Motor Neuron Gene Therapy: Lessons from Spinal Muscular Atrophy for Amyotrophic Lateral Sclerosis
 Andrew P. Tosolini
Andrew P. Tosolini James N. Sleigh
James N. Sleigh- Sobell Department of Motor Neuroscience and Movement Disorders, Institute of Neurology, University College London, London, United Kingdom
Spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS) are severe nervous system diseases characterized by the degeneration of lower motor neurons. They share a number of additional pathological, cellular, and genetic parallels suggesting that mechanistic and clinical insights into one disorder may have value for the other. While there are currently no clinical ALS gene therapies, the splice-switching antisense oligonucleotide, nusinersen, was recently approved for SMA. This milestone was achieved through extensive pre-clinical research and patient trials, which together have spawned fundamental insights into motor neuron gene therapy. We have thus tried to distil key information garnered from SMA research, in the hope that it may stimulate a more directed approach to ALS gene therapy. Not only must the type of therapeutic (e.g., antisense oligonucleotide vs. viral vector) be sensibly selected, but considerable thought must be applied to the where, which, what, and when in order to enhance treatment benefit: to where (cell types and tissues) must the drug be delivered and how can this be best achieved? Which perturbed pathways must be corrected and can they be concurrently targeted? What dosing regime and concentration should be used? When should medication be administered? These questions are intuitive, but central to identifying and optimizing a successful gene therapy. Providing definitive solutions to these quandaries will be difficult, but clear thinking about therapeutic testing is necessary if we are to have the best chance of developing viable ALS gene therapies and improving upon early generation SMA treatments.
Introduction
Spinal muscular atrophy and amyotrophic lateral sclerosis are two devastating neurological conditions with the common pathological hallmark of motor neuron degeneration, ultimately leading to muscle wasting and death. While etiology, age of onset, progression, and survival outcomes can drastically differ between the diseases, they share a number mechanistic parallels; thus, experimental and clinical insights into the one disorder may prove useful for the other.
It is an exciting time for the SMA community as the first treatment, an ASO gene therapy called nusinersen, was approved in the US by the FDA on 23rd December, 2016. Nusinersen subsequently received marketing authorisation in the EU from the EMA in June, 2017. This starkly contrasts with the situation for ALS, where clinically viable gene therapies are currently non-existent, while recent trials of chemically diverse drugs have failed to live up to expectations piqued by mouse experiments. Although there are numerous complications in treating ALS that do not pertain to SMA, a number of fundamental lessons have been learnt from the gamut of pre-clinical research and clinical trials of SMA gene therapies that could prove useful in galvanizing a targeted approach to ALS gene therapy design and development.
In this review, we will first provide introductions to SMA, ALS, and commonalities between the two, and follow this with an overview of gene therapies tested in clinical trials for both diseases. We use the term gene therapy to encompass both virus-mediated gene transfer and ASO gene targeting. Rather than provide an exhaustive review of all SMA and ALS gene therapy research, which has been collectively well covered (Federici and Boulis, 2012; Nizzardo et al., 2012; Mulcahy et al., 2014; Scarrott et al., 2015; Singh N.N. et al., 2017), we will then outline some of the major issues that SMA gene therapy has encountered, try to distil key, emergent concepts, and frame this in the context of ALS in order to provide possible future experimental directions.
Diseases of Motor Neurons: Genetics, Classifications, And Mechanisms
Spinal Muscular Atrophy
Spinal muscular atrophy (SMA) is a monogenic neuromuscular disorder affecting ≈1 in 8,500–12,500 newborns, and is the most common genetic cause of infant mortality (Verhaart et al., 2017a,b). Patients present with severe muscle weakness and atrophy, predominantly in proximal (e.g., trunk) muscles, due to degeneration of LMNs of the spinal cord ventral horn. Pathology in additional cells and tissues can be observed in more severe manifestations of the disease, which has considerable implications for treatment (Hamilton and Gillingwater, 2013). SMA is caused by reduced levels of SMN protein (Lefebvre et al., 1995), which is found in the nucleus and cytoplasm of almost all cells, and plays a vital, canonical, housekeeping role in spliceosome assembly (Fischer et al., 1997; Liu et al., 1997; Pellizzoni et al., 2002), amongst other functions (Singh R.N. et al., 2017). Specifically, as part of the multi-protein SMN complex, SMN directs the efficient cytoplasmic assemblage of small nuclear RNAs (snRNAs) with Sm protein rings leading to the formation of small nuclear ribonucleoproteins (snRNPs) (Gruss et al., 2017). After nuclear-import, snRNPs function in the catalytic removal of introns from pre-mRNA transcripts in the process of splicing (Pellizzoni et al., 2002). Despite having a good understanding of this and other functions of SMN, the precise cause of the largely selective LMN death remains to be fully resolved. It is likely that a combination of multiple mechanisms account for this vulnerability, including mis-splicing of LMN-specific genes, SMN levels being lower in LMNs than other cell types, and disturbance of a possible non-canonical, LMN-specific SMN function (Figure 1A) (Tisdale and Pellizzoni, 2015; Jablonka and Sendtner, 2017; Tu et al., 2017).

FIGURE 1. Spinal muscular atrophy mechanisms and genetics. (A) Multiple hypothetical mechanisms have been suggested to cause or contribute to the selective LMN degeneration of SMA, the three most plausible of which are that: #1 SMN reduction impairs splicing fidelity of a LMN-specific gene(s). E, exon; I, intron; #2 less SMN is available in LMNs than other cells due to a negative feedback loop, more prominent in LMNs, in which low SMN levels impair further SMN2 exon 7 inclusion (see B) exacerbating SMN reduction. Δ7, truncated SMN mRNA; FL, full-length SMN mRNA; FL-SMN, full-length SMN protein; snRNPs, small nuclear ribonucleoproteins; and #3 a non-canonical, LMN-specific function of SMN is perturbed, for instance anterograde axonal transport of mRNA. (B) A single copy of SMN1 produces enough SMN protein for motor neurons to thrive and for SMA disease carriers to remain healthy. Due to a single base pair distinction (C-to-T transition in exon 7), exon 7 is mis-spliced out of ≈90% of SMN2 transcripts, resulting in a truncated, less stable, and only partially functional protein. This figure has been adapted from Neuromuscular Disorders, 23, Sleigh et al., 2013 Spinal muscular atrophy at the crossroads of basic science and therapy, 96, Copyright (2013), with permission from Elsevier.
Survival motor neuron is encoded by two almost identical genes, SMN1 and its paralogue SMN2 (Figure 1B) (Lefebvre et al., 1995). A single functioning SMN1 allele produces sufficient protein for LMNs to remain healthy – as demonstrated by the ≈1 in 40–60 people with one functional copy of SMN1 (i.e., SMA carriers) showing no clinical phenotype. However, due to a single nucleotide distinction (synonymous C-to-T alteration 6 nucleotides into exon 7), exon 7 of SMN2 is aberrantly spliced ≈90% of the time (creating truncated, non-functional SMNΔ7 protein) and is therefore capable of producing only ≈10% of the full-length SMN made by SMN1 (Figure 1B) (Lorson et al., 1999; Monani et al., 1999). Thus, when protein production from SMN1 is impaired, as it is in SMA patients, SMN2 can only partially compensate. SMN is highly conserved throughout evolution, permitting modeling of reduced SMN function in diverse organisms (Grice et al., 2011; Patten et al., 2014; Duque et al., 2015); however, all models naturally only possess an ortholog of SMN1, but not SMN2. To better mimic disease genetics, many transgenic mice have been engineered to express diminished SMN levels (Sleigh et al., 2011), the most frequently used being the SMNΔ7 mouse, which combines human SMN2 and SMNΔ7 transgenes on a null Smn background (Le et al., 2005). Relative genomic instability of the region (5q13) is believed to be the reason for the recent evolutionary duplication of the SMN locus (Rochette et al., 2001), and may also account for there being considerable variability in SMN2 copy number within the population. The number of SMN2 genes a patient with SMA possesses has important ramifications for disease severity, as more SMN2 copies can produce more SMN, which correlates with diminished symptom severity (Lefebvre et al., 1997). Although not predictive at the individual level, in a population, SMN2 copy number thus inversely correlates with SMA severity (McAndrew et al., 1997), which is categorised into four principal post-natal types (I-IV) based on age of onset and motor milestones achieved (Munsat and Davies, 1992).
Manifesting at or before 6 months and radically limiting life expectancy (<2 years), type I SMA (a.k.a. Werdnig-Hoffmann disease) is the most severe and frequently diagnosed form of SMA, and prevents children from ever being able to sit unaided. Infant death is usually caused by respiratory complications, although with specialized care, lifespan can be artificially extended for long periods. Type II SMA (Intermediate/Dubowitz Syndrome) presents between 7 and 18 months, permits unaided sitting but not walking, and has survival probabilities of ≈93% and ≈52% at 20 and 40 years, respectively (Farrar et al., 2013). Type III SMA (Kugelberg-Welander disease) limits motor function and has an onset > 18 months, but before adolescence, while type IV SMA (adult-onset) typically manifests in the second or third decade of life with mild-to-moderate muscle weakness, but generally no respiratory issues.
Amyotrophic Lateral Sclerosis
With a lifetime risk of ≈1 in 400 (Alonso et al., 2009), ALS, also called MND and Lou Gehrig’s disease, is a fatal, progressive, mostly adult-onset disorder of both LMNs and UMNs. Neurodegeneration is observed in the cortex, corticospinal tracts, brainstem, and spinal ventral horn neurons and is accompanied by neuroinflammation (Brown and Al-Chalabi, 2017). Starting focally and spreading, this causes major symptoms of muscle weakness and fasciculations with subsequent atrophy, leading to death usually through respiratory failure within 3 years of diagnosis (Chiò et al., 2009). ALS also shares neuropathological and genetic features with FTD (Morita et al., 2006; Neumann et al., 2006; Vance et al., 2006), with approximately half of ALS patients showing some level of cognitive impairment (Ringholz et al., 2005). This has led to ALS and FTD being considered as part of the same clinicopathological spectrum (Ling et al., 2013). ALS patients display considerable symptom heterogeneity inclusive of age, site of disease onset, the rate and pattern of spread, and relative LMN/UMN involvement (Kiernan et al., 2011). ALS can therefore be sub-categorized based on several clinical and neuropathological criteria (Al-Chalabi and Hardiman, 2013; Bäumer et al., 2014). Given this variability, the ALS diagnosis is challenging, particularly during early disease stages (Byrne et al., 2012). The mean time from first noticeable symptom to clinical diagnosis is consequently (≈1 year). Contributing to this delay, no diagnostic or prognostic biomarkers are yet in regular clinical use for ALS, which also impacts the assessment of therapeutic efficacy in patient trials (Rosenfeld and Strong, 2015). Nevertheless, a number of prospective biomarkers have recently been identified, including the neurotrophin receptor p75NTR extracellular domain in urine (Shepheard et al., 2017) and neurofilament chains in plasma (Lu et al., 2015) and CSF (Oeckl et al., 2016).
ALS has traditionally been classified into clinically indistinguishable sporadic (sALS) and familial (fALS) forms; sALS occurs without family history of the disease and represents the majority of cases (≈90%), whereas fALS contributes ≈10% of patients and is genetically inherited, predominantly in an autosomal, dominant fashion. The pathological and clinical variability of the disease has led to the idea that, in addition to being on a continuum with FTD, ALS itself may not be a single disorder, but a syndrome (Turner et al., 2013). Consistent with this, aberrations in over 25 genetic loci have been reproducibly linked with the ALS phenotype (Brown and Al-Chalabi, 2017), with new genes constantly being identified (Freischmidt et al., 2015; Brenner et al., 2016; Mackenzie et al., 2017). The four most common mutations are large, intronic, hexanucleotide repeat (G4C2) expansions in chromosome 9 open reading frame 72 (C9orf72) (DeJesus-Hernandez et al., 2011; Renton et al., 2011), and dominant mutations in superoxide dismutase 1 (SOD1) (Rosen et al., 1993), transactive-region DNA binding protein (TARDBP encoding TDP-43) (Sreedharan et al., 2008), and fused in sarcoma (FUS, a.k.a. translocated in liposarcoma, TLS) (Kwiatkowski et al., 2009; Vance et al., 2009). Mutations in C9orf72 are the most common genetic cause of ALS hitherto identified, accounting for ≈40% of fALS and ≈7% of sALS (in populations of European ancestry) (Renton et al., 2014). The exact function of the encoded protein remains unclear, but it appears that it may be important in membrane trafficking and autophagy (Nassif et al., 2017). Encoding a Cu/Zn dismutase enzyme that provides defense against toxic superoxide free radicals, SOD1 was the first gene linked to ALS (Rosen et al., 1993), and its mutation is responsible for ≈12% and ≈1% of fALS and sALS patients, respectively (Renton et al., 2014). As a consequence of its early identification and the rapid generation of the SOD1G93A mouse model (Gurney et al., 1994), research into SOD1 has shaped much of the ALS research landscape. Nevertheless, many other cellular and animal models are now available for different genetic forms of the disease (van Damme et al., 2017). TARDBP and FUS encode nucleic acid-binding proteins that predominantly reside in the nucleus, and are involved in multiple aspects of RNA processing, such as transcription and splicing. Mutations in these two genes each account for ≈4% of fALS and ≈1% of sALS patients (Renton et al., 2014). Despite significant progress in our understanding of the molecular pathogenesis linked to these four genes, it has not been fully resolved as to whether pathology is solely caused by a toxic gain-of-function or whether there are also loss-of-function effects (Lee et al., 2012; Bunton-Stasyshyn et al., 2015; Scekic-Zahirovic et al., 2016; Moens et al., 2017).
Causative genetic mutations have been identified in only ≈68% and ≈11% of fALS and sALS patients, respectively (Renton et al., 2014). This lack of an obvious genetic cause in most ALS patients, along with incomplete penetrance in several fALS pedigrees (Cirulli et al., 2015; Freischmidt et al., 2015), suggests that ALS may most frequently arise from additive effects of an assortment of predispositions and insults (Al-Chalabi and Hardiman, 2013). Indeed, rare variants in many other genes have been identified as ALS risk factors (van Rheenen et al., 2016), as have particular environmental stimuli (Martin et al., 2017a). Moreover, twin studies indicate that sALS heritability in the absence of a family history of the disease is still ≈60% (Al-Chalabi et al., 2010). Together, these data indicate that ALS may develop through a multi-step process in which aging is a critical component (Al-Chalabi et al., 2014), involving varying degrees of heritability and diverse, but inter-related, functional pathways that, upon dysregulation, yield motor neuron degeneration (Turner et al., 2013). This makes the strict fALS/sALS distinction an artificial dichotomy (Talbot, 2011), and presents obvious and considerable hurdles for the identification and development of viable therapeutic strategies for the disease.
Intrinsic motor neuron defects and non-cell autonomous toxicities in associated cell types (e.g., glia, interneurons) contribute to ALS (Ilieva et al., 2009; Ramírez-Jarquín et al., 2014; Puentes et al., 2016), but similar to SMA, the exact mechanisms underpinning motor neuron death, and their relative vulnerability-resistance axis (Nijssen et al., 2017), remain to be elucidated. Nevertheless, given the known functions of major ALS genes, altered RNA processing, nuclear protein mishandling/protein quality control, and impaired cytoskeletal dynamics appear to be three inter-related central themes (Bäumer et al., 2010; Brown and Al-Chalabi, 2017). Congruously, the vast majority of both sALS and fALS patients display cytoplasmic depositions of aggregated proteins, the main component of which is TDP-43 (Neumann et al., 2006), albeit with varied cellular distributions (Al-Sarraj et al., 2011). However, these inclusions conspicuously lack TDP-43 in SOD1-linked (Mackenzie et al., 2007) and FUS-linked (Vance et al., 2009) ALS. Additional defects in diverse cellular processes have been implicated in ALS including excitotoxicity, oxidative stress, altered oligodendrocyte function, axonal transport defects, mitochondrial malfunction, and neurotrophic factor deficits (reviewed in Kiernan et al., 2011; Taylor et al., 2016). It remains unclear as to which, if any, of these phenomena play a primary role in disease pathogenesis, rather than simply being non-specific consequences of a dysfunctional system. Moreover, it should be noted that most of these pathologies were identified using SOD1G93A mice, which have their limitations for modeling all forms of ALS (Kiernan et al., 2011; Turner et al., 2013). To overcome this, numerous transgenic mouse models of ALS have been developed (van Damme et al., 2017), and strict guidelines for their use in pre-clinical therapeutic trials have been created to limit irreproducibility (Ludolph et al., 2010).
SMA and ALS: A Common Mechanism?
SMA and ALS share a propensity for LMN degeneration leading to muscle wasting and atrophy. A number of key cellular and molecular parallels between the two diseases have also been reported (Cauchi, 2014; Gama-Carvalho et al., 2017; Hensel and Claus, 2017). The causative gene in SMA encodes a widely expressed, multi-functional protein important for fundamental cellular processes including pre-mRNA splicing (Fischer et al., 1997; Liu et al., 1997; Pellizzoni et al., 2002), transcription (Pellizzoni et al., 2001), and mRNA transport and stability (Rossoll et al., 2003; Zhang et al., 2003). While the importance of protein quality control to ALS should not be underestimated, some of the major genetic contributors to the disease, perhaps with the exception of C9ORF72, are also found ubiquitously and perform similar functions vital to RNA processing and maturation (Bäumer et al., 2010). For instance, both TDP-43 and FUS are involved in splicing (Zhou et al., 2002; Polymenidou et al., 2011) and transcription (Uranishi et al., 2001), and, along with SOD1, are thought to be important for the transport and stability of mRNA (Fujii and Takumi, 2005; Lu et al., 2007; Strong et al., 2007). Recently, dominant mutations in T-cell restricted intracellular antigen 1 (TIA1), which is an RNA-binding protein involved in SMN2 exon 7 splicing (Singh et al., 2011), were shown to play a causative role in ALS (Mackenzie et al., 2017), while TIA1 knockout modifies phenotypes of mild male SMA mice (Howell et al., 2017). Furthermore, wild-type TDP-43 and FUS interact with SMN (Wang I.-F. et al., 2002; Yamazaki et al., 2012; Groen et al., 2013; Sun et al., 2015), and all three proteins have been implicated in the formation of stress granules (Hua and Zhou, 2004; Andersson et al., 2008; Colombrita et al., 2009), as has C9ORF72 (Maharjan et al., 2017).
Comparable in structure to stress granules, Gemini of Cajal bodies (a.k.a. gems) are membrane-free, nuclear conglomerates of SMN and associated proteins (Liu and Dreyfuss, 1996), the number of which correlates with SMN availability and is thus inversely related to SMA severity (Lefebvre et al., 1997; Feng et al., 2005). Numerous studies have shown that gem distribution/number is also affected in SOD1-, FUS-, and TDP-43-associated ALS patient tissue, mice, and cellular models (Shan et al., 2010; Gertz et al., 2012; Kariya et al., 2012; Yamazaki et al., 2012; Ishihara et al., 2013; Tsuiji et al., 2013; Sun et al., 2015), although this pattern was not observed in sALS patient fibroblasts (Kariya et al., 2014b). Nonetheless, SMN protein levels are reduced in sALS patient spinal cords (Turner et al., 2014) and pre-symptomatically in SOD1G93A mouse spinal cords, while a 50% reduction in SMN exacerbates the SOD1G93A phenotype (Turner et al., 2009). Furthermore, one study showed that ALS patients on average possess fewer SMN2 copies, while having one SMN1 copy is associated with increased ALS susceptibility (Veldink et al., 2005), although this link is not straightforward (Blauw et al., 2012; Corcia et al., 2012; Wang X.-B. et al., 2014). Nevertheless, SMN upregulation protects against mutant SOD1-induced cell death in an immortalized motor neuronal cell line (NSC-34) (Zou et al., 2007), rescues mutant FUS-mediated axonal defects in primary cortical neurons (Groen et al., 2013), and can improve survival of iPSC-derived motor neurons differentiated from SOD1 and TDP-43 ALS patient fibroblasts (Rodriguez-Muela et al., 2017). Moreover, neuronal SMN overexpression aids motor neuron survival and delays symptom onset in SOD1G85A, SOD1G93A, and TDP-43A315T mice (Kariya et al., 2012; Turner et al., 2014; Perera et al., 2016). Survival of both SOD1 models was unaffected, but female TDP-43 mice displayed a significant extension. SMN may in fact serve as a general survival factor for motor neurons, as it is required to facilitate neuromuscular regeneration post-sciatic nerve crush in adult mice (Kariya et al., 2014a). In addition, low SMN levels in healthy iPSC-derived motor neurons correlate with greater cell death, and SMN upregulation promotes increased survival of control motor, but not cortical, neurons (Rodriguez-Muela et al., 2017).
These commonalities between SMA and ALS suggest that a shared mechanism could underlie at least certain aspects of the two diseases. Perhaps the most likely cause of the link is sequestration of SMN and/or splicing factors into cytoplasmic inclusions by mutant ALS gene products, resulting in defective RNA homeostasis. Indeed, ALS-associated mutations in FUS can enhance its association with SMN and impinge upon its axonal localization (Groen et al., 2013; Sun et al., 2015). Additionally, mutant FUS and C9orf72 expansion can affect splicing factor distribution (Gerbino et al., 2013; Lee et al., 2013; Mori et al., 2013; Yu et al., 2015; Reber et al., 2016), as can homozygous overexpression of human wild-type FUS in mice (Mirra et al., 2017). Widespread splicing defects are unlikely to account for the motor neuron selectivity observed in SMA (Bäumer et al., 2009); however, the early and specific mis-splicing of a few crucial motor neuron-expressed genes may be particularly relevant to disease pathogenesis (Zhang et al., 2013; Sleigh et al., 2014). Splicing impairments have also been reported in ALS models and patient tissue (Chabot and Shkreta, 2016; Conlon et al., 2016); it is thus plausible that early splicing perturbations in a common set of critical genes could explain some of the shared pathomechanisms of SMA and ALS. Indeed, considerable overlap in alternative splicing events between SMA models and human FUS-expressing mice were recently reported, including in a number of ALS-pertinent genes (Mirra et al., 2017). These discoveries and the mechanistic intersection of ALS with SMA, suggest that gene therapy strategies able to augment SMN levels may be beneficial to both fALS and sALS patients, perhaps not in isolation, but as part of a combinatorial approach.
Clinical Gene Therapy for Motor Neuron Diseases
SMA: SMN Restoration Is Key
The genetic lesion underlying SMA causes diminished SMN protein levels; in theory, treatment is thus simple – replenish SMN. Small molecule, SMN2 splice-modifying drugs, such as RG7916 (Roche) and LMI070 (Novartis), that augment SMN and SMN-independent, neuroprotection strategies are being pursued (Scoto et al., 2017), but SMN gene therapies are currently proving more clinically promising. In the last decade, we have rapidly transitioned from several early ineffective SMA patient trials (Fuller et al., 2010), to the recent regulatory approval of nusinersen for the treatment of SMA types I-IV.
Nusinersen (a.k.a. Spinraza, IONIS-SMNRx, ISIS-SMNRx, ISIS 396443, and ASO-10-27) is an ASO developed through work of numerous laboratories and a collaboration between Biogen Idec and Ionis Pharmaceuticals (formerly Isis Pharmaceuticals). ASOs are short (15–25 nucleotides), synthetic, single-stranded DNA or RNA sequences that specifically bind to target pre-mRNA or mRNA sequences, impacting gene expression. ASOs that specifically modulate splicing are also called SSOs. Importantly for diseases affecting the nervous system, ASOs distribute widely when injected into the CSF, do not require carrier molecules, and have relatively long half-lives (Geary et al., 2015). Nusinersen, which is delivered via single intrathecal injections directly into the CSF (i.e., by lumbar puncture) (Chiriboga et al., 2016; Haché et al., 2016), is a 2′-O-(2-methoxyethyl) modified ASO complementary to the ISS-N1 found in intron 7 of SMN2 pre-mRNA (Singh et al., 2006; Hua et al., 2008). Specific ASO/pre-mRNA hybridisation restricts exon 7 mis-splicing, thereby increasing the amount of functional SMN made by SMN2 (Figure 2A). A considerable amount of work in SMA mice provided substantial evidence for in vivo efficacy of nusinersen (Singh N.N. et al., 2017), leading to a series of stratified clinical trials (Chiriboga et al., 2016; Finkel et al., 2016, 2017). A pre-specified interim analysis from a randomized, double-blind, sham procedure-controlled phase III trial in SMA type I patients called ENDEAR (ClinicalTrials.gov Identifier: NCT02193074), provided enough evidence in mid-2016 that nusinersen caused statistically significant improvements in motor function (Finkel et al., 2017), prompting submission to the FDA of the new drug application. Interim analyses from a second phase III trial with type II SMA patients called CHERISH (NCT02292537) and an open-label study in pre-symptomatic infants named NURTURE (NCT02386553) have also proven encouraging.
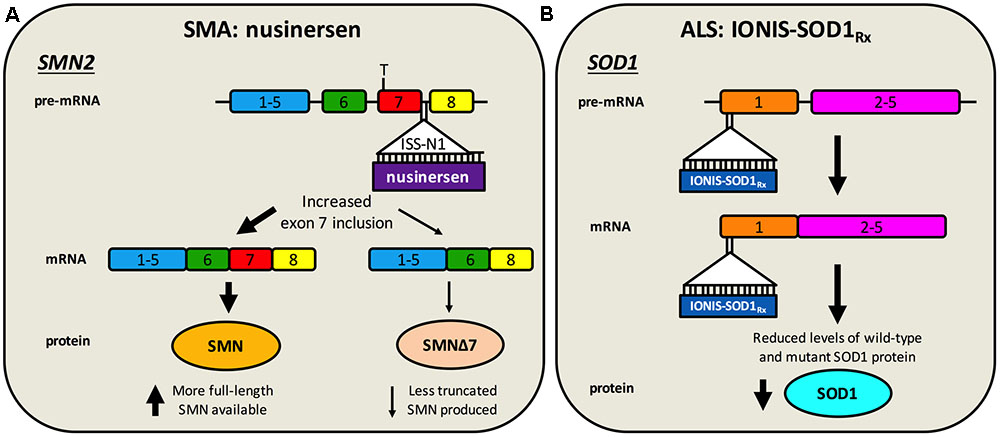
FIGURE 2. Antisense oligonucleotide targeting of SMN2 splicing and SOD1 translation. (A) Nusinersen is a SSO complementary to an intronic sequence in SMN2 called ISS-N1, which is the main inhibitory element for exon 7 splicing. Hybridisation of nusinersen to ISS-N1 within pre-mRNA causes more frequent inclusion of exon 7 in mature SMN2 transcripts, leading to an increase in the production of full-length SMN protein. Nusinersen is FDA- and EMA- approved for the treatment of SMA. (B) IONIS-SOD1Rx specifically targets a 20 nucleotide-long sequence within exon 1 of SOD1, resulting in binding to both pre-mRNA and mature RNA causing inhibition of wild-type and mutant SOD1 expression. ALS patients are currently being recruited for a phase Ib/IIa trial of IONIS-SOD1Rx.
A second gene therapy called AVXS-101 (a.k.a. scAAV9.CB.SMN and ChariSMA), which delivers the SMN1 gene using non-replicating self-complementary adeno-associated virus serotype 9 (scAAV9), has also shown significant pre-clinical potential (Mulcahy et al., 2014). A major advantage of this therapy over nusinersen is that AAV9 can cross the BBB in mice, cats, and non-human primates, permitting intravenous delivery (Duque et al., 2009; Foust et al., 2009; Samaranch et al., 2012). Moreover, AAV9 displays neuronal tropism and can mediate stable, long-term expression with a single administration, which is important given immunogenicity issues associated with viruses (Lorain et al., 2008). This contrasts with the multiple, invasive intrathecal injections of nusinersen, which can have adverse side effects (Haché et al., 2016). Marketed by AveXis, AVXS-101 has completed testing in type I SMA patients in an open-label, dose-escalation phase I clinical trial (NCT02122952). The treatment is safe and well tolerated, and caused improvements in survival, attainment of motor milestones, and motor function when compared with historical SMA type I cohorts (Mendell et al., 2017). Two further open-label phase III trials with type I patients in the US and EU are planned. SMN1 expression is driven by a hybrid cytomegalovirus enhancer/chicken β-actin (CAG) promoter, and AVXS-101 is being injected intravenously.
ALS: A Gene Therapy on the Horizon?
No drugs are currently being tested in late-stage clinical trials for ALS, and until recently, the only FDA-/EMA-approved drug for the disease was the orally available Riluzole, which prolongs patient survival by ≈3 months (Bellingham, 2011). Riluzole can influence ion channel function, neurotransmission, and growth factor secretion, but inhibition of glutamate release from pre-synaptic nerve terminals counteracting motor neuron excitotoxicity appears to be the most disease-relevant benefit (Bellingham, 2011). In May 2017, the FDA surprisingly approved Edaravone (a.k.a. MCI-186 and Radicava), which is a free radical scavenger shown to modestly slow symptom progression in SOD1G93A mice (Ito et al., 2008). In an open-label phase II study without comparator arm involving 19 ALS patients, intravenous administration of Edaravone was shown to be safe and reduce oxidative stress (Yoshino and Kimura, 2006); however, in a subsequent double-blind, placebo-controlled study, drug efficacy was not demonstrated (Abe et al., 2014). Nevertheless, a phase III trial with narrow inclusion criteria showed Edaravone modestly delayed disease progression in a limited subset of ALS patients (Writing Group and Edaravone (MCI-186) ALS 19 Study Group, 2017). Edaravone is unlikely to be effective in a wider ALS population and there is a sizeable administration burden, limiting excitement for the drug (Hardiman and van den Berg, 2017). Palliative care incorporating dietary and respiratory support, speech and language therapy, and specialist physiotherapy can also improve survival in ALS (Martin et al., 2017b), and provides arguably a greater benefit to quality of life than current pharmaceutical intervention (Hardiman et al., 2011).
In over 20 years since the approval of Riluzole, more than 20 additional compounds have been tested in over 50 randomized, controlled trials, involving in excess of 13,000 ALS patients, with little clinical success (Mitsumoto et al., 2014; Petrov et al., 2017). Consistent with ALS complexity, the tested drugs possess a broad range of proposed mechanisms of action, including anti-inflammation and anti-oxidation. Rather than targeting the underlying genetics, these compounds were trialled for their ability to support the ailing ALS nervous system and restrict the insidious progression of disease. These unsuccessful therapeutics include three neurotrophic factors delivered as recombinant proteins - BDNF, CNTF, and insulin-like growth factor-1 (IGF-1) (Bartus and Johnson, 2017a). Also known as neurotrophins, neurotrophic factors are target-secreted (e.g., from muscles) proteins essential for the growth, development and survival of several nerve types, including motor neurons (Huang and Reichardt, 2001). Delivering neurotrophins has been pursued as a therapy for ALS because their expression can decline with time in models and patients (Krakora et al., 2012). While injection of recombinant proteins is not gene therapy per se, when adapted for delivery by viruses, these and other neurotrophic factors have shown promising results in pre-clinical ALS models (Henriques et al., 2010; Nizzardo et al., 2012) (Table 1). Additionally, intramuscular injections of plasmids encoding multiple isoforms of hepatocyte growth factor (HGF, drug named VM202) or a transcription factor able to increase expression of vascular endothelial growth factor (VEGF, drug named SB-509) have been trialled in ALS patients (Scarrott et al., 2015). Both plasmids had favorable safety profiles in phase I/II studies (VM202, NCT02039401 and SB-509, NCT00748501) (Sufit et al., 2017), but whether these drugs will be tested further remains unclear (Scarrott et al., 2015).

TABLE 1. Virus-mediated, neuroprotection gene therapies tested in animal models of ALS.
Without detailed knowledge of disease etiology and underlying cellular pathologies, neuroprotection is potentially the only viable method for tackling a complex syndrome like ALS. However, with increased understanding of gain- and loss-of-function mechanisms of genetic forms of ALS, a second category of knockdown gene therapies encompassing ASOs and RNAi has emerged. These have principally been tested in SOD1, but also C9ORF72, rodent models (Table 2). These oligonucleotide-mediated therapeutics are designed to specifically target and reduce levels of toxic, mutant proteins (e.g., C9ORF72, SOD1, TDP-43, FUS) and are showing promise in mice. While they may have a narrow applicability window due to small percentages of genetically determined ALS, given pathological commonalities, such as cytoplasmic TDP-43 sequestration (Neumann et al., 2006) and possible involvement of wild-type SOD1 misfolding in disease (Bosco et al., 2010), there is scope for broader application. Moreover, many sALS patients possess mutations in genes linked to fALS. Targeting the cause of disease in this manner is likely to have the greatest therapeutic impact, and obviates the requirement for co-treatment of multiple downstream pathways. The ASO IONIS-SOD1Rx (a.k.a. ISIS 333611 and BIIB067) targets both wild-type and mutant SOD1 mRNA for degradation (Figure 2B). Importantly for this strategy, Sod1 knockout mice develop normally and do not show motor neuron loss, although their response to axonal injury is impaired (Reaume et al., 1996), and there is evidence that SOD1 loss-of-function may modify ALS severity (Saccon et al., 2013). IONIS-SOD1Rx administration into CSF of SOD1G93A rats resulted in reduced SOD1 protein in spinal cord (Winer et al., 2013). IONIS-SOD1Rx was thus tested in 24 ALS patients in a randomized, placebo-controlled phase I trial (Miller et al., 2013). In this first-in-human clinical study of intrathecal ASO delivery, ≈12 h infusion of IONIS-SOD1Rx was shown to be safe and well tolerated. A phase Ib/IIa trial (NCT02623699) is currently recruiting ALS patients to further evaluate safety, tolerability, and pharmacokinetics of IONIS-SOD1Rx.
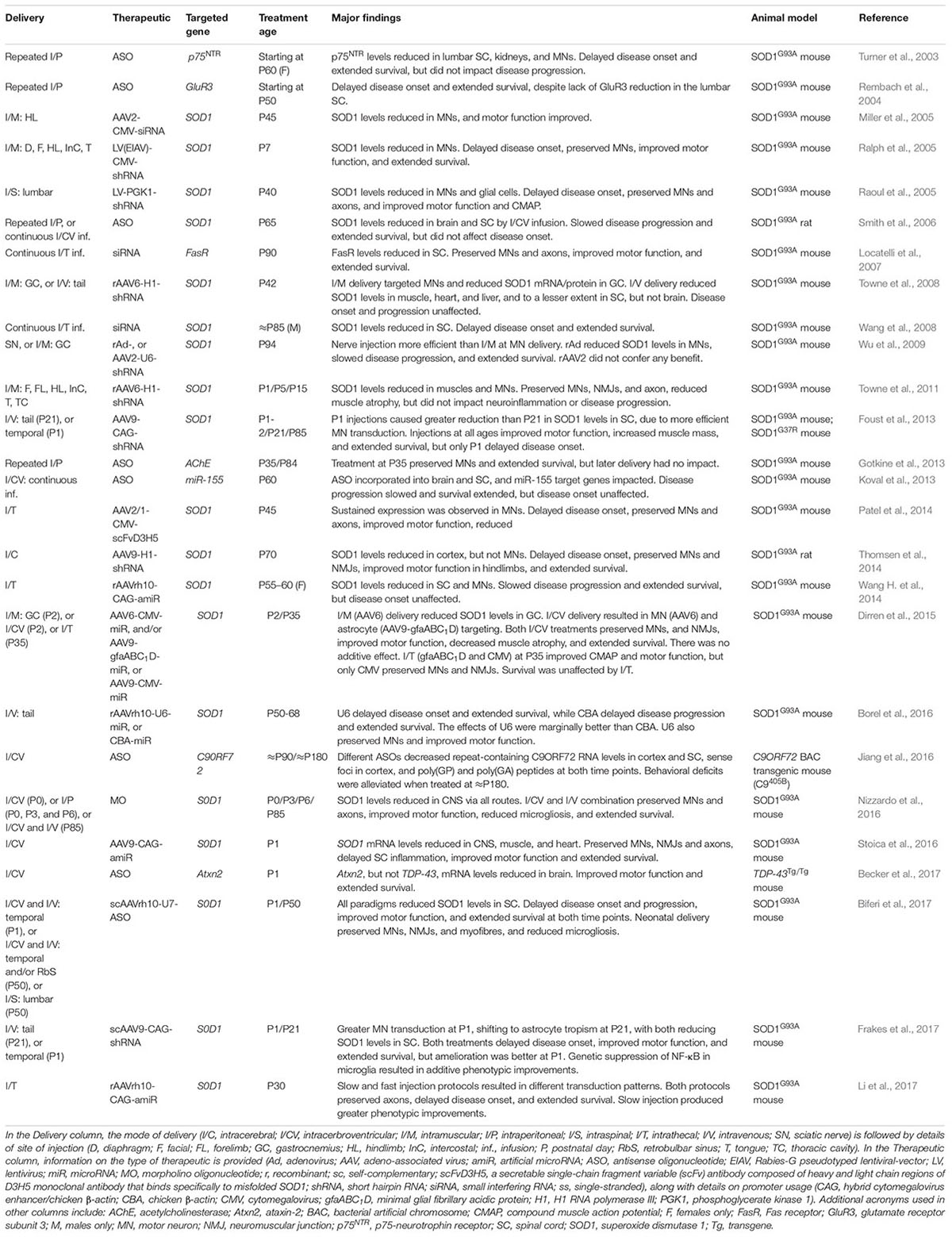
TABLE 2. Knockdown strategy gene therapies tested in animal models of ALS.
SMA Gene Therapy Lessons for ALS
There are numerous possible intersecting explanations for the plethora of failed ALS clinical trials (Mitsumoto et al., 2014), not limited to (1) most pre-clinical research has been conducted in SOD1G93A mice, which do not accurately model the entire ALS spectrum; (2) poor experimental design and execution of pre-clinical work (Scott et al., 2008); (3) focusing on mouse survival as an indicator of drug potential (Genç and Özdinler, 2014); (4) pharmacological issues such as insufficient dose or access/bioavailability to targeted tissue; (5) timing of intervention (Benatar, 2007); (6) patient heterogeneity, poor trial stratification, and scarcity of biomarkers; and (7) incomplete understanding of disease mechanism(s).
Similar difficulties have, at least partially, been overcome by the SMA research community in order for nusinersen to receive regulatory approval. Being a monogenic condition, treating SMA is undoubtedly simpler than the challenge posed by the broad-ranging heterogeneity of ALS. Nonetheless, the clinical approval of nusinersen was a significant milestone not just for SMA, but gene therapy as a whole. That is not to say that the job is complete for SMA, as the clinical response to nusinersen is wide-ranging and includes non-responders (Finkel et al., 2017). However, over the last decade, a great deal has been learnt from the pre-clinical development of SMA gene therapies and the clinical trials of nusinersen and AVXS-101. Given the clinical and mechanistic overlap between the diseases, these lessons learned from SMA may be useful for ALS, particularly when considering the array of gene therapies pre-clinically tested in ALS rodent models (Tables 1, 2) and thus likely to be in the clinical drug pipeline. We have therefore summarised this information to emphasize some key points for ALS gene therapy development.
Careful Therapeutic Targeting Is Required
A clear understanding of where a therapy is needed is mandatory for clinical success. SMA is primarily a LMN disorder; however, in severe cases, pervasive pathology has been reported. For instance, congenital heart problems (Rudnik-Schöneborn et al., 2008), bone complications (Khatri et al., 2008), and vascular defects (Somers et al., 2016) are known to occur in some type I SMA patients. Cell intrinsic SMN depletion causes similar and additional pathologies in mice (Hamilton and Gillingwater, 2013). A sliding scale of vulnerability to SMN reduction has therefore been suggested; at one end, LMNs are the first cell type disturbed by diminished SMN, and as levels are decreased further, more cell types and tissues become affected (Sleigh et al., 2011). This appears to be the case in mouse models, but may be sub-clinical in the majority of SMA patients, as non-motor neuronal involvement is less common. Nevertheless, concerns persist that treating SMA patients with nusinersen, which is delivered directly to the CSF to target LMNs, may alter disease trajectory and reveal novel pathologies caused by chronic SMN deficiency outside the CNS (Tizzano and Finkel, 2017).
There is thus discussion, for both ASOs and viruses, as to which single delivery route provides the best therapeutic outcome for SMA (Hua et al., 2011; Glascock et al., 2012b; Porensky et al., 2012; Nizzardo et al., 2014; Zhou et al., 2015). Major determinants of this are the BBB and the BSCB, which not only restrict access of systemically delivered therapies to the brain and spinal cord, but can also confine drugs within the CNS when directly administered. Targeting therapies to NMJs for uptake and retrograde transport along motor axons can circumvent these barriers (Tosolini et al., 2013; Mohan et al., 2014; Tosolini and Morris, 2016), but this is compromised by denervation. Nonetheless, once inside the CNS, ASOs are able to distribute widely (Rigo et al., 2014). Accordingly, intracerebroventricular injection of SMN ASOs results in robust SMN upregulation in the CNS (Hua et al., 2010; Passini et al., 2011; Porensky et al., 2012; Rigo et al., 2014), while intravenous or subcutaneous administration triggers a more systemic increase outside the CNS (Hua et al., 2011; Keil et al., 2014). Viral vectors are better than ASOs at overcoming BBB permeability problems (Foust et al., 2010; Valori et al., 2010; Dominguez et al., 2011; Meyer et al., 2015), but distinctions in therapeutic outcome between routes still apply (Glascock et al., 2012a,b). In addition, modifying the delivery speed of intrathecal injections can alter AAV tropism; slower injections (i.e., over 8 min) result in preferential transduction of the spinal cord, whereas faster injections (i.e., over 30 s) preferably transduce the brain and peripheral tissue (Li et al., 2017). Notwithstanding, there remains little doubt that using multiple injection modes to provide body-wide SMN augmentation provides the best phenotypic rescue in SMA mice (Nizzardo et al., 2014; Osman et al., 2014), while limiting the onset of delayed, non-neuronal pathologies such as tail necrosis (Foust et al., 2010; Dominguez et al., 2011). The importance of careful therapeutic targeting of SMN to all required cells has been confirmed using numerous transgenic mice where SMN has been overexpressed in SMA models in a tissue-specific fashion (Gogliotti et al., 2012; Martinez et al., 2012).
Cautious selection of administration route is not the only way to affect the voyage of gene therapy through the body. A number of additional therapy-dependent tactics can be employed to optimize delivery. Despite limited packaging capacity (≈4.5 kb for single-stranded and ≈2.4 kb for self-complementary AAV), AAV has become the most promising vector for gene delivery in neurological disease; it establishes stable nuclear episomes, thus reducing the risk of integrating into the host genome and causing insertional mutagenesis, it can transduce both dividing and non-mitotic cells, and it maintains exogenous gene expression for extended periods (Murlidharan et al., 2014). With approximately twice the capacity of AAV, LV has also been employed as a proof-of-concept vector in pre-clinical models of SMA (Azzouz et al., 2004a) and ALS (Tables 1, 2); however, given that LV can randomly insert into the host genome, there are major safety issues associated with its clinical application (Imbert et al., 2017). The advantages of AAV led to scAAV9 being chosen for SMN1 delivery in the AveXis gene therapy, AVXS-101. AAV serotypes possess divergent capsid proteins that bind to distinct host cell surface receptors and co-receptors, thereby determining the cells a virus can transduce (i.e., the tropism) and how efficiently it can spread (Murlidharan et al., 2014). Multiple AAV serotypes have been used in SMA mice (Foust et al., 2010; Passini et al., 2010; Tsai et al., 2012), but serotype 9 was selected for AVXS-101 because of its comparatively strong tropism toward LMNs throughout the spinal cord in a range of species (Foust et al., 2009; Bevan et al., 2011; Federici et al., 2012). While numerous AAV serotypes and administration routes have also been tested in ALS mice (Tables 1, 2), it remains unclear which serotype will prove to be most effective. AAV9 and AAVrh10 are good candidates, but natural and engineered serotypes have recently been identified with improved tropism toward ALS-pertinent tissues (Deverman et al., 2016; Chan et al., 2017). Viral transgene expression can also be restricted using promoters with selective and defined expression patterns (Kügler, 2016). Combining knowledge of viral tropism with promoter selectivity thus provides a potential method for exquisite, cell type-specific targeting (von Jonquieres et al., 2013). Disease state (Chen et al., 2012) and age (Foust et al., 2009; Tosolini and Morris, 2016) also impact viral tropism, likely through alterations in host cell receptor availability, while promoter usage likewise changes with pathology and time; hence, it will be vital to test permutations of virus serotype/promoter in relevant pre-clinical models of ALS throughout all stages of disease.
Unlike AAV, ASOs do not readily penetrate tissues, while their cellular uptake and transition to the nucleus, their site of action, is limited. Indeed, it is estimated that <1% of ASOs reach their desired target, as the majority distribute to unwanted organs such as the liver (Godfrey et al., 2017). This mandates repeated therapeutic injections that can cause adverse events (Haché et al., 2016) and toxic ASO accumulation (Godfrey et al., 2017). Consequently, several chemical modifications to the ASO phosphate backbone have been tested and shown to improve safety and pharmacological properties (Evers et al., 2015). Moreover, a number of drug distribution systems compatible with systemic delivery are being developed that enhance ASO transport to disease-pertinent tissues and therefore ease administration and reduce the required dose. These ASO vectorisation strategies can be divided into (1) viral approaches that use harmless, non-replicating viruses, such as AAV, and (2) non-viral strategies that utilise different positively charged molecules such as lipids or peptides (Lehto et al., 2012). Viruses present immunogenicity issues and are not suitable for all nucleic acid-based molecules; nevertheless, to aid cellular targeting and cytoplasm-to-nucleus transport, viral vectors have been engineered to encode modified snRNAs that incorporate specific ASO sequences. Once inside target cells, these ASOs are imported into the nucleus where they accumulate as part of snRNPs (Imbert et al., 2017). When packaged into viral vectors, snRNAs containing ASOs targeting SMN2 have shown potential for SMA (Meyer et al., 2009; Dal Mas et al., 2015; Odermatt et al., 2016). As has the non-viral approach of conjugating ASOs to CPPs (Hammond et al., 2016; Shabanpoor et al., 2017). CPPs are 5–30 amino acid long, positively charged peptides that transport various macromolecules across cell membranes (Lehto et al., 2012). Highlighting the significance of therapeutic targeting, intravenous injection of an SMN2 SSO conjugated to a CNS-targeting CPP caused the greatest extension in SMA mouse survival reported to date – from 12 to 456 days compared to only 54 days for the “naked” oligonucleotide (Hammond et al., 2016). Importantly, these CPP-oligonucleotides are capable of delivery to the CNS of both neonatal and adult mice (Hammond et al., 2016; Shabanpoor et al., 2017); however, the safety, tolerability, and pharmacokinetics of CPP-conjugated ASOs are yet to be tested in additional organisms.
AAV and LV have separately been combined with RNAi-based strategies for SOD1 knockdown (Table 2), but only recently have ASO vectorisation strategies been tried in ALS models. Compared with non-encapsulated oligonucleotides, loading SOD1-specific ASOs into lipid particles caused a much greater reduction in SOD1 protein in HEK293 cells (Chen et al., 2017). Moreover, direct intravascular delivery of non-ASO-loaded nanoparticles resulted in brain accumulation in wild-type zebrafish, indicating promise for future work in ALS mice (Chen et al., 2017). SOD1 pre-mRNA-targeting ASOs were also embedded in modified snRNAs and engineered into AAVrh10, which was then co-injected into the blood and brain of SOD1G93A mice either at birth or pre-symptomatically at 50 days (Biferi et al., 2017). The ASO skips SOD1 exon 2 generating a premature stop codon, which resulted in ≈70% reduction of SOD1 protein levels in the spinal cord 112 days post-perinatal administration. The gene therapy caused vast improvements in neuromuscular function, restricted weight loss, and, when given early, resulted in the greatest SOD1G93A survival extension yet reported to ≈250 days (Biferi et al., 2017).
Amyotrophic lateral sclerosis patients and models do not show the same cell and tissue vulnerability-resistance continuum as SMA; however, it is clear that pathology is not limited to LMNs and UMNs, probably contributing to the gamut of unsuccessful ALS clinical trials (Bartus and Johnson, 2017a). Indeed, the ALS/FTD clinicopathological overlap (Morita et al., 2006; Neumann et al., 2006; Vance et al., 2006) has been replicated in ALS mice, indicating the importance of cortical cells and synapses to the disease (Fogarty et al., 2015, 2016). Furthermore, the non-cell autonomous toxicities emanating from cells such as microglia (Puentes et al., 2016), and the prion-like cell-to-cell spread of pathological aggregates, also indicate that targeting therapies to motor neurons as well as additional cells and tissues is likely to be required in order to generate meaningful improvements in ALS prognosis. Indeed, disease onset and mortality are delayed in mutant SOD1 mice when microglial activation is pharmacologically restricted prior to disease onset (Kriz et al., 2002; van den Bosch et al., 2002), while disease progression is slowed in mutant SOD1 mice in which SOD1 is removed from microglia, oligodendrocytes, or astrocytes (Boillée et al., 2006; Yamanaka et al., 2008; Kang et al., 2013). Moreover, expression of mutant SOD1 in motor neurons alone is insufficient to fully recapitulate the mutant SOD1 mouse phenotype (Lino et al., 2002; Pramatarova et al., 2001), while deletion of SOD1 from motor neurons and interneurons of SOD1 mutant mice only delays disease onset (Wang et al., 2009).
Spinal muscular atrophy research has highlighted the importance of comparing different therapy injection sites and testing novel technologies to improve targeting of therapeutics not only to the required tissues, but also to the correct resident cells and subcellular locations. Viral targeting of rapidly dividing cells affected by ALS (e.g., astrocytes), subtleties in motor neuron subtype vulnerability (Nijssen et al., 2017), along with the indication that different subcellular motor neuron compartments, such as the NMJ, may require differential support and treatment (Moloney et al., 2014), add to the complexity of the challenge ahead. It is therefore imperative that during ALS therapy design, careful consideration is given to what cells and tissues need to be targeted and how exactly this can be most effectively achieved.
Combinatorial Treatment Is Key
It is conceivable that carefully crafted gene therapy combinations targeting multiple disease mechanisms could provide additive effects in MNDs. Indeed, AAV1-follistatin treatment significantly boosted muscle and body weight of SMA mice suboptimally dosed with SMN2 ASO (Feng et al., 2016), while in isolation, follistatin upregulation or myostatin reduction have little effect (Sumner et al., 2009; Rindt et al., 2012). Co-delivery of recombinant follistatin with an SMN2-inducing compound also resulted in a small additive enhancement in motor function, but not survival (Harris and Butchbach, 2015). Similarly, genetic upregulation of SMA modifier plastin 3 has no effect on untreated SMA mouse lifespan (Ackermann et al., 2013), but can drastically improve survival in ASO-dosed animals (Hosseinibarkooie et al., 2016). A range of other pathological mechanisms could be concomitantly targeted in SMA (Bowerman et al., 2017). Combinations of drugs directed at just the SMN pathway have also proven more effective than individual treatments (Kwon et al., 2011; Liu et al., 2013), while the impact of ASOs targeting SMN2 has been enhanced by co-treatment with a compound (Osman et al., 2017), co-targeting the SMN2-repressing long non-coding RNA, SMN-AS1, for ASO-mediated knockdown (d’Ydewalle et al., 2017; Woo et al., 2017), and co-masking additional negative SMN2 splicing elements (Pao et al., 2013). Furthermore, a holistic approach to treatment appears to be important, as providing nutritional support to SMA mice can advance therapeutic efficacy (Narver et al., 2008; Butchbach et al., 2014).
These SMA studies suggest that previously unsuccessful therapeutics may provide additive benefits when used in combination, highlighting the importance of simultaneously treating multiple disease pathways. Given the pathology observed in ALS, it will also be vital to co-modify pathways in different cell types. To facilitate this, viral vectors engineered with selective promoters could be united with CPP-conjugated ASOs in order to synergistically target multiple genes in related and independent pathways across distinct and highly specific cell populations. This was recently confirmed in mutant SOD1G93A mice by concomitantly targeting distinct disease mechanisms in motor neurons, astrocytes and microglia (Frakes et al., 2017). In isolation, genetic suppression of the NF-κB pathway in microglia or shRNA-mediated knockdown of SOD1 in motor neurons and astrocytes via systemic AAV9 administration resulted in similar improvements in survival, disease onset, and progression of mutant SOD1 mice. However, the combined targeting of these two pathomechanisms across three major cell types resulted in an additive amelioration in all assessed phenotypes. The median mutant lifespan was expanded from 137 to 188 days with a maximum survival of 204 days, which is one of the best extensions reported to date (Frakes et al., 2017). In a separate study, joint lentiviral targeting of three distinct disease pathways aiming to reduce excitotoxicity resulted in a synergistic neuroprotective effect in SOD1G93A mice (Benkler et al., 2016), while simultaneous delivery of multiple neurotrophic factors can also induce modest additive effects over individual therapies (Haase et al., 1997; Krakora et al., 2013; Dadon-Nachum et al., 2015), although not consistently (Dodge et al., 2010). Like SMA, it is thus clear that prognosis can be improved in ALS models by attempting a multifaceted gene therapy approach. It is somewhat surprising that there have not yet been any published studies that test concurrent delivery of neuroprotective and knockdown strategies in vivo for ALS. ASO-mediated depletion of toxic mutant proteins is likely to be most critical in ALS, and should be joined with a gamut of neuroprotective accessory therapies such as neurotrophic factors (Henriques et al., 2010), SMN upregulation (e.g., via nusinersen or AVXS-101) (Kariya et al., 2012; Turner et al., 2014), and preservation of NMJ innervation (Miyoshi et al., 2017), in order to determine the most efficacious drug combinations.
Consider Dose Number and Drug Concentrations
In addition to delivering gene therapies to the intended cellular and subcellular site(s), effectors must be expressed/released at a therapeutically viable concentration. Providing multiple SMN2 ASO doses results in greater phenotypic amelioration in SMA mice (Hua et al., 2011; Zhou et al., 2015; Hammond et al., 2016), while increased concentrations of ASOs or AAVs have a similar positive effect (Meyer et al., 2015; Hammond et al., 2016; Hosseinibarkooie et al., 2016). These results unsurprisingly indicate that delivering greater quantities of therapy, results in higher SMN upregulation, and therefore a better phenotypic rescue of SMA mice. There will inevitably be a point at which SMN overexpression becomes detrimental to cells; however, two-fold genetic overexpression of SMN in the nervous system of control mice appears to be safe (Turner et al., 2014; Perera et al., 2016). Nevertheless, this may not be the case for all genes (Denovan-Wright et al., 2008), and thus care must be taken when artificially increasing protein abundance, especially considering that secreted proteins can elicit autocrine and paracrine effects (Baumgartner and Shine, 1997).
Caution must also be exercised with target gene reduction strategies as there may be disparate, time-dependent consequences between protein reduction below a physiological threshold and complete absence (Rossi et al., 2015). That being said, toxicities associated with ASO or AAV accumulation are likely to arise before viability is perturbed by excessive modulation of a gene. Hence, there is a fine balance between administering sufficient gene therapy to ensure correct targeting in effective quantities without causing systemic toxic accumulation and adverse side effects. Upon intravenous injection, ASOs accumulate in the liver, kidneys, and lymph nodes, amongst other places, and can cause hybridisation-independent toxicities (Godfrey et al., 2017), while AAVs can become similarly enriched (Zincarelli et al., 2008). The methods to enhance delivery to disease-susceptible cells and tissues discussed above (e.g., ASO-CPP conjugation) will undoubtedly aid in this battle, and should be optimized in ALS mice, along with ASO chemistries, to enrich the appropriately targeted therapeutic load. Moreover, multiple ASO doses and increasing concentrations of gene therapies must be tested in relevant models to identify the most effective and safe therapeutic regimes. Due to the immune response, repeated AAV dosing is not practical (although immunomodulation is an option) (Lorain et al., 2008), but ASOs can be administered at multiple time points. However, ASO concentration must also be optimized to escape host immune responses (Wang et al., 2008). It should also be remembered that once an AAV has been delivered, relatively little can be done to regulate transgene expression, but ASOs can be neutralized by sequestration using complimentary decoy ASOs (Rigo et al., 2014; Hua et al., 2015).
Therapeutic Timing Is Critical
As disease progresses, the number of impacted proteins and processes will likely increase as a result of time as pathways diverge from the initial cause of pathology. This provides a rationale for why selectively targeting individual disease pathways toward the end of a cascade may provide only limited benefit. Moreover, the greater the duration over which a disease develops, the more anatomical, circuit, and cellular damage will ensue leading to greater loss-of-function and thus a more significant challenge of recovery (Ramírez-Jarquín et al., 2014; Bartus and Johnson, 2017b). For example, early NMJ denervation followed by LMN die-back will severely, if not totally, restrict the ability of the LMN to respond to extracellular neurotrophic signaling. The successful treatment of any disorder is thus more likely to occur when a therapy is administered during early pathogenesis rather than at later time points and, in particular, at disease end stage. Whilst intuitive, this highlights the importance of earlier diagnosis, especially for ALS.
Indeed, the earlier SMN levels are augmented in SMA mice via SMN gene therapy, the better the therapeutic outcome (Foust et al., 2010; Hua et al., 2010, 2011; Porensky et al., 2012; Bogdanik et al., 2015; Zhou et al., 2015). Age-dependent differences in BBB permeability, neuropil density, and cell tropisms may contribute (Foust et al., 2009, 2010); however, the early temporal requirement for SMN has been corroborated using mice with inducible SMN alleles (Le et al., 2011; Lutz et al., 2011; Kariya et al., 2014a), and is consistent with human and mouse SMN being most highly expressed in the CNS perinatally (Jablonka and Sendtner, 2017), and the most common and severe form of SMA (type I) manifesting before 6 months of age. Accordingly, artificially reducing SMN levels in young adult mice has fewer repercussions than when SMN is diminished at earlier time points (Le et al., 2011; Kariya et al., 2014a). All of this suggests that there is a therapeutic window of opportunity during early development in which SMN gene therapy is likely to have the greatest chance of success. One study indicates that in mice, the postnatal window of highest SMN requirement coincides with neuromuscular maturation, and that NMJ disruption causes SMN upregulation in motor neurons (Kariya et al., 2014a). Accordingly, nusinersen is being tested in the open-label NURTURE trial in pre-symptomatic newborns genetically diagnosed with SMA. Interim analyses appear promising, and when complete are likely to provide compelling evidence of the importance of treating SMA as early as possible. It is perhaps for this reason, that a number of small molecule drugs that have proven useful in SMA mice dosed from birth, have failed in clinical trials in which patients are treated post-symptom onset (Fuller et al., 2010). Nevertheless, post-symptomatic restoration of SMN using an inducible allele has been shown to reverse overt neuromuscular pathology and significantly improve SMA mouse lifespan (Lutz et al., 2011). This was corroborated using systemic administration of SMN2-targeting ASOs in a mild SMA mouse model (Bogdanik et al., 2015), and in SMA mice sub-optimally dosed with a small molecule SMN2 splice-modifying drug and subsequently re-treated with the same compound or AAV1-follistatin gene therapy (Feng et al., 2016).
Early therapeutic interventions in mutant SOD1 mice also often result in greater impact on disease. For example, pre-symptomatic injection of VEGF-expressing LV into SOD1G93A mouse muscles resulted in greater delay in disease onset and progression compared to injection at paralysis onset (Azzouz et al., 2004b). The same is true for AAV-mediated IGF-1 delivery (Kaspar et al., 2003) and SOD1 silencing (Foust et al., 2013; Biferi et al., 2017), and recombinant VEGF injections into the brain of mutant SOD1 rats (Storkebaum et al., 2005). Significantly, these studies show, like in SMA, that survival of mutant SOD1 rodents can be extended even when therapies are delivered post-symptomatically. There is also evidence to indicate that treatment during the perinatal stage (e.g., postnatal day 1, P1) can cause even greater improvements in SOD1G93A lifespan (Foust et al., 2013; Biferi et al., 2017), perhaps due in part to sub-clinical embryonic/perinatal defects (van Zundert et al., 2012) and differential age-dependent gene therapy tropism (Foust et al., 2009); however, testing intervention timing in these models needs to be therapeutically appropriate, as ALS is an adult-onset neurodegenerative disease currently with no biomarkers. The pre-symptomatic treatment of fALS patients with a known ALS-causing genetic mutation is a possibility, in the same vein as SMA patients in the NURTURE trial of nusinersen, but treatment in humans at an age akin to P1 is highly unlikely. Nevertheless, perinatal therapy administration paradigms provide useful proof-of-concept information, and are invaluable in gene therapy optimisation.
Amyotrophic lateral sclerosis greatly impacts the neuro-muscular system and its proteome, which could cause disease-specific, time-constrained alterations that affect therapeutic efficacy; for instance, viral tropism could be impacted through differential receptor expression (Tosolini and Morris, 2016). Rodent work indicates that there is likely to be an optimal period for ALS therapy delivery, but with a broader therapeutic window, in which disease progression can at least be slowed, if not halted or even partially reversed. It is therefore paramount that potential therapeutics are tested at a range of time points in ALS rodents and in large animals including non-human primates in order to improve therapeutic timing strategy.
Conclusion
Typified by extensive heterogeneity, the ALS disease spectrum poses a daunting challenge for developing effective treatments. This diversity, along with numerous other factors, has resulted in an overabundance of unsuccessful clinical trials. Many of these involved compounds targeting likely secondary pathogenic pathways with only limited therapeutic potential. However, over the last two decades, gene therapy using ASOs or viral vectors have emerged as the most promising strategy for treating nervous system disorders. The predominantly LMN disease SMA has benefited from this burgeoning field, with the recent regulatory approval of the ASO nusinersen. Through developing ASO- and virus-mediated drugs for SMA, much has been learnt about gene therapy design and development that could help to alleviate the impact of other MNDs. Pre-clinical SMA research has made it clear that gene therapies must be efficiently delivered to pertinent sites of pathology, at concentrations within the therapeutic range, and at appropriate times in order to increase the chances of success. Tantamount to this is the parallel modification of multiple disease pathways across cell and tissue types. Similar conclusions are also beginning to emerge from pre-clinical ALS models. It is thus by no coincidence that the greatest amelioration of the SOD1G93A mouse phenotype to date was driven by the combined targeting of two pathomechanisms across multiple cell types, and the dual-administration of an AAV-guided SOD1-specific ASO into the blood and brain. Given the current lack of diagnostic and prognostic biomarkers for ALS and reliance upon the SOD1G93A mouse, successful translation to patients will be tricky. Nevertheless, by considering issues outlined in this review and thinking clearly about treatment logistics, a viable ALS gene therapy is unlikely to be far from the clinic.
Author Contributions
APT and JNS wrote the manuscript, and have approved submission of this work.
Funding
This work was funded by a Wellcome Trust Sir Henry Wellcome Postdoctoral Fellowship [103191/A/13/Z to JNS], and a postdoctoral position [APT] supported by a Wellcome Trust Senior Investigator Award [107116/Z/15/Z] to Giampietro Schiavo (Institute of Neurology, University College London).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This paper is dedicated to the memory of Paul Victor Tosolini, who, like all those living with ALS, showed endless courage and never gave up. The authors would like to thank Nicol Birsa, Pietro Fratta, and Giampietro Schiavo (Institute of Neurology, University College London) for critical reading of the manuscript.
Abbreviations
AAV, adeno-associated virus; ALS, amyotrophic lateral sclerosis; ASO, antisense oligonucleotide; BBB, blood-brain barrier; BDNF, brain-derived neurotrophic factor; BSCB, blood-spinal cord barrier; C9ORF72, chromosome 9 open reading frame 72; CNS, central nervous system; CNTF, ciliary-derived neurotrophic factor; CPP, cell-penetrating peptide; CSF, cerebrospinal fluid; EMA, European Medicines Agency; fALS, familial ALS; FDA, Food and Drug Administration; FTD, frontotemporal dementia; FUS, fused in sarcoma; HGF, hepatocyte growth factor; IGF-1, insulin-like growth factor; iPSC, induced pluripotent stem cell; ISS-N1, intronic splicing silencer N1; LMN, lower motor neuron; LV, lentivirus; MND, motor neuron disease; NMJ, neuromuscular junction; RNAi, RNA interference; sALS, sporadic ALS; scAAV, self-complementary AAV; SMA, spinal muscular atrophy; SMN, survival motor neuron; snRNA, small nuclear RNA; snRNP, small nuclear ribonucleoprotein; SOD1, superoxide dismutase 1; SSO, splice-switching oligonucleotide; TARDBP, transactive-region DNA binding protein; TLS, translocated in liposarcoma; UMN, upper motor neuron; VEGF, vascular endothelial growth factor.
References
Abe, K., Itoyama, Y., Sobue, G., Tsuji, S., Aoki, M., Doyu, M., et al. (2014). Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 610–617. doi: 10.3109/21678421.2014.959024
Ackermann, B., Kröber, S., Torres-Benito, L., Borgmann, A., Peters, M., Hosseini Barkooie, S. M., et al. (2013). Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 22, 1328–1347. doi: 10.1093/hmg/dds540
Acsadi, G., Anguelov, R. A., Yang, H., Toth, G., Thomas, R., Jani, A., et al. (2002). Increased survival and function of SOD1 mice after glial cell-derived neurotrophic factor gene therapy. Hum. Gene Ther. 13, 1047–1059. doi: 10.1089/104303402753812458
Al-Chalabi, A., Calvo, A., Chio, A., Colville, S., Ellis, C. M., Hardiman, O., et al. (2014). Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 13, 1108–1113. doi: 10.1016/S1474-4422(14)70219-4
Al-Chalabi, A., Fang, F., Hanby, M. F., Leigh, P. N., Shaw, C. E., Ye, W., et al. (2010). An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatr. 81, 1324–1326. doi: 10.1136/jnnp.2010.207464
Al-Chalabi, A., and Hardiman, O. (2013). The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 9, 617–628. doi: 10.1038/nrneurol.2013.203
Allodi, I., Comley, L., Nichterwitz, S., Nizzardo, M., Simone, C., Benitez, J. A., et al. (2016). Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep. 6:25960. doi: 10.1038/srep25960
Alonso, A., Logroscino, G., Jick, S. S., and Hernán, M. A. (2009). Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur. J. Neurol. 16, 745–751. doi: 10.1111/j.1468-1331.2009.02586.x
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi: 10.1007/s00401-011-0911-2
Andersson, M. K., Ståhlberg, A., Arvidsson, Y., Olofsson, A., Semb, H., Stenman, G., et al. (2008). The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 9:37. doi: 10.1186/1471-2121-9-37
Ayers, J. I., Fromholt, S., Sinyavskaya, O., Siemienski, Z., Rosario, A. M., Li, A., et al. (2015). Widespread and efficient transduction of spinal cord and brain following neonatal AAV injection and potential disease modifying effect in ALS mice. Mol. Ther. 23, 53–62. doi: 10.1038/mt.2014.180
Azzouz, M., Hottinger, A., Paterna, J. C., Zurn, A. D., Aebischer, P., and Büeler, H. (2000). Increased motoneuron survival and improved neuromuscular function in transgenic ALS mice after intraspinal injection of an adeno-associated virus encoding Bcl-2. Hum. Mol. Genet. 9, 803–811. doi: 10.1093/hmg/9.5.803
Azzouz, M., Le, T., Ralph, G. S., Walmsley, L., Monani, U. R., Lee, D. C. P., et al. (2004a). Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J. Clin. Invest. 114, 1726–1731.
Azzouz, M., Ralph, G. S., Storkebaum, E., Walmsley, L. E., Mitrophanous, K. A., Kingsman, S. M., et al. (2004b). VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 429, 413–417.
Bartus, R. T., and Johnson, E. M. (2017a). Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 1: where have we been and what have we learned? Neurobiol. Dis. 97, 156–168. doi: 10.1016/j.nbd.2016.03.027
Bartus, R. T., and Johnson, E. M. (2017b). Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 2: where do we stand and where must we go next? Neurobiol. Dis. 97, 169–178. doi: 10.1016/j.nbd.2016.03.026
Bäumer, D., Ansorge, O., Almeida, M., and Talbot, K. (2010). The role of RNA processing in the pathogenesis of motor neuron degeneration. Expert Rev. Mol. Med. 12:e21. doi: 10.1017/S1462399410001523
Bäumer, D., Lee, S., Nicholson, G., Davies, J. L., Parkinson, N. J., Murray, L. M., et al. (2009). Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLOS Genet. 5:e1000773. doi: 10.1371/journal.pgen.1000773
Bäumer, D., Talbot, K., and Turner, M. R. (2014). Advances in motor neurone disease. J. R. Soc. Med. 107, 14–21. doi: 10.1177/0141076813511451
Baumgartner, B. J., and Shine, H. D. (1997). Targeted transduction of CNS neurons with adenoviral vectors carrying neurotrophic factor genes confers neuroprotection that exceeds the transduced population. J. Neurosci. 17, 6504–6511.
Becker, L. A., Huang, B., Bieri, G., Ma, R., Knowles, D. A., Jafar-Nejad, P., et al. (2017). Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371. doi: 10.1038/nature22038
Bellingham, M. C. (2011). A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci. Ther. 17, 4–31. doi: 10.1111/j.1755-5949.2009.00116.x
Benatar, M. (2007). Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol. Dis. 26, 1–13. doi: 10.1016/j.nbd.2006.12.015
Benkler, C., Barhum, Y., Ben-Zur, T., and Offen, D. (2016). Multifactorial gene therapy enhancing the glutamate uptake system and reducing oxidative stress delays symptom onset and prolongs survival in the SOD1-G93A ALS mouse model. J. Mol. Neurosci. 58, 46–58. doi: 10.1007/s12031-015-0695-2
Bevan, A. K., Duque, S., Foust, K. D., Morales, P. R., Braun, L., Schmelzer, L., et al. (2011). Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 19, 1971–1980. doi: 10.1038/mt.2011.157
Biferi, M. G., Cohen-Tannoudji, M., Cappelletto, A., Giroux, B., Roda, M., Astord, S., et al. (2017). A new AAV10-U7-mediated gene therapy prolongs survival and restores function in an ALS mouse model. Mol. Ther. 25, 2038–2052. doi: 10.1016/j.ymthe.2017.05.017
Blauw, H. M., Barnes, C. P., van Vught, P. W. J., van Rheenen, W., Verheul, M., Cuppen, E., et al. (2012). SMN1 gene duplications are associated with sporadic ALS. Neurology 78, 776–780. doi: 10.1212/WNL.0b013e318249f697
Bogdanik, L. P., Osborne, M. A., Davis, C., Martin, W. P., Austin, A., Rigo, F., et al. (2015). Systemic, postsymptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 112, E5863–E5872. doi: 10.1073/pnas.1509758112
Boillée, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Bordet, T., Lesbordes, J. C., Rouhani, S., Castelnau-Ptakhine, L., Schmalbruch, H., Haase, G., et al. (2001). Protective effects of cardiotrophin-1 adenoviral gene transfer on neuromuscular degeneration in transgenic ALS mice. Hum. Mol. Genet. 10, 1925–1933. doi: 10.1093/hmg/10.18.1925
Borel, F., Gernoux, G., Cardozo, B., Metterville, J. P., Toro Cabreja, G. C., Song, L., et al. (2016). Therapeutic rAAVrh10 mediated SOD1 silencing in adult SOD1G93A mice and nonhuman primates. Hum. Gene Ther. 27, 19–31. doi: 10.1089/hum.2015.122
Bosco, D. A., Morfini, G., Karabacak, N. M., Song, Y., Gros-Louis, F., Pasinelli, P., et al. (2010). Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403. doi: 10.1038/nn.2660
Bowerman, M., Becker, C. G., Yáñez-Muñoz, R. J., Ning, K., Wood, M. J. A., Gillingwater, T. H., et al. (2017). Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Model. Mech. 10, 943–954. doi: 10.1242/dmm.030148
Brenner, D., Müller, K., Wieland, T., Weydt, P., Böhm, S., Lulé, D., et al. (2016). NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139:e28. doi: 10.1093/brain/aww033
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377, 162–172. doi: 10.1056/NEJMra1603471
Bucher, T., Colle, M.-A., Wakeling, E., Dubreil, L., Fyfe, J., Briot-Nivard, D., et al. (2013). scAAV9 intracisternal delivery results in efficient gene transfer to the central nervous system of a feline model of motor neuron disease. Hum. Gene Ther. 24, 670–682. doi: 10.1089/hum.2012.218
Bunton-Stasyshyn, R. K. A., Saccon, R. A., Fratta, P., and Fisher, E. M. C. (2015). SOD1 function and its implications for amyotrophic lateral sclerosis pathology: new and renascent themes. Neuroscientist 21, 519–529. doi: 10.1177/1073858414561795
Butchbach, M. E. R., Singh, J., Gurney, M. E., and Burghes, A. H. M. (2014). The effect of diet on the protective action of D156844 observed in spinal muscular atrophy mice. Exp. Neurol. 256, 1–6. doi: 10.1016/j.expneurol.2014.03.005
Byrne, S., Elamin, M., Bede, P., and Hardiman, O. (2012). Absence of consensus in diagnostic criteria for familial neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatr. 83, 365–367. doi: 10.1136/jnnp-2011-301530
Cauchi, R. J. (2014). Gem depletion: amyotrophic lateral sclerosis and spinal muscular atrophy crossover. CNS Neurosci. Ther. 20, 574–581. doi: 10.1111/cns.12242
Chabot, B., and Shkreta, L. (2016). Defective control of pre-messenger RNA splicing in human disease. J. Cell Biol. 212, 13–27. doi: 10.1083/jcb.201510032
Chan, K. Y., Jang, M. J., Yoo, B. B., Greenbaum, A., Ravi, N., Wu, W.-L., et al. (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20, 1172–1179. doi: 10.1038/nn.4593
Chen, L., Watson, C., Morsch, M., Cole, N. J., Chung, R. S., Saunders, D. N., et al. (2017). Improving the delivery of SOD1 antisense oligonucleotides to motor neurons using calcium phosphate-lipid nanoparticles. Front. Neurosci. 11:476. doi: 10.3389/fnins.2017.00476
Chen, Y. H., Claflin, K., Geoghegan, J. C., and Davidson, B. L. (2012). Sialic acid deposition impairs the utility of AAV9, but not peptide-modified AAVs for brain gene therapy in a mouse model of lysosomal storage disease. Mol. Ther. 20, 1393–1399. doi: 10.1038/mt.2012.100
Chiò, A., Logroscino, G., Hardiman, O., Swingler, R., Mitchell, D., Beghi, E., et al. (2009). Prognostic factors in ALS: a critical review. Amyotroph. Lateral Scler. 10, 310–323. doi: 10.3109/17482960802566824
Chiriboga, C. A., Swoboda, K. J., Darras, B. T., Iannaccone, S. T., Montes, J., De Vivo, D. C., et al. (2016). Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 86, 890–897. doi: 10.1212/WNL.0000000000002445
Cirulli, E. T., Lasseigne, B. N., Petrovski, S., Sapp, P. C., Dion, P. A., Leblond, C. S., et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. doi: 10.1126/science.aaa3650
Colombrita, C., Zennaro, E., Fallini, C., Weber, M., Sommacal, A., Buratti, E., et al. (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x
Conlon, E. G., Lu, L., Sharma, A., Yamazaki, T., Tang, T., Shneider, N. A., et al. (2016). The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 5:e17820. doi: 10.7554/eLife.17820
Corcia, P., Ingre, C., Blasco, H., Press, R., Praline, J., Antar, C., et al. (2012). Homozygous SMN2 deletion is a protective factor in the Swedish ALS population. Eur. J. Hum. Genet. 20, 588–591. doi: 10.1038/ejhg.2011.255
Dadon-Nachum, M., Ben-Yaacov, K., Ben-Zur, T., Barhum, Y., Yaffe, D., Perlson, E., et al. (2015). Transplanted modified muscle progenitor cells expressing a mixture of neurotrophic factors delay disease onset and enhance survival in the SOD1 mouse model of ALS. J. Mol. Neurosci. 55, 788–797. doi: 10.1007/s12031-014-0426-0
Dal Mas, A., Rogalska, M. E., Bussani, E., and Pagani, F. (2015). Improvement of SMN2 pre-mRNA processing mediated by exon-specific U1 small nuclear RNA. Am. J. Hum. Genet. 96, 93–103. doi: 10.1016/j.ajhg.2014.12.009
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Denovan-Wright, E. M., Attis, M., Rodriguez-Lebron, E., and Mandel, R. J. (2008). Sustained striatal ciliary neurotrophic factor expression negatively affects behavior and gene expression in normal and R6/1 mice. J. Neurosci. Res. 86, 1748–1757. doi: 10.1002/jnr.21636
Deverman, B. E., Pravdo, P. L., Simpson, B. P., Kumar, S. R., Chan, K. Y., Banerjee, A., et al. (2016). Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204–209. doi: 10.1038/nbt.3440
Dirren, E., Aebischer, J., Rochat, C., Towne, C., Schneider, B. L., and Aebischer, P. (2015). SOD1 silencing in motoneurons or glia rescues neuromuscular function in ALS mice. Ann. Clin. Transl. Neurol. 2, 167–184. doi: 10.1002/acn3.162
Dodge, J. C., Haidet, A. M., Yang, W., Passini, M. A., Hester, M., Clarke, J., et al. (2008). Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol. Ther. 16, 1056–1064. doi: 10.1038/mt.2008.60
Dodge, J. C., Treleaven, C. M., Fidler, J. A., Hester, M., Haidet, A., Handy, C., et al. (2010). AAV4-mediated expression of IGF-1 and VEGF within cellular components of the ventricular system improves survival outcome in familial ALS mice. Mol. Ther. 18, 2075–2084. doi: 10.1038/mt.2010.206
Dominguez, E., Marais, T., Chatauret, N., Benkhelifa-Ziyyat, S., Duque, S., Ravassard, P., et al. (2011). Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 20, 681–693. doi: 10.1093/hmg/ddq514
Duque, S., Joussemet, B., Riviere, C., Marais, T., Dubreil, L., Douar, A.-M., et al. (2009). Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 17, 1187–1196. doi: 10.1038/mt.2009.71
Duque, S. I., Arnold, W. D., Odermatt, P., Li, X., Porensky, P. N., Schmelzer, L., et al. (2015). A large animal model of spinal muscular atrophy and correction of phenotype. Ann. Neurol. 77, 399–414. doi: 10.1002/ana.24332
d’Ydewalle, C., Ramos, D. M., Pyles, N. J., Ng, S.-Y., Gorz, M., Pilato, C. M., et al. (2017). The antisense transcript SMN-AS1 regulates SMN expression and is a novel therapeutic target for spinal muscular atrophy. Neuron 93, 66–79. doi: 10.1016/j.neuron.2016.11.033
Eleftheriadou, I., Manolaras, I., Irvine, E. E., Dieringer, M., Trabalza, A., and Mazarakis, N. D. (2016). αCAR IGF-1 vector targeting of motor neurons ameliorates disease progression in ALS mice. Ann. Clin. Transl. Neurol. 3, 752–768. doi: 10.1002/acn3.335
Evers, M. M., Toonen, L. J. A., and van Roon-Mom, W. M. C. (2015). Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 87, 90–103. doi: 10.1016/j.addr.2015.03.008
Farrar, M. A., Vucic, S., Johnston, H. M., du Sart, D., and Kiernan, M. C. (2013). Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J. Pediatr. 162, 155–159. doi: 10.1016/j.jpeds.2012.05.067
Federici, T., and Boulis, N. M. (2012). Gene therapy for amyotrophic lateral sclerosis. Neurobiol. Dis. 48, 236–242. doi: 10.1016/j.nbd.2011.08.018
Federici, T., Taub, J. S., Baum, G. R., Gray, S. J., Grieger, J. C., Matthews, K. A., et al. (2012). Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 19, 852–859. doi: 10.1038/gt.2011.130
Feng, W., Gubitz, A. K., Wan, L., Battle, D. J., Dostie, J., Golembe, T. J., et al. (2005). Gemins modulate the expression and activity of the SMN complex. Hum. Mol. Genet. 14, 1605–1611. doi: 10.1093/hmg/ddi168
Feng, Z., Ling, K. K. Y., Zhao, X., Zhou, C., Karp, G., Welch, E. M., et al. (2016). Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet. 25, 964–975. doi: 10.1093/hmg/ddv629
Finkel, R. S., Chiriboga, C. A., Vajsar, J., Day, J. W., Montes, J., De Vivo, D. C., et al. (2016). Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026. doi: 10.1016/S0140-6736(16)31408-8
Finkel, R. S., Mercuri, E., Darras, B. T., Connolly, A. M., Kuntz, N. L., Kirschner, J., et al. (2017). Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732. doi: 10.1056/NEJMoa1702752
Fischer, U., Liu, Q., and Dreyfuss, G. (1997). The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 90, 1023–1029.
Fogarty, M. J., Klenowski, P. M., Lee, J. D., Drieberg-Thompson, J. R., Bartlett, S. E., Ngo, S. T., et al. (2016). Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 6:37968. doi: 10.1038/srep37968
Fogarty, M. J., Noakes, P. G., and Bellingham, M. C. (2015). Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neurosci. 35, 643–647. doi: 10.1523/JNEUROSCI.3483-14.2015
Foust, K. D., Nurre, E., Montgomery, C. L., Hernandez, A., Chan, C. M., and Kaspar, B. K. (2009). Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65. doi: 10.1038/nbt.1515
Foust, K. D., Salazar, D. L., Likhite, S., Ferraiuolo, L., Ditsworth, D., Ilieva, H., et al. (2013). Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol. Ther. 21, 2148–2159. doi: 10.1038/mt.2013.211
Foust, K. D., Wang, X., McGovern, V. L., Braun, L., Bevan, A. K., Haidet, A. M., et al. (2010). Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 28, 271–274. doi: 10.1038/nbt.1610
Frakes, A. E., Braun, L., Ferraiuolo, L., Guttridge, D. C., and Kaspar, B. K. (2017). Additive amelioration of ALS by co-targeting independent pathogenic mechanisms. Ann. Clin. Transl. Neurol. 4, 76–86. doi: 10.1002/acn3.375
Franz, C. K., Federici, T., Yang, J., Backus, C., Oh, S. S., Teng, Q., et al. (2009). Intraspinal cord delivery of IGF-I mediated by adeno-associated virus 2 is neuroprotective in a rat model of familial ALS. Neurobiol. Dis. 33, 473–481. doi: 10.1016/j.nbd.2008.12.003
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Müller, K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636. doi: 10.1038/nn.4000
Fujii, R., and Takumi, T. (2005). TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci. 118, 5755–5765. doi: 10.1242/jcs.02692
Fuller, H. R., Barišić, M., Šešo-Šimić, .,Špeljko, T., Morris, G. E., and Šimić, G. (2010). Treatment strategies for spinal muscular atrophy. Transl. Neurosci. 1, 308–321. doi: 10.2478/v10134-010-0045-4
Gama-Carvalho, M., Garcia-Vaquero, L. M., Pinto, R. F., Besse, F., Weis, J., Voigt, A., et al. (2017). Linking amyotrophic lateral sclerosis and spinal muscular atrophy through RNA-transcriptome homeostasis: a genomics perspective. J. Neurochem. 141, 12–30. doi: 10.1111/jnc.13945
Geary, R. S., Norris, D., Yu, R., and Bennett, C. F. (2015). Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 87, 46–51. doi: 10.1016/j.addr.2015.01.008
Genç, B., and Özdinler, P. H. (2014). Moving forward in clinical trials for ALS: motor neurons lead the way please. Drug Discov. Today 19, 441–449. doi: 10.1016/j.drudis.2013.10.014
Gerbino, V., Carrì, M. T., Cozzolino, M., and Achsel, T. (2013). Mislocalised FUS mutants stall spliceosomal snRNPs in the cytoplasm. Neurobiol. Dis. 55, 120–128. doi: 10.1016/j.nbd.2013.03.003
Gertz, B., Wong, M., and Martin, L. J. (2012). Nuclear localization of human SOD1 and mutant SOD1-specific disruption of survival motor neuron protein complex in transgenic amyotrophic lateral sclerosis mice. J. Neuropathol. Exp. Neurol. 71, 162–177. doi: 10.1097/NEN.0b013e318244b635
Glascock, J. J., Osman, E. Y., Wetz, M. J., Krogman, M. M., Shababi, M., and Lorson, C. L. (2012a). Decreasing disease severity in symptomatic, Smn-/-;SMN2+/+, spinal muscular atrophy mice following scAAV9-SMN delivery. Hum. Gene Ther. 23, 330–335. doi: 10.1089/hum.2011.166
Glascock, J. J., Shababi, M., Wetz, M. J., Krogman, M. M., and Lorson, C. L. (2012b). Direct central nervous system delivery provides enhanced protection following vector mediated gene replacement in a severe model of spinal muscular atrophy. Biochem. Biophys. Res. Commun. 417, 376–381. doi: 10.1016/j.bbrc.2011.11.121
Godfrey, C., Desviat, L. R., Smedsrød, B., Piétri-Rouxel, F., Denti, M. A., Disterer, P., et al. (2017). Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol Med 9, 545–557. doi: 10.15252/emmm.201607199
Gogliotti, R. G., Quinlan, K. A., Barlow, C. B., Heier, C. R., Heckman, C. J., and Didonato, C. J. (2012). Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J. Neurosci. 32, 3818–3829. doi: 10.1523/JNEUROSCI.5775-11.2012
Gotkine, M., Rozenstein, L., Einstein, O., Abramsky, O., Argov, Z., and Rosenmann, H. (2013). Presymptomatic treatment with acetylcholinesterase antisense oligonucleotides prolongs survival in ALS (G93A-SOD1) mice. Biomed. Res. Int. 2013:845345. doi: 10.1155/2013/845345
Grice, S. J., Sleigh, J. N., Liu, J.-L., and Sattelle, D. B. (2011). Invertebrate models of spinal muscular atrophy: insights into mechanisms and potential therapeutics. Bioessays 33, 956–965. doi: 10.1002/bies.201100082
Groen, E. J. N., Fumoto, K., Blokhuis, A. M., Engelen-Lee, J., Zhou, Y., van den Heuvel, D. M. A., et al. (2013). ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum. Mol. Genet. 22, 3690–3704. doi: 10.1093/hmg/ddt222
Gruss, O. J., Meduri, R., Schilling, M., and Fischer, U. (2017). UsnRNP biogenesis: mechanisms and regulation. Chromosoma 126, 577–593. doi: 10.1007/s00412-017-0637-6
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Haase, G., Kennel, P., Pettmann, B., Vigne, E., Akli, S., Revah, F., et al. (1997). Gene therapy of murine motor neuron disease using adenoviral vectors for neurotrophic factors. Nat. Med. 3, 429–436. doi: 10.1038/nm0497-429
Haase, G., Pettmann, B., Vigne, E., Castelnau-Ptakhine, L., Schmalbruch, H., and Kahn, A. (1998). Adenovirus-mediated transfer of the neurotrophin-3 gene into skeletal muscle of pmn mice: therapeutic effects and mechanisms of action. J. Neurol. Sci. 160(Suppl. 1), S97–S105. doi: 10.1016/S0022-510X(98)00207-X
Haché, M., Swoboda, K. J., Sethna, N., Farrow-Gillespie, A., Khandji, A., Xia, S., et al. (2016). Intrathecal injections in children with spinal muscular atrophy: nusinersen clinical trial experience. J. Child Neurol. 31, 899–906. doi: 10.1177/0883073815627882
Hamilton, G., and Gillingwater, T. H. (2013). Spinal muscular atrophy: going beyond the motor neuron. Trends Mol. Med. 19, 40–50. doi: 10.1016/j.molmed.2012.11.002
Hammond, S. M., Hazell, G., Shabanpoor, F., Saleh, A. F., Bowerman, M., Sleigh, J. N., et al. (2016). Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 113, 10962–10967. doi: 10.1073/pnas.1605731113
Hardiman, O., and van den Berg, L. H. (2017). Edaravone: a new treatment for ALS on the horizon? Lancet Neurol. 16, 490–491. doi: 10.1016/S1474-4422(17)30163-1
Hardiman, O., van den Berg, L. H., and Kiernan, M. C. (2011). Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 639–649. doi: 10.1038/nrneurol.2011.153
Harris, A. W., and Butchbach, M. E. R. (2015). The effect of the DcpS inhibitor D156844 on the protective action of follistatin in mice with spinal muscular atrophy. Neuromuscul. Disord. 25, 699–705. doi: 10.1016/j.nmd.2015.05.008
Henriques, A., Pitzer, C., Dittgen, T., Klugmann, M., Dupuis, L., and Schneider, A. (2011). CNS-targeted viral delivery of G-CSF in an animal model for ALS: improved efficacy and preservation of the neuromuscular unit. Mol. Ther. 19, 284–292. doi: 10.1038/mt.2010.271
Henriques, A., Pitzer, C., and Schneider, A. (2010). Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: where do we stand? Front. Neurosci. 4:32. doi: 10.3389/fnins.2010.00032
Hensel, N., and Claus, P. (2017). The actin cytoskeleton in SMA and ALS: how does it contribute to motoneuron degeneration? Neuroscientist doi: 10.1177/1073858417705059 [Epub ahead of print].
Hosseinibarkooie, S., Peters, M., Torres-Benito, L., Rastetter, R. H., Hupperich, K., Hoffmann, A., et al. (2016). The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am. J. Hum. Genet. 99, 647–665. doi: 10.1016/j.ajhg.2016.07.014
Howell, M. D., Ottesen, E. W., Singh, N. N., Anderson, R. L., Seo, J., Sivanesan, S., et al. (2017). TIA1 is a gender-specific disease modifier of a mild mouse model of spinal muscular atrophy. Sci. Rep. 7:7183. doi: 10.1038/s41598-017-07468-2
Hua, Y., Liu, Y. H., Sahashi, K., Rigo, F., Bennett, C. F., and Krainer, A. R. (2015). Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev. 29, 288–297. doi: 10.1101/gad.256644.114
Hua, Y., Sahashi, K., Hung, G., Rigo, F., Passini, M. A., Bennett, C. F., et al. (2010). Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 24, 1634–1644. doi: 10.1101/gad.1941310
Hua, Y., Sahashi, K., Rigo, F., Hung, G., Horev, G., Bennett, C. F., et al. (2011). Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126. doi: 10.1038/nature10485
Hua, Y., Vickers, T. A., Okunola, H. L., Bennett, C. F., and Krainer, A. R. (2008). Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 82, 834–848. doi: 10.1016/j.ajhg.2008.01.014
Hua, Y., and Zhou, J. (2004). Survival motor neuron protein facilitates assembly of stress granules. FEBS Lett. 572, 69–74. doi: 10.1016/j.febslet.2004.07.010
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772. doi: 10.1083/jcb.200908164
Imbert, M., Dias-Florencio, G., and Goyenvalle, A. (2017). Viral Vector-Mediated Antisense therapy for genetic diseases. Genes 8:E51. doi: 10.3390/genes8020051
Ishihara, T., Ariizumi, Y., Shiga, A., Kato, T., Tan, C.-F., Sato, T., et al. (2013). Decreased number of Gemini of coiled bodies and U12 snRNA level in amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 4136–4147. doi: 10.1093/hmg/ddt262
Ito, H., Wate, R., Zhang, J., Ohnishi, S., Kaneko, S., Ito, H., et al. (2008). Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 213, 448–455. doi: 10.1016/j.expneurol.2008.07.017
Jablonka, S., and Sendtner, M. (2017). Developmental regulation of SMN expression: pathophysiological implications and perspectives for therapy development in spinal muscular atrophy. Gene Ther. 24, 506–513. doi: 10.1038/gt.2017.46
Jiang, J., Zhu, Q., Gendron, T. F., Saberi, S., McAlonis-Downes, M., Seelman, A., et al. (2016). Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550. doi: 10.1016/j.neuron.2016.04.006
Kang, S. H., Li, Y., Fukaya, M., Lorenzini, I., Cleveland, D. W., Ostrow, L. W., et al. (2013). Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 16, 571–579. doi: 10.1038/nn.3357
Kariya, S., Obis, T., Garone, C., Akay, T., Sera, F., Iwata, S., et al. (2014a). Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J. Clin. Invest. 124, 785–800. doi: 10.1172/JCI72017
Kariya, S., Re, D. B., Jacquier, A., Nelson, K., Przedborski, S., and Monani, U. R. (2012). Mutant superoxide dismutase 1 (SOD1), a cause of amyotrophic lateral sclerosis, disrupts the recruitment of SMN, the spinal muscular atrophy protein to nuclear Cajal bodies. Hum. Mol. Genet. 21, 3421–3434. doi: 10.1093/hmg/dds174
Kariya, S., Sampson, J. B., Northrop, L. E., Luccarelli, C. M., Naini, A. B., Re, D. B., et al. (2014b). Nuclear localization of SMN and FUS is not altered in fibroblasts from patients with sporadic ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 581–587. doi: 10.3109/21678421.2014.907319
Kaspar, B. K., Lladó, J., Sherkat, N., Rothstein, J. D., and Gage, F. H. (2003). Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301, 839–842. doi: 10.1126/science.1086137
Keil, J. M., Seo, J., Howell, M. D., Hsu, W. H., Singh, R. N., and DiDonato, C. J. (2014). A short antisense oligonucleotide ameliorates symptoms of severe mouse models of spinal muscular atrophy. Mol. Ther. Nucleic Acids 3:e174. doi: 10.1038/mtna.2014.23
Khatri, I. A., Chaudhry, U. S., Seikaly, M. G., Browne, R. H., and Iannaccone, S. T. (2008). Low bone mineral density in spinal muscular atrophy. J .Clin. Neuromuscul. Dis. 10, 11–17. doi: 10.1097/CND.0b013e318183e0fa
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Klein, S. M., Behrstock, S., McHugh, J., Hoffmann, K., Wallace, K., Suzuki, M., et al. (2005). GDNF delivery using human neural progenitor cells in a rat model of ALS. Hum. Gene Ther. 16, 509–521. doi: 10.1089/hum.2005.16.509
Koval, E. D., Shaner, C., Zhang, P., du Maine, X., Fischer, K., Tay, J., et al. (2013). Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 22, 4127–4135. doi: 10.1093/hmg/ddt261
Krakora, D., Macrander, C., and Suzuki, M. (2012). Neuromuscular junction protection for the potential treatment of amyotrophic lateral sclerosis. Neurol. Res. Int. 2012:379657. doi: 10.1155/2012/379657
Krakora, D., Mulcrone, P., Meyer, M., Lewis, C., Bernau, K., Gowing, G., et al. (2013). Synergistic effects of GDNF and VEGF on lifespan and disease progression in a familial ALS rat model. Mol. Ther. 21, 1602–1610. doi: 10.1038/mt.2013.108
Kriz, J., Nguyen, M. D., and Julien, J.-P. (2002). Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 10, 268–278. doi: 10.1006/nbdi.2002.0487
Kügler, S. (2016). Tissue-specific promoters in the CNS. Methods Mol. Biol. 1382, 81–91. doi: 10.1007/978-1-4939-3271-9_6
Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Kwon, D. Y., Motley, W. W., Fischbeck, K. H., and Burnett, B. G. (2011). Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Hum. Mol. Genet. 20, 3667–3677. doi: 10.1093/hmg/ddr288
Le, T. T., McGovern, V. L., Alwine, I. E., Wang, X., Massoni-Laporte, A., Rich, M. M., et al. (2011). Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 20, 3578–3591. doi: 10.1093/hmg/ddr275
Le, T. T., Pham, L. T., Butchbach, M. E. R., Zhang, H. L., Monani, U. R., Coovert, D. D., et al. (2005). SMNΔ7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845–857. doi: 10.1093/hmg/ddi078
Lee, E. B., Lee, V. M.-Y., and Trojanowski, J. Q. (2012). Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50.
Lee, Y.-B., Chen, H.-J., Peres, J. N., Gomez-Deza, J., Attig, J., Stalekar, M., et al. (2013). Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 5, 1178–1186. doi: 10.1016/j.celrep.2013.10.049
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Lefebvre, S., Burlet, P., Liu, Q., Bertrandy, S., Clermont, O., Munnich, A., et al. (1997). Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16, 265–269. doi: 10.1038/ng0797-265
Lehto, T., Kurrikoff, K., and Langel, Ü. (2012). Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 9, 823–836. doi: 10.1517/17425247.2012.689285
Lepore, A. C., Haenggeli, C., Gasmi, M., Bishop, K. M., Bartus, R. T., Maragakis, N. J., et al. (2007). Intraparenchymal spinal cord delivery of adeno-associated virus IGF-1 is protective in the SOD1G93A model of ALS. Brain Res. 1185, 256–265. doi: 10.1016/j.brainres.2007.09.034
Li, D., Liu, C., Yang, C., Wang, D., Wu, D., Qi, Y., et al. (2017). Slow intrathecal injection of rAAVrh10 enhances its transduction of spinal cord and therapeutic efficacy in a mutant SOD1 model of ALS. Neuroscience 365, 192–205. doi: 10.1016/j.neuroscience.2017.10.001
Lin, H., Hu, H., Duan, W., Liu, Y., Tan, G., Li, Z., et al. (2016). Intramuscular delivery of scAAV9-hIGF1 prolongs survival in the hSOD1G93A ALS mouse model via upregulation of D-amino acid oxidase. Mol. Neurobiol. (in press). doi: 10.1007/s12035-016-0335-z
Ling, S.-C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Lino, M. M., Schneider, C., and Caroni, P. (2002). Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22, 4825–4832.
Liu, H.-C., Ting, C.-H., Wen, H.-L., Tsai, L.-K., Hsieh-Li, H.-M., Li, H., et al. (2013). Sodium vanadate combined with L-ascorbic acid delays disease progression, enhances motor performance, and ameliorates muscle atrophy and weakness in mice with spinal muscular atrophy. BMC Med. 11:38. doi: 10.1186/1741-7015-11-38
Liu, Q., and Dreyfuss, G. (1996). A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 15, 3555–3565.
Liu, Q., Fischer, U., Wang, F., and Dreyfuss, G. (1997). The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 90, 1013–1021. doi: 10.1016/S0092-8674(00)80367-0
Locatelli, F., Corti, S., Papadimitriou, D., Fortunato, F., Del Bo, R., Donadoni, C., et al. (2007). Fas small interfering RNA reduces motoneuron death in amyotrophic lateral sclerosis mice. Ann. Neurol. 62, 81–92. doi: 10.1002/ana.21152
Lorain, S., Gross, D.-A., Goyenvalle, A., Danos, O., Davoust, J., and Garcia, L. (2008). Transient immunomodulation allows repeated injections of AAV1 and correction of muscular dystrophy in multiple muscles. Mol. Ther. 16, 541–547. doi: 10.1038/sj.mt.6300377
Lorson, C. L., Hahnen, E., Androphy, E. J., and Wirth, B. (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 96, 6307–6311. doi: 10.1073/pnas.96.11.6307
Lu, C.-H., Macdonald-Wallis, C., Gray, E., Pearce, N., Petzold, A., Norgren, N., et al. (2015). Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology 84, 2247–2257. doi: 10.1212/WNL.0000000000001642
Lu, L., Zheng, L., Viera, L., Suswam, E., Li, Y., Li, X., et al. (2007). Mutant Cu/Zn-superoxide dismutase associated with amyotrophic lateral sclerosis destabilizes vascular endothelial growth factor mRNA and downregulates its expression. J. Neurosci. 27, 7929–7938. doi: 10.1523/JNEUROSCI.1877-07.2007
Ludolph, A. C., Bendotti, C., Blaugrund, E., Chio, A., Greensmith, L., Loeffler, J.-P., et al. (2010). Guidelines for preclinical animal research in ALS/MND: a consensus meeting. Amyotroph. Lateral Scler. 11, 38–45. doi: 10.3109/17482960903545334
Lutz, C. M., Kariya, S., Patruni, S., Osborne, M. A., Liu, D., Henderson, C. E., et al. (2011). Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Invest. 121, 3029–3041. doi: 10.1172/JCI57291
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95, 808–816.e9. doi: 10.1016/j.neuron.2017.07.025
Mackenzie, I. R. A., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434. doi: 10.1002/ana.21147
Maharjan, N., Künzli, C., Buthey, K., and Saxena, S. (2017). C9ORF72 regulates stress granule formation and its deficiency impairs stress granule assembly, hypersensitizing cells to stress. Mol. Neurobiol. 54, 3062–3077. doi: 10.1007/s12035-016-9850-1
Mancuso, R., Martínez-Muriana, A., Leiva, T., Gregorio, D., Ariza, L., Morell, M., et al. (2016). Neuregulin-1 promotes functional improvement by enhancing collateral sprouting in SOD1G93A ALS mice and after partial muscle denervation. Neurobiol. Dis. 95, 168–178. doi: 10.1016/j.nbd.2016.07.023
Martin, S., Al Khleifat, A., and Al-Chalabi, A. (2017a). What causes amyotrophic lateral sclerosis? F1000Res. 6:371. doi: 10.12688/f1000research.10476.1
Martin, S., Trevor-Jones, E., Khan, S., Shaw, K., Marchment, D., Kulka, A., et al. (2017b). The benefit of evolving multidisciplinary care in ALS: a diagnostic cohort survival comparison. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 569–575. doi: 10.1080/21678421.2017.1349151
Martinez, T. L., Kong, L., Wang, X., Osborne, M. A., Crowder, M. E., van Meerbeke, J. P., et al. (2012). Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J. Neurosci. 32, 8703–8715. doi: 10.1523/JNEUROSCI.0204-12.2012
McAndrew, P. E., Parsons, D. W., Simard, L. R., Rochette, C., Ray, P. N., Mendell, J. R., et al. (1997). Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 60, 1411–1422. doi: 10.1086/515465
Mendell, J. R., Al-Zaidy, S., Shell, R., Arnold, W. D., Rodino-Klapac, L. R., Prior, T. W., et al. (2017). Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722. doi: 10.1056/NEJMoa1706198
Meyer, K., Ferraiuolo, L., Schmelzer, L., Braun, L., McGovern, V., Likhite, S., et al. (2015). Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol. Ther. 23, 477–487. doi: 10.1038/mt.2014.210
Meyer, K., Marquis, J., Trüb, J., Nlend Nlend, R., Verp, S., Ruepp, M.-D., et al. (2009). Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum. Mol. Genet. 18, 546–555. doi: 10.1093/hmg/ddn382
Miller, T. M., Kaspar, B. K., Kops, G. J., Yamanaka, K., Christian, L. J., Gage, F. H., et al. (2005). Virus-delivered small RNA silencing sustains strength in amyotrophic lateral sclerosis. Ann. Neurol. 57, 773–776. doi: 10.1002/ana.20453
Miller, T. M., Pestronk, A., David, W., Rothstein, J., Simpson, E., Appel, S. H., et al. (2013). An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442. doi: 10.1016/S1474-4422(13)70061-9
Mirra, A., Rossi, S., Scaricamazza, S., Di Salvio, M., Salvatori, I., Valle, C., et al. (2017). Functional interaction between FUS and SMN underlies SMA-like splicing changes in wild-type hFUS mice. Sci. Rep. 7:2033. doi: 10.1038/s41598-017-02195-0
Mitsumoto, H., Brooks, B. R., and Silani, V. (2014). Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol. 13, 1127–1138. doi: 10.1016/S1474-4422(14)70129-2
Miyoshi, S., Tezuka, T., Arimura, S., Tomono, T., Okada, T., and Yamanashi, Y. (2017). DOK7 gene therapy enhances motor activity and life span in ALS model mice. EMBO Mol. Med. 9, 880–889. doi: 10.15252/emmm.201607298
Moens, T. G., Partridge, L., and Isaacs, A. M. (2017). Genetic models of C9orf72: what is toxic? Curr. Opin. Genet. Dev. 44, 92–101. doi: 10.1016/j.gde.2017.01.006
Mohajeri, M. H., Figlewicz, D. A., and Bohn, M. C. (1999). Intramuscular grafts of myoblasts genetically modified to secrete glial cell line-derived neurotrophic factor prevent motoneuron loss and disease progression in a mouse model of familial amyotrophic lateral sclerosis. Hum. Gene Ther. 10, 1853–1866. doi: 10.1089/10430349950017536
Mohan, R., Tosolini, A. P., and Morris, R. (2014). Targeting the motor end plates in the mouse hindlimb gives access to a greater number of spinal cord motor neurons: an approach to maximize retrograde transport. Neuroscience 274, 318–330. doi: 10.1016/j.neuroscience.2014.05.045
Moloney, E. B., de Winter, F., and Verhaagen, J. (2014). ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 8:252. doi: 10.3389/fnins.2014.00252
Monani, U. R., Lorson, C. L., Parsons, D. W., Prior, T. W., Androphy, E. J., Burghes, A. H., et al. (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183. doi: 10.1093/hmg/8.7.1177
Mori, K., Lammich, S., Mackenzie, I. R. A., Forné, I., Zilow, S., Kretzschmar, H., et al. (2013). hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 125, 413–423. doi: 10.1007/s00401-013-1088-7
Morita, M., Al-Chalabi, A., Andersen, P. M., Hosler, B., Sapp, P., Englund, E., et al. (2006). A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66, 839–844. doi: 10.1212/01.wnl.0000200048.53766.b4
Mulcahy, P. J., Iremonger, K., Karyka, E., Herranz-Martn, S., Shum, K.-T., Tam, J. K. V., et al. (2014). Gene therapy: a promising approach to treating spinal muscular atrophy. Hum. Gene Ther. 25, 575–586. doi: 10.1089/hum.2013.186
Munsat, T. L., and Davies, K. E. (1992). International SMA consortium meeting. (26-28 June 1992, Bonn, Germany). Neuromuscul. Disord. 2, 423–428. doi: 10.1016/S0960-8966(06)80015-5
Murlidharan, G., Samulski, R. J., and Asokan, A. (2014). Biology of adeno-associated viral vectors in the central nervous system. Front. Mol. Neurosci. 7:76. doi: 10.3389/fnmol.2014.00076
Nanou, A., Higginbottom, A., Valori, C. F., Wyles, M., Ning, K., Shaw, P., et al. (2013). Viral delivery of antioxidant genes as a therapeutic strategy in experimental models of amyotrophic lateral sclerosis. Mol. Ther. 21, 1486–1496. doi: 10.1038/mt.2013.115
Narver, H. L., Kong, L., Burnett, B. G., Choe, D. W., Bosch-Marcé, M., Taye, A. A., et al. (2008). Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann. Neurol. 64, 465–470. doi: 10.1002/ana.21449
Nassif, M., Woehlbier, U., and Manque, P. A. (2017). The enigmatic role of C9ORF72 in autophagy. Front. Neurosci. 11:442. doi: 10.3389/fnins.2017.00442
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nijssen, J., Comley, L. H., and Hedlund, E. (2017). Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 133, 863–885. doi: 10.1007/s00401-017-1708-8
Nizzardo, M., Simone, C., Falcone, M., Riboldi, G., Rizzo, F., Magri, F., et al. (2012). Research advances in gene therapy approaches for the treatment of amyotrophic lateral sclerosis. Cell Mol. Life. Sci. 69, 1641–1650. doi: 10.1007/s00018-011-0881-5
Nizzardo, M., Simone, C., Rizzo, F., Ulzi, G., Ramirez, A., Rizzuti, M., et al. (2016). Morpholino-mediated SOD1 reduction ameliorates an amyotrophic lateral sclerosis disease phenotype. Sci. Rep. 6:21301. doi: 10.1038/srep21301
Nizzardo, M., Simone, C., Salani, S., Ruepp, M.-D., Rizzo, F., Ruggieri, M., et al. (2014). Effect of combined systemic and local morpholino treatment on the spinal muscular atrophy Δ7 mouse model phenotype. Clin. Ther. 36, 340–356.e5. doi: 10.1016/j.clinthera.2014.02.004
Odermatt, P., Trüb, J., Furrer, L., Fricker, R., Marti, A., and Schümperli, D. (2016). Somatic therapy of a mouse SMA model with a U7 snRNA gene correcting SMN2 splicing. Mol. Ther. 24, 1797–1805. doi: 10.1038/mt.2016.152
Oeckl, P., Jardel, C., Salachas, F., Lamari, F., Andersen, P. M., Bowser, R., et al. (2016). Multicenter validation of CSF neurofilaments as diagnostic biomarkers for ALS. Amyotroph. Lateral Scler. Frontotemporal. Degener. 17, 404–413. doi: 10.3109/21678421.2016.1167913
Osman, E. Y., Miller, M. R., Robbins, K. L., Lombardi, A. M., Atkinson, A. K., Brehm, A. J., et al. (2014). Morpholino antisense oligonucleotides targeting intronic repressor Element1 improve phenotype in SMA mouse models. Hum. Mol. Genet. 23, 4832–4845. doi: 10.1093/hmg/ddu198
Osman, E. Y., Washington, C. W., Simon, M. E., Megiddo, D., Greif, H., and Lorson, C. L. (2017). Analysis of azithromycin monohydrate as a single or a combinatorial therapy in a mouse model of severe spinal muscular atrophy. J. Neuromuscul. Dis. 4, 237–249. doi: 10.3233/JND-170230
Pao, P. W., Wee, K. B., Yee, W. C., and Pramono, Z. A. D. (2013). Dual masking of specific negative splicing regulatory elements resulted in maximal exon 7 inclusion of SMN2 gene. Mol. Ther. 22, 854–861. doi: 10.1038/mt.2013.276
Passini, M. A., Bu, J., Richards, A. M., Kinnecom, C., Sardi, S. P., Stanek, L. M., et al. (2011). Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 3:72ra18. doi: 10.1126/scitranslmed.3001777
Passini, M. A., Bu, J., Roskelley, E. M., Richards, A. M., Sardi, S. P., O’Riordan, C. R., et al. (2010). CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 120, 1253–1264. doi: 10.1172/JCI41615
Patel, P., Kriz, J., Gravel, M., Soucy, G., Bareil, C., Gravel, C., et al. (2014). Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol. Ther. 22, 498–510. doi: 10.1038/mt.2013.239
Patten, S. A., Armstrong, G. A. B., Lissouba, A., Kabashi, E., Parker, J. A., and Drapeau, P. (2014). Fishing for causes and cures of motor neuron disorders. Dis. Model. Mech. 7, 799–809. doi: 10.1242/dmm.015719
Pellizzoni, L., Charroux, B., Rappsilber, J., Mann, M., and Dreyfuss, G. (2001). A functional interaction between the survival motor neuron complex and RNA polymerase II. J. Cell Biol. 152, 75–85. doi: 10.1083/jcb.152.1.75
Pellizzoni, L., Yong, J., and Dreyfuss, G. (2002). Essential role for the SMN complex in the specificity of snRNP assembly. Science 298, 1775–1779. doi: 10.1126/science.1074962
Perera, N. D., Sheean, R. K., Crouch, P. J., White, A. R., Horne, M. K., and Turner, B. J. (2016). Enhancing survival motor neuron expression extends lifespan and attenuates neurodegeneration in mutant TDP-43 mice. Hum. Mol. Genet. 25, 4080–4093. doi: 10.1093/hmg/ddw247
Petrov, D., Mansfield, C., Moussy, A., and Hermine, O. (2017). ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 9:68. doi: 10.3389/fnagi.2017.00068
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
Porensky, P. N., Mitrpant, C., McGovern, V. L., Bevan, A. K., Foust, K. D., Kaspar, B. K., et al. (2012). A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 21, 1625–1638. doi: 10.1093/hmg/ddr600
Pramatarova, A., Laganière, J., Roussel, J., Brisebois, K., and Rouleau, G. A. (2001). Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 21, 3369–3374.
Puentes, F., Malaspina, A., van Noort, J. M., and Amor, S. (2016). Non-neuronal cells in ALS: role of glial, immune cells and blood-CNS barriers. Brain Pathol. 26, 248–257. doi: 10.1111/bpa.12352
Ralph, G. S., Radcliffe, P. A., Day, D. M., Carthy, J. M., Leroux, M. A., Lee, D. C. P., et al. (2005). Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 11, 429–433.
Ramírez-Jarquín, U. N., Lazo-Gómez, R., Tovar-Y-Romo, L. B., and Tapia, R. (2014). Spinal inhibitory circuits and their role in motor neuron degeneration. Neuropharmacology 82, 101–107. doi: 10.1016/j.neuropharm.2013.10.003
Raoul, C., Abbas-Terki, T., Bensadoun, J.-C., Guillot, S., Haase, G., Szulc, J., et al. (2005). Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 11, 423–428.
Reaume, A. G., Elliott, J. L., Hoffman, E. K., Kowall, N. W., Ferrante, R. J., Siwek, D. F., et al. (1996). Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 13, 43–47.
Reber, S., Stettler, J., Filosa, G., Colombo, M., Jutzi, D., Lenzken, S. C., et al. (2016). Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J. 35, 1504–1521. doi: 10.15252/embj.201593791
Rembach, A., Turner, B. J., Bruce, S., Cheah, I. K., Scott, R. L., Lopes, E. C., et al. (2004). Antisense peptide nucleic acid targeting GluR3 delays disease onset and progression in the SOD1 G93A mouse model of familial ALS. J. Neurosci. Res. 77, 573–582.
Renton, A. E., Chiò, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rigo, F., Chun, S. J., Norris, D. A., Hung, G., Lee, S., Matson, J., et al. (2014). Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharmacol. Exp. Ther. 350, 46–55. doi: 10.1124/jpet.113.212407
Rindt, H., Buckley, D. M., Vale, S. M., Krogman, M., Rose, F. F., Garcia, M. L., et al. (2012). Transgenic inactivation of murine myostatin does not decrease the severity of disease in a model of Spinal Muscular Atrophy. Neuromuscul. Disord. 22, 277–285. doi: 10.1016/j.nmd.2011.10.012
Ringholz, G. M., Appel, S. H., Bradshaw, M., Cooke, N. A., Mosnik, D. M., and Schulz, P. E. (2005). Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65, 586–590.
Rochette, C. F., Gilbert, N., and Simard, L. R. (2001). SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 108, 255–266.
Rodriguez-Muela, N., Litterman, N. K., Norabuena, E. M., Mull, J. L., Galazo, M. J., Sun, C., et al. (2017). Single-cell analysis of SMN reveals its broader role in neuromuscular disease. Cell Rep. 18, 1484–1498. doi: 10.1016/j.celrep.2017.01.035
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62.
Rosenfeld, J., and Strong, M. J. (2015). Challenges in the understanding and treatment of amyotrophic lateral sclerosis/motor neuron disease. Neurotherapeutics 12, 317–325. doi: 10.1007/s13311-014-0332-8
Rossi, A., Kontarakis, Z., Gerri, C., Nolte, H., Hölper, S., Krüger, M., et al. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. doi: 10.1038/nature14580
Rossoll, W., Jablonka, S., Andreassi, C., Kröning, A.-K., Karle, K., Monani, U. R., et al. (2003). Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 163, 801–812.
Rudnik-Schöneborn, S., Heller, R., Berg, C., Betzler, C., Grimm, T., Eggermann, T., et al. (2008). Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J. Med. Genet. 45, 635–638. doi: 10.1136/jmg.2008.057950
Saccon, R. A., Bunton-Stasyshyn, R. K. A., Fisher, E. M. C., and Fratta, P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136, 2342–2358. doi: 10.1093/brain/awt097
Samaranch, L., Salegio, E. A., San Sebastian, W., Kells, A. P., Foust, K. D., Bringas, J. R., et al. (2012). Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum. Gene Ther. 23, 382–389. doi: 10.1089/hum.2011.200
Scarrott, J. M., Herranz-Martín, S., Alrafiah, A. R., Shaw, P. J., and Azzouz, M. (2015). Current developments in gene therapy for amyotrophic lateral sclerosis. Expert Opin. Biol. Ther. 15, 935–947. doi: 10.1517/14712598.2015.1044894
Scekic-Zahirovic, J., Sendscheid, O., El Oussini, H., Jambeau, M., Sun, Y., Mersmann, S., et al. (2016). Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 35, 1077–1097. doi: 10.15252/embj.201592559
Scoto, M., Finkel, R. S., Mercuri, E., and Muntoni, F. (2017). Therapeutic approaches for spinal muscular atrophy (SMA). Gene Ther. 24, 514–519. doi: 10.1038/gt.2017.45
Scott, S., Kranz, J. E., Cole, J., Lincecum, J. M., Thompson, K., Kelly, N., et al. (2008). Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph. Lateral Scler. 9, 4–15. doi: 10.1080/17482960701856300
Shabanpoor, F., Hammond, S. M., Abendroth, F., Hazell, G., Wood, M. J. A., and Gait, M. J. (2017). Identification of a peptide for systemic brain delivery of a morpholino oligonucleotide in mouse models of spinal muscular atrophy. Nucleic Acid Ther. 27, 130–143. doi: 10.1089/nat.2016.0652
Shan, X., Chiang, P.-M., Price, D. L., and Wong, P. C. (2010). Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 107, 16325–16330. doi: 10.1073/pnas.1003459107
Shepheard, S. R., Wuu, J., Cardoso, M., Wiklendt, L., Dinning, P. G., Chataway, T., et al. (2017). Urinary p75ECD: a prognostic, disease progression, and pharmacodynamic biomarker in ALS. Neurology 88, 1137–1143. doi: 10.1212/WNL.0000000000003741
Singh, N. K., Singh, N. N., Androphy, E. J., and Singh, R. N. (2006). Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 26, 1333–1346.
Singh, N. N., Howell, M. D., Androphy, E. J., and Singh, R. N. (2017). How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. 24, 520–526. doi: 10.1038/gt.2017.34
Singh, N. N., Seo, J., Ottesen, E. W., Shishimorova, M., Bhattacharya, D., and Singh, R. N. (2011). TIA1 prevents skipping of a critical exon associated with spinal muscular atrophy. Mol. Cell. Biol. 31, 935–954. doi: 10.1128/MCB.00945-10
Singh, R. N., Howell, M. D., Ottesen, E. W., and Singh, N. N. (2017). Diverse role of survival motor neuron protein. Biochim. Biophys. Acta 1860, 299–315.
Sleigh, J. N., Barreiro-Iglesias, A., Oliver, P. L., Biba, A., Becker, T., Davies, K. E., et al. (2014). Chondrolectin affects cell survival and neuronal outgrowth in in vitro and in vivo models of spinal muscular atrophy. Hum. Mol. Genet. 23, 855–869. doi: 10.1093/hmg/ddt477
Sleigh, J. N., Gillingwater, T. H., and Talbot, K. (2011). The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy. Dis. Model. Mech. 4, 457–467. doi: 10.1242/dmm.007245
Sleigh, J. N., Grice, S. J., Davies, K. E., and Talbot, K. (2013). Spinal muscular atrophy at the crossroads of basic science and therapy. Neuromuscul. Disord. 23:96.
Smith, R. A., Miller, T. M., Yamanaka, K., Monia, B. P., Condon, T. P., Hung, G., et al. (2006). Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 116, 2290–2296.
Somers, E., Lees, R. D., Hoban, K., Sleigh, J. N., Zhou, H., Muntoni, F., et al. (2016). Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann. Neurol. 79, 217–230. doi: 10.1002/ana.24549
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Stoica, L., Todeasa, S. H., Cabrera, G. T., Salameh, J. S., ElMallah, M. K., Mueller, C., et al. (2016). Adeno-associated virus-delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann. Neurol. 79, 687–700. doi: 10.1002/ana.24618
Storkebaum, E., Lambrechts, D., Dewerchin, M., Moreno-Murciano, M.-P., Appelmans, S., Oh, H., et al. (2005). Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat. Neurosci. 8, 85–92.
Strong, M. J., Volkening, K., Hammond, R., Yang, W., Strong, W., Leystra-Lantz, C., et al. (2007). TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci. 35, 320–327.
Sufit, R. L., Ajroud-Driss, S., Casey, P., and Kessler, J. A. (2017). Open label study to assess the safety of VM202 in subjects with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 269–278. doi: 10.1080/21678421.2016.1259334
Sumner, C. J., Wee, C. D., Warsing, L. C., Choe, D. W., Ng, A. S., Lutz, C., et al. (2009). Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum. Mol. Genet. 18, 3145–3152. doi: 10.1093/hmg/ddp253
Sun, S., Ling, S.-C., Qiu, J., Albuquerque, C. P., Zhou, Y., Tokunaga, S., et al. (2015). ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 6:6171. doi: 10.1038/ncomms7171
Suzuki, M., McHugh, J., Tork, C., Shelley, B., Hayes, A., Bellantuono, I., et al. (2008). Direct muscle delivery of GDNF with human mesenchymal stem cells improves motor neuron survival and function in a rat model of familial ALS. Mol. Ther. 16, 2002–2010. doi: 10.1038/mt.2008.197
Talbot, K. (2011). Familial versus sporadic amyotrophic lateral sclerosis–a false dichotomy? Brain 134, 3429–3431.
Taylor, J. P., Brown, R. H., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Thomsen, G. M., Alkaslasi, M., Vit, J. P., Lawless, G., Godoy, M., Gowing, G., et al. (2017). Systemic injection of AAV9-GDNF provides modest functional improvements in the SOD1G93A ALS rat but has adverse side effects. Gene Ther. 24, 245–252. doi: 10.1038/gt.2017.9
Thomsen, G. M., Gowing, G., Latter, J., Chen, M., Vit, J.-P., Staggenborg, K., et al. (2014). Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J. Neurosci. 34, 15587–15600. doi: 10.1523/JNEUROSCI.2037-14.2014
Tisdale, S., and Pellizzoni, L. (2015). Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 35, 8691–8700. doi: 10.1523/JNEUROSCI.0417-15.2015
Tizzano, E. F., and Finkel, R. S. (2017). Spinal muscular atrophy: a changing phenotype beyond the clinical trials. Neuromuscul. Disord. 27, 883–889. doi: 10.1016/j.nmd.2017.05.011
Tosolini, A. P., Mohan, R., and Morris, R. (2013). Targeting the full length of the motor end plate regions in the mouse forelimb increases the uptake of fluoro-gold into corresponding spinal cord motor neurons. Front. Neurol. 4:58. doi: 10.3389/fneur.2013.00058
Tosolini, A. P., and Morris, R. (2016). Targeting motor end plates for delivery of adenoviruses: an approach to maximize uptake and transduction of spinal cord motor neurons. Sci. Rep. 6:33058. doi: 10.1038/srep33058
Towne, C., Raoul, C., Schneider, B. L., and Aebischer, P. (2008). Systemic AAV6 delivery mediating RNA interference against SOD1: neuromuscular transduction does not alter disease progression in fALS mice. Mol. Ther. 16, 1018–1025. doi: 10.1038/mt.2008.73
Towne, C., Setola, V., Schneider, B. L., and Aebischer, P. (2011). Neuroprotection by gene therapy targeting mutant SOD1 in individual pools of motor neurons does not translate into therapeutic benefit in fALS mice. Mol. Ther. 19, 274–283. doi: 10.1038/mt.2010.260
Tsai, L.-K., Chen, Y.-C., Cheng, W.-C., Ting, C.-H., Dodge, J. C., Hwu, W.-L., et al. (2012). IGF-1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol. Dis. 45, 272–279. doi: 10.1016/j.nbd.2011.06.021
Tsuiji, H., Iguchi, Y., Furuya, A., Kataoka, A., Hatsuta, H., Atsuta, N., et al. (2013). Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol. Med. 5, 221–234. doi: 10.1002/emmm.201202303
Tu, W.-Y., Simpson, J. E., Highley, J. R., and Heath, P. R. (2017). Spinal muscular atrophy: factors that modulate motor neurone vulnerability. Neurobiol. Dis. 102, 11–20. doi: 10.1016/j.nbd.2017.01.011
Turner, B. J., Alfazema, N., Sheean, R. K., Sleigh, J. N., Davies, K. E., Horne, M. K., et al. (2014). Overexpression of survival motor neuron improves neuromuscular function and motor neuron survival in mutant SOD1 mice. Neurobiol. Aging 35, 906–915. doi: 10.1016/j.neurobiolaging.2013.09.030
Turner, B. J., Cheah, I. K., Macfarlane, K. J., Lopes, E. C., Petratos, S., Langford, S. J., et al. (2003). Antisense peptide nucleic acid-mediated knockdown of the p75 neurotrophin receptor delays motor neuron disease in mutant SOD1 transgenic mice. J. Neurochem. 87, 752–763. doi: 10.1046/j.1471-4159.2003.02053.x
Turner, B. J., Parkinson, N. J., Davies, K. E., and Talbot, K. (2009). Survival motor neuron deficiency enhances progression in an amyotrophic lateral sclerosis mouse model. Neurobiol. Dis. 34, 511–517. doi: 10.1016/j.nbd.2009.03.005
Turner, M. R., Hardiman, O., Benatar, M., Brooks, B. R., Chio, A., de Carvalho, M., et al. (2013). Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322. doi: 10.1016/S1474-4422(13)70036-X
Uranishi, H., Tetsuka, T., Yamashita, M., Asamitsu, K., Shimizu, M., Itoh, M., et al. (2001). Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in nuclear factor-kappa B p65-mediated transcription as a coactivator. J. Biol. Chem. 276, 13395–13401. doi: 10.1074/jbc.M011176200
Valori, C. F., Ning, K., Wyles, M., Mead, R. J., Grierson, A. J., Shaw, P. J., et al. (2010). Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2:35ra42. doi: 10.1126/scitranslmed.3000830
van Damme, P., Robberecht, W., and van den Bosch, L. (2017). Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis. Model. Mech. 10, 537–549. doi: 10.1242/dmm.029058
van den Bosch, L., Tilkin, P., Lemmens, G., and Robberecht, W. (2002). Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport 13, 1067–1070. doi: 10.1097/00001756-200206120-00018
van Rheenen, W., Shatunov, A., Dekker, A. M., McLaughlin, R. L., Diekstra, F. P., Pulit, S. L., et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048. doi: 10.1038/ng.3622
van Zundert, B., Izaurieta, P., Fritz, E., and Alvarez, F. J. (2012). Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Cell. Biochem. 113, 3301–3312. doi: 10.1002/jcb.24234
Vance, C., Al-Chalabi, A., Ruddy, D., Smith, B. N., Hu, X., Sreedharan, J., et al. (2006). Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain 129, 868–876. doi: 10.1093/brain/awl030
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Veldink, J. H., Kalmijn, S., van der Hout, A. H., Lemmink, H. H., Groeneveld, G. J., Lummen, C., et al. (2005). SMN genotypes producing less SMN protein increase susceptibility to and severity of sporadic ALS. Neurology 65, 820–825. doi: 10.1212/01.wnl.0000174472.03292.dd
Verhaart, I. E. C., Robertson, A., Leary, R., McMacken, G., König, K., Kirschner, J., et al. (2017a). A multi-source approach to determine SMA incidence and research ready population. J. Neurol. 264, 1465–1473. doi: 10.1007/s00415-017-8549-1
Verhaart, I. E. C., Robertson, A., Wilson, I. J., Aartsma-Rus, A., Cameron, S., Jones, C. C., et al. (2017b). Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet. J. Rare Dis. 12:124. doi: 10.1186/s13023-017-0671-8
von Jonquieres, G., Mersmann, N., Klugmann, C. B., Harasta, A. E., Lutz, B., Teahan, O., et al. (2013). Glial promoter selectivity following AAV-delivery to the immature brain. PLOS ONE 8:e65646. doi: 10.1371/journal.pone.0065646
Wang, H., Ghosh, A., Baigude, H., Yang, C.-S., Qiu, L., Xia, X., et al. (2008). Therapeutic gene silencing delivered by a chemically modified small interfering RNA against mutant SOD1 slows amyotrophic lateral sclerosis progression. J. Biol. Chem. 283, 15845–15852. doi: 10.1074/jbc.M800834200
Wang, H., Yang, B., Qiu, L., Yang, C., Kramer, J., Su, Q., et al. (2014). Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum. Mol. Genet. 23, 668–681. doi: 10.1093/hmg/ddt454
Wang, I.-F., Reddy, N. M., and Shen, C.-K. J. (2002). Higher order arrangement of the eukaryotic nuclear bodies. Proc. Natl. Acad. Sci. U.S.A. 99, 13583–13588. doi: 10.1073/pnas.212483099
Wang, L.-J., Lu, Y.-Y., Muramatsu, S., Ikeguchi, K., Fujimoto, K., Okada, T., et al. (2002). Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J. Neurosci. 22, 6920–6928.
Wang, L., Sharma, K., Grisotti, G., and Roos, R. P. (2009). The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol. Dis. 35, 234–240. doi: 10.1016/j.nbd.2009.05.002
Wang, W., Duan, W., Wang, Y., Wen, D., Liu, Y., Li, Z., et al. (2017). Intrathecal delivery of ssAAV9-DAO extends survival in SOD1G93A ALS mice. Neurochem. Res. 42, 986–996. doi: 10.1007/s11064-016-2131-6
Wang, X.-B., Cui, N.-H., Gao, J.-J., Qiu, X.-P., and Zheng, F. (2014). SMN1 duplications contribute to sporadic amyotrophic lateral sclerosis susceptibility: evidence from a meta-analysis. J. Neurol. Sci. 340, 63–68. doi: 10.1016/j.jns.2014.02.026
Wang, Y., Duan, W., Wang, W., Di Wen, Liu, Y., Liu, Y., et al. (2016). scAAV9-VEGF prolongs the survival of transgenic ALS mice by promoting activation of M2 microglia and the PI3K/Akt pathway. Brain Res. 1648(Pt A), 1–10. doi: 10.1016/j.brainres.2016.06.043
Winer, L., Srinivasan, D., Chun, S., Lacomis, D., Jaffa, M., Fagan, A., et al. (2013). SOD1 in cerebral spinal fluid as a pharmacodynamic marker for antisense oligonucleotide therapy. JAMA Neurol. 70, 201–207. doi: 10.1001/jamaneurol.2013.593
Woo, C. J., Maier, V. K., Davey, R., Brennan, J., Li, G., Brothers, J., et al. (2017). Gene activation of SMN by selective disruption of lncRNA-mediated recruitment of PRC2 for the treatment of spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 114, E1509–E1518. doi: 10.1073/pnas.1616521114
Writing Group and Edaravone (MCI-186) ALS 19 Study Group (2017). Safety, and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 16, 505–512. doi: 10.1016/S1474-4422(17)30115-1
Wu, R., Wang, H., Xia, X., Zhou, H., Liu, C., Castro, M., et al. (2009). Nerve injection of viral vectors efficiently transfers transgenes into motor neurons and delivers RNAi therapy against ALS. Antioxid. Redox. Signal. 11, 1523–1534. doi: 10.1089/ARS.2009.2618
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253. doi: 10.1038/nn2047
Yamazaki, T., Chen, S., Yu, Y., Yan, B., Haertlein, T. C., Carrasco, M. A., et al. (2012). FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2, 799–806. doi: 10.1016/j.celrep.2012.08.025
Yoshino, H., and Kimura, A. (2006). Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph. Lateral Scler. 7, 241–245. doi: 10.1080/17482960600881870
Yu, Y., Chi, B., Xia, W., Gangopadhyay, J., Yamazaki, T., Winkelbauer-Hurt, M. E., et al. (2015). U1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafish. Nucleic Acids Res. 43, 3208–3218. doi: 10.1093/nar/gkv157
Zhang, H. L., Pan, F., Hong, D., Shenoy, S. M., Singer, R. H., and Bassell, G. J. (2003). Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J. Neurosci. 23, 6627–6637.
Zhang, Z., Pinto, A. M., Wan, L., Wang, W., Berg, M. G., Oliva, I., et al. (2013). Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A. 110, 19348–19353. doi: 10.1073/pnas.1319280110
Zhou, H., Meng, J., Marrosu, E., Janghra, N., Morgan, J., and Muntoni, F. (2015). Repeated low doses of morpholino antisense oligomer: an intermediate mouse model of spinal muscular atrophy to explore the window of therapeutic response. Hum. Mol. Genet. 24, 6265–6277. doi: 10.1093/hmg/ddv329
Zhou, Z., Licklider, L. J., Gygi, S. P., and Reed, R. (2002). Comprehensive proteomic analysis of the human spliceosome. Nature 419, 182–185. doi: 10.1038/nature01031
Zincarelli, C., Soltys, S., Rengo, G., and Rabinowitz, J. E. (2008). Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 16, 1073–1080. doi: 10.1038/mt.2008.76
Keywords: adeno-associated virus (AAV), ALS, antisense oligonucleotide (ASO), motor neuron disease (MND), neurodegeneration, neurotrophic factor, SMA, survival motor neuron (SMN)
Citation: Tosolini AP and Sleigh JN (2017) Motor Neuron Gene Therapy: Lessons from Spinal Muscular Atrophy for Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 10:405. doi: 10.3389/fnmol.2017.00405
Received: 19 September 2017; Accepted: 21 November 2017;
Published: 07 December 2017.
Edited by:
Michael J. Schmeisser, Universitätsklinikum Magdeburg, GermanyReviewed by:
Patrick Weydt, University of Bonn, GermanyJochen H. Weishaupt, Universitäts- und Rehabilitationskliniken Ulm, Germany
Copyright © 2017 Tosolini and Sleigh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James N. Sleigh, j.sleigh@ucl.ac.uk