PLPP/CIN Regulates Seizure Activity by the Differential Modulation of Calsenilin Binding to GluN1 and Kv4.2 in Mice
 Ji-Eun Kim
Ji-Eun Kim Hye-Won Hyun
Hye-Won Hyun Su-Ji Min
Su-Ji Min  Tae-Cheon Kang
Tae-Cheon Kang- Department of Anatomy and Neurobiology, Institute of Epilepsy Research, College of Medicine, Hallym University, Chuncheon, South Korea
Calsenilin (CSEN) binds to Kv4.2 (an A-type K+ channel) as well as N-methyl-D-aspartate receptor (NMDAR), and modulates their activities. However, the regulatory mechanisms for CSEN-binding to Kv4.2 or NMDAR remain elusive. Here, we demonstrate the novel role of pyridoxal-5′-phosphate phosphatase/chronophin (PLPP/CIN), one of the cofilin-mediated F-actin regulators, in the CSEN binding to Kv4.2 or GluN1 (an NMDAR subunit). PLPP/CIN dephosphorylated CSEN in competition with casein kinase 1, independent of cofilin dephosphorylation. As compared to wild-type mice, PLPP/CIN transgenic (PLPP/CINTg) mice showed the enhancement of Kv4.2–CSEN binding, but the reduction in CSEN–GluN1 binding. In addition, PLPP/CINTg mice exhibited the higher intensity (severity), duration and progression of seizures, but the longer latency of seizure on-set in response to kainic acid. PLPP/CIN knockout mice reversed these phenomena. Therefore, we suggest that PLPP/CIN-mediated CSEN dephosphorylation may play an important role in the functional coupling of NMDAR and Kv4.2, which regulates the neuronal excitability.
Introduction
Epilepsy is one of the common neurological disorders, suffering about 1% of all people, which is characterized by the periodic occurrence of seizures exhibiting abnormal synchronized neuronal discharges. A delicate balance between excitatory and inhibitory inputs in neurons and impairment of this balance lead to the uncontrolled neuronal hyperexcitability, which initiates neuronal damage and symptomatic seizure activity. Unprovoked recurrent seizures contribute to a cyclical or progressive process of worsening epilepsy and neurological deficits including learning disabilities and memory problems (Elger et al., 2004; Kim J.E. et al., 2007; Kim et al., 2008; Seeburg and Sheng, 2008; Mantegazza et al., 2010; Kim and Kang, 2011). Although the pathophysiology underlying seizure susceptibility is relevant to channelopathy, aberrant synaptic organization, impaired glial function, inflammation, and neuronal loss (Kang et al., 2006; McNamara et al., 2006; Wetherington et al., 2008; Vezzani et al., 2011), the cellular and molecular mechanisms in epilepsy remain unclear.
Pyridoxal-5′-phosphate phosphatase/chronophin (PLPP/CIN) is a serine (S) protein phosphatase, which activates cofilin-mediated F-actin depolymerization (Gohla et al., 2005; Kim et al., 2008, 2016). We have reported that PLPP/CIN expression is up-regulated in the rat hippocampus following pilocarpine-induced status epilepticus (SE), which is closely relevant to increased neuronal excitability (Kim et al., 2008). Recently, we have also reported that PLPP/CIN transgenic (PLPP/CINTg) mice show the enhancement of N-methyl-D-aspartate receptor (NMDAR)-mediated synaptic plasticity, but PLPP/CIN knockout (PLPP/CIN-/-) mice show reduced synaptic strength without the synaptic NMDAR distribution and the heterotrimerization (Kim et al., 2016). Therefore, PLPP/CIN would be a potentially important factor to regulate seizure activity via interaction with NMDAR. However, the PLPP/CIN-mediated signal pathways available to identify seizure susceptibility are less defined.
Calsenilin (CSEN) is first identified as a Ca2+-regulated transcriptional repressor, contains four Ca2+-binding EF hand domains and belongs to the neuronal calcium sensor family (Burgoyne, 2007). CSEN has multifunctional properties to block gene expression via direct binding with the downstream regulatory element sequence in target genes (Gomez-Villafuertes et al., 2005), and to act as a partner for Kv4.x subunits (Morohashi et al., 2002). Thus, CSEN is also named downstream regulatory element antagonist modulator (DREAM) or potassium channel interacting protein 3 (KChIP3). Indeed, CSEN regulates the neuronal excitability by diminishing dendritic excitability and back-propagated action potentials accompanied by Kv4.x channels (Hoffman et al., 1997; An et al., 2000; Cai et al., 2004). Furthermore, CSEN expression is markedly decreased in the hippocampi of experimental animal model and human epilepsy patient (Hong et al., 2003). Interestingly, CSEN is one of the negative modulators of NMDAR (Zhang et al., 2010). Since NMDAR and Kv4.2 reciprocally regulate each other (Kim J. et al., 2007; Jung et al., 2008) and CSEN activity is modulated by the casein kinase 1 (CK1)-mediated phosphorylation (Choi et al., 2003), it is likely that PLPP/CIN-mediated CSEN dephosphorylation would be involved in the pathophysiology of epilepsy by modulating the bindings of CSEN to NMDAR or Kv4.2 channels. Here, we demonstrate the novel role of PLPP/CIN in CSEN dephosphorylation that regulated seizure activity induced by kainic acid (KA) via the modulation of Kv4.2–CSEN or GluN1–CSEN bindings, independent of cofilin-mediated F-actin depolymerization. Therefore, we suggest that PLPP/CIN may play an important role in the functional coupling of Kv4.2 and NMDAR via CSEN, which regulates the neuronal excitability.
Materials and Methods
Experimental Animals and Chemicals
PLPP/CINTg (C57BL/6J background) and PLPP/CIN-/- mice (129/SvEv-C57BL/6J background) were used in the present study. The same genetic background mice were used as control animals for each group (Kim et al., 2016). Animals were provided with a commercial diet and water ad libitum under controlled conditions (22 ± 2°C, 55 ± 5% and a 12:12 light/dark cycle). All reagents were obtained from Sigma–Aldrich (St. Louis, MO, United States), except as noted. The antibody used in the present study is listed in Supplementary Table 1. All experimental protocols were approved by the Animal Care and Use Committee of Hallym University.
Surgery
Under anesthesia with isoflurane (3% induction and 1.5% maintenance in a 65:35 mixture of N2O:O2), animals were positioned over a heated pad, and core temperature was maintained 37–38°C. An infusion guide cannula with osmotic pump connector (3260PGA, Plastics One, Roanoke, VA, United States) was implanted into the ventricle (2 mm depth from bregma), and connected to an osmotic pump (1003D, Alzet, Cupertino, CA, United States) containing (1) vehicle, (2) IC261 (3 μM), or (3) CKI-7 (100 μM). The pump was subcutaneously placed in the interscapular region (Kim and Kang, 2011, 2015; Kim et al., 2014). In some animals, a standard infusion guide cannula (C315GA, Plastics One, Roanoke, VA, United States) was implanted into the same site. Thereafter, the cannula was sealed with a dummy cannula. Animals were also implanted monopolar stainless steel electrode (Plastics One, Roanoke, VA, United States) into the left dorsal hippocampus (2 mm posterior; 1.25 mm lateral; 2 mm depth from bregma). Three days after surgery, freely moving animals were given KA as followed.
Seizure Induction and EEG Recording
After baseline recording for at least 30 min, an internal infusion cannula (C315IA, Plastics One, Roanoke, VA, United States) was inserted into the lumen of the guide cannula to inject KA (0.15 μg/μl) or 4-aminopyridine (4-AP; 0.1 μg/μl) into the ventricle over a 1-min period using a microinjection pump (1 μl/min, KD Scientific, Holliston, MA, United States). Control animals received an equal volume of normal saline instead of KA or 4-AP. EEG signals were recorded with a DAM 80 differential amplifier (0.1–3000 Hz bandpass; World Precision Instruments, United States) and the data were digitized (1000 Hz) and analyzed using LabChart Pro v7 software (AD Instruments, NSW, Australia). Time of seizure onset was defined as the time point showing paroxysmal depolarizing shift, defined as lasting more than 3 s and consisting of a rhythmic discharge between 4 and 10 Hz with amplitude of at least two times higher than the baseline EEG (Kim and Kang, 2011, 2015). Total power was measured during the 2-h recording session and normalized by the baseline value obtained from each animal. Spectrograms were automatically calculated using a Hanning sliding window with 50% overlap by LabChart Pro v7. Diazepam (Valium; Hoffman la Roche, Neuilly-sur-Seine, France; 10 mg/kg, i.p.) was administered 2 h after KA injection and repeated, as needed. After recording, animals were used for anatomical or biochemical experiments.
In Vitro PLPP/CIN Phosphatase Assay
Modified in vitro PLPP/CIN phosphatase assay using full-length recombinant human CSEN (Abcam, Cambridge, United Kingdom) and PLPP/CIN (Abcam, Cambridge, United Kingdom) was performed as described previously (Choi et al., 2003). Phosphorylation of CSEN (10 ng/μl) was performed by incubation with 200 U/μl CK1 (New England BioLabs, Ipswich, MA, United States) in 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 5 mM dithiothreitol and 100 μM ATP at 30°C for 1 h. Thereafter, the sample was portioned the same volume, added PLPP/CIN (10 ng/μl) or 50 mM Tris buffer (control), and incubated at 30°C for 1 h. Crude extracts obtained from the same PLPP/CIN-/- mice were used the same method without CK1 and CSEN treatment. Thereafter, the samples were used co-precipitation and western blot analysis (see below).
Co-immunoprecipitation and Western Blot
As described previously (Kim et al., 2016), animals were quickly decapitated, and their hippocampi were dissected out in the presence of cooled artificial cerebrospinal fluid (in mM: 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 dextrose, 1.5 MgCl2, and 2.5 CaCl2). The hippocampal tissues were lysed in radioimmunoprecipitation assay buffer (RIPA: 50 mM Tris–HCl pH 8.0; 1% Nonidet P-40; 0.5% deoxycholate; 0.1% SDS, Thermo Fisher Scientific, United States) containing protease inhibitor cocktail (Roche Applied Sciences, United States), phosphatase inhibitor cocktail (PhosSTOP®, Roche Applied Science, United States) and 1 mM sodium orthovanadate. Protein concentrations were determined by BCA protein assay (Pierce, United States) and equal amounts of total proteins were precipitated with the appropriate primary antibodies and protein G sepharose at 4°C overnight. Beads were collected by centrifugation, eluted in 2× SDS sample buffer, and boiled at 95°C for 5 min. Next, western blotting was performed according to standard procedures. The rabbit anti-β-actin primary antibody was used as internal reference. The signals were scanned and quantified on ImageQuant LAS 4000 system (GE health, United States). The values of each sample were normalized with the corresponding amount of β-actin.
Immunohistochemistry, FJB Staining, Golgi Impregnation, and Analysis of Spine Morphology
Under urethane anesthesia (1.5 g/kg, i.p.), animals were perfused transcardially with 4% paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4). Brains were post-fixed in the same fixative overnight and then cryoprotected and sectioned at 30 μm with a cryostat. Free-floating coronal sections were incubated in a mixture of primary antibodies in PBS containing 0.3% Triton X-100 and 2% normal chicken serum overnight at room temperature. Sections were also incubated in a mixture of FITC- and Cy3-conjugated secondary antisera (Amersham, Piscataway, NJ, United States, 1:200) for 1 h at room temperature (Kim et al., 2014). Some tissues were used for a conventional Fluoro-Jade B (FJB) staining (Kim et al., 2014; Kim and Kang, 2015; Hyun et al., 2016). To establish the specificity of the immunostaining, a negative control test was carried out with preimmune serum instead of the primary antibody. No immunoreactivity was observed for the negative control in any structures. Golgi impregnation was performed using FD Rapid GolgiStainTM kit (FD NeuroTechnologies, Inc., MD, United States), according to the manufacturer’s instructions. All images and dendritic spine morphology were analyzed using an AxioImage M2 microscope and AxioVision Rel. 4.8 software or a confocal laser scanning microscope (LSM 710, Carl Zeiss Inc., Oberkochen, Germany).
Statistical Analysis
After evaluating the values on normality using Shapiro–Wilk W-test, data were analyzed by Student’s t-test or ANOVA followed by Newman–Keuls post hoc test. A p < 0.05 is considered to be statistically different. Quantitative data were expressed as mean ± standard error of the mean.
Results
PLPP/CIN Decreases Seizure Susceptibility, but Increases the Progression of Seizures and Neuronal Damage in Response to KA
Consistent with our previous study (Kim et al., 2016), average spine width was ∼0.40 μm and head/neck ratio was >2 in wild-type (WT, PLPP/CIN+/+) mice. As compared to WT animals, the spines in PLPP/CINTg mice showed thin necks with very small heads (head/neck ratio: 1.13), while PLPP/CIN-/- mice showed gigantic spines with a head/neck ratio of ∼4 (p < 0.05 vs. WT animals, respectively; Figures 1A,B). Since dendritic spines are the major integral site excitatory input, changed spine structures affect the neuronal excitability and seizure susceptibility (Wong and Guo, 2013). Thus, we measured the seizure susceptibility of PLPP/CINTg and PLPP/CIN-/- mice in response to KA.
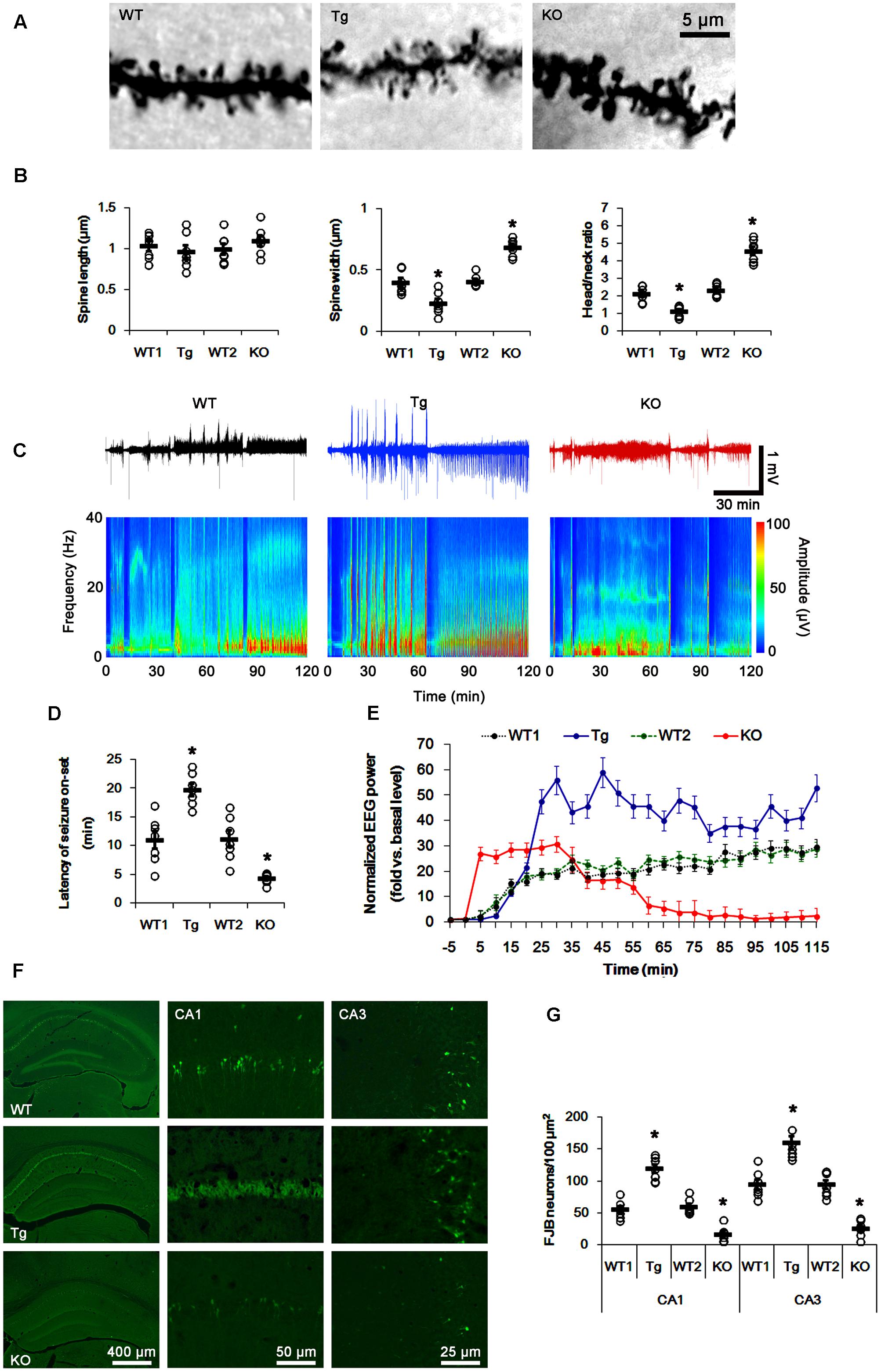
FIGURE 1. Profiles of seizure activity in PLPP/CINTg and PLPP/CIN-/- mice induce by KA. (A,B) Characteristics of dendritic spine morphology in PLPP/CINTg and PLPP/CIN-/- mice. As compared to WT animals, the spines in PLPP/CINTg mice show thin necks with very small heads, while PLPP/CIN-/- mice show gigantic spines. (A) Representative photos of dendritic spines in dentate granule cells. (B) Spine length, spine width, and head/neck ratio of dentate granule cells. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SD, ∗p < 0.05 vs. WT animals; n = 7, respectively). (C–E) Seizure activity in response to KA. PLPP/CINTg mice demonstrate increase seizure intensity, duration, and the latency of seizure on-set, as compared to PLPP/CIN-/- mice. (C) Representative EEG traces and frequency-power spectral temporal maps in response to KA. (D) Quantification of latency of seizure on-set. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. WT animals; n = 7, respectively). (E) Quantification of total EEG power (seizure intensity) in response to KA (mean ± SEM; p < 0.05 vs. WT animals; n = 7, respectively). (F,G) KA-induced neuronal death. One day after KA injection, neuronal death in PLPP/CINTg mice is more severe than that in PLPP/CIN-/- mice, as compared to WT animals. (F) Representative photos of FJB positive degenerating neurons. (G) Quantification of the number of FJB positive neurons in response to KA. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. WT animals; n = 7, respectively).
In WT animals, the first seizure occurred 11.3 min after KA injection, progressing with time to prolonged episodes of high-frequency and high-amplitude discharges by 120 min post-KA injection (Figures 1C–E). EEG analyses revealed no difference in the latency of seizure on-set, duration and intensity (total EEG power) of seizures between two different genetic background WT mice. PLPP/CINTg and PLPP/CIN-/- mice showed the first seizure activity within 18.7 and 5 min after KA injection, respectively (p < 0.05 vs. WT animals, respectively; Figures 1C–E). PLPP/CINTg mice showed increase in seizure intensity (severity) to 1.5∼3-fold of that observed in WT animal. In PLPP/CIN-/- mice, seizure activity reduced ∼60 min after KA injection (p < 0.05 vs. WT animals, respectively; Figures 1C–E).
Having observed that PLPP/CIN deletion interrupted the progression of seizures in response to KA, we investigated SE-induced neuronal damage in the hippocampus 1 day post-KA injection. WT mice displayed typical CA1 and CA3 pyramidal cell loss for FJB staining (Figures 1F,G). As compared to WT animals, PLPP/CINTg mice showed profound neuronal injury in the CA1 and CA3 regions, but PLPP/CIN-/- mice demonstrated the overt attenuation of neuronal damage in these regions (p < 0.05 vs. WT animals, respectively; Figures 1F,G). These findings indicate that PLPP/CIN may increase seizure intensity, duration, and the latency of seizure on-set.
F-Actin Polymerization Affects Seizure Intensity, But Not Latency of Seizure On-Set
To elucidate the role of PLPP/CIN-mediated F-actin depolymerization in response to KA, we measured cofilin phosphorylation level 2 h after KA injection. In control group of in PLPP/CINTg and PLPP/CIN-/- mice, phospho (p)-Cofilin/Cofilin ratio were 0.8- and 1.22-fold of WT animals, respectively (n = 7, respectively, p < 0.05, respectively; Figures 2A,B and Supplementary Figure 3). KA increased PLPP/CIN expression, but reduced cofilin phosphorylation level in all groups except PLPP/CIN-/- mice (n = 7, respectively, p < 0.05, respectively; Figures 2A,B and Supplementary Figure 3). The reduced cofilin phosphorylation in PLPP/CINTg mice was more efficient than WT animals, while that in PLPP/CIN-/- mice was less efficient (n = 7, respectively, p < 0.05, respectively; Figures 2A,B and Supplementary Figure 3). Consistent with our previous study (Kim et al., 2008), these findings indicate that PLPP/CIN may regulate cofilin phosphorylation induced by seizure activity.
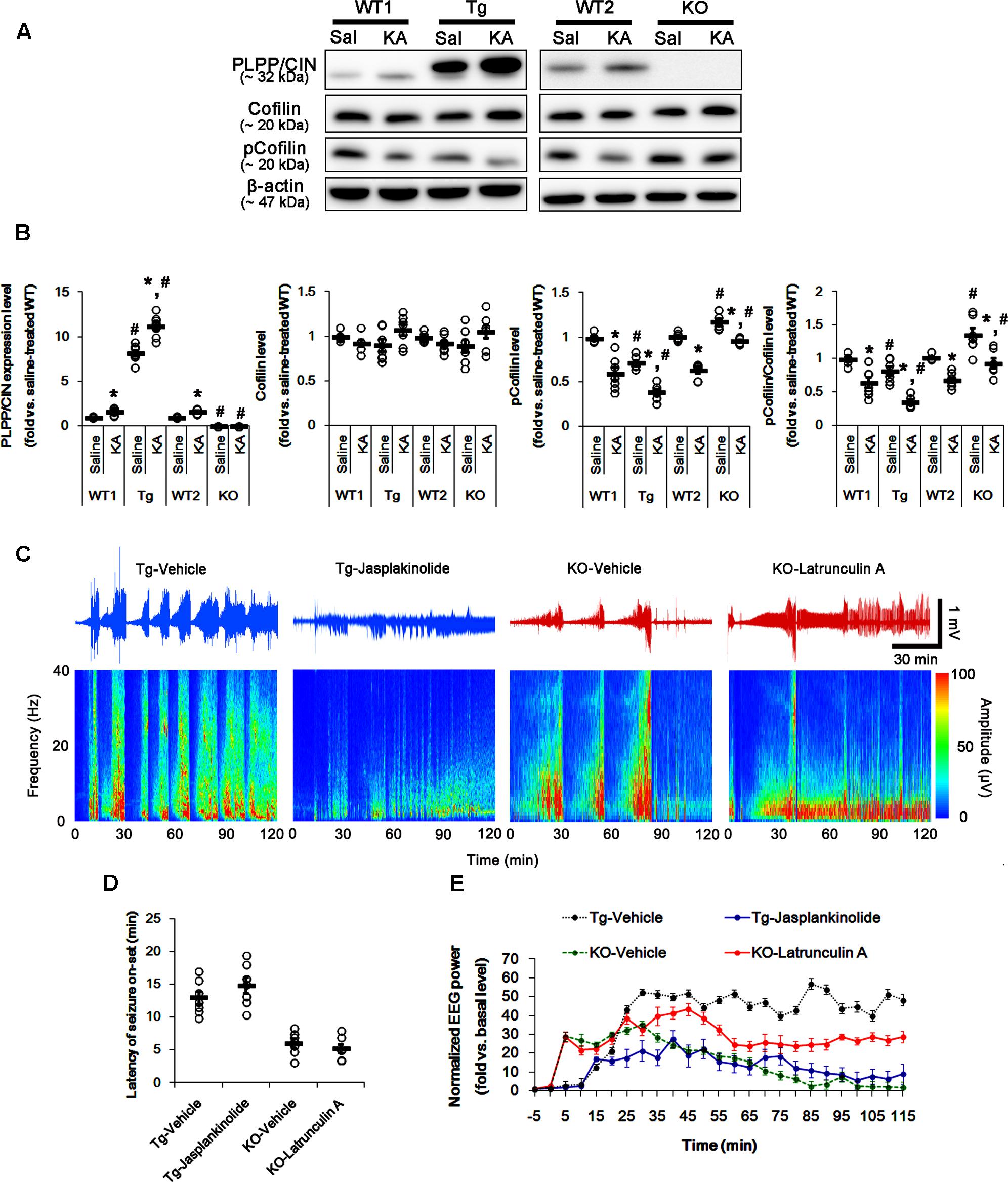
FIGURE 2. The role of F-actin dynamics in seizure activity induced by KA. (A,B) Changed PLPP/CIN expression and cofilin phosphorylation 2 h after KA injection. As compared to WT animals, cofilin phosphorylation is lower in PLPP/CINTg mice, but is higher in PLPP/CIN-/- mice. Western blot shows the up-regulation of PLPP/CIN expression and the reduction in pCofilin level without altered cofilin expression in WT animals. The reduced cofilin phosphorylation in PLPP/CINTg mice is more efficient, while that in PLPP/CIN-/- mice is less, as compared to WT animals. (A) Western blot of PLPP/CIN, cofilin, and pCofilin. (B) Quantification of PLPP/CIN expression, cofilin expression, and cofilin phosphorylation level based on western blot data. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗,#p < 0.05 vs. saline-treated and WT animals, respectively; n = 7, respectively). (C–E) The effect of F-actin dynamics on seizure activity in response to KA. Jasplakinolide (an F-actin stabilizer) decreases seizure intensity (total EEG power) in PLPP/CINTg mice, but latrunculin A (an F-actin depolymerizer) enhances it in PLPP/CIN-/- animals. However, neither jasplakinolide nor latrunculin A affects the latency of seizure on-set in both groups. (C) Representative EEG traces and frequency-power spectral temporal maps in response to KA. (D) Quantification of latency of seizure on-set. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; n = 7, respectively). (E) Quantification of total EEG power (seizure intensity) in response to KA (mean ± SEM; p < 0.05 vs. vehicle; n = 7, respectively).
Next, we explored whether F-actin depolymerization affects KA-induced seizure activity in PLPP/CINTg and PLPP/CIN-/- animals. As compared to vehicle, jasplakinolide (an F-actin stabilizer) significantly attenuated seizure intensity (total EEG power) in PLPP/CINTg mice, while latrunculin A (an F-actin depolymerizer) increased it in PLPP/CIN-/- animals (n = 7, respectively, p < 0.05 vs. vehicle, respectively; Figures 2C–E). However, neither jasplakinolide nor latrunculin A affected the latency of seizure on-set in both groups (Figures 2C–E). These findings indicate that PLPP/CIN-mediated F-actin depolymerization may contribute to the seizure intensity (severity), but not seizure threshold.
PLPP/CIN Regulates CSEN Phosphorylation
Since seizure activity affects CSEN expression in the mouse hippocampus following KA injection (Matsu-ura et al., 2002; Hong et al., 2003), we explored the effect of KA-induced seizure on CSEN expression. In control animals, double immunofluorescent study revealed that PLPP/CIN was colocalized with CSEN in hippocampal neurons, and that PLPP/CIN overexpression or deletion did not affect CSEN expression. However, PLPP/CIN overexpression decreased pCSEN level, but PLPP/CIN deletion increased it (Figures 3A–C and Supplementary Figure 4).
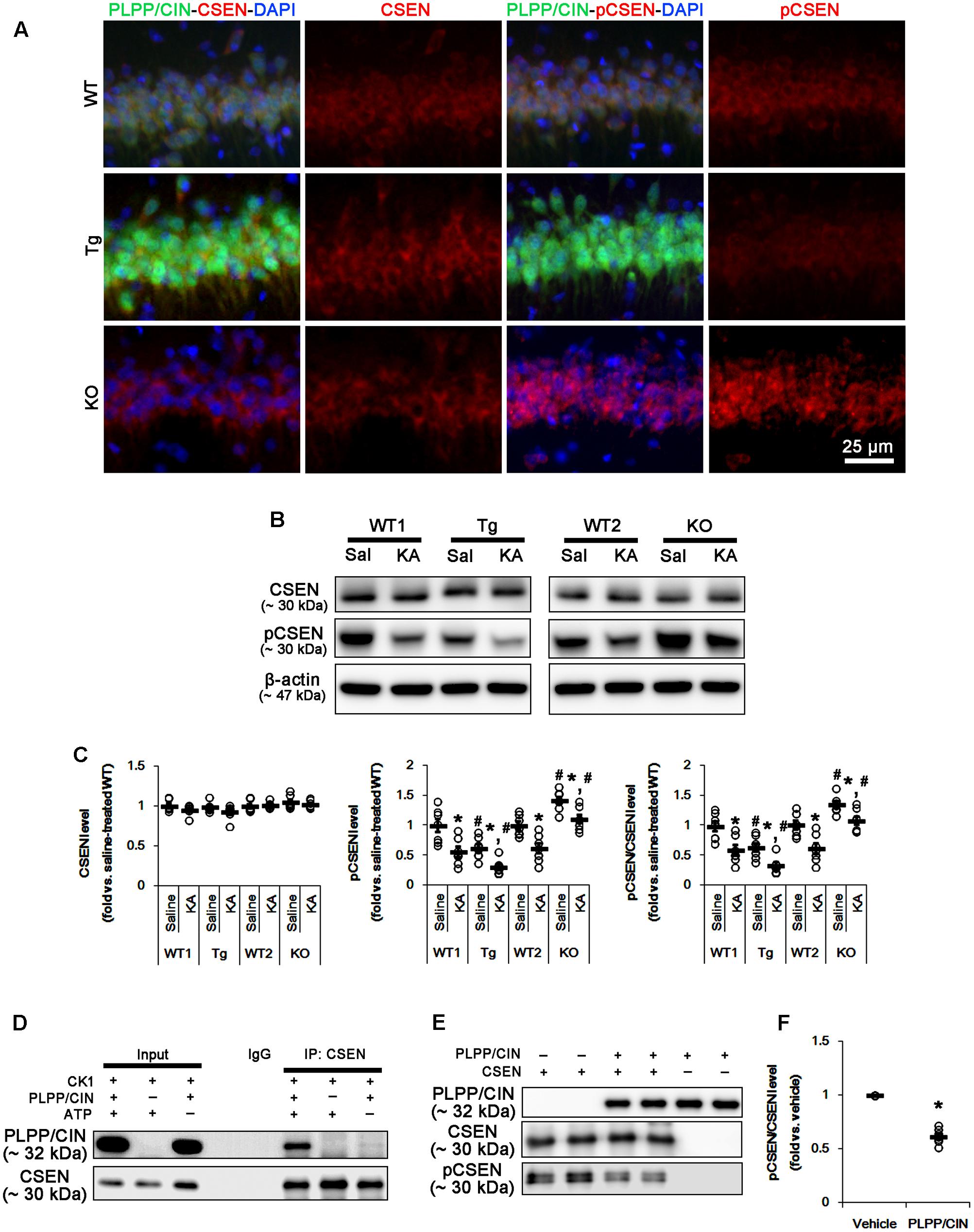
FIGURE 3. PLPP/CIN-mediated CSEN dephosphorylation. (A) Representative double immunofluorescent photos for PLPP/CIN and CSEN. PLPP/CIN is colocalized with CSEN in hippocampal neurons in PLPP/CINTg mice. (B,C) Changed CSEN phosphorylation 2 h after KA injection. Western blot shows the CSEN phosphorylation is lower in PLPP/CINTg mice, but is higher in PLPP/CIN-/- mice. In addition, KA reduces CSEN phosphorylation without altered CSEN expression in WT animals. The reduced CSEN phosphorylation in PLPP/CINTg mice is more efficient, while that in PLPP/CIN-/- mice is less, as compared to WT animals. (B) Western blot of CSEN and pCSEN. (C) Quantification of CSEN expression and its phosphorylation level based on western blot data. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗,#p < 0.05 vs. saline-treated and WT animals, respectively; n = 7, respectively). (D–F) In vitro assay using recombinant proteins. Co-immunoprecipitation and western blot reveal that PLPP/CIN binds to CSEN, and dephosphorylates CSEN (n = 7, p < 0.05 vs. vehicle). (D) Co-immunoprecipitation analysis of PLPP/CIN interaction with CSEN. (E) Western blot of PLPP/CIN, CSEN, and pCSEN. (F) Quantification of CSEN expression and its phosphorylation level based on western blot data. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. vehicle; n = 7).
As compared to WT animals, pCSEN levels in PLPP/CINTg and PLPP/CIN-/- mice were 0.72- and 1.44-fold of WT animal levels, respectively (n = 7, respectively, p < 0.05, respectively; Figures 3B,C and Supplementary Figure 4). Two hours after KA injection, pCSEN level were reduced to 0.59- and 0.62-fold of control level in both WT animals without altered CSEN expression (n = 7, respectively, p < 0.05 vs. control level; Figures 3B,C and Supplementary Figure 4). In PLPP/CINTg and PLPP/CIN-/- mice, pCSEN levels were 0.33- and 1.07-fold of control level in WT animals, respectively (n = 7, respectively, p < 0.05 vs. control level; Figures 3B,C and Supplementary Figure 4).
To investigate whether other protein phosphatases affect pCSEN levels, we examined the expressions and phosphorylation levels of protein phosphatase 1A (PP1A), protein phosphatase 2A (PP2A), and protein phosphatase 2B (PP2B) after KA injection. However, KA injection did not influence on protein phosphatase expressions and phosphorylations (Supplementary Figure 1, 9). Together with increases in PLPP/CIN expression and its activity, our findings demonstrate that PLPP/CIN may dephosphorylate CSEN.
To clarify the direct phosphatase activity of PLPP/CIN on CSEN, we applied in vitro assay using recombinant proteins. Co-immunoprecipitation revealed that PLPP/CIN bound to CSEN, and effectively reduced CSEN phosphorylation to 62% of vehicle level (n = 7, p < 0.05 vs. vehicle; Figures 3D–F and Supplementary Figure 4). Taken together, our findings suggest that PLPP/CIN may directly regulate CSEN phosphorylation.
Seizure Activity Increases the Binding of PLPP/CIN to CSEN
Next, we investigated whether seizure activity affects the binding activity of PLPP/CIN to CSEN in vivo. Under physiological condition (control animals), PLPP/CIN-CSEN binding in PLPP/CINTg mice was 1.33-fold of WT animal level (n = 7, p < 0.05, respectively; Figures 4A,B and Supplementary Figure 5). Two hours after KA injection, the PLPP/CIN-CSEN binding in WT animals was increased to 1.5-fold of control level (n = 7, p < 0.05, Figures 4A,B and Supplementary Figure 5). In PLPP/CINTg mice, the PLPP/CIN-CSEN binding was 1.7-fold of control level in WT animal (n = 7, p < 0.05, Figures 4A,B and Supplementary Figure 5). These findings indicate that seizure activity may increase the binding of PLPP/CIN with CSEN, which may represent activity-dependent CSEN dephosphorylation by PLPP/CIN.
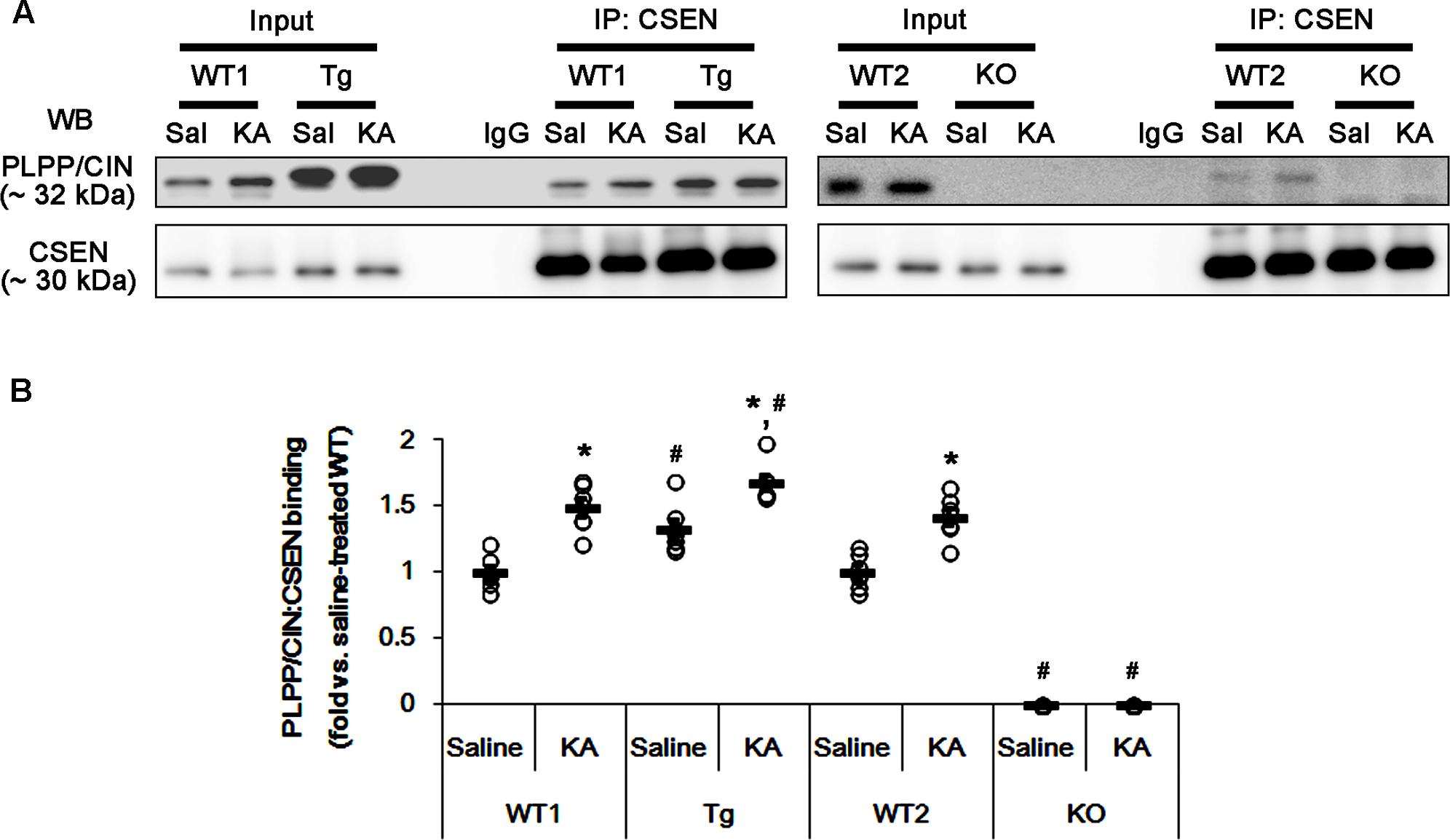
FIGURE 4. The effect of seizure activity on PLPP/CIN-mediated CSEN dephosphorylation in vivo. (A,B) Changed PLPP/CIN-CSEN bindings 2 h after KA injection. Western blot shows that PLPP/CIN-CSEN co-precipitation in PLPP/CINTg mice is higher than WT animals under physiological condition. KA increases the binding of PLPP/CIN to CSEN in WT and PLPP/CINTg mice. (A) Co-immunoprecipitation of PLPP/CIN and CSEN in vivo. (B) Co-immunoprecipitation analysis of PLPP/CIN interaction with CSEN in vivo. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗,#p < 0.05 vs. saline-treated and WT animals, respectively; n = 7, respectively).
PLPP/CIN-Mediated CSEN Dephosphorylation Increases CSEN–Kv4.2 Bindings
Pharmacological blockade and genetic Kv4.2 channel deletion increase neuronal excitability and seizure susceptibility in response to KA (Magee and Carruth, 1999; Cai et al., 2004; Barnwell et al., 2009). Interestingly, CSEN plays an important role in increased functional surface expression, slower inactivation, and faster recovery from inactivation of Kv4.2 channels, which inhibit neuronal hyperactivity by generating A-type K+ current (IA) (Hoffman et al., 1997; An et al., 2000). Therefore, we investigated whether PLPP/CIN-mediated CSEN dephosphorylation affects the interaction of CSEN with Kv4.2, which influences on KA-induced seizures.
In control animals, CSEN co-precipitation with Kv4.2 in PLPP/CINTg and PLPP/CIN-/- mice were 1.27- and 0.79-fold of WT animal levels (n = 7, respectively, p < 0.05, respectively; Figures 5A,B and Supplementary Figure 6). However, the binding of CSEN with Kv4.2 was increased by KA among all groups except PLPP/CIN-/- mice (n = 7, respectively, p < 0.05; Figures 5A,B and Supplementary Figure 6).
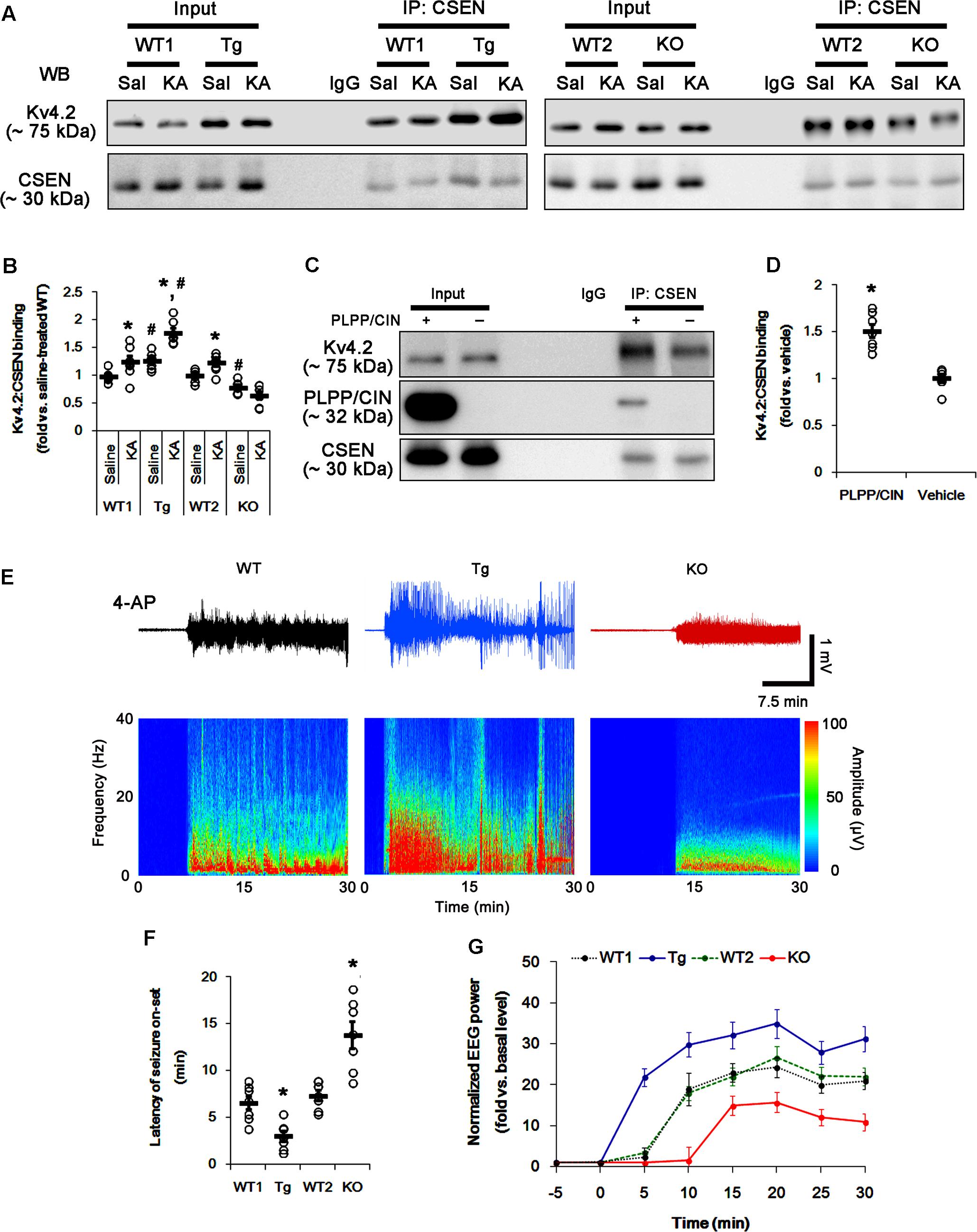
FIGURE 5. The PLPP/CIN-mediated CSEN–Kv4.2 bindings in vivo and in vitro. (A,B) Changed Kv4.2–CSEN bindings 2 h after KA injection. Under physiological condition, western blot shows that Kv4.2–CSEN co-precipitation is higher in PLPP/CINTg mice, but is lower in PLPP/CIN-/- mice, as compared to WT animals. KA increases the binding of CSEN to Kv4.2 in WT and PLPP/CINTg mice. (A) Co-immunoprecipitation of CSEN and Kv4.2 in vivo. (B) Co-immunoprecipitation analysis of CSEN interaction with Kv4.2 in vivo. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗,#p < 0.05 vs. saline-treated and WT animals, respectively; n = 7, respectively). (C,D) In vitro assay using crude extract obtained from PLPP/CIN-/- mouse brain. PLPP/CIN treatment increases the Kv4.2–CSEN binding, as compared to vehicle. (C) Co-immunoprecipitation of CSEN and Kv4.2 in vitro. (D) Co-immunoprecipitation analysis of CSEN interaction with Kv4.2 in vitro. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. vehicle; n = 7, respectively). (E–G) The comparison of responsiveness to 4-AP. As compared to WT animals, PLPP/CINTg mice show the higher responsiveness to 4-AP, but PLPP/CIN-/- animals reveal lower responsiveness, as compared to WT animals. (E) Representative EEG traces and frequency-power spectral temporal maps in response to 4-AP. (F) Quantification of latency of seizure on-set. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. WT animals; n = 7, respectively). (G) Quantification of total EEG power (seizure intensity) in response to 4-AP (mean ± SEM; p < 0.05 vs. WT animals; n = 7, respectively).
To identify the role of PLPP/CIN in the binding of CSEN to Kv4.2, we applied in vitro assay using crude extracts from the PLPP/CIN-/- brains. PLPP/CIN treatment significantly increased CSEN co-precipitation with Kv4.2 to 1.5-fold of vehicle level (n = 7, p < 0.05 vs. vehicle; Figures 5C,D and Supplementary Figure 6). Together with PLPP-mediated CSEN dephosphorylation (Figure 3), these findings suggest that the affinity of intact (dephosphorylated) CSEN to Kv4.2 may higher than that of pCSEN.
To evaluate the role of CSEN–Kv4.2 bindings in seizure activity, we analyzed the responsiveness to 4-AP (an IA blocker; Castro et al., 2001; Cai et al., 2004) in PLPP/CINTg and PLPP/CIN-/- animals. As compared to WT animals, PLPP/CINTg mice showed higher total EEG power (seizure intensity) and shorter latency of seizure on-set in response to 4-AP (n = 7, respectively, p < 0.05; Figures 5E–G). However, PLPP/CIN-/- animals revealed lower responsiveness to 4-AP (n = 7, p < 0.05, respectively; Figures 5E–G). Since the lack of Kv4.2 channel reduces the sensitivity to 4-AP (Castro et al., 2001; Cai et al., 2004) and Kv4.2 channel plays an inhibitory role in KA-induced seizures (Barnwell et al., 2009), these findings indicate that PLPP/CIN overexpression may increase seizure threshold (the latency of seizure on-set) in response to KA through the enhanced CSEN–Kv4.2 interactions.
PLPP/CIN-Mediated Dephosphorylation Inhibits the Binding of CSEN to GluN1 Subunit
Since PLPP/CIN deletion (Kim et al., 2016) and the binding of CSEN to GluN1 subunit (Zhang et al., 2010) negatively regulate NMDAR function, it is likely that PLPP/CIN-mediated CSEN dephosphorylation may reduce KA-induced seizure duration via inhibiting the CSEN-binding to GluN1 subunits. Thus, we measured the binding of CSEN with GluN1. KA injection did not affect GluN1 expression in the hippocampus of all groups (Supplementary Figures 2A,B, 10). Under physiological condition, the bindings of CSEN with GluN1 were 0.76- and 2.23-fold of WT animal level in PLPP/CINTg and PLPP/CIN-/- mice, respectively (n = 7, respectively, p < 0.05 vs. WT animals; Figures 6A,B and Supplementary Figure 7). Two hours after KA injection, the bindings of CSEN to GluN1 were reduced to 0.53- and 0.62-fold of control level in both WT animals, respectively (n = 7, respectively, p < 0.05; Figures 6A,B and Supplementary Figure 7). In PLPP/CINTg and PLPP/CIN-/- mice, the CSEN binding to GluN1 were 0.42- and 1.75-fold of control level in WT animals (n = 7, respectively, p < 0.05, respectively; Figures 6A,B and Supplementary Figure 7). GluN1 co-precipitation with CSEN was similarly observed in PLPP/CINTg and PLPP/CIN-/- mice (Supplementary Figures 2A,C, 10). These findings indicate that PLPP/CIN-mediated CSEN dephosphorylation may play a crucial role in the reduced binding of CSEN to GluN1.
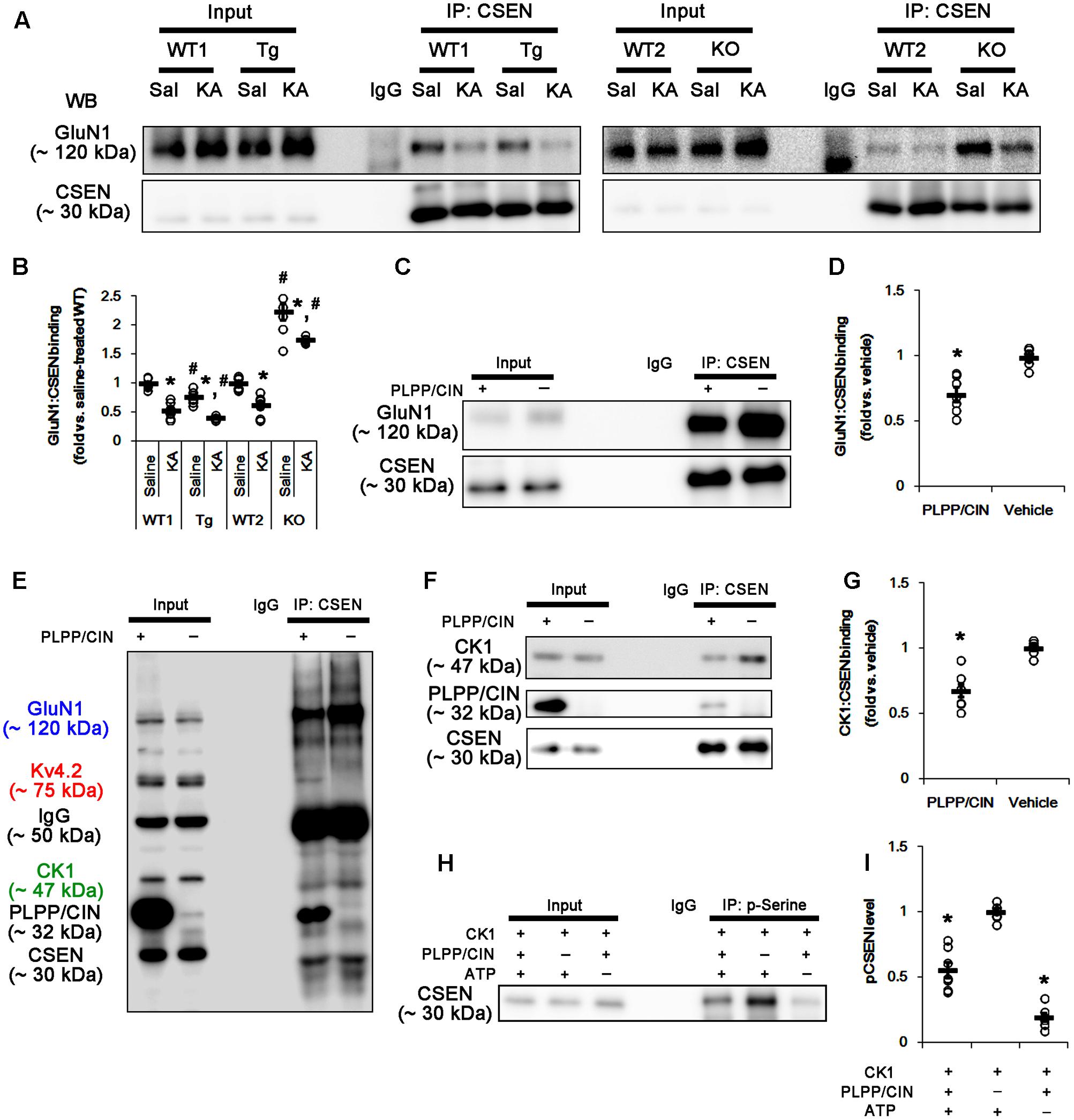
FIGURE 6. The roles of PLPP/CIN in GluN1–CSEN binding and CK1-mediated CSEN phosphorylation in vivo and in vitro. (A,B) Changed GluN1–CSEN bindings 2 h after KA injection. Under physiological condition, western blot shows that GluN1–CSEN co-precipitation is lower in PLPP/CINTg mice, but is higher in PLPP/CIN-/- mice, as compared to WT animals. KA decreases the binding of CSEN to GluN1 in all groups. (A) Co-immunoprecipitation of CSEN and GluN1 in vivo. (B) Co-immunoprecipitation analysis of CSEN interaction with GluN1 in vivo. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗,#p < 0.05 vs. saline-treated and WT animals, respectively; n = 7, respectively). (C,D) In vitro assay using crude extract obtained from PLPP/CIN-/- mouse brain. PLPP/CIN treatment decreases the GluN1–CSEN binding, as compared to vehicle. (C) Co-immunoprecipitation of CSEN and GluN1 in vitro. (D) Co-immunoprecipitation analysis of CSEN interaction with Kv4.2 in vitro. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. vehicle; n = 7, respectively). (E–I) The competitive role of PLPP/CIN in the binding of CK1 to CSEN in vitro. The interactions between PLPP/CIN and CSEN are in competition with CK1. (E) Whole western blot image using crude extract obtained from PLPP/CIN-/- mouse brain. In the presence of PLPP/CIN, the GluN1–CSEN and CK–CSEN bindings are reduced, while Kv4.2–CSEN co-precipitation is increased. (F) Co-immunoprecipitation of CSEN with CK1 or PLPP/CIN using crude extract obtained from PLPP/CIN-/- mouse brain. (G) Co-immunoprecipitation analysis of CSEN interaction with CK1 or PLPP/CIN in vitro. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. vehicle; n = 7, respectively). (H) In vitro assay using recombinant proteins. Co-immunoprecipitation reveals that PLPP/CIN inhibits CK1-mediated CSEN phosphorylation. (I) Co-immunoprecipitation analysis of the role of PLPP/CIN in CK1-mediated CSEN phosphorylation. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. vehicle; n = 7).
In vitro assay using crude extracts of the PLPP/CIN-/- brains also revealed that the binding of CSEN to GluN1 was decreased to 0.71-fold of vehicle level in the presence of PLPP/CIN (n = 7, respectively, p < 0.05; Figures 6C,D and Supplementary Figure 7). However, the binding of CSEN to Kv4.2 was increased (n = 7, respectively, p < 0.05; Figures 5C,D, 6E). Because PLPP/CIN overexpression increases NMDAR functionality without changed NMDAR distribution and the heterotrimerization (Kim et al., 2016), our findings suggest that PLPP/CIN may potentiate NMDAR functionality by inhibiting the CSEN–GluN1 binding.
CSEN Phosphorylation Is Regulated by the Competitive Binding with PLPP/CIN and CK1
Next, we investigated the relationship between PLPP/CIN and CK1, since CK1 phosphorylates CSEN (Choi et al., 2003). In vitro binding assay using crude extracts from the PLPP/CIN-/- brains revealed that PLPP/CIN inhibited the binding of CSEN with CK1 to 0.68-fold of vehicle level (n = 7, respectively, p < 0.05; Figures 6E–G and Supplementary Figure 7). In vitro binding assay using recombinant proteins also demonstrated that PLPP/CIN reduced CK1-mediated CSEN phosphorylation to 0.56-fold of vehicle level (n = 7, respectively, p < 0.05; Figures 6H,I). Taken together, these findings indicate that PLPP/CIN may be one of the counterparts of CK1 for CSEN phosphorylation.
CSEN Phosphorylation Directly Regulates Seizure Activity in Response to KA Independent of Cofilin Phosphorylation
The remaining question is whether CSEN phosphorylation affects seizure activity in response to KA by regulating GluN1–CSEN and Kv4.2–CSEN binding in vivo. Thus, we applied two CK1 inhibitors (IC261 and CKI-7) in PLPP/CIN-/- mice to decrease CSEN phosphorylation. Neither IC261 nor CKI-7 affected pCofilin level. Both CK1 inhibitors inhibited the binding of CK1 to CSEN, and reduced pCSEN level as well as GluN1–CSEN bindings (n = 7, respectively, p < 0.05; Figures 7A–C and Supplementary Figure 8), However, CK1 inhibitions increased Kv4.2–CSEN co-precipitation (n = 7, respectively, p < 0.05; Figures 7A–C and Supplementary Figure 8). In addition, CK1 inhibitors increased the latency of seizure on-set, the seizure duration and seizure-induced neuronal damage in response to KA (n = 7, respectively, p < 0.05 vs. vehicle; Figures 7D–H). These findings indicate that CSEN may directly regulate the seizure susceptibility and its progression by differential modulation of its interaction with Kv4.2 and GluN1 subunit.
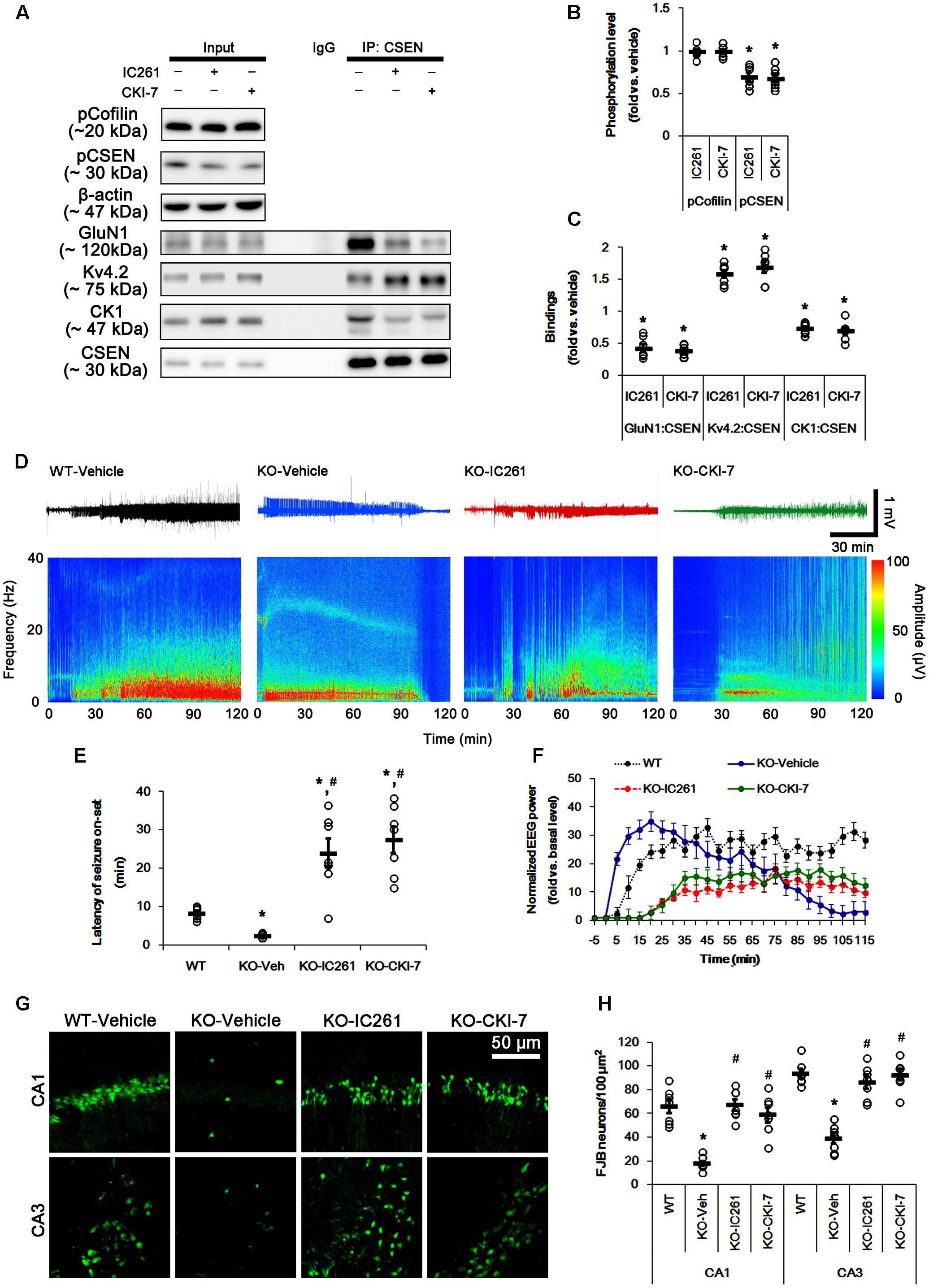
FIGURE 7. Effect of CK1 inhibitors on CSEN phosphorylation, CSEN interaction, and seizure activity in PLPP/CIN-/- mice. (A–C) The effect of CK1 inhibitors on CSEN phosphorylation and CSEN interaction with GluN1 and Kv4.2. IC261 and CKI-7 cannot change pCofilin level. Both CK1 inhibitors decrease pCSEN level, and CK1–CSEN and GluN1–CSEN bindings. However, CK1 inhibitors increase Kv4.2–CSEN co-precipitation. (A) Western blot for pCSEN and pCofilin, and co-immunoprecipitation of CSEN and GluN1, Kv4.2 or CK1 in vivo. (B) Quantification of pCofilin and pCSEN levels based on western blot data. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. vehicle; n = 7). (C) Co-immunoprecipitation analysis of CSEN interaction with GluN1, Kv4.2, and CK1 in vivo. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗p < 0.05 vs. vehicle; n = 7, respectively). (D–F) The effect of CK1 inhibitors on seizure activity in response to KA. CK1 inhibitors increase the latency of seizure on-set and the seizure duration in response to KA. (D) Representative EEG traces and frequency-power spectral temporal maps in response to KA. (E) Quantification of latency of seizure on-set. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗,#p < 0.05 vs. WT animals and vehicle, respectively; n = 7, respectively). (F) Quantification of total EEG power (seizure intensity) in response to KA (mean ± SEM; p < 0.05 vs. WT animals and vehicle; n = 7, respectively). (G,H) Effect of CK1 inhibitors on KA-induced neuronal death. One day after KA injection, both CK1 inhibitors exacerbate neuronal death, as compared to vehicle. (G) Representative photos of FJB positive degenerating neurons. (H) Quantification of the number of FJB positive neurons in response to KA. Open circles indicate each individual value. Horizontal bars indicate mean value (mean ± SEM; ∗,#p < 0.05 vs. WT animals and vehicle, respectively; n = 7, respectively).
Discussion
The present study investigated the involvement of PLPP/CIN in the seizure activity in response to KA in mice. This study demonstrated four major findings: (1) PLPP/CIN-mediated F-actin depolymerization correlated with the seizure intensity (total EEG power) in response to KA. (2) PLPP/CIN dephosphorylated CSEN, reflecting that CSEN is the novel substrate for PLPP/CIN. (3) PLPP/CIN-mediated CSEN dephosphorylation facilitated the Kv4.2–CSEN bindings, which increased seizure threshold (the latency of seizure on-set) in response to KA. (4) PLPP/CIN-mediated CSEN dephosphorylation inhibited the binding of CSEN to GluN1, which prolonged the seizure duration.
PLPP/CIN is firstly discovered as a phosphatase for pyridoxal-5′-phosphate (PLP, an active form of vitamin B6; Turner and Happold, 1961), and later identified as a serine specific phosphatase of the non-thiol-based haloacid dehalogenase superfamily of hydrolases and a modulator for cofilin activity (Gohla et al., 2005; Kim et al., 2008, 2016). In our previous study (Kim et al., 2008), Seizure activity elevates PLPP/CIN expression in the rat hippocampus accompanied by increase in neuronal excitability. In the present study, the genetic PLPP/CIN manipulation changed seizure activity in response to KA. PLP plays a role as a cofactor in γ-aminobutyric acid (GABA) synthesis by glutamate decarboxylase (Miller et al., 1978). Thus, genetic defect of PLP metabolism results in vitamin B6-dependent epilepsy, which is an autosomal recessive epileptic encephalopathy responsive to pharmacological dosages of pyridoxine (a PLP precursor) and resistant to antiepileptic drugs (Stockler et al., 2011). With respect to these reports, altered seizure activity in PLPP/CINTg and PLPP/CIN-/- mice would be affected by PLPP/CIN-mediated alterations in GABAergic inhibitory neurotransmission via changed PLP concentration. However, this hypothesis can be excluded because the PLP concentration in the brain is tightly regulated by two rate-limiting synthetic enzymes, pyridoxal kinase and pyridoxine-5′-phosphate oxidase (Choi et al., 1983; Ubbink et al., 1990). Indeed, pyridoxine could not inhibit seizure activity in rats (Kim et al., 2008). Furthermore, both PLPP/CINTg and PLPP/CIN-/- mice show the similar GABAergic transmission under physiological condition (Kim et al., 2016).
In the present study, we found that F-actin polymerization by jasplakinolide significantly attenuated seizure intensity in PLPP/CINTg mice, but F-actin depolymerization by latrunculin A increased it in PLPP/CIN-/- animals. These findings indicate that PLPP/CIN-mediated F-actin depolymerization itself affects KA-induced seizure intensity. However, neither jasplakinolide nor latrunculin A affected the latency of seizure on-set in both groups. Thus, it is likely that the unknown PLPP/CIN-mediated mechanism may underlie changed seizure susceptibility and its duration. In the present study, PLPP/CIN bound to CSEN and dephosphorylated it in vivo and in vitro. In addition, these interactions between PLPP/CIN and CSEN were in competition with CK1 that phosphorylated CSEN. These findings are the first evidence providing the role of PLPP/CIN as a CSEN dephosphorylating enzyme. Furthermore, PLPP/CIN-mediated CSEN dephosphorylation inhibited the GluN1–CSEN binding, but enhanced the Kv4.2–CSEN binding in vivo and in vitro. These findings also provide the novel biological functions of PLPP/CIN-mediated CSEN dephosphorylation.
CSEN modulates multiple intracellular events related to pain (Cheng et al., 2002), long-term potentiation (An et al., 2000), learning and memory (Lilliehook et al., 2003; Alexander et al., 2009). In particular, CSEN plays an important role in maintenance of Kv4.2 channel functionality (Hoffman et al., 1997; An et al., 2000). Kv4.2 channels are pore-forming subunits of the neuronal somatodendritic IA, which inhibits neuronal hyperexcitability by dampening dendritic excitability and back-propagated action potentials (Hoffman et al., 1997; Cai et al., 2004). CSEN binds to Kv4.2 channels and promotes Kv4.2 channel functionality (An et al., 2000). Indeed, CSEN deletion reduces IA density in hippocampal dentate granule cells in mice (Lilliehook et al., 2003). Furthermore, reductions in Kv4.2 channel expression and a mutation in the gene encoding Kv4.2 (KCND2) are reported in animal models of epilepsy (Tsaur et al., 1992; Monaghan et al., 2008) and patients with temporal lobe epilepsy (Singh et al., 2006). With respect to these previous reports, it is likely that PLPP/CIN-mediated changes in seizure susceptibility would be relevant to the role of CSEN as an auxiliary subunit for Kv4.2 channels, although we did not investigate the Kv4.2 functionality by a direct electrophysiological approach. Indeed, the present study demonstrates that PLPP/CINTg mice showed significantly increases in CSEN co-precipitation with Kv4.2 and responsiveness to 4-AP. In addition, PLPP/CIN-/- mice reversed to these phenomena. Therefore, our findings provide the possibility that PLPP/CIN may decrease seizure susceptibility via the enhancing CSEN–Kv4.2 interactions.
A local increase in dendritic excitability favors the back-propagated action potentials with a subsequent boost in the NMDAR-mediated Ca2+ influx (Frick et al., 2004). Indeed, Kv4.2 regulates Ca2+ signaling through spontaneous NMDAR activation to control synaptic NMDAR expression and plasticity (Jung et al., 2008), which in turn leads to activity-dependent Kv4.2 internalization (inactivation) (Kim J. et al., 2007). Thus, it is likely that Kv4.2 and NMDAR may reciprocally regulate each other under normal condition. In the present study, we found that CSEN co-precipitation with GluN1 was decreased in PLPP/CINTg mice under normal condition. Furthermore, PLPP/CINTg mice showed the prolonged seizure duration and the severe progression of seizure activity in response to KA, accompanied by the rapid dissociation of CSEN from GluN1. In contrast, PLPP/CIN-/- mice exhibited the spontaneous abrogation of KA-induced seizure activity with less reduced CSEN co-precipitation with GluN1. Furthermore, inhibition of CSEN phosphorylation by CK1 inhibitors delayed the latency of seizure on-set in response to KA, accompanied by reduced CSEN–GluN1 binding. Since CSEN (Zhang et al., 2010) and PLPP/CIN deletion (Kim et al., 2016) inhibit NMDAR functionality, our findings suggest that PLPP/CIN may increase KA-induced seizure duration and neuronal death due to the NMDAR activation by the reduction of the CSEN–GluN1 binding. In light of the functional coupling of NMDAR and Kv4.2 (Frick et al., 2004; Kim J. et al., 2007; Jung et al., 2008), the present study clearly demonstrates that PLPP/CIN-mediated CSEN regulation may maintain appropriate neuronal activity under physiological condition (Figure 8A) and have a key role in seizure susceptibility, duration, and progression (Figure 8B).
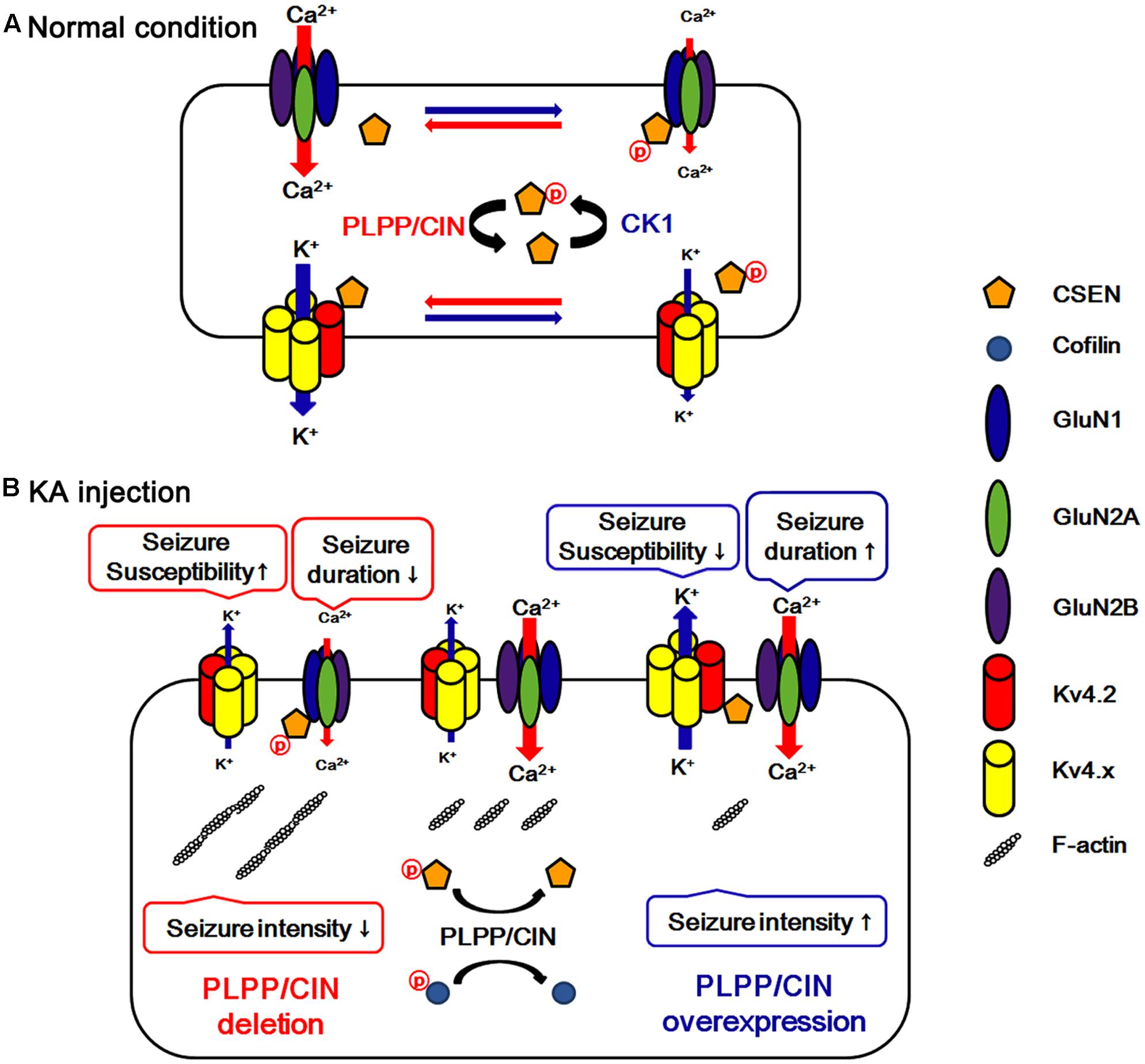
FIGURE 8. Hypothesized role of PLPP/CIN -mediated CSEN regulation in normal condition (A) and seizure susceptibility in response to KA injection (B). PLPP/CIN dephosphorylates CSEN, which has the distinct affinity to bind with Kv4.2 and GluN1, independent of cofilin-mediated F-actin depolymerization. Dephosphorylated CSEN binds to Kv4.2, which facilitates Kv4.2 activity. However, phosphorylated CSEN binds to GluN1, and inhibits NMDAR activity. These PLPP/CIN-mediated functional coupling of NMDAR and Kv4.2 differently affects seizure susceptibility and its duration, accompanied by F-actin depolymerization.
PLPP/CIN-mediated F-actin dynamics play an important role in the maturation of dendritic spines (Kim et al., 2016). Dendritic spines are critical for synaptic transmission, representing the primary location of excitatory synaptic inputs to neurons (Kasai et al., 2010). Thus, dendritic spine pathology (loss of dendritic spines and alterations in spine shape/size) seems to be an important pathophysiological mechanism in epilepsy (Ferhat et al., 2003; Wong, 2005). Indeed, a significant decrease in dendritic spine density is frequently seen in hippocampal pyramidal neurons and dentate granule cells in patients with temporal lobe epilepsy (Freiman et al., 2011) and epilepsy animal models (González-Burgos et al., 2004; Ampuero et al., 2007). However, the ictogenic role of dendritic spine morphology has been still controversial. For example, enhanced LIM kinase 1 (LIMK1, one of counterparts of PLPP/CIN for cofilin phosphorylation) protein translation reduces the seizure susceptibility accompanied by dendritic spine loss as well as paradoxical increased spine volume (Jimenez-Mateos et al., 2012, 2015). In the present study, PLPP/CINTg mice were less susceptible to KA-induced seizures with small and immature dendritic spines. In contrast, PLPP/CIN-/- mice were more sensitive with abnormal gigantic spines. Furthermore, jasplakinolide abolished seizure intensity (not seizure susceptibility or its duration) in PLPP/CINTg mice, and latrunculin A enhanced it in PLPP/CIN-/- animals. Since small spines are less efficient in the compartmentalization of Ca2+ from the dendritic shaft than large spines (Majewska et al., 2000), the present data provide the possibility that the small spine size or F-actin depolymerization may enhance seizure intensity due to increased Ca2+ flux from spines to the dendritic shaft. Further studies are needed to elucidate the correlation between ictogenesis and spine pathology.
Conclusion
We found that PLPP/CIN-mediated F-actin depolymerization increased seizure intensity in response to KA. In addition, PLPP/CIN dephosphorylated CSEN, which differently affected seizure susceptibility and its duration by regulating the binding of CSEN to Kv4.2 and NMDAR. Thus, we suggest that PLPP/CIN may play an important role in the regulation of neuronal excitability via modulating CSEN phosphorylation under physiological and pathological conditions (Figure 8).
Author Contributions
T-CK designed and supervised the project. J-EK performed the experiments described in the manuscript with H-WH, S-JM, D-SL, AJ, and MK. J-EK and T-CK analyzed the data and wrote the manuscript.
Funding
This study was supported by a grant of National Research Foundation of Korea (NRF, grant no. 2015R1A2A2A01003539). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fnmol.2017.00303/full#supplementary-material
References
Alexander, J. C., McDermott, C. M., Tunur, T., Rands, V., Stelly, C., Karhson, D., et al. (2009). The role of calsenilin/DREAM/KChIP3 in contextual fear conditioning. Learn. Mem. 16, 167–177. doi: 10.1101/lm.1261709
Ampuero, E., Dagnino-Subiabre, A., Sandoval, R., Zepeda-Carreño, R., Sandoval, S., Viedma, A., et al. (2007). Status epilepticus induces region-specific changes in dendritic spines, dendritic length and TrkB protein content of rat brain cortex. Brain Res. 1150, 225–238. doi: 10.1016/j.brainres.2007.02.089
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H. P., Mendoza, G., et al. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556. doi: 10.1038/35000592
Barnwell, L. F., Lugo, J. N., Lee, W. L., Willis, S. E., Gertz, S. J., Hrachovy, R. A., et al. (2009). Kv4.2 knockout mice demonstrate increased susceptibility to convulsant stimulation. Epilepsia 50, 1741–1751. doi: 10.1111/j.1528-1167.2009.02086.x
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193. doi: 10.1038/nrn2093
Cai, X., Liang, C. W., Muralidharan, S., Kao, J. P., Tang, C.-M., and Thompson, S. M. (2004). Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron 44, 351–364. doi: 10.1016/j.neuron.2004.09.026
Castro, P. A., Cooper, E. C., Lowenstein, D. H., and Baraban, S. C. (2001). Hippocampal heterotopia lack functional Kv4.2 potassium channels in the methylazoxymethanol model of cortical malformations and epilepsy. J. Neurosci. 21, 6626–6634.
Cheng, H. Y., Pitcher, G. M., Laviolette, S. R., Whishaw, I. Q., Tong, K. I., Kockeritz, L. K., et al. (2002). DREAM is a critical transcriptional repressor for pain modulation. Cell 108, 31–43. doi: 10.1016/S0092-8674(01)00629-8
Choi, E. K., Miller, J. S., Zaidi, N. F., Salih, E., Buxbaum, J. D., and Wasco, W. (2003). Phosphorylation of calsenilin at Ser63 regulates its cleavage by caspase-3. Mol. Cell. Neurosci. 23, 495–506. doi: 10.1016/S1044-7431(03)00072-1
Choi, J. D., Bowers-Komro, M., Davis, M. D., Edmondson, D. E., and McCormick, D. B. (1983). Kinetic properties of pyridoxamine (pyridoxine)-5′-phosphate oxidase from rabbit liver. J. Biol. Chem. 258, 840–845.
Elger, C. E., Helmstaedter, C., and Kurthen, M. (2004). Chronic epilepsy and cognition. Lancet Neurol. 3, 663–672. doi: 10.1016/S1474-4422(04)00906-8
Ferhat, L., Esclapez, M., Represa, A., Fattoum, A., Shirao, T., and Ben-Ari, Y. (2003). Increased levels of acidic calponin during dendritic spine plasticity after pilocarpine-induced seizures. Hippocampus 13, 845–858. doi: 10.1002/hipo.10136
Freiman, T. M., Eismann-Schweimler, J., and Frotscher, M. (2011). Granule cell dispersion in temporal lobe epilepsy is associated with changes in dendritic orientation and spine distribution. Exp. Neurol. 229, 332–338. doi: 10.1016/j.expneurol.2011.02.017
Frick, A., Magee, J., and Johnston, D. (2004). LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat. Neurosci. 7, 126–135. doi: 10.1038/nn1178
Gohla, A., Birkenfeld, J., and Bokoch, G. M. (2005). Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat. Cell Biol. 7, 21–29. doi: 10.1038/ncb1201
Gomez-Villafuertes, R., Torres, B., Barrio, J., Savignac, M., Gabellini, N., Rizzato, F., et al. (2005). Downstream regulatory element antagonist modulator regulates Ca2+ homeostasis and viability in cerebellar neurons. J. Neurosci. 25, 10822–10830. doi: 10.1523/JNEUROSCI.3912-05.2005
González-Burgos, I., López-Vázquez, M. A., and Beas-Zárate, C. (2004). Density, but not shape, of hippocampal dendritic spines varies after a seizure-inducing acute dose of monosodium glutamate in rats. Neurosci. Lett. 363, 22–24. doi: 10.1016/j.neulet.2004.03.035
Hoffman, D. A., Magee, J. C., Colbert, C. M., and Johnston, D. (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875. doi: 10.1038/43119
Hong, Y. M., Jo, D. G., Lee, M. C., Kim, S. Y., and Jung, Y. K. (2003). Reduced expression of calsenilin/DREAM/KChIP3 in the brains of kainic acid-induced seizure and epilepsy patients. Neurosci. Lett. 340, 33–36. doi: 10.1016/S0304-3940(03)00067-3
Hyun, H. W., Ko, A. R., and Kang, T. C. (2016). Mitochondrial translocation of high mobility group box 1 facilitates LIM kinase 2-mediated programmed necrotic neuronal death. Front. Cell Neurosci. 10:99. doi: 10.3389/fncel.2016.00099
Jimenez-Mateos, E. M., Engel, T., Merino-Serrais, P., Fernaud-Espinosa, I., Rodriguez-Alvarez, N., Reynolds, J., et al. (2015). Antagomirs targeting microRNA-134 increase hippocampal pyramidal neuron spine volume in vivo and protect against pilocarpine-induced status epilepticus. Brain Struct. Funct. 220, 2387–2399. doi: 10.1007/s00429-014-0798-5
Jimenez-Mateos, E. M., Engel, T., Merino-Serrais, P., McKiernan, R. C., Tanaka, K., Mouri, G., et al. (2012). Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat. Med. 18, 1087–1094. doi: 10.1038/nm.2834
Jung, S. C., Kim, J., and Hoffman, D. A. (2008). Rapid, bidirectional remodeling of synaptic NMDA receptor subunit composition by A-type K+ channel activity in hippocampal CA1 pyramidal neurons. Neuron 60, 657–671. doi: 10.1016/j.neuron.2008.08.029
Kang, T. C., Kim, D. S., Kwak, S. E., Kim, J. E., Won, M. H., Kim, D. W., et al. (2006). Epileptogenic roles of astroglial death and regeneration in the dentate gyrus of experimental temporal lobe epilepsy. Glia 54, 258–271. doi: 10.1002/glia.20380
Kasai, H., Fukuda, M., Watanabe, S., Hayashi-Takagi, A., and Noguchi, J. (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129. doi: 10.1016/j.tins.2010.01.001
Kim, J., Jung, S. C., Clemens, A. M., Petralia, R. S., and Hoffman, D. A. (2007). Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 54, 933–947. doi: 10.1016/j.neuron.2007.05.026
Kim, J. E., and Kang, T. C. (2011). The P2X7 receptor-pannexin-1 complex decreases muscarinic acetylcholine receptor-mediated seizure susceptibility in mice. J. Clin. Invest. 12, 2037–2047. doi: 10.1172/JCI44818
Kim, J. E., Kim, D. S., Kwak, S. E., Choi, H. C., Song, H. K., Choi, S. Y., et al. (2007). Anti-glutamatergic effect of riluzole: comparison with valproic acid. Neuroscience 147, 136–145. doi: 10.1016/j.neuroscience.2007.04.018
Kim, J. E., Kim, D. W., Kwak, S. E., Kwon, O. S., Choi, S. Y., and Kang, T. C. (2008). Potential role of pyridoxal-5′-phosphate phosphatase/chronophin in epilepsy. Exp. Neurol. 211, 128–140. doi: 10.1016/j.expneurol.2008.01.029
Kim, J. E., Kim, Y. J., Lee, D. S., Kim, J. Y., Ko, A. R., Hyun, H. W., et al. (2016). PLPP/CIN regulates bidirectional synaptic plasticity via GluN2A interaction with postsynaptic proteins. Sci. Rep. 6:26576. doi: 10.1038/srep26576
Kim, J. E., Ryu, H. J., Kim, M. J., and Kang, T. C. (2014). LIM kinase-2 induces programmed necrotic neuronal death via dysfunction of DRP1-mediated mitochondrial fission. Cell Death. Differ. 21, 1036–1049. doi: 10.1038/cdd.2014.17
Kim, Y. J., and Kang, T. C. (2015). The role of TRPC6 in seizure susceptibility and seizure-related neuronal damage in the rat dentate gyrus. Neuroscience 307, 215–230. doi: 10.1016/j.neuroscience.2015.08.054
Lilliehook, C., Bozdagi, O., Yao, J., Gomez-Ramirez, M., Zaidi, N. F., Wasco, W., et al. (2003). Altered Abeta formation and long-term potentiation in a calsenilin knock-out. J. Neurosci. 23, 9097–9106.
Magee, J. C., and Carruth, M. (1999). Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J. Neurophysiol. 82, 1895–1901.
Majewska, A., Brown, E., Ross, J., and Yuste, R. (2000). Mechanisms of calcium decay kinetics in hippocampal spines: role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J. Neurosci. 20, 1722–1734.
Mantegazza, M., Curia, G., Biagini, G., Ragsdale, D. S., and Avoli, M. (2010). Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 9, 413–424. doi: 10.1016/S1474-4422(10)70059-4
Matsu-ura, T., Konishi, Y., Aoki, T., Naranjo, J. R., Mikoshiba, K., and Tamura, T. A. (2002). Seizure-mediated neuronal activation induces DREAM gene expression in the mouse brain. Brain Res. Mol. Brain Res. 109, 198–206. doi: 10.1016/S0169-328X(02)00562-4
McNamara, J. O., Huang, Y. Z., and Leonard, A. S. (2006). Molecular signaling mechanisms underlying epileptogenesis. Sci. STKE. 2006:re12. doi: 10.1126/stke.3562006re12
Miller, L. P., Martin, D. L., Mazumder, A., and Walters, J. R. (1978). Studies on the regulation of GABA synthesis: substrate-promoted dissociation of pyridoxal-5′-phosphate from GAD. J. Neurochem. 30, 361–369. doi: 10.1111/j.1471-4159.1978.tb06538.x
Monaghan, M. M., Menegola, M., Vacher, H., Rhodes, K. J., and Trimmer, J. S. (2008). Altered expression and localization of hippocampal A-type potassium channel subunits in the pilocarpine-induced model of temporal lobe epilepsy. Neuroscience 156, 550–562. doi: 10.1016/j.neuroscience.2008.07.057
Morohashi, Y., Hatano, N., Ohya, S., Takikawa, R., Watabiki, T., Takasugi, N., et al. (2002). Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. J. Biol. Chem. 277, 14965–14975. doi: 10.1074/jbc.M200897200
Seeburg, D. P., and Sheng, M. (2008). Activity-induced Polo-like kinase 2 is required for homeostatic plasticity of hippocampal neurons during epileptiform activity. J. Neurosci. 28, 6583–6591. doi: 10.1523/JNEUROSCI.1853-08.2008
Singh, B., Ogiwara, I., Kaneda, M., Tokonami, N., Mazaki, E., Baba, K., et al. (2006). A Kv4.2 truncation mutation in a patient with temporal lobe epilepsy. Neurobiol. Dis. 24, 245–253. doi: 10.1016/j.nbd.2006.07.001
Stockler, S., Plecko, B., Gospe, S. M. Jr., Coulter-Mackie, M., Connolly, M., van Karnebeek, C., et al. (2011). Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol. Genet. Metab. 104, 48–60. doi: 10.1016/j.ymgme.2011.05.014
Tsaur, M.-L., Sheng, M., Lowenstein, D. H., Jan, Y. N., and Jan, L. Y. (1992). Differential expression of K+ channel mRNAs in the rat brain and down-regulation in the hippocampus following seizures. Neuron 8, 1055–1067. doi: 10.1016/0896-6273(92)90127-Y
Turner, J. M., and Happold, F. C. (1961). Pyridoxamine phosphate-oxidase and pyridoxal phosphate-phosphatase activities in Escherichia coli. Biochem. J. 78, 364–372. doi: 10.1042/bj0780364
Ubbink, J. B., Bissbort, S., Vermaak, W. J., and Delport, R. (1990). Inhibition of pyridoxal kinase by methylxanthines. Enzyme 43, 72–79. doi: 10.1159/000468709
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011). The role of inflammation in epilepsy. Nat. Rev. Neurol. 7, 31–40. doi: 10.1038/nrneurol.2010.178
Wetherington, J., Serrano, G., and Dingledine, R. (2008). Astrocytes in the epileptic brain. Neuron 58, 168–178. doi: 10.1016/j.neuron.2008.04.002
Wong, M. (2005). Modulation of dendritic spines in epilepsy: cellular mechanisms and functional implications. Epilepsy Behav. 7, 569–577. doi: 10.1016/j.yebeh.2005.08.007
Wong, M., and Guo, D. (2013). Dendritic spine pathology in epilepsy: cause or consequence? Neuroscience 251, 141–150. doi: 10.1016/j.neuroscience.2012.03.048
Keywords: cofilin, dendritic spine, DREAM, epilepsy, F-actin, KChIP3
Citation: Kim J-E, Hyun H-W, Min S-J, Lee D-S, Jeon AR, Kim MJ and Kang T-C (2017) PLPP/CIN Regulates Seizure Activity by the Differential Modulation of Calsenilin Binding to GluN1 and Kv4.2 in Mice. Front. Mol. Neurosci. 10:303. doi: 10.3389/fnmol.2017.00303
Received: 22 July 2017; Accepted: 11 September 2017;
Published: 25 September 2017.
Edited by:
Shin-ichi Hisanaga, Tokyo Metropolitan University, JapanReviewed by:
Manuel L. Covarrubias, Thomas Jefferson University, United StatesJaewon Ko, Daegu Gyeongbuk Institute of Science and Technology (DGIST), South Korea
Copyright © 2017 Kim, Hyun, Min, Lee, Jeon, Kim and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tae-Cheon Kang, tckang@hallym.ac.kr