MeCP2 Deficiency in Neuroglia: New Progress in the Pathogenesis of Rett Syndrome
 Xu-Rui Jin
Xu-Rui Jin Xing-Shu Chen
Xing-Shu Chen Lan Xiao
Lan Xiao- 1Department of Histology and Embryology, Faculty of Basic Medicine, Collaborative Program for Brain Research, Third Military Medical University, Chongqing, China
- 2The Cadet Brigade of Clinic Medicine, Third Military Medical University, Chongqing, China
Rett syndrome (RTT) is an X-linked neurodevelopmental disease predominantly caused by mutations of the methyl-CpG-binding protein 2 (MeCP2) gene. Generally, RTT has been attributed to neuron-centric dysfunction. However, increasing evidence has shown that glial abnormalities are also involved in the pathogenesis of RTT. Mice that are MeCP2-null specifically in glial cells showed similar behavioral and/or neuronal abnormalities as those found in MeCP2-null mice, a mouse model of RTT. MeCP2 deficiency in astrocytes impacts the expression of glial intermediate filament proteins such as fibrillary acidic protein (GFAP) and S100 and induces neuron toxicity by disturbing glutamate metabolism or enhancing microtubule instability. MeCP2 deficiency in oligodendrocytes (OLs) results in down-regulation of myelin gene expression and impacts myelination. While MeCP2-deficient microglia cells fail in response to environmental stimuli, release excessive glutamate, and aggravate impairment of the neuronal circuit. In this review, we mainly focus on the progress in determining the role of MeCP2 in glial cells involved in RTT, which may provide further insight into a therapeutic intervention for RTT.
Introduction
Rett Syndrome (RTT) is an X-linked autism spectrum disorder that affects 1 in every 10,000–15,000 newborns in the United States (Chahrour and Zoghbi, 2007). It is specially characterized by a period of seemingly normal development that lasts for 6–18 months after birth. Subsequently, microcephaly and stereotypic hand wringing start to appear (Nomura, 2005) and more special symptoms appear as the age increases, such as a loss of motor coordination, ataxia, gait apraxia, seizures, poor sleep efficiency or parkinsonian features (Roze et al., 2007). In an overwhelming majority (more than 95%) of RTT patients, the syndrome is caused by mutations in a gene called methyl-CpG-binding protein 2 (MeCP2), a transcriptional corepressor that can bind to methylated CpG islands and complex with Sin3 homolog A (Sin3A) and histone deacetylases (HDACs) to regulate gene expression (Lyst and Bird, 2015). Moreover, different mutations are associated with disease severity, and there are approximately 30 types of mutations that can cause RTT. Patients with the R270X mutation and frame shift deletions in a (CCACC)n-rich region present with the most typical symptoms (Bienvenu et al., 2000).
Generally, the symptoms of RTT are attributed to neuronal dysfunction. Substantial evidence shows that neuronal, morphological and functional abnormalities are involved in RTT pathogenesis. For instance, MeCP2-deficient neurons have an abnormal morphology (e.g., fewer dendritic spines and reduced arborization) (Zhou et al., 2006; Smrt et al., 2007; Palmer et al., 2008). The number of synapses is decreased in hippocampi of MeCP2-null mice and conversely, the change was elevated in MeCP2-overexpressing mice (Johnston et al., 2001; Chao et al., 2007; Banerjee et al., 2012). Moreover, the re-expression of MECP2 in MeCP2-null neurons effectively rescued behavioral abnormalities in mice (Luikenhuis et al., 2004).
However, increasing evidence has shown that white matter damage and/or glial cell (i.e., astrocyte, oligodendrocyte and microglia) dysfunction induced by a change in the DNA methylation state is also involved in the pathogenesis of RTT (Ballas et al., 2009; Maezawa and Jin, 2010; Okabe et al., 2012; Durand et al., 2013; Nguyen et al., 2013). Recently, it was reported that MeCP2-null astrocytes are incapable of supporting the normal development of co-cultured wild-type (WT) neurons (Williams et al., 2014). MeCP2-null microglia and astrocytes have been reported to be toxic to neurons through non-cell autonomous mechanisms, including a slower rate of glutamate (Glu) clearance and release of excessive Glu, as well as glial connexin (Maezawa et al., 2009; Maezawa and Jin, 2010). This latter finding proposes a viewpoint that RTT is not simply a disease of neurons alone, but a complex disease in which glial cells might play a vital role in the pathological process. Hence, we will attempt to summarize the progress of glial abnormalities involved in the pathogenesis of RTT in this review.
MeCP2 and RTT
MeCP2 is one of the members of the methyl-CpG-binding domain protein (MBD) family, which is functionally involved in chromatin remodeling or transcriptional regulation. There are two crucial domains in MeCP2:, one is MBD and another is the transcriptional repression domain (TRD), which can recruit different protein partners, such as HDACs and Sin3A, to form a transcriptional repression complex and regulate target gene expression (Du et al., 2015) (Figure 1). The MeECP2 gene consists of four exons (exon 1–4) and three introns (intron 1–3) and is located on the X chromosome. The transcriptional level of MeCP2 exon 1 (E1) is much higher than other exons in the brain, and mutations in MeCP2 E1 are sufficient to cause RTT (Fichou et al., 2009). Furthermore, the MeCP2 isoform has a time-specific expression pattern during brain development. MeCP2 E1 in the mouse hippocampus was detected as early as at E14, whereas MeCP2 E2 was detected at E18 (Olson et al., 2014). Generally, MECP2 was believed to bind to methylated CpG islands; however, a recent study showed that MeCP2 can bind to non-CG methylated DNA and influence the transcription of disease-relevant genes in the adult mouse brain (Chen et al., 2015; Luo and Ecker, 2015) (Figure 1). Those results provide insight into the molecular mechanism of MECP2 in the delayed onset of RTT.
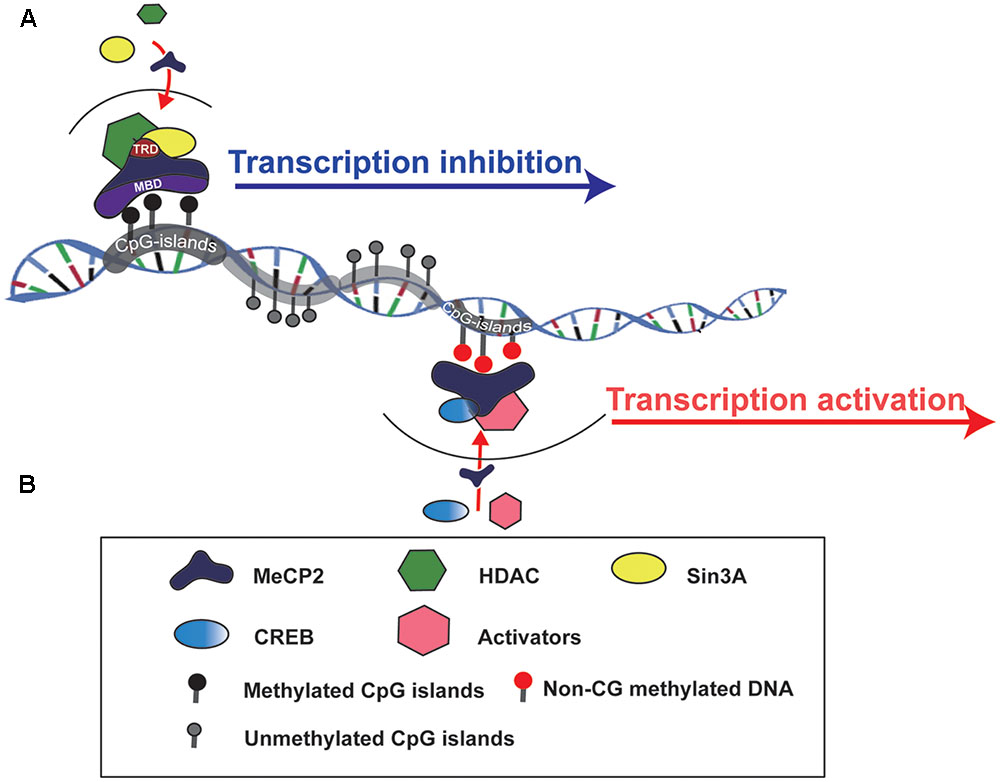
FIGURE 1. Schematic to show how MeCP2 regulates target gene expression. (A) MeCP2 recruits a transcriptional corepressor complex containing Sin3A and histone deacetylase (HDAC) to methylated CpG islands and results in target gene transcription inhibition. TRD, transcriptional repression domain; MBD, methyl-CpG-binding domain. (B) MeCP2 is able to active gene transcription by recruiting CREB and other transcriptional factors to non-CG methylted DNA regions.
Generally, almost 95% of RTT patients carry mutations in the MeCP2 gene, and recent findings demonstrated that two additional genes, cyclin-dependent kinase like 5 (CDKL5) (Evans et al., 2005) and fork head box G1 (FOXG1), can also be involved in the pathogenesis of this syndrome (Mencarelli et al., 2010). Furthermore, CDKL5 has been shown to have the ability to promote the release of MeCP2 from DNA by phosphorylating MeCP2 (Mari et al., 2005; Bertani et al., 2006), while a direct functional relationship between these two molecules in RTT is controversial.
MeCP2 Deficiency and Neuronal Dysfunction
The function of MeCP2 in brain is multifarious, including modulation of neurogenesis, synaptic development and maintenance of neural circuits (Chahrour and Zoghbi, 2007; Banerjee et al., 2012; Lyst and Bird, 2015). It was demonstrated that MeCP2 is essential for neurogenesis in Xenopus embryos, and deficiency of MeCP2 resulted in a decreased number of neuronal precursors (Stancheva et al., 2003). In vitro, MeCP2 mutant mesenchymal stem cells presented impaired neural differentiation and increased the rate of senescence (Squillaro et al., 2012). MeCP2-deficient neurons have decreased numbers of axons and dendrites (Nguyen et al., 2012), and the neurite complexity was reduced in cultured MeCP2-null embryonic primary cortical neurons (Vacca et al., 2016), indicating that MeCP2 plays a crucial role in modulating neuronal differentiation and terminal maturation. Recent studies have revealed that MeCP2 is also involved in neuronal cell fate specification and migration (Feldman et al., 2016). It was found that neural progenitor cells (NPCs) lacking MeCP2 exhibited delayed corticogenesis with respect to abnormal migration of NPCs from the subventricular and ventricular zones into the cortical plate (Bedogni et al., 2016).
Some of the abnormal social behaviors of patients with RTT, such as anxiety and autistic features, are thought to be caused by MeCP2 deficiency in the neurons of certain special brain areas, including the forebrain, hypothalamus and basolateral amygdala (Armstrong et al., 1998; Gemelli et al., 2006; Fyffe et al., 2008; Chao et al., 2010; Williams et al., 2014). MECP2 conditional knockout in glutamatergic neurons, but not in inhibitory neurons, leads to more serious RTT-like symptoms in mice (Meng et al., 2016). It has been shown that the balance between Glu excitatory synapses and GABAergic (gamma-amino butyric acid) inhibitory synapses is disrupted in RTT (Nelson and Valakh, 2015). MeCP2 knockdown reduces the excitatory synapse number and attenuates synaptic scale-up by reducing the expression of metabotropic glutamate receptor 2 (GluR2), and there is a major down-regulation of GABAergic inhibitory synapses in MeCP2 knockout mice (He et al., 2014; Kang et al., 2014). Thus, correcting the abnormal MeCP2 level by adding the gene back or over-expressing is a valid method to study the function of MeCP2 and treatment for RTT. For instance, dendritic abnormalities and behavioral changes can be ameliorated by reactivation of MeCP2 expression in MeCP2-null mice (Armstrong et al., 1998; Stearns et al., 2007; Robinson et al., 2012). Recently, MECP2 gene therapy by intracisternal injection of transgenic adeno-associated virus 9 (AAV9/hMECP2) has been shown to extend survival of MeCP2-deficient (Mecp2-/y) mice without apparent toxicity (Matagne et al., 2017; Sinnett et al., 2017). Moreover, as RTT is always caused by heterozygous mutations in an X-linked gene and there exists a prevalent wild-type MeCP2 allele on the inactive X chromosome, reactivation of the inactive X chromosome-linked wild-type allele may represent another way to rescue MeCP2 deficiency. Recently, several trans-acting X-chromosome inactivation factors (XCIFs) have been identified and the inhibitor of two XCIFs (PDPK1, AURKA) has been demonstrated to exhibit reactive expression of the WT MeCP2 allele on the inactive X chromosome, which can be considered as a potential therapy for RTT (Bhatnagar et al., 2014).
In addition to RTT, evidence has shown that MeCP2 regulates the expression of its downstream genes, such as brain-derived neurotrophic factor (BDNF) (Chen et al., 2003) and ubiquitin-protein ligase E3A (UBE3a), the latter being involved in Angelman syndrome (Makedonski et al., 2005). In addition, MeCP2 also regulates the expression of distal-less homeobox 5/6 (Dlx5/6) genes, which are necessary for spinal skeletal development and are related to definite symptoms of RTT patients, such as scoliosis and microcephaly (Nakashima et al., 2010).
MeCP2 Deficiency and Glial Cell Dysfunction
Glial cells are non-excitable cells that are functional support neurons and maintain the stability of neuronal structure and function. In the central nervous system (CNS), glial cells mainly include astrocytes, OLs and microglia. Previous studies have revealed that MeCP2 is present in a majority of neurons but is absent from glial cells (Shahbazian et al., 2002). However, increasing evidence suggests that the abnormality of MeCP2 also plays an important role in white matter damage and/or glial dysfunction in RTT (Ballas et al., 2009; Maezawa and Jin, 2010; Okabe et al., 2012; Durand et al., 2013; Nguyen et al., 2013). Mice with MeCP2 loss specifically in glial cells presented Rett-like symptoms due to neuronal toxicity via a non-cell autonomous mechanism, and the restoration of MeCP2 in glial cells can rescue some of these defects (Lioy et al., 2011; Nguyen et al., 2013; Cronk et al., 2015). Evidence from magnetic resonance spectroscopy (MRS) indicated an increased glia-to-neuron ratio in the white matter of RTT patients along with ongoing axonal damage and glial abnormalities (Khong et al., 2002). The contribution of MeCP2 deficiency to dysfunction of different types of glial cells varies in RTT pathogenesis.
MeCP2 in Astrocytes
Astrocytes are the most abundant of all glial cell types and are well known for supporting neurons and maintaining brain function. Although the expression level of MeCP2 in astrocytes is lower, it is important for astrocyte differentiation and function. After MECP2 was specifically knocked out in neural stem cells (NSCs), these NSCs tend to differentiate into more astrocytes (Andoh-Noda et al., 2015). While the growth rate of MeCP2-deficient astrocytes is significantly slower and more interleukin (IL)-1β and IL-6 are released in response to immune stimulation in vitro, no obvious morphological change was found in those cells (De Filippis et al., 2012). MeCP2-deficient astrocytes that were differentiated from induced pluripotent stem cell (iPSC) lines from RTT patients have negative regulatory effects on neuronal morphology and function (Williams et al., 2014). Moreover, subsequent studies found that MeCP2-null astrocytes have certain abnormalities in target gene regulation and are toxic to neurons mainly due to abnormal Glu metabolism. Similarly, it was shown that a state of MeCP2 deficiency can spread in the brain of MeCP2-/+ mice via a non-cell autonomous mechanism (Maezawa et al., 2009). This suggests that abnormal astrocytes are able to deteriorate the function of neurons, which speeds up the process of RTT. It was found that preferential re-expression of Mecp2 in astrocytes dramatically improved RTT symptoms and the lifetimes of MeCP2-deficient mice (Kifayathullah et al., 2010; Lioy et al., 2011; Zachariah et al., 2012).
Target Gene Dysregulation in MeCP2-Null Astrocytes
The expression of astroglial marker genes GFAP and S100β was significantly higher in MeCP2-null astrocytes than that in WT astrocytes (Forbes-Lorman et al., 2014). It was noted that MECP2 small interference RNA (siRNA) increased the expression of GFAP in the female amygdala (Forbes-Lorman et al., 2014). These observations suggest that GFAP and S100β genes are suppressed by MeCP2. Although the mechanism by which MeCP2 regulates these two genes is unclear, evidence has shown that MeCP2 E1 can couple with the Sin3A/HDAC complex, which can bind to the GFAP promoter and regulate GFAP transcription. On embryonic day 11.5 (E11.5), the promoter of the GFAP gene is highly methylated, which may facilitate assembly of MeCP2-Sin3A/HDAC complexes and suppress GFAP gene transcription. On E14, the promoter of the GFAP gene is demethylated, which may lead to the disintegration of this complex and results in GFAP gene expression. In the early stages of astrocyte differentiation, MeCP2 also has a vital effect by binding to the promoter of the S100β gene, while this binding is gradually reduced in the later stages, as demethylation of a specific CpG site occurs (Cheng et al., 2011; Forbes-Lorman et al., 2014).
In addition to astroglial marker genes, many other target genes such as solute carrier family member 38, member 1 (Slc38a1), neuronal regeneration-related protein (Nrep), and nuclear receptor subfamily 2, group F, member 2 (Nr2f2) are also regulated by MeCP2. Slc38a1 is a rate-limiting transporter of glutamine (Gln) across the plasma membrane, Nrep is a transcriptional factor that is involved in glial mobility and neoplasia, and Nr2f2 is a transcription factor that is necessary for glial differentiation from NSCs (Mackenzie and Erickson, 2004; Yasui et al., 2013; Delepine et al., 2015). In addition, Nr2f2 can regulate some target genes including those encoding chromogranin B (Chgb), chemokine (C-C motif) ligand 2 (CCL2), and lipocalin 2 (LCN2). Chgb is a highly efficient system that is directly involved in monoamine accumulation. In MeCP2-deficient mice, the expression of Nr2f2 is up-regulated, which may down-regulate Chgb and cause excess monoamine accumulation in the extracellular fluid and impair neurons. This finding suggests that influence of MeCP2 deficiency can be aggravated through a downstream target gene cascade in astrocytes (Kloukina-Pantazidou et al., 2013; Delepine et al., 2015). Interestingly, in response to LPS stimulation, MeCP2-deficient astrocytes released fewer cytokines such as IL-1β and IL-6 (Maezawa et al., 2009).
Microtubule Instability in MeCP2-Null Astrocytes
Cellular morphology, division, migration and intracellular transportation of vesicles are controlled by microtubules (MTs), which assemble from α- and β-tubulin dimers. The acetylation modification of tubulin is deemed to be a characteristic of stable MTs (Palazzo et al., 2003). In Mecp2308/y and Mecp2 p. Arg294∗ iPSC-derived astrocytes, the level of acetylated tubulin is reduced, and histone deacetylase 6 (HDAC6) is overexpressed (Delepine et al., 2016). A similar result was also reported in MeCP2-deficient fibroblasts and MeCP2-null neurons (Gold et al., 2015). MT growth speed of Mecp2308/y astrocytes was higher, and MT polymerization was significantly increased. Moreover, MT-dependent lysosome vesicle mobility was obviously increased in both Mecp2308/y and Mecp2 p.Arg294∗ iPSC-derived astrocytes; however, the percentage of highly directional vesicles was reduced (Delepine et al., 2016). A special type of directional vesicle cellular transport can regulate Glu uptake, which suggests an abnormal Glu uptake rate may be associated with MT-dependent vesicle mobility in RTT (Li et al., 2015). The aforementioned phenomenon was related to the decreased expression of stathmin 2 (STMN2), which could inhibit MT polymerization (Nectoux et al., 2012). Moreover, expression of TUBA1B, which encodes the ubiquitous α-tubulin, is down-regulated in brain tissue of patients with RTT (Abuhatzira et al., 2009). These findings suggest a role of MeCP2 as an activator for MT-associated genes. Molecularly, a recent study revealed a potential mechanism by which sumoylated MeCP2 releases CREB from the repressor complex and increases the transcription of CREB-regulated genes such as BDNF (Tai et al., 2016). Interestingly, sumoylation of MeCP2 was found to be decreased in RTT, and further study is warranted to examine the sumoylation status of MeCP2 in glial cells of RTT. In addition, epothilone D, a brain-penetrating MT-stabilizing natural product, can rescue MT growth velocity, and epothilone D corrects the abnormal behavioral symptoms of Mecp2308/y mice (Delepine et al., 2016). Treatment via targeting MTs seems to be a new approach for RTT therapy. Notably, when co-cultured with MeCP2-deficient fibroblasts, MT stability of WT human fibroblasts is reduced (Delepine et al., 2013). All of this evidence suggests that MT impairment seems to be a common phenomenon of MeCP2 deficiency.
Abnormal Glutamate Metabolism in MeCP2-Deficient Astrocytes
Glutamate is an important signaling molecule in the CNS. At a high extracellular concentration, it is a potent cytotoxin that can induce both neuronal and glial death through excitotoxicity or oxidative stress. Extracellular glutamate concentration is maintained at an appropriate level predominantly by active transport mediated by excitatory amino acid transporters (EAATs) of astrocytes (Lehmann et al., 2009). Normally, when astrocytes are incubated with high levels of extracellular Glu, EAAT1/EAAT2 expression is rapidly decreased. However, when the down-regulation of EAAT1/2 was impaired, EAAT1/EAAT2 expression had no obvious changes in MeCP2-null astrocytes (Okabe et al., 2012). Regarding the underlying mechanism, it was found that HDAC I and II served as repressors for EAAT2 promoter activity (Karki et al., 2014), thus, MeCP2 deficiency may fail to recruit HDACs to inhibit EAAT gene expression upon environment stimulation. Notably, the Glu clearance rate of MeCP2-null astrocytes was lower than that of WT astrocytes in vitro (Okabe et al., 2012). Moreover, the glutamine synthetase (GS) protein was significantly higher in MeCP2-null astrocytes than that in WT astrocytes (Okabe et al., 2012). All of this evidence seems to support the concept that both Glu clearance and production are abnormal in MeCP2-deficient astrocytes, and that these abnormalities may contribute to the pathological process of RTT (Figure 2).
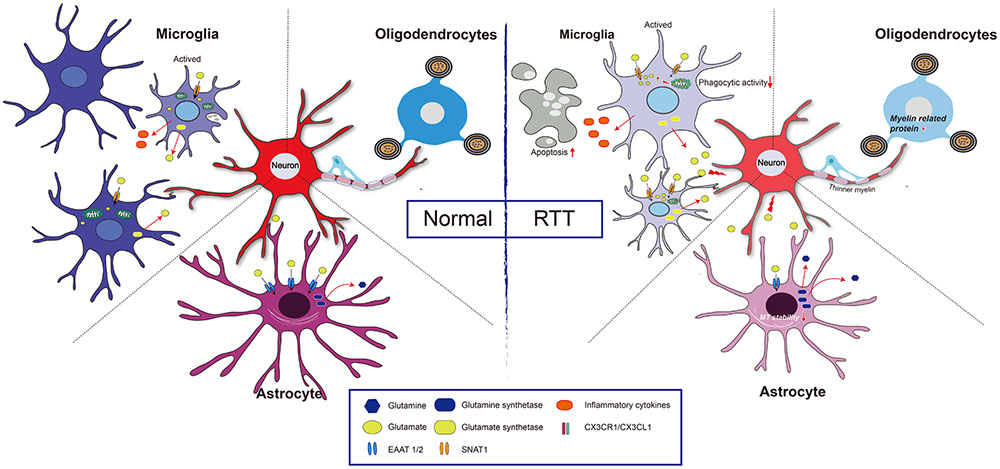
FIGURE 2. Glia and neuron interaction under pathologic condition of RTT. Normal: Extracellular glutamate (Glu) can be uptake by excitatory amino acid transporter 1 and 2 (EAAT1/2) in astrocyte and sodium-coupled neutral amino acid transporter 1 (SNAT1) in microglial cell, and is utilized by glutamine synthetase to reduce its toxicity to neuron. Both astrocytes and microglia can be active upon stimulation, and the active microglia has capacity of phagocytosis. Furthermore, CX3CR1 (a chemokine receptor in microglial cell) and its neuronal ligand CX3CL1 mediates neuron-microglia interaction. Myelinating oligodendrocyte is important to maintain axonal integrity. RTT: MeCP2 deficiency in astrocytes leads to a decreased expression of EAAT1/2, and increased expression of glutamine synthetase, which results in a high concentration of extracellular Glu, and thus impairs neuronal activity. Additionally, those astrocytes present decreases microtubule stability. In microglia, MeCP2 deficiency results in decreased phagocytic activity and overloads of Glu due to upregulation of SNAT1. The MeCP2-deficient in oligodendrocyte results in thinner myelin due to down-regulation of myelin-related genes.
Spread of MeCP2 Deficiency in Astrocytes
An interesting phenomenon is that MeCP2 expression in Mecp2-/+ mice is much lower at 7 months than that at 1 month, when proliferation rates of astrocytes are higher. In vitro, MeCP2 expression was significantly decreased in WT astrocytes that were co-cultured with MeCP2-deficient astrocytes for a long period, indicating that MeCP2 deficiency can spread among astrocytes progressively and eventually lead to a dysfunctional brain (Maezawa et al., 2009). Recent studies have shown that MeCP2 deficiency can spread among astrocytes via connexin, especially connexin 43 (Cx43)-mediated gap junction. When Cx43 was knocked down with siRNA, the spread of MeCP2 deficiency was significantly reduced (Maezawa et al., 2009). This finding suggests that astroglial Cx may be a new target for preventing the phenotype of RTT.
Abnormal Astrocytes May Be the Main Contributor to the Disordered Breathing Pattern
RTT symptoms always include a severely disordered breathing pattern and reduced CO2 sensitivity. In the respiratory control center, highly chemosensitive astrocytes can respond to the physiological decrease in pH with a vigorous increase in intracellular Ca2+ and release of ATP (Gourine et al., 2010). MeCP2 deficiency in astrocytes induced an obvious depression of the ventilatory responses to an increased level of CO2, which is similar to global MeCP2-null mice (Zhang et al., 2011). Additionally, although neurons constitute the respiratory control network and determine the ventilatory response to CO2, MeCP2 mutation selectively in neurons leads to an absolute lower depression of the CO2 response than MeCP2 loss in astrocytes (Fyffe et al., 2008; Garg et al., 2015). After re-expression of MeCP2 in astrocytes, the respiratory phenotype is rescued (Garg et al., 2015), suggesting that the disordered breathing pattern may be caused by abnormal astrocytes in RTT patients. However, recent evidence suggests that RTT-like breathing abnormality was only developed in 20% of Mecp2+/- mice (Johnson et al., 2015). Imaging of calcium signaling in ventral medullary astrocytes reveals that these phenomena may be related to a reduction in the ability of MeCP2-deficient astrocytes to sense PCO2/[H+] and that the CO2-induced [Ca2+]i response is impaired. Additionally, the ATP response of MeCP2-deficient astrocytes is not affected. Normally, ATP propagates astrocytic Ca2+ excitation, depolarizes chemoreceptor neurons, and induces adaptive increases in breathing. This evidence suggests that the most apparent deficiency of the MeCP2-null astrocyte is the ability to sense physiological increases in CO2, while the downstream signaling pathway can still be activated by ATP (Bissonnette and Knopp, 2008; Zhang et al., 2011; Turovsky et al., 2015).
MeCP2 in Microglia
Mecp2 Regulates Microglial Activation
Microglia are considered to be resident myeloid-derived cells distributed throughout the brain and account for over 10% of CNS cells (Kawabori and Yenari, 2015). Microglial activation can occur in response to stimuli such as glucocorticoids, hypoxia, and inflammation (Kiernan et al., 2016). In Mecp2-null mice, microglial cells are relatively small but become larger in response to environmental stimuli compared to those of wild-type mice, and these microglial cells are lost with disease progression. Moreover, the expression of glucocorticoid-induced transcriptional signature genes and a subset of hypoxia-inducible genes were up-regulated in Mecp2-deficient microglia, suggesting the regulatory role of MeCP2 in microglia activation (Cronk et al., 2015) (Figure 2). From another point of view, a gene array study confirmed that the function of differentially expressed genes, as MeCP2 deficiency is strongly related to regulating the activation states of microglia (Zhao et al., 2017). In addition, studies have shown that bone marrow transplantation increases the number of microglia in the brain (Derecki et al., 2012). MeCP2-null mice that were transplanted with WT bone marrow partly restored behavior and functional abnormalities (Derecki et al., 2012). However, another study reached the opposite conclusion that WT marrow transplantation did not positively influence MeCP2-null mice (Wang et al., 2015). The beneficial effect declined when the phagocytic activity of these transplanted cells was inhibited and the ability to clear apoptotic targets was reduced. This result indicated that the phagocytic activity of microglia is impaired in MeCP2-null microglia and that a non-cell autonomous mechanism is involved in spreading the MeCP2-deficient state (Maezawa and Jin, 2010). In contrast to microglia, the MeCP2 knockout of myeloid-derived cells led to the release of more tumor necrosis factor-α (TNF-α) and inflammatory cytokines, such as IL-6, TNF-α, and IL-3 (O’Driscoll et al., 2015).
Mutant MECP2 in Microglia Impairs the Neuronal Circuit
In the developing CNS, microglia have been demonstrated to play an important role in shaping neuronal circuit structure by microglia-synapse interactions or removing excess synapses, and an etiology of disrupted synaptic function can be detected in mouse models of RTT (Johnston et al., 2001; Noutel et al., 2011). Recent research offered a new viewpoint of the effect of MECP2-defective microglia on the neuronal circuit. It was reported that in MECP2-null mice, microglia contribute to RTT pathogenesis by excessively engulfing and thereby eliminating, which is concomitant with synaptic loss at the end stages of the disease (Schafer et al., 2016). Intriguingly, gain or loss of Mecp2 expression specifically in microglia cannot induce the abovementioned phenomena (Schafer et al., 2016), indicating that abnormalities in microglia may accelerate the pathological process by impairing the neuronal circuit as a secondary effect, not the primary cause, of RTT. In addition, the result of another research study also implicitly demonstrated this point that restoring the expression of Mecp2 in myeloid cells by bone marrow transplantation made no sense in the Mecp2-null mouse (Wang et al., 2015).
A recent study showed that ablating CX3CR1, a chemokine receptor of microglia mediating neuron–microglia interaction by pairing with its neuronal ligand CX3CL1, can rescue the negative effect of MeCP2-deficient microglia on neurons. After blocking the interaction, the survival of MECP2-null mice was remarkably improved and behavioral abnormalities were partly restored. These results indicated that blocking intercellular interaction in brains of RTT patients might be a novel therapeutic approach for RTT (Horiuchi et al., 2016).
MeCP2-Deficient Microglia Leads to Neurotoxicity
Activation of glutamate receptors and the expression level of Glu transporters are important for maintaining the plasticity of glutamatergic synapses (Groc et al., 2006; Yuan and Bellone, 2013). MeCP2-deficient microglia released a fivefold higher level of Glu, which was associated with neurotoxic or synaptotoxic activity, as the neuronal abnormality can be partially restored by blocking the NMDA receptor (Maezawa and Jin, 2010). In addition,which can release Glu, was significantly up-regulated in MeCP2-null microglia, and the blockade of Cx32 can partially ameliorate the excessive release of Glu (Maezawa and Jin, 2010).
From the perspective of bioenergetics, MeCP2 loss in microglia led to mitochondrial dysfunction by impairing Glu homeostasis (Jin et al., 2015). In MeCP2-deficient microglia, the overexpression of the Glu transporter sodium-coupled neutral amino acid transporter 1 (SNAT1) promoted microglial uptake of Glu; the excessive Glu was translated into the mitochondria, coupled with the rising mitochondrial reactive oxygen and damaged the mitochondria (Jin et al., 2015). As a consequence, adenosine triphosphate (ATP) production is significantly reduced in MeCP2-deficient microglia, and this ATP reduction leads to a greater potential for the apoptosis of abnormal microglia (Jin et al., 2015) (Figure 2).
MeCP2 in Oligodendroglia
Oligodendrocytes are the myelinating cells in the brain that maintain nerve impulse conduction and provide nutrition for axons (recently reviewed in Bergles and Richardson, 2016). There are several markers that distinguish the oligodendroglial cells at different stages. OL progenitor cells (OPCs) are positive for platelet-derived growth factor receptor α (PDGFR-α) or neural antigen 2 (NG2); immature OLs are O4-positive, and mature OLs express myelin-associated glycoprotein (MAG), proteolipid protein (PLP), CNP and myelin basic protein (MBP) (He and Lu, 2013).
Some studies have found the expression of MeCP2 in oligodendroglial lineage cells (KhorshidAhmad et al., 2016), which may be associated with oxidative damage of white matter that occurred in the early stages of RTT (Durand et al., 2013). In MeCP2-null mice, although the OL morphology is unchanged, the expression of myelin-related genes is partially changed in some specific regions. For example, in the frontal cortex of MeCP2-null mice, no changes in global expression levels of the MBP, MAG and PLP were found, but MAG expression was significantly increased in the corpus callosum, while PLP expression was significantly decreased in the cerebellum (Vora et al., 2010). Furthermore, in Mecp2308/y-mutant mice, GFAP, Cx43, Cx45, Cx40 and Cx32 were unchanged, but the CNP expression level was decreased (Wu et al., 2012). Additionally, cultured OLs with MeCP2 knocked down exhibited an increase in myelin genes, including MBP, PLP, myelin oligodendrocyte glycoprotein (MOG), myelin-associated oligodendrocyte basic protein (MOBP) and Ying Yang 1 (YY1) (KhorshidAhmad et al., 2016), indicating MeCP2 is a negative regulator for myelin protein expression (Sharma et al., 2015). As a co-repressor of MeCP2, HDACs have also been found to be functionally involved in oligodendroglial development (Huang et al., 2015). Moreover, when MeCP2 was specifically knocked out in NG2+ OPCs, the mice displayed more active behaviors, with severe hind limb-clasping phenotypes (Nguyen et al., 2013). The PLP and MBP expression levels were lower in those mice, and thinner myelin was present. After restoring the expression of MeCP2 in the OPCs, the ethological abnormality was significantly corrected in both female and male mice (Nguyen et al., 2013) (Figure 2). It is noteworthy that MeCP2 re-expression in OPCs, compared with that in astrocytes or microglia cells, shows more potent ability to prolong the lifespan of MeCP2stop/y mice (Nguyen et al., 2013). However, the expression of myelin protein (MBP) was mildly or not rescued (CNP, MOG, or PLP) after re-expression of MeCP2 in oligodendrocytes in otherwise MeCP2-null mice, suggesting that a likely non-cell autonomous mechanism regulated the expression of OL-related genes (Nguyen et al., 2013). This mechanism is maybe related to the excessive Glu released by other glial cell types, such as astrocytes or microglia, because excessive Glu might partly restore the development of OLs through activating n-methyl-D-aspartic acid (NMDA) receptors in MeCP2-null brains (Lundgaard et al., 2013). In addition, white matter damage in RTT patients has been related to impaired nuclear factor kappa-B (NF-κB) signaling: there were additional copies of the inhibitor of the kappa light polypeptide gene, kinase gamma (IKBKG) enhancer in B-cells of patients with Xq28 duplications, and three of the five presented white matter anomalies in brain MRI (Philippe et al., 2013).
Perspectives
In this review, we summarized glial abnormalities induced by MeCP2 deficiency and how these cells are toxic to neurons and disturb the neural circuit (Table 1). The mice with MeCP2-null specifically in glial cells showed behavioral and neural synaptic abnormalities that are similar to those in MeCP2-null mice, and re-expression of MeCP2 in glial cells can restore those abnormalities and prolong the life span of MeCP2-null mice. Additionally, we discussed the role of MeCP2 in glial cells to gain a better understanding of the pathology process of RTT and ultimately a new glial-target strategy for RTT treatment. For instance, Glu metabolism abnormalities may be a predominant component of the RTT pathology process, especially in glial cells. Some drugs such as riluzole, a glutamatergic modulator that has been approved for treating amyotrophic lateral sclerosis (ALS) by primarily rectifying extensive Glu levels, may also be utilized to treat RTT. Moreover, the regulatory mechanism of MeCP2 in the oligodendroglial development involved in RTT pathogenesis is understudied.
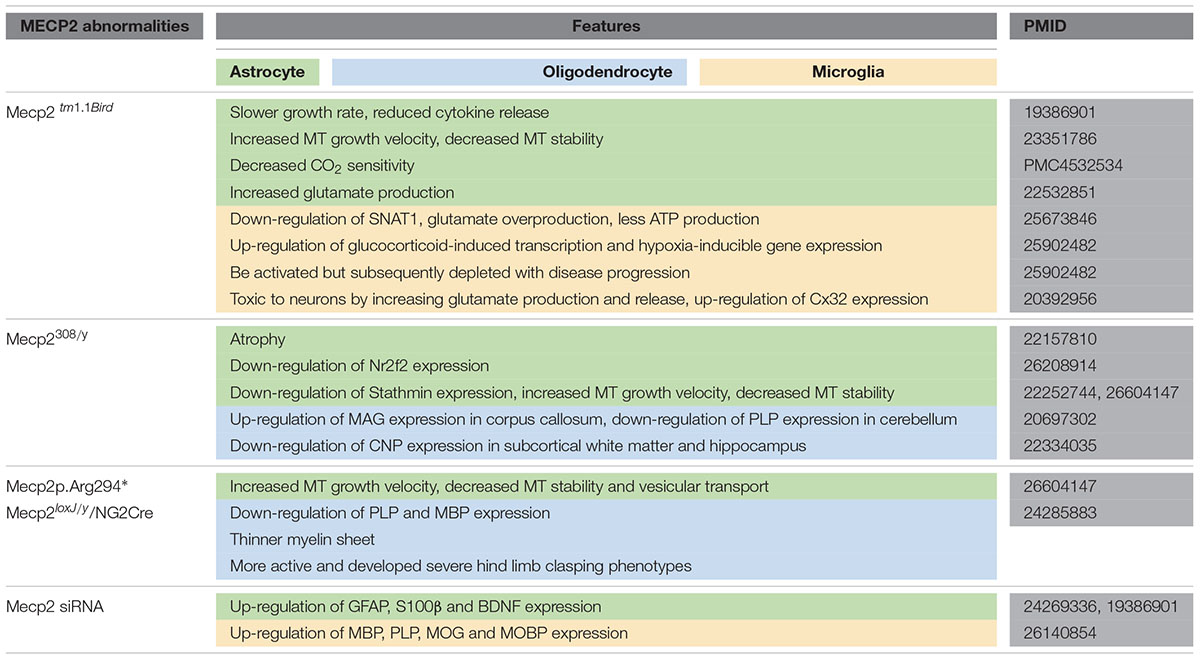
TABLE 1. A summary of the abnormal features in glial cells induced by MeCP2 abnormalities.
Author Contributions
X-RJ and X-SC wrote the manuscript, LX designed and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (31100771), Natural Science Foundation Project of CQ CSTC (2016jcyjA0600, CSTCKJCXLJRC07), and Postdoctoral Foundation of CQ CSTC (2011018).
References
Abuhatzira, L., Shemer, R., and Razin, A. (2009). MeCP2 involvement in the regulation of neuronal alpha-tubulin production. Hum. Mol. Genet. 18, 1415–1423. doi: 10.1093/hmg/ddp048
Andoh-Noda, T., Akamatsu, W., Miyake, K., Matsumoto, T., Yamaguchi, R., Sanosaka, T., et al. (2015). Differentiation of multipotent neural stem cells derived from Rett syndrome patients is biased toward the astrocytic lineage. Mol. Brain 8:31. doi: 10.1186/s13041-015-0121-2
Armstrong, D. D., Dunn, K., and Antalffy, B. (1998). Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J. Neuropathol. Exp. Neurol. 57, 1013–1017. doi: 10.1097/00005072-199811000-00003
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317. doi: 10.1038/nn.2275
Banerjee, A., Castro, J., and Sur, M. (2012). Rett syndrome: genes, synapses, circuits, and therapeutics. Front. Psychiatry 3:34. doi: 10.3389/fpsyt.2012.00034
Bedogni, F., Cobolli Gigli, C., Pozzi, D., Rossi, R. L., Scaramuzza, L., Rossetti, G., et al. (2016). Defects during Mecp2 null embryonic cortex development precede the onset of overt neurological symptoms. Cereb. Cortex 26, 2517–2529. doi: 10.1093/cercor/bhv078
Bergles, D. E., and Richardson, W. D. (2016). Oligodendrocyte development and plasticity. Cold Spring Harb. Perspect. Biol. 8:a020453. doi: 10.1101/cshperspect.a020453
Bertani, I., Rusconi, L., Bolognese, F., Forlani, G., Conca, B., De Monte, L., et al. (2006). Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 281, 32048–32056. doi: 10.1074/jbc.M606325200
Bhatnagar, S., Zhu, X., Ou, J., Lin, L., Chamberlain, L., Zhu, L. J., et al. (2014). Genetic and pharmacological reactivation of the mammalian inactive X chromosome. Proc. Natl. Acad. Sci. U.S.A. 111, 12591–12598. doi: 10.1073/pnas.1413620111
Bienvenu, T., Carrie, A., De Roux, N., Vinet, M. C., Jonveaux, P., Couvert, P., et al. (2000). MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum. Mol. Genet. 9, 1377–1384. doi: 10.1093/hmg/9.9.1377
Bissonnette, J. M., and Knopp, S. J. (2008). Effect of inspired oxygen on periodic breathing in methy-CpG-binding protein 2 (Mecp2) deficient mice. J. Appl. Physiol. 104, 198–204. doi: 10.1152/japplphysiol.00843.2007
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chao, H. T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65. doi: 10.1016/j.neuron.2007.08.018
Chen, L., Chen, K., Lavery, L. A., Baker, S. A., Shaw, C. A., Li, W., et al. (2015). MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 112, 5509–5514. doi: 10.1073/pnas.1505909112
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., et al. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889. doi: 10.1126/science.1086446
Cheng, P. Y., Lin, Y. P., Chen, Y. L., Lee, Y. C., Tai, C. C., Wang, Y. T., et al. (2011). Interplay between SIN3A and STAT3 mediates chromatin conformational changes and GFAP expression during cellular differentiation. PLOS ONE 6:e22018. doi: 10.1371/journal.pone.0022018
Cronk, J. C., Derecki, N. C., Ji, E., Xu, Y., Lampano, A. E., Smirnov, I., et al. (2015). Methyl-CpG binding protein 2 regulates microglia and macrophage gene expression in response to inflammatory stimuli. Immunity 42, 679–691. doi: 10.1016/j.immuni.2015.03.013
De Filippis, B., Fabbri, A., Simone, D., Canese, R., Ricceri, L., Malchiodi-Albedi, F., et al. (2012). Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of Rett syndrome. Neuropsychopharmacology 37, 1152–1163. doi: 10.1038/npp.2011.301
Delepine, C., Meziane, H., Nectoux, J., Opitz, M., Smith, A. B., Ballatore, C., et al. (2016). Altered microtubule dynamics and vesicular transport in mouse and human MeCP2-deficient astrocytes. Hum. Mol. Genet. 25, 146–157. doi: 10.1093/hmg/ddv464
Delepine, C., Nectoux, J., Bahi-Buisson, N., Chelly, J., and Bienvenu, T. (2013). MeCP2 deficiency is associated with impaired microtubule stability. FEBS Lett. 587, 245–253. doi: 10.1016/j.febslet.2012.11.033
Delepine, C., Nectoux, J., Letourneur, F., Baud, V., Chelly, J., Billuart, P., et al. (2015). Astrocyte transcriptome from the Mecp2(308)-truncated mouse model of rett syndrome. Neuromol. Med. 17, 353–363. doi: 10.1007/s12017-015-8363-9
Derecki, N. C., Cronk, J. C., Lu, Z., Xu, E., Abbott, S. B., Guyenet, P. G., et al. (2012). Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109. doi: 10.1038/nature10907
Du, Q., Luu, P. L., Stirzaker, C., and Clark, S. J. (2015). Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics 7, 1051–1073. doi: 10.2217/epi.15.39
Durand, T., De Felice, C., Signorini, C., Oger, C., Bultel-Ponce, V., Guy, A., et al. (2013). F(2)-Dihomo-isoprostanes and brain white matter damage in stage 1 Rett syndrome. Biochimie 95, 86–90. doi: 10.1016/j.biochi.2012.09.017
Evans, J. C., Archer, H. L., Colley, J. P., Ravn, K., Nielsen, J. B., Kerr, A., et al. (2005). Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur. J. Hum. Genet. 13, 1113–1120. doi: 10.1038/sj.ejhg.5201451
Feldman, D., Banerjee, A., and Sur, M. (2016). Developmental dynamics of Rett syndrome. Neural Plast. 2016:6154080. doi: 10.1155/2016/6154080
Fichou, Y., Nectoux, J., Bahi-Buisson, N., Rosas-Vargas, H., Girard, B., Chelly, J., et al. (2009). The first missense mutation causing Rett syndrome specifically affecting the MeCP2_e1 isoform. Neurogenetics 10, 127–133. doi: 10.1007/s10048-008-0161-1
Forbes-Lorman, R. M., Kurian, J. R., and Auger, A. P. (2014). MeCP2 regulates GFAP expression within the developing brain. Brain Res. 1543, 151–158. doi: 10.1016/j.brainres.2013.11.011
Fyffe, S. L., Neul, J. L., Samaco, R. C., Chao, H. T., Ben-Shachar, S., Moretti, P., et al. (2008). Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron 59, 947–958. doi: 10.1016/j.neuron.2008.07.030
Garg, S. K., Lioy, D. T., Knopp, S. J., and Bissonnette, J. M. (2015). Conditional depletion of methyl-CpG-binding protein 2 in astrocytes depresses the hypercapnic ventilatory response in mice. J. Appl. Physiol. 119, 670–676. doi: 10.1152/japplphysiol.00411.2015
Gemelli, T., Berton, O., Nelson, E. D., Perrotti, L. I., Jaenisch, R., and Monteggia, L. M. (2006). Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol. Psychiatry 59, 468–476. doi: 10.1016/j.biopsych.2005.07.025
Gold, W. A., Lacina, T. A., Cantrill, L. C., and Christodoulou, J. (2015). MeCP2 deficiency is associated with reduced levels of tubulin acetylation and can be restored using HDAC6 inhibitors. J. Mol. Med. 93, 63–72. doi: 10.1007/s00109-014-1202-x
Gourine, A. V., Kasymov, V., Marina, N., Tang, F., Figueiredo, M. F., Lane, S., et al. (2010). Astrocytes control breathing through pH-dependent release of ATP. Science 329, 571–575. doi: 10.1126/science.1190721
Groc, L., Gustafsson, B., and Hanse, E. (2006). AMPA signalling in nascent glutamatergic synapses: there and not there! Trends Neurosci. 29, 132–139. doi: 10.1016/j.tins.2006.01.005
He, L., and Lu, Q. R. (2013). Coordinated control of oligodendrocyte development by extrinsic and intrinsic signaling cues. Neurosci. Bull. 29, 129–143. doi: 10.1007/s12264-013-1318-y
He, L. J., Liu, N., Cheng, T. L., Chen, X. J., Li, Y. D., Shu, Y. S., et al. (2014). Conditional deletion of Mecp2 in parvalbumin-expressing GABAergic cells results in the absence of critical period plasticity. Nat. Commun. 5:5036. doi: 10.1038/ncomms6036
Horiuchi, M., Smith, L., Maezawa, I., and Jin, L. W. (2016). CX3CR1 ablation ameliorates motor and respiratory dysfunctions and improves survival of a Rett syndrome mouse model. Brain Behav. Immun. 60, 106–116. doi: 10.1016/j.bbi.2016.02.014
Huang, N., Niu, J., Feng, Y., and Xiao, L. (2015). Oligodendroglial development: new roles for chromatin accessibility. Neuroscientist 21, 579–588. doi: 10.1177/1073858414565467
Jin, L. W., Horiuchi, M., Wulff, H., Liu, X. B., Cortopassi, G. A., Erickson, J. D., et al. (2015). Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: a mechanism for mitochondrial dysfunction and neurotoxicity. J. Neurosci. 35, 2516–2529. doi: 10.1523/JNEUROSCI.2778-14.2015
Johnson, C. M., Cui, N., Zhong, W., Oginsky, M. F., and Jiang, C. (2015). Breathing abnormalities in a female mouse model of Rett syndrome. J. Physiol. Sci. 65, 451–459. doi: 10.1007/s12576-015-0384-5
Johnston, M. V., Jeon, O. H., Pevsner, J., Blue, M. E., and Naidu, S. (2001). Neurobiology of Rett syndrome: a genetic disorder of synapse development. Brain Dev. 23(Suppl. 1), S206–S213. doi: 10.1016/S0387-7604(01)00351-5
Kang, S. K., Kim, S. T., Johnston, M. V., and Kadam, S. D. (2014). Temporal- and location-specific alterations of the GABA recycling system in Mecp2 KO mouse brains. J. Cent. Nerv. Syst. Dis. 6, 21–28. doi: 10.4137/JCNSD.S14012
Karki, P., Webb, A., Smith, K., Johnson, J Jr, Lee, K., Son, D. S., et al. (2014). Yin Yang 1 is a repressor of glutamate transporter EAAT2, and it mediates manganese-induced decrease of EAAT2 expression in astrocytes. Mol. Cell. Biol. 34, 1280–1289. doi: 10.1128/MCB.01176-13
Kawabori, M., and Yenari, M. A. (2015). The role of the microglia in acute CNS injury. Metab. Brain Dis. 30, 381–392. doi: 10.1007/s11011-014-9531-6
Khong, P. L., Lam, C. W., Ooi, C. G., Ko, C. H., and Wong, V. C. (2002). Magnetic resonance spectroscopy and analysis of MECP2 in Rett syndrome. Pediatr. Neurol. 26, 205–209. doi: 10.1016/S0887-8994(01)00385-X
KhorshidAhmad, T., Acosta, C., Cortes, C., Lakowski, T. M., Gangadaran, S., and Namaka, M. (2016). Transcriptional regulation of brain-derived neurotrophic factor (BDNF) by methyl CpG binding protein 2 (MeCP2): a novel mechanism for re-myelination and/or myelin repair involved in the treatment of multiple sclerosis (MS). Mol. Neurobiol. 53, 1092–1107. doi: 10.1007/s12035-014-9074-1
Kiernan, E. A., Smith, S. M., Mitchell, G. S., and Watters, J. J. (2016). Mechanisms of microglial activation in models of inflammation and hypoxia: implications for chronic intermittent hypoxia. J. Physiol. 594, 1563–1577. doi: 10.1113/JP271502
Kifayathullah, L. A., Arunachalam, J. P., Bodda, C., Agbemenyah, H. Y., Laccone, F. A., and Mannan, A. U. (2010). MeCP2 mutant protein is expressed in astrocytes as well as in neurons and localizes in the nucleus. Cytogenet. Genome. Res. 129, 290–297. doi: 10.1159/000315906
Kloukina-Pantazidou, I., Chrysanthou-Piterou, M., Havaki, S., and Issidorides, M. R. (2013). Chromogranin A and vesicular monoamine transporter 2 immunolocalization in protein bodies of human locus coeruleus neurons. Ultrastruct. Pathol. 37, 102–109. doi: 10.3109/01913123.2012.750410
Lehmann, C., Bette, S., and Engele, J. (2009). High extracellular glutamate modulates expression of glutamate transporters and glutamine synthetase in cultured astrocytes. Brain Res. 1297, 1–8. doi: 10.1016/j.brainres.2009.08.070
Li, D., Herault, K., Zylbersztejn, K., Lauterbach, M. A., Guillon, M., Oheim, M., et al. (2015). Astrocyte VAMP3 vesicles undergo Ca2+ -independent cycling and modulate glutamate transporter trafficking. J. Physiol. 593, 2807–2832. doi: 10.1113/JP270362
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., et al. (2011). A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500. doi: 10.1038/nature10214
Luikenhuis, S., Giacometti, E., Beard, C. F., and Jaenisch, R. (2004). Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc. Natl. Acad. Sci. U.S.A. 101, 6033–6038. doi: 10.1073/pnas.0401626101
Lundgaard, I., Luzhynskaya, A., Stockley, J. H., Wang, Z., Evans, K. A., Swire, M., et al. (2013). Neuregulin and BDNF induce a switch to NMDA receptor-dependent myelination by oligodendrocytes. PLOS Biol. 11:e1001743. doi: 10.1371/journal.pbio.1001743
Luo, C., and Ecker, J. R. (2015). Epigenetics. Exceptional epigenetics in the brain. Science 348, 1094–1095. doi: 10.1126/science.aac5832
Lyst, M. J., and Bird, A. (2015). Rett syndrome: a complex disorder with simple roots. Nat. Rev. Genet. 16, 261–275. doi: 10.1038/nrg3897
Mackenzie, B., and Erickson, J. D. (2004). Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers. Arch. 447, 784–795. doi: 10.1007/s00424-003-1117-9
Maezawa, I., and Jin, L. W. (2010). Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 30, 5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010
Maezawa, I., Swanberg, S., Harvey, D., Lasalle, J. M., and Jin, L. W. (2009). Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 29, 5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009
Makedonski, K., Abuhatzira, L., Kaufman, Y., Razin, A., and Shemer, R. (2005). MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum. Mol. Genet. 14, 1049–1058. doi: 10.1093/hmg/ddi097
Mari, F., Azimonti, S., Bertani, I., Bolognese, F., Colombo, E., Caselli, R., et al. (2005). CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 14, 1935–1946. doi: 10.1093/hmg/ddi198
Matagne, V., Ehinger, Y., Saidi, L., Borges-Correia, A., Barkats, M., Bartoli, M., et al. (2017). A codon-optimized Mecp2 transgene corrects breathing deficits and improves survival in a mouse model of Rett syndrome. Neurobiol. Dis. 99, 1–11. doi: 10.1016/j.nbd.2016.12.009
Mencarelli, M. A., Spanhol-Rosseto, A., Artuso, R., Rondinella, D., De Filippis, R., Bahi-Buisson, N., et al. (2010). Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J. Med. Genet. 47, 49–53. doi: 10.1136/jmg.2009.067884
Meng, X., Wang, W., Lu, H., He, L. J., Chen, W., Chao, E. S., et al. (2016). Manipulations of MeCP2 in glutamatergic neurons highlight their contributions to Rett and other neurological disorders. Elife 5:e14199. doi: 10.7554/eLife.14199
Nakashima, N., Yamagata, T., Mori, M., Kuwajima, M., Suwa, K., and Momoi, M. Y. (2010). Expression analysis and mutation detection of DLX5 and DLX6 in autism. Brain Dev. 32, 98–104. doi: 10.1016/j.braindev.2008.12.021
Nectoux, J., Florian, C., Delepine, C., Bahi-Buisson, N., Khelfaoui, M., Reibel, S., et al. (2012). Altered microtubule dynamics in Mecp2-deficient astrocytes. J. Neurosci. Res. 90, 990–998. doi: 10.1002/jnr.23001
Nelson, S. B., and Valakh, V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. doi: 10.1016/j.neuron.2015.07.033
Nguyen, M. V., Du, F., Felice, C. A., Shan, X., Nigam, A., Mandel, G., et al. (2012). MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 32, 10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012
Nguyen, M. V., Felice, C. A., Du, F., Covey, M. V., Robinson, J. K., Mandel, G., et al. (2013). Oligodendrocyte lineage cells contribute unique features to Rett syndrome neuropathology. J. Neurosci. 33, 18764–18774. doi: 10.1523/JNEUROSCI.2657-13.2013
Nomura, Y. (2005). Early behavior characteristics and sleep disturbance in Rett syndrome. Brain Dev. 27(Suppl. 1), S35–S42. doi: 10.1016/j.braindev.2005.03.017
Noutel, J., Hong, Y. K., Leu, B., Kang, E., and Chen, C. (2011). Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron 70, 35–42. doi: 10.1016/j.neuron.2011.03.001
O’Driscoll, C. M., Lima, M. P., Kaufmann, W. E., and Bressler, J. P. (2015). Methyl CpG binding protein 2 deficiency enhances expression of inflammatory cytokines by sustaining NF-kappaB signaling in myeloid derived cells. J. Neuroimmunol. 283, 23–29. doi: 10.1016/j.jneuroim.2015.04.005
Okabe, Y., Takahashi, T., Mitsumasu, C., Kosai, K., Tanaka, E., and Matsuishi, T. (2012). Alterations of gene expression and glutamate clearance in astrocytes derived from an MeCP2-null mouse model of Rett syndrome. PLOS ONE 7:e35354. doi: 10.1371/journal.pone.0035354
Olson, C. O., Zachariah, R. M., Ezeonwuka, C. D., Liyanage, V. R., and Rastegar, M. (2014). Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLOS ONE 9:e90645. doi: 10.1371/journal.pone.0090645
Palazzo, A., Ackerman, B., and Gundersen, G. G. (2003). Cell biology: Tubulin acetylation and cell motility. Nature 421:230. doi: 10.1038/421230a
Palmer, A., Qayumi, J., and Ronnett, G. (2008). MeCP2 mutation causes distinguishable phases of acute and chronic defects in synaptogenesis and maintenance, respectively. Mol. Cell. Neurosci. 37, 794–807. doi: 10.1016/j.mcn.2008.01.005
Philippe, O., Rio, M., Malan, V., Van Esch, H., Baujat, G., Bahi-Buisson, N., et al. (2013). NF-kappaB signalling requirement for brain myelin formation is shown by genotype/MRI phenotype correlations in patients with Xq28 duplications. Eur. J. Hum. Genet. 21, 195–199. doi: 10.1038/ejhg.2012.140
Robinson, L., Guy, J., Mckay, L., Brockett, E., Spike, R. C., Selfridge, J., et al. (2012). Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain 135, 2699–2710. doi: 10.1093/brain/aws096
Roze, E., Cochen, V., Sangla, S., Bienvenu, T., Roubergue, A., Leu-Semenescu, S., et al. (2007). Rett syndrome: an overlooked diagnosis in women with stereotypic hand movements, psychomotor retardation, Parkinsonism, and dystonia? Mov. Disord. 22, 387–389. doi: 10.1002/mds.21276
Schafer, D. P., Heller, C. T., Gunner, G., Heller, M., Gordon, C., Hammond, T., et al. (2016). Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. Elife 5:e15224. doi: 10.7554/eLife.15224
Shahbazian, M. D., Antalffy, B., Armstrong, D. L., and Zoghbi, H. Y. (2002). Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 11, 115–124. doi: 10.1093/hmg/11.2.115
Sharma, K., Singh, J., Pillai, P. P., and Frost, E. E. (2015). Involvement of MeCP2 in regulation of myelin-related gene expression in cultured rat oligodendrocytes. J. Mol. Neurosci. 57, 176–184. doi: 10.1007/s12031-015-0597-3
Sinnett, S. E., Hector, R. D., Gadalla, K. K. E., Heindel, C., Chen, D., Zaric, V., et al. (2017). Improved MECP2 gene therapy extends the survival of MeCP2-null mice without apparent toxicity after intracisternal delivery. Mol. Ther. Methods Clin. Dev. 5, 106–115. doi: 10.1016/j.omtm.2017.04.006
Smrt, R. D., Eaves-Egenes, J., Barkho, B. Z., Santistevan, N. J., Zhao, C., Aimone, J. B., et al. (2007). Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol. Dis. 27, 77–89. doi: 10.1016/j.nbd.2007.04.005
Squillaro, T., Alessio, N., Cipollaro, M., Melone, M. A., Hayek, G., Renieri, A., et al. (2012). Reduced expression of MECP2 affects cell commitment and maintenance in neurons by triggering senescence: new perspective for Rett syndrome. Mol. Biol. Cell 23, 1435–1445. doi: 10.1091/mbc.E11-09-0784
Stancheva, I., Collins, A. L., Van Den Veyver, I. B., Zoghbi, H., and Meehan, R. R. (2003). A mutant form of MeCP2 protein associated with human Rett syndrome cannot be displaced from methylated DNA by notch in Xenopus embryos. Mol. Cell. 12, 425–435. doi: 10.1016/S1097-2765(03)00276-4
Stearns, N. A., Schaevitz, L. R., Bowling, H., Nag, N., Berger, U. V., and Berger-Sweeney, J. (2007). Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience 146, 907–921. doi: 10.1016/j.neuroscience.2007.02.009
Tai, D. J., Liu, Y. C., Hsu, W. L., Ma, Y. L., Cheng, S. J., Liu, S. Y., et al. (2016). MeCP2 SUMOylation rescues Mecp2-mutant-induced behavioural deficits in a mouse model of Rett syndrome. Nat. Commun. 7:10552. doi: 10.1038/ncomms10552
Turovsky, E., Karagiannis, A., Abdala, A. P., and Gourine, A. V. (2015). Impaired CO2 sensitivity of astrocytes in a mouse model of Rett syndrome. J. Physiol. 593, 3159–3168. doi: 10.1113/JP270369
Vacca, M., Tripathi, K. P., Speranza, L., Aiese Cigliano, R., Scalabri, F., Marracino, F., et al. (2016). Effects of Mecp2 loss of function in embryonic cortical neurons: a bioinformatics strategy to sort out non-neuronal cells variability from transcriptome profiling. BMC Bioinformat. 17(Suppl. 2):14. doi: 10.1186/s12859-015-0859-7
Vora, P., Mina, R., Namaka, M., and Frost, E. E. (2010). A novel transcriptional regulator of myelin gene expression: implications for neurodevelopmental disorders. Neuroreport 21, 917–921. doi: 10.1097/WNR.0b013e32833da500
Wang, J., Wegener, J. E., Huang, T. W., Sripathy, S., De Jesus-Cortes, H., Xu, P., et al. (2015). Wild-type microglia do not reverse pathology in mouse models of Rett syndrome. Nature 521, E1–E4. doi: 10.1038/nature14671
Williams, E. C., Zhong, X., Mohamed, A., Li, R., Liu, Y., Dong, Q., et al. (2014). Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum. Mol. Genet. 23, 2968–2980. doi: 10.1093/hmg/ddu008
Wu, W., Gu, W., Xu, X., Shang, S., and Zhao, Z. (2012). Downregulation of CNPase in a MeCP2 deficient mouse model of Rett syndrome. Neurol. Res. 34, 107–113. doi: 10.1179/016164111X13214359296301
Yasui, D. H., Xu, H., Dunaway, K. W., Lasalle, J. M., Jin, L. W., and Maezawa, I. (2013). MeCP2 modulates gene expression pathways in astrocytes. Mol. Autism 4:3. doi: 10.1186/2040-2392-4-3
Yuan, T., and Bellone, C. (2013). Glutamatergic receptors at developing synapses: the role of GluN3A-containing NMDA receptors and GluA2-lacking AMPA receptors. Eur. J. Pharmacol. 719, 107–111. doi: 10.1016/j.ejphar.2013.04.056
Zachariah, R. M., Olson, C. O., Ezeonwuka, C., and Rastegar, M. (2012). Novel MeCP2 isoform-specific antibody reveals the endogenous MeCP2E1 expression in murine brain, primary neurons and astrocytes. PLOS ONE 7:e49763. doi: 10.1371/journal.pone.0049763
Zhang, X., Su, J., Cui, N., Gai, H., Wu, Z., and Jiang, C. (2011). The disruption of central CO2 chemosensitivity in a mouse model of Rett syndrome. Am. J. Physiol. Cell Physiol. 301, C729–C738. doi: 10.1152/ajpcell.00334.2010
Zhao, D., Mokhtari, R., Pedrosa, E., Birnbaum, R., Zheng, D., and Lachman, H. M. (2017). Transcriptome analysis of microglia in a mouse model of Rett syndrome: differential expression of genes associated with microglia/macrophage activation and cellular stress. Mol. Autism 8:17. doi: 10.1186/s13229-017-0134-z
Keywords: Rett syndrome (RTT), MeCP2, astrocyte, oligodendrocyte, microglia
Citation: Jin X-R, Chen X-S and Xiao L (2017) MeCP2 Deficiency in Neuroglia: New Progress in the Pathogenesis of Rett Syndrome. Front. Mol. Neurosci. 10:316. doi: 10.3389/fnmol.2017.00316
Received: 30 April 2017; Accepted: 19 September 2017;
Published: 04 October 2017.
Edited by:
Alexej Verkhratsky, University of Manchester, United KingdomReviewed by:
Xiaolu Zhang, Northern Jiangsu People’s Hospital, ChinaMichele Papa, Università degli Studi della Campania “Luigi Vanvitelli” Caserta, Italy
Copyright © 2017 Jin, Chen and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xing-Shu Chen, xingshuchen2011@163.com Lan Xiao, xiaolan35@hotmail.com