Ubiquitin-dependent proteolysis in yeast cells expressing neurotoxic proteins
 Ralf J. Braun
Ralf J. Braun- Institut für Zellbiologie, Universität Bayreuth, Bayreuth, Germany
Critically impaired protein degradation is discussed to contribute to neurodegenerative disorders, including Parkinson's, Huntington's, Alzheimer's, and motor neuron diseases. Misfolded, aggregated, or surplus proteins are efficiently degraded via distinct protein degradation pathways, including the ubiquitin-proteasome system, autophagy, and vesicular trafficking. These pathways are regulated by covalent modification of target proteins with the small protein ubiquitin and are evolutionary highly conserved from humans to yeast. The yeast Saccharomyces cerevisiae is an established model for deciphering mechanisms of protein degradation, and for the elucidation of pathways underlying programmed cell death. The expression of human neurotoxic proteins triggers cell death in yeast, with neurotoxic protein-specific differences. Therefore, yeast cell death models are suitable for analyzing the role of protein degradation pathways in modulating cell death upon expression of disease-causing proteins. This review summarizes which protein degradation pathways are affected in these yeast models, and how they are involved in the execution of cell death. I will discuss to which extent this mimics the situation in other neurotoxic models, and how this may contribute to a better understanding of human disorders.
Introduction
Ubiquitin is a highly conserved protein with 76 amino acids (Weissman et al., 2011; Finley et al., 2012). It is covalently linked to lysine side chains of substrate proteins by the sequential action of the ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzymes (E2), and substrate-specific ubiquitin ligases (E3) (Weissman et al., 2011; Finley et al., 2012). Deubiquitylating enzymes (DUBs) recycle ubiquitin, replenishing the cellular pool of free ubiquitin. The high variety of ubiquitin modifications, including mono- and polyubiquitylations, destines the degradation, localization, and/or function of substrate proteins. Consequently, numerous cellular processes are regulated by ubiquitylation, including protein degradation, cell death control, and vesicular trafficking (Weissman et al., 2011; Finley et al., 2012). These ubiquitin-dependent processes are highly conserved from yeast to humans (Finley et al., 2012).
Polyubiquitylated proteins are degraded within the proteasome, a cylindrical multiprotein complex with chymotrypsin-, trypsin-, and caspase-like proteolytic activities. This ubiquitin-dependent degradation of proteins via the proteasome is called the ubiquitin-proteasome system (UPS) (Weissman et al., 2011; Finley et al., 2012). The ubiquitylation of many plasma membrane proteins promotes their targeting into endosomes and multivesicular bodies (MVB) leading to their degradation by multiple proteases in the lysosomes (or vacuoles in yeast) (Finley et al., 2012; MacGurn et al., 2012). This MVB pathway of protein degradation (also called endosomal-lysosomal pathway) is a ubiquitin-controlled vesicle-based protein degradation pathway, which is independent from proteasomes. Ubiquitylation can also be involved in the degradation of substrate proteins via autophagy (Kuang et al., 2013; Lu et al., 2014). Autophagy is a cellular process where proteins, protein aggregates, or organelles are enclosed by a double membrane forming autophagosomes, which eventually fuse with lysosomes (vacuoles) for degradation. One mechanism to ensure specificity during autophagy relies on the ubiquitylation of target proteins or organelles and the consequent use of specific adaptors that connect the ubiquitin system with the autophagy pathway (Kuang et al., 2013; Lu et al., 2014). The UPS, the MVB pathway, and autophagy share some components, such as the AAA-ATPase p97/VCP (or Cdc48 in yeast) (Bug and Meyer, 2012; Dargemont and Ossareh-Nazari, 2012), and the E3 ligase Nedd4 (or Rsp5 in yeast) (MacGurn et al., 2012; Fang et al., 2014; Lu et al., 2014), making these proteins to potential key players that decide by which pathway a protein is to be degraded.
Accumulation of aggregated proteins is a common hallmark of many neurodegenerative disorders, and believed to contribute to neuronal dysfunction (Lansbury and Lashuel, 2006). In Parkinson's disease (PD) cytoplasmic Lewy bodies are protein aggregates mainly comprised by the protein α-synuclein (Uversky, 2007), and nuclear protein aggregates of the polyglutamine protein hungtingtin are typical for Huntington's disease (HD) (Ross and Tabrizi, 2011). In Alzheimer's disease (AD) the hydrophobic peptide β-amyloid is produced in cells but accumulates in extracellular plaques (Laferla et al., 2007). The microtubule-associated protein tau (MAPT), and UBB+1, the frameshift variant of human ubiquitin B, is enriched in intracellular inclusions during AD (van Leeuwen et al., 1998; Mandelkow and Mandelkow, 2012). Accumulation of cytoplasmic aggregates of disease-causing proteins, such as TDP-43 or FUS/TLS, occurs during the motor neuron disease amyotrophic lateral sclerosis (ALS) (Andersen and Al-Chalabi, 2011). Most of these disease-associated proteins or aggregates are ubiquitylated. Therefore, the observed accumulation of protein aggregates has been explained by dysfunctional protein degradation pathways, including the UPS and autophagy (Dennissen et al., 2012; Dantuma and Bott, 2014). However, the precise role of ubiquitin-dependent proteolysis and its importance for the progression of the human disorders remains poorly understood.
Yeast is an established model for measuring cytotoxicity and programmed cell death, and for dissecting conserved mechanisms of apoptosis and necrosis (Carmona-Gutierrez et al., 2010). Diverse roles of ubiquitin-dependent protein degradation have been described in distinct yeast cell death scenarios. The ubiquitin-dependent and proteasome-independent routing of misfolded proteins to the MVB pathway for their degradation protects cells from cytotoxicity (Wang et al., 2011). Elevated proteasome capacity extends the replicative life span and fitness of yeast cells, which are more resistant against proteotoxic stress (Kruegel et al., 2011). Decreased proteasome capacity by proteasome inhibition leads to disturbances in the amino acid homeostasis, thereby executing cell death (Suraweera et al., 2012). Impairment of distinct branches of the UPS pathway, including the ER- and the mitochondrion-associated degradation (ERAD/MAD) are sufficient to trigger cell death in yeast, emphasizing their cytoprotective role in the homeostasis of the ER and mitochondria (Braun et al., 2006; Zischka et al., 2006; Heo et al., 2010). However, UPS impairment (due to proteasome inhibition) can also prevent from cell death, e.g., when cell death is triggered by acetic acid and the chemotherapeutic drug cisplatin, respectively (Valenti et al., 2008; Cunha et al., 2013). Thus, ubiquitin-dependent proteolysis is involved in both the execution of and the prevention from yeast cell death.
In recent years, many yeast models have been established to analyze the influence of human neurotoxic protein expression on yeast cell survival, including models for PD, HD, AD, and ALS (Gitler, 2008; Miller-Fleming et al., 2008; Winderickx et al., 2008; Bharadwaj et al., 2010; Braun et al., 2010; Khurana and Lindquist, 2010; Bastow et al., 2011; Mason and Giorgini, 2011). Here, I summarize how ubiquitin-dependent protein degradation is impaired in yeast cell death models expressing neurotoxic proteins, and which role proteolysis plays in the execution of cell death. Further, I will discuss some similarities between the yeast models expressing neurotoxic proteins, and the animal and cell culture disease models.
Yeast Models Expressing Neurotoxic Proteins
Parkinson's Disease (PD)
PD is the most prevalent age-related movement disorder characterized by a progressive loss of dopaminergic neurons in the substantia nigra, leading to the impairment of normal motor function culminating in resting tremor, bradykinesia and rigidity (Lees et al., 2009). In most familiar and sporadic cases, PD is associated with Lewy bodies, i.e., intracellular cytoplasmic aggregates composed of the protein α-synuclein (Uversky, 2007). Missense mutations in the SNCA gene, resulting in the expression of α-synuclein variants (A18T, A29S, A30P, A53T, E46K, H50Q, G51D), as well as duplication and triplication of SNCA, leading to elevated α-synuclein levels, are causative for PD in some familiar forms of the disorder (Fujioka et al., 2014). Numerous α-synuclein disease models expressing wild-type and disease-associated variants have been established, including several yeast models.
In Yeast, α-Synuclein is a Membrane-Associated Protein, which is Degraded Via the UPS, Autophagy, and Potentially the MVB Pathway
When expressed in yeast, α-synuclein binds to vesicles of the secretory pathway, leading to its localization to the plasma membrane (Outeiro and Lindquist, 2003; Dixon et al., 2005; Sharma et al., 2006; Zabrocki et al., 2008). This depends on α-synuclein phosphorylation (Basso et al., 2013; Tenreiro et al., 2014), and can be interrupted by genetic manipulation (e.g., α-synuclein-A30P) (Outeiro and Lindquist, 2003; Dixon et al., 2005; Sharma et al., 2006). Upon high expression levels α-synuclein forms cellular aggregates in a nucleation-dependent manner, which starts at the plasma membrane and eventually leads to cytoplasmic inclusions (Outeiro and Lindquist, 2003). These inclusions co-localize with markers of different vesicles, including Ypt1 (ER-to-Golgi), Ypt31 (late Golgi), Sec4 (secretory vesicles-to-plasma membrane), Ypt6 (endosome-to-Golgi), Vps21 and Ypt52 (early-to-late endosome) and Ypt7 (late endosome-to-vacuole) (Gitler et al., 2008). Thus, α-synuclein is an aggregation-prone membrane-associated protein.
Although α-synuclein is ubiquitylated in yeast (Outeiro and Lindquist, 2003), to which extent the UPS contributes to its degradation in human cells (Xilouri et al., 2013) or more specifically in yeast (see below), remains an open debate. Treatment of yeast cells expressing α-synuclein with the proteasome inhibitor lactacystin resulted in increased α-synuclein aggregation (Zabrocki et al., 2005; Lee et al., 2008). Consistently, expression of α-synuclein in the yeast strain sen3-1, which harbors a mutation in the gene encoding the regulatory proteasome subunit Rpn2, led to increased steady-state levels of α-synuclein (using untagged α-synuclein) and to increased formation of aggregates (using GFP-tagged α-synuclein) (Sharma et al., 2006). These data suggest that α-synuclein is a UPS substrate in yeast.
In other studies with yeast cells expressing GFP-tagged α-synuclein, α-synuclein aggregate clearance was neither affected by treatment with the proteasome inhibitor MG132 (Petroi et al., 2012; Tenreiro et al., 2014), nor by mutation in the gene encoding the regulatory proteasome subunit Rpt6 (cim3-1) (Petroi et al., 2012). Further, the steady-state levels of α-synuclein were not affected by MG132 treatment (Tenreiro et al., 2014). These data argue against the contribution of the UPS in α-synuclein degradation in yeast. Here, α-synuclein aggregate clearance was dependent on autophagy and vacuolar protease activity. Treatment of yeast cells expressing α-synuclein-GFP with the protease inhibitor phenylmethylsulfonyl fluoride (PMSF), an inhibitor of vacuolar proteases, resulted in a significant reduction of this clearance (Petroi et al., 2012). Similarly, genetic interruption of autophagy (Δatg1 or Δatg7) delayed aggregate clearance (Petroi et al., 2012; Tenreiro et al., 2014), and increased the steady-state levels of α-synuclein (Δatg7) (Tenreiro et al., 2014). Consistently, inducing autophagy by rapamycin promoted aggregate removal (Zabrocki et al., 2005), confirming that α-synuclein aggregates are degraded via autophagy.
Aggregate clearance was not limited to autophagy, because aggregate clearance still took place in the absence of autophagy and upon very low UPS activity (cim3-1 Δatg1 strain), suggesting for an additional cellular clearing mechanism independent from autophagy and the UPS (Petroi et al., 2012). Indeed, the yeast E3 ligase Rsp5, and its homolog Nedd4 in mammalian cells, play critical roles in α-synuclein degradation (Tofaris et al., 2011). In yeast, α-synuclein was identified as an Rsp5 target for ubiquitylation, and upon RSP5 mutation (rsp5-1 strain), both the steady-state level of α-synuclein, as well as the number of cells showing α-synuclein aggregation were increased as compared to wild-type strain (Tofaris et al., 2011). Although Rsp5/Nedd4 is critically involved in many cellular processes, including the MVB pathway, ubiquitin-dependent autophagy and the proteasome-dependent degradation of misfolded proteins (MacGurn et al., 2012; Fang et al., 2014; Lu et al., 2014), there are some line of evidence suggesting that the MVB pathway contributes to α-synuclein degradation (Tofaris et al., 2011). Mammalian Nedd4 promotes the degradation of endogenous α-synuclein by lysosomes, and the targeting of α-synuclein to the lysosomes depends on the endosomal sorting complex (ESCRT) (Tofaris et al., 2011). Due to the high conservation of protein degradation pathways between mammalian cells and yeast, the critical contribution of Rsp5 in α-synuclein degradation in yeast might suggest for a potential role of the MVB pathway in α-synuclein degradation, besides the UPS and autophagy.
α-Synuclein Expression in Yeast Leads to Impairment of the UPS, and Vesicular Trafficking
Expression of α-synuclein in yeast leads to UPS impairment. The protein composition of the proteasome is altered, concomitant to a moderate decrease in the chymotrypsin-like enzymatic proteasomal activity (in isolated proteasomes), and to a marked delay in the degradation of short-lived proteins (pulse-chase assay) (Chen et al., 2005). Consequently, the cellular levels of polyubiquitylated proteins increased moderately (Chen et al., 2005). The degradation of the UPS model substrate GFPu, in which the C-terminus of this protein comprises a degron, was delayed upon α-synuclein expression in yeast (Outeiro and Lindquist, 2003). The impairment of UPS-dependent protein degradation upon α-synuclein expression appears to be substrate specific. Whereas, the degradation of the cytosolic proteasome substrate Deg1-β-Gal was unaffected, the degradation of the ER luminal substrate CPY* but not of the ER membrane substrate sec61-2 was severely impaired (Cooper et al., 2006). Thus, α-synuclein expression impairs the UPS and more specifically the degradation of selective substrates of the ER-associated degradation (ERAD) pathway.
α-Synuclein expression also affects vesicular trafficking, including ER to Golgi transport, endocytosis, vesicular recycling back to the plasma membrane, and vacuolar fusion (Cooper et al., 2006; Gitler et al., 2008; Zabrocki et al., 2008; Basso et al., 2013). Since the MVB pathway and autophagy depend both on vesicular fusion processes, it is very likely that α-synuclein expression also affects the protein degradation via these two vesicle-based pathways. Measuring the degradation rates of substrates of autophagy or the MVB pathway upon α-synuclein expression will help to address this issue.
α-Synuclein Expression in Yeast Triggers Cell Death, which is Modulated by the Activities of the UPS, Autophagy, and Ubiquitin-Dependent Vesicular Trafficking
Yeast cells overexpressing wild-type and disease-associated α-synuclein demonstrated growth deficits and age-dependent loss of clonogenic cell survival paralleled by the emergence of morphological markers of apoptosis and necrosis (Willingham et al., 2003; Flower et al., 2005; Witt and Flower, 2006; Büttner et al., 2008, 2013a,b; Lee et al., 2008; Su et al., 2010). The cellular accumulation of ROS and mitochondrial dysfunction are pivotal for the execution of α-synuclein-triggered cell death. The use of the antioxidant N-acetyl cysteine (NAC) or the use of yeast strains deleted for mitochondrial DNA (ρ0 strain) protected from ROS and α-synuclein-triggered cell death (Büttner et al., 2008, 2013a). The translocation of the mitochondrial cell death proteins Nuc1 and cytochrome c, into the nucleus and the cytosol, respectively, was observed to be critical for the execution of cell death (Flower et al., 2005; Büttner et al., 2013b). The ER also contributes to cytotoxicity, because α-synuclein expression results in ER stress and in the induction of the unfolded protein response (UPR) (Cooper et al., 2006).
Since α-synuclein has been proposed to be a UPS substrate and since α-synuclein expression resulted in UPS impairment (especially ERAD), it is likely that the UPS is involved in modulating α-synuclein-triggered cytotoxicity. Moderate α-synuclein expression, which is non-toxic for wild-type yeast cells, resulted in severe growth deficits in yeast cells bearing mutations in the 20S proteasomal barrel (pre1-1001, pre2-1001, doa3-1) (Dixon et al., 2005; Sharma et al., 2006) and in the 19S regulatory particle of the 26S proteasome (sen3-1) (Sharma et al., 2006), or treated with the proteasome inhibitor lactacystin (Lee et al., 2008). Consistently, expression of Rpt5, a component of the 19S regulatory particle, and expression of the ERAD ubiquitin ligase Hrd1 suppressed α-synuclein-triggered cytotoxicity (Liang et al., 2008; Gitler et al., 2009). Bridging high-throughput genetic and transcriptional data with the ResponseNet algorithm predicted the AAA-ATPase Cdc48, also critically involved in ERAD, to be a modulator of α-synuclein-triggered cytotoxicity (Yeger-Lotem et al., 2009). Thus, the UPS in general, and specifically the ERAD pathway appears to be a potent modulator of α-synuclein-triggered cell death.
Since α-synuclein aggregates have been proposed to be substrates of autophagy, it is likely that autophagy, like the UPS, modulates α-synuclein-triggered cytotoxicity. In fact, the in silico combination of high-throughput genetic and transcriptional data predicted the target of rapamycin (TOR) pathway, as a modulator of α-synuclein-triggered cytotoxicity (Yeger-Lotem et al., 2009). Addition of the TOR-inhibitor rapamycin markedly enhanced the growth deficits elicited by α-synuclein (Yeger-Lotem et al., 2009). Since inactivation of the TOR pathway induces autophagy, these data suggested, that enhancing autophagy is harmful but not cytoprotective for cultures expressing α-synuclein. Consistently, pharmacological inhibition of autophagy by treatment with chloroquine markedly extended chronological life span of yeast cells expressing α-synuclein (Sampaio-Marques et al., 2012). Although rapamycin also affects other cellular pathways, it remains possible that autophagy, in contrast to the UPS, plays a detrimental role in α-synuclein-triggered cytotoxicity.
Besides the UPS and autophagy, ubiquitin-dependent vesicle trafficking plays a role in modulating α-synuclein-triggered cytotoxicity. The E3 ligase Rsp5, involved in ubiquitin-dependent vesicle trafficking, was predicted by the ResponseNet algorithm to affect α-synuclein-triggered cytotoxicity (Yeger-Lotem et al., 2009). Indeed, loss-of-function mutations in the gene encoding Rsp5 (rsp5-1 strain) increased α-synuclein-triggered growth deficits, whereas overexpression of Rsp5 was cytoprotective (Tofaris et al., 2011). Chemical genetic screens in wild-type yeast cells established that N-aryl benzimidazole (NAB) promoted endosomal transport and protected cells from cytotoxicity (Tardiff et al., 2013). This was dependent on the deubiquitinase Doa4, the E3 ubiquitin ligase Rsp5, the Rsp5 adaptor Bul1, the DUBs Ubp7, and Ubp11, which can deubiquitylate Rsp5 substrates, the potential Rsp5 substrates (Bap2, Bap3, and Mmp1), and Vps23, which directs Rsp5 substrates for degradation in the vacuole (Tardiff et al., 2013). Notably, promoting ER-Golgi vesicle trafficking had very similar effects: α-synuclein-triggered cytotoxicity was reduced, and this reduction also depended on ubiquitin proteases, namely Ubp3 and its co-factor Bre5 (Cooper et al., 2006; Gitler et al., 2008). Thus, promoting ubiquitin-regulated vesicle trafficking prevents α-synuclein-triggered cytotoxicity.
Yeast α-Synuclein Models are Highly Useful to Elucidate the Role of Diverse Ubiquitin-Related Protein Degradation Pathways in Modulating Cytotoxicity and Neuronal Cell Death
In yeast, α-synuclein is both a substrate and an inhibitor for the UPS, autophagy, and the ubiquitin-dependent vesicular trafficking (Figure 1, Table 1). The activities of the UPS and of MVB pathways appear to play protective roles, whereas increased autophagy potentially contribute to α-synuclein-triggered cytotoxicity. The relevance of the different pathways in modulating α-synuclein-triggered cytotoxicity might depend on α-synuclein itself, e.g., the expression levels, the post-translational modifications, the cellular localizations, folding or distinct aggregation conditions. Similarly, the chronological and replicative aging of the yeast cultures might be decisive. Systematic analyses of these factors will help to get a better understanding of the pathophysiological effects of α-synuclein expression in yeast with respect to the reciprocal effects to ubiquitin-dependent protein degradation.
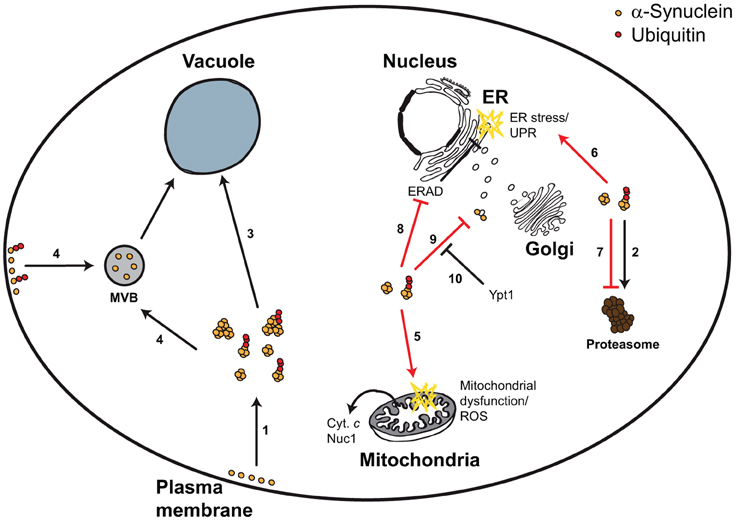
Figure 1. Yeast model for α-synuclein-triggered cytotoxicity. α-Synuclein is a plasma membrane- and vesicle-bound protein that upon high expression levels or upon mutation forms smaller and larger aggregates, which can be ubiquitylated (1). α-Synuclein can be degraded via the UPS (2), autophagy (3), and potentially via the MVB pathway (4). Aggregated α-synuclein triggers mitochondrial dysfunction, ROS, and mitochondrion-dependent cell death (5), as well as ER stress and the UPR (6). These cytotoxic effects can at least partially be explained by α-synuclein-dependent inhibition of the proteasome (7), the ERAD pathway (8), or vesicular trafficking (9). Impaired vesicular trafficking includes (but is not limited to) ER-to-Golgi transport, which can be efficiently restored by Ypt1 expression (10).
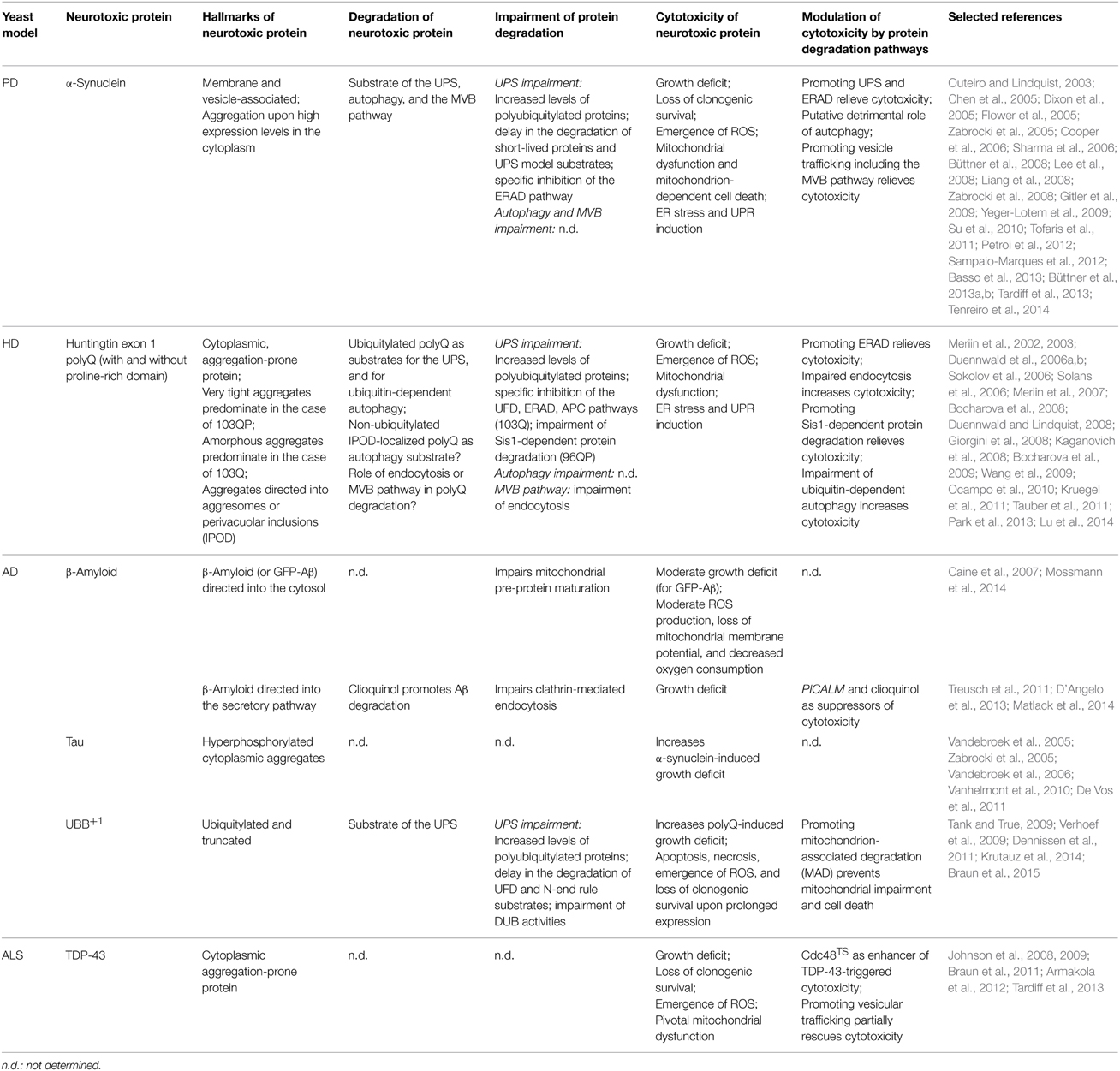
Table 1. Ubiquitin-dependent protein degradation in yeast models expressing neurotoxic proteins.
These studies are very promising, due to the high consistence of the yeast α-synuclein models with other α-synuclein model systems. In mammalian cells, α-synuclein degradation also depends on the UPS, autophagy, and ubiquitin-dependent vesicular trafficking (more specifically the MVB pathway) (Tofaris et al., 2011; Ebrahimi-Fakhari et al., 2012; Xilouri et al., 2013). Likewise, in mammalian cells, α-synuclein interferes with these ubiquitin-modulated protein degradation pathways, and the activities of these pathways are discussed to influence the cytotoxicity of α-synuclein (Ebrahimi-Fakhari et al., 2012; Xilouri et al., 2013). Yeast α-synuclein models have already been very valuable in deciphering novel cellular mechanisms linking ubiquitin-dependent pathways with α-synuclein-triggered cytotoxicity. For instance, the influence of the ubiquitin-modulated ER-Golgi vesicle trafficking on α-synuclein-triggered cytotoxicity was first identified in yeast and then confirmed in flies and worms (Cooper et al., 2006). Likewise, the degradation of α-synuclein via the MVB pathway is conserved from yeast to mammalians (Tofaris et al., 2011), and the protective effect of promoting ubiquitin-dependent endosomal trafficking by NAB was first identified in yeast and later on confirmed in worms, rats, and human PD patient-derived neurons (Chung et al., 2013; Tardiff et al., 2013).
Huntington's Disease (HD)
HD is an autosomal dominant neurodegenerative disorder characterized by a progressive loss of neurons in the striatum and the cortex with a consequent decline of cognitive and motor functions (Ross and Tabrizi, 2011). HD is caused by an abnormal polyglutamine (polyQ) expansion in the protein huntingtin due to an aberrant CAG codon expansion in the exon 1 of the gene encoding huntingtin (Ross and Tabrizi, 2011). This results in an aggregation-prone protein eventually triggering cytotoxicity and neuronal cell loss (Ross and Tabrizi, 2011). Increasing the length of the polyQ expansion accelerates aggregation of huntingtin and strictly correlates with the increase in cytotoxicity and the decrease in disease onset (Ross and Tabrizi, 2011). In order to dissect underlying mechanisms, various HD models have been established, comprising transgenic mouse lines, mammalian cell culture, and yeast (Mason and Giorgini, 2011; Ross and Tabrizi, 2011).
In Yeast, Huntingtin with Disease-Associated Expanded Glutamine Stretches (PolyQ) is a Cytoplasmic Aggregation-Prone Protein, which is Degraded Via the UPS and Autophagy
In yeast, expression of fluorescence protein-tagged huntingtin exon 1 with disease-inducing polyQ expansions (e.g., 103Q) led to very efficient cytoplasmic aggregation, in contrast to fusion proteins with normal glutamine repeats (e.g., 25Q) (Meriin et al., 2002, 2003; Duennwald et al., 2006a,b). Intermediate polyQ length (e.g., 47Q) showed moderate aggregation when expressed in logarithmically growing cells but here strong aggregation occurred delayed upon chronological aging (Cohen et al., 2012). Besides the length of polyQ expansions, aggregation was influenced by the amino acid sequences flanking the polyQ stretch (Duennwald et al., 2006b; Wang et al., 2009), and by the presence of endogenous proteins with prion properties and glutamine repeats (Duennwald et al., 2006a). The N-terminal domain of huntingtin (N17 fragment) which precedes the polyQ stretch is required for recruiting the chaperonin TRiC and the 14-3-3 protein Bmh1 which both promote aggregation in yeast and cell-free systems (Tam et al., 2009; Wang et al., 2009; Duennwald, 2011; Crick et al., 2013). PolyQ followed by the endogenous proline-rich region of huntingtin (103QP) tended to form very tight aggregates (one or two per cell), whereas polyQ lacking the proline-rich region (103Q) preferred to form amorphous aggregates dispersed throughout the cytosol (Duennwald et al., 2006b). The yeast protein Rnq1 co-localized with both 103QP and 103Q aggregates, and its prion conformation [RNQ+] was found to be necessary for aggregation of 103QP and 103Q (Duennwald et al., 2006a).
In line with the appearance of different types of polyQ aggregates, these aggregates are directed to at least two cellular compartments. First, misfolded polyQ (103QP) was actively transported to aggresomes (Wang et al., 2009), which are large juxtanuclear aggregates that co-localize with the centrosomes in mammalian cells and the spindle pole bodies in yeast (Johnston et al., 1998; Wang et al., 2009). In mammalian cells, aggresomes are ubiquitylated, highly dynamic and easily accessible to the UPS (Johnston et al., 1998). They are formed from smaller aggregates, which are actively transported to the centrosome via the microtubule cytoskeleton (Johnston et al., 1998). Both the N17 fragment and the proline-rich domain of huntingtin are required for aggresome formation in yeast (Wang et al., 2009). Alternatively, polyQ aggregates (it is unclear whether 103Q or 103QP has been used) could be transported to perivacuolar inclusions, called “insoluble protein deposits” (IPOD) (Kaganovich et al., 2008). IPODs are non-ubiquitylated insoluble protein aggregates covered by the ubiquitin-like autophagy protein Atg8 (Kaganovich et al., 2008). Therefore, it is tempting to speculate that polyQ aggregates in perivacuolar inclusions are substrates of autophagy, whereas polyQ aggregates in aggresomes are primarily substrates of the UPS.
PolyQ aggregates could be ubiquitylated (96QP) (Lu et al., 2014), and physically interacted with proteins of the UPS, including proteasomal subunits (103QP, 96QP) (Wang et al., 2009; Park et al., 2013). Consistently, polyQ aggregate clearance (103Q) was improved in a yeast strain with elevated UPS capacities (Δubr2), confirming that polyQ aggregates are substrates of the UPS (Kruegel et al., 2011). However, the degradation of ubiquitylated polyQ was not limited to the UPS but could also occur by ubiquitin-dependent autophagy (Lu et al., 2014). Here, polyQ (96QP) was ubiquitylated by the E3 ligase Rsp5, and ubiquitylated polyQ was recognized by the ubiquitin-Atg8 adaptor protein Cue5, enabling the targeting of polyQ aggregates (96QP) to the vacuole for their degradation (Lu et al., 2014). In sum, ubiquitylated polyQ aggregates are degraded via the UPS, and ubiquitin-dependent autophagy.
Yeast strains with mutations affecting the formation of endocytic vesicles demonstrated decreased aggregation of polyQ (47Q, 103Q, 103QP) (Meriin et al., 2007). In contrast, lack of proteins which are pivotal for later steps of endocytosis increased polyQ aggregation (103Q), and when present these proteins co-localized with polyQ aggregates (103Q) (Meriin et al., 2007). These data suggest that these ubiquitin-controlled vesicular processes are involved in the formation of polyQ aggregates (Meriin et al., 2007). However, whether they are also involved in the degradation of polyQ remains elusive.
PolyQ Expression Leads to Impairment of Specific Branches of the UPS Pathway in Yeast
PolyQ expression inhibits the UPS. In the presence of polyQ, increased cellular levels of polyubiquitylated proteins were observed (with 103Q but not with 103QP) (Duennwald and Lindquist, 2008), genes involved in ubiquitin cycle and protein ubiquitylation were up-regulated (with 103Q) (Giorgini et al., 2008; Tauber et al., 2011), and the degradation of cytosolic and ER-associated proteins were impaired (with 103Q but not with 103QP) (Duennwald and Lindquist, 2008). Notably, the polyQ-dependent UPS impairment is substrate specific. Upon polyQ expression (103Q), the degradation of cytosolic ubiquitin-fusion degradation (UFD) substrates (Ub-P-LacZ) was more affected than the degradation of cytosolic substrates of the N-end rule pathway (Ub-R-LacZ) (Duennwald and Lindquist, 2008). Consistently, the degradation of ER-associated proteins via ERAD, which shares many components with the UFD pathway, was drastically affected upon polyQ expression (103Q) (Duennwald and Lindquist, 2008). Here, the degradation of the ER luminal misfolded variant of the carboxypeptidase Y (CPY*) was impaired, as well as the degradation of the misfolded ER membrane protein sec61-2, and others (Duennwald and Lindquist, 2008). PolyQ (both 103Q and 103QP) physically interacted with the AAA-ATPase Cdc48 and its co-factors Ufd1 and Npl4, which are pivotally involved in UFD and ERAD (Duennwald and Lindquist, 2008; Wang et al., 2009). It has been proposed that sequestration of the Cdc48-Ufd1/Npl4 complex by polyQ (103Q) is the cause for the specific impairment of the UFD and ERAD pathways, culminating in ER stress and UPR induction (Duennwald and Lindquist, 2008). Notably, the interaction of polyQ (103QP) with the Cdc48-Ufd1/Npl4 complex was essential for the formation of the polyQ-containing aggresomes (Wang et al., 2009). The formation of these aggresomes is believed to be a protective cellular mechanism to prevent the evenly spread of misfolded proteins within a cell (Johnston et al., 1998). Therefore, the interaction of the Cdc48-Ufd1/Npl4 complex with polyQ might be both protective (at least for 103QP) and detrimental (for 103Q); it prevents from the accumulation of misfolded polyQ but promotes specific dysfunction of the UFD and ERAD pathways.
Expression of polyQ containing the proline-rich domain (97QP) can also lead to UPS dysfunction via an alternative mechanism, which is based on the sequestration of chaperones. PolyQ (97QP) inhibited the UPS-mediated degradation of the misfolded cytosolic variant of carboxypeptidase Y (ΔssCPY*) lacking the signaling sequence (ss) for entering the secretory pathway (Park et al., 2013). Upon polyQ expression (97QP), the steady-state levels and the turnover rates of ΔssCPY* were increased and delayed, respectively (Park et al., 2013). PolyQ-triggered (97QP) UPS inhibition was not due to direct interference with proteasomal function, because the ubiquitin-independent proteasomal degradation of ornithine decarboxylase was not affected by polyQ expression (Park et al., 2013). UPS dysfunction could also not be explained by interference of polyQ (97QP) with the ubiquitylation of ΔssCPY* prior its proteasomal degradation (Park et al., 2013). Instead, polyQ (97QP) interfered with the transfer of the misfolded protein to the proteasome, prior proteasomal degradation, which surprisingly took place in the nucleus (Park et al., 2013). The nuclear transfer of misfolded ΔssCPY* was done by binding to the type II Hsp40 chaperone Sis1, which shuttles into the nucleus, and polyQ (97QP) interfered with this process by sequestering Sis1 in the cytosol (Park et al., 2013). Consistently, Sis1 overexpression restored the degradation of ΔssCPY* in the presence of polyQ. Thus, polyQ (both 103Q and 97QP) interferes with the proteasome-dependent degradation of proteins in the nucleus and the cytosol, by sequestering the Sis1 chaperone (for 97QP) and the Cdc48-Ufd1/Npl4 complex (for 103Q), respectively.
Whether polyQ interferes with autophagy and protein degradation based on ubiquitin-controlled vesicular transport remains to be determined. Since polyQ aggregates (103Q) impaired endocytosis in yeast (Meriin et al., 2003, 2007), it is tempting to speculate that polyQ aggregates (103Q) also affect these vesicle-based protein degradation pathways.
PolyQ Expression in Yeast Triggers Mitochondrial Dysfunction and ER Stress, which is Modulated by Specific UPS Activities
PolyQ constructs encoding 103 glutamine residues (103Q) efficiently triggered growth deficits and morphological markers of apoptosis in yeast (Meriin et al., 2002; Duennwald et al., 2006a; Sokolov et al., 2006; Solans et al., 2006). In contrast, polyQ constructs encoding 25 glutamine residues (25Q) remained non-toxic, and the constructs with intermediate polyQ lengths showed intermediate cytotoxicity (Meriin et al., 2002; Duennwald et al., 2006a; Sokolov et al., 2006; Solans et al., 2006; Ocampo et al., 2010). Besides the number of glutamine expansions, the cytotoxicity of polyQ strongly depended on the proline-rich domain flanking the polyQ stretch (Duennwald et al., 2006b). PolyQ (103QP) remained non-toxic, whereas polyQ lacking the proline-rich region (103Q) induced cytotoxicity (growth deficits) (Duennwald et al., 2006b). In this context, the presence of the prion conformation of the yeast protein Rnq1 was a prerequisite for polyQ-triggered cytotoxicity (103Q), and the presence of other endogenous yeast proteins with glutamine repeats also influenced polyQ-triggered cytotoxicity (103Q) (Duennwald et al., 2006a). Thus, polyQ-triggered cytotoxicity depends on the length of the glutamine repeats, the absence or presence of the proline-rich domain, and the cellular protein interaction network.
Cytotoxic polyQ (103Q) physically interacted with mitochondria and triggers critical mitochondrial dysfunction (Solans et al., 2006; Ocampo et al., 2010). PolyQ expression (103Q but not 103QP) also induced ER stress leading to UPR (Duennwald and Lindquist, 2008), and has also been proposed to lead to lethal impairment of cell cycle progression (Bocharova et al., 2008, 2009). ER and cell cycle impairments are believed to be consequences of the impairment of specific branches of the UPS pathway by polyQ expression (103Q), including the ERAD and anaphase promoting complex (APC) pathways (Bocharova et al., 2008, 2009; Duennwald and Lindquist, 2008). The mitochondrion-associated protein degradation pathway (MAD) shares pivotal components with the ERAD pathway (Heo et al., 2010; Taylor and Rutter, 2011). Therefore, it is tempting to speculate that polyQ expression (103Q) also impairs MAD, contributing to the observed mitochondrial damage.
PolyQ-triggered cytotoxicity was increased by genetic impairment of the ERAD and UPR pathways and by application of ER stress (for 103Q but not for 103QP) (Duennwald and Lindquist, 2008). In contrast, promoting ERAD by expression of Npl4 and Ufd1, which are pivotally involved in ERAD, or constitutive activation of the UPR suppressed polyQ-triggered cytotoxicity (103Q) (Duennwald and Lindquist, 2008). These data are in line with the idea that ERAD dysfunction by polyQ expression (103Q) is critical in the execution of cytotoxicity and cell death. Deletion of the APC substrate ASE1 relieved polyQ-triggered cytotoxicity (103Q), suggesting that preventing the accumulation of Ase1 upon dysfunction of the APC pathway is beneficial (Bocharova et al., 2008). Expression of polyQ flanked by the proline-rich domain (96QP), which is non-toxic under normal conditions, became cytotoxic upon accumulation of the UPS model substrate ΔssCPY* and could be relieved upon Sis1 overexpression (Park et al., 2013). Genetic inactivation of ubiquitin-dependent autophagy (e.g., rsp5-2, Δcue5) also induced cytotoxicity upon polyQ expression (96QP) (Lu et al., 2014). These data suggest that besides ERAD and APC pathways, the Sis1-dependent protein degradation and the ubiquitin-dependent autophagy are pivotal in the modulation of polyQ-triggered cytotoxicity in yeast.
Yeast PolyQ Models are Highly Useful to Elucidate the Role of Diverse Ubiquitin-Related Protein Degradation Pathways in Modulating Cytotoxicity and Neuronal Cell Death
PolyQ expression in yeast leads to aggregation, and aggregates can be allocated to distinct cellular compartments, including aggresomes and perivacuolar inclusions (IPOD) (Figure 2, Table 1). Aggregated polyQ is a substrate of the UPS and autophagy. Besides its role of a substrate of protein degradation, polyQ impairs the UPS (especially ERAD), and also affects proper function of vesicular transport (more specifically endocytosis). This leads to ER stress, mitochondrial dysfunction, and cell death. All these features depend on the length of the polyQ stretch, the absence (or presence) of the N-terminal part (N17 fragment) and the proline-rich domain, respectively, and the cellular protein interaction network.
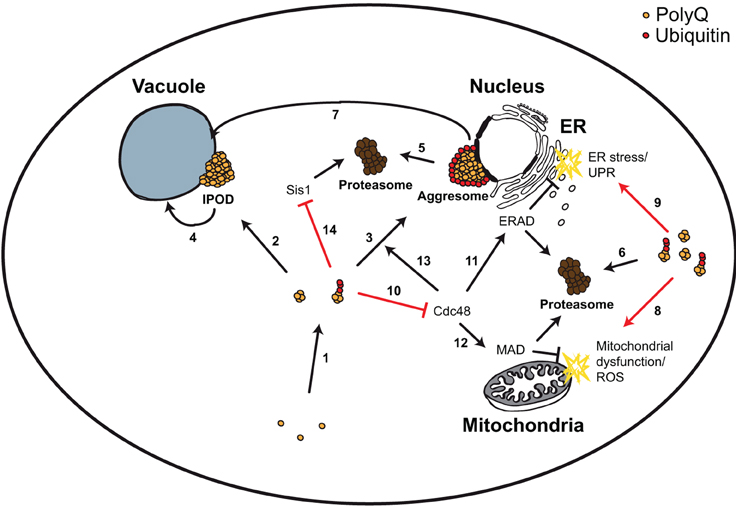
Figure 2. Yeast model for polyQ-triggered cytotoxicity. PolyQ monomers are very aggregation-prone forming smaller and larger aggregates, which can be ubiquitylated (1). These aggregates can be transported to two cellular compartments, the perivacuolar inclusion (IPOD, non-ubiquitylated) (2) or the perinuclear aggresome (ubiquitylated) (3). IPODs are potential substrates of autophagy (4). Aggresomes and other ubiquitylated polyQ aggregates are substrates of the UPS (5, 6), and of ubiquitin-dependent autophagy (via the Cue5 ubiquitin-Atg8 adaptor protein) (7). PolyQ aggregates trigger mitochondrial dysfunction and ROS (8), as well as ER stress and UPR (9). The first was explained by a direct detrimental physical interaction of polyQ aggregates with mitochondria. For the latter it has been shown that polyQ aggregates sequester the AAA-ATPase Cdc48 (10), leading to ERAD dysfunction (11). Sequestering Cdc48 could potentially lead to MAD dysfunction (12) (which shares many components with ERAD) and to impairment of aggresome formation (13) (which depends on Cdc48). Ubiquitylated polyQ aggregates inhibit the degradation of misfolded cytosolic proteins in the nucleus via sequestering the chaperone Sis1 (14).
The data obtained in yeast HD models have been very helpful for a better understanding of polyQ-triggered effects in higher model systems, as well as in humans. For instance, the allocation of different aggregates into distinct cellular compartments (aggresomes and IPOD, respectively) was conserved from yeast to mammalians (Johnston et al., 1998; Kaganovich et al., 2008; Wang et al., 2009). As in yeast, polyQ are substrates of the UPS and autophagy (Lu et al., 2014; Margulis and Finkbeiner, 2014; Martin et al., 2014). PolyQ aggregates block specific branches of the UPS (including the ERAD and the Sis1-dependent pathways) in mammalians as in yeast (Duennwald and Lindquist, 2008; Park et al., 2013; Margulis and Finkbeiner, 2014). Like in yeast cells, polyQ impairs vesicle-based protein degradation, including autophagy (Sapp et al., 1997; Aronin et al., 1999; Meriin et al., 2003, 2007; Martin et al., 2014).
Interestingly, in cultured cells and in flies, ubiquitylation (and sumolyation) of the N-terminal part of huntingtin (N17) at specific residues (K6, K9, and K15) turned out to be an efficient modulator of polyQ aggregation and neurotoxicity (Steffan et al., 2004). Thus, distinct (poly)ubiquitylation patterns could on the one hand dictate the fate of polyQ (e.g., aggregation and degradation) and on the other hand determine its effects on ubiquitin-dependent degradation of other cellular proteins. This issue could further be dissected in yeast HD models.
Of specific interest is the role of p97/VCP, the ortholog of the yeast AAA-ATPase Cdc48. As in yeast, p97/VCP and its cofactors Npl4 and Ufd1 were sequestered by polyQ aggregates in mammalian cells leading to ERAD dysfunction, ER stress, and UPR induction (Duennwald and Lindquist, 2008). Overexpression of Npl4, Ufd1, or p97/VCP rescued from ERAD dysfunction and/or ER stress and UPR (Duennwald and Lindquist, 2008; Leitman et al., 2013). In C. elegans, p97/VCP was involved in the clearance of detrimental polyQ aggregates (Nishikori et al., 2008). Since p97/VCP has also been proposed to play important roles in autophagy and apoptosis (Braun and Zischka, 2008; Krick et al., 2010), it is tempting to speculate that the interaction of polyQ aggregates with this protein complex is critical for the switch from UPS and autophagy dysfunctions to neuronal cell loss.
Alzheimer's Disease (AD)
AD is the most prevalent form of age-related dementia (Querfurth and Laferla, 2010). Synaptic loss and neuronal decline can be observed in affected brain regions, including the hippocampus and the cortex (Querfurth and Laferla, 2010). The accumulation of extracellular senile plaques and intracellular aggregates, composed of β-amyloid, and the intracellular accumulation of neurofibrillary tangles comprising hyperphosphorylated variants of the protein tau and UBB+1, the frameshift variant of ubiquitin B, contribute to AD progression (van Leeuwen et al., 1998; Laferla et al., 2007; Benilova et al., 2012; Mandelkow and Mandelkow, 2012).
Yeast β-Amyloid Models
Several yeast models expressing β-amyloid in the cytosol and in the secretory pathway have been generated (Caine et al., 2007; Treusch et al., 2011; D'Angelo et al., 2013; Matlack et al., 2014; Mossmann et al., 2014) (Table 1). Cytosolic β-amyloid (GFP-Aβ42 or Aβ42-GFP fusion proteins) led to moderate growth deficits and to the induction of the heat shock response in wild-type cells (Caine et al., 2007). Mitochondria isolated from aging-prone yeast cells (Δcoa6) upon prolonged exposure to cytosolic Aβ42 demonstrated signs of mitochondrial impairment, including inhibition of mitochondrial pre-protein maturation, increased ROS production, decreased mitochondrial membrane potential and reduced oxygen consumption (Mossmann et al., 2014). Recent studies suggest that mitochondrial impairment triggers UPS dysfunction and vice versa (Livnat-Levanon et al., 2014; Maharjan et al., 2014; Segref et al., 2014; Braun et al., 2015). Since mitochondrial and UPS dysfunction are believed to contribute to AD progression, it is of high interest, whether cytosolic Aβ42 interacts with ubiquitin-dependent processes.
Whereas, the detrimental effects of cytosolic β-amyloid remained moderate, β-amyloid directed to the secretory pathway resulted in high cytotoxicity concomitant to β-amyloid oligomerization and aggregation (Treusch et al., 2011; D'Angelo et al., 2013; Matlack et al., 2014). Here, β-amyloid impaired the clathrin-mediated endocytic trafficking of a plasma membrane receptor (which is a ubiquitin-modulated process), and expression of endocytic genes rescued β-amyloid-triggered cytotoxicity (Treusch et al., 2011; D'Angelo et al., 2013). A genome-wide screen for modifiers of β-amyloid-triggered cytotoxicity identified the yeast homolog of phosphatidylinositol binding clathrin assembly protein (PICALM), as suppressor of cytotoxicity (Treusch et al., 2011). A screen of ~140,000 compounds for β-amyloid rescue in yeast identified the 8-hydroxyquinoline clioquinol as effective in preventing β-amyloid-triggered growth deficits via promoting β-amyloid degradation and restoring endocytic function (Matlack et al., 2014). The outcome of these two unbiased yeast screens validate the usefulness of yeast AD models, because PICALM is one of the most highly validated AD risk factors (Treusch et al., 2011), and clioquinol is effective in both mouse and C. elegans AD models (Matlack et al., 2014).
Yeast tau Models
Yeast models expressing AD-associated tau have been generated (De Vos et al., 2011) (Table 1). Tau forms oligomers and is hyperphosphorylated when expressed in yeast as it is in AD patients (Vandebroek et al., 2005, 2006; Zabrocki et al., 2005; Vanhelmont et al., 2010). Although tau expression per se remained non-toxic in wild-type cells, it increased α-synuclein-triggered growth deficits (Zabrocki et al., 2005). Therefore, combining several AD risk factors (e.g., tau, β-amyloid, and/or UBB+1) could lead to more effective yeast AD models. This turned out to be necessary in mouse AD models, because mutations in tau are also not very toxic in transgenic mice (Nisbet et al., 2015). Similarly, AD-associated mutations in the β-amyloid precursor protein (APP) or the γ-secretase components presenilin 1 or 2, which are pivotally involved in β-amyloid generation from APP, do also not fully recapitulate the key pathological events of AD (Nisbet et al., 2015). In order to mimic simultaneously more than one pathological aspect of AD double and triple transgenic AD mouse models or AD patient-derived neuronal stem-cell-derived three-dimensional culture systems have been developed (van Tijn et al., 2012; Choi et al., 2014; Nisbet et al., 2015).
Yeast UBB+1 Models
UBB+1-expressing yeast models have been established (Tank and True, 2009; Verhoef et al., 2009; Dennissen et al., 2011; Krutauz et al., 2014; Braun et al., 2015) (Table 1). As in mammalian cells, UBB+1 is a substrate of the UPS in yeast. It is ubiquitylated prior proteasomal degradation and truncated by the DUB Yuh1 (or mammalian ubiquitin carboxy-terminal hydrolase UCH-L3) (De Vrij et al., 2001; van Tijn et al., 2007, 2010; Tank and True, 2009; Verhoef et al., 2009; Dennissen et al., 2011; Braun et al., 2015). Accumulation of UBB+1 impairs the UPS in yeast and mammalian cells, culminating in the accumulation of polyubiquitylated proteins. In both cases, the degradation of UFD substrates (e.g., Ub-G76V-GFP, Ub-P-LacZ), and of N-end rule substrates (e.g., Ub-R-GFP, Ub-R-LacZ) was significantly reduced (Lindsten et al., 2002; van Tijn et al., 2007; Tank and True, 2009; Braun et al., 2015). The inhibitory effect of UBB+1 on the UPS is at least partially due to inhibition of DUBs, especially Ubp6/USP14, which is associated with the 26S proteasome and involved in the disassembly of polyubiquitin tags from substrate proteins (Krutauz et al., 2014).
Despite of the UBB+1-triggered UPS impairment, both yeast and mammalian cells can tolerate UBB+1 without marked signs of cytotoxicity (Hope et al., 2003; van Tijn et al., 2007, 2012; Tank and True, 2009; Yim et al., 2014), and in some cases UBB+1 expression was even protective when mammalian cells were treated with chemicals inducing oxidative stress (Hope et al., 2003; Yim et al., 2014). Consistently, transgenic expression of UBB+1 in mice failed to cause overt neurodegeneration although it did affect spatial reference memory and caused a central dysfunction of respiratory regulation (Fischer et al., 2009; van Tijn et al., 2011; Irmler et al., 2012). In contrast, prolonged expression of high levels of UBB+1 induced cell death and pivotal mitochondrial impairment in neuronal cells and in yeast (De Vrij et al., 2001; Tan et al., 2007; Braun et al., 2015). In yeast the UBB+1-triggered cell death could be prevented by specifically promoting the UPS activity at mitochondria (mitochondrion-associated degradation, MAD), which protected cells from mitochondrial impairment but did not alter the steady-state levels of UBB+1 (Braun et al., 2015). The accumulation of UBB+1 increased the cytotoxicity of polyQ (103Q) in yeast (Tank and True, 2009), which is highly comparable to mammalian systems, where UBB+1 expression accelerates cell death in transgenic HD mouse models (de Pril et al., 2004, 2007, 2010). Thus, on the one hand UBB+1 is a neurotoxic protein by itself, whose cytotoxicity depends both on the cellular UPS capacity and on mitochondrial (dys)function. On the other hand UBB+1 is putatively a potent modifier of cytotoxicity of other misfolded neurotoxic proteins. Especially when combined with further AD risk factors, including β-amyloid and tau, the yeast UBB+1 model could be very valuable for elucidating the molecular connections among AD risk factors, UPS (dys)function, mitochondrial activity, and cell survival.
Amyotrophic Lateral Sclerosis (ALS)
ALS is a frequent degenerative motor neuron disease, resulting in muscle weakness and wasting (Andersen and Al-Chalabi, 2011). Among the most common ALS-associated genes are TARDBP, and FUS, encoding the RNA-binding proteins TDP-43 and FUS/TLS (Andersen and Al-Chalabi, 2011). ALS-associated variants of these proteins demonstrate mislocalization and/or a high tendency for aggregation, and these aggregates are ubiquitylated (Andersen and Al-Chalabi, 2011; Da Cruz and Cleveland, 2011). Yeast models expressing ALS-associated wild-type and mutant TDP-43 and FUS/TLS have been established to further analyze the detrimental roles of these proteins on cell survival (Bastow et al., 2011).
TDP-43 and FUS/TLS efficiently form cytoplasmic aggregates when expressed in yeast cells triggering cytotoxicity and cell death (Johnson et al., 2008; Braun et al., 2011; Kryndushkin et al., 2011; Sun et al., 2011) (Table 1). It is very little known via which pathways these neurotoxic proteins and their aggregates are degraded in yeast, whether these proteins impair ubiquitin-modulated protein degradation, and whether the activities of ubiquitin-modulated proteolysis pivotally affect their cytotoxicity. In the case of TDP-43, a temperature-sensitive variant of the AAA-ATPase Cdc48 was found as an enhancer of TDP-43-triggered cytotoxicity (growth deficits) (Armakola et al., 2012). Promoting vesicular trafficking (ER -> Golgi) by treating TDP-43-expressing yeast cells with NAB relieved TDP-43-triggered cytotoxicity (Tardiff et al., 2012, 2013). Thus, the activities of ubiquitin-modulated vesicular transport and Cdc48-dependent cellular processes may be pivotal in modulating TDP-43-triggered cytotoxicity in yeast and neurons. Cdc48 and its mammalian homolog p97/VCP are involved in many different ubiquitin-modulated processes, including ERAD, MAD, and vesicular transport, such as endocytosis, and autophagy (Bug and Meyer, 2012). Indeed, mutations in p97/VCP lead to cytoplasmic TDP-43 aggregation and cell death in neurons of transgenic mice and flies (Gitcho et al., 2009; Johnson et al., 2010; Ritson et al., 2010; Rodriguez-Ortiz et al., 2013). Yeast TDP-43 models could help to further discriminate the role of the distinct Cdc48/p97/VCP pathways in modulating cell death and neuronal loss.
Conclusions and Outlook
In yeast models expressing neurotoxic proteins, these proteins are on the one hand substrates of distinct protein degradation pathways, and on the other hand the trigger for their impairment. Consistently, the activities of the UPS, autophagy, and other ubiquitin-controlled vesicle-based protein degradation pathways are pivotal for the cytotoxicity of neurotoxic proteins. Best described for yeast PD and HD models, these findings could be confirmed in other neurotoxic model systems, confirming that yeast models expressing neurotoxic proteins are very helpful in elucidating novel paradigms of pathobiology in neurodegenerative disorders. It is very likely, that yeast models for AD (β-amyloid, tau, UBB+1), and ALS (e.g., TDP-43, FUS/TLS), will lead to the identification of further ubiquitin-regulated protein degradation pathways that potentially underlie neuronal dysfunction and cell loss. In this respect, the easy and fast combination of different neurotoxic proteins in one yeast model (e.g., different AD-associated proteins) could be a very straightforward approach.
The role of free ubiquitin homeostasis has been largely unattended when analyzing the effects of ubiquitylated neurotoxic proteins and ubiquitin-dependent protein degradation on neuronal survival. For instance, loss of free ubiquitin was found in schizophrenia (Rubio et al., 2013), and down-regulation of free ubiquitin was determined to be causative for p53 accumulation and apoptosis in hippocampal neurons from rats (Tan et al., 2000). In contrast, free ubiquitin levels increase in cancer cells, which turned out to be pivotal for cell growth (Oh et al., 2013). Consistently, high levels of free ubiquitin confers resistance to inhibitors of protein translation in yeast (Hanna et al., 2003). Modulating free ubiquitin levels in yeast models expressing human neurotoxic proteins could address the role of free ubiquitin homeostasis on the degradation and the cytotoxicity of these proteins.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I am grateful to the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) for grant BR 3706/3-1. This publication was funded by the University of Bayreuth in the funding program Open Access Publishing.
Abbreviations
AD, Alzheimer’s disease; ALS, Amyotrophic lateral sclerosis; DUB, Deubiquitylating enzyme; ERAD, ER-associated degradation; HD, Huntington’s disease; IPOD, Insoluble protein deposit/ perivacuolar aggregates; MAD, Mitochondrion-associated degradation; MVB, Multivesicular bodies; PD, Parkinson’s disease; polyQ, Proteins with abnormal glutamine expansions; ROS, Reactive oxygen species; UFD, Ubiquitin-fusion degradation pathway; UPR, Unfolded protein response; UPS, Ubiquitin-proteasome system.
References
Andersen, P. M., and Al-Chalabi, A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615. doi: 10.1038/nrneurol.2011.150
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Armakola, M., Higgins, M. J., Figley, M. D., Barmada, S. J., Scarborough, E. A., Diaz, Z., et al. (2012). Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat. Genet. 44, 1302–1309. doi: 10.1038/ng.2434
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aronin, N., Kim, M., Laforet, G., and Difiglia, M. (1999). Are there multiple pathways in the pathogenesis of Huntington's disease? Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 995–1003. doi: 10.1098/rstb.1999.0451
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Basso, E., Antas, P., Marijanovic, Z., Goncalves, S., Tenreiro, S., and Outeiro, T. F. (2013). PLK2 modulates alpha-synuclein aggregation in yeast and mammalian cells. Mol. Neurobiol. 48, 854–862. doi: 10.1007/s12035-013-8473-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bastow, E. L., Gourlay, C. W., and Tuite, M. F. (2011). Using yeast models to probe the molecular basis of amyotrophic lateral sclerosis. Biochem. Soc. Trans. 39, 1482–1487. doi: 10.1042/BST0391482
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bharadwaj, P., Martins, R., and Macreadie, I. (2010). Yeast as a model for studying Alzheimer's disease. FEMS Yeast Res. 10, 961–969. doi: 10.1111/j.1567-1364.2010.00658.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bocharova, N. A., Sokolov, S. S., Knorre, D. A., Skulachev, V. P., and Severin, F. F. (2008). Unexpected link between anaphase promoting complex and the toxicity of expanded polyglutamines expressed in yeast. Cell Cycle 7, 3943–3946. doi: 10.4161/cc.7.24.7398
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bocharova, N., Chave-Cox, R., Sokolov, S., Knorre, D., and Severin, F. (2009). Protein aggregation and neurodegeneration: clues from a yeast model of Huntington's disease. Biochemistry Mosc. 74, 231–234. doi: 10.1134/S0006297909020163
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braun, R. J., Büttner, S., Ring, J., Kroemer, G., and Madeo, F. (2010). Nervous yeast: modeling neurotoxic cell death. Trends Biochem. Sci. 35, 135–144. doi: 10.1016/j.tibs.2009.10.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braun, R. J., Sommer, C., Carmona-Gutierrez, D., Khoury, C. M., Ring, J., Buttner, S., et al. (2011). Neurotoxic 43-kDa TAR DNA-binding protein (TDP-43) triggers mitochondrion-dependent programmed cell death in yeast. J. Biol. Chem. 286, 19958–19972. doi: 10.1074/jbc.M110.194852
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braun, R. J., Sommer, C., Leibiger, C., Gentier, R. J. G., Dumit, V. I., Paduch, K., et al. (2015). Accumulation of basic amino acids at mitochondria dictates the cytotoxicity of aberrant ubiquitin. Cell Rep. 10. doi: 10.1016/j.celrep.2015.02.009
Braun, R. J., and Zischka, H. (2008). Mechanisms of Cdc48/VCP-mediated cell death: from yeast apoptosis to human disease. Biochim. Biophys. Acta 1783, 1418–1435. doi: 10.1016/j.bbamcr.2008.01.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braun, R. J., Zischka, H., Madeo, F., Eisenberg, T., Wissing, S., Büttner, S., et al. (2006). Crucial mitochondrial impairment upon CDC48 mutation in apoptotic yeast. J. Biol. Chem. 281, 25757–25767. doi: 10.1074/jbc.M513699200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bug, M., and Meyer, H. (2012). Expanding into new markets—VCP/p97 in endocytosis and autophagy. J. Struct. Biol. 179, 78–82. doi: 10.1016/j.jsb.2012.03.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Büttner, S., Bitto, A., Ring, J., Augsten, M., Zabrocki, P., Eisenberg, T., et al. (2008). Functional mitochondria are required for alpha-synuclein toxicity in aging yeast. J. Biol. Chem. 283, 7554–7560. doi: 10.1074/jbc.M708477200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Büttner, S., Faes, L., Reichelt, W. N., Broeskamp, F., Habernig, L., Benke, S., et al. (2013a). The Ca2+/Mn2+ ion-pump PMR1 links elevation of cytosolic Ca(2+) levels to alpha-synuclein toxicity in Parkinson's disease models. Cell Death Differ. 20, 465–477. doi: 10.1038/cdd.2012.142
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Büttner, S., Habernig, L., Broeskamp, F., Ruli, D., Vögtle, F. N., Vlachos, M., et al. (2013b). Endonuclease G mediates alpha-synuclein cytotoxicity during Parkinson's disease. EMBO J. 32, 3041–3054. doi: 10.1038/emboj.2013.228
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Caine, J., Sankovich, S., Antony, H., Waddington, L., Macreadie, P., Varghese, J., et al. (2007). Alzheimer's Abeta fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Res. 7, 1230–1236. doi: 10.1111/j.1567-1364.2007.00285.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carmona-Gutierrez, D., Eisenberg, T., Büttner, S., Meisinger, C., Kroemer, G., and Madeo, F. (2010). Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ. 17, 763–773. doi: 10.1038/cdd.2009.219
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, Q., Thorpe, J., and Keller, J. N. (2005). Alpha-synuclein alters proteasome function, protein synthesis, and stationary phase viability. J. Biol. Chem. 280, 30009–30017. doi: 10.1074/jbc.M501308200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D'Avanzo, C., et al. (2014). A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 515, 274–278. doi: 10.1038/nature13800
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chung, C. Y., Khurana, V., Auluck, P. K., Tardiff, D. F., Mazzulli, J. R., Soldner, F., et al. (2013). Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987. doi: 10.1126/science.1245296
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cohen, A., Ross, L., Nachman, I., and Bar-Nun, S. (2012). Aggregation of polyQ proteins is increased upon yeast aging and affected by Sir2 and Hsf1: novel quantitative biochemical and microscopic assays. PLoS ONE 7:e44785. doi: 10.1371/journal.pone.0044785
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313, 324–328. doi: 10.1126/science.1129462
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Crick, S. L., Ruff, K. M., Garai, K., Frieden, C., and Pappu, R. V. (2013). Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U.S.A. 110, 20075–20080. doi: 10.1073/pnas.1320626110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cunha, D., Cunha, R., Corte-Real, M., and Chaves, S. R. (2013). Cisplatin-induced cell death in Saccharomyces cerevisiae is programmed and rescued by proteasome inhibition. DNA Repair (Amst). 12, 444–449. doi: 10.1016/j.dnarep.2013.02.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Da Cruz, S., and Cleveland, D. W. (2011). Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 21, 904–919. doi: 10.1016/j.conb.2011.05.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
D'Angelo, F., Vignaud, H., Di Martino, J., Salin, B., Devin, A., Cullin, C., et al. (2013). A yeast model for amyloid-beta aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity. Dis. Model. Mech. 6, 206–216. doi: 10.1242/dmm.010108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dantuma, N. P., and Bott, L. C. (2014). The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front. Mol. Neurosci. 7:70. doi: 10.3389/fnmol.2014.00070
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dargemont, C., and Ossareh-Nazari, B. (2012). Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim. Biophys. Acta 1823, 138–144. doi: 10.1016/j.bbamcr.2011.07.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dennissen, F. J., Kholod, N., Hermes, D. J., Kemmerling, N., Steinbusch, H. W., Dantuma, N. P., et al. (2011). Mutant ubiquitin (UBB+1) associated with neurodegenerative disorders is hydrolyzed by ubiquitin C-terminal hydrolase L3 (UCH-L3). FEBS Lett. 585, 2568–2574. doi: 10.1016/j.febslet.2011.06.037
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dennissen, F. J., Kholod, N., and van Leeuwen, F. W. (2012). The ubiquitin proteasome system in neurodegenerative diseases: culprit, accomplice or victim? Prog. Neurobiol. 96, 190–207. doi: 10.1016/j.pneurobio.2012.01.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
de Pril, R., Fischer, D. F., Maat-Schieman, M. L., Hobo, B., de Vos, R. A., Brunt, E. R., et al. (2004). Accumulation of aberrant ubiquitin induces aggregate formation and cell death in polyglutamine diseases. Hum. Mol. Genet. 13, 1803–1813. doi: 10.1093/hmg/ddh188
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
de Pril, R., Fischer, D. F., Roos, R. A., and van Leeuwen, F. W. (2007). Ubiquitin-conjugating enzyme E2-25K increases aggregate formation and cell death in polyglutamine diseases. Mol. Cell. Neurosci. 34, 10–19. doi: 10.1016/j.mcn.2006.09.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
de Pril, R., Hobo, B., van Tijn, P., Roos, R. A., van Leeuwen, F. W., and Fischer, D. F. (2010). Modest proteasomal inhibition by aberrant ubiquitin exacerbates aggregate formation in a Huntington disease mouse model. Mol. Cell. Neurosci. 43, 281–286. doi: 10.1016/j.mcn.2009.12.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Vos, A., Anandhakumar, J., Van den Brande, J., Verduyckt, M., Franssens, V., Winderickx, J., et al. (2011). Yeast as a model system to study tau biology. Int. J. Alzheimers. Dis. 2011:428970. doi: 10.4061/2011/428970
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Vrij, F. M., Sluijs, J. A., Gregori, L., Fischer, D. F., Hermens, W. T., Goldgaber, D., et al. (2001). Mutant ubiquitin expressed in Alzheimer's disease causes neuronal death. FASEB J. 15, 2680–2688. doi: 10.1096/fj.01-0438com
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dixon, C., Mathias, N., Zweig, R. M., Davis, D. A., and Gross, D. S. (2005). Alpha-synuclein targets the plasma membrane via the secretory pathway and induces toxicity in yeast. Genetics 170, 47–59. doi: 10.1534/genetics.104.035493
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duennwald, M. L. (2011). Polyglutamine misfolding in yeast: toxic and protective aggregation. Prion 5, 285–290. doi: 10.4161/pri.18071
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duennwald, M. L., Jagadish, S., Giorgini, F., Muchowski, P. J., and Lindquist, S. (2006a). A network of protein interactions determines polyglutamine toxicity. Proc. Natl. Acad. Sci. U.S.A. 103, 11051–11056. doi: 10.1073/pnas.0604548103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duennwald, M. L., Jagadish, S., Muchowski, P. J., and Lindquist, S. (2006b). Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. U.S.A. 103, 11045–11050. doi: 10.1073/pnas.0604547103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duennwald, M. L., and Lindquist, S. (2008). Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 22, 3308–3319. doi: 10.1101/gad.1673408
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ebrahimi-Fakhari, D., Wahlster, L., and McLean, P. J. (2012). Protein degradation pathways in Parkinson's disease: curse or blessing. Acta Neuropathol. 124, 153–172. doi: 10.1007/s00401-012-1004-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fang, N. N., Chan, G. T., Zhu, M., Comyn, S. A., Persaud, A., Deshaies, R. J., et al. (2014). Rsp5/Nedd4 is the main ubiquitin ligase that targets cytosolic misfolded proteins following heat stress. Nat. Cell Biol. 16, 1227–1237. doi: 10.1038/ncb3054
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Finley, D., Ulrich, H. D., Sommer, T., and Kaiser, P. (2012). The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192, 319–360. doi: 10.1534/genetics.112.140467
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fischer, D. F., van Dijk, R., van Tijn, P., Hobo, B., Verhage, M. C., van der Schors, R. C., et al. (2009). Long-term proteasome dysfunction in the mouse brain by expression of aberrant ubiquitin. Neurobiol. Aging 30, 847–863. doi: 10.1016/j.neurobiolaging.2008.06.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Flower, T. R., Chesnokova, L. S., Froelich, C. A., Dixon, C., and Witt, S. N. (2005). Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson's disease. J. Mol. Biol. 351, 1081–1100. doi: 10.1016/j.jmb.2005.06.060
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujioka, S., Ogaki, K., Tacik, P. M., Uitti, R. J., Ross, O. A., and Wszolek, Z. K. (2014). Update on novel familial forms of Parkinson's disease and multiple system atrophy. Parkinsonism Relat. Disord. 20(Suppl. 1), S29–S34. doi: 10.1016/S1353-8020(13)70010-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giorgini, F., Moller, T., Kwan, W., Zwilling, D., Wacker, J. L., Hong, S., et al. (2008). Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J. Biol. Chem. 283, 7390–7400. doi: 10.1074/jbc.M708192200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gitcho, M. A., Strider, J., Carter, D., Taylor-Reinwald, L., Forman, M. S., Goate, A. M., et al. (2009). VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem. 284, 12384–12398. doi: 10.1074/jbc.M900992200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gitler, A. D. (2008). Beer and bread to brains and beyond: can yeast cells teach us about neurodegenerative disease? Neurosignals 16, 52–62. doi: 10.1159/000109759
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gitler, A. D., Bevis, B. J., Shorter, J., Strathearn, K. E., Hamamichi, S., Su, L. J., et al. (2008). The Parkinson's disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 145–150. doi: 10.1073/pnas.0710685105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gitler, A. D., Chesi, A., Geddie, M. L., Strathearn, K. E., Hamamichi, S., Hill, K. J., et al. (2009). Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 41, 308–315. doi: 10.1038/ng.300
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hanna, J., Leggett, D. S., and Finley, D. (2003). Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol. Cell. Biol. 23, 9251–9261. doi: 10.1128/MCB.23.24.9251-9261.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Heo, J. M., Livnat-Levanon, N., Taylor, E. B., Jones, K. T., Dephoure, N., Ring, J., et al. (2010). A stress-responsive system for mitochondrial protein degradation. Mol. Cell 40, 465–480. doi: 10.1016/j.molcel.2010.10.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hope, A. D., de Silva, R., Fischer, D. F., Hol, E. M., van Leeuwen, F. W., and Lees, A. J. (2003). Alzheimer's associated variant ubiquitin causes inhibition of the 26S proteasome and chaperone expression. J. Neurochem. 86, 394–404. doi: 10.1046/j.1471-4159.2003.01844.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Irmler, M., Gentier, R. J., Dennissen, F. J., Schulz, H., Bolle, I., Holter, S. M., et al. (2012). Long-term proteasomal inhibition in transgenic mice by UBB(+1) expression results in dysfunction of central respiration control reminiscent of brainstem neuropathology in Alzheimer patients. Acta Neuropathol. 124, 187–197. doi: 10.1007/s00401-012-1003-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnson, B. S., McCaffery, J. M., Lindquist, S., and Gitler, A. D. (2008). A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 6439–6444. doi: 10.1073/pnas.0802082105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnson, B. S., Snead, D., Lee, J. J., McCaffery, J. M., Shorter, J., and Gitler, A. D. (2009). TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339. doi: 10.1074/jbc.M109.010264
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnston, J. A., Ward, C. L., and Kopito, R. R. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898. doi: 10.1083/jcb.143.7.1883
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaganovich, D., Kopito, R., and Frydman, J. (2008). Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095. doi: 10.1038/nature07195
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khurana, V., and Lindquist, S. (2010). Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nat. Rev. Neurosci. 11, 436–449. doi: 10.1038/nrn2809
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krick, R., Bremer, S., Welter, E., Schlotterhose, P., Muehe, Y., Eskelinen, E. L., et al. (2010). Cdc48/p97 and Shp1/p47 regulate autophagosome biogenesis in concert with ubiquitin-like Atg8. J. Cell Biol. 190, 965–973. doi: 10.1083/jcb.201002075
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kruegel, U., Robison, B., Dange, T., Kahlert, G., Delaney, J. R., Kotireddy, S., et al. (2011). Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 7:e1002253. doi: 10.1371/journal.pgen.1002253
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krutauz, D., Reis, N., Nakasone, M. A., Siman, P., Zhang, D., Kirkpatrick, D. S., et al. (2014). Extended ubiquitin species are protein-based DUB inhibitors. Nat. Chem. Biol. 10, 664–670. doi: 10.1038/nchembio.1574
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kryndushkin, D., Wickner, R. B., and Shewmaker, F. (2011). FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell 2, 223–236. doi: 10.1007/s13238-011-1525-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuang, E., Qi, J., and Ronai, Z. (2013). Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem. Sci. 38, 453–460. doi: 10.1016/j.tibs.2013.06.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Laferla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-beta in Alzheimer's disease. Nat. Rev. Neurosci. 8, 499–509. doi: 10.1038/nrn2168
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lansbury, P. T., and Lashuel, H. A. (2006). A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443, 774–779. doi: 10.1038/nature05290
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, I. H., Kim, H. Y., Kim, M., Hahn, J. S., and Paik, S. R. (2008). Dequalinium-induced cell death of yeast expressing alpha-synuclein-GFP fusion protein. Neurochem. Res. 33, 1393–1400. doi: 10.1007/s11064-008-9598-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lees, A. J., Hardy, J., and Revesz, T. (2009). Parkinson's disease. Lancet 373, 2055–2066. doi: 10.1016/S0140-6736(09)60492-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leitman, J., Ulrich Hartl, F., and Lederkremer, G. Z. (2013). Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 4, 2753. doi: 10.1038/ncomms3753
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liang, J., Clark-Dixon, C., Wang, S., Flower, T. R., Williams-Hart, T., Zweig, R., et al. (2008). Novel suppressors of alpha-synuclein toxicity identified using yeast. Hum. Mol. Genet. 17, 3784–3795. doi: 10.1093/hmg/ddn276
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lindsten, K., de Vrij, F. M., Verhoef, L. G., Fischer, D. F., van Leeuwen, F. W., Hol, E. M., et al. (2002). Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J. Cell Biol. 157, 417–427. doi: 10.1083/jcb.200111034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Livnat-Levanon, N., Kevei, E., Kleifeld, O., Krutauz, D., Segref, A., Rinaldi, T., et al. (2014). Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 7, 1371–1380. doi: 10.1016/j.celrep.2014.04.030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lu, K., Psakhye, I., and Jentsch, S. (2014). Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 158, 549–563. doi: 10.1016/j.cell.2014.05.048
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
MacGurn, J. A., Hsu, P. C., and Emr, S. D. (2012). Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 81, 231–259. doi: 10.1146/annurev-biochem-060210-093619
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maharjan, S., Oku, M., Tsuda, M., Hoseki, J., and Sakai, Y. (2014). Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 4:5896. doi: 10.1038/srep05896
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mandelkow, E. M., and Mandelkow, E. (2012). Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2:a006247. doi: 10.1101/cshperspect.a006247
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Margulis, J., and Finkbeiner, S. (2014). Proteostasis in striatal cells and selective neurodegeneration in Huntington's disease. Front. Cell. Neurosci. 8:218. doi: 10.3389/fncel.2014.00218
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martin, D. D., Ladha, S., Ehrnhoefer, D. E., and Hayden, M. R. (2014). Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 38, 26–35 doi: 10.1016/j.tins.2014.09.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mason, R. P., and Giorgini, F. (2011). Modeling Huntington disease in yeast: perspectives and future directions. Prion 5, 269–276. doi: 10.4161/pri.18005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matlack, K. E., Tardiff, D. F., Narayan, P., Hamamichi, S., Caldwell, K. A., Caldwell, G. A., et al. (2014). Clioquinol promotes the degradation of metal-dependent amyloid-beta (Abeta) oligomers to restore endocytosis and ameliorate Abeta toxicity. Proc. Natl. Acad. Sci. USA. 111, 4013–4018. doi: 10.1073/pnas.1402228111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meriin, A. B., Zhang, X., Alexandrov, I. M., Salnikova, A. B., Ter-Avanesian, M. D., Chernoff, Y. O., et al. (2007). Endocytosis machinery is involved in aggregation of proteins with expanded polyglutamine domains. FASEB J. 21, 1915–1925. doi: 10.1096/fj.06-6878com
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meriin, A. B., Zhang, X., He, X., Newnam, G. P., Chernoff, Y. O., and Sherman, M. Y. (2002). Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 157, 997–1004. doi: 10.1083/jcb.200112104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meriin, A. B., Zhang, X., Miliaras, N. B., Kazantsev, A., Chernoff, Y. O., McCaffery, J. M., et al. (2003). Aggregation of expanded polyglutamine domain in yeast leads to defects in endocytosis. Mol. Cell. Biol. 23, 7554–7565. doi: 10.1128/MCB.23.21.7554-7565.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miller-Fleming, L., Giorgini, F., and Outeiro, T. F. (2008). Yeast as a model for studying human neurodegenerative disorders. Biotechnol. J. 3, 325–338. doi: 10.1002/biot.200700217
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mossmann, D., Vögtle, F. N., Taskin, A. A., Teixeira, P. F., Ring, J., Burkhart, J. M., et al. (2014). Amyloid-beta Peptide Induces Mitochondrial Dysfunction by Inhibition of Preprotein Maturation. Cell Metab. 20, 662–669. doi: 10.1016/j.cmet.2014.07.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nisbet, R. M., Polanco, J. C., Ittner, L. M., and Gotz, J. (2015). Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 129, 207–220. doi: 10.1007/s00401-014-1371-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nishikori, S., Yamanaka, K., Sakurai, T., Esaki, M., and Ogura, T. (2008). p97 Homologs from Caenorhabditis elegans, CDC-48.1 and CDC-48.2, suppress the aggregate formation of huntingtin exon1 containing expanded polyQ repeat. Genes Cells 13, 827–838. doi: 10.1111/j.1365-2443.2008.01214.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ocampo, A., Zambrano, A., and Barrientos, A. (2010). Suppression of polyglutamine-induced cytotoxicity in Saccharomyces cerevisiae by enhancement of mitochondrial biogenesis. FASEB J. 24, 1431–1441. doi: 10.1096/fj.09-148601
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oh, C., Park, S., Lee, E. K., and Yoo, Y. J. (2013). Downregulation of ubiquitin level via knockdown of polyubiquitin gene Ubb as potential cancer therapeutic intervention. Sci. Rep. 3:2623. doi: 10.1038/srep02623
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Outeiro, T. F., and Lindquist, S. (2003). Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775. doi: 10.1126/science.1090439
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, S. H., Kukushkin, Y., Gupta, R., Chen, T., Konagai, A., Hipp, M. S., et al. (2013). PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154, 134–145. doi: 10.1016/j.cell.2013.06.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petroi, D., Popova, B., Taheri-Talesh, N., Irniger, S., Shahpasandzadeh, H., Zweckstetter, M., et al. (2012). Aggregate clearance of alpha-synuclein in Saccharomyces cerevisiae depends more on autophagosome and vacuole function than on the proteasome. J. Biol. Chem. 287, 27567–27579. doi: 10.1074/jbc.M112.361865
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Querfurth, H. W., and Laferla, F. M. (2010). Alzheimer's disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ritson, G. P., Custer, S. K., Freibaum, B. D., Guinto, J. B., Geffel, D., Moore, J., et al. (2010). TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. Res. 30, 7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rodriguez-Ortiz, C. J., Hoshino, H., Cheng, D., Liu-Yescevitz, L., Blurton-Jones, M., Wolozin, B., et al. (2013). Neuronal-specific overexpression of a mutant valosin-containing protein associated with IBMPFD promotes aberrant ubiquitin and TDP-43 accumulation and cognitive dysfunction in transgenic mice. Am. J. Pathol. 183, 504–515. doi: 10.1016/j.ajpath.2013.04.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ross, C. A., and Tabrizi, S. J. (2011). Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. doi: 10.1016/S1474-4422(10)70245-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rubio, M. D., Wood, K., Haroutunian, V., and Meador-Woodruff, J. H. (2013). Dysfunction of the ubiquitin proteasome and ubiquitin-like systems in schizophrenia. Neuropsychopharmacology 38, 1910–1920. doi: 10.1038/npp.2013.84
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sampaio-Marques, B., Felgueiras, C., Silva, A., Rodrigues, M., Tenreiro, S., Franssens, V., et al. (2012). SNCA (alpha-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 8, 1494–1509. doi: 10.4161/auto.21275
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sapp, E., Schwarz, C., Chase, K., Bhide, P. G., Young, A. B., Penney, J., et al. (1997). Huntingtin localization in brains of normal and Huntington's disease patients. Ann. Neurol. 42, 604–612. doi: 10.1002/ana.410420411
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Segref, A., Kevei, E., Pokrzywa, W., Schmeisser, K., Mansfeld, J., Livnat-Levanon, N., et al. (2014). Pathogenesis of human mitochondrial diseases is modulated by reduced activity of the ubiquitin/proteasome system. Cell Metab. 19, 642–652. doi: 10.1016/j.cmet.2014.01.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sharma, N., Brandis, K. A., Herrera, S. K., Johnson, B. E., Vaidya, T., Shrestha, R., et al. (2006). alpha-Synuclein budding yeast model: toxicity enhanced by impaired proteasome and oxidative stress. J. Mol. Neurosci. 28, 161–178. doi: 10.1385/JMN:28:2:161
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sokolov, S., Pozniakovsky, A., Bocharova, N., Knorre, D., and Severin, F. (2006). Expression of an expanded polyglutamine domain in yeast causes death with apoptotic markers. Biochim. Biophys. Acta 1757, 660–666. doi: 10.1016/j.bbabio.2006.05.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Solans, A., Zambrano, A., Rodriguez, M., and Barrientos, A. (2006). Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum. Mol. Genet. 15, 3063–3081. doi: 10.1093/hmg/ddl248
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C., Slepko, N., et al. (2004). SUMO modification of Huntingtin and Huntington's disease pathology. Science 304, 100–104. doi: 10.1126/science.1092194
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Su, L. J., Auluck, P. K., Outeiro, T. F., Yeger-Lotem, E., Kritzer, J. A., Tardiff, D. F., et al. (2010). Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson's disease models. Dis. Model. Mech. 3, 194–208. doi: 10.1242/dmm.004267
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sun, Z., Diaz, Z., Fang, X., Hart, M. P., Chesi, A., Shorter, J., et al. (2011). Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 9:e1000614. doi: 10.1371/journal.pbio.1000614
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suraweera, A., Munch, C., Hanssum, A., and Bertolotti, A. (2012). Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell 48, 242–253. doi: 10.1016/j.molcel.2012.08.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tam, S., Spiess, C., Auyeung, W., Joachimiak, L., Chen, B., Poirier, M. A., et al. (2009). The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285. doi: 10.1038/nsmb.1700
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tan, Z., Qu, W., Tu, W., Liu, W., Baudry, M., and Schreiber, S. S. (2000). p53 accumulation due to down-regulation of ubiquitin: relevance for neuronal apoptosis. Cell Death Differ. 7, 675–681. doi: 10.1038/sj.cdd.4400697
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tan, Z., Sun, X., Hou, F. S., Oh, H. W., Hilgenberg, L. G., Hol, E. M., et al. (2007). Mutant ubiquitin found in Alzheimer's disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ. 14, 1721–1732. doi: 10.1038/sj.cdd.4402180
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tank, E. M., and True, H. L. (2009). Disease-associated mutant ubiquitin causes proteasomal impairment and enhances the toxicity of protein aggregates. PLoS Genet. 5:e1000382. doi: 10.1371/journal.pgen.1000382
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tardiff, D. F., Jui, N. T., Khurana, V., Tambe, M. A., Thompson, M. L., Chung, C. Y., et al. (2013). Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science 342, 979–983. doi: 10.1126/science.1245321
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tardiff, D. F., Tucci, M. L., Caldwell, K. A., Caldwell, G. A., and Lindquist, S. (2012). Different 8-hydroxyquinolines protect models of TDP-43 protein, alpha-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. J. Biol. Chem. 287, 4107–4120. doi: 10.1074/jbc.M111.308668
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tauber, E., Miller-Fleming, L., Mason, R. P., Kwan, W., Clapp, J., Butler, N. J., et al. (2011). Functional gene expression profiling in yeast implicates translational dysfunction in mutant huntingtin toxicity. J. Biol. Chem. 286, 410–419. doi: 10.1074/jbc.M110.101527
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taylor, E. B., and Rutter, J. (2011). Mitochondrial quality control by the ubiquitin-proteasome system. Biochem. Soc. Trans. 39, 1509–1513. doi: 10.1042/BST0391509
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tenreiro, S., Reimao-Pinto, M. M., Antas, P., Rino, J., Wawrzycka, D., Macedo, D., et al. (2014). Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson's disease. PLoS Genet. 10:e1004302. doi: 10.1371/journal.pgen.1004302
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tofaris, G. K., Kim, H. T., Hourez, R., Jung, J. W., Kim, K. P., and Goldberg, A. L. (2011). Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 17004–17009. doi: 10.1073/pnas.1109356108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Treusch, S., Hamamichi, S., Goodman, J. L., Matlack, K. E., Chung, C. Y., Baru, V., et al. (2011). Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science 334, 1241–1245. doi: 10.1126/science.1213210
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Uversky, V. N. (2007). Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 103, 17–37. doi: 10.1111/j.1471-4159.2007.04764.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Valenti, D., Vacca, R. A., Guaragnella, N., Passarella, S., Marra, E., and Giannattasio, S. (2008). A transient proteasome activation is needed for acetic acid-induced programmed cell death to occur in Saccharomyces cerevisiae. FEMS Yeast Res. 8, 400–404. doi: 10.1111/j.1567-1364.2008.00348.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vandebroek, T., Terwel, D., Vanhelmont, T., Gysemans, M., Van Haesendonck, C., Engelborghs, Y., et al. (2006). Microtubule binding and clustering of human Tau-4R and Tau-P301L proteins isolated from yeast deficient in orthologues of glycogen synthase kinase-3beta or cdk5. J. Biol. Chem. 281, 25388–25397. doi: 10.1074/jbc.M602792200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vandebroek, T., Vanhelmont, T., Terwel, D., Borghgraef, P., Lemaire, K., Snauwaert, J., et al. (2005). Identification and isolation of a hyperphosphorylated, conformationally changed intermediate of human protein tau expressed in yeast. Biochemistry 44, 11466–11475. doi: 10.1021/bi0506775
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vanhelmont, T., Vandebroek, T., De Vos, A., Terwel, D., Lemaire, K., Anandhakumar, J., et al. (2010). Serine-409 phosphorylation and oxidative damage define aggregation of human protein tau in yeast. FEMS Yeast Res. 10, 992–1005. doi: 10.1111/j.1567-1364.2010.00662.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van Leeuwen, F. W., de Kleijn, D. P., van den Hurk, H. H., Neubauer, A., Sonnemans, M. A., Sluijs, J. A., et al. (1998). Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science 279, 242–247. doi: 10.1126/science.279.5348.242
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van Tijn, P., Dennissen, F. J., Gentier, R. J., Hobo, B., Hermes, D., Steinbusch, H. W., et al. (2012). Mutant ubiquitin decreases amyloid beta plaque formation in a transgenic mouse model of Alzheimer's disease. Neurochem. Int. 61, 739–748. doi: 10.1016/j.neuint.2012.07.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van Tijn, P., de Vrij, F. M., Schuurman, K. G., Dantuma, N. P., Fischer, D. F., van Leeuwen, F. W., et al. (2007). Dose-dependent inhibition of proteasome activity by a mutant ubiquitin associated with neurodegenerative disease. J. Cell Sci. 120, 1615–1623. doi: 10.1242/jcs.03438
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van Tijn, P., Hobo, B., Verhage, M. C., Oitzl, M. S., van Leeuwen, F. W., and Fischer, D. F. (2011). Alzheimer-associated mutant ubiquitin impairs spatial reference memory. Physiol. Behav. 102, 193–200. doi: 10.1016/j.physbeh.2010.11.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van Tijn, P., Verhage, M. C., Hobo, B., van Leeuwen, F. W., and Fischer, D. F. (2010). Low levels of mutant ubiquitin are degraded by the proteasome in vivo. J. Neurosci. Res. 88, 2325–2337. doi: 10.1002/jnr.22396
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verhoef, L. G., Heinen, C., Selivanova, A., Halff, E. F., Salomons, F. A., and Dantuma, N. P. (2009). Minimal length requirement for proteasomal degradation of ubiquitin-dependent substrates. FASEB J. 23, 123–133. doi: 10.1096/fj.08-115055
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, S., Thibault, G., and Ng, D. T. (2011). Routing misfolded proteins through the multivesicular body (MVB) pathway protects against proteotoxicity. J. Biol. Chem. 286, 29376–29387. doi: 10.1074/jbc.M111.233346
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Meriin, A. B., Zaarur, N., Romanova, N. V., Chernoff, Y. O., Costello, C. E., et al. (2009). Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J. 23, 451–463. doi: 10.1096/fj.08-117614
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weissman, A. M., Shabek, N., and Ciechanover, A. (2011). The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell Biol. 12, 605–620. doi: 10.1038/nrm3173
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Willingham, S., Outeiro, T. F., Devit, M. J., Lindquist, S. L., and Muchowski, P. J. (2003). Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 302, 1769–1772. doi: 10.1126/science.1090389
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Winderickx, J., Delay, C., De Vos, A., Klinger, H., Pellens, K., Vanhelmont, T., et al. (2008). Protein folding diseases and neurodegeneration: lessons learned from yeast. Biochim. Biophys. Acta 1783, 1381–1395. doi: 10.1016/j.bbamcr.2008.01.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Witt, S. N., and Flower, T. R. (2006). alpha-Synuclein, oxidative stress and apoptosis from the perspective of a yeast model of Parkinson's disease. FEMS Yeast Res. 6, 1107–1116. doi: 10.1111/j.1567-1364.2006.00135.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xilouri, M., Brekk, O. R., and Stefanis, L. (2013). alpha-Synuclein and protein degradation systems: a reciprocal relationship. Mol. Neurobiol. 47, 537–551. doi: 10.1007/s12035-012-8341-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yeger-Lotem, E., Riva, L., Su, L. J., Gitler, A. D., Cashikar, A. G., King, O. D., et al. (2009). Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat. Genet. 41, 316–323. doi: 10.1038/ng.337
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yim, N., Ryu, S. W., Han, E. C., Yoon, J., Choi, K., and Choi, C. (2014). Mutant ubiquitin UBB+1 induces mitochondrial fusion by destabilizing mitochondrial fission-specific proteins and confers resistance to oxidative stress-induced cell death in astrocytic cells. PLoS ONE 9:e99937. doi: 10.1371/journal.pone.0099937
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zabrocki, P., Bastiaens, I., Delay, C., Bammens, T., Ghillebert, R., Pellens, K., et al. (2008). Phosphorylation, lipid raft interaction and traffic of alpha-synuclein in a yeast model for Parkinson. Biochim. Biophys. Acta 1783, 1767–1780. doi: 10.1016/j.bbamcr.2008.06.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zabrocki, P., Pellens, K., Vanhelmont, T., Vandebroek, T., Griffioen, G., Wera, S., et al. (2005). Characterization of alpha-synuclein aggregation and synergistic toxicity with protein tau in yeast. FEBS J. 272, 1386–1400. doi: 10.1111/j.1742-4658.2005.04571.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zischka, H., Braun, R. J., Marantidis, E. P., Büringer, D., Bornhövd, C., Hauck, S. M., et al. (2006). Differential analysis of Saccharomyces cerevisiae mitochondria by free flow electrophoresis. Mol. Cell. Proteomics 5, 2185–2200. doi: 10.1074/mcp.T600018-MCP200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: ubiquitylation, ubiquitin-proteasome system, autophagy, ubiquitin-dependent vesicular trafficking, neurodegeneration, cell death, Saccharomyces cerevisiae
Citation: Braun RJ (2015) Ubiquitin-dependent proteolysis in yeast cells expressing neurotoxic proteins. Front. Mol. Neurosci. 8:8. doi: 10.3389/fnmol.2015.00008
Received: 20 October 2014; Accepted: 24 February 2015;
Published: 12 March 2015.
Edited by:
Fred Van Leeuwen, Maastricht University, NetherlandsReviewed by:
Nico P. Dantuma, Karolinska Institute, SwedenZhiqun Tan, University of California, Irvine, USA
Copyright © 2015 Braun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf J. Braun, Institut für Zellbiologie, Universität Bayreuth, 95440 Bayreuth, Germany ralf.braun@uni-bayreuth.de