Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities?
 Christopher P. Webster
Christopher P. Webster Emma F. Smith
Emma F. Smith Pamela J. Shaw
Pamela J. Shaw Kurt J. De Vos
Kurt J. De Vos- Sheffield Institute for Translational Neuroscience (SITraN), Department of Neuroscience, University of Sheffield, Sheffield, UK
Protein homeostasis (proteostasis), the correct balance between production and degradation of proteins, is essential for the health and survival of cells. Proteostasis requires an intricate network of protein quality control pathways (the proteostasis network) that work to prevent protein aggregation and maintain proteome health throughout the lifespan of the cell. Collapse of proteostasis has been implicated in the etiology of a number of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), the most common adult onset motor neuron disorder. Here, we review the evidence linking dysfunctional proteostasis to the etiology of ALS and discuss how ALS-associated insults affect the proteostasis network. Finally, we discuss the potential therapeutic benefit of proteostasis network modulation in ALS.
Introduction
The proteostasis network is a complex regulatory network that maintains proteostasis. The proteostasis network consists of several pathways that control protein biosynthesis, folding, trafficking, and clearance (degradation) and responds to specific protein stress pathways such as the unfolded protein response (UPR) in the endoplasmic reticulum (ER), the mitochondrial UPR and the cytosolic heat shock response (Figure 1).
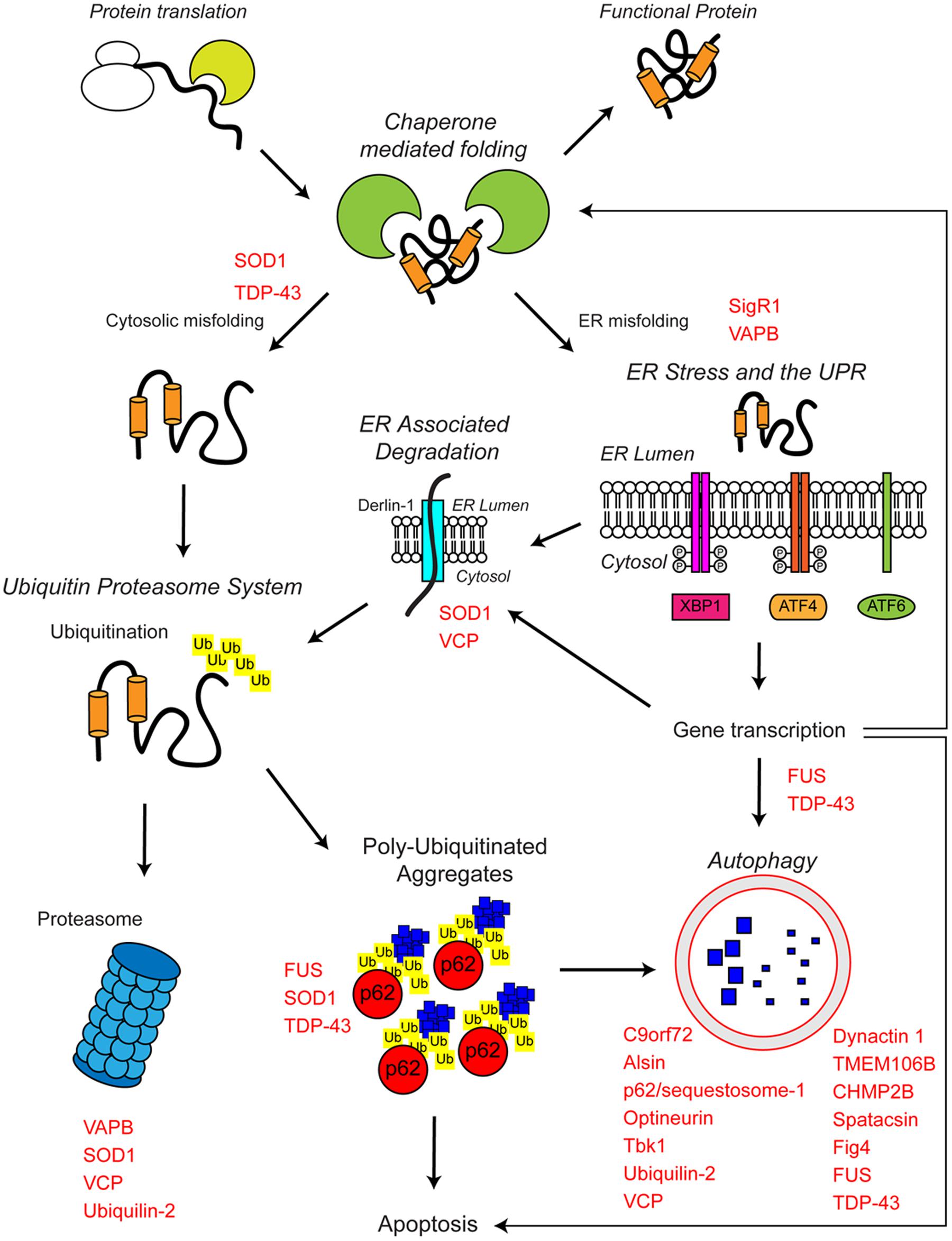
FIGURE 1. The proteostasis network and ALS. Protein folding occurs co-translationally at the ribosome with the aid of molecular chaperones, including Hsp70. Correct folding is essential for protein function. Protein folding and refolding continues in the cytosol and the endoplasmic reticulum (ER) lumen. Chronic misfolding in the cytosol leads to targeting of misfolded substrates to the ubiquitin proteasome system (UPS). Poly-ubiquitin chains target substrates for degradation by the proteasome. Overwhelming of the UPS can lead to poly-ubiquitinated aggregate formation, which are cleared by the autophagosome–lysosome pathway. Chronic misfolding in the ER leads to the induction of ER stress and activation of the unfolded protein response (UPR). The UPR leads to altered gene transcription, upregulating ER associated degradation (ERAD) and autophagy. The proteostasis network seeks to restore protein homeostasis, but failure of the pathway leads to the aggregation of potentially toxic species. Disruption of the proteostasis network is prevalent in the pathogenesis of ALS. A large number of ALS-associated genes (indicated in red) directly or indirectly regulate the proteostasis network. In addition, some ALS-associated proteins such as FUS, TDP-43, and SOD1 are also substrates of these pathways. For further details please refer to the main text.
Proteins are constantly turned over to ensure a steady supply of functional proteins. Newly synthesized proteins fold into their specific three-dimensional shape co-translationally as the nascent polypeptide chain emerges from the ribosome. The specific three-dimensional structure of a protein, which is in part determined by its amino acid sequence, is crucial to its function. A number cytosolic and ER resident folding factors aid the complex process of protein folding, such as chaperones and co-chaperones of the heat shock protein (Hsp) family, peptidyl prolyl cis/trans isomerases, and oxidoreductases (reviewed in Braakman and Bulleid, 2011; Kim et al., 2013). Nevertheless, the nature of protein folding and the cellular environment is such that unfolding and misfolding are relatively common events. Upon protein misfolding, specific protein stress pathways such as the heat shock response and UPR are activated to boost chaperone levels and aid refolding or to stimulate removal of terminally misfolded proteins to prevent protein aggregation and proteotoxic stress.
Eukaryotic cells have two major pathways of protein degradation: the proteasome and the lysosome. The proteasome is a multimeric ATP-dependent protease complex that selectively recognizes ubiquitinated substrates. Degradation by the proteasome requires protein unfolding and relies on chaperones to prevent proteins from aggregating (reviewed in Hershko and Ciechanover, 1998). Autophagy denotes the delivery of cytoplasmic components to the lysosome. Autophagy can be separated into three types depending on how the substrate is delivered to the lysosome: chaperone mediated autophagy (CMA), microautophagy and macroautophagy, herein termed autophagy (reviewed in Bento et al., 2016). Aggregated proteins are mostly removed by autophagy, a process that is also called aggrephagy (reviewed in Lamark and Johansen, 2012).
Ultimately proteostasis collapse due to failure of the proteostasis network to refold, degrade or effectively sequester and compartmentalize aggregation-prone, misfolded and potentially toxic protein species is deleterious to cells. Neuronal cells appear to be particularly vulnerable to disturbances in proteostasis because they are long-lived post-mitotic cells that are not able to dilute out protein aggregates during cell divisions (Son et al., 2012). Furthermore, as the ability of cells to maintain proteostasis declines with aging it is not surprising that aberrant protein folding and aggregate deposition in neurons is a common feature of age-associated neurodegenerative disease.
Here, we review the evidence linking dysfunctional proteostasis to the etiology of amyotrophic lateral sclerosis (ALS) and discuss how ALS-associated insults affect the proteostasis network.
Loss of Protein Homeostasis in ALS
ALS is a progressive adult onset motor neuron disease characterized by selective degeneration of upper and lower motor neurons in the motor cortex, brainstem, and spinal cord. The progressive deterioration of the motor system leads to muscle wasting, paralysis and eventual premature death, most commonly due to respiratory failure. Death occurs on average within approximately 3 years of symptom onset (reviewed in Kiernan et al., 2011). The causes of ALS are numerous and complex, but remain incompletely understood. Proposed mechanisms include, among others, oxidative stress, mitochondrial dysfunction, defective axonal transport, RNA toxicity, excitotoxicity, neuroinflammation, and loss of protein homeostasis (reviewed in Ferraiuolo et al., 2011; De Vos and Hafezparast, 2017). While most ALS cases have no clear genetic basis (sporadic ALS), approximately 10% of ALS is inherited, usually in an autosomal dominant fashion (familial ALS). Studies of familial ALS cases have revealed multiple ALS-associated genes. While these genes play significant roles in a range of essential cellular processes including RNA processing, mitochondrial function and endosomal trafficking, a significant number of these have been linked to different aspects of the proteostasis network (Table 1 and Figure 1).
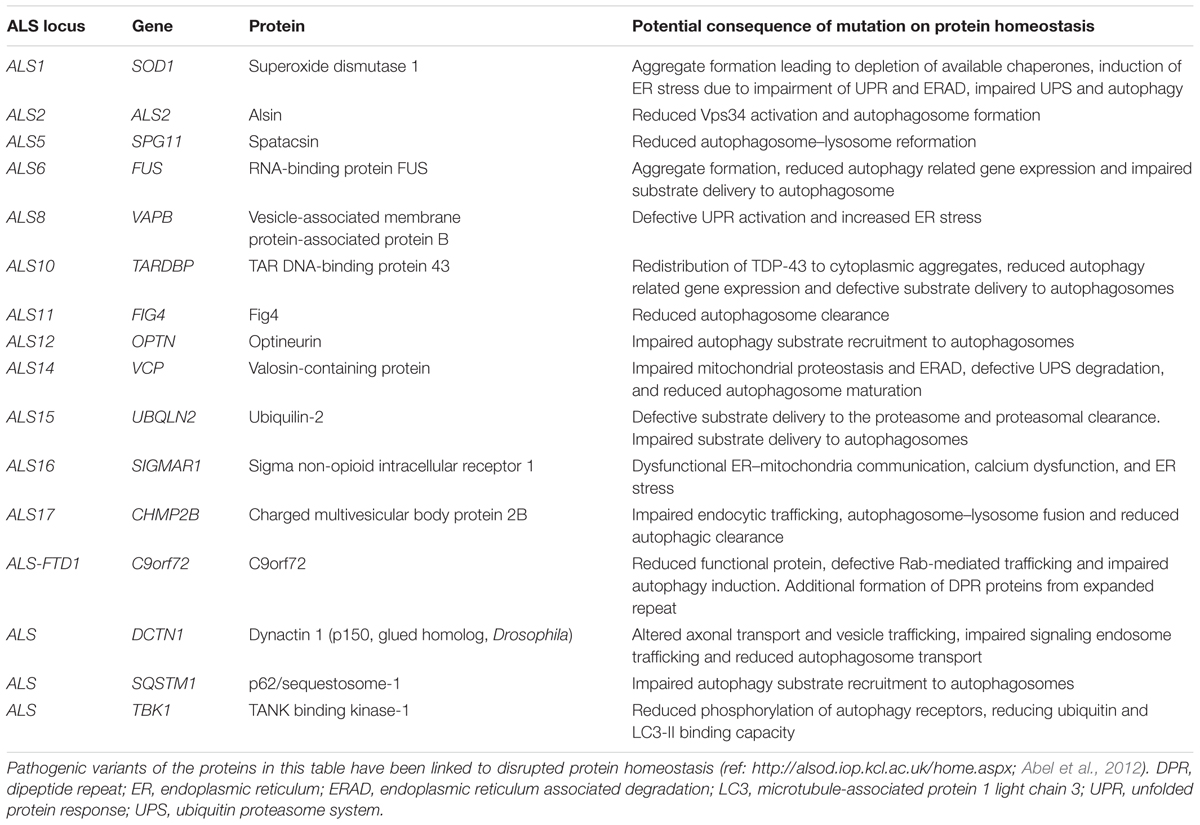
TABLE 1. Potential impact of ALS-associated genes on protein quality control.
There is a significant clinical, neuropathological, and genetic overlap between ALS and frontotemporal dementia (FTD), a common form of early onset dementia that is characterized by changes in behavior and personality or language dysfunction. Up to 15% of ALS cases are clinically diagnosed with FTD and approximately 50% of FTD cases display motor symptoms (reviewed in Swinnen and Robberecht, 2014).
Neuropathological Evidence for Dysfunctional Proteostasis in ALS
Intracellular proteinaceous inclusions are a hallmark neuropathological feature of ALS. Inclusions are found in both degenerating neurons and surrounding glia (Piao et al., 2003; Nishihira et al., 2008; Zhang et al., 2008) and are found not only in the brainstem and spinal cord, but also in the cerebellum, hippocampus, and the frontal and temporal lobes (reviewed in Al-Chalabi et al., 2012). The most common inclusions are of ubiquitinated proteins, which are found in both the upper and lower motor neurons (Neumann et al., 2006), and are suggestive of defects in protein turnover (Blokhuis et al., 2013). Based on their morphology, these ubiquitinated inclusions are subdivided into skein-like inclusions that are filamentous in structure, and rounded Lewy body-like inclusions (Leigh et al., 1988; Lowe et al., 1988; Kato et al., 1989). ALS-associated ubiquitinated inclusions are typically positive for p62/sequestosome-1 (King et al., 2011), a ubiquitin binding protein involved in autophagy (see below). In the vast majority of sporadic and familial ALS cases ubiquitinated protein inclusions are positive for Tar DNA-binding protein of 43 kDa (TDP-43) (Arai et al., 2006; Neumann et al., 2006). TDP-43 was also identified as the pathological protein in frontotemporal lobar degeneration (FTLD)-related ubiquitinated inclusions, supporting the idea that ALS and FTD reside on the same spectrum of disease (Arai et al., 2006; Neumann et al., 2006; reviewed in Swinnen and Robberecht, 2014). Not only are aggregates of wild type TDP-43 found in nearly all cases of disease, but mutations in the TARDPB gene that encodes TDP-43 are also causative for ALS (Kabashi et al., 2008; Sreedharan et al., 2008). ALS-associated mutations in TARDBP lead to cytoplasmic TDP-43 mislocalisation and its aberrant incorporation into neurotoxic ubquitinated cytoplasmic aggregates (Barmada et al., 2010). Inhibition of the proteasome or autophagy leads to the aggregation of TDP-43 (Urushitani et al., 2010; Wang et al., 2010) (see below).
Although the majority of ubiquitinated inclusions are immunoreactive for TDP-43, notable exceptions are the inclusions found in ALS patients with mutations in SOD1 or FUS, which are negative for ubiquitinated TDP-43 but immunoreactive for mutant aggregated Cu/Zn superoxide dismutase (SOD1) and fused in sarcoma protein (FUS) respectively (Watanabe et al., 2001; Wang et al., 2002; Mackenzie et al., 2007; Kwiatkowski et al., 2009; Vance et al., 2009). Similar to TDP-43, mutant FUS demonstrates abnormal cytoplasmic redistribution and aggregation (Kwiatkowski et al., 2009; Vance et al., 2009; Dormann et al., 2010). Further familial ALS-associated mutant proteins that are prone to aggregation are valosin containing protein (VCP), dynactin-1 (DCTN1), optineurin (OPTN) and ubiquilin-2 (UBQLN2) (Levy et al., 2006; Maruyama et al., 2010; Deng et al., 2011; Koppers et al., 2012).
C9orf72-related ALS presents an outlier to classical ALS pathology. In C9orf72-related ALS TDP-43 proteinopathy is present, but additional inclusions are p62/sequestosome-1 and ubiquitin positive, yet devoid of TDP-43 (Al-Sarraj et al., 2011; Cooper-Knock et al., 2012; Mackenzie et al., 2014). In addition, the GGGGCC repeat expansion in the C9ORF72 gene gives rise to five species of dipeptide protein (DPR) inclusions (GA, GR, GP, PR, and PA) by repeat-associated non-AUG translation (Mann et al., 2013; Mori et al., 2013; Mackenzie et al., 2014).
Other ALS-associated inclusions include Bunina bodies (found post-mortem in approximately 86% of sporadic ALS patients) and hyaline conglomerate inclusions (Bunina, 1962; Kato et al., 1989; Murayama et al., 1989). Bunina bodies are comprised of cystatin C, transferrin, peripherin, and sortilin-related receptor CNS expressed 2 (SORCS2) and are found in the surviving lower motor neurons within the brain stem and spinal cord (Okamoto et al., 1993; Piao et al., 2003; Mizuno et al., 2006, 2011; Mori et al., 2015). They also contain small organelle fragments such as vesicles and ER (Okamoto et al., 2008; Kimura et al., 2014). Bunina bodies and TDP-43 positive inclusions have been shown to co-localize, and TDP-43 inclusion prevalence increased with Bunina body presence suggesting a synergy between the two inclusion types (Mori et al., 2010, 2014). Hyaline conglomerate inclusions are comprised of phosphorylated and non-phosphorylated neurofilaments (Hirano et al., 1984; Munoz et al., 1988; Hays et al., 2006) and their formation is possibly linked to defective axonal transport (Munoz et al., 1988; Ackerley et al., 2000, 2003).
The typical occurrence of these protein aggregates in ALS patients strongly suggest a collapse of proteostasis in ALS. These proteinaceous inclusions are replicated in a number ALS-associated animal models, including mutant SOD1, FUS, and TDP-43 transgenic mice. The fact that these models are able to effectively replicate some of the most prevalent neuropathological features of ALS suggests that the proteinaceous inclusions and aggregates are major contributors to disease pathogenesis.
Genetic Evidence for Dysfunctional Proteostasis in ALS
As mentioned above, about 10% of ALS cases are inherited, usually in an autosomal dominant fashion (reviewed in Renton et al., 2014). A number of familial ALS-associated proteins are known to be involved in the proteostasis network, including C9orf72, VCP, p62/sequestosome-1, ubiquilin-2, optineurin, dynactin, and TANK binding kinase 1 (TBK1) (Table 1 and Figure 1). The role of these genes in proteostasis is discussed in detail in the following sections.
Altered Chaperone Function in ALS
Molecular chaperones assist protein folding and help maintain proteins in their native folded state. In addition, they function in proteostasis to facilitate protein unfolding and disaggregation, and the targeting of terminally misfolded proteins for degradation. In relation to neurodegeneration, perhaps the key function of chaperones is to prevent protein aggregation under conditions of stress. Altered chaperone function has been implicated in ALS (Figure 2).
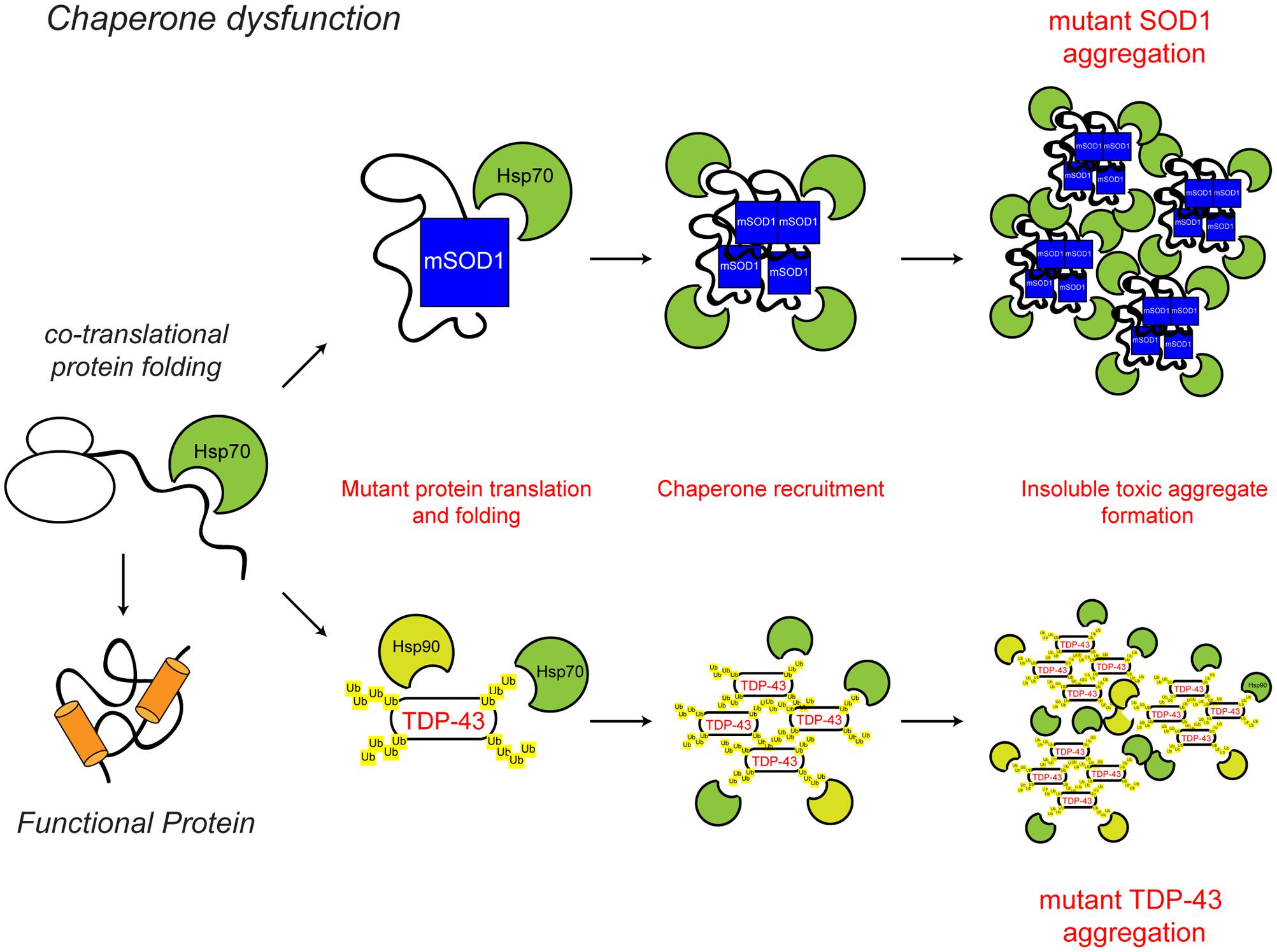
FIGURE 2. Chaperone dysfunction in ALS. Protein folding occurs co-translationally at the ribosome. Correct folding and re-folding continues in the cytoplasm or ER with the help of molecular chaperones and leads to correctly folded, fully functional proteins. Chaperone dysfunction has been implicated in ALS. Aggregating ALS mutant SOD1 and mislocalised TDP-43 interact with chaperones of the heat shock protein family, namely Hsp70 and Hsp90. While the recruitment of chaperones to the aggregates is likely a protective mechanism, their sequestration potentially depletes the levels of available chaperones, decreasing chaperone folding activity, therefore leading to toxicity. The ALS-associated protein aggregates and other ALS-associated defects to the chaperone system are indicated in red. For further details please refer to the main text.
The presence of chaperones, such as heat shock cognate protein of 70 kDa (Hsc70) and heat shock protein of 90 kDa (Hsp90) in detergent-insoluble neuronal aggregates in the SOD1G93A transgenic mouse model of familial ALS and post-mortem human sporadic ALS cases indicates that altered chaperone function may contribute to disease pathogenesis (Watanabe et al., 2001; Basso et al., 2009; Bergemalm et al., 2010). In cell based assays and mouse models, ALS mutant SOD1G93A and G85R show increased interaction with a number of chaperones, including Hsp70, suggesting that ALS-associated mutant species may result in a depletion of available chaperones and chaperone activity, therefore leading to cellular toxicity (Tummala et al., 2005; Ganesan et al., 2007). Furthermore, reduced levels of the chaperone alpha-B-crystallin (CRYAB) and increased incorporation of other molecular chaperones, including Hsc70, into the insoluble aggregate fraction are features of a faster progressing phenotype in SOD1G93A transgenic mice (Marino et al., 2015).
As discussed above, redistribution of wild type or mutant TDP-43 to the cytoplasm and its aberrant inclusion into ubiquitinated cytoplasmic aggregates is a hallmark of ALS (Arai et al., 2006; Neumann et al., 2006). Interestingly, TDP-43 aggregates have been shown to interact with Hsp70 and Hsp90 under conditions of heat shock stress or reactive oxygen species (ROS) insult (Chang et al., 2013; Udan-Johns et al., 2014; Budini et al., 2015). The finding that activating Hsp70 reduces the insoluble aggregates of TDP-43, suggests that chaperone dysregulation could be a contributing factor to TDP-43 aggregate formation and toxicity (Chang et al., 2013). In agreement with this, knock-down of the molecular chaperones Hsp70 and Hsp90, leads to increased aggregation of TDP-43 (Zhang et al., 2010), while increasing levels of the small heat shock protein B8 (HspB8) increased the solubility of mutant TDP-43 and reduced toxicity (Crippa et al., 2010). Similarly, upregulation of the small heat shock protein CG14207 was able to reduce neurotoxicity of full length TDP-43 and the C-terminal fragment of TDP-43 in a Drosophila model of ALS (Gregory et al., 2012). Overexpression of Hsc70 in Drosophila also prevents the aggregation of FUS protein into insoluble fractions (Miguel et al., 2012).
Thus, increased chaperone levels may provide a protective mechanism in ALS. The reported upregulation of HspB1 and HspB8 chaperone expression in the lumbar spinal cord of 39 ALS cases (4 of which were familial ALS cases) compared to 19 control samples may be an indication of such a protective response (Anagnostou et al., 2010).
ER Stress, the UPR and ALS
Cells use stress sensors and inducible pathways to respond to a loss of proteostatic control. One such pathway is the UPR, an adaptive response to the accumulation of misfolded proteins in the lumen of the ER (i.e., ER stress) (Figure 3). ER stress induced by accumulation of unfolded proteins activates three UPR pathways mediated by three ER membrane resident stress sensors, protein kinase RNA-like ER kinase (PERK), inositol-requiring protein 1 (IRE1) and activating transcription factor 6 (ATF6) (reviewed in Hetz, 2012). The UPR tries to restore proteostasis by (i) attenuation of translation, (ii) induction of chaperones to aid protein folding, and (iii) upregulation of degradation pathways such as ER-associated degradation (ERAD) and autophagy to remove misfolded proteins. If ER stress is short-lived, the UPR restores proteostasis and the cell survives. If in contrast ER stress persists, as for example in ALS, the UPR triggers apoptosis and the cell is lost.
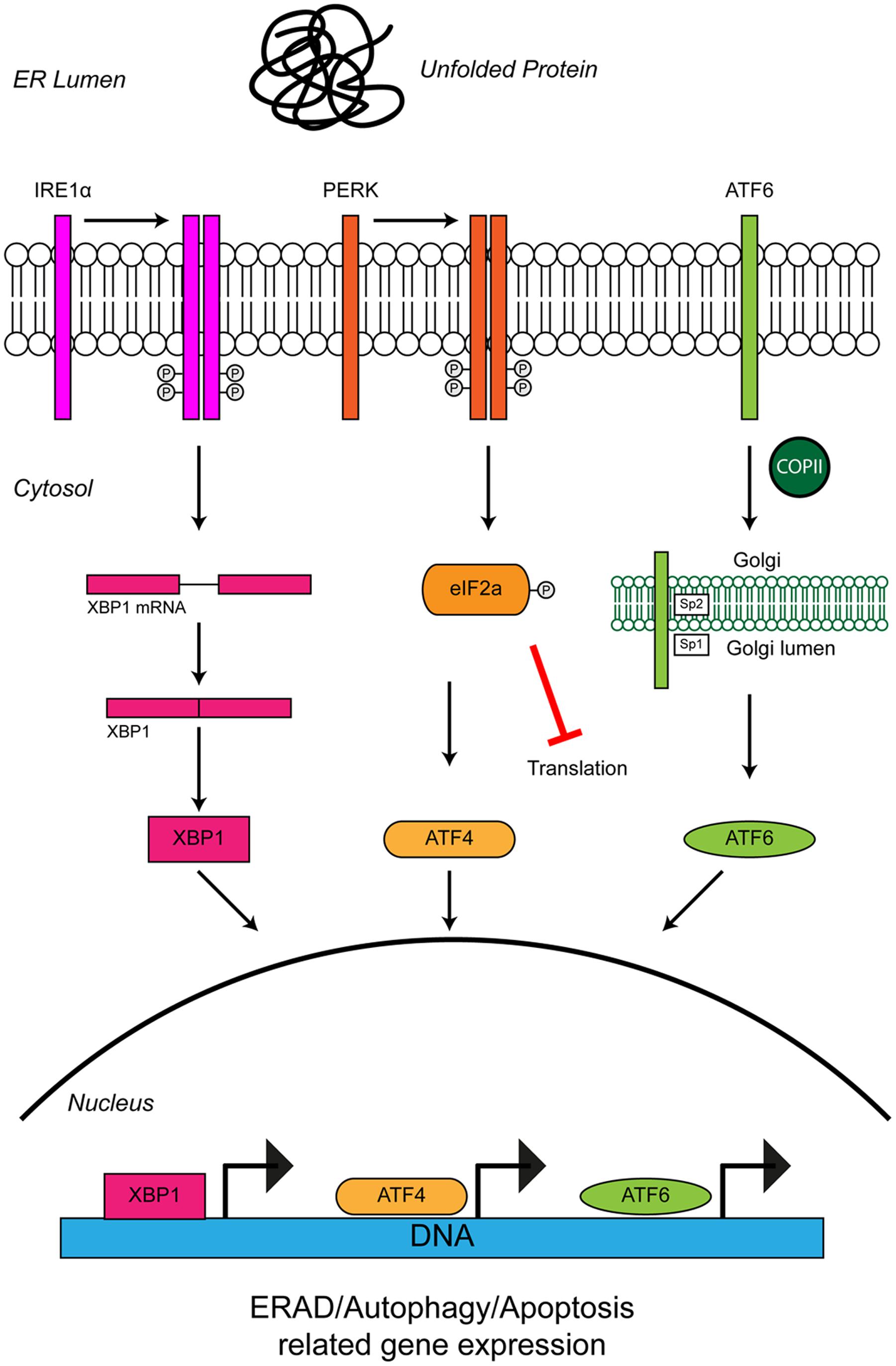
FIGURE 3. Unfolded protein response in the ER leads to ER stress. The accumulation of protein aggregates is sensed by three ER-stress transducers: IRE1α, PERK, and ATF6. ER stress causes IRE1α dimerisation, which activates its intrinsic RNAse activity and leads to alternative splicing of XBP1 mRNA. Spliced XBP1 forms a functional transcription factor. XBP1 increases expression of chaperone related genes and those involved in ERAD. PERK also dimerises due to ER stress. PERK dimerization leads to phosphorylation of the eukaryotic initiation factor eIF2α, thus inhibiting general protein synthesis. Inhibition of protein synthesis allows the translation of stress response transcription factor, ATF4. ATF4 increases expression of genes related to autophagy and apoptosis. Via the action of coat protein complex II (COPII), ATF6 translocates from the ER membrane to the Golgi during ER stress where it is processed by the Site 1 (Sp1) and Site 2 (Sp2) proteases. Cleavage produces a functional cytosolic fragment of ATF6. The ATF6 transcription factor induces expression of genes related to ERAD, but also XBP1, thereby promoting UPR. Chronic ER stress and UPR activation indicates the cell has failed to respond to ER stress. Under such conditions all three ER stress transducers lead to the increased expression of CHOP, which promotes apoptosis. For further details please refer to the main text.
Multiple lines of evidence from ALS patients and models suggest that ER stress may be a contributing factor to the development of ALS (Figure 4). Amorphous, granular material indicative of unfolded proteins has been shown to accumulate in the ER of motor neurons of sporadic ALS patients (Sasaki, 2010). In line with ER stress and induction of the UPR, the ER chaperone binding immunoglobulin protein/glucose-regulated protein 78 (BiP/GRP-78) was upregulated in motor neurons of sporadic ALS patients (Sasaki, 2010) and the levels of all three UPR stress transducers were found to be elevated in cerebrospinal fluid (CSF) and spinal cord of sporadic ALS patients (Atkin et al., 2008). In addition, a number of UPR-related chaperones, including protein disulfide isomerase (PDI) were present in CSF and throughout the motor neurons of sporadic ALS patients (Atkin et al., 2006, 2008). PDI has also been found in association with SOD1-inclusions in SOD1G93A transgenic mice, and it has been suggested that PDI is involved in the removal of these aggregates (Atkin et al., 2006). In SOD1G93A transgenic mice elevated UPR stress sensor levels have been observed prior to disease onset, as early as postnatal day 5, suggesting ER stress may be an early pathogenic event in the development of ALS (Atkin et al., 2008; Saxena et al., 2009). In vitro, expression of mutant SOD1A4V in the NSC-34 motor neuron-like cell line induces ER stress and apoptosis (Walker et al., 2010).
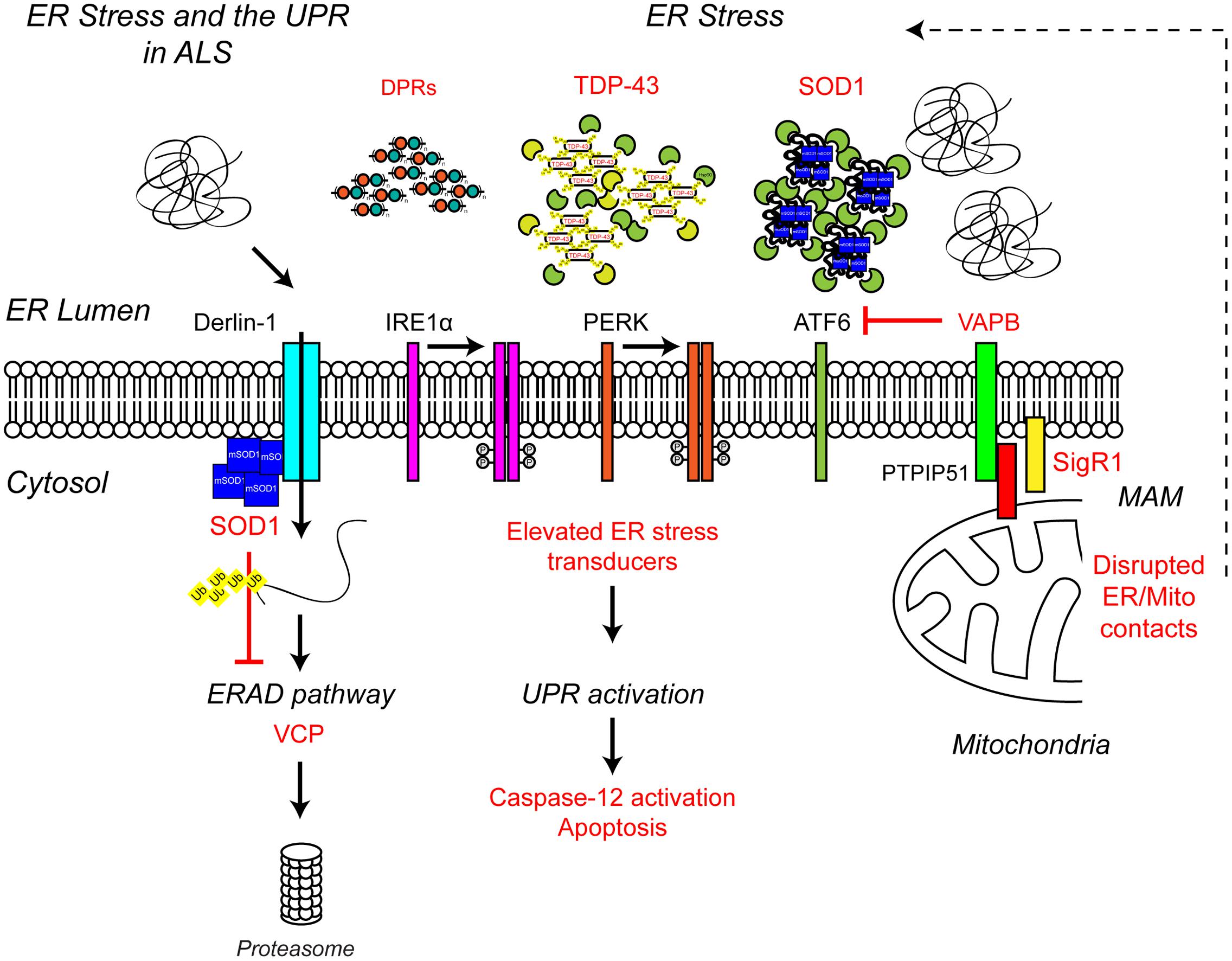
FIGURE 4. Endoplasmic reticulum stress the UPR and ALS. ALS-associated genes have been implicated in ER stress and the UPR. The ALS-associated genes and their positions within the UPR pathway are indicated in red, as are other ALS-associated defects to the UPR. Briefly, ALS-associated protein aggregates, including C9orf72-related DPR proteins, TDP-43 and SOD1, are sensed by the ER-stress transducers leading to chronic activation of the UPR, caspase-12 cleavage and apoptosis, while disruption of ER/mitochondria contact sites leads to dysfunctional calcium homeostasis and, in turn, elevated ER stress. Finally, mutant SOD1 aggregates interact with Derlin-1, a member of the ERAD pathway, and disrupts the proteasome-dependent degradation of misfolded ER proteins, thus promoting further ER stress. For further details please refer to the main text.
Interestingly, exposure of primary motor neurons to the CSF of sporadic ALS patients leads to ER stress, activation of the UPR and neuronal degeneration (Vijayalakshmi et al., 2009, 2011). How CSF elicits ER stress and degeneration is not clear but the process appears to involve activation of Caspase-12. Caspase-12 is known to mediate ER stress-induced apoptosis, and is activated by the UPR (Nakagawa et al., 2000; Martinez et al., 2010).
Chemical induction of ER stress in NSC-34 cells led to increased TDP-43 cleavage (Suzuki et al., 2011), while overexpression of ALS-associated TDP-43A315T and Q331K mutants in Neuro2a cells led to ER stress and activation of a number of UPR stress response pathways, including increased CHOP protein levels, increased nuclear XBP1 and increased activation of ATF6 (Walker et al., 2013). This activation of the UPR by mutant TDP-43 could further promote the cytoplasmic mislocalisation of TDP-43, and therefore toxicity (Walker et al., 2013). Similarly, expression of ALS mutant FUS and C9orf72-associated poly-GA DPRs have been shown to induce ER stress in NSC-34 cells and primary neurons, respectively (Farg et al., 2012; Zhang Y.J. et al., 2014).
Several ALS-associated proteins appear to directly interfere with pathways required for proteostasis and/or UPR. ALS mutant SOD1 has been shown to interact with Derlin-1, an ER protein essential for ERAD, and perturb the ERAD pathway (Nishitoh et al., 2008). Dysfunctional ERAD induces ER stress which eventually triggers apoptosis via the ASK1 pathway (Nishitoh et al., 2008). Mutations in vesicle-associated membrane protein-associated protein B (VAPB), an integral ER protein that has been implicated in UPR activation via the IRE1 and ATF6 pathways cause ALS8 (Nishimura et al., 2004; Kanekura et al., 2006; Gkogkas et al., 2008; Suzuki et al., 2009). Overexpression of ALS mutant VAPBP56S has been shown to induce ER stress (Suzuki et al., 2009). However, VAPB expression is down-regulated in ALS8 patient-derived iPSC neurons due to reduced expression of the VAPBP56S mutant (Mitne-Neto et al., 2011), therefore it is more likely ALS-associated loss of function of VAPB may predispose motor neurons to ER stress (Kanekura et al., 2006; Gkogkas et al., 2008; Suzuki et al., 2009). Indeed, since knock-down of VAPB inhibits activation of the IRE/XBP1 pathway in response to chemical ER stressors, VAPB appears to promote the UPR in reaction to ER stress under physiological conditions (Kanekura et al., 2006). How VAPB regulates the UPR is not yet clear but may involve its role in maintaining ER/mitochondria contacts via interaction with the mitochondrial outer membrane protein PTPIP51 (De Vos et al., 2012).
ER/mitochondria contacts allow mitochondria and ER to communicate directly with each other via the exchange of calcium signals (reviewed in Paillusson et al., 2016). Under physiological conditions, mitochondrial calcium activates the rate-limiting enzymes of the Krebs cycle and thereby increases oxidative phosphorylation and ATP synthesis to match local energy demand. In turn, energized mitochondria influence ER calcium homeostasis and redox dependent ER processes such as oxidative protein folding (reviewed in Walczak et al., 2012). Disruption of ER/mitochondria contacts has been shown to induce ER stress and the UPR (Simmen et al., 2005), possibly by disturbing the variety of ER chaperones, such as BiP, calnexin, calreticulin, ERp44, ERp57, and Sigma non-opioid intracellular receptor 1 (Sig1R) that are present in mitochondria-associated ER membranes (MAMs) (Hayashi and Su, 2007). Reduced levels of VAPB have been reported in the spinal cord of sporadic ALS cases, suggesting that ER/mitochondria contacts and the UPR may be impaired as a consequence (Anagnostou et al., 2010). Consistent with this possibility, neuronal overexpression of wild type human VAPB has been shown to slow disease and increase survival in SOD1G93A transgenic mice (Kim et al., 2016) but whether this is related to the restoration of the UPR and ER/mitochondria contacts remains to be determined. Interestingly disruption of ER/mitochondria contacts appears to be a common phenomenon in ALS with reduced ER/mitochondria contact sites found in mutant SOD1, Sig1R, TDP-43, and FUS-related ALS (Lautenschlager et al., 2013; Stoica et al., 2014, 2016; Watanabe et al., 2016).
Mutations in the SIGMAR1 gene that encodes Sig1R cause a juvenile form of ALS (ALS16) (Al-Saif et al., 2011). Sig1R1 is an ER protein that resides at ER/mitochondria contacts where it interacts with BiP and regulates calcium exchange by acting as a ligand-operated receptor chaperone for the inositol 1,4,5-trisphosphate receptor 3 (IP3R3) (Hayashi and Su, 2007). ALS-associated loss of function of Sig1R has been linked to dysfunctional ER/mitochondria communication calcium dysfunction and ER stress (Prause et al., 2013; Vollrath et al., 2014).
Mutations in the ATPase VCP cause of 1–2% of familial ALS cases as well as inclusion body myopathy (IBM) with Paget’s disease (PDB) and FTD (Johnson et al., 2010; Koppers et al., 2012). Among other functions, VCP is involved in protein degradation, ERAD, ER stress and autophagy (reviewed in Meyer and Weihl, 2014), and pathogenic mutations have been shown to impair mitochondrial proteostasis, attenuate ERAD and lead to an impaired stress response (Fang et al., 2015; Wang et al., 2016). Mutant VCP has been linked to altered TDP-43 metabolism in Drosophila (Ritson et al., 2010) and spinal cord motor neurons of mutant VCP transgenic mice exhibit TDP-43 pathology (Custer et al., 2010). Mutant VCP expression also leads to redistribution of wild type TDP-43 from the nucleus to the cytoplasm in vitro and in vivo (Gitcho et al., 2009; Custer et al., 2010; Ritson et al., 2010). Thus, the typical TDP-43 pathology observed in ALS may be a direct consequence of dysfunctional proteostasis.
Proteasome Dysfunction and ALS
The characteristic pathological protein accumulations seen in ALS are indicative of defective protein clearance. In eukaryotes, the majority of misfolded proteins are degraded by the UPS in which poly-ubiquitin-tagged substrates are directed to and degraded by the 26S proteasome, a multimeric ATP-dependent protease complex comprised of the core 20S protease capped with two 19S regulatory subunits (reviewed in Eytan et al., 1989; Driscoll and Goldberg, 1990; Hoffman et al., 1992; Voges et al., 1999; Ciechanover and Kwon, 2015). Evidence from both familial and sporadic cases of ALS indicates proteasome dysfunction may be involved in disease pathogenesis (Figure 5).
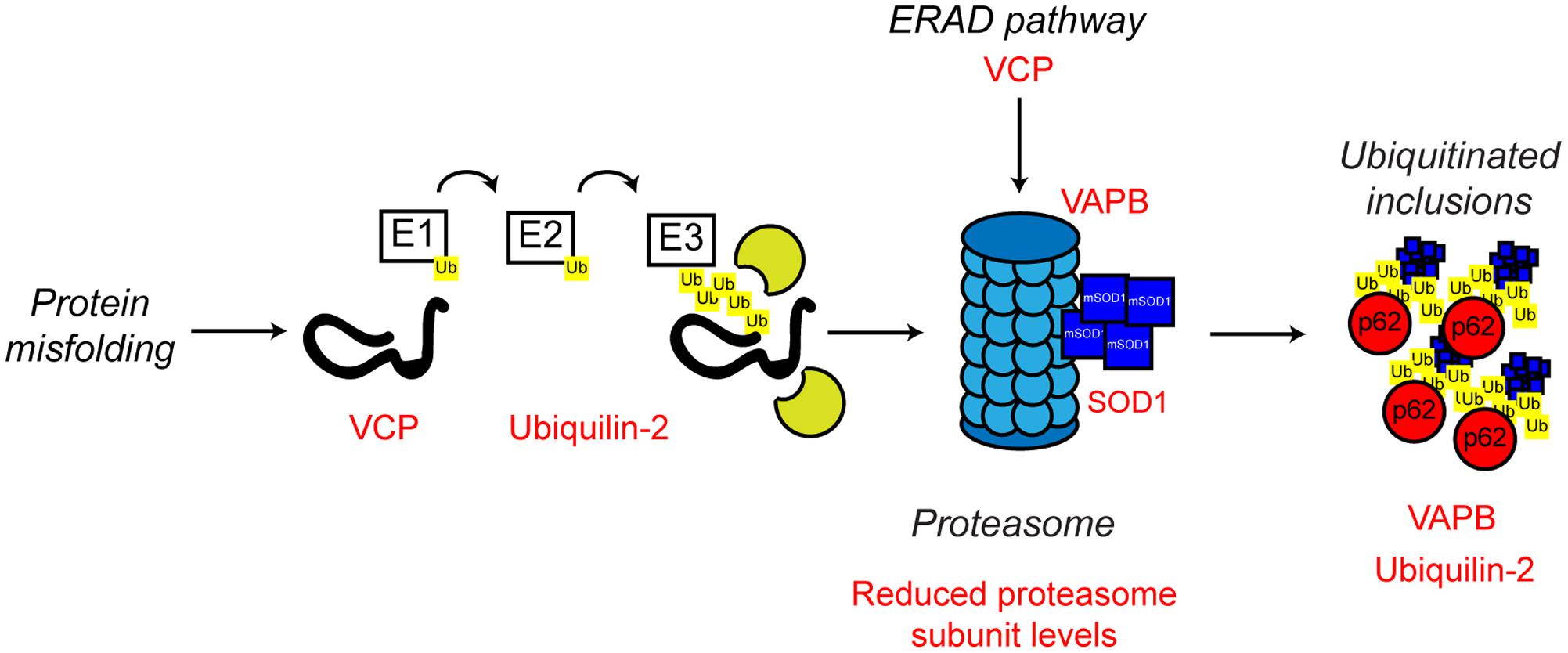
FIGURE 5. Proteasome dysfunction in ALS. The ubiquitin proteasome system is responsible for the degradation of poly-ubiquitinated protein substrates. Misfolded proteins are poly-ubiquitinated by the action of the E1, E2, and E3 ubiquitin ligases. Proteasome dysfunction has been implicated in ALS. Altered substrate delivery to the proteasome, mutant protein interaction with the proteasome, as in the case of mutant SOD1, and reduced proteasome function have all been implicated in ALS pathogenesis, ultimately leading to poly-ubiquitinated protein aggregate formation. The ALS-associated genes and their positions in the UPS are indicated in red, as are other ALS-associated defects. Interestingly, not only can mutant SOD1 interact with the 19S subunit of the proteasome, but mutant SOD1 is also a substrate for proteasome clearance. For further details please refer to the main text.
ALS mutant SOD1 has been shown to directly interact with the 19S regulatory subunits of the proteasome, which could contribute to the observed proteasomal inhibition seen in multiple models of ALS (Urushitani et al., 2002; Kabashi et al., 2004; Cheroni et al., 2005, 2009). Furthermore, reduced expression of UPS components in the spinal cord of SOD1G93A transgenic mice has been reported (Basso et al., 2009; Marino et al., 2015), and ALS mutant SOD1 itself is poly-ubiquitinated and cleared by the proteasome (Niwa et al., 2002; Urushitani et al., 2004). Possibly, age-related reductions in UPS activity or high demand, leads to the formation of cytotoxic mutant SOD1 inclusions (Kitamura et al., 2014).
Other familial ALS-associated proteins also support the role of a defective UPS in the development of ALS. In addition to its role in ERAD and ER stress discussed above, VCP is involved in substrate delivery to the proteasome (Dai and Li, 2001; Meyer and Weihl, 2014) and regulation of proteasome activity (Wojcik et al., 2004; Clemen et al., 2015). ALS-associated missense mutations in VCP have been shown to disrupt VCP-proteasome interaction, possibly resulting in defective proteasomal clearance of ubiquitinated proteins resulting in aggregation (Barthelme et al., 2015).
Mutations in UBQLN2 which encodes the ubiquitin-like protein, ubiquilin-2, are associated with X-linked ALS and ALS-dementia (Deng et al., 2011). Ubiquilin-2 is a member of the ubiquilin family, which regulates the degradation of ubiquitinated proteins. Patients with ubiquilin-2 mutations display the classical ALS-associated ubiquitinated protein aggregates, which, interestingly, are also positive for ubiquilin-2 (Deng et al., 2011). Like VCP, ubiquilin-2 is able to bind poly-ubiquitin chains and aids in substrate delivery to the proteasome (Ko et al., 2004). ALS-associated ubiquilin-2 mutants are defective in proteasome binding, resulting in defective substrate delivery to the proteasome and the accumulation of poly-ubiquitinated proteasome substrates (Chang and Monteiro, 2015). Ubiquilin-2 has also been implicated in an autophagy independent pathway that clears protein aggregates via the proteasome. In this pathway, ubiquilin-2 shuttles Hsp70-bound aggregated proteins to the proteasome to allow degradation. ALS mutant ubiquilin-2 has lost the ability to bind to Hsp70 and this sensitizes cells to protein stress (Hjerpe et al., 2016).
As discussed above, ALS mutant VAPB is linked with ER stress and defective UPR. However, there is also evidence that ALS mutant VAPB may impair the UPS. VAPBP56S is found in cytoplasmic aggregates, which by itself could be an indication of defective proteasome function (Suzuki et al., 2009; Moumen et al., 2011). Expression of mutant VAPBP56S in cells promotes the formation of ubiquitinated aggregates, as well as the accumulation of other proteasomal substrates (Chen et al., 2010; Moumen et al., 2011). Similarly, ubiquitinated inclusions have been found in the spinal cord motor neurons of VAPBP56S transgenic mice (Tudor et al., 2010). Furthermore, mutant VAPBP56S has been demonstrated to interact with the 20S subunit of the proteasome, suggesting proteasome sequestering and trapping, and therefore UPS dysfunction, could be a contributing factor in ALS8 (Moumen et al., 2011).
Impaired proteasome function has also been reported in sporadic ALS cases (Kabashi et al., 2012). Levels of the 20S proteasome subunit were significantly reduced in motor neurons of sporadic ALS cases compared to controls, and proteasomal activity was found to be impaired in the spinal cord (Kabashi et al., 2012). As sporadic ALS cases make up the vast majority of clinically diagnosed ALS cases, this finding suggests that defective proteasomal activity may explain ALS pathology and be a contributing factor in a high percentage of ALS cases. In this context, it is noteworthy that conditional knockout of the proteasome subunit Rpt3 in motor neurons in mice results in motor neuron degeneration and locomotor defects accompanied by the accumulation of a number of ALS-related proteins, including TDP-43, FUS and optineurin in intraneuronal inclusions (Tashiro et al., 2012). Such inclusions are obviously reminiscent of the classical ALS pathology, suggesting that proteasomal dysfunction alone may be sufficient to drive the development of ALS.
Autophagy
Autophagy is an intracellular lysosomal degradation pathway responsible for the bulk clearance of cytoplasmic components such as misfolded proteins and damaged organelles. Autophagy requires the formation of a double membrane structure called the autophagosome, which encapsulates autophagic substrates prior to their transport to, and fusion with the lysosome (reviewed in Bento et al., 2016). The autophagic process can be divided into four distinct stages: (i) translocation and initiation, (ii) elongation and recruitment, (iii) completion, and (iv) lysosome fusion and degradation. The stages of autophagy are detailed in Figure 6.
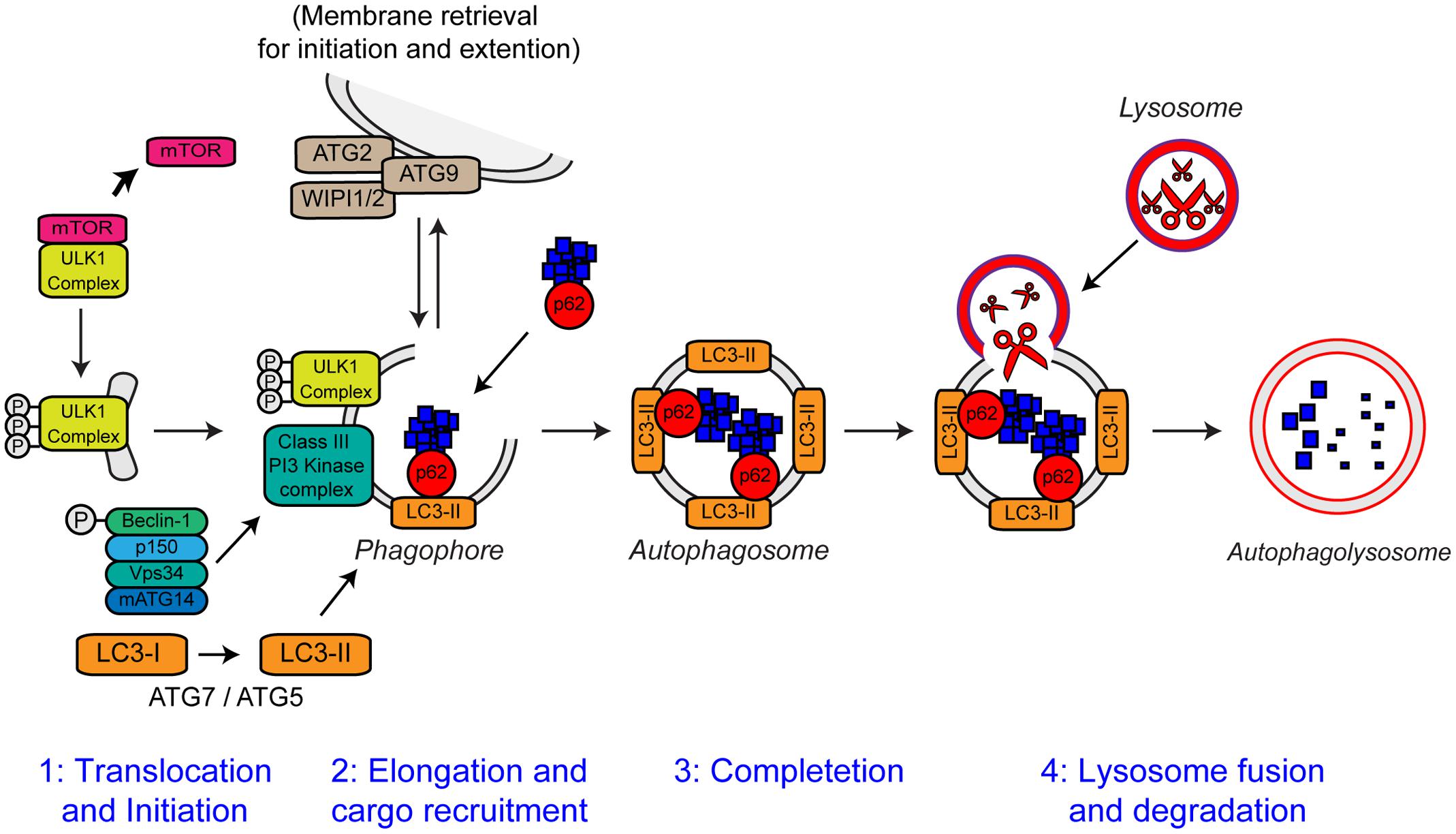
FIGURE 6. The steps of autophagy. The four stages of autophagy are indicated. (1) Translocation of the ULK1 initiation complex to the phagophore is the first step in autophagy initiation. Inhibition of mTOR, releases the ULK1 complex allowing activation and translocation of the complex. (2) Elongation of the phagophore membrane is mediated by the Class III PI3 kinase complex. Cargo recruitment to the growing phagophore is mediated by the autophagy receptors, p62/sequestosome-1 and optineurin. Autophagy receptors bind both poly-ubiquitin chains on autophagy substrates via the ubiquitin-like (Ubl) domains and LC3-II on the growing phagophore via LC3-interacting regions (LIRs). (3) After substrate recruitment and closure, completed autophagosomes are transported to allow fusion with the lysosome. (4) Autophagosome–lysosome fusion allows the degradation of the autophagic substrates by the action of acid hydrolases, present within the lysosome. Degradation allows the recycling of nutrients back to the cytosol. For further details please refer to the main text. Figure adapted from Webster et al. (2016b) under the terms of the Creative Commons Attribution License (CC BY).
Autophagy is essential for neuronal health. Inhibition of autophagy in neurons by neuronal-specific knockout of essential autophagy genes such as Atg7, Atg5, and RB1CC1 (FIP200) causes neurodegeneration in mice in absence of any other contributory factors (Hara et al., 2006; Komatsu et al., 2006; Liang et al., 2010). Loss of autophagy in these mice is accompanied by progressive deficits in motor function, including abnormal limb-clasping reflexes (also observed in ALS mutant SOD1 transgenic mice) and a reduction in coordinated movement. Furthermore, reminiscent of many neurodegenerative diseases including ALS, loss of basal autophagy causes accumulation of neuronal ubiquitin-positive cytoplasmic inclusion bodies (Hara et al., 2006; Komatsu et al., 2006; Liang et al., 2010). Basal neuronal autophagy appears to be especially required for the maintenance of axons as loss of autophagy causes axonal dystrophy (Komatsu et al., 2006).
Defective Autophagy in ALS
Growing evidence supports a role of defective autophagy in the pathogenesis of ALS. As discussed above, ubiquitinated inclusion bodies are a neuropathological hallmark of ALS, suggesting autophagy may be compromised. Both SOD1 and TDP-43 are autophagy substrates, and compromised autophagy causes accumulation of mutant SOD1 and TDP-43 (Kabuta et al., 2006; Hetz et al., 2009; Wang et al., 2010; Brady et al., 2011; Barmada et al., 2014). Consistent with a block in autophagic flux, the levels of LC3-II are increased in SOD1G93A and H46R transgenic mice (Li et al., 2008; Hadano et al., 2010) as well as C9orf72 knockout mice (O’Rourke et al., 2016). However, it has to be noted that increased LC3-II levels may also be the result increased induction of autophagy in response to protein aggregation. Induction of autophagy using trehalose enhanced SOD1 clearance in NSC-34 cells and protected SOD1G93A transgenic mice (Castillo et al., 2013). Genetic induction of autophagy by XBP-1 knockout also extended lifespan in SOD1G93A transgenic mice (Hetz et al., 2009; Matus et al., 2009). Conversely, heterozygous deletion of Beclin-1 exacerbated disease in SOD1G127X transgenic mice (Tokuda et al., 2016). Autophagy enhancers increased TDP-43 turnover and prevented cell death in ALS mutant TDP-43 expressing cell lines but in vivo data are not yet available (Barmada et al., 2014).
There appears to be a regulatory feedback loop between TDP-43 and autophagy. TDP-43 has been shown to regulate the transcription of the essential autophagy gene Atg7 (Bose et al., 2011). Loss of TDP-43 decreases Atg7 mRNA levels, in turn causing impairment of autophagy and accumulation of ubiquitinated proteins and p62/sequestosome-1. TDP-43, as well as FUS, regulate transcription of HDAC6 which, along with its binding partner p62/sequestosome-1, plays an important role in the clearance of protein aggregates by aggrephagy (reviewed in Lamark and Johansen, 2012), such that loss of TDP-43 or FUS dramatically reduces the level of HDAC6 mRNA (Fiesel et al., 2010; Kim et al., 2010; Xia et al., 2015). Hence cytoplasmic aggregation of TDP-43/FUS, and associated loss of function, decreases Atg7 and HDAC6 expression and consequently autophagy while, conversely, reduced autophagy promotes TDP-43/FUS aggregation, in a perpetuating toxic loop.
The Function of ALS Genes in Autophagy
Several familial ALS genes function at different stages in the autophagy pathway (Figure 7).
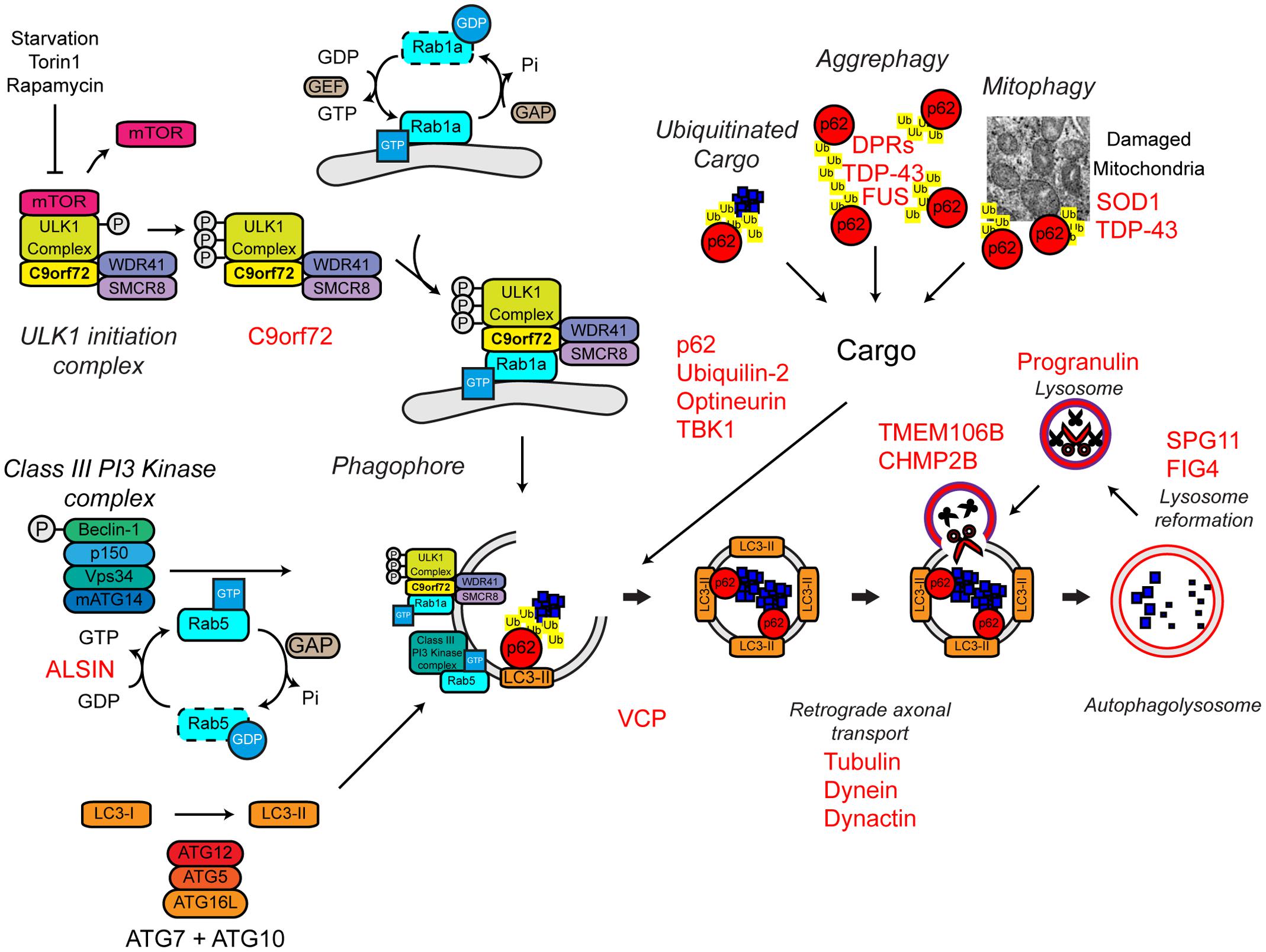
FIGURE 7. Autophagy dysfunction and ALS. Many ALS-associated genes, indicated in red, are implicated in the autophagy pathway. The location of gene names indicates the likely part of the pathway affected in ALS. Protein aggregates are a common feature of ALS pathology. The autophagy cargoes detailed here include a number of ALS-associated proteins, namely SOD1 and TDP-43, both of which are considered autophagy substrates. Mutant forms of these proteins, alterations to the pathway as a whole, or aberrant production of autophagy substrates, potentially in the case of C9orf72-related DPR proteins, may disrupt efficient substrate clearance, overwhelming the autophagy pathway and further promoting autophagy dysfunction. Importantly, TDP-43 is also important for autophagy gene transcription, thus participating as both a substrate and a regulator of the autophagy. For further details please refer to the main text. Figure adapted from Webster et al. (2016b) under the terms of the Creative Commons Attribution License (CC BY).
Initiation
The most common genetic cause of ALS and FTD is a hexanucleotide repeat expansion of GGGGCC in the first intron of the C9ORF72 gene (collectively termed C9ALS/FTD) (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The pathogenic mechanisms behind this repeat expansion have been reviewed elsewhere, but may include toxic gain-of-function, namely via RNA toxicity and DPR formation, or alternatively loss-of-function via C9ORF72 haploinsufficiency (reviewed in Cooper-Knock et al., 2015; Haeusler et al., 2016). With evidence for all three mechanisms in patients and a range of models, it is likely that all three mechanisms are at play. We and others have recently identified the C9orf72 protein as a regulator of autophagy (Amick et al., 2016; Sellier et al., 2016; Sullivan et al., 2016; Webster et al., 2016a; reviewed in Webster et al., 2016b; Yang et al., 2016). We found that C9orf72 is an effector of Rab1a that facilitates trafficking of the ULK1 initiation complex to the phagophore during autophagy initiation (Webster et al., 2016a) while others demonstrated that a complex of C9orf72, SMCR8, and WDR41 acts as a guanine nucleotide exchange factor for RAB8a and RAB39b further down the autophagy pathway during autophagosome maturation (Sellier et al., 2016). Furthermore, the C9orf72/SMCR8/WDR41 complex was shown to interact with the autophagy receptor optineurin (itself a Rab8a interactor) and TBK1, a well-known regulator of autophagy (Sellier et al., 2016). Both optineurin and TBK1 have been shown to be mutated in ALS cases (Table 1). Loss of C9orf72 in neurons inhibited autophagy and caused accumulation of p62/sequestosome-1 (Sellier et al., 2016; Webster et al., 2016a) while C9ALS/FTD patient-derived iNeurons exhibited reduced basal autophagy levels compared to controls (Webster et al., 2016a). Thus, these data predict that C9orf72 haploinsufficiency impairs autophagy. Consistent with compromised autophagy, C9ALS/FTD patients characteristically exhibit ubiquitin and p62/sequestosome-1 positive, TDP-43 negative, inclusions in the cerebellum and hippocampus (Al-Sarraj et al., 2011; Cooper-Knock et al., 2012; Mahoney et al., 2012).
In addition to C9orf72, another Rab GTPase-associated protein associated with autophagy is involved in ALS. Alsin is a GDP/GTP exchange factor for the small GTPase Rab5 (Topp et al., 2004). Rab5 is involved in autophagy initiation via activation of the Vps34 complex and the recruitment of the ATG12–ATG5 conjugation system required for autophagosome formation (Ravikumar et al., 2008; Figure 7). Pathogenic missense mutations in ALS2 lead to loss of Alsin function, reduced Vps34 activation, defective autophagosome formation, and ultimately the development of ALS (Hadano et al., 2001, 2010; Otomo et al., 2011). Consistent with a role in autophagosome formation, genetic ablation of Alsin in SOD1H46R transgenic mice exacerbated disease and enhanced accumulation of a range of autophagy substrates including mutant SOD1 aggregates, poly-ubiquitinated proteins, and p62/sequestosome-1 (Hadano et al., 2010).
Delivery of substrates to the autophagosome
Autophagy receptors deliver cargoes to the forming autophagosome by binding to both ubiquitinated substrates and LC3-II via ubiquitin-associated (UBA) and LC3-interacting region (LIR) domains, respectively. Autophagy receptors are crucial for effective substrate clearance (reviewed in Stolz et al., 2014). Mutations in the autophagy receptors, ubiquilin-2, optineurin, and p62/sequestosome-1 have been shown to cause ALS (Fecto et al., 2011; Hirano et al., 2013; Teyssou et al., 2013; Chen et al., 2014; Majcher et al., 2015; Figure 7). These mutations appear to be loss-of function mutations that impair delivery of substrates to the autophagosome.
Mutations in ubiquilin-2, which have been found in both sporadic and familial ALS, lead to the incorporation of ubiquilin-2 into cytoplasmic aggregates in the brain and spinal cord (Deng et al., 2011). These aggregates are also positive for other ALS-related proteins, including TDP-43 and ubiquitin. Furthermore, ubiquilin-2 has been shown to interact directly with TDP-43 (Cassel and Reitz, 2013), suggesting that the mislocalisation of TDP-43 and its incorporation into cytoplasmic aggregates could be a product of defective autophagic clearance. Wild type ubiquilin-2 co-localizes with optineurin on p62/sequestosome-1 and ULK1 positive vesicles during autophagy induction (Osaka et al., 2015). However, the ALS-associated mutant ubiquilin-2 is absent from these optineurin positive vesicles, indicating a potential loss of function mechanism, and supporting the idea that ALS-associated mutations in ubiquilin-2 cause autophagic clearance defects (Osaka et al., 2015).
ALS-associated mutations within the OPTN gene lead to the increased formation of cytoplasmic aggregates, which are immunoreactive for optineurin itself (Maruyama et al., 2010; Ito et al., 2011; Korac et al., 2013; Weishaupt et al., 2013). The E478G mutation is found within the ubiquitin binding domain of optineurin, suggesting that, at least in the case of this mutant, substrate binding and therefore autophagosomal delivery may be compromised (Maruyama et al., 2010). It has also been reported that mutant optineurin can sequester and “trap” wild type proteins leading to defective autophagosome maturation, and therefore defective clearance (Shen et al., 2015).
ALS-associated mutations in p62/sequestosome-1 map to the LIR domain (Chen et al., 2014). One of these mutations, L341V, was shown to disrupt interaction with LC3-II (Goode et al., 2016), supporting the idea that p62/sequestosome-1 mutations disturb the delivery of autophagic substrates to the autophagosome. However, the effect of other mutations on the function of p62/sequestosome-1 and their effects on autophagy are yet to be determined.
The activities of p62/sequestosome-1 and optineurin are, in part, regulated by TBK1. Phosphorylation by TBK1 increases interaction of p62/sequestosome-1 and optineurin with LC3-II and/or ubiquitin chains, and increases autophagic clearance (Weidberg and Elazar, 2011; Wild et al., 2011; Matsumoto et al., 2015; Richter et al., 2016). As mentioned above, TBK1 also interacts with the C9orf72/SMCR8/WDR41 complex and phosphorylates SMCR8, but the significance of this phosphorylation is not yet clear (Sellier et al., 2016). Haploinsufficiency of TBK1 has been shown to cause ALS, likely by compromising its regulatory function in autophagy (Freischmidt et al., 2015).
Maturation
As previously discussed, VCP mutations have been found in both sporadic and familial ALS (Table 1). Although, VCP was initially identified as being involved in proteasome substrate delivery – see above (Dai and Li, 2001), there is also evidence supporting a role in autophagy. Reduced VCP activity leads to impaired maturation of autophagosomes and thus accumulation of ubiquitin and p62/sequestosome-1-positive aggregates (Ju et al., 2009; Tresse et al., 2010). VCP is also linked to two other ALS-associated proteins, TDP-43 and FUS. Both TDP-43 and FUS are involved in stress granule formation, cytoplasmic sites of stalled mRNA translation that form in response to stress (Buchan and Parker, 2009; Colombrita et al., 2009; Dormann et al., 2010; Bentmann et al., 2012). Mutant TDP-43 and FUS readily accumulate into stress granules (Bosco et al., 2010; Baron et al., 2013; Vance et al., 2013; Walker et al., 2013) and VCP and autophagy have been linked to stress granule clearance (Buchan et al., 2013).
FIG4 regulates the cellular levels of phosphatidylinositol-3,5-bisphosphate (PIP2), a signaling lipid required in autophagy (Ferguson et al., 2009). FIG4 mutations lead to ALS potentially by a loss of function mechanism (Chow et al., 2009). FIG4 knockout in mice leads to the accumulation of p62/sequestosome-1 and LC3-II, suggesting reduced autophagosome clearance could be at play in FIG4-related ALS (Ferguson et al., 2009).
Dynactin, a multimeric protein complex, interacts with cytoplasmic dynein to bring about retrograde transport of cargos (Waterman-Storer et al., 1997). Multiple point mutations in the p150 subunit of dynactin (DCTN1; dynactin-1) are found in a number of neurodegenerative diseases, including ALS (Puls et al., 2003; Münch et al., 2004, 2005; Vilariño-Güell et al., 2009). The ALS-associated mutations in dynactin may disrupt dynein–dynactin interactions, leading to aggregation of mutant dynactin and its respective cargos (Levy et al., 2006). Autophagosome transport in neurons is regulated by the dynein–dynactin complex and therefore disruption of this complex may lead to aberrant autophagosome transport and protein aggregation (Ravikumar et al., 2005; Maday et al., 2012; Maday and Holzbaur, 2014). In support of this, knock down of dynactin-1 in Caenorhabditis elegans causes motor defects, axonal degeneration and impaired transport and subsequent accumulation of autophagosomes (Ikenaka et al., 2013). Furthermore, reduced expression of dynactin-1 has been reported in sporadic ALS, suggesting alterations to the dynactin complex and disrupted autophagosome transport could be a common pathogenic mechanism in ALS (Jiang et al., 2007; Kuzma-Kozakiewicz et al., 2013).
Degradation
Missense mutations in the charged multivesicular body protein 2B (CHMP2B) gene is associated with FTD and predominantly lower motor neuron ALS (Skibinski et al., 2005; Parkinson et al., 2006; Cox et al., 2010). The CHMP2B protein is part of the endosomal sorting complex required for transport-III (ESCRT-III) (Bodon et al., 2011), which sorts ubiquitinated protein substrates from endosomes to the lysosomes by the formation of multivesicular bodies (MVBs) (reviewed in Williams and Urbe, 2007). Efficient autophagic degradation requires the formation of functional MVBs (Filimonenko et al., 2007). Interestingly, loss of ESCRT-III members or expression of mutant CHMP2B leads to the accumulation of autophagosome markers such as p62/sequestosome-1 and LC3-II consistent with reduced lysosomal clearance (Filimonenko et al., 2007; Lee et al., 2007; Urwin et al., 2010). Indeed impaired fusion events between the autophagosome and the lysosome has been demonstrated in HEK293 cells overexpressing mutant CHMP2B (Urwin et al., 2010).
Further evidence of defective autophagosome–lysosome fusion and clearance comes from the study of patients harboring mutations in SPG11, which encodes the Spatacsin protein. Deletions and frameshift mutations in SPG11, lead to hereditary spastic paraplegia (HSP) as well as juvenile recessive ALS (Orlacchio et al., 2010). Autophagosome accumulation in mutant SPG11 patient fibroblast cells indicates that loss of spatacsin results in reduced clearance, potentially due to reduced lysosome biogenesis (Chang et al., 2014; Renvoisé et al., 2014). Data from mutant SPG11 patient-derived iPSC neurons also indicates that loss of spatacsin may lead to impaired axonal transport, suggesting a convergence of pathogenic mechanisms (Pérez-Brangulí et al., 2014).
Modulating Proteostasis to Treat ALS
The evidence discussed above strongly suggests that protein aggregation due to collapse of proteostasis may contribute to the etiology of ALS. Thus, restoring proteostasis to reduce aggregated protein burden has emerged as an attractive therapeutic target. Several pathways in the proteostasis network are amenable to pharmaceutical intervention and have been targeted in experimental models. To date, treatments have focused on either increasing protein folding to prevent protein aggregation or removal of aggregated proteins (Table 2).
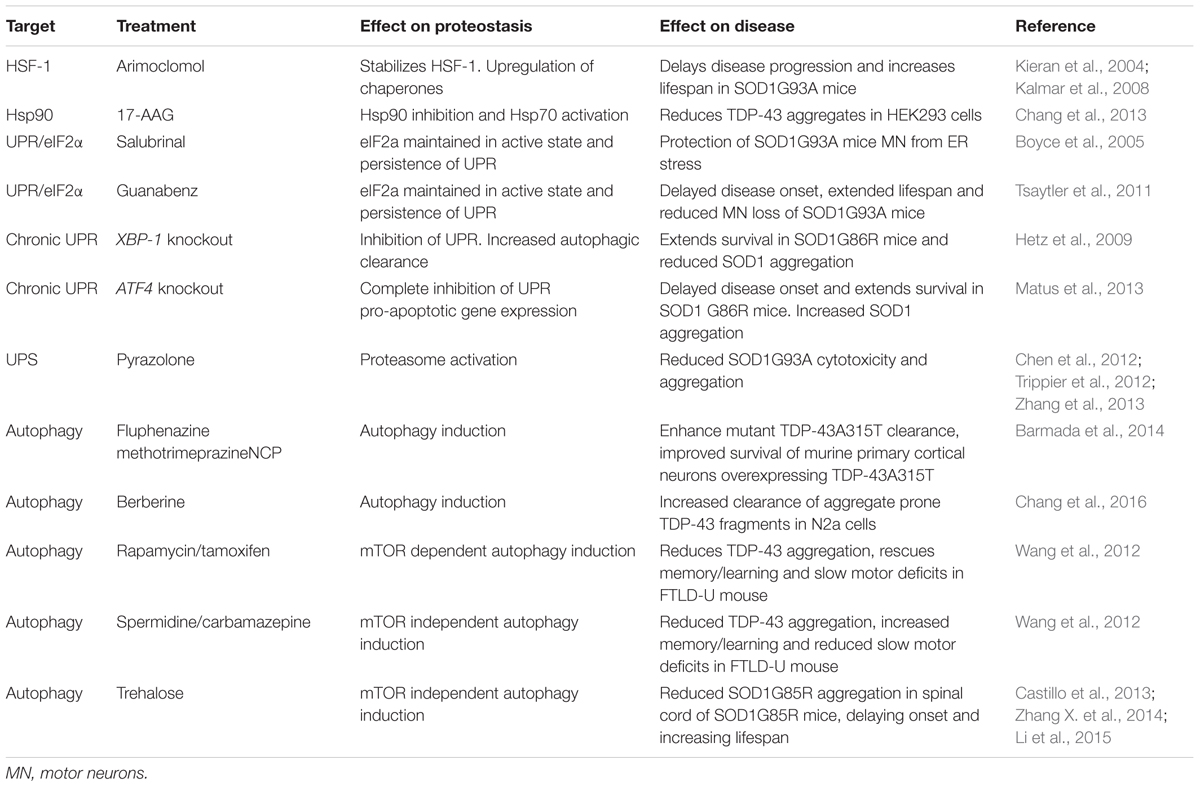
TABLE 2. Restoring protein homeostasis as a treatment for ALS.
Upregulation of Chaperone Function
As discussed above, molecular chaperones play an essential role in the proteostasis network. They are upregulated in response to protein stress (e.g., by the UPR), and thus are amenable to regulation. Furthermore, evidence suggests that inhibition of chaperone activity may contribute to ALS.
Hydroxylamine derivatives, such as Arimoclomol stabilize the transcription factor heat shock factor protein 1 (HSF-1) in its active state leading to its prolonged activation and the upregulation of a number of heat shock family chaperones, including Hsp60, Hsp70, and Hsp90 (Vígh et al., 1997). Arimoclomol has been shown to delay disease progression, extend lifespan, increase muscle function, and prevent aggregation of mutant SOD1 in SOD1G93A mouse models of ALS (Kieran et al., 2004; Kalmar et al., 2008, 2012). Importantly, HSF-1 does not bind its corresponding DNA elements in the absence of cellular stress. Thus, the prolonged activation of HSF-1 by hydroxylamine derivatives only enhances the heat shock response in already stressed cells, therefore mitigating the potentially toxic induction of the heat shock response and chaperone upregulation in otherwise healthy cell populations (Hargitai et al., 2003).
In a similar fashion to Arimoclomol, treatment with the Hsp90 inhibitor 17-AAG activates Hsp70 reducing the number of pathological TDP-43 aggregates in HEK293T cells overexpressing aggregation-prone TDP-43 C-terminal fragments (Chang et al., 2013).
Treatments Targeting the UPR
ER stress appears to be involved in ALS pathogenesis. Accordingly, the UPR which alleviates ER stress under physiological conditions may be an attractive therapeutic target.
The PERK branch of the UPR leads to phosphorylation of the eukaryotic translational initiation factor eIF2α which decreases global protein synthesis and up-regulates the translation of selected stress-induced mRNAs such as the transcription factor ATF4. ATF4 induces expression of genes involved in amino acid metabolism, resistance to oxidative stress, and the proapoptotic transcription factor CHOP (Figure 3). The ER stress inhibitors salubrinal and guanabenz prevent eIF2α dephosphorylation and keep eIF2α in its inactive state (Boyce et al., 2005; Tsaytler et al., 2011). Salubrinal treatment protected SOD1G93A transgenic mouse motor neurons from ER stress (Saxena et al., 2009) and treatment of SOD1G93A transgenic mice with guanabenz delayed onset of disease, attenuated motor neuron loss and significantly extended lifespan (Jiang et al., 2014; Wang et al., 2014). In contrast, others have shown that guanabenz treatment accelerated disease progression in SOD1G93A transgenic mice (Vieira et al., 2015). The reason for these opposing findings is not clear, but may be attributable to differences in study design (discussed in Vieira et al., 2015). Both guanabenz and salubrinal attenuated ER stress and reduced paralysis and neurodegeneration in mutant TDP-43 Caenorhabditis elegans and zebrafish models of ALS (Vieira et al., 2015).
Perhaps counter-intuitively, conditional knockout of XBP-1 was shown to extend the survival of female SOD1G86R transgenic mice (Hetz et al., 2009). However, in this model, abrogation of the UPR coincided with increased autophagic clearance of SOD1 aggregates, suggesting that a lower aggregated protein burden elicits the protective effect of XBP-1 knockout (Hetz et al., 2009). Similarly, knockout of ATF4 also delayed disease onset and prolonged life span in SOD1G86R transgenic mice. ATF4 deficiency completely prevented the induction of pro-apoptotic genes, but increased SOD1 aggregation suggesting that, in this case, prevention of UPR-induced apoptosis mediated protection (Matus et al., 2013).
Increasing Degradation of Misfolded Proteins and Aggregates
While increasing protein folding may help to reduce the pathological protein accumulations in ALS, an alternative approach is to promote their degradation by activation of the UPS and/or autophagy.
Proteasome Activation
Pyrazolone-containing small molecules have been shown to block ALS mutant SOD1 mediated cytotoxicity and aggregation in a number of studies (Chen et al., 2012; Trippier et al., 2012; Zhang et al., 2013). The mechanism of action of pyrazolone was shown to be by activation of the proteasome, suggesting that proteasome activators may be of therapeutic benefit in ALS (Trippier et al., 2014). However, the protective action of pyrazolones has only been tested in mutant SOD1-related ALS models, and as such more studies are needed to test the efficacy of these small molecules in other ALS models.
Enhancing Autophagy
Most attempts to modulate autophagy as a treatment for ALS have been aimed at enhancing the removal of aggregated proteins by stimulating autophagic flux. In vitro, three potent inducers of autophagy, fluphenazine, methotrimeprazine and 10-(4′-(N-diethylamino)butyl)-2-chlorophenoxazine, efficiently enhance ALS mutant TDP-43A315T aggregate clearance and improved survival of murine primary cortical neurons overexpressing TDP-43A315T as well as iPSC-derived motor neurons and astrocytes carrying the pathogenic M337V TDP-43 mutation (Barmada et al., 2014). Similarly, the traditional medicinal herb and known inducer of autophagy, berberine, increased clearance of EGFP-tagged aggregation-prone C-terminal TDP-43 fragments, and reduced insoluble TDP-43 aggregates in transfected Neuro2a cells (Chang et al., 2016). In vivo, a number of mTOR-dependent (rapamycin, tamoxifen) and independent (spermidine, carbamazepine) autophagy enhancers have been shown to reduce TDP-43 aggregation, rescue learning/memory and slow motor deficits in an FTLD-U mouse model with transgenic overexpression of TDP-43 in the hippocampus, cortex, and striatum (Wang et al., 2012). Hence induction of autophagy may be of therapeutic benefit to clear ALS-associated TDP-43 aggregates.
Activating autophagy may also be beneficial in the removal of mutant SOD1 aggregates. Trehalose, an mTOR independent activator of autophagy, has been shown to reduce the aggregation of SOD1G85R in the spinal cord of SOD1G85R transgenic mice, an effect that was accompanied by increased lifespan and delayed disease onset in these mice (Castillo et al., 2013). Similar results were reported in SOD1G93A transgenic mice (Zhang X. et al., 2014), although the efficacy of trehalose may decline as the disease progresses (Li et al., 2015). In contrast to trehalose, mTOR dependent induction of autophagy using rapamycin has either no effect (Staats et al., 2013) or exacerbates disease in SOD1G93A transgenic mice (Zhang et al., 2011). This effect of rapamycin may stem from its function as an immunosuppressant, because rapamycin treatment did increase survival in SOD1G93A transgenic mice lacking mature lymphocytes (Staats et al., 2013).
Discussion and Outlook
Collapse of proteostasis and resulting protein aggregation is a universal feature of ALS as discussed above. Most evidence suggest that protein aggregation is deleterious for cells and, by extension, this may also be the case in ALS. In agreement, boosting clearance of protein aggregates and/or preventing protein aggregation appears to be of benefit in animal models of ALS. However, it has been suggested that protein aggregation may be a protective mechanism. By compartmentalization of the toxic misfolded proteins the cell prevents them from causing damage, e.g., to mitochondria or to proteostasis by sequestering chaperones and blocking the UPS. Furthermore, compartmentalization may facilitate clearance by aggrephagy. Thus, some caution is warranted when devising treatments to prevent protein aggregation.
Proteostasis is a universal process in every cell, not just neurons. Before embarking on long term treatment with for example autophagy enhancers, possible side effects need to be considered. For example, evidence from the cancer field indicates that in most cases autophagy facilitates tumorigenesis (reviewed in White, 2015). Similarly, inhibition of the PERK branch of the UPR may seem a good idea in principle but balancing the UPR appears to be critical for neuronal integrity, and long-term PERK inhibition may not be feasible due to considerable side effects (Scheper and Hoozemans, 2013). Furthermore, is important to take into account the genetic background of individual ALS patients when considering employing modulators of protein quality control as a treatment of disease. For example, treating patients with a known autophagosome clearance defect with autophagy enhancers would not be likely to be beneficial and may even be deleterious.
Nevertheless, results from ALS models and models of other neurodegenerative diseases such as Huntington’s disease indicate that treatments targeting proteostasis and in particular autophagy have great potential. Further understanding of the genetics of ALS and the mechanisms of proteostasis dysfunction associated with ALS will be vital if proteostasis modulators are to be used effectively as neuroprotective therapies.
Author Contributions
CW, ES, and KDV: wrote the manuscript. CW: prepared the figures. PS and KDV: edited the manuscript and figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by grants from the Medical Research Council (MRC; MR/K005146/1 and MR/M013251/1 to KDV) and Alzheimer’s Society [260 (AS-PG-15-023)]. ES is supported by a Motor Neurone Disease Association Prize Studentship (DeVos/Oct13/870-892 to KDV). PS is supported as an NIHR Senior Investigator and by funding award from the UK Medical Research Council, the National Institute for Health Research, the Motor Neurone Disease Association and the EU Horizon 2020 program.
References
Abel, O., Powell, J. F., Andersen, P. M., and Al-Chalabi, A. (2012). ALSoD: a user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 33, 1345–1351. doi: 10.1002/humu.22157
Ackerley, S., Grierson, A. J., Brownlees, J., Thornhill, P., Anderton, B. H., Nigel Leigh, P., et al. (2000). Glutamate slows axonal transport of neurofilaments in transfected neurons. J. Cell Biol. 150, 165–175. doi: 10.1083/jcb.150.1.165
Ackerley, S., Thornhill, P., Grierson, A. J., Brownlees, J., Anderton, B. H., Leigh, P. N., et al. (2003). Neurofilament heavy chain side arm phosphorylation regulates axonal transport of neurofilaments. J. Cell Biol. 161, 489–495. doi: 10.1083/jcb.200303138
Al-Chalabi, A., Jones, A., Troakes, C., King, A., Al-Sarraj, S., and van den Berg, L. H. (2012). The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 124, 339–352. doi: 10.1007/s00401-012-1022-4
Al-Saif, A., Al-Mohanna, F., and Bohlega, S. (2011). A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 70, 913–919. doi: 10.1002/ana.22534
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). P62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi: 10.1007/s00401-011-0911-2
Amick, J., Roczniak-Ferguson, A., and Ferguson, S. M. (2016). C9orf72 binds SMCR8, localizes to lysosomes and regulates mTORC1 signaling. Mol. Biol. Cell 27, 3040–3051. doi: 10.1091/mbc.E16-01-0003
Anagnostou, G., Akbar, M. T., Paul, P., Angelinetta, C., Steiner, T. J., and de Belleroche, J. (2010). Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging 31, 969–985. doi: 10.1016/j.neurobiolaging.2008.07.005
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Atkin, J. D., Farg, M. A., Turner, B. J., Tomas, D., Lysaght, J. A., Nunan, J., et al. (2006). Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 281, 30152–30165. doi: 10.1074/jbc.M603393200
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Barmada, S. J., Serio, A., Arjun, A., Bilican, B., Daub, A., Ando, D. M., et al. (2014). Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 10, 677–685. doi: 10.1038/nchembio.1563
Barmada, S. J., Skibinski, G., Korb, E., Rao, E. J., Wu, J. Y., and Finkbeiner, S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 30, 639–649. doi: 10.1523/JNEUROSCI.4988-09.2010
Baron, D. M., Kaushansky, L. J., Ward, C. L., Sama, R. R. K., Chian, R.-J., Boggio, K. J., et al. (2013). Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 8, 1–18. doi: 10.1186/1750-1326-8-30
Barthelme, D., Jauregui, R., and Sauer, R. T. (2015). An ALS disease mutation in Cdc48/p97 impairs 20S proteasome binding and proteolytic communication. Protein Sci. 24, 1521–1527. doi: 10.1002/pro.2740
Basso, M., Samengo, G., Nardo, G., Massignan, T., D’Alessandro, G., Tartari, S., et al. (2009). Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS ONE 4:e8130. doi: 10.1371/journal.pone.0008130
Bentmann, E., Neumann, M., Tahirovic, S., Rodde, R., Dormann, D., and Haass, C. (2012). Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094. doi: 10.1074/jbc.M111.328757
Bento, C. F., Renna, M., Ghislat, G., Puri, C., Ashkenazi, A., Vicinanza, M., et al. (2016). Mammalian autophagy: how does it work? Annu. Rev. Biochem. 85, 685–713. doi: 10.1146/annurev-biochem-060815-014556
Bergemalm, D., Forsberg, K., Srivastava, V., Graffmo, K. S., Andersen, P. M., Brannstrom, T., et al. (2010). Superoxide dismutase-1 and other proteins in inclusions from transgenic amyotrophic lateral sclerosis model mice. J. Neurochem. 114, 408–418. doi: 10.1111/j.1471-4159.2010.06753.x
Blokhuis, A. M., Groen, E. J. N., Koppers, M., van den Berg, L. H., and Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794. doi: 10.1007/s00401-013-1125-6
Bodon, G., Chassefeyre, R., Pernet-Gallay, K., Martinelli, N., Effantin, G., Hulsik, D. L., et al. (2011). Charged multivesicular body protein 2B (CHMP2B) of the Endosomal Sorting Complex Required for Transport-III (ESCRT-III) polymerizes into helical structures deforming the plasma membrane. J. Biol. Chem. 286, 40276–40286. doi: 10.1074/jbc.M111.283671
Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J., et al. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19, 4160–4175. doi: 10.1093/hmg/ddq335
Bose, J. K., Huang, C. C., and Shen, C. K. (2011). Regulation of autophagy by neuropathological protein TDP-43. J. Biol. Chem. 286, 44441–44448. doi: 10.1074/jbc.M111.237115
Boyce, M., Bryant, K. F., Jousse, C., Long, K., Harding, H. P., Scheuner, D., et al. (2005). A Selective Inhibitor of eIF2α Dephosphorylation protects cells from ER stress. Science 307, 935. doi: 10.1126/science.1101902
Braakman, I., and Bulleid, N. J. (2011). Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99. doi: 10.1146/annurev-biochem-062209-093836
Brady, O. A., Meng, P., Zheng, Y., Mao, Y., and Hu, F. (2011). Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 116, 248–259. doi: 10.1111/j.1471-4159.2010.07098.x
Buchan, J. R., Kolaitis, R.-M., Taylor, J. P., and Parker, R. (2013). Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153, 1461–1474. doi: 10.1016/j.cell.2013.05.037
Buchan, J. R., and Parker, R. (2009). Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36, 932–941. doi: 10.1016/j.molcel.2009.11.020
Budini, M., Romano, V., Quadri, Z., Buratti, E., and Baralle, F. E. (2015). TDP-43 loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N prion-like domain. Hum. Mol. Genet. 24, 9–20. doi: 10.1093/hmg/ddu415
Bunina, T. L. (1962). On intracellular inclusions in familial amyotrophic lateral sclerosis. Zh. Nevropatol. Psikhiatr. Im. S S Korsakova 62, 1293–1299.
Cassel, J. A., and Reitz, A. B. (2013). Ubiquilin-2 (UBQLN2) binds with high affinity to the C-terminal region of TDP-43 and modulates TDP-43 levels in H4 cells: characterization of inhibition by nucleic acids and 4-aminoquinolines. Biochim. Biophys. Acta 1834, 964–971. doi: 10.1016/j.bbapap.2013.03.020
Castillo, K., Nassif, M., Valenzuela, V., Rojas, F., Matus, S., Mercado, G., et al. (2013). Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9, 1308–1320. doi: 10.4161/auto.25188
Chang, C.-F., Lee, Y.-C., Lee, K.-H., Lin, H.-C., Chen, C.-L., Shen, C.-K. J., et al. (2016). Therapeutic effect of berberine on TDP-43-related pathogenesis in FTLD and ALS. J. Biomed. Sci. 23, 72. doi: 10.1186/s12929-016-0290-z
Chang, H. Y., Hou, S. C., Way, T. D., Wong, C. H., and Wang, I. F. (2013). Heat-shock protein dysregulation is associated with functional and pathological TDP-43 aggregation. Nat. Commun. 4, 2757. doi: 10.1038/ncomms3757
Chang, J., Lee, S., and Blackstone, C. (2014). Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Invest. 124, 5249–5262. doi: 10.1172/JCI77598
Chang, L., and Monteiro, M. J. (2015). Defective proteasome delivery of polyubiquitinated proteins by ubiquilin-2 proteins containing ALS mutations. PLoS ONE 10:e0130162. doi: 10.1371/journal.pone.0130162
Chen, H.-J., Anagnostou, G., Chai, A., Withers, J., Morris, A., Adhikaree, J., et al. (2010). Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J. Biol. Chem. 285, 40266–40281. doi: 10.1074/jbc.M110.161398
Chen, T., Benmohamed, R., Kim, J., Smith, K., Amante, D., Morimoto, R. I., et al. (2012). ADME-guided design and synthesis of aryloxanyl pyrazolone derivatives to block mutant superoxide dismutase 1 (SOD1) cytotoxicity and protein aggregation: potential application for the treatment of amyotrophic lateral sclerosis. J. Med. Chem. 55, 515–527. doi: 10.1021/jm2014277
Chen, Y., Zheng, Z. Z., Chen, X., Huang, R., Yang, Y., Yuan, L., et al. (2014). SQSTM1 mutations in Han Chinese populations with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 35, e727–e729. doi: 10.1016/j.neurobiolaging.2013.09.008
Cheroni, C., Marino, M., Tortarolo, M., Veglianese, P., De Biasi, S., Fontana, E., et al. (2009). Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, 82–96. doi: 10.1093/hmg/ddn319
Cheroni, C., Peviani, M., Cascio, P., Debiasi, S., Monti, C., and Bendotti, C. (2005). Accumulation of human SOD1 and ubiquitinated deposits in the spinal cord of SOD1G93A mice during motor neuron disease progression correlates with a decrease of proteasome. Neurobiol. Dis. 18, 509–522. doi: 10.1016/j.nbd.2004.12.007
Chow, C. Y., Landers, J. E., Bergren, S. K., Sapp, P. C., Grant, A. E., Jones, J. M., et al. (2009). Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 84, 85–88. doi: 10.1016/j.ajhg.2008.12.010
Ciechanover, A., and Kwon, Y. T. (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 47, e147. doi: 10.1038/emm.2014.117
Clemen, C. S., Marko, M., Strucksberg, K. H., Behrens, J., Wittig, I., Gartner, L., et al. (2015). VCP and PSMF1: antagonistic regulators of proteasome activity. Biochem. Biophys. Res. Commun. 463, 1210–1217. doi: 10.1016/j.bbrc.2015.06.086
Colombrita, C., Zennaro, E., Fallini, C., Weber, M., Sommacal, A., Buratti, E., et al. (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x
Cooper-Knock, J., Hewitt, C., Highley, J. R., Brockington, A., Milano, A., Man, S., et al. (2012). Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 135, 751–764. doi: 10.1093/brain/awr365
Cooper-Knock, J., Kirby, J., Highley, R., and Shaw, P. J. (2015). The Spectrum of C9orf72-mediated neurodegeneration and amyotrophic lateral sclerosis. Neurotherapeutics 12, 326–339. doi: 10.1007/s13311-015-0342-1
Cox, L. E., Ferraiuolo, L., Goodall, E. F., Heath, P. R., Higginbottom, A., Mortiboys, H., et al. (2010). Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 5:e9872. doi: 10.1371/journal.pone.0009872
Crippa, V., Sau, D., Rusmini, P., Boncoraglio, A., Onesto, E., Bolzoni, E., et al. (2010). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 19, 3440–3456. doi: 10.1093/hmg/ddq257
Custer, S. K., Neumann, M., Lu, H., Wright, A. C., and Taylor, J. P. (2010). Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet. 19, 1741–1755. doi: 10.1093/hmg/ddq050
Dai, R. M., and Li, C. C. (2001). Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat. Cell Biol. 3, 740–744. doi: 10.1038/35087056
De Vos, K. J., and Hafezparast, M. (2017). Neurobiology of axonal transport defects in motor neuron diseases: opportunities for translational research? Neurobiol. Dis. doi: 10.1016/j.nbd.2017.02.004 [Epub ahead of print].
De Vos, K. J., Mórotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deng, H.-X., Chen, W., Hong, S.-T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 29, 2841–2857. doi: 10.1038/emboj.2010.143
Driscoll, J., and Goldberg, A. L. (1990). The proteasome (multicatalytic protease) is a component of the 1500-kDa proteolytic complex which degrades ubiquitin-conjugated proteins. J. Biol. Chem. 265, 4789–4792.
Eytan, E., Ganoth, D., Armon, T., and Hershko, A. (1989). ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc. Natl. Acad. Sci. U.S.A. 86, 7751–7755. doi: 10.1073/pnas.86.20.7751
Fang, L., Hemion, C., Pinho Ferreira Bento, A. C., Bippes, C. C., Flammer, J., and Neutzner, A. (2015). Mitochondrial function in neuronal cells depends on p97/VCP/Cdc48-mediated quality control. Front. Cell Neurosci. 9:16. doi: 10.3389/fncel.2015.00016
Farg, M. A., Soo, K. Y., Walker, A. K., Pham, H., Orian, J., Horne, M. K., et al. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 33, 2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi: 10.1001/archneurol.2011.250
Ferguson, C. J., Lenk, G. M., and Meisler, M. H. (2009). Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum. Mol. Genet. 18, 4868–4878. doi: 10.1093/hmg/ddp460
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630. doi: 10.1038/nrneurol.2011.152
Fiesel, F. C., Voigt, A., Weber, S. S., Van den Haute, C., Waldenmaier, A., Gorner, K., et al. (2010). Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 29, 209–221. doi: 10.1038/emboj.2009.324
Filimonenko, M., Stuffers, S., Raiborg, C., Yamamoto, A., Malerød, L., Fisher, E. M. C., et al. (2007). Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 179, 485–500. doi: 10.1083/jcb.200702115
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Müller, K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636. doi: 10.1038/nn.4000
Ganesan, S., Rohde, G., Eckermann, K., Sroka, K., Schaefer, M. K. E., Dohm, C. P., et al. (2007). Mutant SOD1 detoxification mechanisms in intact single cells. Cell Death Differ. 15, 312–321. doi: 10.1038/sj.cdd.4402262
Gitcho, M. A., Strider, J., Carter, D., Taylor-Reinwald, L., Forman, M. S., Goate, A. M., et al. (2009). VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem. 284, 12384–12398. doi: 10.1074/jbc.M900992200
Gkogkas, C., Middleton, S., Kremer, A. M., Wardrope, C., Hannah, M., Gillingwater, T. H., et al. (2008). VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 17, 1517–1526. doi: 10.1093/hmg/ddn040
Goode, A., Butler, K., Long, J., Cavey, J., Scott, D., Shaw, B., et al. (2016). Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 12, 1094–1104. doi: 10.1080/15548627.2016.1170257
Gregory, J. M., Barros, T. P., Meehan, S., Dobson, C. M., and Luheshi, L. M. (2012). The aggregation and neurotoxicity of TDP-43 and its ALS-associated 25 kDa fragment are differentially affected by molecular chaperones in Drosophila. PLoS ONE 7:e31899. doi: 10.1371/journal.pone.0031899
Hadano, S., Hand, C. K., Osuga, H., Yanagisawa, Y., Otomo, A., Devon, R. S., et al. (2001). A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 29, 166–173. doi: 10.1038/ng1001-166
Hadano, S., Otomo, A., Kunita, R., Suzuki-Utsunomiya, K., Akatsuka, A., Koike, M., et al. (2010). Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1H46R-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS ONE 5:e9805. doi: 10.1371/journal.pone.0009805
Haeusler, A. R., Donnelly, C. J., and Rothstein, J. D. (2016). The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 17, 383–395. doi: 10.1038/nrn.2016.38
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Hargitai, J., Lewis, H., Boros, I., Rácz, T., Fiser, A., Kurucz, I., et al. (2003). Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem. Biophys. Res. Commun. 307, 689–695. doi: 10.1016/S0006-291X(03)01254-3
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Hays, A. P., Naini, A., He, C. Z., Mitsumoto, H., and Rowland, L. P. (2006). Sporadic amyotrophic lateral sclerosis and breast cancer: hyaline conglomerate inclusions lead to identification of SOD1 mutation. J. Neurol. Sci. 242, 67–69. doi: 10.1016/j.jns.2005.11.016
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., et al. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. doi: 10.1101/gad.1830709
Hirano, A., Donnenfeld, H., Sasaki, S., and Nakano, I. (1984). Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 43, 461–470. doi: 10.1097/00005072-198409000-00001
Hirano, M., Nakamura, Y., Saigoh, K., Sakamoto, H., Ueno, S., Isono, C., et al. (2013). Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology 80, 458–463. doi: 10.1212/WNL.0b013e31827f0fe5
Hjerpe, R., Bett, J. S., Keuss, M. J., Solovyova, A., McWilliams, T. G., Johnson, C., et al. (2016). UBQLN2 mediates autophagy-independent protein aggregate clearance by the proteasome. Cell 166, 935–949. doi: 10.1016/j.cell.2016.07.001
Hoffman, L., Pratt, G., and Rechsteiner, M. (1992). Multiple forms of the 20 S multicatalytic and the 26 S ubiquitin/ATP-dependent proteases from rabbit reticulocyte lysate. J. Biol. Chem. 267, 22362–22368.
Ikenaka, K., Kawai, K., Katsuno, M., Huang, Z., Jiang, Y. M., Iguchi, Y., et al. (2013). dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS ONE 8:e54511. doi: 10.1371/journal.pone.0054511
Ito, H., Nakamura, M., Komure, O., Ayaki, T., Wate, R., Maruyama, H., et al. (2011). Clinicopathologic study on an ALS family with a heterozygous E478G optineurin mutation. Acta Neuropathol. 122, 223–229. doi: 10.1007/s00401-011-0842-y
Jiang, H. Q., Ren, M., Jiang, H. Z., Wang, J., Zhang, J., Yin, X., et al. (2014). Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 277, 132–138. doi: 10.1016/j.neuroscience.2014.03.047
Jiang, Y. M., Yamamoto, M., Tanaka, F., Ishigaki, S., Katsuno, M., Adachi, H., et al. (2007). Gene expressions specifically detected in motor neurons (dynactin 1, early growth response 3, acetyl-CoA transporter, death receptor 5, and cyclin C) differentially correlate to pathologic markers in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 66, 617–627. doi: 10.1097/nen.0b013e318093ece3
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
Ju, J. S., Fuentealba, R. A., Miller, S. E., Jackson, E., Piwnica-Worms, D., Baloh, R. H., et al. (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888. doi: 10.1083/jcb.200908115
Kabashi, E., Agar, J. N., Strong, M. J., and Durham, H. D. (2012). Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 367–371. doi: 10.3109/17482968.2012.686511
Kabashi, E., Agar, J. N., Taylor, D. M., Minotti, S., and Durham, H. D. (2004). Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 89, 1325–1335. doi: 10.1111/j.1471-4159.2004.02453.x
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kabuta, T., Suzuki, Y., and Wada, K. (2006). Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J. Biol. Chem. 281, 30524–30533. doi: 10.1074/jbc.M603337200
Kalmar, B., Edet-Amana, E., and Greensmith, L. (2012). Treatment with a coinducer of the heat shock response delays muscle denervation in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 378–392. doi: 10.3109/17482968.2012.660953
Kalmar, B., Novoselov, S., Gray, A., Cheetham, M. E., Margulis, B., and Greensmith, L. (2008). Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J. Neurochem. 107, 339–350. doi: 10.1111/j.1471-4159.2008.05595.x
Kanekura, K., Nishimoto, I., Aiso, S., and Matsuoka, M. (2006). Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J. Biol. Chem. 281, 30223–30233. doi: 10.1074/jbc.M605049200
Kato, T., Katagiri, T., Hirano, A., Kawanami, T., and Sasaki, H. (1989). Lewy body-like hyaline inclusions in sporadic motor neuron disease are ubiquitinated. Acta Neuropathol. 77, 391–396. doi: 10.1007/BF00687373
Kieran, D., Kalmar, B., Dick, J. R., Riddoch-Contreras, J., Burnstock, G., and Greensmith, L. (2004). Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 10, 402–405. doi: 10.1038/nm1021
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kim, J. Y., Jang, A., Reddy, R., Yoon, W. H., and Jankowsky, J. L. (2016). Neuronal overexpression of human VAPB slows motor impairment and neuromuscular denervation in a mouse model of ALS. Hum. Mol. Genet. 25, 4661–4673. doi: 10.1093/hmg/ddw294
Kim, S. H., Shanware, N. P., Bowler, M. J., and Tibbetts, R. S. (2010). Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 285, 34097–34105. doi: 10.1074/jbc.M110.154831
Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M., and Hartl, F. U. (2013). Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355. doi: 10.1146/annurev-biochem-060208-092442
Kimura, T., Jiang, H., Konno, T., Seto, M., Iwanaga, K., Tsujihata, M., et al. (2014). Bunina bodies in motor and non-motor neurons revisited: a pathological study of an ALS patient after long-term survival on a respirator. Neuropathology 34, 392–397. doi: 10.1111/neup.12105
King, A., Maekawa, S., Bodi, I., Troakes, C., and Al-Sarraj, S. (2011). Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP-43 proteinopathies but are only rarely additionally immunopositive for phosphorylation-dependent TDP-43. Neuropathology 31, 239–249. doi: 10.1111/j.1440-1789.2010.01171.x
Kitamura, A., Inada, N., Kubota, H., Matsumoto, G., Kinjo, M., Morimoto, R. I., et al. (2014). Dysregulation of the proteasome increases the toxicity of ALS-linked mutant SOD1. Genes Cells 19, 209–224. doi: 10.1111/gtc.12125
Ko, H. S., Uehara, T., Tsuruma, K., and Nomura, Y. (2004). Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 566, 110–114. doi: 10.1016/j.febslet.2004.04.031
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J.-I., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Koppers, M., van Blitterswijk, M. M., Vlam, L., Rowicka, P. A., van Vught, P. W., Groen, E. J., et al. (2012). VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 33, 837.e7–13. doi: 10.1016/j.neurobiolaging.2011.10.006
Korac, J., Schaeffer, V., Kovacevic, I., Clement, A. M., Jungblut, B., Behl, C., et al. (2013). Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 126, 580–592. doi: 10.1242/jcs.114926
Kuzma-Kozakiewicz, M., Chudy, A., Kazmierczak, B., Dziewulska, D., Usarek, E., and Baranczyk-Kuzma, A. (2013). Dynactin deficiency in the CNS of humans with sporadic ALS and mice with genetically determined motor neuron degeneration. Neurochem. Res. 38, 2463–2473. doi: 10.1007/s11064-013-1160-7
Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (New York, NY) 323, 1205–1208. doi: 10.1126/science.1166066
Lamark, T., and Johansen, T. (2012). Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012:736905. doi: 10.1155/2012/736905
Lautenschlager, J., Prell, T., Ruhmer, J., Weidemann, L., Witte, O. W., and Grosskreutz, J. (2013). Overexpression of human mutated G93A SOD1 changes dynamics of the ER mitochondria calcium cycle specifically in mouse embryonic motor neurons. Exp. Neurol. 247, 91–100. doi: 10.1016/j.expneurol.2013.03.027
Lee, J. A., Beigneux, A., Ahmad, S. T., Young, S. G., and Gao, F. B. (2007). ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 17, 1561–1567. doi: 10.1016/j.cub.2007.07.029
Leigh, P. N., Anderton, B. H., Dodson, A., Gallo, J. M., Swash, M., and Power, D. M. (1988). Ubiquitin deposits in anterior horn cells in motor neurone disease. Neurosci. Lett. 93, 197–203. doi: 10.1016/0304-3940(88)90081-X
Levy, J. R., Sumner, C. J., Caviston, J. P., Tokito, M. K., Ranganathan, S., Ligon, L. A., et al. (2006). A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J. Cell Biol. 172, 733–745. doi: 10.1083/jcb.200511068
Li, L., Zhang, X., and Le, W. (2008). Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 4, 290–293. doi: 10.4161/auto.5524
Li, Y., Guo, Y., Wang, X., Yu, X., Duan, W., Hong, K., et al. (2015). Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 298, 12–25. doi: 10.1016/j.neuroscience.2015.03.061
Liang, C. C., Wang, C., Peng, X., Gan, B., and Guan, J. L. (2010). Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 285, 3499–3509. doi: 10.1074/jbc.M109.072389
Lowe, J., Lennox, G., Jefferson, D., Morrell, K., McQuire, D., Gray, T., et al. (1988). A filamentous inclusion body within anterior horn neurones in motor neurone disease defined by immunocytochemical localisation of ubiquitin. Neurosci. Lett. 94, 203–210. doi: 10.1016/0304-3940(88)90296-0
Mackenzie, I. R., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434. doi: 10.1002/ana.21147
Mackenzie, I. R. A., Frick, P., and Neumann, M. (2014). The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 127, 347–357. doi: 10.1007/s00401-013-1232-4
Maday, S., and Holzbaur, E. L. F. (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 30, 71–85. doi: 10.1016/j.devcel.2014.06.001
Maday, S., Wallace, K. E., and Holzbaur, E. L. F. (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 196, 407–417. doi: 10.1083/jcb.201106120
Mahoney, C. J., Beck, J., Rohrer, J. D., Lashley, T., Mok, K., Shakespeare, T., et al. (2012). Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain 135, 736–750. doi: 10.1093/brain/awr361
Majcher, V., Goode, A., James, V., and Layfield, R. (2015). Autophagy receptor defects and ALS-FTLD. Mol. Cell. Neurosci. 66, 43–52. doi: 10.1016/j.mcn.2015.01.002
Mann, D. M., Rollinson, S., Robinson, A., Bennion Callister, J., Thompson, J. C., Snowden, J. S., et al. (2013). Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 1, 68. doi: 10.1186/2051-5960-1-68
Marino, M., Papa, S., Crippa, V., Nardo, G., Peviani, M., Cheroni, C., et al. (2015). Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol. Aging 36, 492–504. doi: 10.1016/j.neurobiolaging.2014.06.026
Martinez, J. A., Zhang, Z., Svetlov, S. I., Hayes, R. L., Wang, K. K., and Larner, S. F. (2010). Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis 15, 1480–1493. doi: 10.1007/s10495-010-0526-4
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Matsumoto, G., Shimogori, T., Hattori, N., and Nukina, N. (2015). TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 24, 4429–4442. doi: 10.1093/hmg/ddv179
Matus, S., Lopez, E., Valenzuela, V., Nassif, M., and Hetz, C. (2013). Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS ONE 8:e66672. doi: 10.1371/journal.pone.0066672
Matus, S., Nassif, M., Glimcher, L. H., and Hetz, C. (2009). XBP-1 deficiency in the nervous system reveals a homeostatic switch to activate autophagy. Autophagy 5, 1226–1228. doi: 10.4161/auto.5.8.10247
Meyer, H., and Weihl, C. C. (2014). The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J. Cell Sci. 127, 3877–3883. doi: 10.1242/jcs.093831
Miguel, L., Avequin, T., Delarue, M., Feuillette, S., Frebourg, T., Campion, D., et al. (2012). Accumulation of insoluble forms of FUS protein correlates with toxicity in Drosophila. Neurobiol. Aging 33, e1001–e1015. doi: 10.1016/j.neurobiolaging.2011.10.008
Mitne-Neto, M., Machado-Costa, M., Marchetto, M. C., Bengtson, M. H., Joazeiro, C. A., Tsuda, H., et al. (2011). Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum. Mol. Genet. 20, 3642–3652. doi: 10.1093/hmg/ddr284
Mizuno, Y., Amari, M., Takatama, M., Aizawa, H., Mihara, B., and Okamoto, K. (2006). Transferrin localizes in Bunina bodies in amyotrophic lateral sclerosis. Acta Neuropathol. 112, 597–603. doi: 10.1007/s00401-006-0122-4
Mizuno, Y., Fujita, Y., Takatama, M., and Okamoto, K. (2011). Peripherin partially localizes in Bunina bodies in amyotrophic lateral sclerosis. J. Neurol. Sci. 302, 14–18. doi: 10.1016/j.jns.2010.12.023
Mori, F., Kakita, A., Takahashi, H., and Wakabayashi, K. (2014). Co-localization of Bunina bodies and TDP-43 inclusions in lower motor neurons in amyotrophic lateral sclerosis. Neuropathology 34, 71–76. doi: 10.1111/neup.12044
Mori, F., Miki, Y., Tanji, K., Kakita, A., Takahashi, H., Utsumi, J., et al. (2015). Sortilin-related receptor CNS expressed 2 (SorCS2) is localized to Bunina bodies in amyotrophic lateral sclerosis. Neurosci. Lett. 608, 6–11. doi: 10.1016/j.neulet.2015.09.030
Mori, F., Tanji, K., Miki, Y., Kakita, A., Takahashi, H., and Wakabayashi, K. (2010). Relationship between Bunina bodies and TDP-43 inclusions in spinal anterior horn in amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 36, 345–352. doi: 10.1111/j.1365-2990.2010.01081.x
Mori, K., Arzberger, T., Grässer, F. A., Gijselinck, I., May, S., Rentzsch, K., et al. (2013). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893. doi: 10.1007/s00401-013-1189-3
Moumen, A., Virard, I., and Raoul, C. (2011). Accumulation of Wildtype and ALS-linked mutated VAPB impairs activity of the proteasome. PLoS ONE 6:e26066. doi: 10.1371/journal.pone.0026066
Münch, C., Rosenbohm, A., Sperfeld, A. D., Uttner, I., Reske, S., Krause, B. J., et al. (2005). Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 58, 777–780. doi: 10.1002/ana.20631
Münch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.WNL.0000134608.83927.B1
Munoz, D. G., Greene, C., Perl, D. P., and Selkoe, D. J. (1988). Accumulation of phosphorylated neurofilaments in anterior horn motoneurons of amyotrophic lateral sclerosis patients. J. Neuropathol. Exp. Neurol. 47, 9–18. doi: 10.1097/00005072-198801000-00002
Murayama, S., Ookawa, Y., Mori, H., Nakano, I., Ihara, Y., Kuzuhara, S., et al. (1989). Immunocytochemical and ultrastructural study of Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis. Acta Neuropathol. 78, 143–152. doi: 10.1007/BF00688202
Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B. A., et al. (2000). Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-[beta]. Nature 403, 98–103. doi: 10.1038/47513
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, NY) 314, 130–133. doi: 10.1126/science.1134108
Nishihira, Y., Tan, C. F., Onodera, O., Toyoshima, Y., Yamada, M., Morita, T., et al. (2008). Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol. 116, 169–182. doi: 10.1007/s00401-008-0385-z
Nishimura, A. L., Mitne-Neto, M., Silva, H. C. A., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. doi: 10.1086/425287
Nishitoh, H., Kadowaki, H., Nagai, A., Maruyama, T., Yokota, T., Fukutomi, H., et al. (2008). ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 22, 1451–1464. doi: 10.1101/gad.1640108
Niwa, J., Ishigaki, S., Hishikawa, N., Yamamoto, M., Doyu, M., Murata, S., et al. (2002). Dorfin ubiquitylates mutant SOD1 and prevents mutant SOD1-mediated neurotoxicity. J. Biol. Chem. 277, 36793–36798. doi: 10.1074/jbc.M206559200
Okamoto, K., Hirai, S., Amari, M., Watanabe, M., and Sakurai, A. (1993). Bunina bodies in amyotrophic lateral sclerosis immunostained with rabbit anti-cystatin C serum. Neurosci. Lett. 162, 125–128. doi: 10.1016/0304-3940(93)90576-7
Okamoto, K., Mizuno, Y., and Fujita, Y. (2008). Bunina bodies in amyotrophic lateral sclerosis. Neuropathology 28, 109–115. doi: 10.1111/j.1440-1789.2007.00873.x
Orlacchio, A., Babalini, C., Borreca, A., Patrono, C., Massa, R., Basaran, S., et al. (2010). SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 133, 591–598. doi: 10.1093/brain/awp325
O’Rourke, J. G., Bogdanik, L., Yáñez, A., Lall, D., Wolf, A. J., Muhammad, A. K. M. G., et al. (2016). C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329. doi: 10.1126/science.aaf1064
Osaka, M., Ito, D., Yagi, T., Nihei, Y., and Suzuki, N. (2015). Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum. Mol. Genet. 24, 1617–1629. doi: 10.1093/hmg/ddu575
Otomo, A., Kunita, R., Suzuki-Utsunomiya, K., Ikeda, J. E., and Hadano, S. (2011). Defective relocalization of ALS2/alsin missense mutants to Rac1-induced macropinosomes accounts for loss of their cellular function and leads to disturbed amphisome formation. FEBS Lett. 585, 730–736. doi: 10.1016/j.febslet.2011.01.045
Paillusson, S., Stoica, R., Gomez-Suaga, P., Lau, D. H., Mueller, S., Miller, T., et al. (2016). There’s something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 39, 146–157. doi: 10.1016/j.tins.2016.01.008
Parkinson, N., Ince, P. G., Smith, M. O., Highley, R., Skibinski, G., Andersen, P. M., et al. (2006). ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67, 1074–1077. doi: 10.1212/01.wnl.0000231510.89311.8b
Pérez-Brangulí, F., Mishra, H. K., Prots, I., Havlicek, S., Kohl, Z., Saul, D., et al. (2014). Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum. Mol. Genet. 23, 4859–4874. doi: 10.1093/hmg/ddu200
Piao, Y. S., Wakabayashi, K., Kakita, A., Yamada, M., Hayashi, S., Morita, T., et al. (2003). Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol. 13, 10–22. doi: 10.1111/j.1750-3639.2003.tb00002.x
Prause, J., Goswami, A., Katona, I., Roos, A., Schnizler, M., Bushuven, E., et al. (2013). Altered localization, abnormal modification and loss of function of sigma receptor-1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 1581–1600. doi: 10.1093/hmg/ddt008
Puls, I., Jonnakuty, C., LaMonte, B. H., Holzbaur, E. L. F., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456. doi: 10.1038/ng1123
Ravikumar, B., Acevedo-Arozena, A., Imarisio, S., Berger, Z., Vacher, C., O’Kane, C. J., et al. (2005). Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 37, 771–776. doi: 10.1038/ng1591
Ravikumar, B., Imarisio, S., Sarkar, S., O’Kane, C. J., and Rubinsztein, D. C. (2008). Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J. Cell Sci. 121, 1649–1660. doi: 10.1242/jcs.025726
Renton, A. E., Chio, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Renvoisé, B., Chang, J., Singh, R., Yonekawa, S., FitzGibbon, E. J., Mankodi, A., et al. (2014). Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann. Clin. Transl. Neurol. 1, 379–389. doi: 10.1002/acn3.64
Richter, B., Sliter, D. A., Herhaus, L., Stolz, A., Wang, C., Beli, P., et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U.S.A. 113, 4039–4044. doi: 10.1073/pnas.1523926113
Ritson, G. P., Custer, S. K., Freibaum, B. D., Guinto, J. B., Geffel, D., Moore, J., et al. (2010). TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 30, 7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010
Sasaki, S. (2010). Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 346–355. doi: 10.1097/NEN.0b013e3181d44992
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Scheper, W., and Hoozemans, J. J. (2013). A new PERKspective on neurodegeneration. Sci. Transl. Med. 5, 206fs37. doi: 10.1126/scitranslmed.3007641
Sellier, C., Campanari, M. L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Shen, W. C., Li, H. Y., Chen, G. C., Chern, Y., and Tu, P. H. (2015). Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 11, 685–700. doi: 10.4161/auto.36098
Simmen, T., Aslan, J. E., Blagoveshchenskaya, A. D., Thomas, L., Wan, L., Xiang, Y., et al. (2005). PACS-2 controls endoplasmic reticulum–mitochondria communication and Bid-mediated apoptosis. EMBO J. 24, 717–729. doi: 10.1038/sj.emboj.7600559
Skibinski, G., Parkinson, N. J., Brown, J. M., Chakrabarti, L., Lloyd, S. L., Hummerich, H., et al. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808. doi: 10.1038/ng1609
Son, J. H., Shim, J. H., Kim, K.-H., Ha, J.-Y., and Han, J. Y. (2012). Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 44, 89–98. doi: 10.3858/emm.2012.44.2.031
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science (New York, NY) 319, 1668–1672. doi: 10.1126/science.1154584
Staats, K. A., Hernandez, S., Schonefeldt, S., Bento-Abreu, A., Dooley, J., Van Damme, P., et al. (2013). Rapamycin increases survival in ALS mice lacking mature lymphocytes. Mol. Neurodegener. 8:31. doi: 10.1186/1750-1326-8-31
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K.-F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5, 3996. doi: 10.1038/ncomms4996
Stoica, R., Paillusson, S., Gomez-Suaga, P., Mitchell, J. C., Lau, D. H., Gray, E. H., et al. (2016). ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 17, 1326–1342. doi: 10.15252/embr.201541726
Stolz, A., Ernst, A., and Dikic, I. (2014). Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 16, 495–501. doi: 10.1038/ncb2979
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4, 51. doi: 10.1186/s40478-016-0324-5
Suzuki, H., Kanekura, K., Levine, T. P., Kohno, K., Olkkonen, V. M., Aiso, S., et al. (2009). ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 108, 973–985. doi: 10.1111/j.0022-3042.2008.05857.x
Suzuki, H., Lee, K., and Matsuoka, M. (2011). TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J. Biol. Chem. 286, 13171–13183. doi: 10.1074/jbc.M110.197483
Swinnen, B., and Robberecht, W. (2014). The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 10, 661–670. doi: 10.1038/nrneurol.2014.184
Tashiro, Y., Urushitani, M., Inoue, H., Koike, M., Uchiyama, Y., Komatsu, M., et al. (2012). Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J. Biol. Chem. 287, 42984–42994. doi: 10.1074/jbc.M112.417600
Teyssou, E., Takeda, T., Lebon, V., Boillée, S., Doukouré, B., Bataillon, G., et al. (2013). Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 125, 511–522. doi: 10.1007/s00401-013-1090-0
Tokuda, E., Brannstrom, T., Andersen, P. M., and Marklund, S. L. (2016). Low autophagy capacity implicated in motor system vulnerability to mutant superoxide dismutase. Acta Neuropathol. Commun. 4, 6. doi: 10.1186/s40478-016-0274-y
Topp, J. D., Gray, N. W., Gerard, R. D., and Horazdovsky, B. F. (2004). Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 279, 24612–24623. doi: 10.1074/jbc.M313504200
Tresse, E., Salomons, F. A., Vesa, J., Bott, L. C., Kimonis, V., Yao, T. P., et al. (2010). VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227. doi: 10.4161/auto.6.2.11014
Trippier, P. C., Benmohamed, R., Kirsch, D. R., and Silverman, R. B. (2012). Substituted pyrazolones require N2 hydrogen bond donating ability to protect against cytotoxicity from protein aggregation of mutant superoxide dismutase 1. Bioorg. Med. Chem. Lett. 22, 6647–6650. doi: 10.1016/j.bmcl.2012.08.114
Trippier, P. C., Zhao, K. T., Fox, S. G., Schiefer, I. T., Benmohamed, R., Moran, J., et al. (2014). Proteasome activation is a mechanism for pyrazolone small molecules displaying therapeutic potential in amyotrophic lateral sclerosis. ACS Chem. Neurosci. 5, 823–829. doi: 10.1021/cn500147v
Tsaytler, P., Harding, H. P., Ron, D., and Bertolotti, A. (2011). Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91. doi: 10.1126/science.1201396
Tudor, E. L., Galtrey, C. M., Perkinton, M. S., Lau, K. F., De Vos, K. J., Mitchell, J. C., et al. (2010). Amyotrophic lateral sclerosis mutant vesicle-associated membrane protein-associated protein-B transgenic mice develop TAR-DNA-binding protein-43 pathology. Neuroscience 167, 774–785. doi: 10.1016/j.neuroscience.2010.02.035
Tummala, H., Jung, C., Tiwari, A., Higgins, C. M., Hayward, L. J., and Xu, Z. (2005). Inhibition of chaperone activity is a shared property of several Cu,Zn-superoxide dismutase mutants that cause amyotrophic lateral sclerosis. J. Biol. Chem. 280, 17725–17731. doi: 10.1074/jbc.M501705200
Udan-Johns, M., Bengoechea, R., Bell, S., Shao, J., Diamond, M. I., True, H. L., et al. (2014). Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet. 23, 157–170. doi: 10.1093/hmg/ddt408
Urushitani, M., Kurisu, J., Tateno, M., Hatakeyama, S., Nakayama, K., Kato, S., et al. (2004). CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J. Neurochem. 90, 231–244. doi: 10.1111/j.1471-4159.2004.02486.x
Urushitani, M., Kurisu, J., Tsukita, K., and Takahashi, R. (2002). Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem. 83, 1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x
Urushitani, M., Sato, T., Bamba, H., Hisa, Y., and Tooyama, I. (2010). Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. J. Neurosci. Res. 88, 784–797. doi: 10.1002/jnr.22243
Urwin, H., Authier, A., Nielsen, J. E., Metcalf, D., Powell, C., Froud, K., et al. (2010). Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum. Mol. Genet. 19, 2228–2238. doi: 10.1093/hmg/ddq100
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science (New York, NY) 323, 1208–1211. doi: 10.1126/science.1165942
Vance, C., Scotter, E. L., Nishimura, A. L., Troakes, C., Mitchell, J. C., Kathe, C., et al. (2013). ALS mutant FUS disrupts nuclear localisation and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 22, 2676–2688. doi: 10.1093/hmg/ddt117
Vieira, F. G., Ping, Q., Moreno, A. J., Kidd, J. D., Thompson, K., Jiang, B., et al. (2015). Guanabenz treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS ONE 10:e0135570. doi: 10.1371/journal.pone.0135570
Vígh, L., Literáti, P. N., Horváth, I., Török, Z., Balogh, G., Glatz, A., et al. (1997). Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein- inducing activity and cytoprotective effects. Nat. Med. 3, 1150–1154. doi: 10.1038/nm1097-1150
Vijayalakshmi, K., Alladi, P. A., Ghosh, S., Prasanna, V. K., Sagar, B. C., Nalini, A., et al. (2011). Evidence of endoplasmic reticular stress in the spinal motor neurons exposed to CSF from sporadic amyotrophic lateral sclerosis patients. Neurobiol. Dis. 41, 695–705. doi: 10.1016/j.nbd.2010.12.005
Vijayalakshmi, K., Alladi, P. A., Sathyaprabha, T. N., Subramaniam, J. R., Nalini, A., and Raju, T. R. (2009). Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces degeneration of a cultured motor neuron cell line. Brain Res. 1263, 122–133. doi: 10.1016/j.brainres.2009.01.041
Vilariño-Güell, C., Wider, C., Soto-Ortolaza, A. I., Cobb, S. A., Kachergus, J. M., Keeling, B. H., et al. (2009). Characterization of DCTN1 genetic variability in neurodegeneration. Neurology 72, 2024–2028. doi: 10.1212/WNL.0b013e3181a92c4c
Voges, D., Zwickl, P., and Baumeister, W. (1999). The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068. doi: 10.1146/annurev.biochem.68.1.1015
Vollrath, J. T., Sechi, A., Dreser, A., Katona, I., Wiemuth, D., Vervoorts, J., et al. (2014). Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell Death Dis. 5, e1290. doi: 10.1038/cddis.2014.243
Walczak, C. P., Bernardi, K. M., and Tsai, B. (2012). Endoplasmic reticulum-dependent redox reactions control endoplasmic reticulum-associated degradation and pathogen entry. Antioxid. Redox Signal. 16, 809–818. doi: 10.1089/ars.2011.4425
Walker, A. K., Farg, M. A., Bye, C. R., McLean, C. A., Horne, M. K., and Atkin, J. D. (2010). Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 133, 105–116. doi: 10.1093/brain/awp267
Walker, A. K., Soo, K. Y., Sundaramoorthy, V., Parakh, S., Ma, Y., Farg, M. A., et al. (2013). ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 8:e81170. doi: 10.1371/journal.pone.0081170
Wang, I. F., Guo, B. S., Liu, Y. C., Wu, C. C., Yang, C. H., Tsai, K. J., et al. (2012). Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. U.S.A. 109, 15024–15029. doi: 10.1073/pnas.1206362109
Wang, J., Xu, G., and Borchelt, D. R. (2002). High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol. Dis. 9, 139–148. doi: 10.1006/nbdi.2001.0471
Wang, L., Popko, B., Tixier, E., and Roos, R. P. (2014). Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 71, 317–324. doi: 10.1016/j.nbd.2014.08.010
Wang, T., Xu, W., Qin, M., Yang, Y., Bao, P., Shen, F., et al. (2016). Pathogenic mutations in the valosin-containing protein/p97(VCP) N-domain inhibit the SUMOylation of VCP and lead to impaired stress response. J. Biol. Chem. 291, 14373–14384. doi: 10.1074/jbc.M116.729343
Wang, X., Fan, H., Ying, Z., Li, B., Wang, H., and Wang, G. (2010). Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci. Lett. 469, 112–116. doi: 10.1016/j.neulet.2009.11.055
Watanabe, M., Dykes-Hoberg, M., Culotta, V. C., Price, D. L., Wong, P. C., and Rothstein, J. D. (2001). Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol. Dis. 8, 933–941. doi: 10.1006/nbdi.2001.0443
Watanabe, S., Ilieva, H., Tamada, H., Nomura, H., Komine, O., Endo, F., et al. (2016). Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 8, 1421–1437. doi: 10.15252/emmm.201606403
Waterman-Storer, C. M., Karki, S. B., Kuznetsov, S. A., Tabb, J. S., Weiss, D. G., Langford, G. M., et al. (1997). The interaction between cytoplasmic dynein and dynactin is required for fast axonal transport. Proc. Natl. Acad. Sci. U.S.A. 94, 12180–12185. doi: 10.1073/pnas.94.22.12180
Webster, C. P., Smith, E. F., Bauer, C. S., Moller, A., Hautbergue, G. M., Ferraiuolo, L., et al. (2016a). The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 35, 1656–1676. doi: 10.15252/embj.201694401
Webster, C. P., Smith, E. F., Grierson, A. J., and De Vos, K. J. (2016b). C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases doi: 10.1080/21541248.2016.1240495 [Epub ahead of print].
Weidberg, H., and Elazar, Z. (2011). TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 4, e39. doi: 10.1126/scisignal.2002355
Weishaupt, J. H., Waibel, S., Birve, A., Volk, A. E., Mayer, B., Meyer, T., et al. (2013). A novel optineurin truncating mutation and three glaucoma-associated missense variants in patients with familial amyotrophic lateral sclerosis in Germany. Neurobiol. Aging 34, 1516.e9–15. doi: 10.1016/j.neurobiolaging.2012.09.007
White, E. (2015). The role for autophagy in cancer. J. Clin. Invest. 125, 42–46. doi: 10.1172/JCI73941
Wild, P., Farhan, H., McEwan, D. G., Wagner, S., Rogov, V. V., Brady, N. R., et al. (2011). Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (New York, NY) 333, 228–233. doi: 10.1126/science.1205405
Williams, R. L., and Urbe, S. (2007). The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell Biol. 8, 355–368. doi: 10.1038/nrm2162
Wojcik, C., Yano, M., and DeMartino, G. N. (2004). RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J. Cell Sci. 117, 281–292. doi: 10.1242/jcs.00841
Xia, Q., Wang, H., Zhang, Y., Ying, Z., and Wang, G. (2015). Loss of TDP-43 inhibits amyotrophic lateral sclerosis-linked mutant SOD1 aggresome formation in an HDAC6-dependent manner. J. Alzheimers Dis. 45, 373–386. doi: 10.3233/JAD-150150
Yang, M., Liang, C., Swaminathan, K., Herrlinger, S., Lai, F., Shiekhattar, R., et al. (2016). A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2:e1601167. doi: 10.1126/sciadv.1601167
Zhang, H., Tan, C. F., Mori, F., Tanji, K., Kakita, A., Takahashi, H., et al. (2008). TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 115, 115–122. doi: 10.1007/s00401-007-0285-7
Zhang, X., Chen, S., Song, L., Tang, Y., Shen, Y., Jia, L., et al. (2014). MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 10, 588–602. doi: 10.4161/auto.27710
Zhang, X., Li, L., Chen, S., Yang, D., Wang, Y., Zhang, X., et al. (2011). Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7, 412–425. doi: 10.4161/auto.7.4.14541
Zhang, Y., Benmohamed, R., Huang, H., Chen, T., Voisine, C., Morimoto, R. I., et al. (2013). Arylazanylpyrazolone derivatives as inhibitors of mutant superoxide dismutase 1 dependent protein aggregation for the treatment of amyotrophic lateral sclerosis. J. Med. Chem. 56, 2665–2675. doi: 10.1021/jm400079a
Zhang, Y. J., Gendron, T. F., Xu, Y. F., Ko, L. W., Yen, S. H., and Petrucelli, L. (2010). Phosphorylation regulates proteasomal-mediated degradation and solubility of TAR DNA binding protein-43 C-terminal fragments. Mol. Neurodegener. 5:33. doi: 10.1186/1750-1326-5-33
Keywords: protein homeostasis, protein aggregation, amyotrophic lateral sclerosis (ALS), motor neuron disease, autophagy, chaperonins, unfolded protein response (UPR), proteostasis
Citation: Webster CP, Smith EF, Shaw PJ and De Vos KJ (2017) Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities? Front. Mol. Neurosci. 10:123. doi: 10.3389/fnmol.2017.00123
Received: 24 February 2017; Accepted: 11 April 2017;
Published: 02 May 2017.
Edited by:
Angelo Poletti, Università degli Studi di Milano, ItalyReviewed by:
Mauricio Fernando Budini, Universidad de Chile, ChileCaterina Bendotti, Istituto di Ricerche Farmacologiche Mario Negri, Italy
Copyright © 2017 Webster, Smith, Shaw and De Vos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kurt J. De Vos, k.de_vos@sheffield.ac.uk Pamela J. Shaw, pamela.shaw@sheffield.ac.uk