Chromatin Remodeling BAF (SWI/SNF) Complexes in Neural Development and Disorders
 Godwin Sokpor
Godwin Sokpor Yuanbin Xie
Yuanbin Xie Joachim Rosenbusch
Joachim Rosenbusch Tran Tuoc
Tran Tuoc- 1Institute of Neuroanatomy, University Medical Center, Georg-August-University Goettingen, Goettingen, Germany
- 2DFG Center for Nanoscale Microscopy and Molecular Physiology of the Brain, Goettingen, Germany
The ATP-dependent BRG1/BRM associated factor (BAF) chromatin remodeling complexes are crucial in regulating gene expression by controlling chromatin dynamics. Over the last decade, it has become increasingly clear that during neural development in mammals, distinct ontogenetic stage-specific BAF complexes derived from combinatorial assembly of their subunits are formed in neural progenitors and post-mitotic neural cells. Proper functioning of the BAF complexes plays critical roles in neural development, including the establishment and maintenance of neural fates and functionality. Indeed, recent human exome sequencing and genome-wide association studies have revealed that mutations in BAF complex subunits are linked to neurodevelopmental disorders such as Coffin-Siris syndrome, Nicolaides-Baraitser syndrome, Kleefstra's syndrome spectrum, Hirschsprung's disease, autism spectrum disorder, and schizophrenia. In this review, we focus on the latest insights into the functions of BAF complexes during neural development and the plausible mechanistic basis of how mutations in known BAF subunits are associated with certain neurodevelopmental disorders.
Introduction
The developmental processes that lead to formation of the central nervous system (CNS) are intricately regulated to ensure proper cellular architecture and functionality. Mammalian neural structures are functionally made up of neural cells that develop under strict molecular and cellular instructions to confer cell subtype differentiation. During CNS development, cell fate acquisition is initiated by the decline in neural stem cell self-renewal and amplification potentials to yield fate-committed progenitors that ultimately undergo neurogenic or gliogenic differentiation as reviewed in Guillemot (2007), Kriegstein and Alvarez-Buylla (2009), Taverna et al. (2014) and Tuoc et al. (2014). Highly ordered gene expression programs regulate the establishment of various cell fate status during neural development. Many factors, including epigenetic and chromatin regulators act in concert to determine and sustain cell-specific transcriptional programs. The establishment of specific epigenetic landscapes provides strong evidence that link chromatin state with gene expression profiles. Thus, regions of DNA compaction (heterochromatin) are transcription-repressed sites, whereas less compact or open regions (euchromatin) have active transcriptional activity (Hirabayashi and Gotoh, 2010; Juliandi et al., 2010; Coskun et al., 2012; Ronan et al., 2013; Narayanan and Tuoc, 2014; Watson and Tsai, 2016; Yao et al., 2016). Chromatin regulators are capable of reordering chromatin state (heterochromatin/euchromatin) and hence are critical determinants of access to genomic loci by the transcriptional machinery (Hirabayashi and Gotoh, 2010; Juliandi et al., 2010; Coskun et al., 2012; Ronan et al., 2013; Narayanan and Tuoc, 2014; Watson and Tsai, 2016; Yao et al., 2016). Consequently, a coordinated deployment of chromatin regulators, along with the recruitment of transcription factors, are essential for establishing gene expression patterns in response to various cellular cues and physiological states in health and disease.
During development, signals within and outside cells cause genome-wide chromatin dynamics that correspond with instructive gene expression patterns for cell lineage specifications. For instance, during cortical neurogenesis, various chromatin remodeling machinery are actively involved in directing the expression of numerous neurogenic gene targets, leading to progenitor proliferation and/or differentiation (Juliandi et al., 2010; Ronan et al., 2013; Narayanan and Tuoc, 2014).
Chromatin structure can be regulated at various levels, including covalent modification (e.g., methylation, acetylation, ubiquitination, and phosphorylation) of DNA, RNA, histones, and remodeling of nucleosome by ATP—dependent chromatin remodeling factors, as well as activities of non-coding RNAs (Strahl and Allis, 2000; Goldberg et al., 2007; Kouzarides, 2007). Epigenetic factors act together to instruct transcriptional regulation through direct interaction with transcription factors at genomic loci. ATP-dependent chromatin factors are particularly known powerful regulators of chromatin state and DNA accessibility to transcription factors that are critical for transcriptional regulation and gene expression outcomes (Ronan et al., 2013; Narayanan and Tuoc, 2014).
In our review, we will concentrate on the multi-subunit mammalian SWItch/Sucrose Non-Fermentable (mSWI/SNF), also known as BRG1/BRM associated factor (BAF) chromatin modifier family that are capable of utilizing ATP energy to alter nucleosomal units in chromatin structure, culminating in heterochromatin-euchromatin state inter-convertibility. The mSWI/SNF complexes play central roles in epigenetic regulatory mechanisms that impact on developmental processes. Emerging findings implicate the mSWI/SNF complex in orchestrating rate-limiting epigenetic regulation of various pre- and post-natal neural developmental events (Matsumoto et al., 2006; Lessard et al., 2007; Wu et al., 2007; Weider et al., 2012; Li et al., 2013; Ninkovic et al., 2013; Tuoc et al., 2013a,b, 2016; Vogel-Ciernia et al., 2013; Yu et al., 2013; Narayanan and Tuoc, 2014; Bischof et al., 2015; Narayanan et al., 2015; Wiegreffe et al., 2015; Bachmann et al., 2016; Nguyen et al., 2016). However, similar to other biological systems, abnormal chromatin regulation can sometimes occur. Chromatin remodelers are thus amenable to alterations and mutations that can lead to aberrant chromatin modifications capable of engendering developmental disorders and homeostatic disturbances during neural development. Of special interest, due to their prominent neurodevelopmental functions, misregulation or mutations of mSWI/SNF complex subunits cause neurological disorders with syndromic and non-syndromic cognitive dysfunction phenotypes (Ronan et al., 2013). Notably, Coffin-Siris syndrome, Schizophrenia, Nicolaides-Baraitser syndrome, and autism spectrum disorders are characterized human diseases caused by defective mSWI/SNF complexes. As such, the link between mSWI/SNF complexes and neural development in health and disease is increasingly attracting investigative efforts aimed at elucidating the mechanisms involved.
This review focuses on the emerging role of the mSWI/SNF complex as chromatin regulation factor, and its involvement in neurodevelopmental processes such as neural stem cell/precursor generation, proliferation, and neuronal subtype specification, differentiation, and migration. To that end, we will detail proven findings of the functions of the mSWI/SNF complex in various neural perturbations.
Biochemical Characteristics of the mSWI/SNF (BAF) Complexes
The mSWI/SNF (BAF) complex was originally identified in yeast (Neigeborn and Carlson, 1984; Wang et al., 1996). The BAF complex comprises at least 15 subunits of ~2 MDa in size, and thus more multimeric than the yeast version (Lessard et al., 2007; Wu et al., 2007). Typically, there is enrichment of BAF complex at promoters (Ho et al., 2009a,b, 2011), and particularly much more variable at (super)enhancers (Bossen et al., 2015; Barutcu et al., 2016; Alver et al., 2017; Wang et al., 2017) of active genes involved in cellular processes such as proliferation, cell identity, differentiation, and motility.
The complex is usually made up of exchangeable ATPase core(s), and other core subunits including scaffolding proteins around which the entire complex is assembled. Brahma (BRM) and BRM/SWI2 related gene 1 (BRG1) are the only known two catalytic ATPase subunits out of the 29 possible SWI2/SNF2-like ATPases encoded by the mammalian genome that are incorporated into the BAF complex (Neigeborn and Carlson, 1984; Wang et al., 1996; Lessard et al., 2007; Wu et al., 2007; Kadoch et al., 2013; Narayanan et al., 2015; Bachmann et al., 2016). The core subunits include ubiquitously expressed BAF47, BAF155, and BAF170 (Phelan et al., 1999) (see Table 1 and Figure 1 for a list of BAF subunits). Many core and variant BAF subunits contain DNA- and histone-binding domains such as zinc finger, AT-hook (including high mobility group [HMG] proteins), AT-rich interaction domain (ARID), plant homeodomain (PHD), helicase/SANT-associated (HSA) domain, “switching-defective protein 3 (Swi3), adaptor 2 (Ada2), nuclear receptor co-repressor (N-CoR), transcription factor (TF)IIIB” [SANT] domain, as well as chromo- and bromodomains, which afford transcription factor recruitment specificity, genome targeting, and protein-protein interactions. As such, the BAF complex has an autonomous capacity to specifically regulate gene expression patterns in a cell lineage restricted manner. In vivo, the BAF complex is dynamic and exhibits considerable plurality. The existence of several subunit variants and polymorphic sites is believed to make it possible to distinctively assemble hundreds of BAF complexes in a combinative fashion (Lessard et al., 2007; Wu et al., 2007; Kadoch et al., 2013; Bachmann et al., 2016).
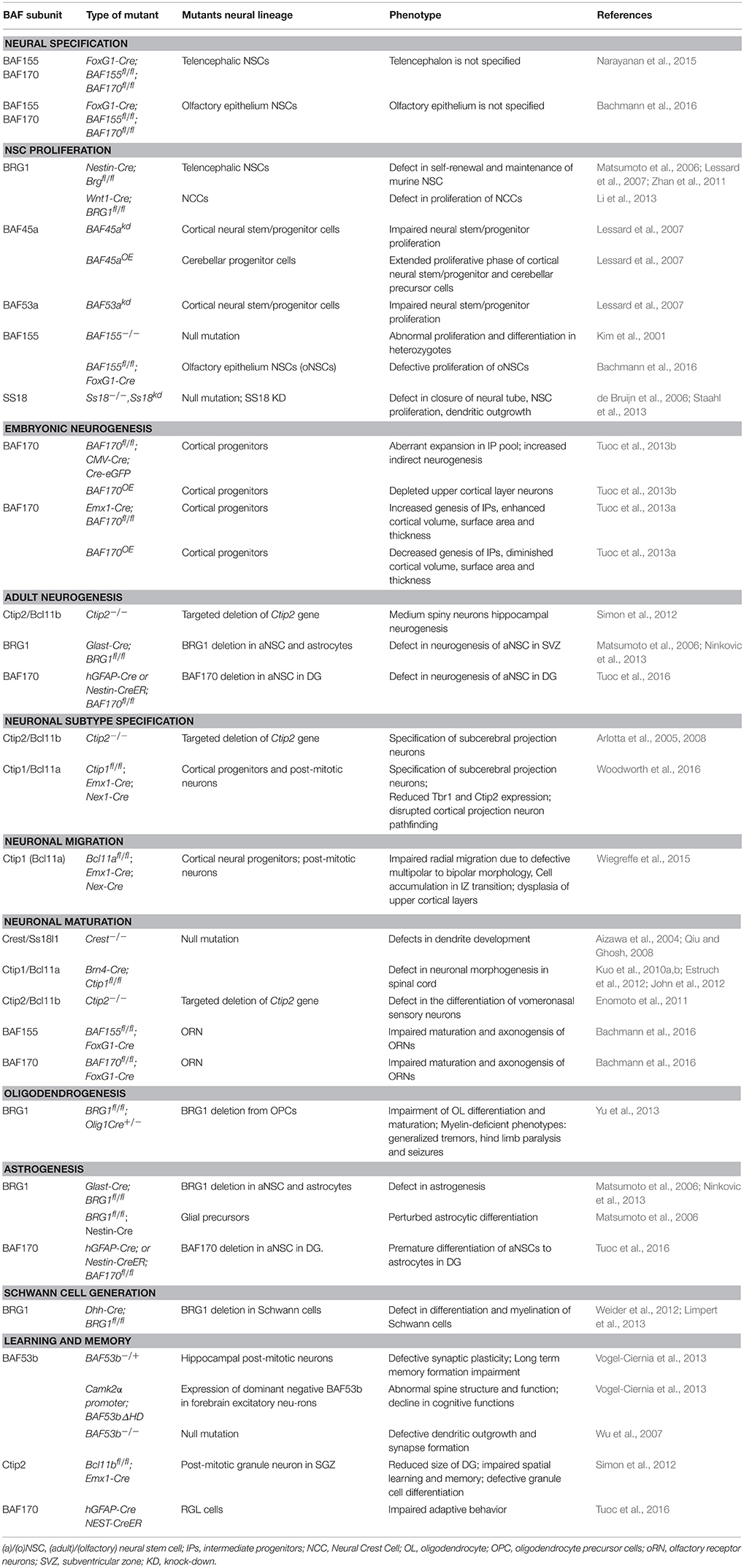
Table 1. BAF subunits and their role in aspects of neural development.
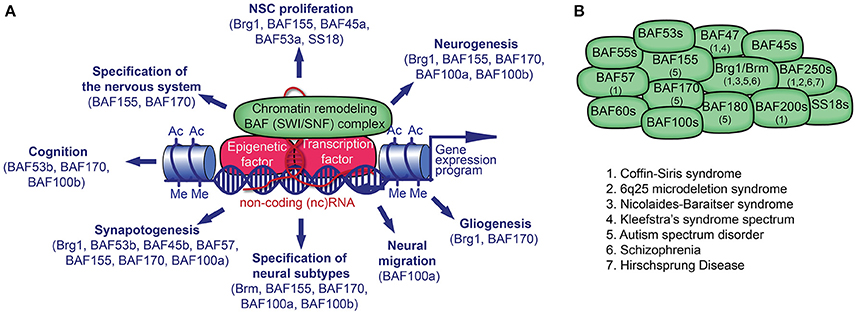
Figure 1. Chromatin remodeling BAF (mSWI/SNF) complex in neural development and disorders. (A) The BAF complex, epigenetic factors (including non-coding [nc] RNA), and transcription factors (TF) control gene expression. TFs and ncRNAs bind to specific DNA sequences. The recruitment of BAF complexes and other epigenetic factors on the genome leads to altered epigenetic marks (e.g., histone acetylation, Ac; histone methylation, Me) and chromatin structure in order to activate or repress a specific gene expression program in cell lineages. Many BAF subunits as indicated, regulate distinct processes of neural development. (B) The presence of known BAF subunits in different BAF complexes in neural cells is indicated. The mutation of genes encoding for the noted BAF subunits has been reported in various neurological disorders.
Mechanistically, a BAF complex remodels chromatin using its ATPase core subunits to hydrolyze ATP and hence generate energy for nucleosomal unwrapping, mobilization, ejection, or histone dimer exchange (Cairns, 1998; Phelan et al., 1999; Whitehouse et al., 1999; Saha et al., 2002; Gutiérrez et al., 2007; Tang et al., 2010). This process relaxes condensed chromatin (heterochromatin) to increase accessibility of transcription factor binding to activate gene expression (Hara and Sancar, 2002; Gong et al., 2006; Ho et al., 2009a; Hu et al., 2011; Tolstorukov et al., 2013). The converse holds true whereby BAF complex disengagement leads to polycomb repressor complex-driven re-establishment of heterochromatin signatures associated with gene repression (Boyer et al., 2005; Ho et al., 2011; Kadoch et al., 2017).
Various studies consolidate the notion that BAF complex subunits are widely expressed, and the specific combinatorial assemblies and subunit switching contribute to the formation of cell lineage specific BAF complexes capable of instructing specific cell fates In vivo (Lessard et al., 2007; Wu et al., 2007; Tuoc et al., 2013a; Bachmann et al., 2016). For instance, the epigenetic landscape in embryonic stem (ES) cells include specific BAF complex needed for maintenance of their proliferative (self-renewal) capacity and pluripotency. The ES cell BAF (esBAF) complex so established is characteristically composed of BRG1, BAF60a/b, BAF155, and BAF250a, and excluding their site-specific polymorphic variants, which are BRM, BAF60c, BAF170, and BAF250b, respectively (Kaeser et al., 2008; Ho et al., 2009a; Kidder et al., 2009). Similarly, during neural development, the BAF complex participates in stereotypic patterns that lead to formation of neural tissue, thus indicating some form of customization of BAF complex therein. After receiving the appropriate stimuli, ES cells differentiate into neural stem cells (NSCs). Accordingly, reconstitution of the esBAF produces neuronal progenitor BAF (npBAF) complex, which subsequently switch to neuron-specific BAF (nBAF) complex during differentiation (Lessard et al., 2007; Wu et al., 2007; Bachmann et al., 2016). The significance of both npBAF and nBAF complexes in neural development is discussed further in the next two sections.
Function of BAF Complex(es) in Development of the Nervous System
The ubiquitous expression of the chromatin remodeling BAF complex reflects its extensive involvement in controlling embryogenesis (Smith-Roe and Bultman, 2013; Alexander et al., 2015; Nguyen et al., 2016). The BAF complex has been shown in many studies to be required in the maintenance of neural development and the establishment of fully functional nervous system. Such studies explain the critical role of BAF complex in altering chromatin state to mainly influence gene expression patterns that greatly impact on neurodevelopmental events, such as specification of brain structures, neurogenesis (cell proliferation, fate specification, differentiation), cell migration and maturation, and functional integration of neurons. Plausible modulatory mechanisms that may explain how BAF complex particularly influences neurodevelopmental events among other biological processes likely include its spatiotemporal antagonistic interaction with the polycomb complex (Ho et al., 2011; Kadoch et al., 2017) and microRNA-mediated re-composition of its subunit(s) in neural tissues during development (Yoo and Crabtree, 2009). Interestingly both BAF and polycomb complexes interactively target the Wnt signaling factors. While the BAF complex may exhibit selective (inhibition and activation) regulatory effect on Wnt gene targets through direct interaction with β-catenin (Barker et al., 2001; Ronan et al., 2013; Son and Crabtree, 2014; Vasileiou et al., 2015), the polycomb complex antagonizes Wnt signaling to suppress neurogenic fate acquisition during neural development (Hirabayashi et al., 2009). Implying, at least in part, that in the absence of BAF complex, generation of neurons may be fundamentally disturbed during development of the nervous system. The next few sections in this review detail how BAF complex singularly or concertedly drives various aspects of neural development.
BAF Complex Is Indispensable in Neural Tissue Specification
Lethality associated with complete knockout models of BAF complexes has posed major challenges in assessing the biological significance of loss of BAF subunits during development. Recently it was found that conditional knockout mouse models of BAF complex subunits, BAF155 and BAF170, resulted in dissociation of other BAF complex subunits, and hence disassembly of the entire complex. The free subunits were then degraded by the ubiquitin-proteasome system resulting in loss of BAF complex functionality (Narayanan et al., 2015). By applying the above mouse model system, the activity of chromatin remodeling BAF complex has been shown to be vital for the overall formation of nervous system structures, because loss of their functions caused severe neural tissue agenesis (Narayanan et al., 2015; Bachmann et al., 2016; Nguyen et al., 2016).
Following loss of BAF complex due to FoxG1-Cre-mediated double conditional deletion of the scaffolding subunits, BAF155 and BAF170, in the telencephalon, the entire forebrain and related structures including the olfactory bulb failed to develop (Bachmann et al., 2016). Similarly, though Emx1-Cre-mediated ablation of BAF complex also had severe effects on specification of cortical structure, the cortex formed thereof was flimsy and markedly malformed (Narayanan et al., 2015).
Induction of FoxG1-Cre activity via tamoxifen injection 3 days after the formation of primary NSCs caused loss of Sox2+ cells in olfactory placode and epithelium of BAF155/BAF170_dcKO mutants that were immunohistologically examined at developmental stages E9.5, E10.5, and E11.5. Consistently, there was an absolute lack of HuCD+, Tuj+, and Ctip2+ neurons in the olfactory placode/epithelium, hence confirming that olfactory epithelium (OE) was virtually not specified. Given that Pax6-dependent transcriptional activity was greatly reduced in dcKO of BAF155 and BAF 170 in olfactory NSCs, it has been mechanistically postulated that the BAF complex is essential for conventional activation of Pax6-dependent transcriptional activity in neural precursor cells (Ninkovic et al., 2013; Tuoc et al., 2013a; Bachmann et al., 2016).
NSC Proliferation Is Tightly Modulated by Activities of BAF Complex
The mammalian nervous system consists of billions of neurons and glia that are predominantly derived from multipotent NSCs. NSCs initially exit as neuroepithelial cells (NECs), which later in early neural development (~E9.5–E10.5) transform into the so called multi-/unipotent radial glia cells (RGCs) located in the germinal zone of the brain and spinal cord. During development of the brain and spinal cord, RGCs proliferate via symmetric mitotic cell division to amplify their stemness pool from which subsequent neuronal and glial cell type diversity originates (Guillemot, 2007; Kriegstein and Alvarez-Buylla, 2009; Taverna et al., 2014; Tuoc et al., 2014).
Epigenetic alterations, including chromatin remodeling at specific genomic locations, have been correlated with differential expression of transcription factors, leading to temporal regulation of specific genes that define cell autonomous paradigms needed to establish NSC fate (Hirabayashi and Gotoh, 2010; Juliandi et al., 2010; Coskun et al., 2012; Ronan et al., 2013; Narayanan and Tuoc, 2014; Watson and Tsai, 2016; Yao et al., 2016). These alterations are necessary to maintain NSCs in the cell cycle so as to generate the appropriate number of progeny that will later differentiate to afford the right number of neurons and glia needed for normal neural tissue development. The BAF complex has emerged as a key determinant in the selective activation of developmental signals that ensure NSCs proliferate within a developmental time window before switching to differentiation mode.
Several experiments have led to the concept of the existence of specific npBAF complex (Lessard et al., 2007; Bachmann et al., 2016). The npBAF complex is exclusively functional in neural stem/progenitor cells, and is composed of either a BRG1 or BRM core ATPase, a BAF155::BAF155 homodimer or BAF155::BAF170 heterodimer, and BAF250 paralogs (BAF250A or BAF250B). This is in contrast to esBAF complex that is made up of only BRG1 ATPase with functionally non-redundant BAF155 and BAF170 subunits. In both cases however, subunits such as BAF45a/d, BAF53a, and BAF55a remain unchanged, and during neural induction are capable of specifying self-renewal and proliferation (Lessard et al., 2007; Staahl et al., 2013; Bachmann et al., 2016). Thus, esBAF complex is plausibly reconstituted in NSCs to form npBAF complex in order to confer multipotency, while maintaining proliferative ability. However, as neural development advances, say neural progenitors differentiating into neurons, npBAF complex switches subunits to form neuron-specific nBAF complex (Lessard et al., 2007; Bachmann et al., 2016), which is discussed further in the next section.
In a recent study, it was found that olfactory neural stem cells (oNSCs) express a specific BAF complex (onscBAF) that is essential for their proliferation and maintenance during the development of the OE, a peripheral aspect of the olfactory system (Bachmann et al., 2016). Even though it was apparent (as per Ascl1 and Ngn1 progenitor-specific, and HuC/D, Tuj, and Lhx2 immature neuron-specific immunostainings) that neurogenesis at early (~E10.5) but not later (~E13.5) stages in FoxG1 BAF155 mutants was not significantly affected, there was striking reduction in Pax6+, Nestin+, and Sox2+ oNSCs in the OE basal layer. By applying various cell cycle markers, it was ascertained that progenitor proliferation and cell cycle exit index was greatly reduced in BAF155 mutants. Interestingly, proliferation of apically located Pax6+, Sox2+, Otx2+, REEP6+, and K18+ glial-like sustentacular cells were unaffected by loss of BAF155 in the olfactory placode or epithelium (Bachmann et al., 2016). Explanatorily, loss of BAF155 subunit during prenatal development of the mouse OE caused precocious regression of NSCs proliferative capacity, leading to reduced generation of neural precursors and hence neurons. Consequently, neurogenesis was impaired at later developmental stages and resulted in dysgenesis of the OE. The function of BAF155 in NSCs to a large extent seem to be evolutionally conserved as its homolog, psa-4, together with psa-1 (BRG1) in worms are known to be important for asymmetric division of NSCs (Bultman et al., 2000; Sawa et al., 2000; Kim et al., 2001).
Direct observation of neurosphere formation in culture indicated impaired proliferative and self-renewal capacity in NSCs lacking BRG1. To that end, In vivo Cre-directed deletion of BRG1 under Nestin promoter in mouse NSCs at E10.5 led to reduced brain size, consequent to diminished proliferation and depletion of neural precursor population. Perinatal lethality was thus inevitable (Matsumoto et al., 2006; Lessard et al., 2007). Compelling evidence obtained from knockdown investigations indicates that other BAF subunits, such as BAF45a/d, BAF53a, and BAF55a are also indispensable for NSC proliferation and self-regeneration (Lessard et al., 2007; Staahl et al., 2013).
Various signaling pathways known to drive cell proliferation are engaged during BAF complex-mediated transcription regulation of neural development (Zhan et al., 2011; Vasileiou et al., 2015). For instance, BRG1-containing npBAF interacts with components of the Wnt signaling pathway to promote neural progenitor proliferation in the telencephalon (Vasileiou et al., 2015). Also during neural patterning, npBAF complex activates aspects of the notch signaling cascade leading to proliferation of CNS precursor cells, while suppressing the sonic hedgehog signaling pathway (Zhan et al., 2011).
Role of BAF Complex in Neurogenesis
During embryonic and early post-natal neural development, proliferative and multipotent NSCs acquire unipotent fate and ultimately differentiate to generate neurons in the process of neurogenesis. In the mammalian embryonic brain, two major neurogenic (germinal) zones, ventricular and subventricular zones (VZ/SVZ), are the main sites of neurogenesis. The VZ abuts the lateral ventricles whereas the SVZ is atop the VZ (Guillemot, 2007; Kriegstein and Alvarez-Buylla, 2009; Taverna et al., 2014; Tuoc et al., 2014). Some neurogenic centers persist in the adult brain to ensure what is known as adult neurogenesis (Zhao et al., 2008; Ming and Song, 2011).
Neuronal generation, identity establishment, and migration to final functional areal position are under multiple regulatory factors including chromatin remodelers. In this section, we will describe the crucial involvement of the chromatin remodeling BAF complex in orchestrating the transition of neural stem/progenitor cells to fully differentiated neurons, and their final localization during early and late neural development.
BAF Complex Drives Embryonic Neurogenesis
During CNS embryogenesis, such as cortical development, multipotent neural stem cell-like RGCs located in the VZ are able to undergo symmetric mitotic divisions to amplify their pool, or asymmetrically to generate lineage-restricted progeny (Noctor et al., 2004; Guillemot, 2007; Kriegstein and Alvarez-Buylla, 2009; Taverna et al., 2014; Tuoc et al., 2014). Early during embryonic neurogenesis, RGCs divide to directly produce neurons, and later indirectly to produce neurogenic precursors known as intermediate progenitors (IPs) (Haubensak et al., 2004; Miyata et al., 2004) in lissencephalics (lacking cortical folding) and gyrencephalics (with convoluted cortex), as well as outer radial glia (oRG) progenitors that reside in the SVZ and outer SVZ, respectively (Haubensak et al., 2004; Miyata et al., 2004; Hansen et al., 2010; Wang et al., 2011; Borrell and Gotz, 2014; Wong et al., 2015).
As these neuronal stem cell-like precursors acquire neuronal fate, they switch from proliferative to neurogenic cell division mode in the telencephalon. As a result, there is a corresponding shift in epigenetic paradigms including competitive subunit dynamics and BAF complex re-composition, leading to npBAF complex transition to nBAF complex (Lessard et al., 2007; Wu et al., 2007; Bachmann et al., 2016). So far, three key subunit substitutions have been observed in the npBAF-nBAF complex transition: BAF45a/d are swapped for BAF45b/c, BAF53a for BAF53b, and BAF55a for BAF55b; as well as change in expression level of BAF155 and BAF170 (Lessard et al., 2007; Wu et al., 2007; Bachmann et al., 2016).
Mechanistically, Yoo et al. (2009) have ascribed the BAF complex subunit reconstitution phenomenon during neural development (especially for BAF53a/b transition) to activities of two important microRNAs (miR9* and miR124) that are selectively expressed in post-mitotic neurons. That is, this microRNA-mediated repression of BAF53a may be responsible for the activation of its variant BAF53b exclusively found in the nBAF complex. In that direction, they affirmed previous findings that miR9* and miR124 are antagonistic targets of the repressor-element-1-silencing transcription factor [REST] (Conaco et al., 2006). This suggests a possible mechanism of BAF53a/b switching in which REST is recruited to derepress the direct suppressive effect of miR9* and miR124 on BAF53a. While these findings extend our understanding of BAF complex dynamics, they are provocative enough to warrant further investigations aimed at dissecting the switching mechanisms of other BAF subunits which lead to npBAF-nBAF transition that critically coincides with cell cycle exit of multipotent neuronal precursors to become more differentiated.
The BAF complex has also been shown to be an influential factor in determining the mode of embryonic neurogenesis; that is whether by direct generation of neurons from RGCs (direct neurogenesis) or indirectly from IPs (Tuoc et al., 2013a,b). Indirect neurogenesis has been shown to result in expansion of neuronal progenitor pool, and hence contribute to evolutionary cortical expansion and complexity. During development of nervous tissues, BAF complex interacts with the transcription factor, Pax6 that principally regulates neurogenesis. Competitive dynamics of BAF155 and BAF170 subunit incorporation into npBAF complex regulates chromatin state and tendency of recruiting the transcription repressor, REST, to Pax6 gene. Notably, In vitro studies show that, while BAF170 subunit has REST binding motif in its C-terminus, BAF155 bears divergent amino acid sequence in its C-terminus, and hence unable to bind and recruit REST (Tuoc et al., 2013a). Early in neocorticogenesis (E12.5–E14.5), BAF170-incorporated BAF complex repressively interacts with Pax6 by creating a heterochromatin state, which restricts the expression of Pax6 target genes such as Tle1, Cux1, and Tbr2, and consequently promotes direct neurogenesis. However, beyond E14.5, BAF170 expression decreases while BAF155 expression is augmented. This promotes euchromatin state and activates transcription of the aforementioned Pax6 target genes; thereby leading to enhancement of indirect neurogenesis and hence increase in basal progenitor pool (Tuoc et al., 2013a,b). In vivo conditional loss of BAF170 in neocortical progenitors using an Emx1-Cre causes aberrant expansion in IP pool with attendant increased propensity toward upper cortical layer fate. These happen to be effects converse to Pax6 deletion. On the other hand, BAF170 gain of function has phenotypic effects including upper cortical layer neuron depletion similar to Pax6 null (Tuoc et al., 2013a).
Control of Adult Neurogenesis by BAF Complex
After birth, neurogenesis persists in limited regions of the mammalian CNS, constituting the so-called adult neurogenesis (Zhao et al., 2008; Ming and Song, 2011). Adult neurogenic regions are referred to as stem cell niches because they contain NSCs in specialized microenvironment (Zhao et al., 2008; Ming and Song, 2011). There are two major NSC niches defined so far in adult mouse brain: subgranular zone (SGZ) of the dentate gyrus (DG) and the subependymal zone (SEZ) of the lateral ventricles, from where neuroblasts migrate rostrally to populate the olfactory bulb (OB) and also supplies neurons to striatal areas throughout life (Zhao et al., 2008; Ming and Song, 2011).
Few important studies have shown that components of the BAF complex promote adult neurogenesis. In the most recent study, BAF170 subunit was found to be expressed in radial glia-like (RGL) cells and other adult neurogenic cell type in the hippocampal formation. Conditional loss of BAF170 in adult murine brain led to depletion of RGL precursor pool while promoting astrocytic fate. The resultant imbalance in adult neurogenesis was correlated with cognitive dysfunction (Tuoc et al., 2016).
In another study, it was found that expression of the BAF subunit, Ctip2 (Bcl11b), by post-mitotic granule neurons was important for the post-natal development of the dentate gyrus and hippocampus. By using forebrain specific Nex- and Emx1-Cre mediated ablation of Ctip2, it was shown that loss of Ctip2 expression in the adult hippocampus and dentate gyrus caused reduction in neural progenitor pool, neuron generation, and impairment in neuronal differentiation. It was thus revealed that while Ctip2 expression by post-mitotic granule cells is required for their own differentiation, it exerts a paracrine effect by also regulating proliferation of progenitors in a feedback mechanistic fashion. A dual phase-specific Ctip2 functionality is hence proposed to be operational during adult neurogenesis in the hippocampal formation. Interestingly, re-expression of a direct transcriptional downstream effector of Ctip2, Desmoplakin (functions as cell adhesion molecule), is capable of reversing the said mutagenic phenotypes attributable to Ctip2 ablation in the adult hippocampus and dentate gyrus (Simon et al., 2012). Similarly, by using tetracycline-dependent induction of forebrain-specific Cre-mediated deletion of Ctip2 under the calcium/calmodulin-dependent protein kinase type II alpha chain (CaMKIIα) promoter, it has been corroborated that the post-natal expression of Ctip2 is crucial for the survival, differentiation, and circuit integration of granule cell neurons generated in the adult stages during hippocampal development (Simon et al., 2016). Altogether, the studies suggest that sustained post-natal expression of Ctip2 by differentiated granule cells is essential for the post-natal development of the hippocampus, which, as already mentioned, is a major center of cognition in the mammalian brain.
Evidence of the role of chromatin remodeling BAF complex in adult neurogenesis has also been deepened by findings that BRG1, but not BRM-containing BAF complex essentially interacts with Pax6 to specify neuronal identity of adult neural progenitors resident in the subependymal stem cell niche. The study showed that by directly interacting with Pax6, the BRG1-ATPase core of BAF complex is able to potentiate the recruitment of neurogenic transcription factors such as Sox11, Nfib, and Pou3f4 to Pax6 gene targets, thereby forming regulatory feedback network in orchestrating fate specification during adult neurogenesis. Adult NSCs lose neuronal fate tendency and become bias toward glial cell lineage upon deletion of BRG1 just as in the case of Pax6 ablation (Ninkovic et al., 2013; Petrik et al., 2015).
Genesis of Neuronal Subtypes Is Regulated by SWI/SNF (BAF) Complex
A vast number of neurons with phenomenal diversity are formed during development of the nervous system. These neurons receive specific instructions in order to acquire unique neuronal identity. The cerebral cortex for example is made up of millions of projection/pyramidal neurons (PNs) broadly organized into several functional domains (Custo Greig et al., 2013; Harb et al., 2016). When PNs are newly generated from their precursors in the VZ/SVZ, they are differentially naive as they share widely similar and overlapping characteristics. However, through selective transcriptional activation and repression of a number of fate specifying genes and fine regulatory mechanisms, specific PN identities are acquired, yielding the phenomenal diversity in PN population in the post-natal cortex (Custo Greig et al., 2013).
Several studies elaborate on the pivotal involvement of the BAF complex in the specification and generation of neuronal subtype identity during neural development, and they also give accounts of various aberrant phenotypes attributable to the ablation of specific BAF complex subunits. Using an in vivo mouse model, it has been shown that the BRM-ATPase based BAF complex containing BAF155 and BAF170 subunits is able to bind to Pax6 in a time-regulated manner to modulate the expression of related gene targets such as Cux1, Tle1, and Tbr2. These genes are required for the generation and specification of upper layer neuronal identity (Tuoc et al., 2013a,b).
Fascinatingly, two BAF complex paralogous subunits, BAF100a (Ctip1/Bcl11a) and BAF100b (Ctip2/Bcl11b), have also been identified as crucial molecular determinants of PN subtype specification. While Ctip1 is strongly expressed by post-mitotic callosal and corticothalamic PNs, Ctip2 is solely expressed in subcerebral PNs and post-mitotic striatal medium spiny neurons during embryonic stages of cortical development. As such, Ctip1 has been reported to modulate precision of neocorticogenesis via regulation of factors that define subclass identity of deep-layer PNs, whereas Ctip2 expression is critical for corticospinal PN terminal differentiation and circuitry (Arlotta et al., 2005, 2008; Woodworth et al., 2016). Ablation of Ctip1 leads to preponderance of subcerebral PN development in sensory cortical areas over genesis of deep-layer callosal and corticothalamic PNs, hence indicating Ctip1 importance in establishing sensory areas in the cortex. In vivo Ctip1 gain-of-function (GOF) however, suppresses generation and axonogenesis of subcerebral PNs (Woodworth et al., 2016).
Various findings point to the fact that Ctip2 expression is controlled by other transcription factors in directing PN specification and terminal differentiation. For instance, the transcription factor Fezf2 promotes the expression of Ctip2 to synergistically control differentiation of neurons that project to subcerebral targets, while adequately restricting the expression of other neuronal subtype identity-specifying transcription factors (e.g., Sox5, Satb2, and Trb1) (Arlotta et al., 2005; Molyneaux et al., 2005; Chen et al., 2008; Bedogni et al., 2010). It implies that co-repression of Ctip2 and Fezf2 is necessary to permit other neuronal subtype generation. For example, repression by Sox5 is critical for establishing neocortical layer 5/6 (lower layer) PN identity (Kwan et al., 2008; Lai et al., 2008; Shim et al., 2012), whereas repression by Satb2 yields upper layer PN identity (Alcamo et al., 2008). Suppression of Ctip2 by Tbr1 is also known to determine layer 6 (corticothalamic) PN subclass (Bedogni et al., 2010; Han et al., 2011). Neuronal subtype identity is however a continuous process that may involve further modulatory/tweaking factors even at late embryonic stages (Azim et al., 2009).
Loss of Ctip2 in the brain interferes with normal differentiation of subcerebral PN. Mainly, axonal growth, trimming, and pathfinding is affected in the absence of Ctip2 expression (Arlotta et al., 2005). In the hippocampus, null mutation of Ctip2 causes dispersion of neurons that under normal conditions are kept in the confinement of the granular layer. These neurons also display impaired dendritogenesis and decreased expression of calbindin (Wu et al., 2007).
BAF Complex Orchestrates Neuronal Migration, Maturation, and Dendritic Morphogenesis
Accompanying the generation of neurons from progenitors during neurogenesis, immature neurons usually undergo different forms of migration (e.g., radial, tangential, and multipolar) from their birthplaces to their final functional location in the developing brain (Evsyukova et al., 2013). In the developing neocortex, newly born neurons predominantly migrate radially under glial guidance out of the VZ or SVZ to form the laminated cortical plate.
Various factors regulate neuronal migration in the developing cortex (Evsyukova et al., 2013). Although much is yet to be known about the role of chromatin remodeling factors in neuronal migration, Wiegreffe et al. (2015) have elegantly provided striking evidence showing that the chromatin remodeling BAF complex subunit, BAF100a (Ctip1/Bcl11a), is crucial in regulating radial migration of PNs in the developing cortex. Importantly they indicated that Ctip1 expression by migrating neurons (Leid et al., 2004) in the intermediate zone (IZ) is required for multipolar to bipolar morphology transition, which is a critical stage in the process of radial migration (Wiegreffe et al., 2015). After electroporating Cre-GFP into BAF100a-flox/flox brains at E14.5, more GFP-positive multipolar neurons compared to bipolar neurons were found in the IZ of E17.5 neocortex. The multipolar cell accumulation in the IZ may signify failed or disrupted morphological switch to continue their radial migration to cortical layers. Most of the aberrantly migrating neurons due to Bcl11a deletion displayed disorientation with respect to the pial surface, which is an opposing phenotype to normal radially migrating neurons (Wiegreffe et al., 2015). At post-natal stages, BAF100a-deficient cortex displayed discernable dysplasia, especially in the upper layers with resultant reduction in inter-hemispheric connection. Mechanistically, it was revealed that BAF100a directly regulated its downstream effector Sema3c to control PN polarity and radial migration that ensured proper formation of upper cortical layers during neocorticogenesis (Wiegreffe et al., 2015).
After gaining subtype identity, immature neurons undergo specific differentiation processes to become fully matured and functional. During neural maturation, neurites are specified to become either axons or dendrites with which neurons make synaptic connections with adjoining neurons to afford circuit integration. Improper axonogenesis (axonal growth and pathfinding) and dendritogenesis (dendritic outgrowth, arborization, and refinement) results in anomalous neuronal morphology and synaptic connections that underscore several neurodevelopmental disorders. Many factors, including chromatin regulators play vital roles during neural morphogenesis (axonogenesis and dendritogenesis) (Whitford et al., 2002; Wu et al., 2007; Bachmann et al., 2016).
The BAF complex is important in neuronal morphogenesis during neural development (Whitford et al., 2002; Wu et al., 2007; John et al., 2012; Weinberg et al., 2013; Choi et al., 2015; Bachmann et al., 2016). BAF complex controls specification of neurites to differentiate into axons, usually one per neuron, and subsequent axonal fasciculation and pathfinding (Weinberg et al., 2013; Bachmann et al., 2016). For example, sensory neurons found in the dorsal root ganglion (DRG) extend axons that synapse with second order neurons in the dorsal spinal cord to allow relay of somatosensory information. It has been revealed that the BAF complex subunit, BAF100a, is required for terminal maturation and morphogenesis of dorsal spinal neurons during development of the spinal cord (John et al., 2012). Loss of BAF100a expression in spinal neurons was shown to cause central axon dysgenesis, leading to denervation of the dorsal horn by primary somatosensory neurons in the DRG.
Post-mitotic neuron specific BAF53b subunit (Olave et al., 2002) has been shown to play a key role in activity-dependent dendritic morphogenesis. Homozygous knockout of BAF53b subunit in mice largely led to early post-natal lethality, with just about 10% surviving up to adult stages, albeit they displayed behavioral disturbances such as hyperactivity (Lessard et al., 2007; Wu et al., 2007). Cultured cortical neurons lacking BAF53b (BAF53b−/−) exhibited striking activity-dependent dendritogenic incompetence, which phenotype could only be rescued by exogenous BAF53b GOF but not its homolog, BAF53a (Wu et al., 2007). To mutually exclude the functional role of BAF53b from its homolog, BAF53a, in orchestrating dendritogenesis, two chimeric protein candidates, BAF53a with subdomain 2 of BAF53b and its reverse were generated. Intriguingly, the chimeric BAF53a protein containing subdomain 2 of BAF53b, but not its reverse candidate was able to affect activity-dependent dendrite outgrowth, hence suggesting the divergent subdomain 2 of BAF53b as indispensable for activity-dependent dendrite formation (Wu et al., 2007). That notwithstanding, phenotypes similar to BAF53b−/− were observed when other subunits of the nBAF complex (i.e., BRG1, BAF45b, and BAF57) were knocked down using RNA interference (Lessard et al., 2007; Wu et al., 2007), suggesting multiple BAF subunit involvement in neurite development. The outcome of investigations in invertebrate models recapitulates BAF53b as a critical player in neuronal dendritogenesis. Loss of function of BAP55 (homolog of BAF53a/b) in Drosophila melanogaster interfered with normal dendritic targeting of olfactory projection neurons, which phenotype was rescuable with expression of mammalian BAF53a/b using GH146-Gal4 driver (Tea and Luo, 2011). Additionally, loss of BAP55 and other Drosophila homologs of BRM, BAF60c and BAF47, perturbed formation of sensory neural dendrites in the peripheral nervous system (Parrish et al., 2006). Plausible mechanisms underlying the role of BAF complex in orchestrating dendritic elaboration include the loss of BAF53b, which causes reduction in the entire BAF complex occupancy at promoters of dendritogenic genes such as GAP43 and Ephexin1, hence culminating in alterations in their expression (Wu et al., 2007). BAF complex is also known to mechanistically engage the calcium-mediated transcription activation function of calcium-responsive transactivator (CREST), which links activity responsive Ca2+ signaling and chromatin reorganization by BAF complex known to lead to dendritic outgrowth, arborization, and refinement (Aizawa et al., 2004; Wu et al., 2007).
Even though early olfactory receptor neuron (ORN) population identified by Lhx2+, Tuj+, and HuCD+ immunostaining did not change significantly in BAF155 knockout mice, axonogenesis was markedly defective at early embryonic stage (E13.5) (Bachmann et al., 2016). Likewise, the high expression of BAF170 in ORNs is essential for their maturation, because diminished number of OMP+ mature ORN was found in BAF170-deficit OEs (Bachmann et al., 2016). Traditionally, immature ORNs located in the developing OE project axons that synapse with the dendrites of mitral cells in the olfactory bulb during differentiation. The afferent pathway so formed then permits second order axonal projection from mitral cells to primary olfactory centers in the cortex. To confirm interrupted axonal pathfinding from ORNs to telencephalic regions due to the absence of BAF155 subunit, Dil crystal injection of OE was used to reveal axonal dysgenesis in BAF155 deficit ORNs as opposed to obvious visualization of Dil-labeled olfactory neuronal tract in controls (Bachmann et al., 2016). The observation thus consolidates the idea that BAF155 subunit is important for neuronal axonogenesis in the olfactory system. The significance of BAF complex in directing axonal pathfinding is also observed in Caenorhabditis elegans as deletion of the nematode homolog of BAF60 (ham-3) disturbed axonal projection during differentiation of some serotonin-producing neurons (Weinberg et al., 2013).
Thus, the BAF complex plays crucial roles in neuronal morphogenesis in different parts of the CNS (e.g., dorsal root ganglion, forebrain, and olfactory system) in both invertebrates and vertebrates.
BRG1-Containing BAF Complex Regulates Gliogenesis
Gliogenesis is a neurodevelopmental process that leads to the generation of non-neuronal glial cell types including astrocytes and oligodendrocytes from multipotent NSCs. The production of astrocytes (astrogenesis) and oligodendrocytes (oligodendrogenesis) have been extensively studied in the developing brain and spinal cord (Rowitch and Kriegstein, 2010; Gallo and Deneen, 2014). In many structures of the CNS such as the cortex, differentiation of NSCs initially primed to generate neurons at early stages become progressively restricted in the course of cortical development. Typically, NSCs in the cortex undergo intrinsic changes that make them acquire glial fate at later stages of neural development, with astrogenesis peaking at P0–P2 and oligodendrogenesis at late post-natal stages around P14 in mice (Freeman, 2010; Rowitch and Kriegstein, 2010; Gallo and Deneen, 2014). Epigenetic cell-type-specific regulation of gene expression is implied in changes in NSCs that determine neuro/gliogenic fate. These include transcription factor expression selectivity engendered by epigenetic alterations such as chromatin remodeling that control NSC fate (Hirabayashi and Gotoh, 2010; Juliandi et al., 2010; Coskun et al., 2012; Narayanan and Tuoc, 2014; Yao et al., 2016).
Although the interest in astrocyte function has increased dramatically in recent years, our understanding of astrocyte development, especially under the control of epigenetic factors, has lagged behind as compared to other brain cell types. In addition, the molecular mechanisms underlying astrocyte specification and expansion are largely unknown. Phenotypic analyses of early loss of BRG1 in BRG1cKO mutants with Nestin-Cre indicated that the loss of this core BAF subunit led to a severe defect in the maintenance of NSC pool and consequently also in the production of neurons and glia cells (Matsumoto et al., 2006; Lessard et al., 2007). However, it is unknown whether chromatin remodeling via the BAF-complex regulatory function might control embryonic astrogenesis. In post-natal brain, BAF complex is apparently required in permitting neurogenic tendencies of NSCs, while suppressing their gliogenic commitments. Hence, when BRG1 (Ninkovic et al., 2013) or BAF170 (Tuoc et al., 2016) were abolished in adult NSCs, there was premature onset of gliogenesis.
Oligodendrocyte identity specification is critical to establishing myelination schemes during CNS development. The chromatin remodeling BAF complex also finely regulates generation and maturation of oligodendrocytes. The BRG1 ATPase core of the BAF complex is reported to be vital for the generation of oligodendrocyte lineage. Multi-stage chromatin immunoprecipitation (ChIP) sequencing studies reveal that co-occupancy of BRG1 and the oligodendrocyte-lineage specifying basic-helix-loop-helix transcription factor, Olig2, at enhancer elements synergistically regulate recruitment of transcription factors to gene loci central to oligodendrogenesis (Yu et al., 2013; Bischof et al., 2015). Matsumoto et al. (2006) went further to elucidate how BRG1 interacts with Olig2 to specify oligodendrocytic fate in neural progenitors at early cortical developmental stages. They reported that at about E14 in the mouse cortex, at which stage oligodendrocyte precursors are yet to colonize the cortex, BRG1 was found to interact with Olig2 promoter, leading to Olig2 transcriptional repression. As such, nestin-dependent deletion of BRG1 in early neural progenitor cells resulted in premature expression of Olig2 in the cortex, although such precocious Olig2 expressing cells could not differentiate into oligodendrocyte precursor cells (OPCs). That notwithstanding, BRG1 was found to interact with the said proximal Olig2 promoter in the ganglionic eminence where neural progenitors normally transform into Olig2+ OPCs, implying that the BRG1 component of the BAF complex is a repressor of Olig2 expression and an indirect suppressor of OPC specification in the early developing cortex. However, it is needed for the differentiation and maturation of OPCs (Matsumoto et al., 2006; Yu et al., 2013; Bischof et al., 2015).
BAF Complex Function in Cognition: Learning and Memory
Learning and memory (formation and storage) are cognitive functions that are underscored by persistent alterations in memory-related neural circuits due to transient environmental stimuli that result in behavioral changes (Day and Sweatt, 2010; Fischer, 2014; Watson and Tsai, 2016). The hippocampal formation is involved in cognitive functions such as memory formation, fear conditioning, and learning (Goncalves et al., 2016). The evidence thereof is mostly centered on hippocampal and DG neurogenesis (adult neurogenesis), dendritogenesis of hippocampal neurons, and various hippocampal-dependent activity experimentations in which synaptogenesis and synaptic alterations such as long-term potentiation (LTP) have been used to study memory formation and maintenance (Goncalves et al., 2016). Overall generation and integration of new neurons into complex neural circuits at pre-/post-natal developmental stages are also of great importance in memory formation and behavior (Deng et al., 2010; Urban and Guillemot, 2014). Given that the hippocampus is a major center of cognitive function in the brain, neurogenic activities that lead to its formation is of critical value to learning and memory. Hippocampal structures, such as the hippocampus proper and the DG are malformed in the presence of defective neurogenesis, which are strongly correlated with behavioral imbalances.
Various regulatory factors play importance roles in learning and memory. For instance, long-term memory processing requires fine interplay of intracellular signaling with transcriptional, translational, and epigenetic factors to regulate gene expression in cognitive function development (Deng et al., 2010; Urban and Guillemot, 2014; Goncalves et al., 2016). Epigenetic mechanisms such as methylation, acetylation, phosphorylation, ribosylation, and chromatin remodeling state (heterochromatin or euchromatin) regulate memory formation and maintenance (Day and Sweatt, 2010; Fischer, 2014; Watson and Tsai, 2016). Accumulating evidence from recent studies indicates the essentiality of the chromatin remodeling BAF complexes in memory formation and learning. Dysfunction of subunits in both npBAF and nBAF complexes in the CNS has been posited to underline various psychiatric perturbations (Koga et al., 2009; Neale et al., 2012; Yoo et al., 2017).
As already stated, BAF170 subunit in BRG1-containing BAF complex is expressed in RGL progenitors in the DG of the adult brain. For this reason, when hGFAP-Cre and Nestin-CreER-mediated conditional loss of BAF170 in RGL cells was performed, it caused abnormal proliferation and localization of RGL cells, with subsequent depletion of progenitor pools in the SGZ of the DG. Due to the lack of BAF170 in RGL progenitors, neurogenesis in DG was skewed to terminal astrogenesis. By using the Morris water maze test, BAF170 mutant mice were observed to display noticeable impairment in adaptive behavior (Tuoc et al., 2016).
The nBAF subunit, BAF100b (Ctip2/Bcl11b), is exclusively expressed in post-mitotic granule neurons in the SGZ of the DG but not in progenitors. Its expression is important for embryonic and adult hippocampal neurogenesis leading to proper functioning of the hippocampus in memory and learning. Newly born neurons lacking Ctip2 locate ectopically of the granule cell layer in the hippocampus. These Ctip2-deficient neurons characteristically display reduced expression of calbindin 1 (Calb1), which plays important roles in neuro-behavioral and memory formation (Simon et al., 2012; Soontornniyomkij et al., 2012). On the whole, loss of Ctip2 during development of the hippocampus leads to dramatic reduction in size and neuronal pool of the DG, and impairment of spatial memory formation (Simon et al., 2012, 2016; Lennon et al., 2017).
Another nBAF subunit, BAF53b (Vogel-Ciernia et al., 2013; Choi et al., 2015; Yoo et al., 2017) is also shown to play a strong role in cognitive processes in mice. Upon RNA sequencing of the dorsal hippocampus of BAF53b+/− mutant mice, it was revealed that genes involved in memory formation (including cytoskeletal reorganizers and chromatin modifiers) were misregulated when compared with wild-type mice (Vogel-Ciernia et al., 2013). Consistently, alterations in the neuron-specific BAF complex subunit BAF53b expression in amygdala neurons modulated learning and memory formation in adult mice. Specifically, when Yoo et al. (2017) applied viral vector-mediated methods to knockdown or overexpress BAF53b functionality within the lateral amygdala in experimental auditory fear conditioning schemes, they found that inductive expression of BAF53b accompanied learning activities to augment memory consolidation and related neuroplasticity. Likewise, disrupted normal levels of BAF53b expression by genetic manipulations leading to BAF53b heterozygosity (BAF53b+/−) in mouse CaMKIIa+ post-mitotic neurons, caused aberrant neuronal morphology acquisition such as defective spine formation, severe defects in hippocampal synaptic plasticity, and long-term memory impairment (Vogel-Ciernia et al., 2013).
Similar to phenotypes of BAF53b mutant mice, mutation in CREST (also known as SS18L1, a newly identified nBAF subunit) causes defects in learning and memory. Mechanistically, CREST acts together with the co-activator, CREB-binding protein (CBP), to regulate expression of immediate early genes including c-fos. Such early genes play significant roles in brain plasticity, activity dependent memory formation, and other complex behaviors.
All together, these findings suggest that BAF complex-driven chromatin remodeling in neuron generation, differentiation, synaptic development, and plasticity in related structures such as the hippocampus is essential for cognitive functions.
Role of BAF Complex in Neurodevelopmental Disorders
Given that the BAF complex plays crucial roles in neural development, it is clear to rationalize how its misregulation culminates in both syndromic and non-syndromic neurological problems. As such, exome sequencing analyses have implicated de novo BAF complex mutations as the underlying cause of some cognitive and psychiatric disturbances reported in humans (Figure 1, Table 2). Notably, while some polymorphic forms of BAF complex subunits cause more general and/or overlapping defective neurological phenotypes, others have been strongly linked to mutually exclusive intellectual disabilities and related non-neurological problems. Common and most characterized of these BAF complex-related neural disorders include the following discussed syndromes and conditions.
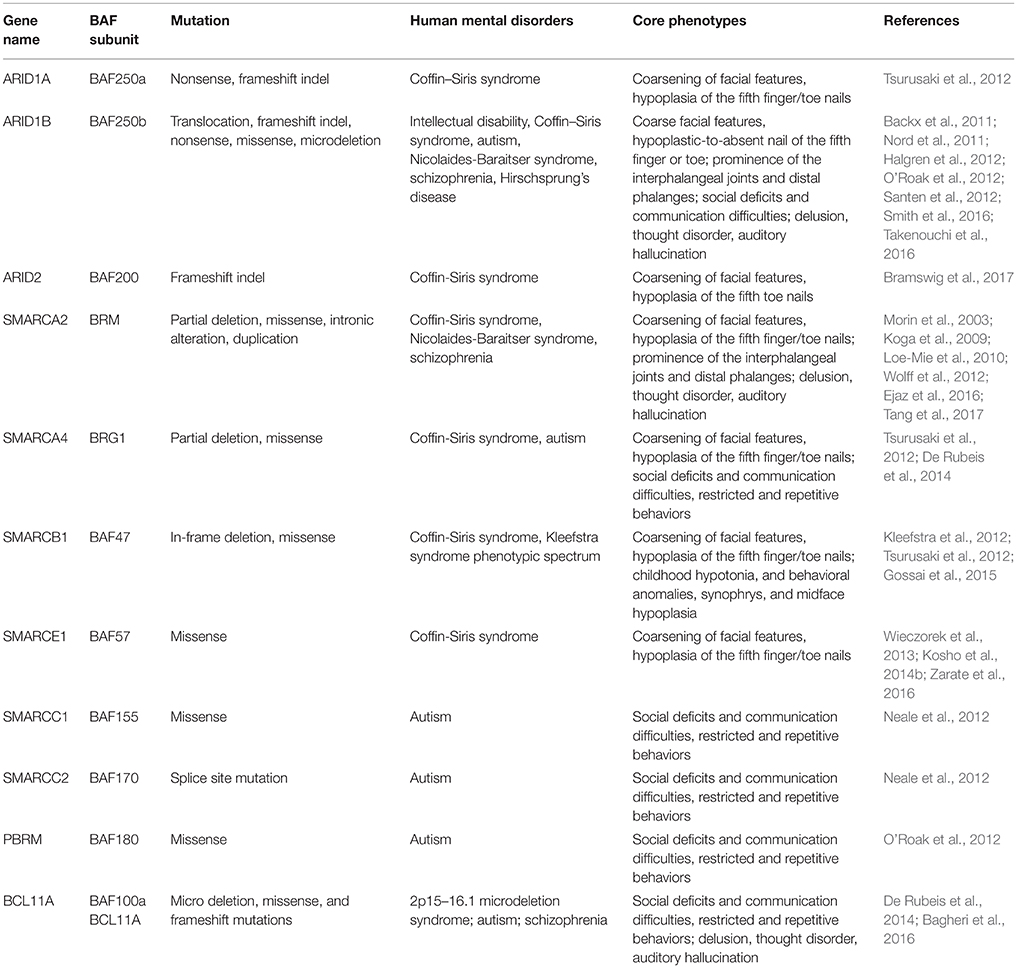
Table 2. Mutated components of BAF complex in human mental disorders.
Coffin-Siris Syndrome (CSS)
Drs. Grange S. Coffin and Evelyn Siris were the first to report CSS (OMIM #135900) in 1970 (Kosho et al., 2014a). It is a rarely occurring congenital abnormality with clinical characteristics such as intellectual disability (ID), progressive coarsening of the face, hypertrichosis, frequent infections, feeding difficulties, and hypoplasia of the 5th distal phalanges and fingernails (Levy and Baraitser, 1991; Schrier et al., 2012). Schrier et al. (2012) summarized 80 previously reported cases for both common and discriminating clinical hallmarks, and brought forward a differential diagnosis between CSS and other related syndromes, including Nicolaides-Baraitser syndrome (NCBRS) and Deafness, Onychodystrophy, Osteodystrophy, Mental Retardation, and Seizures (DOORS) syndrome. In recent years, comprehensive human exome sequencing and genome-wide association studies have revealed that among roughly 180 CSS cases reported, about 110 patients carry a mutation in genes encoding BAF complex subunits. These genes include BAF250A (ARID1A), BAF250B (ARID1B), BRM (SMARCA2), BRG1 (SMARCA4), BAF47 (SMARCB1), BAF57 (SMARCE1), and BAF200 (ARID2) (Santen et al., 2012; Tsurusaki et al., 2012; Bramswig et al., 2015, 2017; Miyake et al., 2016).
BAF250a/BAF250b
Up to date, eight CSS patients have been reported to harbor heterozygous mutations in BAF250a (Tsurusaki et al., 2012; Kosho et al., 2013, 2014a). Such mutations result in truncated BAF250a subunit protein, and are proposed to exert a loss-of-function mechanism. BAF250b, a homolog of BAF250a subunit, exists mutually exclusive to BAF250a in BAF complexes. It has been found that more than 70 patients with CSS (Kosho et al., 2014a; Mari et al., 2015), and >30 patients presenting non-syndromic ID carry BAF250b abnormalities (Kosho et al., 2014a), including cytogenetic abnormalities and point mutations (Table 2). BAF250b abnormalities account for 68–86% of CSS cases (Santen et al., 2013; Wieczorek et al., 2013; Tsurusaki et al., 2014). All of the reported BAF250b mutations involved in CSS resulted in related protein truncation, and are proposed to exert haploinsufficiency. CSS Patients from BAF250b abnormalities present variable developmental delays or ID, together with impaired speech. While BAF250a mutation is known to result in severer CSS clinical features with associated rigorous physical complications, BAF250b mutation-dependent CSS precludes typical characteristics such as coarsening of facial features, and with rather milder digital hypoplasia. The detailed comparison of clinical features of CSS patients from BAF complex subunit mutations has been reported (Kosho et al., 2014a).
BAF250a and BAF250b contain two characterized functional protein domains, an ARID in the center, and a domain of unknown function 3518 (DUF3518) at the C-terminus (Figure 2). ARID binds to AT-rich DNA with no obvious sequence specificity (Wang et al., 2004; Wilsker et al., 2004) and DUF3518 is required for interactions with BRG1 and BRM subunits in BAF complexes. Therefore, it is most likely that missense mutations in ARID will impair the ability of BAF250a/BAF250b to bind to chromatin DNA resulting in compromised BAF complex function, and that the missense mutations in DUF3518 may compromise the association of BAF250a (ARID1A) and BAF250b (ARID1B) with BRG1/BRM to form functional BAF complex (Sim et al., 2015). In line with this notion, it has been reported that the interactions of BAF250a with DNA are essential for SWI/SNF occupancy at promoters, and interruptions of which leads to embryonic lethality in mice (Chandler et al., 2013). Together, missense mutations from either domain would most probably exert a negative impact on BAF complex function. However, the mechanism behind the huge phenotypic variations is still not known. Interestingly, it has been proposed that the majority of nonsense and frameshift mutations result in mRNA degradation through nonsense-mediated mRNA decay (NMD) mechanism (Sim et al., 2015). Therefore, truncating mutations that escape from NMD in general leads to divergent and more serious phenotype than that found in NMD. However, further investigations are needed to clarify whether specific frameshift mutations in BAF250a/BAF250b result in NMD or truncated protein.
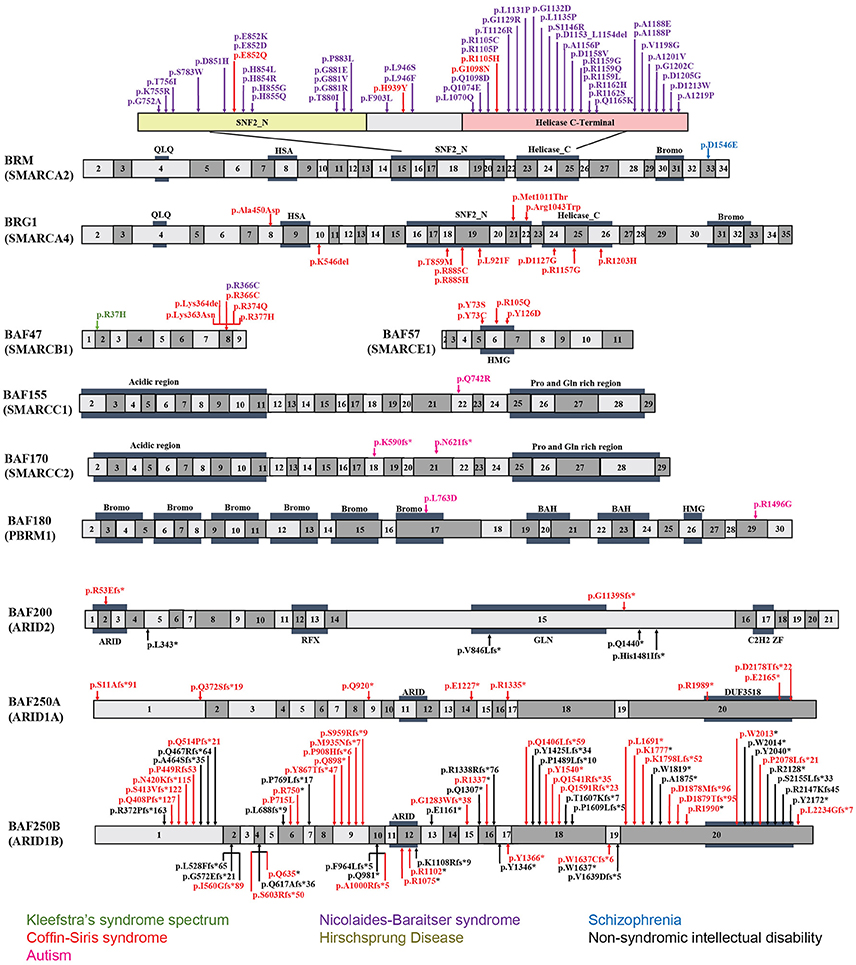
Figure 2. Exon-structure of the genes encoding for BAF subunits and sites of the pathogenic mutations in neurodevelopmental disorders. The specific functional domains are also shown: ARID, A-T rich interaction domain; QLQ, Gln, Leu, Gln motif; HSA, small helicase/SANT-associated domain; SNF, sucrose/non-fermenting domain; BROMO, bromodomain; HMG, high-mobility group domain; RFX, RFX-like DNA binding domain; C2H2 ZF, C2H2 zinc fingers.
BRM/BRG1
BRM mutations, including four missense (Santen et al., 2013) and two duplication mutations (Miyake et al., 2016) have been observed in seven patients with CSS. As shown in Figure 2, the missense mutations were localized in the SNF2-ATPase domain. Therefore, these mutations of BRM most likely result in impairing the proper functioning of BAF complex by influencing ATP-hydrolysis. Remarkably, phenotypes from BRM mutations resulted-CSS highly overlap with that from NCBRS (Wieczorek et al., 2013), which sometimes make it difficult to make a clear diagnosis. In addition, it has been proposed that CSS could also derive from over-dosage of BRM, because two patients with a duplicated region including BRM presented typical CSS phenotype (Miyake et al., 2016).
Heterozygous mutations in BRG1 have been observed in 12 CSS reported cases (Kosho et al., 2014a). As presented in Figure 2, the missense and in-frame deletions within SNF2-ATPase domain were also found in BRG1 mutants in CSS patients. Most likely, the resulted non-truncating BRG1 proteins from the mutations were responsible for the phenotype, through GOF or dominant-negative effects (Kosho et al., 2014a). Although these two subunits have 74% similarity in amino acids sequence, BRG1 mutations were exclusively linked with CSS, while BRM mutations were seen in both CSS and NCBRS (Wieczorek et al., 2013; Kosho et al., 2014a; Miyake et al., 2016).
BAF47/BAF57
To date, five pathogenic mutations have been found in BAF47 in 13 CSS patients (Santen et al., 2013; Wieczorek et al., 2013; Tsurusaki et al., 2014). Interestingly, the mutations were all localized at three amino acid residues at positions 364, 374, and 377 within exons 8 and 9 (Figure 2), and the p.Lys364del mutation was frequently identified in nine patients, indicating the importance of these residues in normal BAF complex functionality. These non-missense or in-frame deletions result in truncated BAF47 protein products that lead to CSS phenotype through either dominant-negative or GOF mechanism. Recently, the emerging studies on genotype-phenotype correlation have found that CSS patients with BAF47 mutations display the most striking physical features and rigorous problems associated with developmental delay, but with least distal limb anomalies (Mari et al., 2015).
Six CSS cases have been associated with BAF57 pathogenic mutations, including pTyr73 Cys, pTyr73Ser, pArg105Gln, and pTyr126Asp (Tsurusaki et al., 2012; Santen et al., 2013; Wieczorek et al., 2013; Zarate et al., 2016). All of these cases have been reported to present with developmental delay in addition to moderate to severe ID. Moreover, they all have the typical features of CSS (Zarate et al., 2016). Notably, all these mutations are localized in the high-mobility-group (HMG) DNA binding domain (Figure 2). Although all mutations are located in the same domain, the resulted phenotypes display vast variation. For instance, abnormal brain structure was seen in only three out of six mutated cases. Therefore, how the missense mutations influence BAF complex function need further investigation.
BAF200
Mutation of BAF200 subunits was recently found to be associated with CSS. Recently, four patients with BAF200 mutations were reported with non-syndromic ID, delayed development, and other clinical features (Shang et al., 2015). In another case, a patient with a duplication of BAF200, spanning exon 1–3, phenotypically displayed impaired language development and short stature (Zahir et al., 2016). More recently, two individuals bearing de novo BAF200 frameshift mutations with typical CSS-like phenotypes including ID, coarse faces, and fifth toe nail hypoplasia were found (Bramswig et al., 2017). Based on the similarity of clinical phenotypes from the above patients, it has been proposed that BAF200 mutations result in CSS as well (Bramswig et al., 2017).
6q25 Microdeletion Syndrome
The 6q25 microdeletion syndrome is an uncommon congenital and genetic disease characterized by ID with developmental delay, microcephaly, hearing impairment, distinct dysmorphic features, visual impairment, and corpus callosum agenesis (Santen et al., 2014; Ronzoni et al., 2016). Mutations of BAF250b were reported to be linked with this syndrome (Pirola et al., 1998). The case presenting with agenesis of corpus callosum (ACC) had a deletion of 5.5–7.6 Mb, including BAF250b and 10 other genes. To date, more than 24 cases with clear deletion information have been reported. Most of the patients carry deletions in BAF250b and several other genes, with deletion sizes of about 58 kb–14.5 Mb. Phenotypes are very variable, and dependent on the size and site of the deletion (Sim et al., 2015). It has been proposed that the haploinsufficiency of the affected genes results in defects in normal brain development and is responsible for the phenotype. The essential region for the phenotype was mapped to a 6q25.3 region harboring the protein-coding genes ZDHHC14 and BAF250b (Michelson et al., 2012). Other studies (Halgren et al., 2012; Santen et al., 2014) further suggested that BAF250b was the critical gene responsible for the core phenotypes of the syndrome. Recently, Ronzoni et al., reported an evidence that loss of BAF250b mainly underpins 6q25 microdeletion syndrome (Ronzoni et al., 2016). In that study, a patient carrying a small 6q25 deletion containing only BAF250b exhibited the typical features of the syndrome including developmental delay, speech impairment, and agenesis of the corpus callosum (Hoyer et al., 2012; Mignot et al., 2016; Ronzoni et al., 2016).
Nicolaides-Baraitser Syndrome (NCBRS)
NCBRS (OMIM #601358) is a disorder that shares remarkable clinical features with CSS. The syndrome is characterized by ID with impaired speech, sparse scalp hair, prominence of the interphalangeal joints and distal phalanges due to decreased subcutaneous fat, characteristic coarse facial features, microcephaly, and seizures (Sousa et al., 2009; Van Houdt et al., 2012). The hallmark differences between NCBRS and CSS come from features of the hands and feet. The representative NCBRS patients show prominent interphalangeal joints and distal phalanges, whereas those with CSS present hypoplasia/aplasia of the fifth fingernails with or without involvement of the terminal phalanges (Wieczorek et al., 2013).
Recently, exome sequencing studies have suggested that NCBRS is associated with BRM (SMARCA2) mutation. Among the 70 mutations reported in NCBRS patients, one is an in-frame 6-bp deletion, two are in-frame multi-exon deletions affecting the ATPase domain, and 62 are missense mutations. Of the 62 missense mutations, 60 are mapped to the ATPase domain, one is proximal but outside the ATPase domain, and the other is in the bromo domain (Van Houdt et al., 2012; Sousa et al., 2014; Ejaz et al., 2016). Notably, deletions encompassing human BRM locus have not been found in NCBRS (Christ et al., 1999). Because null BRM KO mice do not show major developmental defects (Koga et al., 2009; Magnani and Cabot, 2009) and none of the identified mutations cause truncated proteins, it has been proposed that the mutations in BRM cause GOF or dominant-negative effects. Hence, these mutations possibly interfere with the ability of ATP hydrolysis by native BRM protein (Van Houdt et al., 2012; Ejaz et al., 2016). In addition to BRM, missense mutations in BAF47 (p.Arg366Cys) was also reported to present with NCBRS (Wieczorek et al., 2013).
Kleefstra's Syndrome Spectrum (KSS)
Kleefstra's syndrome (OMIM #610253) was known as a recognizable mental retardation syndrome caused by heterozygous mutation in the euchromatin histone methyl transferase 1 (EHMT1) gene (Kleefstra et al., 2006, 2009). However, not all the patients presenting core phenotypes of Kleefstra's syndrome carry an EHMT mutation. Indeed, de novo mutations in four epigenetic regulator-encoding genes, MLL3, MBD5, NR113, and BAF47 (SMARCB1) were also identified from four of nine EHMT1 mutation-negative patients (Kleefstra et al., 2012). These patients exhibit core features of Kleefstra syndrome; nevertheless they have very heterogeneous phenotypes, which are therefore referred to as Kleefstra syndrome spectrum (KSS). Common phenotypic features among these patients include ID, childhood hypotonia, behavioral anomalies, synophrys, and midface hypoplasia. Investigation with drosophila model revealed direct interactions between MLL3, MBD5, NR113, BAF47 (SMARCB1), and EHMT1 (Kleefstra et al., 2012). The findings suggested that mutations in any protein within the complex of MLL3, MBD5, NR113, BAF47, and EHMT1 could lead to KSS.
Autism Spectrum Disorder (ASD)
Autism is a neurodevelopmental disorder characterized by deficits in social skills, speech problems, and restricted and repetitive behaviors. It is well established that a combination of genetic and environmental factors cause autism (Chaste and Leboyer, 2012). However, the underlying genetic basis is unclear for at least 70% of cases. Recent exome sequencing analyses of autism patients have revealed mutations in the following BAF complex components: BAF155, BAF170, BAF180, and BAF250b (Neale et al., 2012; O'Roak et al., 2012). A de novo mutation of genes encoding for the above BAF subunits were identified in patients who showed severe social communication problems with low verbal intelligence quotient (IQ) and nonverbal IQ (Neale et al., 2012; O'Roak et al., 2012).
Recently, mutations in BCL11A that encodes BAF100a subunit were also linked to autism. Three patients with microdeletions of 2p15–p16.1 who presented with developmental delay also showed common features of ASD, including moderate to severe ID, facial dysmorphism, and hypotonia (Basak et al., 2015). Although the 2p15–p16.1 microdeletion encompasses ~15 protein-coding genes and six pseudogenes, BCL11A is the only common candidate gene in three reported patients with ASD (Basak et al., 2015).
Schizophrenia
Schizophrenia (SZ; OMIM #181500) is a chronic, severe, and disabling brain disorder that affects ~0.3–0.7% of the world's population. It is characterized by delusion, thought disorder, auditory hallucination, reduced emotional expression and social engagement, disorganized speech, and lacking motivation. The causes of SZ include genetic and/or environmental factors (Zai et al., 2017). In addition to genetic mutations, epigenetic alteration has also been shown to be involved in SZ (Tsankova et al., 2007). BRM was associated with SZ in a screening study of Japanese population (Koga et al., 2009). Mechanistically, the risk alleles of intronic polymorphisms (rs3763627 and rs3793490) were correlated with decreased expression of BRM in post-mortem prefrontal cortex (Koga et al., 2009). Another risk allele, a missense polymorphism (rs2296212, D1546E), located in a highly conserved functional region in mammalian species, reduces the nuclear localization of BRM (Koga et al., 2009). BRM knockout mice exhibit impaired social interaction and pre-pulse inhibition, which suggest that the impaired functions of BRM might play a role in SZ. In cultured mouse primary cortical neurons, knockdown of BRM resulted in impaired dendritic spine growth and abnormal morphology, which is considered an intermediate phenotype of SZ (Bellon, 2007; Lewis and Gonzalez-Burgos, 2007).
In addition to BRM, intronic SNPs in BCL11A were significantly associated with schizophrenic patients (Basak et al., 2015). Further studies will be necessary to elucidate the influence of the intronic SNPs on BCL11A expression and function.
Hirschsprung's Disease (HSCR)
HSCR is a congenital intestinal disorder characterized by defective migration of enteric neural cells. Recently, a de novo heterozygous frameshift mutation in exon 20 of BAF250b (c.5789delC p.Pro1930Leufs*44) was identified in a patient with HSCR symptoms (Takenouchi et al., 2016). The patient also displayed some CSS features, including coarse facial features, impaired development, epilepsy, and hypoplasia of the corpus callosum and cerebellum. In addition, the patient exhibited congenital cataract disorder and defects in enteric nervous system, which are the cardinal pathologic characteristics of HSCR (Takenouchi et al., 2016). Interestingly, studies in drosophila model confirmed that dysregulation of the ARID1 subunit leads to impaired enteric neural cell migration (Eroglu et al., 2014).
Conclusion and Future Perspectives
It is becoming clear that the BAF complex plays an essential role in mammalian neural development through assembly of developmental stage-specific BAF complexes in NSCs, and post-mitotic neural cells. Therefore, it is not surprising that mutations in BAF subunits are frequently observed in neurological disorders. These disorders are often caused by mutations in many genes encoding BAF subunits, which reflect the fact that different BAF subunits function together to regulate chromatin dynamics and gene expression program. Some potential mechanisms underlying the BAF complex mutations, which cause neurodevelopmental disorders such as GOF or dominant-negative effects, have been proposed. Nevertheless, how these mutations lead to specific neurological phenotypes in patients remains largely unknown. Moreover, most patients with BAF subunit mutations present vast variation of neurological impairments, but the underlining pathological mechanisms still remain an open question. The use of cellular models (e.g., human induced pluripotent stem cells [hiPSCs], cultured brain organoids) and animal models, coupled with new methodologies to generate site-specific editing (e.g., the clustered regularly interspaced short palindromic repeats [CRISPR]/CRISPR-associated protein-9 [Cas9] system) will help to address some of these unsolved questions in the near future (Figure 3). In addition, further studies in structural biology will be necessary to help us understand how the mutations lead to conformational changes of BAF subunits, as well as the deformation of proper functional BAF complexes.
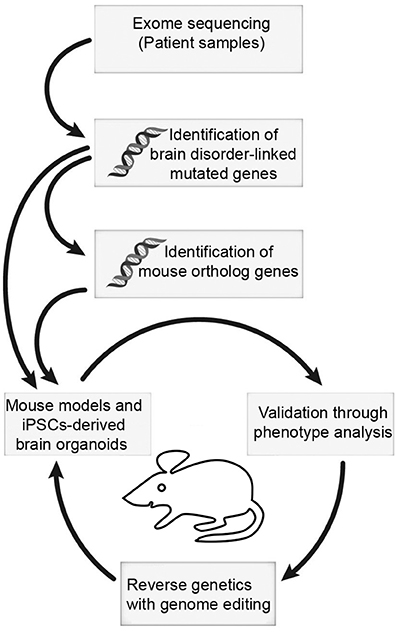
Figure 3. The mouse and iPSC-derived mini-brain organoid pipeline for modeling human brain disorders. Candidates of mutations involved in brain disorders are identified using exome sequencing with DNA samples from patients. When mouse orthologs of candidate genes are identified, they can be edited. Subsequently, phenotypic studies are used to assess validity of the model. Upon validation, reverse genetics approach with genome editing will be applied to restore phenotypes.
Author Contributions
GS, YX, JR, and TT all contributed to writing and editing the manuscript.
Funding
This work was supported by the Research Program at the Faculty of Medicine, Georg-August University Göttingen, TU432/1-1, TU432/1-3 DFG grants, DFG-CNMPB and Schram-Stiftung to TT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer IP and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We apologize to colleagues whose work were not included in this review owing to space limitation or unintentional oversight. We thank J. Staiger for his support, and A. Steixner for preparing illustrations. We acknowledge support by the Open Access Publication Funds of the Göttingen University.
References
Aizawa, H., Hu, S. C., Bobb, K., Balakrishnan, K., Ince, G., Gurevich, I., et al. (2004). Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303, 197–202. doi: 10.1126/science.1089845
Alcamo, E. A., Chirivella, L., Dautzenberg, M., Dobreva, G., Farinas, I., Grosschedl, R., et al. (2008). Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 57, 364–377. doi: 10.1016/j.neuron.2007.12.012
Alexander, J. M., Hota, S. K., He, D., Thomas, S., Ho, L., Pennacchio, L. A., et al. (2015). Brg1 modulates enhancer activation in mesoderm lineage commitment. Development 142, 1418–1430. doi: 10.1242/dev.109496
Alver, B. H., Kim, K. H., Lu, P., Wang, X., Manchester, H. E., Wang, W., et al. (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8:14648. doi: 10.1038/ncomms14648
Arlotta, P., Molyneaux, B. J., Chen, J., Inoue, J., Kominami, R., and Macklis, J. D. (2005). Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45, 207–221. doi: 10.1016/j.neuron.2004.12.036
Arlotta, P., Molyneaux, B. J., Jabaudon, D., Yoshida, Y., and Macklis, J. D. (2008). Ctip2 controls the differentiation of medium spiny neurons and the establishment of the cellular architecture of the striatum. J. Neurosci. 28, 622–632. doi: 10.1523/JNEUROSCI.2986-07.2008
Azim, E., Shnider, S. J., Cederquist, G. Y., Sohur, U. S., and Macklis, J. D. (2009). Lmo4 and Clim1 progressively delineate cortical projection neuron subtypes during development. Cereb. Cortex 19(Suppl. 1), i62–i69. doi: 10.1093/cercor/bhp030
Bachmann, C., Nguyen, H., Rosenbusch, J., Pham, L., Rabe, T., Patwa, M., et al. (2016). mSWI/SNF (BAF) complexes are indispensable for the neurogenesis and development of embryonic olfactory epithelium. PLoS Genet. 12:e1006274. doi: 10.1371/journal.pgen.1006274
Backx, L., Seuntjens, E., Devriendt, K., Vermeesch, J., and Van Esch, H. (2011). A balanced translocation t(6;14)(q25.3;q13.2) leading to reciprocal fusion transcripts in a patient with intellectual disability and agenesis of corpus callosum. Cytogenet. Genome Res. 132, 135–143. doi: 10.1159/000321577
Bagheri, H., Badduke, C., Qiao, Y., Colnaghi, R., Abramowicz, I., Alcantara, D., et al. (2016). Identifying candidate genes for 2p15p16.1 microdeletion syndrome using clinical, genomic, and functional analysis. JCI Insight 1:e85461. doi: 10.1172/jci.insight.85461
Barker, N., Hurlstone, A., Musisi, H., Miles, A., Bienz, M., and Clevers, H. (2001). The chromatin remodelling factor Brg-1 interacts with beta-catenin to promote target gene activation. EMBO J. 20, 4935–4943. doi: 10.1093/emboj/20.17.4935
Barutcu, A. R., Lajoie, B. R., Fritz, A. J., McCord, R. P., Nickerson, J. A., van Wijnen, A. J., et al. (2016). SMARCA4 regulates gene expression and higher-order chromatin structure in proliferating mammary epithelial cells. Genome Res. 26, 1188–1201. doi: 10.1101/gr.201624.115
Basak, A., Hancarova, M., Ulirsch, J. C., Balci, T. B., Trkova, M., Pelisek, M., et al. (2015). BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J. Clin. Invest. 125, 2363–2368. doi: 10.1172/JCI81163
Bedogni, F., Hodge, R. D., Elsen, G. E., Nelson, B. R., Daza, R. A., Beyer, R. P., et al. (2010). Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl. Acad. Sci. U.S.A. 107, 13129–13134. doi: 10.1073/pnas.1002285107
Bellon, A. (2007). New genes associated with schizophrenia in neurite formation: a review of cell culture experiments. Mol. Psychiatry 12, 620–629. doi: 10.1038/sj.mp.4001985
Bischof, M., Weider, M., Kuspert, M., Nave, K. A., and Wegner, M. (2015). Brg1-dependent chromatin remodelling is not essentially required during oligodendroglial differentiation. J. Neurosci. 35, 21–35. doi: 10.1523/JNEUROSCI.1468-14.2015
Borrell, V., and Gotz, M. (2014). Role of radial glial cells in cerebral cortex folding. Curr. Opin. Neurobiol. 27, 39–46. doi: 10.1016/j.conb.2014.02.007
Bossen, C., Murre, C. S., Chang, A. N., Mansson, R., Rodewald, H. R., and Murre, C. (2015). The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat. Immunol. 16, 775–784. doi: 10.1038/ni.3170
Boyer, L. A., Lee, T. I., Cole, M. F., Johnstone, S. E., Levine, S. S., Zucker, J. P., et al. (2005). Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956. doi: 10.1016/j.cell.2005.08.020
Bramswig, N. C., Caluseriu, O., Lüdecke, H.-J., Bolduc, F. V., Noel, N. C. L., Wieland, T., et al. (2017). Heterozygosity for ARID2 loss-of-function mutations in individuals with a Coffin–Siris syndrome-like phenotype. Hum. Genet. 136, 297–305. doi: 10.1007/s00439-017-1757-z
Bramswig, N. C., Ludecke, H. J., Alanay, Y., Albrecht, B., Barthelmie, A., Boduroglu, K., et al. (2015). Exome sequencing unravels unexpected differential diagnoses in individuals with the tentative diagnosis of Coffin-Siris and Nicolaides-Baraitser syndromes. Hum. Genet. 134, 553–568. doi: 10.1007/s00439-015-1535-8
Bultman, S., Gebuhr, T., Yee, D., La Mantia, C., Nicholson, J., Gilliam, A., et al. (2000). A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 6, 1287–1295. doi: 10.1016/S1097-2765(00)00127-1
Cairns, B. R. (1998). Chromatin remodeling machines: similar motors, ulterior motives. Trends Biochem. Sci. 23, 20–25. doi: 10.1016/S0968-0004(97)01160-2
Chandler, R. L., Brennan, J., Schisler, J. C., Serber, D., Patterson, C., and Magnuson, T. (2013). ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol. Cell. Biol. 33, 265–280. doi: 10.1128/MCB.01008-12
Chaste, P., and Leboyer, M. (2012). Autism risk factors: genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 14, 281–292.
Chen, B., Wang, S. S., Hattox, A. M., Rayburn, H., Nelson, S. B., and McConnell, S. K. (2008). The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 105, 11382–11387. doi: 10.1073/pnas.0804918105
Choi, K. Y., Yoo, M., and Han, J. H. (2015). Toward understanding the role of the neuron-specific BAF chromatin remodeling complex in memory formation. Exp. Mol. Med. 47:e155. doi: 10.1038/emm.2014.129
Christ, L. A., Crowe, C. A., Micale, M. A., Conroy, J. M., and Schwartz, S. (1999). Chromosome breakage hotspots and delineation of the critical region for the 9p-deletion syndrome. Am. J. Hum. Genet. 65, 1387–1395. doi: 10.1086/302606
Conaco, C., Otto, S., Han, J. J., and Mandel, G. (2006). Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl. Acad. Sci. U.S.A. 103, 2422–2427. doi: 10.1073/pnas.0511041103
Coskun, V., Tsoa, R., and Sun, Y. E. (2012). Epigenetic regulation of stem cells differentiating along the neural lineage. Curr. Opin. Neurobiol. 22, 762–767. doi: 10.1016/j.conb.2012.07.001
Custo Greig, L. F., Woodworth, M. B., Galazo, M. J., Padmanabhan, H., and Macklis, J. D. (2013). Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 14, 755–769. doi: 10.1038/nrn3586
Day, J. J., and Sweatt, J. D. (2010). DNA methylation and memory formation. Nat. Neurosci. 13, 1319–1323. doi: 10.1038/nn.2666
de Bruijn, D. R., Peters, W. J., Chuva de Sousa Lopes, S. M., van Dijk, A. H., Willemse, M. P., Pfundt, R., et al. (2006). Targeted disruption of the synovial sarcoma-associated SS18 gene causes early embryonic lethality and affects PPARBP expression. Hum. Mol. Genet. 15, 2936–2944. doi: 10.1093/hmg/ddl235
Deng, W., Aimone, J. B., and Gage, F. H. (2010). New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 11, 339–350. doi: 10.1038/nrn2822
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional, and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
Ejaz, R., Babul-Hirji, R., and Chitayat, D. (2016). The evolving features of Nicolaides–Baraitser syndrome – a clinical report of a 20-year follow-up. Clin. Case Rep. 4, 351–355. doi: 10.1002/ccr3.425
Enomoto, T., Ohmoto, M., Iwata, T., Uno, A., Saitou, M., Yamaguchi, T., et al. (2011). Bcl11b/Ctip2 controls the differentiation of vomeronasal sensory neurons in mice. J. Neurosci. 31, 10159–10173. doi: 10.1523/JNEUROSCI.1245-11.2011
Eroglu, E., Burkard, T. R., Jiang, Y., Saini, N., Homem, C. C., Reichert, H., et al. (2014). SWI/SNF complex prevents lineage reversion and induces temporal patterning in neural stem cells. Cell 156, 1259–1273. doi: 10.1016/j.cell.2014.01.053
Estruch, S. B., Buzon, V., Carbo, L. R., Schorova, L., Luders, J., and Estebanez-Perpina, E. (2012). The oncoprotein BCL11A binds to orphan nuclear receptor TLX and potentiates its transrepressive function. PLoS ONE 7:e37963. doi: 10.1371/journal.pone.0037963
Evsyukova, I., Plestant, C., and Anton, E. S. (2013). integrative mechanisms of oriented neuronal migration in the developing brain. Annu. Rev. Cell Dev. Biol. 29, 299–353. doi: 10.1146/annurev-cellbio-101512-122400
Fischer, A. (2014). Epigenetic memory: the Lamarckian brain. EMBO J. 33, 945–967. doi: 10.1002/embj.201387637
Freeman, M. R. (2010). Specification and morphogenesis of astrocytes. Science 330, 774–778. doi: 10.1126/science.1190928
Gallo, V., and Deneen, B. (2014). Glial development: the crossroads of regeneration and repair in the CNS. Neuron 83, 283–308. doi: 10.1016/j.neuron.2014.06.010
Goldberg, A. D., Allis, C. D., and Bernstein, E. (2007). Epigenetics: a landscape takes shape. Cell 128, 635–638. doi: 10.1016/j.cell.2007.02.006
Goncalves, J. T., Schafer, S. T., and Gage, F. H. (2016). Adult neurogenesis in the hippocampus: from stem cells to behavior. Cell 167, 897–914. doi: 10.1016/j.cell.2016.10.021
Gong, F., Fahy, D., and Smerdon, M. J. (2006). Rad4-Rad23 interaction with SWI/SNF links ATP-dependent chromatin remodeling with nucleotide excision repair. Nat. Struct. Mol. Biol. 13, 902–907. doi: 10.1038/nsmb1152
Gossai, N., Biegel, J. A., Messiaen, L., Berry, S. A., and Moertel, C. L. (2015). Report of a patient with a constitutional missense mutation in SMARCB1, Coffin–Siris phenotype, and schwannomatosis. Am. J. Med. Genet. A 167, 3186–3191. doi: 10.1002/ajmg.a.37356
Guillemot, F. (2007). Cell fate specification in the mammalian telencephalon. Prog. Neurobiol. 83, 37–52. doi: 10.1016/j.pneurobio.2007.02.009
Gutiérrez, J., Paredes, R., Cruzat, F., Hill, D. A., van Wijnen, A. J., Lian, J. B., et al. (2007). Chromatin remodeling by SWI/SNF results in nucleosome mobilization to preferential positions in the rat osteocalcin gene promoter. J. Biol. Chem. 282, 9445–9457. doi: 10.1074/jbc.M609847200
Halgren, C., Kjaergaard, S., Bak, M., Hansen, C., El-Schich, Z., Anderson, C. M., et al. (2012). Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin. Genet. 82, 248–255. doi: 10.1111/j.1399-0004.2011.01755.x
Han, W., Kwan, K. Y., Shim, S., Lam, M. M., Shin, Y., Xu, X., et al. (2011). TBR1 directly represses Fezf2 to control the laminar origin and development of the corticospinal tract. Proc. Natl. Acad. Sci. U.S.A. 108, 3041–3046. doi: 10.1073/pnas.1016723108
Hansen, D. V., Lui, J. H., Parker, P. R., and Kriegstein, A. R. (2010). Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561. doi: 10.1038/nature08845
Hara, R., and Sancar, A. (2002). The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol. Cell. Biol. 22, 6779–6787. doi: 10.1128/MCB.22.19.6779-6787.2002
Harb, K., Magrinelli, E., Nicolas, C. S., Lukianets, N., Frangeul, L., Pietri, M., et al. (2016). Area-specific development of distinct projection neuron subclasses is regulated by postnatal epigenetic modifications. Elife 5:e09531. doi: 10.7554/eLife.09531
Haubensak, W., Attardo, A., Denk, W., and Huttner, W. B. (2004). Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 101, 3196–3201. doi: 10.1073/pnas.0308600100
Hirabayashi, Y., and Gotoh, Y. (2010). Epigenetic control of neural precursor cell fate during development. Nat. Rev. Neurosci. 11, 377–388. doi: 10.1038/nrn2810
Hirabayashi, Y., Suzki, N., Tsuboi, M., Endo, T. A., Toyoda, T., Shinga, J., et al. (2009). Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63, 600–613. doi: 10.1016/j.neuron.2009.08.021
Ho, L., Jothi, R., Ronan, J. L., Cui, K., Zhao, K., and Crabtree, G. R. (2009a). An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc. Natl. Acad. Sci. U.S.A. 106, 5187–5191. doi: 10.1073/pnas.0812888106
Ho, L., Miller, E. L., Ronan, J. L., Ho, W. Q., Jothi, R., and Crabtree, G. R. (2011). esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat. Cell Biol. 13, 903–913. doi: 10.1038/ncb2285
Ho, L., Ronan, J. L., Wu, J., Staahl, B. T., Chen, L., Kuo, A., et al. (2009b). An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. U.S.A. 106, 5181–5186. doi: 10.1073/pnas.0812889106
Hoyer, J., Ekici, A. B., Endele, S., Popp, B., Zweier, C., Wiesener, A., et al. (2012). Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 90, 565–572. doi: 10.1016/j.ajhg.2012.02.007
Hu, G., Schones, D. E., Cui, K., Ybarra, R., Northrup, D., Tang, Q., et al. (2011). Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res. 21, 1650–1658. doi: 10.1101/gr.121145.111
John, A., Brylka, H., Wiegreffe, C., Simon, R., Liu, P., Juttner, R., et al. (2012). Bcl11a is required for neuronal morphogenesis and sensory circuit formation in dorsal spinal cord development. Development 139, 1831–1841. doi: 10.1242/dev.072850
Juliandi, B., Abematsu, M., and Nakashima, K. (2010). Chromatin remodeling in neural stem cell differentiation. Curr. Opin. Neurobiol. 20, 408–415. doi: 10.1016/j.conb.2010.04.001
Kadoch, C., Hargreaves, D. C., Hodges, C., Elias, L., Ho, L., Ranish, J., et al. (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45, 592–601. doi: 10.1038/ng.2628
Kadoch, C., Williams, R. T., Calarco, J. P., Miller, E. L., Weber, C. M., Braun, S. M., et al. (2017). Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 49, 213–222. doi: 10.1038/ng.3734
Kaeser, M. D., Aslanian, A., Dong, M. Q., Yates, J. R. III., and Emerson, B. M. (2008). BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol. Chem. 283, 32254–32263. doi: 10.1074/jbc.M806061200
Kidder, B. L., Palmer, S., and Knott, J. G. (2009). SWI/SNF-Brg1 regulates self-renewal and occupies core pluripotency-related genes in embryonic stem cells. Stem Cells 27, 317–328. doi: 10.1634/stemcells.2008-0710
Kim, J. K., Huh, S. O., Choi, H., Lee, K. S., Shin, D., Lee, C., et al. (2001). Srg3, a mouse homolog of yeast SWI3, is essential for early embryogenesis and involved in brain development. Mol. Cell. Biol. 21, 7787–7795. doi: 10.1128/MCB.21.22.7787-7795.2001
Kleefstra, T., Brunner, H. G., Amiel, J., Oudakker, A. R., Nillesen, W. M., Magee, A., et al. (2006). Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 79, 370–377. doi: 10.1086/505693
Kleefstra, T., Kramer, J. M., Neveling, K., Willemsen, M. H., Koemans, T. S., Vissers, L. E., et al. (2012). Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am. J. Hum. Genet. 91, 73–82. doi: 10.1016/j.ajhg.2012.05.003
Kleefstra, T., van Zelst-Stams, W. A., Nillesen, W. M., Cormier-Daire, V., Houge, G., Foulds, N., et al. (2009). Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J. Med. Genet. 46, 598–606. doi: 10.1136/jmg.2008.062950
Koga, M., Ishiguro, H., Yazaki, S., Horiuchi, Y., Arai, M., Niizato, K., et al. (2009). Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum. Mol. Genet. 18, 2483–2494. doi: 10.1093/hmg/ddp166
Kosho, T., Miyake, N., and Carey, J. C. (2014a). Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 241–251. doi: 10.1002/ajmg.c.31415
Kosho, T., Okamoto, N., and Coffin-Siris Syndrome International, C. (2014b). Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am. J. Med. Genet. C Sem. Med. Genet. 166, 262–275. doi: 10.1002/ajmg.c.31407
Kosho, T., Okamoto, N., Ohashi, H., Tsurusaki, Y., Imai, Y., Hibi-Ko, Y., et al. (2013). Clinical correlations of mutations affecting six components of the SWI/SNF complex: detailed description of 21 patients and a review of the literature. Am. Jo. Med. Genet. A 161, 1221–1237. doi: 10.1002/ajmg.a.35933
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Kriegstein, A., and Alvarez-Buylla, A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184. doi: 10.1146/annurev.neuro.051508.135600
Kuo, T. Y., Chen, C. Y., and Hsueh, Y. P. (2010a). Bcl11A/CTIP1 mediates the effect of the glutamate receptor on axon branching and dendrite outgrowth. J. Neurochem. 114, 1381–1392. doi: 10.1111/j.1471-4159.2010.06852.x
Kuo, T. Y., Hong, C. J., Chien, H. L., and Hsueh, Y. P. (2010b). X-linked mental retardation gene CASK interacts with Bcl11A/CTIP1 and regulates axon branching and outgrowth. J. Neurosci. Res. 88, 2364–2373. doi: 10.1002/jnr.22407
Kwan, K. Y., Lam, M. M., Krsnik, Z., Kawasawa, Y. I., Lefebvre, V., and Sestan, N. (2008). SOX5 postmitotically regulates migration, postmigratory differentiation, and projections of subplate and deep-layer neocortical neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 16021–16026. doi: 10.1073/pnas.0806791105
Lai, T., Jabaudon, D., Molyneaux, B. J., Azim, E., Arlotta, P., Menezes, J. R., et al. (2008). SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron 57, 232–247. doi: 10.1016/j.neuron.2007.12.023
Leid, M., Ishmael, J. E., Avram, D., Shepherd, D., Fraulob, V., and Dolle, P. (2004). CTIP1 and CTIP2 are differentially expressed during mouse embryogenesis. Gene Expr. Patterns 4, 733–739. doi: 10.1016/j.modgep.2004.03.009
Lennon, M. J., Jones, S. P., Lovelace, M. D., Guillemin, G. J., and Brew, B. J. (2017). Bcl11b-a critical neurodevelopmental transcription factor-roles in health and disease. Front. Cell. Neurosci. 11:89. doi: 10.3389/fncel.2017.00089
Lessard, J., Wu, J. I., Ranish, J. A., Wan, M., Winslow, M. M., Staahl, B. T., et al. (2007). An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55, 201–215. doi: 10.1016/j.neuron.2007.06.019
Levy, P., and Baraitser, M. (1991). Coffin-Siris syndrome. J. Med. Genet. 28, 338–341. doi: 10.1136/jmg.28.5.338
Lewis, D. A., and Gonzalez-Burgos, G. (2007). Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology 33, 141–165. doi: 10.1038/sj.npp.1301563
Li, W., Xiong, Y., Shang, C., Twu, K. Y., Hang, C. T., Yang, J., et al. (2013). Brg1 governs distinct pathways to direct multiple aspects of mammalian neural crest cell development. Proc. Natl. Acad. Sci. U.S.A. 110, 1738–1743. doi: 10.1073/pnas.1218072110
Limpert, A. S., Bai, S., Narayan, M., Wu, J., Yoon, S. O., Carter, B. D., et al. (2013). NF-kappaB forms a complex with the chromatin remodeler BRG1 to regulate Schwann cell differentiation. J. Neurosci. 33, 2388–2397. doi: 10.1523/JNEUROSCI.3223-12.2013
Loe-Mie, Y., Lepagnol-Bestel, A.-M., Maussion, G., Doron-Faigenboim, A., Imbeaud, S., Delacroix, H., et al. (2010). SMARCA2 and other genome-wide supported schizophrenia-associated genes: regulation by REST/NRSF, network organization and primate-specific evolution. Hum. Mol. Genet. 19, 2841–2857. doi: 10.1093/hmg/ddq184
Magnani, L., and Cabot, R. A. (2009). Manipulation of SMARCA2 and SMARCA4 transcript levels in porcine embryos differentially alters development and expression of SMARCA1, SOX2, NANOG, and EIF1. Reproduction 137, 23–33. doi: 10.1530/REP-08-0335
Mari, F., Marozza, A., Mencarelli, M. A., Lo Rizzo, C., Fallerini, C., Dosa, L., et al. (2015). Coffin-Siris and Nicolaides-Baraitser syndromes are a common well recognizable cause of intellectual disability. Brain Dev. 37, 527–536. doi: 10.1016/j.braindev.2014.08.009
Matsumoto, S., Banine, F., Struve, J., Xing, R., Adams, C., Liu, Y., et al. (2006). Brg1 is required for murine neural stem cell maintenance and gliogenesis. Dev. Biol. 289, 372–383. doi: 10.1016/j.ydbio.2005.10.044
Michelson, M., Ben-Sasson, A., Vinkler, C., Leshinsky-Silver, E., Netzer, I., Frumkin, A., et al. (2012). Delineation of the interstitial 6q25 microdeletion syndrome: refinement of the critical causative region. Am. J. Med. Genet. A 158A, 1395–1399. doi: 10.1002/ajmg.a.35361
Mignot, C., Moutard, M. L., Rastetter, A., Boutaud, L., Heide, S., Billette, T., et al. (2016). ARID1B mutations are the major genetic cause of corpus callosum anomalies in patients with intellectual disability. Brain. 139:e64. doi: 10.1093/brain/aww181
Ming, G. L., and Song, H. (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702. doi: 10.1016/j.neuron.2011.05.001
Miyake, N., Abdel-Salam, G., Yamagata, T., Eid, M. M., Osaka, H., Okamoto, N., et al. (2016). Clinical features of SMARCA2 duplication overlap with Coffin–Siris syndrome. Am. J. Med. Genet. A 170, 2662–2670. doi: 10.1002/ajmg.a.37778
Miyata, T., Kawaguchi, A., Saito, K., Kawano, M., Muto, T., and Ogawa, M. (2004). Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development 131, 3133–3145. doi: 10.1242/dev.01173
Molyneaux, B. J., Arlotta, P., Hirata, T., Hibi, M., and Macklis, J. D. (2005). Fezl is required for the birth and specification of corticospinal motor neurons. Neuron 47, 817–831. doi: 10.1016/j.neuron.2005.08.030
Morin, G., Villemain, L., Baumann, C., Mathieu, M., Blanc, N., and Verloes, A. (2003). Nicolaides–Baraitser syndrome: confirmatory report of a syndrome with sparse hair, mental retardation, and short stature and metacarpals. Clin. Dysmorphol. 12, 237–240. doi: 10.1097/00019605-200310000-00005
Narayanan, R., Pirouz, M., Kerimoglu, C., Pham, L., Wagener, R. J., Kiszka, K. A., et al. (2015). Loss of BAF (mSWI/SNF) complexes causes global transcriptional and chromatin state changes in forebrain development. Cell Rep. 13, 1842–1854. doi: 10.1016/j.celrep.2015.10.046
Narayanan, R., and Tuoc, T. C. (2014). Roles of chromatin remodeling BAF complex in neural differentiation and reprogramming. Cell Tissue Res. 356, 575–584. doi: 10.1007/s00441-013-1791-7
Neale, B. M., Kou, Y., Liu, L., Ma'ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Neigeborn, L., and Carlson, M. (1984). Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics 108, 845–858.
Nguyen, H., Sokpor, G., Pham, L., Rosenbusch, J., Stoykova, A., Staiger, J. F., et al. (2016). Epigenetic regulation by BAF (mSWI/SNF) chromatin remodeling complexes is indispensable for embryonic development. Cell Cycle 15, 1317–1324. doi: 10.1080/15384101.2016.1160984
Ninkovic, J., Steiner-Mezzadri, A., Jawerka, M., Akinci, U., Masserdotti, G., Petricca, S., et al. (2013). The BAF complex interacts with Pax6 in adult neural progenitors to establish a neurogenic cross-regulatory transcriptional network. Cell Stem Cell. 13, 403–418. doi: 10.1016/j.stem.2013.07.002
Noctor, S. C., Martinez-Cerdeno, V., Ivic, L., and Kriegstein, A. R. (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 7, 136–144. doi: 10.1038/nn1172
Nord, A. S., Roeb, W., Dickel, D. E., Walsh, T., Kusenda, M., O'Connor, K. L., et al. (2011). Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet. 19, 727–731. doi: 10.1038/ejhg.2011.24
Olave, I., Wang, W., Xue, Y., Kuo, A., and Crabtree, G. R. (2002). Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes Dev. 16, 2509–2517. doi: 10.1101/gad.992102
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Parrish, J. Z., Kim, M. D., Jan, L. Y., and Jan, Y. N. (2006). Genome-wide analyses identify transcription factors required for proper morphogenesis of Drosophila sensory neuron dendrites. Genes Dev. 20, 820–835. doi: 10.1101/gad.1391006
Petrik, D., Latchney, S. E., Masiulis, I., Yun, S., Zhang, Z., Wu, J. I., et al. (2015). Chromatin remodeling factor brg1 supports the early maintenance and late responsiveness of nestin-lineage adult neural stem and progenitor cells. Stem Cells 33, 3655–3665. doi: 10.1002/stem.2215
Phelan, M. L., Sif, S., Narlikar, G. J., and Kingston, R. E. (1999). Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 3, 247–253. doi: 10.1016/S1097-2765(00)80315-9
Pirola, B., Bortotto, L., Giglio, S., Piovan, E., Janes, A., Guerrini, R., et al. (1998). Agenesis of the corpus callosum with Probst bundles owing to haploinsufficiency for a gene in an 8 cM region of 6q25. J. Med. Genet. 35, 1031–1033. doi: 10.1136/jmg.35.12.1031
Qiu, Z., and Ghosh, A. (2008). A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron 60, 775–787. doi: 10.1016/j.neuron.2008.09.040
Ronan, J. L., Wu, W., and Crabtree, G. R. (2013). From neural development to cognition: unexpected roles for chromatin. Nat. Rev. Genet. 14, 347–359. doi: 10.1038/nrg3413
Ronzoni, L., Tagliaferri, F., Tucci, A., Baccarin, M., Esposito, S., and Milani, D. (2016). Interstitial 6q25 microdeletion syndrome: ARID1B is the key gene. Am. J. Med. Genet. A 170, 1257–1261. doi: 10.1002/ajmg.a.37553
Rowitch, D. H., and Kriegstein, A. R. (2010). Developmental genetics of vertebrate glial-cell specification. Nature 468, 214–222. doi: 10.1038/nature09611
Saha, A., Wittmeyer, J., and Cairns, B. R. (2002). Chromatin remodeling by RSC involves ATP-dependent DNA translocation. Genes Dev. 16, 2120–2134. doi: 10.1101/gad.995002
Santen, G. W. E., Aten, E., Sun, Y., Almomani, R., Gilissen, C., Nielsen, M., et al. (2012). Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 44, 379–380. doi: 10.1038/ng.2217
Santen, G. W. E., Aten, E., Vulto-van Silfhout, A. T., Pottinger, C., van Bon, B. W. M., van Minderhout, I. J. H. M., et al. (2013). Coffin–siris syndrome and the BAF complex: genotype–phenotype study in 63 patients. Hum. Mutat. 34, 1519–1528. doi: 10.1002/humu.22394
Santen, G. W., Clayton-Smith, J., and ARID1B-CSS consortium. (2014). The ARID1B phenotype: what we have learned so far. Am. J. Med. Genet. C Semin. Med. Genet. 166, 276–289. doi: 10.1002/ajmg.c.31414
Sawa, H., Kouike, H., and Okano, H. (2000). Components of the SWI/SNF complex are required for asymmetric cell division in C. elegans. Mol. Cell 6, 617–624. doi: 10.1016/S1097-2765(00)00060-5
Schrier, S. A., Bodurtha, J. N., Burton, B., Chudley, A. E., Chiong, M. A. D., D'avanzo, M. G., et al. (2012). The coffin-siris syndrome: a proposed diagnostic approach and assessment of 15 overlapping cases. Am. J. Med. Genet. A 158A, 1865–1876. doi: 10.1002/ajmg.a.35415
Shang, L., Cho, M. T., Retterer, K., Folk, L., Humberson, J., Rohena, L., et al. (2015). Mutations in ARID2 are associated with intellectual disabilities. Neurogenetics 16, 307–314. doi: 10.1007/s10048-015-0454-0
Shim, S., Kwan, K. Y., Li, M., Lefebvre, V., and Sestan, N. (2012). Cis-regulatory control of corticospinal system development and evolution. Nature 486, 74–79. doi: 10.1038/nature11094
Sim, J. C. H., White, S. M., and Lockhart, P. J. (2015). ARID1B-mediated disorders: mutations and possible mechanisms. Intract. Rare Dis. Res. 4, 17–23. doi: 10.5582/irdr.2014.01021
Simon, R., Baumann, L., Fischer, J., Seigfried, F. A., De Bruyckere, E., Liu, P., et al. (2016). Structure-function integrity of the adult hippocampus depends on the transcription factor Bcl11b/Ctip2. Genes Brain Behav. 15, 405–419. doi: 10.1111/gbb.12287
Simon, R., Brylka, H., Schwegler, H., Venkataramanappa, S., Andratschke, J., Wiegreffe, C., et al. (2012). A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. EMBO J. 31, 2922–2936. doi: 10.1038/emboj.2012.142
Smith, J. A., Holden, K. R., Friez, M. J., Jones, J. R., and Lyons, M. J. (2016). A novel familial autosomal dominant mutation in ARID1B causing neurodevelopmental delays, short stature, and dysmorphic features. Am. J. Med. Geneti. A 170, 3313–3318. doi: 10.1002/ajmg.a.37945
Smith-Roe, S. L., and Bultman, S. J. (2013). Combined gene dosage requirement for SWI/SNF catalytic subunits during early mammalian development. Mamm. Genome 24, 21–29. doi: 10.1007/s00335-012-9433-z
Son, E. Y., and Crabtree, G. R. (2014). The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 333–349. doi: 10.1002/ajmg.c.31416
Soontornniyomkij, V., Risbrough, V. B., Young, J. W., Soontornniyomkij, B., Jeste, D. V., and Achim, C. L. (2012). Hippocampal calbindin-1 immunoreactivity correlate of recognition memory performance in aged mice. Neurosci. Lett. 516, 161–165. doi: 10.1016/j.neulet.2012.03.092
Sousa, S. B., Abdul-Rahman, O. A., Bottani, A., Cormier-Daire, V., Fryer, A., Gillessen-Kaesbach, G., et al. (2009). Nicolaides–Baraitser syndrome: delineation of the phenotype. Am. J. Med. Genet. A 149A, 1628–1640. doi: 10.1002/ajmg.a.32956
Sousa, S. B., Hennekam, R. C., and the Nicolaides–Baraitser Syndrome International Consortium (2014). Phenotype and genotype in Nicolaides–Baraitser syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 166, 302–314. doi: 10.1002/ajmg.c.31409
Staahl, B. T., Tang, J., Wu, W., Sun, A., Gitler, A. D., Yoo, A. S., et al. (2013). Kinetic analysis of npBAF to nBAF switching reveals exchange of SS18 with CREST and integration with neural developmental pathways. J. Neurosci. 33, 10348–10361. doi: 10.1523/JNEUROSCI.1258-13.2013
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi: 10.1038/47412
Takenouchi, T., Yoshihashi, H., Sakaguchi, Y., Uehara, T., Honda, M., Takahashi, T., et al. (2016). Hirschsprung disease as a yet undescribed phenotype in a patient with ARID1B mutation. Am. J. Med. Genet. A 170, 3249–3252. doi: 10.1002/ajmg.a.37861
Tang, L., Nogales, E., and Ciferri, C. (2010). Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog. Biophys. Mol. Biol. 102, 122–128. doi: 10.1016/j.pbiomolbio.2010.05.001
Tang, S., Hughes, E., Lascelles, K., EuroEPINOMICS RES Myoclonic Astatic Epilepsy Working Group, Simpson, M. A., and Pal, D. K. (2017). New SMARCA2 mutation in a patient with Nicolaides–Baraitser syndrome and myoclonic astatic epilepsy. Am. J. Med. Genet. A 173, 195–199. doi: 10.1002/ajmg.a.37935
Taverna, E., Gotz, M., and Huttner, W. B. (2014). The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu. Rev. Cell Dev. Biol. 30, 465–502. doi: 10.1146/annurev-cellbio-101011-155801
Tea, J. S., and Luo, L. (2011). The chromatin remodeling factor Bap55 functions through the TIP60 complex to regulate olfactory projection neuron dendrite targeting. Neural Dev. 6:5. doi: 10.1186/1749-8104-6-5
Tolstorukov, M. Y., Sansam, C. G., Lu, P., Koellhoffer, E. C., Helming, K. C., Alver, B. H., et al. (2013). Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc. Natl. Acad. Sci. U.S.A. 110, 10165–10170. doi: 10.1073/pnas.1302209110
Tsankova, N., Renthal, W., Kumar, A., and Nestler, E. J. (2007). Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8, 355–367. doi: 10.1038/nrn2132
Tsurusaki, Y., Okamoto, N., Ohashi, H., Kosho, T., Imai, Y., Hibi-Ko, Y., et al. (2012). Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 44, 376–378. doi: 10.1038/ng.2219
Tsurusaki, Y., Okamoto, N., Ohashi, H., Mizuno, S., Matsumoto, N., Makita, Y., et al. (2014). Coffin–Siris syndrome is a SWI/SNF complex disorder. Clin. Genet. 85, 548–554. doi: 10.1111/cge.12225
Tuoc, T. C., Boretius, S., Sansom, S. N., Pitulescu, M. E., Frahm, J., Livesey, F. J., et al. (2013a). Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev. Cell 25, 256–269. doi: 10.1016/j.devcel.2013.04.005
Tuoc, T. C., Narayanan, R., and Stoykova, A. (2013b). BAF chromatin remodeling complex: cortical size regulation and beyond. Cell Cycle 12, 2953–2959. doi: 10.4161/cc.25999
Tuoc, T. C., Pavlakis, E., Tylkowski, M. A., and Stoykova, A. (2014). Control of cerebral size and thickness. Cell. Mol. Life Sci. 71, 3199–3218. doi: 10.1007/s00018-014-1590-7
Tuoc, T., Dere, E., Radyushkin, K., Pham, L., Nguyen, H., Tonchev, A. B., et al. (2016). Ablation of BAF170 in developing and postnatal dentate gyrus affects neural stem cell proliferation, differentiation, and learning. Mol. Neurobiol. 54, 4618–4635. doi: 10.1007/s12035-016-9948-5
Urban, N., and Guillemot, F. (2014). Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front. Cell. Neurosci. 8:396. doi: 10.3389/fncel.2014.00396
Van Houdt, J. K. J., Nowakowska, B. A., Sousa, S. B., van Schaik, B. D. C., Seuntjens, E., Avonce, N., et al. (2012). Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat. Genet. 44, 445–449. doi: 10.1038/ng.1105
Vasileiou, G., Ekici, A. B., Uebe, S., Zweier, C., Hoyer, J., Engels, H., et al. (2015). Chromatin-remodeling-factor ARID1B represses Wnt/beta-catenin signaling. Am. J. Hum. Genet. 97, 445–456. doi: 10.1016/j.ajhg.2015.08.002
Vogel-Ciernia, A., Matheos, D. P., Barrett, R. M., Kramar, E. A., Azzawi, S., Chen, Y., et al. (2013). The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat. Neurosci. 16, 552–561. doi: 10.1038/nn.3359
Wang, W., Cote, J., Xue, Y., Zhou, S., Khavari, P. A., Biggar, S. R., et al. (1996). Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 15, 5370–5382.
Wang, X., Lee, R. S., Alver, B. H., Haswell, J. R., Wang, S., Mieczkowski, J., et al. (2017). SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 49, 289–295. doi: 10.1038/ng.3746
Wang, X., Nagl, N. G., Wilsker, D., Van Scoy, M., Pacchione, S., Yaciuk, P., et al. (2004). Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem. J. 383(Pt 2), 319–325. doi: 10.1042/bj20040524
Wang, X., Tsai, J. W., LaMonica, B., and Kriegstein, A. R. (2011). A new subtype of progenitor cell in the mouse embryonic neocortex. Nat. Neurosci. 14, 555–561. doi: 10.1038/nn.2807
Watson, L. A., and Tsai, L. H. (2016). In the loop: how chromatin topology links genome structure to function in mechanisms underlying learning and memory. Curr. Opin. Neurobiol. 43, 48–55. doi: 10.1016/j.conb.2016.12.002
Weider, M., Kuspert, M., Bischof, M., Vogl, M. R., Hornig, J., Loy, K., et al. (2012). Chromatin-remodeling factor Brg1 is required for Schwann cell differentiation and myelination. Dev. Cell 23, 193–201. doi: 10.1016/j.devcel.2012.05.017
Weinberg, P., Flames, N., Sawa, H., Garriga, G., and Hobert, O. (2013). The SWI/SNF chromatin remodeling complex selectively affects multiple aspects of serotonergic neuron differentiation. Genetics 194, 189–198. doi: 10.1534/genetics.112.148742
Whitehouse, I., Flaus, A., Cairns, B. R., White, M. F., Workman, J. L., and Owen-Hughes, T. (1999). Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature 400, 784–787. doi: 10.1038/23506
Whitford, K. L., Dijkhuizen, P., Polleux, F., and Ghosh, A. (2002). Molecular control of cortical dendrite development. Annu. Rev. Neurosci. 25, 127–149. doi: 10.1146/annurev.neuro.25.112701.142932
Wieczorek, D., Bögershausen, N., Beleggia, F., Steiner-Haldenstätt, S., Pohl, E., Li, Y., et al. (2013). A comprehensive molecular study on Coffin–Siris and Nicolaides–Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet. 22, 5121–5135. doi: 10.1093/hmg/ddt366
Wiegreffe, C., Simon, R., Peschkes, K., Kling, C., Strehle, M., Cheng, J., et al. (2015). Bcl11a (Ctip1) controls migration of cortical projection neurons through regulation of sema3c. Neuron 87, 311–325. doi: 10.1016/j.neuron.2015.06.023
Wilsker, D., Patsialou, A., Zumbrun, S. D., Kim, S., Chen, Y., Dallas, P. B., et al. (2004). The DNA-binding properties of the ARID-containing subunits of yeast and mammalian SWI/SNF complexes. Nucleic Acids Res. 32, 1345–1353. doi: 10.1093/nar/gkh277
Wolff, D., Endele, S., Azzarello-Burri, S., Hoyer, J., Zweier, M., Schanze, I., et al. (2012). In-frame deletion and missense mutations of the C-terminal helicase domain of SMARCA2 in three patients with nicolaides-baraitser syndrome. Mol. Syndromol. 2, 237–244. doi: 10.1159/000337323
Wong, F. K., Fei, J.-F., Mora-Bermúdez, F., Taverna, E., Haffner, C., Fu, J., et al. (2015). Sustained Pax6 expression generates primate-like basal radial glia in developing mouse neocortex. PLoS Biol. 13:e1002217. doi: 10.1371/journal.pbio.1002217
Woodworth, M. B., Greig, L. C., Liu, K. X., Ippolito, G. C., Tucker, H. O., and Macklis, J. D. (2016). Ctip1 regulates the balance between specification of distinct projection neuron subtypes in deep cortical layers. Cell Rep. 15, 999–1012. doi: 10.1016/j.celrep.2016.03.064
Wu, J. I., Lessard, J., Olave, I. A., Qiu, Z., Ghosh, A., Graef, I. A., et al. (2007). Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56, 94–108. doi: 10.1016/j.neuron.2007.08.021
Yao, B., Christian, K. M., He, C., Jin, P., Ming, G. L., and Song, H. (2016). Epigenetic mechanisms in neurogenesis. Nat. Rev. Neurosci. 17, 537–549. doi: 10.1038/nrn.2016.70
Yoo, A. S., and Crabtree, G. R. (2009). ATP-dependent chromatin remodeling in neural development. Curr. Opin. Neurobiol. 19, 120–126. doi: 10.1016/j.conb.2009.04.006
Yoo, A. S., Staahl, B. T., Chen, L., and Crabtree, G. R. (2009). MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature 460, 642–646. doi: 10.1038/nature08139
Yoo, M., Choi, K.-Y., Kim, J., Kim, M., Shim, J., Choi, J.-H., et al. (2017). BAF53b, a neuron-specific nucleosome remodeling factor, is induced after learning and facilitates long-term memory consolidation. J. Neurosci. 37:3686. doi: 10.1523/JNEUROSCI.3220-16.2017
Yu, Y., Chen, Y., Kim, B., Wang, H., Zhao, C., He, X., et al. (2013). Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 152, 248–261. doi: 10.1016/j.cell.2012.12.006
Zahir, F. R., Tucker, T., Mayo, S., Brown, C. J., Lim, E. L., Taylor, J., et al. (2016). Intragenic CNVs for epigenetic regulatory genes in intellectual disability: survey identifies pathogenic and benign single exon changes. Am. J. Med. Genet. A 170, 2916–2926. doi: 10.1002/ajmg.a.37669
Zai, G., Robbins, T. W., Sahakian, B. J., and Kennedy, J. L. (2017). A review of molecular genetic studies of neurocognitive deficits in schizophrenia. Neurosci. Biobehav. Rev. 72, 50–67. doi: 10.1016/j.neubiorev.2016.10.024
Zarate, Y. A., Bhoj, E., Kaylor, J., Li, D., Tsurusaki, Y., Miyake, N., et al. (2016). SMARCE1, a rare cause of Coffin–Siris Syndrome: clinical description of three additional cases. Am. J. Med. Genet. A 170, 1967–1973. doi: 10.1002/ajmg.a.37722
Zhan, X., Shi, X., Zhang, Z., Chen, Y., and Wu, J. I. (2011). Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc. Natl. Acad. Sci. U.S.A. 108, 12758–12763. doi: 10.1073/pnas.1018510108
Keywords: epigenetic regulation, chromatin remodeling, BAF (mSWI/SNF) complex, neural development, neurodevelopmental disorder
Citation: Sokpor G, Xie Y, Rosenbusch J and Tuoc T (2017) Chromatin Remodeling BAF (SWI/SNF) Complexes in Neural Development and Disorders. Front. Mol. Neurosci. 10:243. doi: 10.3389/fnmol.2017.00243
Received: 01 June 2017; Accepted: 18 July 2017;
Published: 03 August 2017.
Edited by:
Simone Di Giovanni, Imperial College London, United KingdomReviewed by:
Ilaria Palmisano, Imperial College London, United KingdomGiacomo Masserdotti, Helmholtz Zentrum München (HZ), Germany
Copyright © 2017 Sokpor, Xie, Rosenbusch and Tuoc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tran Tuoc, tran.tuoc@med.uni-goettingen.de
†These authors have contributed equally to this work.