non-proteolytic ubiquitination regulates protein synthesis during LTM formation. Additionally, recent evidence suggests that the USP14 is a critical regulator of LTM formation in the amygdala (Jarome et al., 2014). USP14 acts as a negative regulator of protein turnover (Lee et al., 2010) and regulates presynaptic plasticity in vitro (Wilson et al., 2002; Walters et al., 2008; Bhattacharyya et al., 2012), suggesting that USP14 regulates memory formation through a mechanism independent of protein degradation. These studies highlight that both proteolytic and non-proteolytic functions of the UPS regulate LTM formation in the amygdala and hippocampus, suggesting that overall changes in ubiquitin-proteasome activity correlates with the increased demand for de novo protein synthesis during memory consolidation.
The Linking of Protein Degradation and Synthesis Through NMDA-CaMKII Signaling
In addition to the potential overlap between protein degradation and synthesis in the brain during LTM formation, both of these mechanisms are likely regulated by a similar signaling pathway: NMDA receptor (NMDAR) dependent changes in CaMKII activity. As stated above, similar to protein degradation and synthesis, NMDAR activity at the time of training has been shown to be critical for the formation of different types of memories in various brain regions. NMDAR activity is thought to regulate the changes in protein synthesis necessary for LTM formation by altering the activity of a number of downstream signaling pathways (Johansen et al., 2011; Jarome and Helmstetter, 2013). One such signaling molecule is CaMKII which is thought to be involved in changes in gene transcription and protein synthesis through its regulation of CREB (Wayman et al., 2008), a critical regulator of LTM formation and stability in the neurons (Josselyn et al., 2001; Han et al., 2007, 2008, 2009). Indeed, numerous studies have suggested that CaMKII is critical for LTM formation and that this requirement for CaMKII signaling overlaps with the requirement for protein degradation and synthesis during memory consolidation (Barros et al., 1999; Rodrigues et al., 2004; Von Hertzen and Giese, 2005; Halt et al., 2012; Da Silva et al., 2013). Collectively, these results suggest that changes in NMDAR and CaMKII activity correlate with changes in both protein degradation and protein synthesis during LTM formation in neurons.
In addition to overlapping with an increased need for protein degradation, recent evidence suggests that NMDAR and CAMKII activity can regulate UPS-mediated protein degradation during LTM in neurons (Jarome et al., 2011, 2013). Inhibiting the activity of NR2B-containing NMDARs, a manipulation which impairs LTM (Rodrigues et al., 2001), prevents learning-dependent increases in degradation-specific polyubiquitination in the amygdala (Jarome et al., 2011). This supports the previously identified in vitro relationship between NMDAR and ubiquitin-proteasome activity (Bingol and Schuman, 2006; Bingol et al., 2010) and suggests that NMDAR activity regulates changes in protein degradation in neurons during memory consolidation. Additionally, inhibiting CaMKII signaling in the amygdala during LTM formation prevents learning-induced increases in proteasome activity (Jarome et al., 2013), suggesting that CaMKII signaling is critical for changes in protein degradation in neurons during memory consolidation. Interestingly, pharmacological manipulation of CaMKII also prevented learning-induced increases in the phosphorylation of the proteasome regulatory subunit Rpt6, which is critical for changes in proteasome activity, synaptic plasticity and dendritic spine growth in vitro (Djakovic et al., 2009, 2012; Hamilton et al., 2012), suggesting that CaMKII may regulate protein degradation through its actions on the proteasome ATPase subunits. Importantly, manipulation of protein kinase A (PKA), another NMDAR-dependent signaling molecule that is critical for LTM formation in neurons (Schafe and Ledoux, 2000; Tronson et al., 2006), did not alter the changes in proteasome phosphorylation and activity during memory consolidation. This demonstrates that PKA, which can regulate protein degradation in vitro (Upadhya et al., 2006; Zhang et al., 2007), does not regulate protein degradation during memory formation and suggests that not all NMDAR-dependent signaling pathways regulate ubiquitin-proteasome activity during LTM formation. Collectively, these results suggest that NMDA-CaMKII-dependent changes in ubiquitin-proteasome mediated protein degradation are critical for LTM formation in neurons, suggesting that protein degradation and synthesis could be linked during memory formation by CaMKII signaling, however, it is still unknown if CaMKII actually does regulate protein synthesis during memory formation.
What Comes First, Degradation or Synthesis?
A majority of the studies discussed here reveal a strong correlation between protein degradation and synthesis during LTM formation. This leads to one important question: Which comes first? While the exact relationship between protein degradation and synthesis during memory formation currently remains equivocal, the available evidence suggests that protein degradation likely regulates protein synthesis. For example, fear conditioning leads to an increase in polyubiquitinated proteins being targeted for degradation by the proteasome (Jarome et al., 2011). While a majority of the proteins being targeted by the proteasome for degradation remain unknown, the RNAi-induced Silencing Complex (RISC) factor MOV10 has been identified as a target of the proteasome during increases in activity-dependent protein degradation in vitro (Banerjee et al., 2009) and following behavioral training and retrieval in vivo (Jarome et al., 2011). Increases in the degradation of MOV10 are associated with increased protein synthesis in vitro, suggesting that the proteasome could regulate protein synthesis during LTM formation through the removal of translational repressor proteins such as various RISC factors. However, it is currently unknown if the selective degradation of MOV10, or any RISC factor, is critical for memory formation in neurons. Nonetheless, studies such as these provide indirect evidence that protein degradation by the UPS could regulate protein synthesis during memory formation in the brain.
Some of the best evidence that protein degradation may be upstream of protein synthesis during memory storage comes from studies examining memory reconsolidation following retrieval. For example, inhibiting proteasome activity can prevent the memory impairments that normally result from post-retrieval blockade of protein synthesis in the hippocampus, amygdala, and nucleus accumbens (Lee et al., 2008; Jarome et al., 2011; Ren et al., 2013) as well as during LTF in aplysia (Lee et al., 2012), suggesting that protein degradation is upstream of protein synthesis during memory reconsolidation. This remains some of the best evidence directly linking protein degradation to protein synthesis during memory storage, but it is possible that the rescue of memory impairments in the face of protein synthesis inhibition may occur as an indirect consequence of blocking protein degradation rather than a direct effector. Additionally, some studies find that proteasome inhibitors impair memory reconsolidation when administered on their own (Artinian et al., 2008; Rodriguez-Ortiz et al., 2011), suggesting that this relationship between protein degradation and synthesis may not exist for all types of memories. However, in cell cultures protein degradation has been shown to regulate mTOR activity (Ghosh et al., 2008), a translational control pathway critical for memory formation and storage (Parsons et al., 2006b; Gafford et al., 2011), though it is unknown if this relationship exists in vivo during memory formation. Collectively, the current evidence suggests that protein degradation may determine the requirement for protein synthesis during memory storage, though this relationship has never been directly proven.
While it is currently not known if protein degradation regulates protein synthesis during memory formation, one recent study found that protein degradation regulates protein synthesis during LTP, a proposed cellular analog of memory (Dong et al., 2014). Inhibiting proteasome activity enhances the induction but impairs the maintenance of LTP (Dong et al., 2008). Surprisingly, the enhanced induction of LTP following proteasome inhibition can be blocked by inhibiting the activity of several proteins involved in mTOR-mediated protein synthesis, suggesting that the proteasome targets translational activators during LTP induction. However, proteasome inhibitors cause an increase in translational repressors during L-LTP, suggesting that the proteasome acts to enhance protein synthesis during LTP maintenance. Collectively, these results demonstrate for the first time that protein degradation can directly regulate protein synthesis during activity-dependent synaptic plasticity. While it is currently unknown if the proteasome targets similar proteins during memory formation, this study provides some of the best evidence to date that protein degradation may regulate protein synthesis during learning-dependent synaptic plasticity.
Regulation of Memory Storage by Protein Degradation and Synthesis
While the functional significance of the observed overlap between protein degradation and synthesis during LTM formation is unknown, it is possible that a coordinated balance between protein degradation and synthesis may be necessary for memory formation and storage in neurons (Jarome and Helmstetter, 2013). This theory has been described extensively elsewhere, but in this model CaMKII-dependent increases in proteasome activity leads to reductions in a number of different proteins that normally repress transcription and translation or prevent alterations to the postsynaptic structure. Consistent with this, the proteasome targets the RISC factor MOV10 and synaptic scaffold Shank following behavioral training and memory retrieval (Lee et al., 2008; Jarome et al., 2011), and inhibiting proteasome activity can prevent learning-induced changes in GluR2 expression at synapses (Ren et al., 2013). Thus, the changes in protein degradation could be necessary to remove repressor proteins that prevent the dynamic changes to the postsynaptic structure that characterize memory storage in neurons (Ostroff et al., 2010) while protein synthesis could produce the proteins that are necessary for these changes. In addition, an attractive new addition to this model is that protein degradation and synthesis could regulate dynamic changes in the atypical protein kinase C isoform PKMζ during memory consolidation. A majority of studies have shown that inhibiting PKMζ impairs LTM even after the consolidation process has completed (Pastalkova et al., 2006; Shema et al., 2007; Kwapis et al., 2009, 2012), though a few exceptions have been noted (Parsons and Davis, 2011; Volk et al., 2013), suggesting that PKMζ regulates the maintenance of memories in neurons (Sacktor, 2012; Kwapis and Helmstetter, 2013). Interestingly, one recent study (Vogt-Eisele et al., 2014) found that protein degradation can regulate PKMζ levels during memory storage through targeting of KIBRA (Kidney/BRAin protein). Since behavioral training is thought to increase PKMζ levels in the brain (Sacktor, 2012), this suggests that a balance between protein degradation and synthesis may control LTM formation and storage through regulation of PKMζ levels. Whether a balance between protein degradation and synthesis is necessary for learning-dependent changes in PKMζ levels will be of interest in future studies.
Protein Degradation and Synthesis as Independent Processes During Memory Storage
While the evidence discussed here suggests that protein degradation and protein synthesis may be interconnected processes necessary for memory formation and storage, an alternative theory is that these are actually opposing processes and that memory formation requires an appropriate balance between them. For example, both proteasome and protein synthesis inhibitors impair LTP when applied individually, but actually rescue these deficits when applied simultaneously (Fonseca et al., 2006). This would suggest that memory impairments that result from blocking protein degradation or synthesis individually are caused by an inappropriate balance between the two processes. While an intriguing theory, few studies have directly tested this in vivo during memory formation as most times proteasome inhibitors are given alone and not in combination with protein synthesis inhibitors. Despite this, some studies suggest that an altered balance between protein degradation and synthesis cannot account for memory impairments following disruption of the UPS. For example, proteasome inhibitors infused into the amygdala result in similar memory impairments for a fear conditioning task as those produced by the broad spectrum protein synthesis inhibitor anisomycin and simultaneous infusion of both inhibitors does not rescue these impairments (Jarome et al., 2011). This suggests that an inappropriate balance between protein degradation and synthesis likely cannot account for memory deficits observed following infusions of either inhibitor individually. However, simultaneous blockade of protein degradation and synthesis can rescue memory impairments that results from blocking protein synthesis individually following memory retrieval (Lee et al., 2008; Jarome et al., 2011), though in these cases proteasome inhibitors have no effect on memory on their own. This would lend more toward the theory that protein degradation is upstream of protein synthesis during memory storage following retrieval. Thus, while the idea that protein degradation and protein synthesis are independent processes and a balance between them is the primary factor that underlies memory formation is intriguing, very few studies have directly tested this to date.
Summary and Future Directions
The studies reviewed here suggest a clear correlation between protein degradation and protein synthesis in LTM formation in the brain. However, we are currently far from understanding the exact relationship between these two opposing processes during in memory. As discussed above, the current evidence suggests that protein degradation and synthesis are both important regulators of LTM for auditory delay and trace fear conditioning, contextual fear conditioning, inhibitory avoidance, conditioned taste aversion, MWM, and object recognition memories and simultaneously occur in multiple brain regions including the amygdala, hippocampus, prefrontal cortex, and insular cortex. However, it is unknown if this relationship holds for all brain regions and behavioral tasks. For example, contextual and trace fear memories also require the retrosplinal cortex for their long-term storage (Kwapis et al., 2014), though it is unknown if protein degradation and protein synthesis are required. Future research should determine which behavioral tasks and brain regions require protein degradation and protein synthesis to better determine the extent of overlap in these processes. More importantly, future research should focus on determining if and how protein degradation regulates activity driven protein synthesis during memory formation in the already identified instances in which these two processes locally correlate. Such information is critical to determine not only if protein degradation and synthesis are dissociable during memory formation, but also what the functional role of the changes in protein degradation is. Furthermore, identifying the targets of the proteasome during increased protein degradation levels will also help determine the functional role of protein degradation during memory formation. Understanding the exact relationship between protein degradation and synthesis is a critical step in understanding the molecular neurobiology of LTM formation and storage in neurons.
Additionally, future studies should focus on how CaMKII signaling regulates increases in proteasome activity during memory formation and whether this occurs through Rpt6 phosphorylation. Considering that CaMKII is the only intracellular signaling molecule known to regulate protein degradation during LTM formation (Jarome et al., 2013) and is thought to regulate protein synthesis, it is critical to understand how changes in CaMKII activity contribute to the appropriate regulation of protein degradation and synthesis during LTM formation in neurons. Thus, CaMKII might serve as an attractive target to better understand the role of protein degradation in LTM formation and its relationship to de novo protein synthesis. Finally, future studies should focus on identifying the non-proteolytic functions of the UPS during memory formation and storage. Currently, only two non-proteolytic roles for ubiquitin-proteasome activity exist (Pavlopoulos et al., 2011), though it is likely that many more remain to be discovered. Considering that one of these non-proteolytic functions has been suggested to regulate some protein synthesis, the degradation-independent functions of the UPS may prove to be among the most important regulators of translational regulation by the UPS.
Conclusions
Here, we reviewed a number of studies suggesting that changes in protein degradation correlate with changes in protein synthesis during memory formation and storage in neurons. Additionally, we discussed evidence demonstrating that protein degradation is regulated by NMDAR activity and the intracellular signaling molecule CaMKII, two well-known regulators of memory formation that are thought to regulate protein synthesis, suggesting that protein degradation and synthesis may be linked through CaMKII signaling. Finally, we reviewed evidence demonstrating that protein degradation may be upstream of protein synthesis during LTM formation, suggesting that protein degradation may supersede protein synthesis during the memory consolidation process. Collectively, the studies outlined in this review suggest that protein degradation and protein synthesis are not likely two independent regulators of LTM formation but rather directly interact to modify active synapses and store memory for a variety of different behavioral tasks.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by NIMH grants R01-06558 (Fred J. Helmstetter).
References
Artinian, J., McGauran, A. M., De Jaeger, X., Mouledous, L., Frances, B., and Roullet, P. (2008). Protein degradation, as with protein synthesis, is required during not only long-term spatial memory consolidation but also reconsolidation. Eur. J. Neurosci. 27, 3009–3019. doi: 10.1111/j.1460-9568.2008.06262.x
Bahar, A., Samuel, A., Hazvi, S., and Dudai, Y. (2003). The amygdalar circuit that acquires taste aversion memory differs from the circuit that extinguishes it. Eur. J. Neurosci. 17, 1527–1530. doi: 10.1046/j.1460-9568.2003.02551.x
Bailey, D. J., Kim, J. J., Sun, W., Thompson, R. F., and Helmstetter, F. J. (1999). Acquisition of fear conditioning in rats requires the synthesis of mRNA in the amygdala. Behav. Neurosci. 113, 276–282. doi: 10.1037/0735-7044.113.2.276
Banerjee, S., Neveu, P., and Kosik, K. S. (2009). A coordinated local translational control point at the synapse involving relief from silencing and MOV10 degradation. Neuron 64, 871–884. doi: 10.1016/j.neuron.2009.11.023
Barros, D. M., Izquierdo, L. A., Sant'anna, M. K., Quevedo, J., Medina, J. H., McGaugh, J. L., et al. (1999). Stimulators of the cAMP cascade reverse amnesia induced by intra-amygdala but not intrahippocampal KN-62 administration. Neurobiol. Learn. Mem. 71, 94–103. doi: 10.1006/nlme.1998.3830
Bedford, L., Paine, S., Sheppard, P. W., Mayer, R. J., and Roelofs, J. (2010). Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 20, 391–401. doi: 10.1016/j.tcb.2010.03.007
Bhattacharyya, B. J., Wilson, S. M., Jung, H., and Miller, R. J. (2012). Altered neurotransmitter release machinery in mice deficient for the deubiquitinating enzyme Usp14. Am. J. Physiol. Cell Physiol. 302, C698–C708. doi: 10.1152/ajpcell.00326.2010
Bingol, B., and Schuman, E. M. (2006). Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441, 1144–1148. doi: 10.1038/nature04769
Bingol, B., and Sheng, M. (2011). Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron 69, 22–32. doi: 10.1016/j.neuron.2010.11.006
Bingol, B., Wang, C. F., Arnott, D., Cheng, D., Peng, J., and Sheng, M. (2010). Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140, 567–578. doi: 10.1016/j.cell.2010.01.024
Blum, S., Runyan, J. D., and Dash, P. K. (2006). Inhibition of prefrontal protein synthesis following recall does not disrupt memory for trace fear conditioning. BMC Neurosci. 7:67. doi: 10.1186/1471-2202-7-67
Bourtchouladze, R., Abel, T., Berman, N., Gordon, R., Lapidus, K., and Kandel, E. R. (1998). Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn. Mem. 5, 365–374.
Chain, D. G., Casadio, A., Schacher, S., Hegde, A. N., Valbrun, M., Yamamoto, N., et al. (1999). Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron 22, 147–156. doi: 10.1016/S0896-6273(00)80686-8
Chen, D. Y., Bambah-Mukku, D., Pollonini, G., and Alberini, C. M. (2012). Glucocorticoid receptors recruit the CaMKIIalpha-BDNF-CREB pathways to mediate memory consolidation. Nat. Neurosci. 15, 1707–1714. doi: 10.1038/nn.3266
Czerniawski, J., Ree, F., Chia, C., and Otto, T. (2012). Dorsal versus ventral hippocampal contributions to trace and contextual conditioning: differential effects of regionally selective NMDA receptor antagonism on acquisition and expression. Hippocampus 22, 1528–1539. doi: 10.1002/hipo.20992
Da Silva, W. C., Cardoso, G., Bonini, J. S., Benetti, F., and Izquierdo, I. (2013). Memory reconsolidation and its maintenance depend on L-voltage-dependent calcium channels and CaMKII functions regulating protein turnover in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 110, 6566–6570. doi: 10.1073/pnas.1302356110
Davis, H. P., and Squire, L. R. (1984). Protein synthesis and memory: a review. Psychol. Bull. 96, 518–559. doi: 10.1037/0033-2909.96.3.518
Debiec, J., Ledoux, J. E., and Nader, K. (2002). Cellular and systems reconsolidation in the hippocampus. Neuron 36, 527–538. doi: 10.1016/S0896-6273(02)01001-2
D'Hooge, R., and De Deyn, P. P. (2001). Applications of the Morris water maze in the study of learning and memory. Brain Res. Brain Res. Rev. 36, 60–90. doi: 10.1016/S0165-0173(01)00067-4
Djakovic, S. N., Marquez-Lona, E. M., Jakawich, S. K., Wright, R., Chu, C., Sutton, M. A., et al. (2012). Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J. Neurosci. 32, 5126–5131. doi: 10.1523/JNEUROSCI.4427-11.2012
Djakovic, S. N., Schwarz, L. A., Barylko, B., Demartino, G. N., and Patrick, G. N. (2009). Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 284, 26655–26665. doi: 10.1074/jbc.M109.021956
Dong, C., Bach, S. V., Haynes, K. A., and Hegde, A. N. (2014). Proteasome modulates positive and negative translational regulators in long-term synaptic plasticity. J. Neurosci. 34, 3171–3182. doi: 10.1523/JNEUROSCI.3291-13.2014
Dong, C., Upadhya, S. C., Ding, L., Smith, T. K., and Hegde, A. N. (2008). Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learn. Mem. 15, 335–347. doi: 10.1101/lm.984508
Ehlers, M. D. (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242. doi: 10.1038/nn1013
Escobar, M. L., Alcocer, I., and Chao, V. (1998). The NMDA receptor antagonist CPP impairs conditioned taste aversion and insular cortex long-term potentiation in vivo. Brain Res. 812, 246–251. doi: 10.1016/S0006-8993(98)00931-7
Felsenberg, J., Dombrowski, V., and Eisenhardt, D. (2012). A role of protein degradation in memory consolidation after initial learning and extinction learning in the honeybee (Apis mellifera). Learn. Mem. 19, 470–477. doi: 10.1101/lm.026245.112
Fioravante, D., and Byrne, J. H. (2011). Protein degradation and memory formation. Brain Res. Bull. 85, 14–20. doi: 10.1016/j.brainresbull.2010.11.002
Fonseca, R., Vabulas, R. M., Hartl, F. U., Bonhoeffer, T., and Nagerl, U. V. (2006). A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 52, 239–245. doi: 10.1016/j.neuron.2006.08.015
Gafford, G. M., Parsons, R. G., and Helmstetter, F. J. (2011). Consolidation and reconsolidation of contextual fear memory requires mammalian target of rapamycin-dependent translation in the dorsal hippocampus. Neuroscience 182, 98–104. doi: 10.1016/j.neuroscience.2011.03.023
Garcia-DeLaTorre, P., Rodriguez-Ortiz, C. J., Arreguin-Martinez, J. L., Cruz-Castaneda, P., and Bermudez-Rattoni, F. (2009). Simultaneous but not independent anisomycin infusions in insular cortex and amygdala hinder stabilization of taste memory when updated. Learn. Mem. 16, 514–519. doi: 10.1101/lm.1356509
Ghosh, P., Wu, M., Zhang, H., and Sun, H. (2008). mTORC1 signaling requires proteasomal function and the involvement of CUL4-DDB1 ubiquitin E3 ligase. Cell Cycle 7, 373–381. doi: 10.4161/cc.7.3.5267
Gilmartin, M. R., and Helmstetter, F. J. (2010). Trace and contextual fear conditioning require neural activity and NMDA receptor-dependent transmission in the medial prefrontal cortex. Learn. Mem. 17, 289–296. doi: 10.1101/lm.1597410
Gilmartin, M. R., Kwapis, J. L., and Helmstetter, F. J. (2012). Trace and contextual fear conditioning are impaired following unilateral microinjection of muscimol in the ventral hippocampus or amygdala, but not the medial prefrontal cortex. Neurobiol. Learn. Mem. 97, 452–464. doi: 10.1016/j.nlm.2012.03.009
Gilmartin, M. R., Kwapis, J. L., and Helmstetter, F. J. (2013a). NR2A- and NR2B-containing NMDA receptors in the prelimbic medial prefrontal cortex differentially mediate trace, delay, and contextual fear conditioning. Learn. Mem. 20, 290–294. doi: 10.1101/lm.030510.113
Gilmartin, M. R., and McEchron, M. D. (2005a). Single neurons in the dentate gyrus and CA1 of the hippocampus exhibit inverse patterns of encoding during trace fear conditioning. Behav. Neurosci. 119, 164–179. doi: 10.1037/0735-7044.119.1.164
Gilmartin, M. R., and McEchron, M. D. (2005b). Single neurons in the medial prefrontal cortex of the rat exhibit tonic and phasic coding during trace fear conditioning. Behav. Neurosci. 119, 1496–1510. doi: 10.1037/0735-7044.119.6.1496
Gilmartin, M. R., Miyawaki, H., Helmstetter, F. J., and Diba, K. (2013b). Prefrontal activity links nonoverlapping events in memory. J. Neurosci. 33, 10910–10914. doi: 10.1523/JNEUROSCI.0144-13.2013
Gupta, S., Kim, S. Y., Artis, S., Molfese, D. L., Schumacher, A., Sweatt, J. D., et al. (2010). Histone methylation regulates memory formation. J. Neurosci. 30, 3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010
Halt, A. R., Dallapiazza, R. F., Zhou, Y., Stein, I. S., Qian, H., Juntti, S., et al. (2012). CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 31, 1203–1216. doi: 10.1038/emboj.2011.482
Hamilton, A. M., Oh, W. C., Vega-Ramirez, H., Stein, I. S., Hell, J. W., Patrick, G. N., et al. (2012). Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 74, 1023–1030. doi: 10.1016/j.neuron.2012.04.031
Hamilton, A. M., and Zito, K. (2013). Breaking it down: the ubiquitin proteasome system in neuronal morphogenesis. Neural Plast. 2013, 196848. doi: 10.1155/2013/196848
Han, J. H., Kushner, S. A., Yiu, A. P., Cole, C. J., Matynia, A., Brown, R. A., et al. (2007). Neuronal competition and selection during memory formation. Science 316, 457–460. doi: 10.1126/science.1139438
Han, J. H., Kushner, S. A., Yiu, A. P., Hsiang, H. L., Buch, T., Waisman, A., et al. (2009). Selective erasure of a fear memory. Science 323, 1492–1496. doi: 10.1126/science.1164139
Han, J. H., Yiu, A. P., Cole, C. J., Hsiang, H. L., Neve, R. L., and Josselyn, S. A. (2008). Increasing CREB in the auditory thalamus enhances memory and generalization of auditory conditioned fear. Learn. Mem. 15, 443–453. doi: 10.1101/lm.993608
Hegde, A. N. (2010). The ubiquitin-proteasome pathway and synaptic plasticity. Learn. Mem. 17, 314–327. doi: 10.1101/lm.1504010
Hegde, A. N., Inokuchi, K., Pei, W., Casadio, A., Ghirardi, M., Chain, D. G., et al. (1997). Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89, 115–126. doi: 10.1016/S0092-8674(00)80188-9
Helmstetter, F. J. (1992a). The amygdala is essential for the expression of conditional hypoalgesia. Behav. Neurosci. 106, 518–528. doi: 10.1037/0735-7044.106.3.518
Helmstetter, F. J. (1992b). Contribution of the amygdala to learning and performance of conditional fear. Physiol. Behav. 51, 1271–1276. doi: 10.1016/0031-9384(92)90320-2
Helmstetter, F. J., Parsons, R. G., and Gafford, G. M. (2008). Macromolecular synthesis, distributed synaptic plasticity, and fear conditioning. Neurobiol. Learn. Mem. 89, 324–337. doi: 10.1016/j.nlm.2007.09.002
Hitchcock, J. M., and Davis, M. (1991). Efferent pathway of the amygdala involved in conditioned fear as measured with the fear-potentiated startle paradigm. Behav. Neurosci. 105, 826–842. doi: 10.1037/0735-7044.105.6.826
Hoeffer, C. A., Cowansage, K. K., Arnold, E. C., Banko, J. L., Moerke, N. J., Rodriguez, R., et al. (2011). Inhibition of the interactions between eukaryotic initiation factors 4E and 4G impairs long-term associative memory consolidation but not reconsolidation. Proc. Natl. Acad. Sci. U.S.A. 108, 3383–3388. doi: 10.1073/pnas.1013063108
Izquierdo, I., Schroder, N., Netto, C. A., and Medina, J. H. (1999). Novelty causes time-dependent retrograde amnesia for one-trial avoidance in rats through NMDA receptor- and CaMKII-dependent mechanisms in the hippocampus. Eur. J. Neurosci. 11, 3323–3328. doi: 10.1046/j.1460-9568.1999.00742.x
Jarome, T. J., and Helmstetter, F. J. (2013). The ubiquitin-proteasome system as a critical regulator of synaptic plasticity and long-term memory formation. Neurobiol. Learn. Mem. 105, 107–116. doi: 10.1016/j.nlm.2013.03.009
Jarome, T. J., Kwapis, J. L., Hallengren, J. J., Wilson, S. M., and Helmstetter, F. J. (2014). The ubiquitin-specific protease 14 (USP14) is a critical regulator of long-term memory formation. Learn. Mem. 21, 9–13. doi: 10.1101/lm.032771.113
Jarome, T. J., Kwapis, J. L., Ruenzel, W. L., and Helmstetter, F. J. (2013). CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front. Behav. Neurosci. 7:115. doi: 10.3389/fnbeh.2013.00115
Jarome, T. J., Kwapis, J. L., Werner, C. T., Parsons, R. G., Gafford, G. M., and Helmstetter, F. J. (2012). The timing of multiple retrieval events can alter GluR1 phosphorylation and the requirement for protein synthesis in fear memory reconsolidation. Learn. Mem. 19, 300–306. doi: 10.1101/lm.024901.111
Jarome, T. J., and Lubin, F. D. (2013). Histone lysine methylation: critical regulator of memory and behavior. Rev. Neurosci. 24, 375–387. doi: 10.1515/revneuro-2013-0008
Jarome, T. J., Werner, C. T., Kwapis, J. L., and Helmstetter, F. J. (2011). Activity dependent protein degradation is critical for the formation and stability of fear memory in the amygdala. PLoS ONE 6:e24349. doi: 10.1371/journal.pone.0024349
Jin, Y. N., Chen, P. C., Watson, J. A., Walters, B. J., Phillips, S. E., Green, K., et al. (2012). Usp14 deficiency increases tau phosphorylation without altering tau degradation or causing tau-dependent deficits. PLoS ONE 7:e47884. doi: 10.1371/journal.pone.0047884
Jobim, P. F., Pedroso, T. R., Christoff, R. R., Werenicz, A., Maurmann, N., Reolon, G. K., et al. (2012a). Inhibition of mTOR by rapamycin in the amygdala or hippocampus impairs formation and reconsolidation of inhibitory avoidance memory. Neurobiol. Learn. Mem. 97, 105–112. doi: 10.1016/j.nlm.2011.10.002
Jobim, P. F., Pedroso, T. R., Werenicz, A., Christoff, R. R., Maurmann, N., Reolon, G. K., et al. (2012b). Impairment of object recognition memory by rapamycin inhibition of mTOR in the amygdala or hippocampus around the time of learning or reactivation. Behav. Brain Res. 228, 151–158. doi: 10.1016/j.bbr.2011.12.004
Johansen, J. P., Cain, C. K., Ostroff, L. E., and Ledoux, J. E. (2011). Molecular mechanisms of fear learning and memory. Cell 147, 509–524. doi: 10.1016/j.cell.2011.10.009
Josselyn, S. A., Shi, C., Carlezon, W. A. Jr. Neve, R. L., Nestler, E. J., and Davis, M. (2001). Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J. Neurosci. 21, 2404–2412.
Kaang, B. K., and Choi, J. H. (2012). Synaptic protein degradation in memory reorganization. Adv. Exp. Med. Biol. 970, 221–240. doi: 10.1007/978-3-7091-0932-8_10
Kim, J. J., and Fanselow, M. S. (1992). Modality-specific retrograde amnesia of fear. Science 256, 675–677. doi: 10.1126/science.1585183
Koh, M. T., Thiele, T. E., and Bernstein, I. L. (2002). Inhibition of protein kinase A activity interferes with long-term, but not short-term, memory of conditioned taste aversions. Behav. Neurosci. 116, 1070–1074. doi: 10.1037/0735-7044.116.6.1070
Kowalski, J. R., and Juo, P. (2012). The role of deubiquitinating enzymes in synaptic function and nervous system diseases. Neural Plast. 2012:892749. doi: 10.1155/2012/892749
Kwapis, J. L., and Helmstetter, F. J. (2013). Does PKM(zeta) maintain memory? Brain Res. Bull. 105C, 36–45. doi: 10.1016/j.brainresbull.2013.09.005
Kwapis, J. L., Jarome, T. J., Gilmartin, M. R., and Helmstetter, F. J. (2012). Intra-amygdala infusion of the protein kinase Mzeta inhibitor ZIP disrupts foreground context fear memory. Neurobiol. Learn. Mem. 98, 148–153. doi: 10.1016/j.nlm.2012.05.003
Kwapis, J. L., Jarome, T. J., Lee, J. L., Gilmartin, M. R., and Helmstetter, F. J. (2014). Extinguishing trace fear engages the retrosplenial cortex rather than the amygdala. Neurobiol. Learn. Mem. 113, 41–54. doi: 10.1016/j.nlm.2013.09.007
Kwapis, J. L., Jarome, T. J., Lonergan, M. E., and Helmstetter, F. J. (2009). Protein kinase Mzeta maintains fear memory in the amygdala but not in the hippocampus. Behav. Neurosci. 123, 844–850. doi: 10.1037/a0016343
Kwapis, J. L., Jarome, T. J., Schiff, J. C., and Helmstetter, F. J. (2011). Memory consolidation in both trace and delay fear conditioning is disrupted by intra-amygdala infusion of the protein synthesis inhibitor anisomycin. Learn. Mem. 18, 728–732. doi: 10.1101/lm.023945.111
Ledoux, J. E. (2000). Emotion circuits in the brain. Annu. Rev. Neurosci. 23, 155–184. doi: 10.1146/annurev.neuro.23.1.155
Lee, B. H., Lee, M. J., Park, S., Oh, D. C., Elsasser, S., Chen, P. C., et al. (2010). Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184. doi: 10.1038/nature09299
Lee, J. L. (2008). Memory reconsolidation mediates the strengthening of memories by additional learning. Nat. Neurosci. 11, 1264–1266. doi: 10.1038/nn.2205
Lee, J. L. (2010). Memory reconsolidation mediates the updating of hippocampal memory content. Front. Behav. Neurosci. 4:168. doi: 10.3389/fnbeh.2010.00168
Lee, S. H., Choi, J. H., Lee, N., Lee, H. R., Kim, J. I., Yu, N. K., et al. (2008). Synaptic protein degradation underlies destabilization of retrieved fear memory. Science 319, 1253–1256. doi: 10.1126/science.1150541
Lee, S. H., Kwak, C., Shim, J., Kim, J. E., Choi, S. L., Kim, H. F., et al. (2012). A cellular model of memory reconsolidation involves reactivation-induced destabilization and restabilization at the sensorimotor synapse in Aplysia. Proc. Natl. Acad. Sci. U.S.A. 109, 14200–14205. doi: 10.1073/pnas.1211997109
Leon, W. C., Bruno, M. A., Allard, S., Nader, K., and Cuello, A. C. (2010). Engagement of the PFC in consolidation and recall of recent spatial memory. Learn. Mem. 17, 297–305. doi: 10.1101/lm.1804410
Levenson, J. M., O'Riordan, K. J., Brown, K. D., Trinh, M. A., Molfese, D. L., and Sweatt, J. D. (2004). Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40545–40559. doi: 10.1074/jbc.M402229200
Liang, K. C., Hon, W., Tyan, Y. M., and Liao, W. L. (1994). Involvement of hippocampal NMDA and AMPA receptors in acquisition, formation and retrieval of spatial memory in the Morris water maze. Chin. J. Physiol. 37, 201–212.
Lin, C. H., Yeh, S. H., Lu, H. Y., and Gean, P. W. (2003). The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J. Neurosci. 23, 8310–8317.
Lopez-Salon, M., Alonso, M., Vianna, M. R., Viola, H., Mello e Souza, T., Izquierdo, I., et al. (2001). The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur. J. Neurosci. 14, 1820–1826. doi: 10.1046/j.0953-816x.2001.01806.x
Lubin, F. D., Roth, T. L., and Sweatt, J. D. (2008). Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 28, 10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008
Mabb, A. M., and Ehlers, M. D. (2010). Ubiquitination in postsynaptic function and plasticity. Annu. Rev. Cell Dev. Biol. 26, 179–210. doi: 10.1146/annurev-cellbio-100109-104129
Maddox, S. A., and Schafe, G. E. (2011). Epigenetic alterations in the lateral amygdala are required for reconsolidation of a Pavlovian fear memory. Learn. Mem. 18, 579–593. doi: 10.1101/lm.2243411
Milekic, M. H., Pollonini, G., and Alberini, C. M. (2007). Temporal requirement of C/EBPbeta in the amygdala following reactivation but not acquisition of inhibitory avoidance. Learn. Mem. 14, 504–511. doi: 10.1101/lm.598307
Moguel-Gonzalez, M., Gomez-Palacio-Schjetnan, A., and Escobar, M. L. (2008). BDNF reverses the CTA memory deficits produced by inhibition of protein synthesis. Neurobiol. Learn. Mem. 90, 584–587. doi: 10.1016/j.nlm.2008.06.003
Nader, K., Schafe, G. E., and Le Doux, J. E. (2000). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406, 722–726. doi: 10.1038/35021052
Ostroff, L. E., Cain, C. K., Bedont, J., Monfils, M. H., and Ledoux, J. E. (2010). Fear and safety learning differentially affect synapse size and dendritic translation in the lateral amygdala. Proc. Natl. Acad. Sci. U.S.A. 107, 9418–9423. doi: 10.1073/pnas.0913384107
Ota, K. T., Monsey, M. S., Wu, M. S., and Schafe, G. E. (2010). Synaptic plasticity and NO-cGMP-PKG signaling regulate pre- and postsynaptic alterations at rat lateral amygdala synapses following fear conditioning. PLoS ONE 5:e11236. doi: 10.1371/journal.pone.0011236
Parsons, R. G., and Davis, M. (2011). Temporary disruption of fear-potentiated startle following PKMzeta inhibition in the amygdala. Nat. Neurosci. 14, 295–296. doi: 10.1038/nn.2745
Parsons, R. G., Gafford, G. M., Baruch, D. E., Riedner, B. A., and Helmstetter, F. J. (2006a). Long-term stability of fear memory depends on the synthesis of protein but not mRNA in the amygdala. Eur. J. Neurosci. 23, 1853–1859. doi: 10.1111/j.1460-9568.2006.04723.x
Parsons, R. G., Gafford, G. M., and Helmstetter, F. J. (2006b). Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J. Neurosci. 26, 12977–12983. doi: 10.1523/JNEUROSCI.4209-06.2006
Pastalkova, E., Serrano, P., Pinkhasova, D., Wallace, E., Fenton, A. A., and Sacktor, T. C. (2006). Storage of spatial information by the maintenance mechanism of LTP. Science 313, 1141–1144. doi: 10.1126/science.1128657
Pavlopoulos, E., Trifilieff, P., Chevaleyre, V., Fioriti, L., Zairis, S., Pagano, A., et al. (2011). Neuralized1 activates CPEB3: a function for nonproteolytic ubiquitin in synaptic plasticity and memory storage. Cell 147, 1369–1383. doi: 10.1016/j.cell.2011.09.056
Phillips, R. G., and Ledoux, J. E. (1992). Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav. Neurosci. 106, 274–285. doi: 10.1037/0735-7044.106.2.274
Pick, J. E., Malumbres, M., and Klann, E. (2013a). The E3 ligase APC/C-Cdh1 is required for associative fear memory and long-term potentiation in the amygdala of adult mice. Learn. Mem. 20, 11–20. doi: 10.1101/lm.027383.112
Pick, J. E., Wang, L., Mayfield, J. E., and Klann, E. (2013b). Neuronal expression of the ubiquitin E3 ligase APC/C-Cdh1 during development is required for long-term potentiation, behavioral flexibility, and extinction. Neurobiol. Learn. Mem. 100, 25–31. doi: 10.1016/j.nlm.2012.11.005
Quevedo, J., Vianna, M. R., Martins, M. R., Barichello, T., Medina, J. H., Roesler, R., et al. (2004). Protein synthesis, PKA, and MAP kinase are differentially involved in short- and long-term memory in rats. Behav. Brain Res. 154, 339–343. doi: 10.1016/j.bbr.2004.03.001
Reis, D. S., Jarome, T. J., and Helmstetter, F. J. (2013). Memory formation for trace fear conditioning requires ubiquitin-proteasome mediated protein degradation in the prefrontal cortex. Front. Behav. Neurosci. 7:150. doi: 10.3389/fnbeh.2013.00150
Ren, Z. Y., Liu, M. M., Xue, Y. X., Ding, Z. B., Xue, L. F., Zhai, S. D., et al. (2013). A critical role for protein degradation in the nucleus accumbens core in cocaine reward memory. Neuropsychopharmacology 38, 778–790. doi: 10.1038/npp.2012.243
Rieser, E., Cordier, S. M., and Walczak, H. (2013). Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem. Sci. 38, 94–102. doi: 10.1016/j.tibs.2012.11.007
Rodrigues, S. M., Farb, C. R., Bauer, E. P., Ledoux, J. E., and Schafe, G. E. (2004). Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J. Neurosci. 24, 3281–3288. doi: 10.1523/JNEUROSCI.5303-03.2004
Rodrigues, S. M., Schafe, G. E., and Ledoux, J. E. (2001). Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J. Neurosci. 21, 6889–6896.
Rodriguez-Ortiz, C. J., Balderas, I., Garcia-DeLaTorre, P., and Bermudez-Rattoni, F. (2012). Taste aversion memory reconsolidation is independent of its retrieval. Neurobiol. Learn. Mem. 98, 215–219. doi: 10.1016/j.nlm.2012.08.002
Rodriguez-Ortiz, C. J., Balderas, I., Saucedo-Alquicira, F., Cruz-Castaneda, P., and Bermudez-Rattoni, F. (2011). Long-term aversive taste memory requires insular and amygdala protein degradation. Neurobiol. Learn. Mem. 95, 311–315. doi: 10.1016/j.nlm.2010.12.010
Rosen, J. B., Hitchcock, J. M., Sananes, C. B., Miserendino, M. J., and Davis, M. (1991). A direct projection from the central nucleus of the amygdala to the acoustic startle pathway: anterograde and retrograde tracing studies. Behav. Neurosci. 105, 817–825. doi: 10.1037/0735-7044.105.6.817
Rossato, J. I., Bevilaqua, L. R., Myskiw, J. C., Medina, J. H., Izquierdo, I., and Cammarota, M. (2007). On the role of hippocampal protein synthesis in the consolidation and reconsolidation of object recognition memory. Learn. Mem. 14, 36–46. doi: 10.1101/lm.422607
Sacchetti, B., Baldi, E., Tassoni, G., and Bielavska, E. (2001). CAMKII inhibition in the parabrachial nuclei elicits conditioned taste aversion in rats. Neurobiol. Learn. Mem. 75, 253–261. doi: 10.1006/nlme.2000.3978
Sacktor, T. C. (2012). Memory maintenance by PKMζ–an evolutionary perspective. Mol. Brain 5, 31. doi: 10.1186/1756-6606-5-31
Schafe, G. E., Atkins, C. M., Swank, M. W., Bauer, E. P., Sweatt, J. D., and Ledoux, J. E. (2000). Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J. Neurosci. 20, 8177–8187.
Schafe, G. E., and Ledoux, J. E. (2000). Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J. Neurosci. 20, RC96.
Shema, R., Sacktor, T. C., and Dudai, Y. (2007). Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science 317, 951–953. doi: 10.1126/science.1144334
Taubenfeld, S. M., Milekic, M. H., Monti, B., and Alberini, C. M. (2001). The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat. Neurosci. 4, 813–818. doi: 10.1038/90520
Tronson, N. C., Wiseman, S. L., Olausson, P., and Taylor, J. R. (2006). Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat. Neurosci. 9, 167–169. doi: 10.1038/nn1628
Upadhya, S. C., Ding, L., Smith, T. K., and Hegde, A. N. (2006). Differential regulation of proteasome activity in the nucleus and the synaptic terminals. Neurochem. Int. 48, 296–305. doi: 10.1016/j.neuint.2005.11.003
Vogt-Eisele, A., Kruger, C., Duning, K., Weber, D., Spoelgen, R., Pitzer, C., et al. (2014). KIBRA (Kidney/BRAin protein) regulates learning and memory and stabilizes Protein kinase Mζ. J. Neurochem. 128, 686–700. doi: 10.1111/jnc.12480
Volk, L. J., Bachman, J. L., Johnson, R., Yu, Y., and Huganir, R. L. (2013). PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 493, 420–423. doi: 10.1038/nature11802
Von Hertzen, L. S., and Giese, K. P. (2005). Alpha-isoform of Ca2+/calmodulin-dependent kinase II autophosphorylation is required for memory consolidation-specific transcription. Neuroreport 16, 1411–1414. doi: 10.1097/01.wnr.0000175244.51084.bb
Walker, D. L., and Davis, M. (2000). Involvement of NMDA receptors within the amygdala in short- versus long-term memory for fear conditioning as assessed with fear-potentiated startle. Behav. Neurosci. 114, 1019–1033. doi: 10.1037/0735-7044.114.6.1019
Walters, B. J., Campbell, S. L., Chen, P. C., Taylor, A. P., Schroeder, D. G., Dobrunz, L. E., et al. (2008). Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol. Cell. Neurosci. 39, 539–548. doi: 10.1016/j.mcn.2008.07.028
Warburton, E. C., Barker, G. R., and Brown, M. W. (2013). Investigations into the involvement of NMDA mechanisms in recognition memory. Neuropharmacology 74, 41–47. doi: 10.1016/j.neuropharm.2013.04.013
Wayman, G. A., Lee, Y. S., Tokumitsu, H., Silva, A. J., and Soderling, T. R. (2008). Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron 59, 914–931. doi: 10.1016/j.neuron.2008.08.021
Wilson, S. M., Bhattacharyya, B., Rachel, R. A., Coppola, V., Tessarollo, L., Householder, D. B., et al. (2002). Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat. Genet. 32, 420–425. doi: 10.1038/ng1006
Yasoshima, Y., Morimoto, T., and Yamamoto, T. (2000). Different disruptive effects on the acquisition and expression of conditioned taste aversion by blockades of amygdalar ionotropic and metabotropic glutamatergic receptor subtypes in rats. Brain Res. 869, 15–24. doi: 10.1016/S0006-8993(00)02397-0
Yeh, S. H., Mao, S. C., Lin, H. C., and Gean, P. W. (2006). Synaptic expression of glutamate receptor after encoding of fear memory in the rat amygdala. Mol. Pharmacol. 69, 299–308. doi: 10.1124/mol.105.017194
Keywords: ubiquitin, proteasome, fear conditioning, protein degradation, protein synthesis, amygdala, hippocampus
Citation: Jarome TJ and Helmstetter FJ (2014) Protein degradation and protein synthesis in long-term memory formation. Front. Mol. Neurosci. 7:61. doi: 10.3389/fnmol.2014.00061
Received: 30 April 2014; Paper pending published: 16 May 2014;
Accepted: 09 June 2014; Published online: 26 June 2014.
Edited by:
Ashok Hegde, Wake Forest School of Medicine, USAReviewed by:
Jorge Medina, Universidad de Buenos Aires, ArgentinaDiasynou Fioravante, University of California, Davis, USA
Copyright © 2014 Jarome and Helmstetter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fred J. Helmstetter, Department of Psychology, University of Wisconsin-Milwaukee, Garland Hall, PO Box 413, Milwaukee, WI 53201, USA e-mail: fjh@uwm.edu