Decreased Levels of Foldase and Chaperone Proteins Are Associated with an Early-Onset Amyotrophic Lateral Sclerosis
 Melania Filareti1,2†
Melania Filareti1,2†  Silvia Luotti1†
Silvia Luotti1†  Laura Pasetto1 Mauro Pignataro1 Katia Paolella1 Paolo Messina1 Elisabetta Pupillo1 Massimiliano Filosto3
Laura Pasetto1 Mauro Pignataro1 Katia Paolella1 Paolo Messina1 Elisabetta Pupillo1 Massimiliano Filosto3  Christian Lunetta4
Christian Lunetta4  Jessica Mandrioli5 Giuseppe Fuda6 Andrea Calvo6 Adriano Chiò6 Massimo Corbo2
Jessica Mandrioli5 Giuseppe Fuda6 Andrea Calvo6 Adriano Chiò6 Massimo Corbo2  Caterina Bendotti1 Ettore Beghi1
Caterina Bendotti1 Ettore Beghi1  Valentina Bonetto1*
Valentina Bonetto1*- 1Istituto Di Ricerche Farmacologiche Mario Negri, Istituti di Ricovero e Cura a Carattere Scientifico (IRCCS), Milan, Italy
- 2Department of Neurorehabilitation Sciences, Casa Cura Policlinico, Milan, Italy
- 3Center for Neuromuscular Diseases and Neuropathies, Unit of Neurology, ASST Spedali Civili and University of Brescia, Brescia, Italy
- 4NEuroMuscular Omnicentre, Fondazione Serena Onlus, Milan, Italy
- 5Department of Neuroscience, Azienda Ospedaliero Universitaria di Modena, Ospedale Civile S. Agostino-Estense, Modena, Italy
- 6ALS Center, Department of Neuroscience Rita Levi Montalcini, University of Torino, Torino, Italy
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by a progressive upper and lower motor neuron degeneration. One of the peculiar clinical characteristics of ALS is the wide distribution in age of onset, which is probably caused by different combinations of intrinsic and exogenous factors. We investigated whether these modifying factors are converging into common pathogenic pathways leading either to an early or a late disease onset. This would imply the identification of phenotypic biomarkers, that can distinguish the two populations of ALS patients, and of relevant pathways to consider in a therapeutic intervention. Toward this aim a differential proteomic analysis was performed in peripheral blood mononuclear cells (PBMC) from a group of 16 ALS patients with an age of onset ≤55 years and a group of 16 ALS patients with an age of onset ≥75 years, and matched healthy controls. We identified 43 differentially expressed proteins in the two groups of patients. Gene ontology analysis revealed that there was a significant enrichment in annotations associated with protein folding and response to stress. We next validated a selected number of proteins belonging to this functional group in 85 patients and 83 age- and sex-matched healthy controls using immunoassays. The results of the validation study confirmed that there was a decreased level of peptidyl-prolyl cis-trans isomerase A (also known as cyclophilin A), heat shock protein HSP 90-alpha, 78 kDa glucose-regulated protein (also known as BiP) and protein deglycase DJ-1 in PBMC of ALS patients with an early onset. Similar results were obtained in PBMC and spinal cord from two SOD1G93A mouse models with an early and late disease onset. This study suggests that a different ability to upregulate proteins involved in proteostasis, such as foldase and chaperone proteins, may be at the basis of a different susceptibility to ALS, putting forward the development of therapeutic approaches aiming at boosting the protein quality control system.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by a progressive upper and lower motor neuron degeneration. ALS is a highly heterogeneous disorder, with phenotypic variability involving a number of clinical aspects such as site of onset, age of onset, rate of progression, nonmotor involvement and response to therapy (Beghi et al., 2011). This makes it difficult to decipher pathogenesis and to develop therapeutic strategies, also because biomarkers that reflect heterogeneity and may be useful for stratification are missing. A combination of genetic and environmental/exogenous modifying factors are believed to underlie this variability. The identification of the pathways affected by the modifying factors is of great interest, as they may be targets of therapeutic intervention.
Age is a known risk factor for ALS. The majority of ALS patients have disease onset between 55 and 75 years of age, however patients with an age of onset <55 years and >75 years do exist and represent, respectively, the 22% and the 16% of all patients, as recently reported in a large multicenter Italian study (Calvo et al., 2016). Several factors, both genetic and environmental/exogenous, have been reported to modify age of onset in patients and animal models of ALS. FUS mutations have been frequently associated with early-onset disease in familial and sporadic cases (Chiò et al., 2009; Hübers et al., 2015). Earlier onset of ALS has been also observed in soldiers and professional soccer players (Haley, 2003; Chiò et al., 2005; Pupillo et al., 2012). In these patients, an anticipated onset might be linked to heavy physical exercise, repeated trauma, and/or exposure to as yet unknown toxic agents in genetically predisposed individuals (Beghi and Morrison, 2005; Pupillo et al., 2012; Beghi, 2013). A mutant SOD1G93A mouse model of ALS on a 129Sv genetic background has an earlier age of onset than a SOD1G93A mouse model on a C57BL6 genetic background despite an equal expression of the mutant protein (Marino et al., 2015). These mice have also faster disease progression and an intrinsic marked down-regulation of specific pathways involved in mitochondrial function and protein quality control (Nardo et al., 2013, 2016; Marino et al., 2015). Conversely, ApoE2 polymorphism and lower EPHA4 expression have been correlated with delayed age of onset in ALS patients (Li et al., 2004; Van Hoecke et al., 2012). Moreover, in mutant SOD1 mice female showed a later onset than male and this delay further increased with exercise (Veldink et al., 2003).
Summarizing, different combinations of intrinsic and exogenous factors, that can be hardly discerned, may lead to an anticipated or a delayed onset of the disease. We wanted to test whether or not modifying factors are converging into common pathogenic pathways leading either to an early or a late disease onset. This would imply the identification of phenotypic biomarkers, that can distinguish the two populations of ALS patients, and of relevant pathways to consider in a therapeutic intervention. Toward this aim a significant cohort of incident ALS patients with either early (<55 years of age) or late (>75 years of age) disease onset was recruited from population-based Italian registries, and several analyses were performed. We found that an epigenetic rearrangement such as whole-blood DNA methylation could not distinguish the two groups of ALS patients (Tremolizzo et al., 2014). However, difference in plasma amino acid levels were observed in relation to the age of onset (Cecchi et al., 2014). In this work, we identified protein phenotypic biomarkers in peripheral blood mononuclear cells (PBMC) of the same cohort of patients indicating that protein levels in PBMC could be good biological correlates of the age of onset in ALS. The same phenotypic biomarkers were also analyzed in the two mutant SOD1 mouse models of ALS with early and late disease onset indicating common changes between the sporadic and SOD1-linked forms. These biomarkers are proteins involved in the maintenance of protein homeostasis, or proteostasis. Proteostasis is attained through several quality-control systems that assist protein folding, clear misfolded proteins and respond to protein aggregation, whose major players are molecular chaperones. Interestingly, impaired proteostasis has emerged as a key contributor to the pathogenesis of ALS (Ruegsegger and Saxena, 2016).
Materials and Methods
Antibodies
The antibodies for Western and dot blot, used for human and mouse samples, were: rabbit polyclonal anti-peptidyl-prolyl cis-trans isomerase A (PPIA) antibody (1:2500 dilution; Millipore), rabbit polyclonal anti-heat shock protein HSP 90-alpha (HSP90) (1:3000 dilution; Stressgen), rabbit polyclonal anti-78 kDa glucose-regulated protein (GRP78) (1:500 dilution; Santa Cruz Biotechnology Inc.), mouse monoclonal anti-endoplasmic reticulum protein 57 (ERp57) (1:2500 dilution; Stressgen), rabbit monoclonal anti-protein deglycase DJ-1 (DJ-1) (1:1500 dilution; Abcam), mouse monoclonal anti-heat shock cognate 71 kDa protein (HSC70) (1:1000 dilution; Santa Cruz Biotechnology Inc.), rabbit polyclonal anti-TDP-43 (1:2500 dilution; Proteintech), goat anti-mouse or anti-rabbit peroxidase-conjugated secondary antibodies (1:5000; Santa Cruz Biotechnology Inc.).
Subjects
Following approval of the protocol by the ethics committees (Comitato Etico Interaziendale A.O.U. San Giovanni Battista di Torino – A.O. “C.T.O. Maria Adelaide di Torino”, Torino, Italy; Comitato etico degli Spedali Civili di Brescia, Brescia, Italy; Comitato Etico Provinciale di Modena, Modena, Italy) written informed consent was obtained from all participating subjects. Patients, enrolled in three Italian population-based registries (Lombardia, Piemonte, Emilia Romagna), were newly diagnosed definite, probable or possible ALS, according to the El Escorial criteria (Brooks, 1994). To be eligible, patients had to be ≤55 years of age (early ALS) or ≥75 years of age (late ALS) at diagnosis. Controls were residency-, sex-, and age-matched (±5 years), randomly chosen within the same hospital of the patient among subjects admitted for surgery for a non-spontaneously evolving disease. Subjects enrolled for the discovery phase study were 16 early ALS, 16 late ALS and 32 controls (early and late controls), with demographic and clinical characteristics described in Table 1. Subjects enrolled in the validation study were 85 ALS patients and 83 controls with demographic and clinical characteristics described in Table 2. All cases were sporadic. A wide, although partial screening for ALS-causative mutations was performed (Tremolizzo et al., 2014). Two early ALS cases carried a TARDP mutation (M359V, G368S), one late ALS case carried a SOD1 mutation (D90A) and another one an OPTN mutation (L500P).
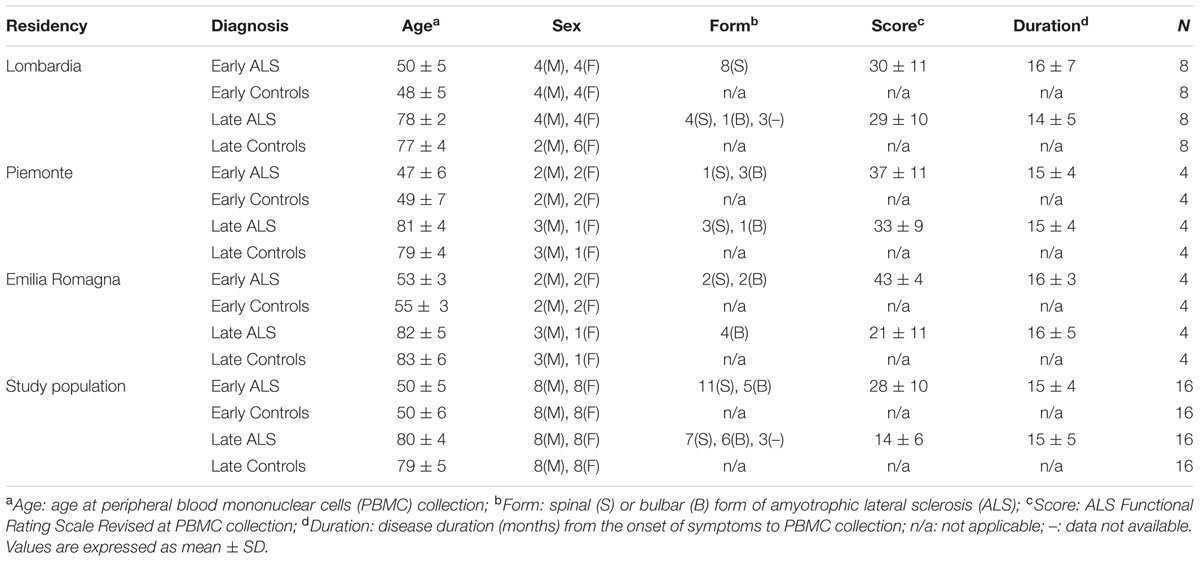
TABLE 1. Demographic and clinical characteristics of the enrolled population in the proteomic study.
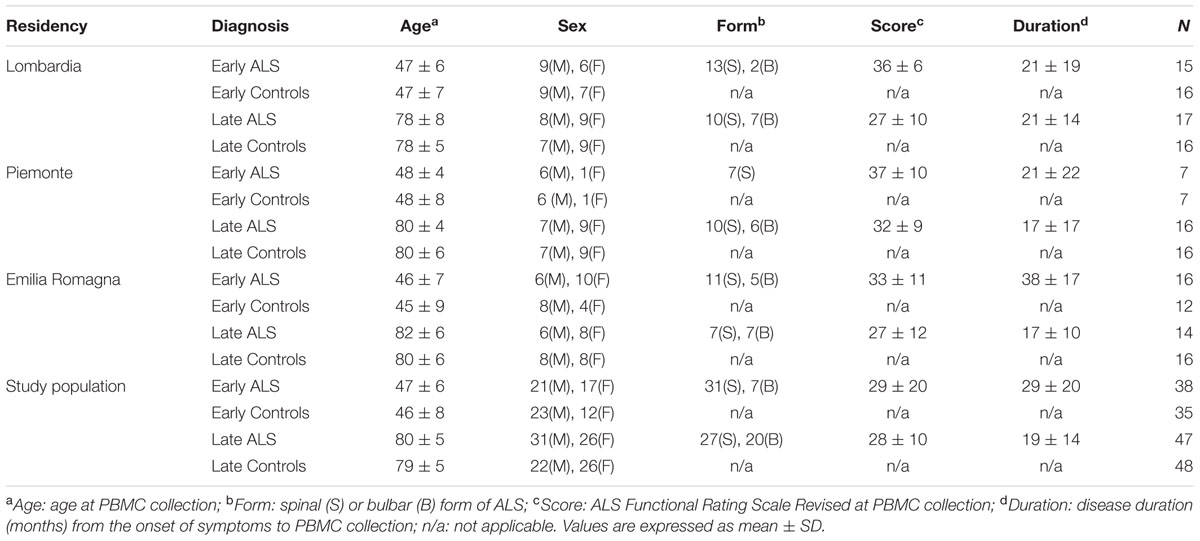
TABLE 2. Demographic and clinical characteristics of the enrolled population in the validation study.
Mice
Procedures involving animals and their care were approved by the Institutional Animal Care and Use Committee and were conducted in conformity with the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; Authorisation n.19/2008-A issued March 6, 2008 by Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorization for persons conducting animal experiments (Quality Management System Certificate – UNI EN ISO 9001:2008 – Reg. No. 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition) and EU directives and guidelines (EEC Council Directive 2010/63/UE). The Statement of Compliance (Assurance) with the Public Health Service (PHS) Policy on Human Care and Use of Laboratory Animals has been recently reviewed (9/9/2014) and will expire on September 30, 2019 (Animal Welfare Assurance #A5023-01). Animals were bred and maintained at the IRCCS-Istituto di Ricerche Farmacologiche Mario Negri, Milan under standard conditions, temperature 21 ± 1°C, relative humidity 55 ± 10%, 12h light schedule, food and water ad libitum. Transgenic SOD1G93A mice expressing about 20 copies of mutant human SOD1 with a Gly93Ala substitution (B6SJL-TgSOD1G93A-1Gur) were originally obtained from Jackson Laboratories and maintained on a C57BL/6JOlaHsd (C57) genetic background at Harlan Italy S.R.L., Bresso, Milan, Italy. By crossbreeding C57 SOD1G93A mice with 129S2/SvHsd (129Sv) mice for >15 generations, SOD1G93A mice on a 129Sv homogenous background were generated (Marino et al., 2015). For biochemical analyses, female mice were deeply anesthetized with ketamine hydrochloride (IMALGENE, 150 mg/kg, Alcyon Italia S.p.A.) and medetomidine hydrochloride (DOMITOR, 2 mg/kg, Alcyon Italia S.p.A.) by intraperitoneal injection and euthanized by decapitation at symptom onset: for 129Sv SOD1G93A mice at 14 weeks of age, and for C57 SOD1G93A mice at 17 weeks of age. The corresponding age-matched nontransgenic mice were used as controls.
PBMC Isolation and Protein Extraction
Peripheral blood mononuclear cells were isolated from peripheral venous blood of human subjects essentially as previously described (Nardo et al., 2011). Briefly, samples of blood were collected in EDTA pre-coated tubes (BD Vacutainer K2EDTA) and PBMC were isolated from EDTA blood by Ficoll-Hypaque (Ficoll-PlaqueTM Plus, GE Healthcare) density gradient centrifugation at 800 × g for 30 min at 18–20°C. Mononuclear cells were collected from the interface and washed three times with RPMI 1640 medium (EuroClone). Platelets were eliminated by an additional wash and centrifugation at 200 × g for 10 min. PBMC were stored as pellets at –80°C. PBMC from mice were isolated from blood sampled by intracardiac puncture and collected in EDTA pre-coated vials (BD Vacutainer K2EDTA). PBMC were isolated from EDTA-blood by Lympholite (Lympholyte-Mammal, Cedarlane) density gradient centrifugation at 1000 × g for 25 min at 18–20°C. Mononuclear cells were harvested from the interface and washed once at 800 × g for 10 min with RPMI 1640 (EuroClone) supplemented with 2.5 mM EDTA, and stored as pellets at –80°C. Before analysis human and mouse PBMC were lysed in 20 mM Tris-HCl pH 7.5, 0.1% NP40, and 0.1% SDS supplemented with Protease Inhibitors (Sigma), boiled for 5 min and centrifuged at 16.000 × g for 10 min at 4°C. Supernatants were analyzed by Western and dot blot analyses. Proteins were quantified by the BCA protein assay (Pierce).
Protein Extraction from the Spinal Cord of the Mice
The spinal cord was flushed from the vertebral column and sectioned into cervical, thoracic and lumbar segments. The samples were immediately frozen on dry-ice and stored at –80°C until analysis. Lumbar spinal cords were homogenized by sonication in 1% boiling SDS. Protein homogenates were further boiled for 10 min and centrifuged at 13.500 × g for 5 min at 4°C. Supernatants were analyzed by Western and dot blot analyses. Proteins were quantified by the BCA protein assay (Pierce).
Two-Dimensional Difference in Gel Electrophoresis (2D DIGE)
Peripheral blood mononuclear cells proteins were prepared for 2D DIGE analysis as follows: four pools of 20 μg from 16 early ALS, 16 early controls, 16 late ALS, and 16 late controls were methanol-precipitated overnight. Proteins were then dissolved in 30 mM Tris-HCl pH 8.5, 7 M urea, 2 M thiourea, CHAPS 4% (w/v) and Cydye-labeled according to the manufacturer’s instructions (GE Healthcare) with minor modifications. Briefly, 20 μg of each pool was labeled with 200 pmol of Cy3 or Cy5 dye for 30 min in ice in the dark. To exclude preferential labeling of the dyes, each sample was also reverse labeled. As an internal standard, aliquots of each pool were mixed and labeled with Cy2 dye. Labeled samples were then resuspended in Destreak SolutionTM (GE Healthcare) with IPG buffer pH 3-11 NL 1% v/v (GE Healthcare) and loaded into 11 cm-IPG strips pI range 3-11NL (GE Healthcare). Isoelectrofocusing was done on an IPGphor apparatus (GE Healthcare) with the following protocol: 300 Vhrs at 30 V, 50 Vhrs at 200 V, 1500 Vhrs at 2000 V, 2800 Vhrs of a linear gradient up to 3500 V, 4800 Vhrs at 3500 V, 8500 Vhr of a linear gradient up to 8000 V, and 30000 Vhr at 8000 V. SDS-PAGE was done using Precast Tris-HCl 10% polyacrylamide SDS gel (Biorad). Eight 2D gels were run with the four experimental groups. Each gel contained two experimental groups, one Cy3-labeled, the other Cy5-labeled plus the Cy2-labeled internal standard. Gel images were captured by the laser scanner Molecular Imager FX (Bio-Rad). Image analysis was done with Progenesis Same Spot software (Nonlinear Dynamics). For each spot the normalized volume was standardized against internal standard spot normal volume. The values for each spot in each group were expressed as the mean of the Cy3- and Cy5-labeled analyses. Values for early and late ALS were further normalized to the means values of early and late controls, respectively. Differential spots were considered only those that had a fold change (normalized late ALS/ normalized early ALS) ≤0.8 or ≥1.3.
Protein Identification
Differential protein spots were located and excised from 2D gels with the EXQuestTM spot cutter (Bio-Rad). Spots were processed and gel-digested with modified trypsin from bovine pancreas (Roche) and identified by mass spectrometry (MS), as previously described (Nardo et al., 2011). Peptide mass fingerprinting and tandem MS/MS were done on a 4800 MALDI TOF/TOF mass spectrometer (Applied Biosystems). The mass spectra were internally calibrated with trypsin autolysis fragments. The five most abundant precursor ions, out of the exclusion mass list (ions from human keratin and trypsin), were selected for MS/MS analysis. The combined MS and MS/MS data were submitted by GPS Explorer v.3.6 software (Applied Biosystems) to the MASCOT database search engine (Version 2.1, Matrix Science) and searched with the following parameters: Uniprot_Swissprot 2012x database over all Homo sapiens protein sequences deposited, no fixed modifications, as possible modifications carboamidomethylation of cysteine and oxidation of methionine, 1 missed trypsin cleavage, a mass tolerance of ±0.1 Da for the peptide masses and ±0.3 Da for the MS/MS fragment ion masses. A protein was regarded as identified if the MASCOT protein score, based on the combined MS and MS/MS data, was above the 5% significance threshold for the database (Pappin et al., 1993).
Gene Ontology (GO) Analysis
Identified proteins were classified on the basis of gene ontology (GO) annotations provided by PANTHER1 (Mi et al., 2013). PANTHER overrepresentation test was performed with the list of the identified differential proteins against the whole Homo sapiens reference list and against a list of about 800 proteins extracted from published works of 2D-based proteomics of lymphocytes/monocytes (Verhoeckx et al., 2004; Rosengren et al., 2005; Xie et al., 2005; Azkargorta et al., 2006; Han et al., 2006; Huang et al., 2006; Ramirez-Boo et al., 2006; Salonen et al., 2006; Zada et al., 2006; Henrich et al., 2007; Lai et al., 2007; Rakkola et al., 2007; Skopeliti et al., 2007; Vergara et al., 2008). PANTHER GO-Slim Biological Process and GO-Slim Molecular Function were used as annotation data sets (version 11.1, released 2016-07-15).
Dot Blot Analysis
Dot blot was used in the validation study after verification that the antibody detected specific bands in WB (Supplementary Figure S1). Proteins (3 μg) were directly loaded onto nitrocellulose Trans-Blot transfer 0.2–0.45 μm (Bio-Rad) membranes, depositing each sample on the membrane by vacuum filtration, as described previously (Nardo et al., 2011). An internal standard which is a pool of all samples in the analysis (healthy controls and patients) was deposited in triplicates. Dot blot membranes were blocked with 3% (w/v) BSA (Sigma) and 0.1% (v/v) Tween 20 in Tris-buffered saline, pH 7.5, incubated with primary antibodies, then with peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology Inc.). Blots were developed with LuminataTM Forte Western Chemiluminescent HRP Substrate (Millipore) on the ChemiDoc XRS system (Bio-Rad). Densitometry was done with Progenesis PG240 v2006 software (Nonlinear Dynamics). The immunoreactivity of the different proteins was normalized to Ponceau Red staining (Fluka) and to the internal standard of each membrane.
Statistical Analysis
Immunoreactivity values for each protein analyzed in early and late ALS were normalized to the means values of early and late controls, respectively. The statistical analysis of the protein expression in the two groups was done by Student’s t-test using PRISM software (GraphPad, San Diego, CA, USA, version 6.01).
Results
Identification of Candidate Phenotypic Biomarkers of ALS in PBMC by 2D-DIGE-Based Proteomics
Figure 1A schematically shows the strategy we used to identify and validate phenotypic protein biomarkers of ALS in PBMC. In the discovery phase, PBMC of ALS patients with ≤55 years of age (early ALS, EA) or ≥75 years of age (late ALS) at diagnosis and matched early and late controls were analyzed by 2D DIGE. The analysis was done with 16 pooled samples for each of the four experimental groups (Table 1). Spot volume values for early and late ALS were normalized to the mean values of early and late controls, respectively. Spots were considered differential if fold change (normalized late ALS/normalized early ALS) was ≤0.8 or ≥1.3. A total of 68 differential spots were detected (Figure 1B). From these spots 43 unique proteins were identified by MS as candidate protein biomarkers (Table 3). Some of the proteins were identified in multiple spots at different pI and Mw, which are fragments or post-translationally modified isoforms of the same protein (Supplementary Table S1). We grouped the differential proteins on the basis of their most recognized function and on GO annotations (Table 3). The overrepresentation test performed with the list of the differential proteins against the Homo sapiens reference list indicated isomerase activity (GO:0016853), associated with PPIA and ERp57, among the most significantly enriched (p = 0.03; fold = 7.8) molecular functions. Protein folding (GO:0006457) and response to stress (GO:0006950) were among the most significantly enriched (p < 0.01; fold>4) biological processes identified. The overrepresentation test performed also against a reference list of lymphocyte/monocyte proteins, identified in 2D-based proteomic studies, indicated response to stress as one of the most significantly enriched (p = 0.03; fold = 2.5) biological processes. This suggested that proteins associated with these molecular functions and biological processes (Table 3), such as PPIA, HSP90, GRP78, ERp57, and DJ-1, were attracting candidate phenotypic biomarkers to further validate in a larger and well-characterized cohort of patients with early and late disease onset.
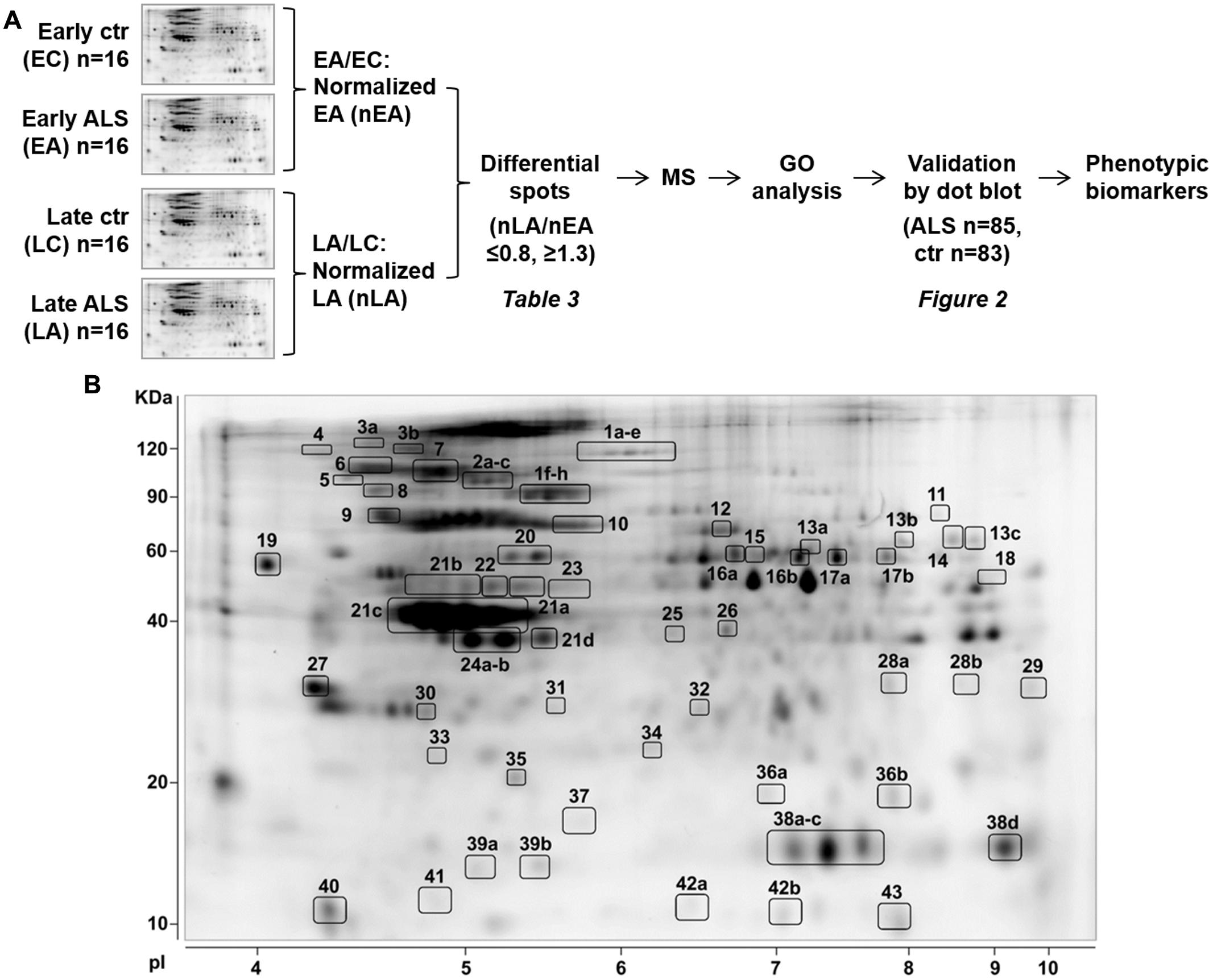
FIGURE 1. Identification of phenotypic biomarkers in PBMC of early and late ALS patients. (A) Scheme of the strategy to identify phenotypic protein biomarkers in PBMC of patients early onset ALS (EA) and late-onset ALS (LA) and matched controls, early (EC), and late (LC). 2D-DIGE analysis was done with 16 pooled samples for each experimental group and spot volume values for EA and LA were normalized to EC and LC, respectively (nEA, nLA). Spots were considered differential if fold change (nLA/nEA) was ≤0.8 or ≥1.3 and identified by mass spectrometry (MS, Supplementary Table S1). Differential proteins were classified on the basis of GO annotations and a selected number of proteins associated with “protein folding and response to stress” biological function was validated by dot blot immunoassays. (B) Representative Sypro-Ruby stained 2D gel of the PBMC proteome. The numbered spots correspond to the differential proteins listed in Table 3 and Supplementary Table S1.
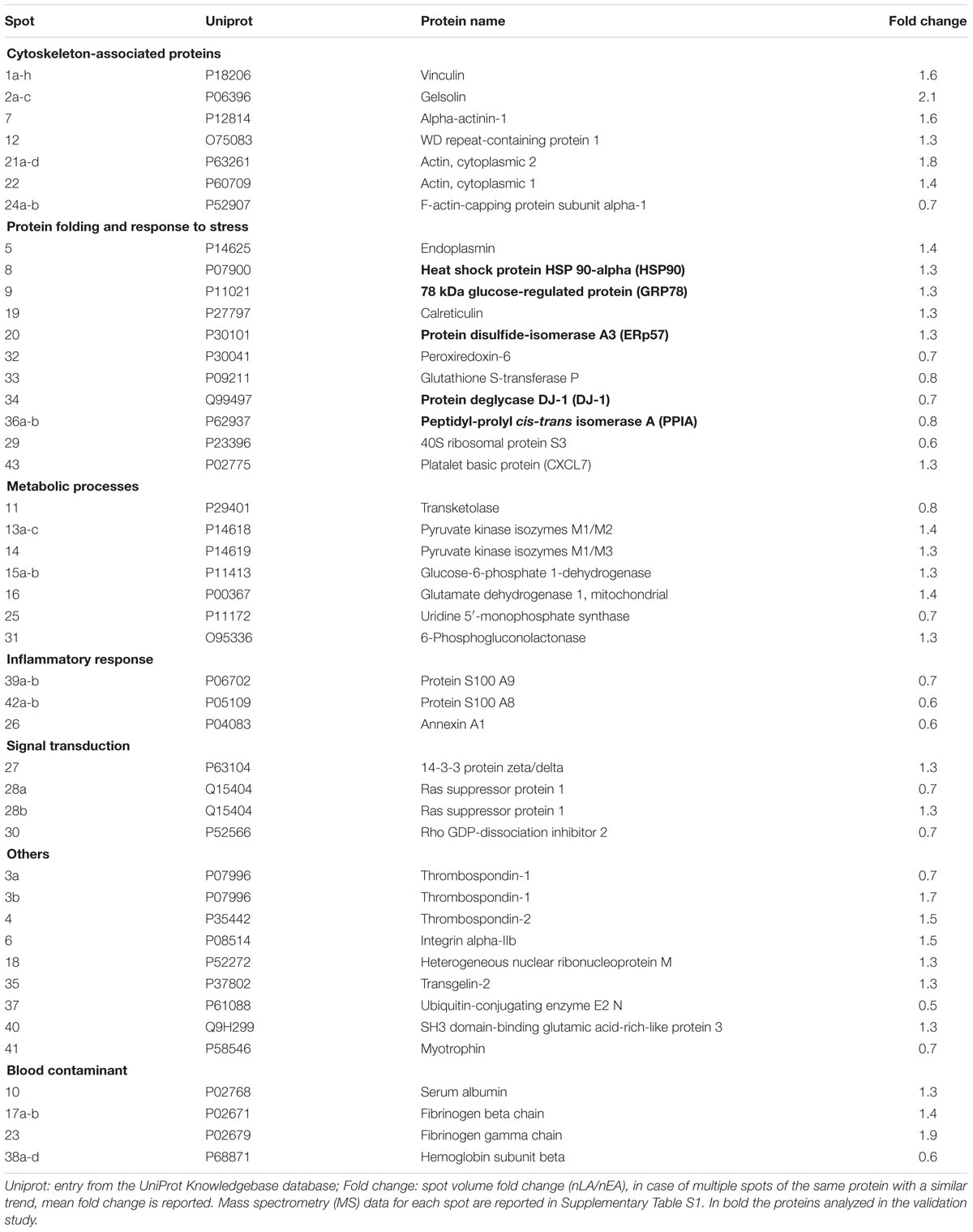
TABLE 3. Differential spots.
Validation of Candidate Phenotypic Biomarkers
In the validation study, we analyzed total protein levels of candidate phenotypic biomarkers, PPIA, HSP90, GRP78, ERp57, and DJ-1, by dot blot analysis in PBMC samples from an independent set of ALS patients (n = 85), n = 38 early ALS and n = 47 late ALS, and matched controls (n = 83), n = 35 early controls and n = 48 late controls (Table 2). Immunoreactivity values for each protein in early and late ALS patients were normalized to the mean values of early and late controls, respectively, similarly to the proteomic analysis. We found that PPIA, HSP90, GRP78, and DJ-1 were significantly lower in early ALS than in late ALS, and therefore can be considered phenotypic biomarkers (Figures 2A–C,E), while ERp57 did not change (Figure 2D). Moreover, PPIA, HSP90, GRP78, and DJ-1 were similarly regulated, with a decreased level in early ALS compared to early controls and an opposite tendency in late ALS (Supplementary Figures S2A–C,E), suggesting that early ALS patients have some defects in the response to stress pathways. In the same validation analysis, we measured another chaperone protein, HSC70, and TDP-43, both not differential in the 2D-DIGE analysis, but up-regulated in PBMC of a cohort of ALS patients with a mean age of onset of 62 ± 10 years, previously reported in a work by our laboratory (Nardo et al., 2011). We found that HSC70 could not distinguish early from late ALS (Figure 2F), but it was up-regulated in late ALS compared to controls (Supplementary Figure S2F). We confirmed that total TDP-43 protein levels in PBMC underscores disease (Figure 2G), in fact it was higher in both early and late ALS compared to controls, but could not discriminate early ALS from late ALS (Figure 2H). It has to be noted that in our assay the total TDP-43 protein level comprise the 43-kDa full-length and fragmented forms (Supplementary Figure S1A).
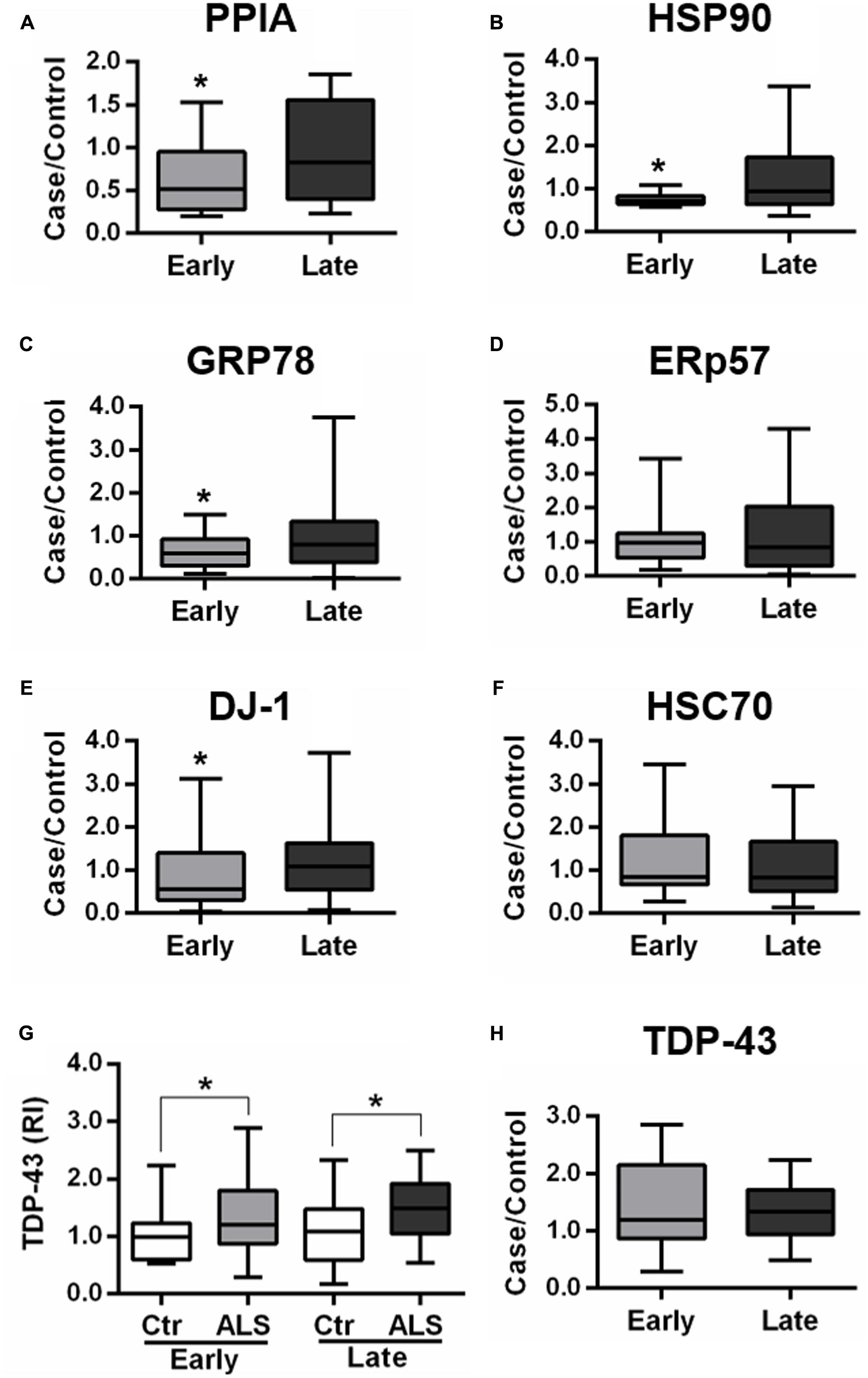
FIGURE 2. Validation of candidate phenotypic biomarkers in PBMC of early and late ALS patients. (A–H) PPIA, HSP90, GRP78, ERp57, DJ-1, HSC70, and TDP-43 were analyzed by dot blot immunoassays in PBMC samples from an independent set of ALS patients (n = 85), n = 38 EA and n = 47 LA, and matched controls (n = 83), n = 35 EC and n = 48 LC. Immunoreactivity was normalized to protein loading, as assessed by Ponceau Red staining, and then to the mean values of matched controls (A–F,H). We found that PPIA, HSP90, GRP78, and DJ-1 were significantly lower in early ALS than in late ALS (A–C,E) and that TDP-43 was higher in both early and late ALS compared to matched controls (G). ∗p < 0.05, by Student’s t-test.
The experimental set up of the analysis has strengths and limitations. The major strengths are the population base and the enrolment of patients with newly diagnosed ALS. Our patients are a fairly representative sample of an incident ALS population. The major limitation is the small sample size. The analysis may thus be underpowered for some differences to achieve statistical significance. Then, we did not adjust our data for multiple comparisons. Thus, we cannot exclude that some of the associations are chance findings. However, due to the exploratory nature of the analysis, we deliberately decided not to use the Bonferroni correction for multiple testing.
Analysis of Phenotypic Biomarkers in Two SOD1G93A Mouse Models with Different Age of Onset
We also analyzed total protein levels of the candidate phenotypic biomarkers, PPIA, HSP90, GRP78, ERp57, and DJ-1 in PBMC of two SOD1G93A mouse models, 129Sv SOD1G93A and C57 SOD1G93A mice and corresponding non-transgenic controls by dot blot analysis. These mice despite having equal SOD1G93A expression have a different disease phenotype, because of the different genetic background (Marino et al., 2015). In particular, 129Sv and C57 SOD1G93A mice have symptom onset at 14 and 17 weeks of age, respectively, thus representing our mouse models of early (129Sv) and late (C57) disease onset. Similarly, to the human samples, immunoreactivity values for each protein in early and late mouse models were normalized to the mean values of the corresponding non-transgenic controls. We found that PPIA and HSP90 were significantly lower in early SOD1G93A mice than in late SOD1G93A mice, as much as in the human samples (Figures 3A,B), and there was a significant lower level of the two proteins in the early SOD1G93A mice compared to their non-transgenic controls (Supplementary Figures S3A,B). DJ-1 had a similar behavior but it did not reach statistical significance (Figure 3E and Supplementary Figure S3E). GRP78 was instead significantly higher in early SOD1G93A mice than in late SOD1G93A mice (Figure 3C), in contrast with the human samples, with an increased level in the early SOD1G93A mice compared to controls (Supplementary Figure S3C). ERp57 was not significantly different in late and early SOD1G93A mice, as much as in the human samples (Figure 3D), but it was significantly lower in late SOD1G93A mice compared to controls (Supplementary Figure S3D). Finally, TDP-43 was not significantly different in early and late SOD1G93A mice, as in the human samples (Figure 3F), but was not different compared to corresponding nontransgenic controls, in contrast with the human samples (Supplementary Figure S3F).
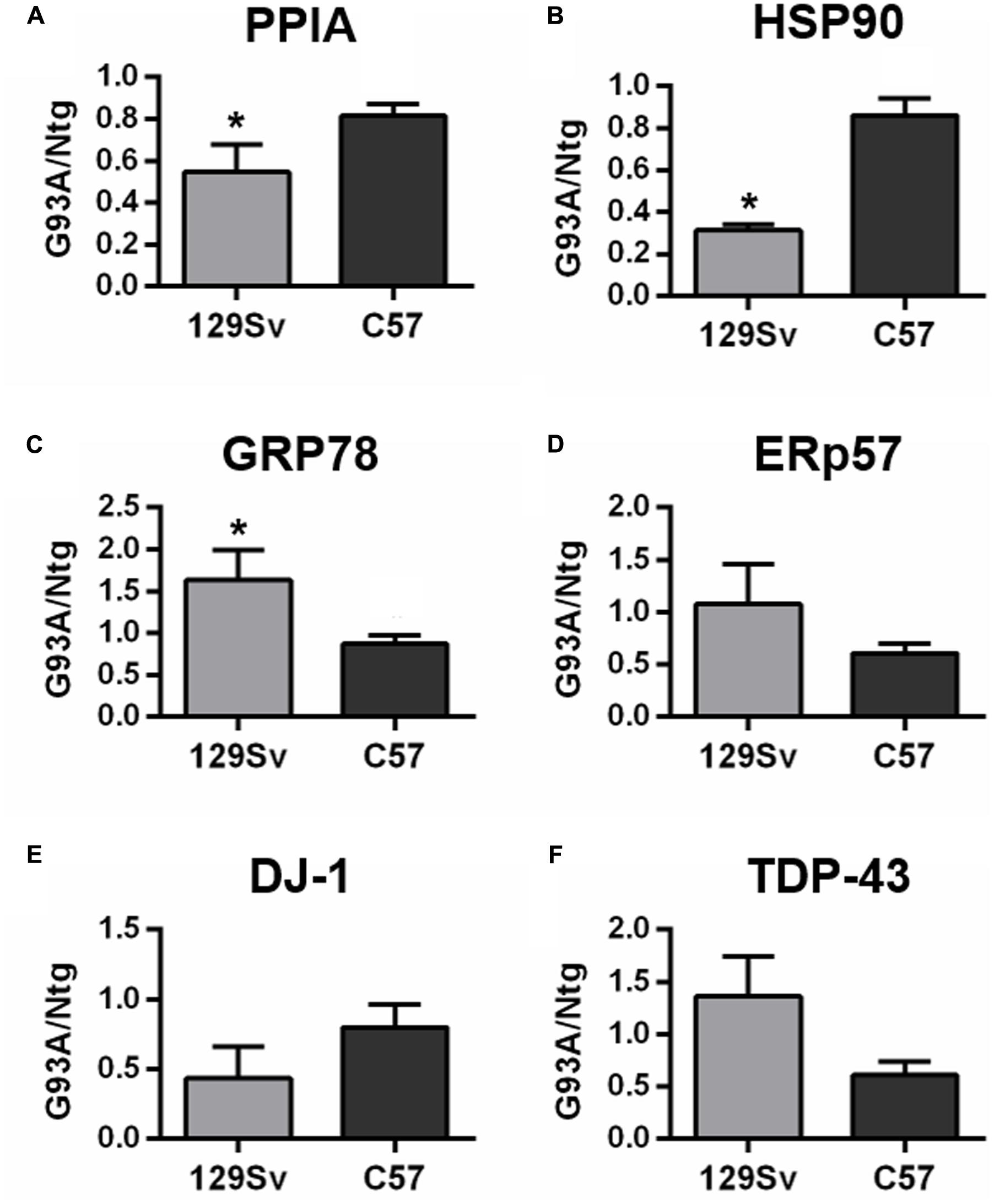
FIGURE 3. Analysis of phenotypic biomarkers in PBMC of SOD1G93A mouse models with early (129Sv) and late (C57) disease onset. (A–F) PPIA, HSP90, GRP78, ERp57, DJ-1, and TDP-43 were analyzed by dot blot immunoassays in PBMC samples (n = 5 per group) from 129Sv SOD1G93A and C57 SOD1G93A (G93A) mice at disease onset, respectively, at 14 and 17 weeks of age, and matched non-transgenic controls (Ntg). Immunoreactivity was normalized to protein loading, as assessed by Ponceau Red staining, and then to the mean values of matched controls. We found that PPIA, HSP90 were significantly lower in early ALS than in late ALS (A,B) similarly to human samples. ∗p < 0.05, by Student’s t-test.
Total protein levels of PPIA, HSP90, GRP78, DJ-1, and TDP-43 were also measured in lumbar spinal cord homogenates from the early and late SOD1G93A mice at disease onset and relative age-matched nontransgenic controls by dot blot analysis (Figure 4 and Supplementary Figure S4). PPIA, HSP90, GRP78 and DJ-1 were significantly lower in early SOD1G93A mice than in late SOD1G93A mice (Figures 4A–C,E) and were similarly regulated to PBMC isolated from patients (Supplementary Figures S4A–E), indicating that also early SOD1G93A mice have some defects in the response to stress pathways, as already reported (Nardo et al., 2013, 2016; Marino et al., 2015). ERp57 and TDP-43 were not different in the two conditions as in the human samples (Figures 4D,F). These data confirmed a previous work where no differences in total soluble TDP-43 were found between the SOD1G93A mice and respective Ntg littermates and between the two mouse strains (Marino et al., 2015). In conclusion, we observed a similar regulation of these proteins, in PBMC of patients and PBMC and spinal cord of mice (Table 4).
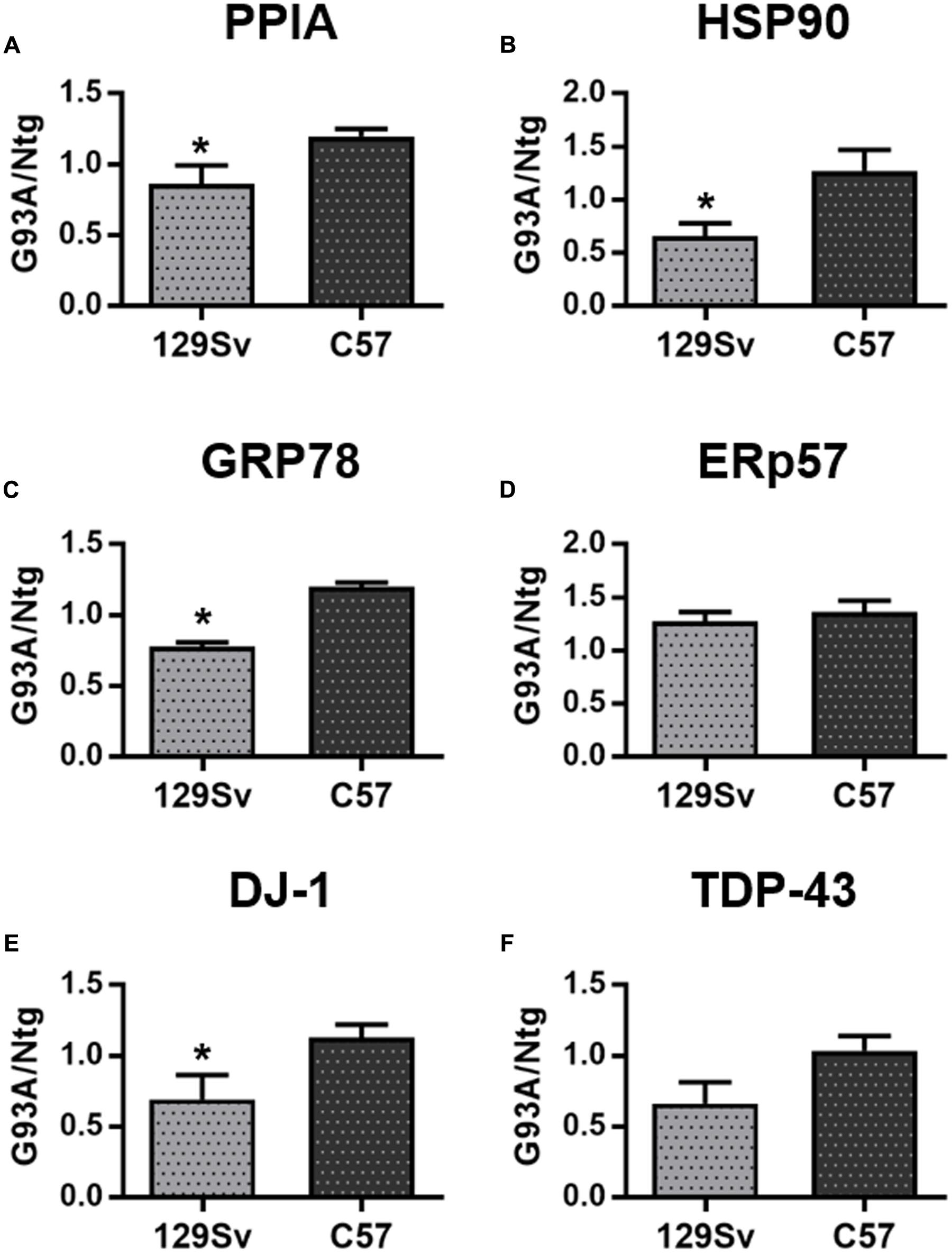
FIGURE 4. Analysis of phenotypic biomarkers in lumbar spinal cord of SOD1G93A mouse models with early (129Sv) and late (C57) disease onset. (A–F) PPIA, HSP90, GRP78, ERp57, DJ-1, and TDP-43 were analyzed by dot blot immunoassays in lumbar spinal cord samples (n = 5 per group) from 129Sv SOD1G93A and C57 SOD1G93A (G93A) mice at disease onset, respectively, at 14 and 17 weeks of age, and matched non-transgenic controls (Ntg). Immunoreactivity was normalized to protein loading, as assessed by Ponceau Red staining, and then to the mean values of matched controls. We found that PPIA, HSP90, GRP78, and DJ-1 were significantly lower in early ALS than in late ALS (A–C,E) similarly to PBMC from patients. ∗p < 0.05, by Student’s t-test.
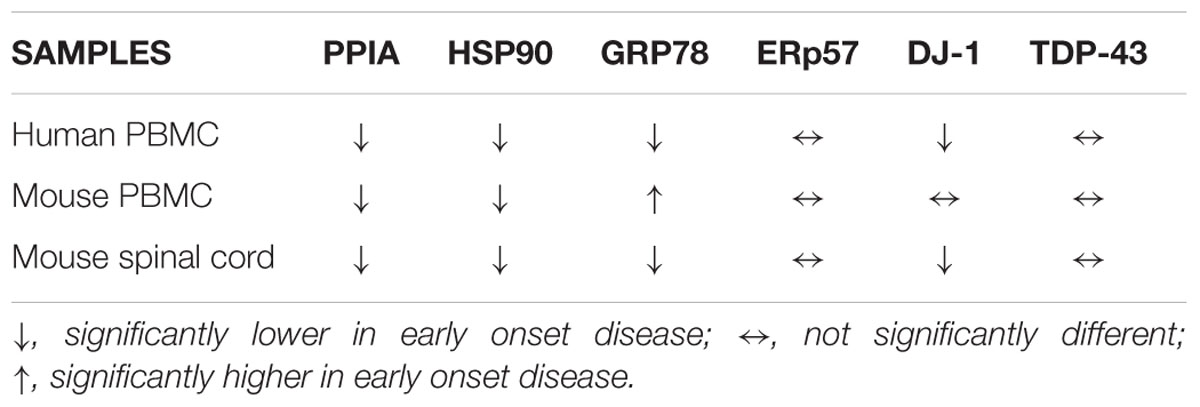
TABLE 4. Summary of the biomarker analysis in early and late ALS.
Discussion
The molecular determinants of ALS heterogeneity are still poorly understood. An anticipated or a delayed onset of the disease may be caused by various combinations of intrinsic and exogenous factors converging into common pathogenic pathways that underlie different susceptibility to ALS and eventually lead to a different prognosis. Here we report the identification of protein biomarkers in PBMC that can discriminate ALS patients with an early disease onset from those with a late disease onset. The identified phenotypic biomarkers have in common a high expression in CNS where they have an important role in protein folding, as foldases and/or classical molecular chaperones, and are upregulated by a variety of stressors. They participate at different levels and in different ways in the maintenance of proteostasis, whose deficit has shown to generate protein misfolding and aggregation and facilitate the development of several neurodegenerative diseases, including ALS (Ruegsegger and Saxena, 2016). These phenotypic biomarkers are: PPIA, HSP90, GRP78, and DJ-1.
Polyclonal anti-peptidyl-prolyl cis-trans isomerase A is ubiquitously expressed, with the highest expression in neurons and motor neurons (Lauranzano et al., 2015). It is a peptidylprolyl cis/trans isomerase and, as a foldase, it accelerates the rate-limiting steps along the folding pathway, but it also acts as a classical molecular chaperone (Fischer et al., 1989; Freskgard et al., 1992). We originally identified it as a candidate protein biomarker in PBMC of ALS patients with classical disease onset, where it is upregulated also in association with disease progression (Nardo et al., 2011). We next found that mutant SOD1 and TDP-43 are substrates of PPIA (Lauranzano et al., 2015). In fact, in absence of PPIA increased levels of mutant SOD1 and TDP-43 were recovered in the aggregates isolated from the spinal cord of SOD1G93A mice that had also an anticipation of the onset and an acceleration of the disease progression.
HSP90 is one of the most abundant and conserved cytosolic heat shock proteins. It is responsible for the correct folding of a number of newly synthesized proteins and for the refolding of misfolded proteins associated with neurodegenerative diseases, including TDP-43 (Basso et al., 2009; Daturpalli et al., 2013; Carlomagno et al., 2014; Schirmer et al., 2016). Accordingly, we found it entrapped in the aggregates isolated from the spinal cord of SOD1G93A mice already at a presymptomatic stage of the disease and from spinal cord tissues of sporadic ALS patients (Basso et al., 2009).
GRP78 is a major ER chaperone and a key regulator of the unfolded protein response. Disturbance of ER homeostasis is a common feature of ALS and defects in ER chaperones caused motor dysfunction in experimental models of ALS (Atkin et al., 2008; Saxena et al., 2009; Woehlbier et al., 2016). Loss of one copy of SIL1, a GRP78 cofactor, anticipated disease onset and reduced life span in the SOD1G93A mouse model (Filézac de L’Etang et al., 2015), however, the precise role of GRP78 in ALS remains to be determined. In Alzheimer’s disease, studies in vitro indicated that it may be part of the defense mechanisms of the cell by inhibiting the generation of amyloid-β peptides (Yang et al., 1998; Hoshino et al., 2007).
DJ-1 is a multifunctional stress response protein with a strong and homogenous expression in all CNS regions (Bader et al., 2005). DJ-1 mutations cause early onset autosomal recessive Parkinson’s disease (Bonifati et al., 2003). It is a chaperone for alpha synuclein and was shown to be protective against oxidative stress by upregulating HSP70 (Shendelman et al., 2004; Zhou et al., 2006; Batelli et al., 2008). In the SOD1G93A mouse model increased oxidized/inactive DJ-1 forms were found in brain tissues and knocking out DJ-1 has led to an accelerated disease course and shortened survival time (Lev et al., 2009, 2015).
Interestingly, we found that PPIA, HSP90, GRP78, and DJ-1 are present at lower levels in PBMC from patients with an early onset disease, and have a similar behavior in PBMC and spinal cord of an early onset SOD1G93A mouse model, in which defects in protein quality control have been previously observed (Marino et al., 2015). These proteins, both in patient PBMC and mouse samples, are less upregulated in early ALS than in late ALS, and for PPIA this is evident already at a presymptomatic stage of the disease in the soluble fraction of the mouse spinal cord (Marino et al., 2015). The bases of these differences in early and late-onset disease are solely genetic and yet unknown in the case of the SOD1G93A mouse models, but exogenous factors cannot be excluded in the patients. For example, it has been reported that exposure to certain toxins lead to GRP78 downregulation (Yang et al., 2000; Namba et al., 2010).
Conclusion
These data suggest that a failure in the response to stress and a reduced ability to upregulate protective proteins may increase susceptibility to ALS. This is in agreement with the fact that vulnerable motor neurons have a high threshold for induction of the protective heat shock response and a higher sensitivity to ER stress (Batulan et al., 2003; Saxena et al., 2009). A protein signature comprising a panel of proteins involved in proteostasis may underline a subset of patients more susceptible to the disease and may help to stratify patients for more targeted ALS clinical trials. These observations further strengthen the notion that proteostasis maintenance by the ubiquitin–proteasome system, autophagy and ER, is a central issue in ALS, and therapeutic approaches aiming at boosting the protein quality control system might be a promising therapeutic strategy. Arimoclomol, a small molecule that acts as a co-inducer of the heat shock response by prolonging HSF1 activation and upregulating a number of proteins such as HSP60, HSP70, HSP90, and GRP94, has shown to be effective in several experimental models of motor neuron degeneration including the SOD1G93A mouse (Kalmar et al., 2014). A Phase II clinical trial in ALS patients has shown that the drug is safe and well tolerated (Cudkowicz et al., 2008) and is now under investigation in a PhaseII/III clinical trial in SOD1 positive familial ALS patients (ClinicalTrials.gov identifier: NCT00706147). Colchicine, a FDA-approved drug that was identified by a high-throughput screening as enhancer of the expression of HSPB8, a key player of the protein quality control system, has demonstrated to facilitate the removal of TDP-43 aggregates by autophagy (Crippa et al., 2016).
Finally, this work confirmed that PBMC are valuable clinical samples since they reflect traits of the disease observed in the central nervous system and that total TDP-43 protein level in PBMC can discriminate ALS patients from healthy controls, as observed in a previous work using the same assay (Nardo et al., 2011). We also further confirmed that the experimental setting in which both 129Sv and C57 SOD1G93A mouse strains are examined is useful to study disease susceptibility and test novel therapeutic approaches aimed at addressing disease heterogeneity.
Ethics Statement
This study was carried out in accordance with the GCP recommendations with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Ethics Committees of the participating institutions.
Procedures involving animals and their care were conducted in conformity with the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; Authorisation n.19/2008-A issued March 6, 2008 by Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorization for persons conducting animal experiments (Quality Management System Certificate – UNI EN ISO 9001:2008 – Reg. No. 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition) and EU directives and guidelines (EEC Council Directive 2010/63/UE). The Statement of Compliance (Assurance) with the Public Health Service (PHS) Policy on Human Care and Use of Laboratory Animals has been recently reviewed (9/9/2014) and will expire on September 30, 2019 (Animal Welfare Assurance #A5023-01).
Author Contributions
Study concept and design: EB, CB, VB. Acquisition, analysis, or interpretation of data: all authors. Drafting the manuscript: VB. Critical revision of the manuscript: EB, CB.
Funding
This work was supported by grants from The Ministry of Health (D31D07000010001, Ricerca Finalizzata 2007) and from the “Fondazione Regionale per la Ricerca Biomedica” (TRANS-ALS project).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Maddalena Fratelli and Dr. Marco Bolis for advice on GO analysis, Tania Massignan for help in analyzing proteomic data and the Fondazione Vialli e Mauro.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fnmol.2017.00099/full#supplementary-material
Footnotes
References
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Azkargorta, M., Arizmendi, J. M., Elortza, F., Alkorta, N., Zubiaga, A. M., and Fullaondo, A. (2006). Differential proteome profiles in E2F2-deficient T lymphocytes. Proteomics 6(Suppl. 1), S42–S50. doi: 10.1002/pmic.200500438
Bader, V., Ran Zhu, X., Lübbert, H., and Stichel, C. C. (2005). Expression of DJ-1 in the adult mouse CNS. Brain Res. 1041, 102–111. doi: 10.1016/j.brainres.2005.02.006
Basso, M., Samengo, G., Nardo, G., Massignan, T., D’Alessandro, G., Tartari, S., et al. (2009). Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS ONE 4:e8130. doi: 10.1371/journal.pone.0008130
Batelli, S., Albani, D., Rametta, R., Polito, L., Prato, F., Pesaresi, M., et al. (2008). DJ-1 modulates alpha-synuclein aggregation state in a cellular model of oxidative stress: relevance for Parkinson’s disease and involvement of HSP70. PLoS ONE 3:e1884. doi: 10.1371/journal.pone.0001884
Batulan, Z., Shinder, G. A., Minotti, S., He, B. P., Doroudchi, M. M., Nalbantoglu, J., et al. (2003). High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J. Neurosci. Off. J. Soc. Neurosci. 23, 5789–5798.
Beghi, E. (2013). Are professional soccer players at higher risk for ALS? Amyotroph. Lateral Scler. Front. Degener. 14, 501–506. doi: 10.3109/21678421.2013.809764
Beghi, E., Chiò, A., Couratier, P., Esteban, J., Hardiman, O., Logroscino, G., et al. (2011). The epidemiology and treatment of ALS: focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph. Lateral Scler. 12, 1–10. doi: 10.3109/17482968.2010.502940
Beghi, E., and Morrison, K. E. (2005). ALS and military service. Neurology 64, 6–7. doi: 10.1212/01.WNL.0000150535.90358.2D
Bonifati, V., Rizzu, P., Squitieri, F., Krieger, E., Vanacore, N., van Swieten, J. C., et al. (2003). DJ-1(PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 24, 159–160. doi: 10.1007/s10072-003-0108-0
Brooks, B. R. (1994). El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J. Neurol Sci. 124(Suppl.), 96–107.
Calvo, A., Moglia, C., Lunetta, C., Marinou, K., Ticozzi, N., Ferrante, G. D., et al. (2016). Factors predicting survival in ALS: a multicenter Italian study. J. Neurol. 264, 54–63. doi: 10.1007/s00415-016-8313-y
Carlomagno, Y., Zhang, Y., Davis, M., Lin, W.-L., Cook, C., Dunmore, J., et al. (2014). Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS ONE 9:e90452. doi: 10.1371/journal.pone.0090452
Cecchi, M., Messina, P., Airoldi, L., Pupillo, E., Bandettini di Poggio, M., Calvo, A., et al. (2014). Plasma amino acids patterns and age of onset of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 15, 371–375. doi: 10.3109/21678421.2014.920032
Chiò, A., Benzi, G., Dossena, M., Mutani, R., and Mora, G. (2005). Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain J. Neurol. 128, 472–476. doi: 10.1093/brain/awh373
Chiò, A., Restagno, G., Brunetti, M., Ossola, I., Calvo, A., Mora, G., et al. (2009). Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol. Aging 30, 1272–1275. doi: 10.1016/j.neurobiolaging.2009.05.001
Crippa, V., D’Agostino, V. G., Cristofani, R., Rusmini, P., Cicardi, M. E., Messi, E., et al. (2016). Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep. 6:22827. doi: 10.1038/srep22827
Cudkowicz, M. E., Shefner, J. M., Simpson, E., Grasso, D., Yu, H., Zhang, H., et al. (2008). Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve 38, 837–844. doi: 10.1002/mus.21059
Daturpalli, S., Waudby, C. A., Meehan, S., and Jackson, S. E. (2013). Hsp90 inhibits α-synuclein aggregation by interacting with soluble oligomers. J. Mol. Biol. 425, 4614–4628. doi: 10.1016/j.jmb.2013.08.006
Filézac de L’Etang, A., Maharjan, N., Cordeiro Braña, M., Ruegsegger, C., Rehmann, R., Goswami, A., et al. (2015). Marinesco-Sjögren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat. Neurosci. 18, 227–238. doi: 10.1038/nn.3903
Fischer, G., Wittmann-Liebold, B., Lang, K., Kiefhaber, T., and Schmid, F. X. (1989). Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337, 476–478. doi: 10.1038/337476a0
Freskgard, P. O., Bergenhem, N., Jonsson, B. H., Svensson, M., and Carlsson, U. (1992). Isomerase and chaperone activity of prolyl isomerase in the folding of carbonic anhydrase. Science 258, 466–468.
Han, Y. H., Xia, L., Song, L. P., Zheng, Y., Chen, W. L., Zhang, L., et al. (2006). Comparative proteomic analysis of hypoxia-treated and untreated human leukemic U937 cells. Proteomics 6, 3262–3274. doi: 10.1002/pmic.200500754
Henrich, S., Cordwell, S. J., Crossett, B., Baker, M. S., and Christopherson, R. I. (2007). The nuclear proteome and DNA-binding fraction of human Raji lymphoma cells. Biochim. Biophys. Acta 1774, 413–432. doi: 10.1016/j.bbapap.2006.12.011
Hoshino, T., Nakaya, T., Araki, W., Suzuki, K., Suzuki, T., and Mizushima, T. (2007). Endoplasmic reticulum chaperones inhibit the production of amyloid-β peptides. Biochem. J. 402, 581–589. doi: 10.1042/BJ20061318
Huang, S. Y., Chen, Y. H., Teng, S. H., Chen, I. C., Ho, L. L., and Tu, C. F. (2006). Protein expression of lymphocytes in HLA-DR transgenic pigs by a proteomic approach. Proteomics 6, 5815–5825. doi: 10.1002/pmic.200600054
Hübers, A., Just, W., Rosenbohm, A., Müller, K., Marroquin, N., Goebel, I., et al. (2015). De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol. Aging 36, .e1–e6. doi: 10.1016/j.neurobiolaging.2015.08.005
Kalmar, B., Lu, C.-H., and Greensmith, L. (2014). The role of heat shock proteins in Amyotrophic Lateral Sclerosis: the therapeutic potential of Arimoclomol. Pharmacol. Ther. 141, 40–54. doi: 10.1016/j.pharmthera.2013.08.003
Lai, C. C., Jou, M. J., Huang, S. Y., Li, S. W., Wan, L., Tsai, F. J., et al. (2007). Proteomic analysis of up-regulated proteins in human promonocyte cells expressing severe acute respiratory syndrome coronavirus 3C-like protease. Proteomics 7, 1446–1460. doi: 10.1002/pmic.200600459
Lauranzano, E., Pozzi, S., Pasetto, L., Stucchi, R., Massignan, T., Paolella, K., et al. (2015). Peptidylprolyl isomerase A governs TARDBP function and assembly in heterogeneous nuclear ribonucleoprotein complexes. Brain 138, 974–991. doi: 10.1093/brain/awv005
Lev, N., Barhum, Y., Lotan, I., Steiner, I., and Offen, D. (2015). DJ-1 knockout augments disease severity and shortens survival in a mouse model of ALS. PLoS ONE 10:e0117190. doi: 10.1371/journal.pone.0117190
Lev, N., Ickowicz, D., Barhum, Y., Melamed, E., and Offen, D. (2009). DJ-1 changes in G93A-SOD1 transgenic mice: implications for oxidative stress in ALS. J. Mol. Neurosci. 38, 94–102. doi: 10.1007/s12031-008-9138-7
Li, Y.-J., Pericak-Vance, M. A., Haines, J. L., Siddique, N., McKenna-Yasek, D., Hung, W.-Y., et al. (2004). Apolipoprotein E is associated with age at onset of amyotrophic lateral sclerosis. Neurogenetics 5, 209–213. doi: 10.1007/s10048-004-0193-0
Marino, M., Papa, S., Crippa, V., Nardo, G., Peviani, M., Cheroni, C., et al. (2015). Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol. Aging 36, 492–504. doi: 10.1016/j.neurobiolaging.2014.06.026
Mi, H., Muruganujan, A., Casagrande, J. T., and Thomas, P. D. (2013). Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566. doi: 10.1038/nprot.2013.092
Namba, T., Hoshino, T., Suemasu, S., Takarada-Iemata, M., Hori, O., Nakagata, N., et al. (2010). Suppression of expression of endoplasmic reticulum chaperones by Helicobacter pylori and its role in exacerbation of non-steroidal anti-inflammatory drug-induced gastric lesions. J. Biol. Chem. 285, 37302–37313. doi: 10.1074/jbc.M110.148882
Nardo, G., Iennaco, R., Fusi, N., Heath, P. R., Marino, M., Trolese, M. C., et al. (2013). Transcriptomic indices of fast and slow disease progression in two mouse models of amyotrophic lateral sclerosis. Brain 136, 3305–3332. doi: 10.1093/brain/awt250
Nardo, G., Pozzi, S., Pignataro, M., Lauranzano, E., Spano, G., Garbelli, S., et al. (2011). Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE 6:e25545. doi: 10.1371/journal.pone.0025545
Nardo, G., Trolese, M. C., Tortarolo, M., Vallarola, A., Freschi, M., Pasetto, L., et al. (2016). New insights on the mechanisms of disease course variability in ALS from mutant SOD1 mouse models. Brain Pathol. Zurich Switz. 26, 237–247. doi: 10.1111/bpa.12351
Pappin, D. J., Hojrup, P., and Bleasby, A. J. (1993). Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 3, 327–332.
Pupillo, E., Messina, P., Logroscino, G., Zoccolella, S., Chiò, A., Calvo, A., et al. (2012). Trauma and amyotrophic lateral sclerosis: a case-control study from a population-based registry. Eur. J. Neurol. 19, 1509–1517. doi: 10.1111/j.1468-1331.2012.03723.x
Rakkola, R., Matikainen, S., and Nyman, T. A. (2007). Proteome analysis of human macrophages reveals the upregulation of manganese-containing superoxide dismutase after toll-like receptor activation. Proteomics 7, 378–384. doi: 10.1002/pmic.200600582
Ramirez-Boo, M., Garrido, J. J., Ogueta, S., Calvete, J. J., Gomez-Diaz, C., and Moreno, A. (2006). Analysis of porcine peripheral blood mononuclear cells proteome by 2-DE and MS: analytical and biological variability in the protein expression level and protein identification. Proteomics 6(Suppl. 1), S215–S225. doi: 10.1002/pmic.200500386
Rosengren, A. T., Nyman, T. A., and Lahesmaa, R. (2005). Proteome profiling of interleukin-12 treated human T helper cells. Proteomics 5, 3137–3141. doi: 10.1002/pmic.200401151
Ruegsegger, C., and Saxena, S. (2016). Proteostasis impairment in ALS. Brain Res. 1648, 571–579. doi: 10.1016/j.brainres.2016.03.032
Salonen, J. M., Valmu, L., Ronnholm, G., Kalkkinen, N., and Vihinen, M. (2006). Proteome analysis of B-cell maturation. Proteomics 6, 5152–5168. doi: 10.1002/pmic.200600156
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype–selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Schirmer, C., Lepvrier, E., Duchesne, L., Decaux, O., Thomas, D., Delamarche, C., et al. (2016). Hsp90 directly interacts, in vitro, with amyloid structures and modulates their assembly and disassembly. Biochim. Biophys. Acta 1860, 2598–2609. doi: 10.1016/j.bbagen.2016.07.033
Shendelman, S., Jonason, A., Martinat, C., Leete, T., and Abeliovich, A. (2004). DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2:e362. doi: 10.1371/journal.pbio.0020362
Skopeliti, M., Kratzer, U., Altenberend, F., Panayotou, G., Kalbacher, H., Stevanovic, S., et al. (2007). Proteomic exploitation on prothymosin alpha-induced mononuclear cell activation. Proteomics 7, 1814–1824. doi: 10.1002/pmic.200600870
Tremolizzo, L., Messina, P., Conti, E., Sala, G., Cecchi, M., Airoldi, L., et al. (2014). Whole-blood global DNA methylation is increased in amyotrophic lateral sclerosis independently of age of onset. Amyotroph. Lateral Scler. Front. Degener. 15, 98–105. doi: 10.3109/21678421.2013.851247
Van Hoecke, A., Schoonaert, L., Lemmens, R., Timmers, M., Staats, K. A., Laird, A. S., et al. (2012). EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 18, 1418–1422. doi: 10.1038/nm.2901
Veldink, J. H., Bär, P. R., Joosten, E. A. J., Otten, M., Wokke, J. H. J., and van den Berg, L. H. (2003). Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul. Disord. 13, 737–743. doi: 10.1016/S0960-8966(03)00104-4
Vergara, D., Chiriaco, F., Acierno, R., and Maffia, M. (2008). Proteomic map of peripheral blood mononuclear cells. Proteomics 8, 2045–2051. doi: 10.1002/pmic.200700726
Verhoeckx, K. C., Bijlsma, S., de Groene, E. M., Witkamp, R. F., van der Greef, J., and Rodenburg, R. J. (2004). A combination of proteomics, principal component analysis and transcriptomics is a powerful tool for the identification of biomarkers for macrophage maturation in the U937 cell line. Proteomics 4, 1014–1028. doi: 10.1002/pmic.200300669
Woehlbier, U., Colombo, A., Saaranen, M. J., Pérez, V., Ojeda, J., Bustos, F. J., et al. (2016). ALS-linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 35, 845–865. doi: 10.15252/embj.201592224
Xie, J., Techritz, S., Haebel, S., Horn, A., Neitzel, H., Klose, J., et al. (2005). A two-dimensional electrophoretic map of human mitochondrial proteins from immortalized lymphoblastoid cell lines: a prerequisite to study mitochondrial disorders in patients. Proteomics 5, 2981–2999. doi: 10.1002/pmic.200401191
Yang, G. H., Li, S., and Pestka, J. J. (2000). Down-regulation of the endoplasmic reticulum chaperone GRP78/BiP by vomitoxin (Deoxynivalenol). Toxicol. Appl. Pharmacol. 162, 207–217. doi: 10.1006/taap.1999.8842
Yang, Y., Turner, R. S., and Gaut, J. R. (1998). The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J. Biol. Chem. 273, 25552–25555. doi: 10.1074/jbc.273.40.25552
Zada, A. A., Geletu, M. H., Pulikkan, J. A., Muller-Tidow, C., Reddy, V. A., Christopeit, M., et al. (2006). Proteomic analysis of acute promyelocytic leukemia: PML-RARalpha leads to decreased phosphorylation of OP18 at serine 63. Proteomics 6, 5705–5719. doi: 10.1002/pmic.200600307
Keywords: protein folding, response to stress, chaperone, biomarkers, foldase
Citation: Filareti M, Luotti S, Pasetto L, Pignataro M, Paolella K, Messina P, Pupillo E, Filosto M, Lunetta C, Mandrioli J, Fuda G, Calvo A, Chiò A, Corbo M, Bendotti C, Beghi E and Bonetto V (2017) Decreased Levels of Foldase and Chaperone Proteins Are Associated with an Early-Onset Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 10:99. doi: 10.3389/fnmol.2017.00099
Received: 30 January 2017; Accepted: 23 March 2017;
Published: 06 April 2017.
Edited by:
Paola Rusmini, Università degli Studi di Milano, ItalyReviewed by:
Laura Ferraiuolo, University of Sheffield, UKAnat Ben-Zvi, Ben-Gurion University of the Negev, Beersheba, Israel
Copyright © 2017 Filareti, Luotti, Pasetto, Pignataro, Paolella, Messina, Pupillo, Filosto, Lunetta, Mandrioli, Fuda, Calvo, Chiò, Corbo, Bendotti, Beghi and Bonetto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Bonetto, valentina.bonetto@marionegri.it
†These authors have contributed equally to this work.