DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes
 Francisco J. del Castillo
Francisco J. del Castillo Ignacio del Castillo
Ignacio del Castillo- 1Servicio de Genética, Hospital Universitario Ramón y Cajal, IRYCIS, Madrid, Spain
- 2Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid, Spain
The inner ear is a very complex sensory organ whose development and function depend on finely balanced interactions among diverse cell types. The many different kinds of inner ear supporting cells play the essential roles of providing physical and physiological support to sensory hair cells and of maintaining cochlear homeostasis. Appropriately enough, the gene most commonly mutated among subjects with hereditary hearing impairment (HI), GJB2, encodes the connexin-26 (Cx26) gap-junction channel protein that underlies both intercellular communication among supporting cells and homeostasis of the cochlear fluids, endolymph and perilymph. GJB2 lies at the DFNB1 locus on 13q12. The specific kind of HI associated with this locus is caused by recessively-inherited mutations that inactivate the two alleles of the GJB2 gene, either in homozygous or compound heterozygous states. We describe the many diverse classes of genetic alterations that result in DFNB1 HI, such as large deletions that either destroy the GJB2 gene or remove a regulatory element essential for GJB2 expression, point mutations that interfere with promoter function or splicing, and small insertions or deletions and nucleotide substitutions that target the GJB2 coding sequence. We focus on how these alterations disrupt GJB2 and Cx26 functions and on their different effects on cochlear development and physiology. We finally discuss the diversity of clinical features of DFNB1 HI as regards severity, age of onset, inner ear malformations and vestibular dysfunction, highlighting the areas where future research should be concentrated.
Introduction
Hearing is an extraordinarily complex process that involves many cell types and diverse cellular and molecular structures and mechanisms. The genetic causes of hereditary hearing impairment (HI) are, in consequence, very heterogeneous. Over one hundred genes underlying nonsyndromic hearing impairment (NSHI) have been identified so far1 and it is estimated that many more genes remain to be identified. Unsurprisingly, most of these genes encode proteins that participate in different aspects of the physiology of the cochlear sensory hair cells, including auditory mechanotransduction and synaptic transmission mechanisms (Richardson et al., 2011; Safieddine et al., 2012). Yet, the gene most frequently mutated among subjects with NSHI in many populations is not at all expressed in hair cells. This gene, GJB2, lies at the DFNB1 locus on chromosome 13q12, and codes for connexin-26 (Cx26), a gap junction protein that is essential for the function of cochlear supporting cells.
DFNB1 owes its name to the first mapped locus for nonsyndromic deafness (DFN) with autosomal recessive inheritance (B; Guilford et al., 1994), and the acronym has eventually come to represent the specific type of HI caused by mutations at this locus (MIM# 220290). The identification of the first GJB2 mutations causing DFNB1 HI (Kelsell et al., 1997) was soon followed by screenings that revealed a high frequency of GJB2 mutations among subjects with NSHI (Denoyelle et al., 1997; Zelante et al., 1997). Since then, high prevalence of DFNB1 HI was demonstrated in most populations (Chan and Chang, 2014), and so searching for mutations affecting GJB2 quickly became the mainstay for the genetic diagnosis of NSHI. The interest on DFNB1 HI spurred many diverse studies to explore the underlying pathogenic mechanisms, ranging from functional assays of wild-type and mutant proteins to generation and analysis of DFNB1 mouse models. Here we review our current knowledge on the molecular pathology and clinical features of DFNB1 HI.
Genetic Alterations That Result in DFNB1 Hearing Impairment
GJB2 is 5513 bp long and contains two exons (193 bp and 2141 bp long, respectively) separated by a 3179-bp intron (Kiang et al., 1997). Transcription is initiated from a single start site and leads to the synthesis of a 2334-nucleotide mRNA (GenBank NM_004004.5), which is considered canonical. The 678-bp sequence that codes for Cx26 is completely contained within GJB2 exon 2. A genome-wide search for alternative transcriptional start sites found an expressed sequence tag (EST) that suggested the existence of an alternative 184-bp first exon, located within the only GJB2 intron (Kimura et al., 2006), which would lead to the production of a 2318-nucleotide mRNA (GenBank XM_011535049.2). This EST (GenBank DA975033.1) was found in a cDNA library of synovial membrane tissue from rheumatoid arthritis patients. Further research is needed to determine whether this mRNA is expressed in the inner ear, a relevant issue given that this alternative first exon could be a novel target for genetic screening in patients with NSHI (Parzefall et al., 2017).
Cx26 belongs to the connexin family of integral membrane proteins, which act as subunits of a hexameric annular assembly, termed connexon, which forms a pore through the lipid bilayer. Connexons can be composed by connexins of either the same or different type (homo- or heteromeric connexons, respectively; Kumar and Gilula, 1996). Because of this combinatorial capability, pathogenic mutations in connexin genes can produce a variety of clinical outcomes, and so it occurs to GJB2. Some missense mutations, inherited in a dominant form, result in Cx26 mutant subunits that exert dominant negative effects on the activity of wild-type Cx26 alone or on Cx26 and connexin-30 (Cx30) together. They cause the DFNA3 type of autosomal dominant nonsyndromic HI (NSHI; Denoyelle et al., 1998; Forge et al., 2003b; Marziano et al., 2003; Zhang et al., 2011). Other dominant missense mutations result in Cx26 mutant subunits that impact the activity of both wild-type Cx26 and connexin-43 (Rouan et al., 2001). They cause different syndromes associating HI with skin disorders, such as Vohwinkel (Maestrini et al., 1999) or Bart-Pumphrey (Richard et al., 2004) syndromes, keratitis-ichthyosis-deafness (KID; Richard et al., 2002; van Steensel et al., 2002), hystrix-like ichthyosis and deafness (HID; van Geel et al., 2002), palmoplantar keratoderma with deafness (PPK-D; Richard et al., 1998) or sensorineural deafness with erythematous plaques, cutaneous orthokeratotic hyperkeratosis and parakeratosis in the oral and esophageal mucosa (Brown et al., 2003).
However, a majority of GJB2 mutations are recessively inherited, and result in DFNB1 NSHI when they are in homozygous or compound heterozygous states (Kelsell et al., 1997). About 200 GJB2 mutations have been reported to cause DFNB1 HI2 (Stenson et al., 2017). They can be classified into many different types: large deletions that remove the whole GJB2 gene (Feldmann et al., 2009; Bliznetz et al., 2014, 2017), large deletions that remove regulatory sequences that are needed for the expression of GJB2 but keep the gene intact (del Castillo et al., 2002, 2005; Wilch et al., 2010; Tayoun et al., 2016; Table 1), and a plethora of small-scale alterations, including nonsense, missense and splice-site point mutations, as well as frameshifting small insertions and deletions (del Castillo and del Castillo, 2011). The frequencies of these mutations are diverse, with different mutant alleles overrepresented in different populations. The genetic epidemiology of DFNB1 HI has been extensively reviewed (Chan and Chang, 2014), and so we will focus on the molecular mechanisms by which these mutations alter the functions of the GJB2 gene and the Cx26 protein.
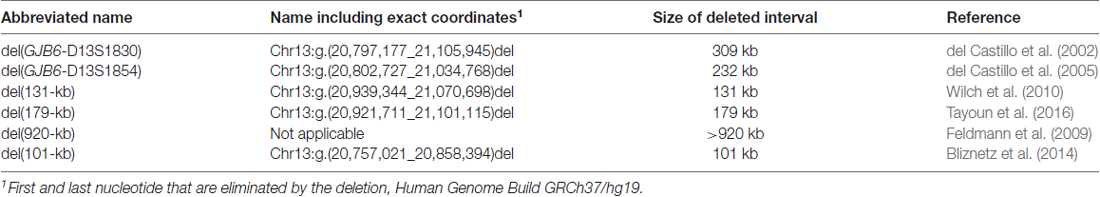
Table 1. Large deletions at the DFNB1 locus on 13q12.
Mutations That Alter GJB2 Expression
GJB2 expression takes place in specific cell types in many different tissues. With the significant exception of hair cells, GJB2 is expressed by nearly all cell types within the human cochlea, including supporting cells in the sensory epithelium, fibrocytes and mesenchymal cells in the lateral wall, basal and intermediate cells of the stria vascularis and type I neurons in the spiral ganglion (Figure 1; Liu et al., 2009).
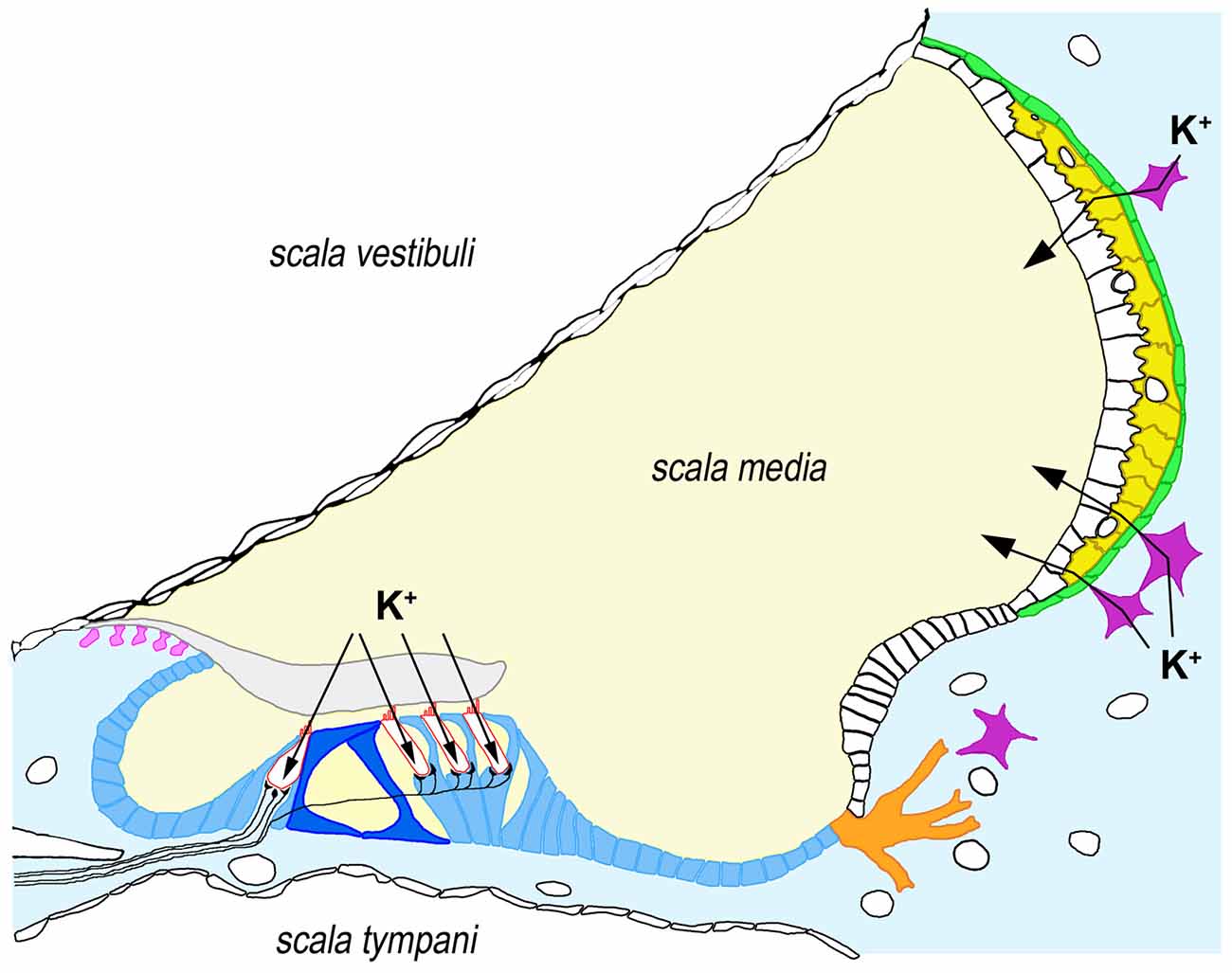
Figure 1. Expression of connexin-26 (Cx26) and connexin-30 (Cx30) in the human cochlea. Cx26 and Cx30 are synthesized by all supporting cells types within the organ of Corti (blue), including inner and outer pillar cells (dark blue), as well as by root cells (orange), interdental cells (pink), fibrocytes from the underlying connective tissue (light blue) and basal (green) and intermediate cells (yellow) from the stria vascularis. For the sake of clarity, we have depicted only four fibrocytes (purple) in connective tissue. Arrows indicate the pathways for K+ influx in hair cells (bordered in red) and K+ secretion into endolymph through the stria vascularis. It must be noted that Cx26 and Cx30 seem to form distinct, homomeric plaques even when co-expressed in the same cell, although these plaques are closely associated. Some immuhistochemical staining experiments suggest that co-expression may not occur in all the cell types indicated above (e.g., Cx26 may not be synthesized in inner pillar cells and strial intermediate cells, whereas Cx30 may not be synthesized in strial basal cells), which might reflect specific functional associations with additional ion transporters (Liu et al., 2009, 2016).
Interestingly, a study performed on the closely related murine Gjb2 promoter (81% sequence identity) revealed that Gjb2 and the neighboring Gjb6 gene (encoding mouse Cx30) are transcriptionally co-regulated in cochlear supporting cells upon activation of NF-κB (Ortolano et al., 2008), which is likely caused by intracellular Ca2+ oscillations induced by connexin signaling (see “Mutations Affecting the Function of Cx26 Hemichannels” section below; Rodriguez et al., 2012). This mechanism may be not directly extrapolatable to the expression of the human ortologs, since the murine promoter contains an NF-κB binding site that is absent in the human promoter. Further studies are needed to investigate a putative co-regulation of GJB2 and GJB6 in human cochlea.
Mutations Affecting the GJB2 Promoter
The 128-bp-long basal promoter of GJB2 lies just upstream of the canonical first exon (Kiang et al., 1997; Tu and Kiang, 1998). The promoter includes a TATA box and two GC boxes (Figure 2), which are bound by the Sp1 and Sp3 transcription factors, as demonstrated by in vitro experiments and by the fact that the promoter activity is drastically reduced by engineered mutations in either of the GC boxes (Tu and Kiang, 1998).
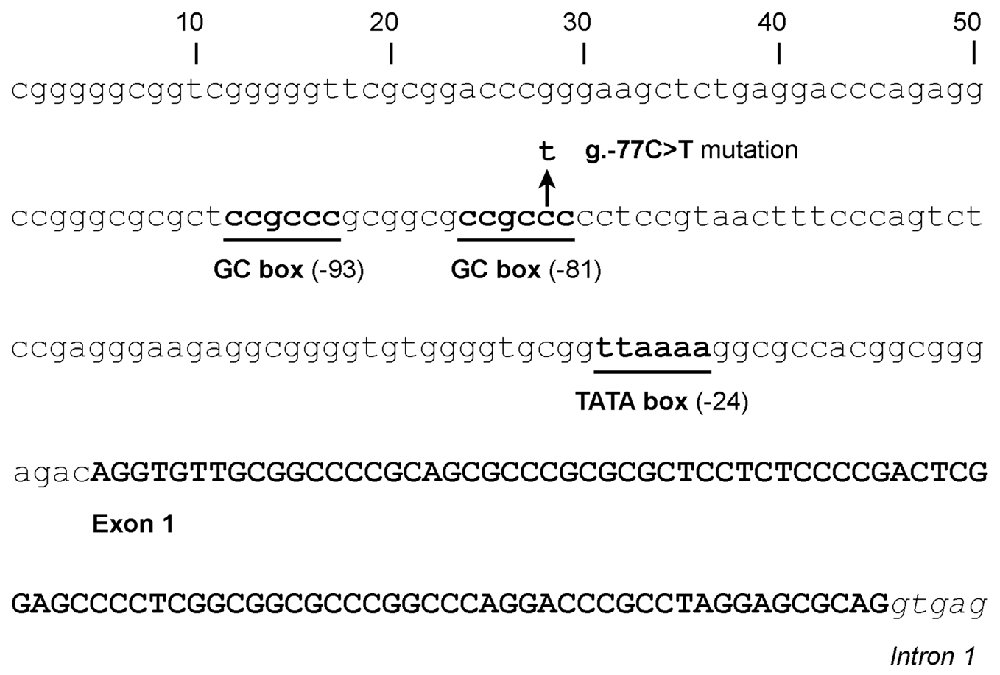
Figure 2. Sequence of the GJB2 basal promoter and first exon, showing the location of the GC boxes (at -81 and -93), the TATA motif (at -24) and the transcription start site of the first canonical exon. The arrow indicates the position of the g.-77C>T promoter mutation.
So far only one human mutation has been reported to affect the GJB2 promoter. It was found in the compound heterozygous state with mutation p.(Val84Met) in a Portuguese hearing-impaired subject (Matos et al., 2007). This promoter mutation, g.-77C>T (also known as -3438C>T and c.-259C>T), affects the GC box at -81 (CCGCCC > CCGCTC). Taking into account the effects of the in vitro engineered mutations in the GC boxes, it was expected that this naturally occurring mutation would impair the binding of Sp1 and Sp3. In fact, the mutant promoter exhibited a drastically reduced activity in reporter-gene experiments performed in different cell lines (Matos et al., 2007).
Deletions Removing a Distal Enhancer
When mutation screening of the GJB2 coding region in subjects with autosomal recessive NSHI became a general practice, it was soon evident that there was an overrepresentation of affected subjects in whom only one heterozygous pathogenic mutation could be found. It was hypothesized that the missing mutations might affect non-coding regulatory sequences or other genes. Research on those unelucidated cases led to the identification of pathogenic large deletions within the DFNB1 locus, which however keep intact the structure of the GJB2 gene. They can be classified in two different types: those that truncate the neighboring GJB6 gene and those that also keep intact its structure.
Two deletions were reported to truncate the GJB6 gene, while keeping GJB2 intact. The first identified was del(GJB6-D13S1830), which removes a 309-kb interval that includes the first five exons of GJB6 and the whole CRYL1 gene (Table 1, Figure 3; Lerer et al., 2001; del Castillo et al., 2002, 2003; Pallares-Ruiz et al., 2002). Few years later, another deletion of the same type was characterized. This deletion, named del(GJB6-D13S1854), removes a 232-kb interval, with the proximal breakpoint within GJB6 intron 5, and the distal breakpoint in intron 4 of CRYL1 (Table 1, Figure 3; del Castillo et al., 2005).
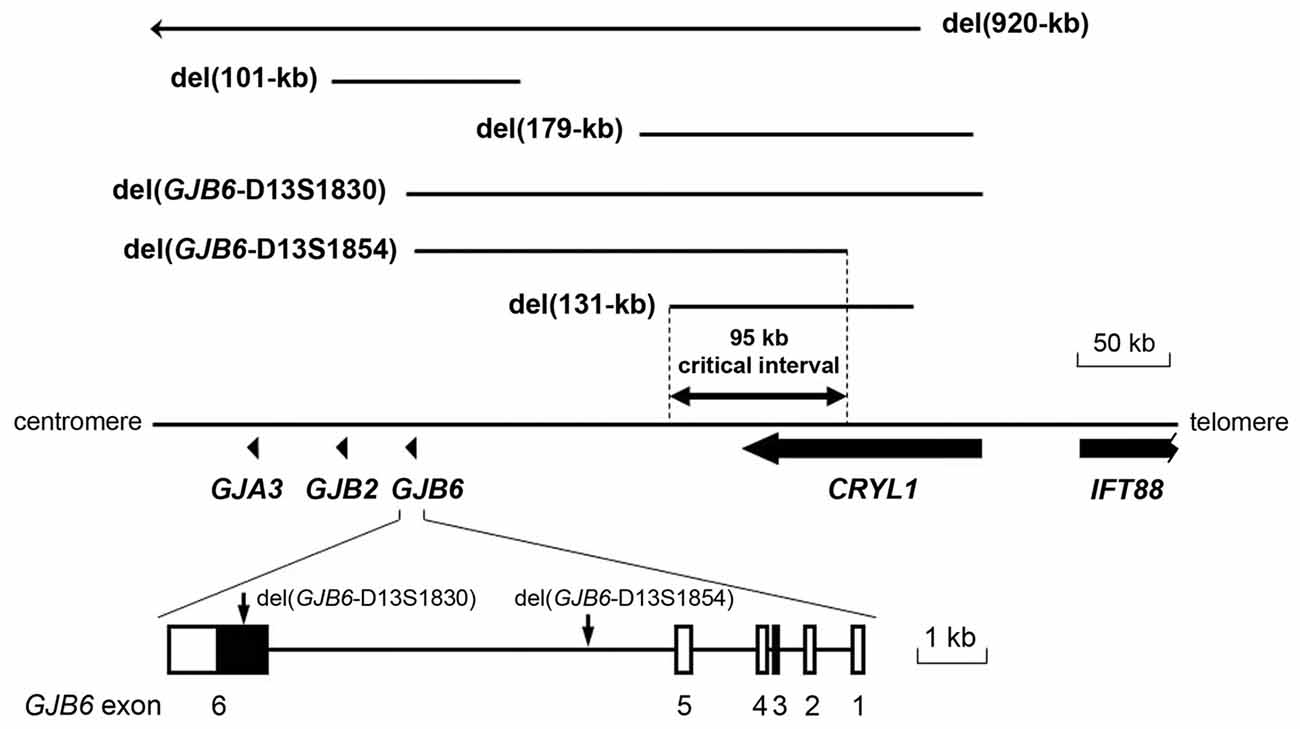
Figure 3. Large deletions on chromosome 13q12 that are responsible for DFNB1 hearing impairment (HI). The proximal breakpoint of del131-kb and the distal breakpoint of del(GJB6-D13S1854) delimit the 95.4-kb sequence stretch where the cis-acting element regulating the expression of GJB2 must be situated. The two proximal breakpoints of deletions del(GJB6-D13S1830) and del(GJB6-D13S1854; vertical arrows) lie within GJB6.
These findings suggested that mutations in GJB2 and GJB6 could follow a digenic pattern of inheritance. Results from some studies in humans and rodents fitted well into this hypothesis. First, Cx26 and Cx30 share the same spatial pattern of expression within the cochlea, with apparent co-localization, and can form heteromeric connexons and heterotypic gap-junction channels (Dahl et al., 1996; Lautermann et al., 1998; Forge et al., 2003a,b), although these conclusions have been partly challenged by a recent study using super-resolution structured illumination fluorescence microscopy (Liu et al., 2016). Second, some mutant Cx26 subunits involved in autosomal dominant NSHI are able to exert dominant negative effects on the activity of wild-type Cx30 (Forge et al., 2003b; Marziano et al., 2003). Third, a mutation in GJB6 was found in a case of autosomal dominant NSHI (Grifa et al., 1999). Finally, Gjb6 knock-out mice were generated by replacing the complete Gjb6 coding sequence with a cassette containing the lacZ reporter and the neo selection gene (Teubner et al., 2003). These mice, termed Gjb6tm1Kwi/tm1Kwi, have severe deafness and show a complete lack of endocochlear potential (Teubner et al., 2003) due to disruption of the endothelial barrier of capillaries embedded within the stria vascularis (Cohen-Salmon et al., 2007). Furthermore, intercellular transfer of metabolites such as glucose among supporting cells in the organ of Corti of Gjb6tm1Kwi/tm1Kwi mice is reduced, though electrical coupling is preserved (Chang et al., 2008). Finally, Gjb2+/− Gjb6+/tm1Kwi double heterozygous mice have moderate HI and a significantly reduced endocochlear potential (Michel et al., 2003; Mei et al., 2017), although this is in contrast with the severe or profound HI observed in a majority of human double heterozygotes carrying del(GJB6-D13S1830) and a pathogenic GJB2 mutation (Snoeckx et al., 2005). A strong support for the hypothesis of the digenic inheritance would have come from the finding of truncating point mutations in GJB6 in cases of human autosomal recessive NSHI, but this type of mutations have not been reported to date.
An alternative explanation hypothesized that GJB2 expression in the cochlea would require a cis-acting regulatory element, which would lie within the interval that is removed by the deletions, and whose absence would abolish the expression of the structurally normal GJB2 allele that is located downstream. In fact, Cx26 expression in the ductal epithelium of sweat glands was shown to be dramatically reduced in a double heterozygote for GJB2 c.35delG and del(GJB6-D13S1830; Common et al., 2005). In other studies, qualitative allele-specific RT-PCR was used to investigate GJB2 expression in buccal epithelium cells. In three double heterozygotes for del(GJB6-D13S1830) and a GJB2 mutation), no expression could be detected from the wild-type GJB2 allele that was in cis with del(GJB6-D13S1830; Rodriguez-Paris and Schrijver, 2009). Similar results were obtained when studying three double heterozygous subjects for del(GJB6-D13S1854) and a GJB2 mutation (Rodriguez-Paris et al., 2011).
The hypothesis of the cis-acting regulatory element received further support from the identification and characterization of the second type of DFNB1 deletions, i.e., those keeping the structure of both GJB2 and GJB6 intact (Wilch et al., 2006, 2010; Tayoun et al., 2016). The first reported deletion of this type was del(ch13:20,939,344-21,070,698; Wilch et al., 2006, 2010). The deletion interval was 131-kb long. Its proximal breakpoint was located between GJB6 (more than 100 kb upstream of its 5′ end) and CRYL1 (Wilch et al., 2010; Table 1, Figure 3). It was found in a family whose affected subjects were double heterozygous, the other allele carrying the c.35delG GJB2 mutation. Interestingly, qualitative allele-specific RT-PCR, which was performed on buccal epithelium cells from these subjects, revealed a dramatic reduction of the expression from the wild-type GJB2 and GJB6 alleles in cis with the deletion. In addition to supporting the existence of the cis-acting regulatory element, these results suggest that it could be implicated in the co-regulation of the expression of GJB2 and GJB6. A second deletion of the same type has been reported recently (Tayoun et al., 2016). It removes a 179-kb interval, with the proximal breakpoint between GJB6 and CRYL1. It was found in a subject with moderate NSHI carrying the p.(Val37Ile) in the other allele.
A re-examination of the mice models also supports the hypothesis of the cis-acting regulatory element. The Gjb6tm1Kwi mouse is not only a Gjb6 knockout, as the engineered mutation also decreases the transcription of the contiguous Gjb2 gene (Ortolano et al., 2008; Lynn et al., 2011), a polar effect that is likely due to the insertion of the long lacZ-neo cassette. Boulay et al. (2013) recently generated an independent Gjb6 knock-out strain (termed Gjb6Δ/Δ) that carries a complete deletion of the Gjb6 coding sequence, but no inserted material, aside from a single loxP site, which minimizes the polar effect on Gjb2. Indeed, while in the two Gjb6 knock-out strains cochlear Gjb2 mRNA and Cx26 protein levels are reduced compared to wild-type controls, Gjb6Δ/Δ mice have Gjb2 mRNA levels similar to those of Gjb6+/tm1Kwi heterozygotes (which are not deaf) and five times the amount of Cx26 shown by their Gjb6tm1Kwi/tm1Kwi counterparts (Boulay et al., 2013). Critically, Gjb6Δ/Δ mice have no HI whatever (Boulay et al., 2013), implying that the impaired hearing observed in Gjb2+/− Gjb6+/tm1Kwi double heterozygotes is solely caused by the reduced levels of Cx26.
Altogether, currently available evidences from human subjects and mice models strongly support the existence of the regulatory element, but it still remains to be identified. It should lie within the critical 95.4 kb interval that has been established by overlapping all deletion intervals that are known to date (Figure 3).
Mutations That Affect RNA Splicing
Given the simple structure of GJB2 (only one intron), few splice-site mutations causing HI have been reported to date in this gene (Denoyelle et al., 1999; Green et al., 1999; Mani et al., 2009; Gandía et al., 2013). Since GJB2 exon 2 contains the entire coding region, these splice-site mutations have no effect on protein coding.
Mutation c.-23+1G>A affects the donor splice site of intron 1. In a lymphoblastoid cell line derived from a heterozygous individual, cDNA sequencing did not reveal any transcript from the allele with this mutation. It was concluded that either the c.-23+1G>A allele was not transcribed or that the transcript was quickly degraded (Shahin et al., 2002).
Mutation c.-22-2A>C abolishes the acceptor splice site of intron 1. It was first reported in three Spanish siblings who were compound heterozygous for c.-22-2A>C and c.35delG, and presented with mild postlingual HI (Gandía et al., 2013). Later on, this same genotype was reported in an Italian subject with moderate HI (Stanghellini et al., 2014). The effects of the mutation were investigated by RT-PCR on RNA extracted from saliva from one of the siblings of the Spanish family. The acceptor splice site was actually abolished, and so normally processed transcripts from this allele were absent. However, the RT-PCR assays detected residual amounts of an abnormally processed transcript. It was generated by using an alternative acceptor splice site, which resulted in the insertion of a 38-bp intronic sequence into the 5′-UTR, but keeping an intact coding region. Therefore, Cx26 could still be translated from this transcript, although its residual amounts are likely to be insufficient to support a normal function, and this could explain the mild phenotype observed in subjects with this mutation (Gandía et al., 2013).
Given the late onset of the mild HI associated with c.-22-2A>C, it was hypothesized that this mutation could contribute to age-related hearing loss. The presence of the mutation was investigated in a large cohort of Italian subjects with age-related hearing loss and ethnically-matched control subjects with normal hearing (Rubinato et al., 2016). The mutation was found in homozygous, heterozygous, or compound heterozygous state with other known pathogenic missense mutations. There was no significant difference in allelic or genotypic frequencies between patient and control groups (Rubinato et al., 2016). Taking everything into consideration, it seems that c.-22-2A>C would be a hypomorphic recessive allele, like p.(Met34Thr) and p.(Val37Ile), with mild phenotypic effects or no effect at all, depending on the pathogenic potential of the accompanying allele.
Finally, exonic mutation c.-23G>T, lying adjacent to the splice donor site of intron 1, was found in the compound heterozygous state with p.(Trp24*) in an Indian subject with severe to profound NSHI (Mani et al., 2009). Although in silico analysis predicted that it could abolish the use of that donor site, this hypothesis still needs experimental confirmation.
Mutations in the GJB2 Coding Region
A Cx26 monomer is a 226-amino acid long polypeptide that consists of four transmembrane helices (TM1–TM4) linked by two extracellular loops (E1 and E2) and one shorter intracellular loop, with N- and C-termini on the cytosolic side of the membrane (Figure 4A). Newly-synthesized Cx26 monomers undergo conformational maturation and assembly into connexons when moving along the secretory pathway for delivery at the plasma membrane. Once there, Cx26 connexons may remain as so-called hemichannels, allowing for transport of diverse small molecules (less than 1 kDa) between the cytosol and the extracellular space. Alternatively, a connexon may contact, in a head to head disposition, another connexon on the plasma membrane of an adjacent cell to form an intercellular channel. The juxtaposition of hundreds of intercellular channels in plaques forms gap junctions (Kumar and Gilula, 1996), directly linking the cytoplasms of the two adjacent cells and creating a functional syncytium. Mutations in the GJB2 coding sequence may disrupt any of these maturation processes or they may interfere with hemichannel or intercellular channel function.
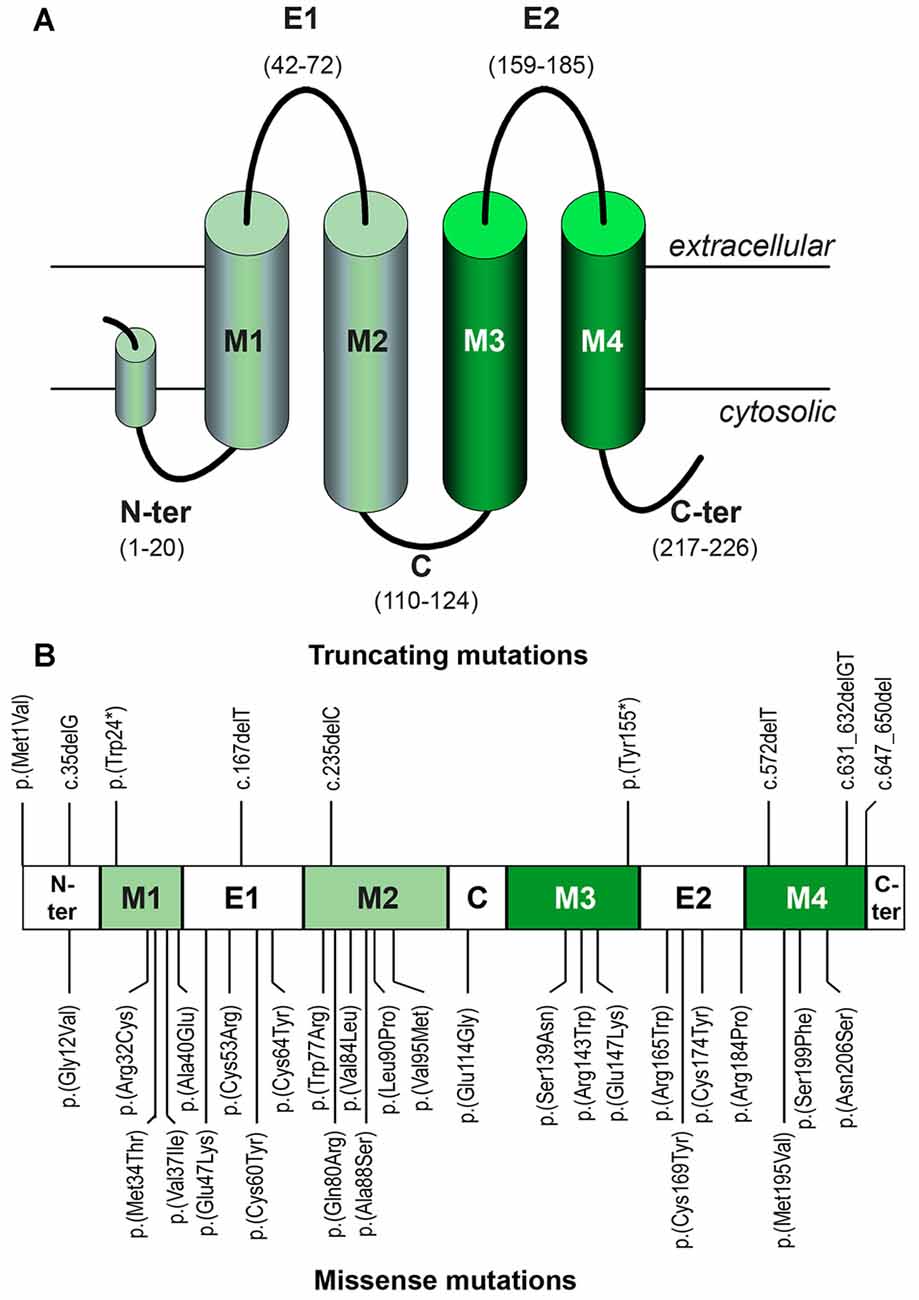
Figure 4. (A) Topological organization of the Cx26 monomer with its different structural elements. Helices are represented as cylinders. Helices in light green line the hydrophilic pore of the channel, whereas helices in dark green are exposed to the hydrophobic environment of the lipid bilayer. Note the projection into the cytosol of helices M2 and M3. (B) Location of the truncating and missense mutations mentioned in the text within the different structural elements.
Mutations Truncating Cx26
Many pathogenic GJB2 mutations generate premature stop codons, either directly (nonsense mutations; e.g., p.(Trp24*)) or because small insertions, duplications or deletions cause frameshifts (e.g., c.35delG, also known as p.(Gly12Valfs*2)). As the coding sequence of the GJB2 gene is completely contained in the last exon, mRNAs harboring premature stop codons are expected to escape the nonsense-mediated decay surveillance pathway (reviewed in Lykke-Andersen and Jensen, 2015). Thus, truncated proteins will actually be synthesized, as shown in HeLa cell assays of the c.235delC (p.(Leu79Cysfs*3)), p.(Tyr155*), c.572delT (p.(Phe191Serfs*5)) and c.631-632delGT (p.(Cys211Leufs*5)) mutations (Figure 4B; Choung et al., 2002; Xiao et al., 2011). Nearly all of the known truncating mutations (such as c.35delG (Denoyelle et al., 1997), c.167delT (Zelante et al., 1997), p.(Trp77*) (Kelsell et al., 1997) and c.235delC (Fuse et al., 1999)) result in proteins lacking one or several of the transmembrane segments and intervening loops, which hampers Cx26 folding and oligomerization, resulting in retention at the endoplasmic reticulum (ER) and ultimately causing a total loss of function. This is the case even with just a partial truncation of the TM4 helix and loss of the C-terminal stretch (mutation c.631-632delGT, p.(Cys211Leufs*5); Lim et al., 2003; Xiao et al., 2011). Interestingly, ER retention of the truncated subunits may induce in some cases the unfolded protein response (reviewed in Lindholm et al., 2017) which in turn may eventually lead to apoptosis of cells expressing those GJB2 mutant alleles, though this hypothesis is yet to be experimentally tested. Only one frameshift mutation in which truncation does not affect the transmembrane helices has been reported so far: c.647-650del (p.(Arg216Ilefs*7); Figure 4B). Although it is a pathogenic mutation (Prasad et al., 2000), its effects have not been functionally assayed yet. It is thus uncertain whether truncation of the C-terminus only would also result in ER retention or it would simply interfere with channel function, since it is believed that the C-terminal hydrophilic stretch, whose crystal structure is not yet solved, may participate in chemical gating or complete pore closure (Maeda et al., 2009).
The p.(Met1Val) mutation (Estivill et al., 1998), which results in no Cx26 protein synthesis at all (p.0 null allele; Thönnissen et al., 2002), although not properly a truncating mutation, should also be included in this category.
Missense Mutations
All the remaining DFNB1 pathogenic mutations result in amino acid substitutions. These mutations fall in two categories: those affecting residues involved in folding, oligomerization or structural stability of Cx26 and those that impair one or several aspects of Cx26 function, although any particular mutation may have several different effects. The landmark publication of the crystal structure of a Cx26 connexon (Maeda et al., 2009) provided a framework for integrating at the molecular level data obtained in diverse functional assays of these mutations.
Mutations affecting the structure of the Cx26 monomer
In the solved crystal structure, the monomer forms a compact bundle of four helices in which adjacent helices are antiparallel. Helices TM1 and TM2 from each monomer face the interior of the connexon channel, while helices TM3 and TM4 face the hydrophobic environment of the membrane. The N-terminus forms a short helix that is also embedded in the lipid bilayer (Figure 4A). Maeda et al. (2009) identified four groups of residues—two hydrophilic and two hydrophobic cores—that stabilized the structure of the monomer by means of intramolecular interactions (salt bridges or hydrogen bonds) among them. It is reckoned that most amino acid substitutions affecting such residues would result in misfolding and retention of the mutant polypeptide in the ER. Mutations at those residues include: p.(Arg32Cys; Prasad et al., 2000), p.(Arg32His; Mustapha et al., 2001), p.(Arg32Leu; Wu et al., 2002), p.(Gln80Arg; Uyguner et al., 2003), p.(Glu147Lys; Frei et al., 2004), p.(Ser199Phe; Green et al., 1999), p.(Arg143Trp; Brobby et al., 1998), p.(Asn206Ser; Marlin et al., 2001), p.(Asn206Thr; Wattanasirichaigoon et al., 2004), p.(Ser139Asn; Marlin et al., 2001), p.(Ala40Glu; Feldmann et al., 2004a), p.(Val43Met; Hwa et al., 2003), p.(Trp77Arg; Carrasquillo et al., 1997) and p.(Met195Val; Tsukada et al., 2010). Indeed, intracellular retention of the mutant polypeptide in expression experiments performed in transfected cells has been demonstrated for p.(Arg32His; Xiao et al., 2011), p.(Trp77Arg; Martin et al., 1999; Bruzzone et al., 2003), p.(Met195Val; Kim et al., 2016) and p.(Ser199Phe; Ambrosi et al., 2013). However, not all mutations at those residues cause intracellular retention, as shown for p.(Arg143Trp; Wang et al., 2003) and p.(Asn206Ser; Ambrosi et al., 2013). Apparently, those two particular amino acid substitutions can be accommodated within the monomer structure without total disruption of the intramolecular-stabilizing interactions.
Additional stabilization of the monomer is provided by three intramolecular disulfide bonds (Cys53-Cys180, Cys60-Cys174 and Cys64-Cys169) connecting the E1 and E2 loops (Maeda et al., 2009); indeed, formation of those disulfide bonds is an essential part of the Cx26 conformational maturation process. Unsurprisingly, all known mutations replacing any of these cysteine residues result in DFNB1 NSHI, as they probably result in misfolding and intracellular retention of the mutant polypeptides, though to our knowledge none have been investigated in functional assays. Examples include p.(Cys53Arg; Dahl et al., 2001), p.(Cys60Tyr; Taniguchi et al., 2015), p.(Cys64Tyr; Putcha et al., 2007), p.(Cys169Tyr; Azaiez et al., 2004) and p.(Cys174Arg; Gardner et al., 2006).
Mutations affecting connexon assembly and oligomerization
Another set of residues participates in intermolecular interactions within the connexon, influencing both correct folding and oligomerization. Most of the mutations affecting those residues are involved in dominantly inherited disorders, because the effect of the amino acid substitution on intermonomer contacts impairs connexon assembly with wild-type Cx26 subunits (Rouan et al., 2001). However, missense mutations at these residues may also result in DFNB1 NSHI, as it happens with p.(Arg184Pro; Denoyelle et al., 1997). Thus, p.(Arg184Pro)-Cx26 is completely incapable of oligomerization in HeLa cell assays (Thönnissen et al., 2002) and so unable to interact with other connexin subunits, which explains why the mutation has no dominant negative effect on wild-type Cx26.
Mutations affecting the hydrophilic channel pore
Missense mutations may also target residues of the hydrophilic channel of the connexon. This channel is lined on the cytosolic end by the protrusions of helices TM2 and TM3 of each monomer, with net positive charge, and on the extracellular end by the residues in the N-terminal half of the E1 loop (Figure 4A). The N-terminus helix and the TM1 helix create a funnel in the center of the channel that is involved in voltage-sensitive channel gating (Maeda et al., 2009), with closure due to inside positive potential (Verselis et al., 1994). DFNB1 mutations at these residues, such as p.(Gly12Val; Rabionet et al., 2000), p.(Met34Thr; Kelsell et al., 1997), p.(Val37Ile; Abe et al., 2000) and p.(Arg143Trp), reduce channel permeability (Martin et al., 1999; Thönnissen et al., 2002; Bruzzone et al., 2003; Bicego et al., 2006; Zonta et al., 2014a; García et al., 2015). The replacement of Met-34 with Thr and of Val-37 with Ile have only slight structural effects and thus p.(Met34Thr) and p.(Val37Ile) have weak pathogenic potentials. It must be remembered, though, that the specific amino acid substitution in each case may have very different functional consequences. For example, both p.(Gly12Val)-Cx26 and p.(Gly12Arg)-Cx26 have altered channel permeabilities. However, whereas p.(Gly12Val) only causes DFNB1 NSHI, p.(Gly12Arg) underlies KID syndrome due to a gain of function mechanism in which the mutant subunit becomes able to oligomerize with Cx43 (García et al., 2015). Finally, mutations that kink transmembrane helix TM2 by introducing proline residues alter the orientation of its cytosolic protrusion, which impairs channel permeability. The best known example is p.(Leu90Pro; Denoyelle et al., 1999; D’Andrea et al., 2002; Thönnissen et al., 2002; Bruzzone et al., 2003).
Mutations affecting the function of Cx26 hemichannels
Cx26 hemichannels and gap-junction intercellular channels play distinct physiological roles in cochlear cells. Cx26 hemichannels mediate paracrine and autocrine signaling that is essential for the acquisition of hearing during cochlear development and in the function of the mature cochlea. One of the crucial morphogenetic events in cochlear development is the acquisition of Ca2+ signaling in all cell types (recently reviewed by Mammano and Bortolozzi, 2017). In supporting cells, developmental Ca2+ signaling is mediated by ATP-induced spontaneous oscillations in the cytosolic concentration of free Ca2+, which in turn propagate intercellular Ca2+ waves. The underlying mechanism is as follows: ATP binding to plasma membrane P2Y receptors provokes phospholipase C-dependent synthesis of inositol 1,4,5-trisphosphate (IP3), which directs Ca2+ release from ER stores. The increase in concentration of intracellular free Ca2+ opens Cx26 hemichannels in the apical plasma membrane, which release cytosolic ATP to the endolymph, thereby propagating the Ca2+ oscillation to neighboring cells, which helps coordinate cellular activity (Beltramello et al., 2005; Piazza et al., 2007; Anselmi et al., 2008). In mice, these intercellular Ca2+ waves control Cx26 and Cx30 expression at the transcriptional level (Rodriguez et al., 2012) and they participate in the final development of cochlear structures, such as the formation of the mature inner sulcus by crenation (osmotic shrinkage of cells) and the functional maturation of inner and outer hair cells (Zhu et al., 2013; Ceriani et al., 2016; Johnson et al., 2017). In fact, supporting cells in the greater epithelial ridge (GER, the precursor of the mature inner sulcus) spontaneously release ATP through connexin hemichannels before hearing onset by the mechanism described above. This spontaneous ATP release coordinates cochlear morphogenetic events with auditory pathway maturation. Autocrine ATP signaling in GER supporting cells triggers both Cl− efflux, which induces crenation, and K+ efflux, which causes depolarization of inner hair cells and their subsequent exocytosis of glutamate, leading to the firing of action potentials by spiral ganglion neurons (Tritsch and Bergles, 2010; Wang et al., 2015). Such action potentials evoke coordinated bursts of activity from central auditory neurons in a precise, stereotyped pattern, which is believed to help consolidate synapses in developing circuits all along the auditory pathway (Tritsch et al., 2010). The spontaneous ATP release by supporting cells ceases at hearing onset. A similar role in ATP paracrine signaling in the adult cochlea was also proposed for Cx26 hemichannels (Anselmi et al., 2008), including modulation of outer hair cell electromotility (Zhao et al., 2005), but further experimentation showed that ATP release induced by Ca2+ signaling in the mature cochlea is predominantly dependent on pannexin1 and not Cx26 (Chen et al., 2015). Some DFNB1 missense mutations interfere with this hemichannel function, either by reducing the stability of hemichannels at the plasma membrane (p.(Asn206Ser); Ambrosi et al., 2013) or by replacing the Glu-47 residue that is critical for hemichannel closure at high extracellular Ca2+ concentration (p.(Glu47Lys); Marlin et al., 2001; Zonta et al., 2014b).
Cx26 hemichannels may also play a second, intriguing role in cochlear physiology. Recently, Cx26 hemichannels have been implicated in CO2 chemosensitivity in the central nervous system (Huckstepp et al., 2010a,b). Indeed, CO2 directly binds Cx26 at the beginning of the M3 helix, probably by carbamylation of Lys-125 (Figure 4), which induces opening of the Cx26 hemichannel and subsequent ATP release (Meigh et al., 2013). Auditory transduction involves high energy consumption in the cochlea and concomitant CO2 production, and thus CO2 may evoke Cx26 hemichannel-dependent ATP signaling for coordination of supporting cell activity. At least two DFNB1 mutations interfere with CO2-mediated hemichannel opening, which would impair signaling: p.(Met34Thr), which reduces the ability of the hemichannel to open in response to CO2, and p.(Ala88Ser; Frei et al., 2002), with a reduced affinity for CO2 (de Wolf et al., 2016).
Mutations affecting the function of Cx26 gap-junction channels
Auditory transduction depends on three related homeostatic processes: (1) maintenance of extracellular fluid (endolymph and perilymph) composition; (2) preservation of the functionality of hair cells; and (3) production of the endocochlear potential. All three processes rely on extensive gap-junction coupling among nearly all cells in the cochlea, with the exceptions of marginal cells in the stria vascularis and hair cells in the organ of Corti. In humans, recent reports show that Cx26 forms small gap-junction plaques that are clearly separate from, but closely associated to, the larger Cx30 plaques all along the cochlea (Liu et al., 2016, 2017), suggesting that each class of plaque would entirely consist of a single connexin subunit (i.e., each plaque would consist of identical homomeric connexons). How this spatial organization is achieved and what are its pathophysiological implications remain to be determined.
It has been proposed that Cx26 gap junctions participate in the organ of Corti in spatial buffering (also termed “sinking”) of excitotoxic substances—K+ and glutamate. Indeed, it was hypothesized that K+ was dispersed away from the hair cells and transported back to the endolymph by a gap-junction mediated pathway (Santos-Sacchi and Dallos, 1983; Kikuchi et al., 1995). Later on, the hair cell degeneration observed in one Gjb2 knockout mouse (Cohen-Salmon et al., 2002) suggested that the major mechanism underlying DFNB1 NSHI might be the loss of such pathway, but recent experiments in mice showing that Cx26 is dispensable for spatial buffering have ruled out this hypothesis (reviewed in Zhao, 2017).
In contrast, Cx26 gap junctions do have an important role in the establishment and maintenance of the endocochlear potential by the stria vascularis, a two-layered epithelium in the lateral wall of the cochlea that generates the endocochlear potential by active transport of K+ from perilymph into endolymph (Wangemann et al., 1995; Takeuchi et al., 2000; Marcus et al., 2002; Lang et al., 2007). Within the stria, Cx26 and Cx30 gap junctions convey K+ ions actively taken up from the perilymph by fibrocytes and basal cells to intermediate cells which release K+ into the intrastrial space. The effects of Cx26 ablation in the stria vascularis have not been explored directly, as mouse models with conditional KO of Gjb2 in this structure have not been generated to date. However, some insights have been provided by Cx30 KO models. Gjb6tm1Kwi/tm1Kwi mice (with critically decreased Gjb2 expression and no Cx30) show no endocochlear potential (Teubner et al., 2003), Gjb2+/− Gjb6+/tm1Kwi double heterozygous mice (also with reduced Gjb2 expression from the Gjb6+/tm1Kwi allele) have a significantly reduced endocochlear potential (Michel et al., 2003; Mei et al., 2017) and Gjb6Δ/Δ mice are not deaf, indicating a normal endocochlear potential (see “Deletions Removing a Distal Enhancer” section). There are two alternative explanations for these results as regards endocochlear potential generation: either Cx26 is essential and Cx30 is dispensable or each of the two connexins may be able to compensate for the lack of the other. These hypotheses must be tested experimentally.
In addition to these homeostatic roles, recent research has unveiled two roles of Cx26 gap junctions in cochlear development. Coordination of gene expression among cells in the developing cochlea is achieved by gap-junction-mediated transfer of microRNAs (miRNAs) and specific second messengers (such as IP3). Interestingly, in the inner ear, only Cx26 gap junctions are permeable to miRNAs (Zhu et al., 2015). Disruption of Cx26 gap junctions in mice blocks miRNA intercellular transfer and results in aberrant organ of Corti development that causes deafness (Kudo et al., 2003; Zhu et al., 2015; Zong et al., 2016). To date, disruption of miRNA permeability has only been demonstrated for the PPK-D mutation p.(Arg75Trp; Zong et al., 2016), but any mutations that reduce channel permeability may interfere with miRNA transfer. Of particular interest are mutations located in the extracellular loops that do not affect connexon intracellular sorting and result in constricted channel pores, such as p.(Arg165Trp; Rickard et al., 2001; Xiao et al., 2011). Future research should address the effects of any mutations on miRNA transfer.
As regards intercellular traffic of second messengers, it is well established that IP3 is one of the major players in inner ear morphogenesis, participating in the gap-junction-mediated propagation of intercellular Ca2+ waves between coupled cells (Mammano and Bortolozzi, 2017). Among other processes, IP3 transfer through gap junctions propagates the signal for cytochrome C-dependent apoptosis from the triggering cell to cells within the cell-death spreading zone, a crucial event in inner ear morphogenesis (Decrock et al., 2012). Mutation p.(Val84Leu; Kelley et al., 1998) specifically impairs Cx26-gap junction IP3 permeability (Beltramello et al., 2005; Decrock et al., 2012), while being completely undistinguishable from wild-type Cx26 in all other aspects tested in functional assays (Bruzzone et al., 2003; Wang et al., 2003). Impairment of IP3 permeability likely happens in other mutations that disrupt gap-junction channel transfer of most substances, as demonstrated for p.(Val95Met; Kelley et al., 1998; Beltramello et al., 2005). However, it seems that impairment of IP3 permeability alone is sufficient to cause HI, underscoring the importance of this particular role of Cx26.
Harmless variants
The last group of missense variants in GJB2 does not affect any residues known to be critical for the structure of Cx26. Given the available genetic data, they are considered harmless variants, among them p.(Val27Ile; Kelley et al., 1998), p.(Phe83Leu; Scott et al., 1998), p.(Arg127His; Estivill et al., 1998), p.(Val153Ile; Rickard et al., 2001) and p.(Ile203Thr; Kudo et al., 2000). However, caution is needed when analyzing those genetic data, as indicated by the case of the p.(Glu114Gly) mutation (Fuse et al., 1999). p.(Glu114Gly) nearly always appears in cis with p.(Val27Ile) in a complex allele, as indicated by the notation [p.(Val27Ile); p.(Glu114Gly)]. Variant p.(Glu114Gly) has usually been considered harmless because [p.(Val27Ile); p.(Glu114Gly)] /[p.(Val27Ile); p.(Glu114Gly)] homozygotes had been observed in a large number of normal hearing control individuals (e.g., 18.7% of controls from the Korean population; Choi et al., 2011). However, functional assays of p.(Glu114Gly) alone indicate defective gap junction channel and hemichannel functions that are largely compensated in the complex allele [p.(Val27Ile); p.(Glu114Gly)] (Choi et al., 2011). Thus, p.(Glu114Gly) might underlie DFNB1 HI in those rare cases in which this mutation does not present in cis with p.(Val27Ile).
Overall, accumulating evidence on Cx26 mutations in the GJB2 coding sequence paints a very complex picture in which particular mutations may have several very different pathological effects, according to the specific Cx26 structural or functional defect(s) that they cause. Truncating mutations, by interfering with most of Cx26 roles in the ear, are supposed to have the most dramatic effects, but even relatively subtle defects that only impair a single function (e.g., p.(Val84Leu)) result in HI. Future research should strive to understand better the pathogenic processes caused by the most common mutations, including: (1) agreeing on a standard set of functional assays that addresses as many Cx26 functional aspects as possible; and (2) paying particular attention to inner ear developmental defects.
Mutation Screening for the Genetic Diagnosis of DFNB1 HI
Genetic diagnosis of inherited NSHI is complicated, in terms of cost-effectiveness, by the extreme genetic heterogeneity of this condition. So the high prevalence of DFNB1 HI in most populations (Chan and Chang, 2014), together with the simple structure of the GJB2 gene, which facilitated the design and application of different molecular tests, made screening for GJB2 mutations the mainstay of genetic testing of inherited NSHI.
Strategies for DFNB1 genetic testing have evolved over the years, following the technical advances of Human Molecular Genetics. As specific GJB2 mutations are overrepresented in different populations and ethnicities (Chan and Chang, 2014), earliest strategies were based on the detection of some of those mutations through specific tests, eventually followed by Sanger DNA sequencing. Given the increasing multiethnicity of many populations, those approaches were considered insufficient (Chan et al., 2011), and DNA sequencing of the whole coding region and splice sites of GJB2 was recommended instead (Hoefsloot et al., 2013). This should be complemented by tests detecting deletions in the DFNB1 locus.
The advent of massively-parallel sequencing (MPS) facilitated novel strategies. Currently, the most cost-effective approach is using comprehensive panels of genes involved in inherited HI, which can provide sequencing data as well as detection of copy number variants (Shearer and Smith, 2015). Other laboratories use whole-exome sequencing followed by targeted analysis of genes known to be involved in HI (Zazo-Seco et al., 2017).
Clinical Features of DFNB1 HI
A summary of the clinical features of DFNB1 HI is presented in Table 2.

Table 2. Summary of clinical features of DFNB1 hearing impairment (HI).
Age of Onset and Severity
DFNB1 HI is mostly prelingual, but postlingual onset has also been reported, usually in association with specific mutations. In most subjects it is stable, but progression has also been documented, usually in cases with late onset. Severity is greatly variable, ranging from mild to profound even among subjects with the same genotype. Audiogram shapes are ordinarily flat or down-sloping.
Although DFNB1 HI is mostly prelingual, it should not be assumed that the onset is actually congenital in all cases. This issue is of concern to programmes for newborn audiological hearing screening, because infants with normal responses at birth could develop a severe HI within the next few months. An early study reported two children who were homozygous for c.35delG in GJB2. One of them had normal auditory brainstem responses at birth, but was diagnosed with a profound deafness at age 15 months. The other child had normal hearing at age 5 months, but was diagnosed with severe HI at age 9 months (Green et al., 2000). In another study, nine children who had passed the newborn hearing screening received a diagnosis of NSHI later in life and were shown to carry two GJB2 mutant alleles. The frequency of non penetrance at birth was estimated to be at least 3.8% (Norris et al., 2006). More recently, 14 deaf children with two GJB2 mutant alleles were reported to have passed the newborn hearing screening, indicating that the frequency of non penetrance at birth could be at least 6.9% (Minami et al., 2013). In fact, it has been postulated that an early but not always congenital onset of DFNB1 HI could be followed by a quick progression of the hearing loss (Gopalarao et al., 2008). Orzan and Murgia (2007) reported that 26 out of 47 children with profound DFNB1 HI had normal hearing at age 3 months, and this was followed by a sudden and severe deterioration. Also, a c.35delG homozygous boy who was diagnosed with HI at age 8 years suffered a sudden progression at age 23 years (Kokotas et al., 2008). Finally, another study reported an 8-year-old boy who was compound heterozygous for c.35delG and c.299-300delAT, whose late-onset hearing loss had started suddenly and progressed rapidly (Kenna et al., 2010).
Given the large numbers of subjects with a molecular diagnosis of DFNB1 HI, correlations between genotype and severity of the hearing loss have been studied in detail. In the largest cross-sectional study so far, genetic and audiological data were gathered from over 1500 affected subjects from 16 countries (Snoeckx et al., 2005). The severity of the HI was widely diverse even for the same genotype. However, when mutations were classified according to the severity of their molecular effects, some correlations were revealed. Genotypes including two truncating mutations (those mutations leading to premature stop codons) resulted in significantly more severe hearing losses than genotypes containing two non-truncating mutations (those mutations leading to amino acid substitutions; Snoeckx et al., 2005). Truncating/non-truncating genotypes resulted in phenotypes of intermediate severity between those two groups. Interestingly, a few genotypes resulted in HI with specific degrees of severity. Thus, profound HI was observed in the majority of cases with the c.35delG/p.(Arg143Trp) or c.35delG/del(GJB6-D13S1830) genotypes (Snoeckx et al., 2005). In contrast, mild or moderate HI was common in subjects carrying p.(Leu90Pro), p.(Met34Thr), or p.(Val37Ile) in combination with a mutation of the truncating type (Snoeckx et al., 2005). Further studies in other cohorts have confirmed these conclusions (Azaiez et al., 2004; Liu et al., 2005; Marlin et al., 2005; Primignani et al., 2009; Bartsch et al., 2010; Chan et al., 2010; Kenna et al., 2010; Tsukada et al., 2010; Burke et al., 2016).
There have been many contradictory reports on the pathogenicity of two missense mutations, p.(Met34Thr) and p.(Val37Ile) (Kelley et al., 1998; Griffith et al., 2000; Kudo et al., 2000; Marlin et al., 2001; Feldmann et al., 2004b). Controversy arose because of their high carrier rates is some populations and because they have been found in hearing-impaired subjects but also in subjects with normal hearing, as part of homozygous or compound heterozygous genotypes, the other allele being a clearly pathogenic mutation. All available data are consistent with the interpretation that these two alleles are hypomorphic, i.e., they have low penetrance and weak pathogenic potential. Accordingly, it is expected that their associated phenotypes range from normal hearing to late onset, progressive, mild to moderate hearing loss (Cucci et al., 2000; Houseman et al., 2001; Dahl et al., 2006; Huculak et al., 2006; Schrijver and Chang, 2006; Pollak et al., 2007; Tsukada et al., 2010; Zhao et al., 2011; Kim et al., 2013; Chai et al., 2015; Huang et al., 2015; Du et al., 2016).
The wide variability in severity of DFNB1 HI could be attributed at least partly to the influence of genetic modifiers. A large whole-genome association study investigated the phenotypic variability of c.35delG homozygotes by grouping cases in two classes according to the severity of the phenotype (mild/moderate HI vs. profound HI; Hilgert et al., 2009). The analysis was performed in two steps. First, a set of 255 samples was investigated by using a pooling-based strategy, followed by individual genotyping of the top 250 most significantly associated SNPs, in the same sample set. In a second step, those SNPs that still had significant P-values were genotyped in an independent set of samples. After this replication assay, nine SNPs still had significant P-values. Results from this study suggest that the variability in the severity of the HI that is observed in c.35delG homozygotes is not caused by one major genetic modifier, and that those nine SNPs might individually contribute just small modifying effects (Hilgert et al., 2009).
Temporal Bone
High-resolution computed tomography (CT) scans of the temporal bone have been used to investigate putative inner ear malformations in subjects with DFNB1 HI. Early studies did not reveal temporal bone anomalies (Cohn et al., 1999; Denoyelle et al., 1999). Subsequent studies have coincided in establishing that temporal bone malformations in subjects with DFNB1 HI would not be frequent, their prevalence being typically lower than 10% (Kenna et al., 2001, 2011; Preciado et al., 2004; Azaiez and Smith, 2007; Lee et al., 2009). Findings included unilateral or bilateral enlarged vestibular aqueduct, and bilateral Mondini dysplasia. In contrast with these results, one study found temporal bone anomalies in up to 72% of subjects with DFNB1 HI. These anomalies included hypoplastic cochlea, hypoplastic modiolus, dilated endolymphatic fossa, or enlarged vestibular aqueduct (Propst et al., 2006). This discrepancy has been attributed to differences in the composition of the cohorts of studied subjects (severity of hearing loss, genotypes…) as well as in methodology (radiologic image acquisition and interpretation; Propst et al., 2007; Kenna et al., 2011).
Histopathology of temporal bones in DFNB1 HI has been scarcely explored. In the only report published so far, samples from subjects with congenital profound HI were selected from the archives of a repository of temporal bones for genetic testing. Only one of the selected samples carried two mutant GJB2 alleles (c.35delG/p.(Glu101Gly) compound heterozygote). Microscopic analysis of cochlear sections from this subject revealed some gross anomalies of the inner ear: agenesis of the stria vascularis, a detached tectorial membrane, and extensive degeneration of hair cells. In contrast, no degeneration of neural structures (spiral ganglion cells and eighth cranial nerve) was observed. Damage to those cochlear structures could be the result of Cx26 deficiency. However, it must be taken into account that the subject had a history of diabetes mellitus, coronary atherosclerosis, hypertension and chronic renal failure, pathologies that might have contributed to those degenerative processes (Jun et al., 2000).
Vestibular Function
Subjects with DFNB1 HI usually do not complain of vertigo or dizziness, so their vestibular function is not routinely explored, and reports on this issue are sparse. In a study, vestibular-evoked myogenic potentials (VEMP) were absent bilaterally in two subjects carrying two pathogenic GJB2 mutations. This finding is suggestive of a saccular dysfunction. However, the patients did not report vertigo or dizziness, probably as a consequence of central compensation (Todt et al., 2005). Also, VEMP and caloric responses could not be elicited in 3 out of 5 subjects with two pathogenic GJB2 mutations (Zagólski, 2007). In another study on seven subjects with DFNB1 HI, vestibular dysfunction was bilateral in one subject and unilateral in four subjects, as revealed by either VEMP recording or the caloric test (Kasai et al., 2010). In a series of 23 subjects with two mutant DFNB1 alleles, only 2 had unilaterally abnormal caloric responses, but 17 showed decreased VEMP amplitudes (Tsukada et al., 2015), which again would suggest a saccular dysfunction. No patient in this series complained of vertigo or dizziness (Tsukada et al., 2015). In another study, a survey was used to assess symptoms of vestibular dysfunction in subjects with two GJB2 mutant alleles, and 127 out of 235 participants (54%) reported dizziness and vertigo (Dodson et al., 2011).
Taking into account the available data, it seems that vestibular dysfunction may be more common in DFNB1 HI than previously recognized, but its manifestations might be so subtle that they could go easily undetected, unless revealed by VEMP recording. Vestibular testing of larger series of DFNB1 hearing-impaired subjects is needed to clarify this issue.
Outcome of Cochlear Implantation
Several studies have compared the outcome of cochlear implantation in subjects with DFNB1 HI vs. subjects with non-DFNB1 HI. Speech perception skills improved clearly after implantation in the two groups. In some studies, differences between the two groups were not significant (Green et al., 2002; Bauer et al., 2003; Cullen et al., 2004; Taitelbaum-Swead et al., 2006; Connell et al., 2007), whereas in other studies the DFNB1 HI group showed better outcomes (Fukushima et al., 2002; Matsushiro et al., 2002; Sinnathuray et al., 2004). These differences are likely due to the heterogeneous compositions of the non-DFNB1 HI groups, which include HI of different, sometimes unknown, etiologies. In contrast, reading performance was consistently better in the DFNB1 HI group (Green et al., 2002; Bauer et al., 2003).
Audiologic Phenotype of Carriers
In agreement with the recessive inheritance pattern of DFNB1 HI, heterozygous carriers of DFNB1 mutations do not show any obvious hearing anomaly. However, some studies have revealed subtle audiological alterations in some subjects. In a cohort of heterozygous carriers for different GJB2 mutations (c.35delG, p.(Trp77Arg), p.(Val37Ile)), conventional pure-tone audiometry and auditory brainstem responses were normal. However, testing for distortion-product oto-acoustic emissions revealed significantly lower amplitudes in carriers than in non-carrier controls (Engel-Yeger et al., 2002, 2003). These results were not replicated in a different cohort (Groh et al., 2013). Disparities in the results could be atributed in part to the heterogeneous composition of the non-carrier groups. In other studies, pure-tone audiometry revealed significant hearing losses for frequencies higher than 6 kHz in c.35delG heterozygous carriers (Franzé et al., 2005; Groh et al., 2013). Recently, a study on subjects who were heterozygous for the splice-site mutation c.-23+1G>A revealed an earlier onset of age-related hearing loss (at about age 40 years) that in the control group (Barashkov et al., 2014). Studies on these issues are still sparse and some of them provide disparate results, indicating that investigation of larger series of carriers of different mutations is needed to establish firm conclusions.
Author Contributions
FJC and IC conceived the study, wrote the text and prepared Tables and Figures.
Funding
This work has received funding from Spanish Instituto de Salud Carlos III (grants PI14/01162 [to IC] and PI16/00077 [to FJC]; Plan Estatal de I+D+I 2013–2016, with co-funding from the European Regional Development Fund).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Abe, S., Usami, S. I., Shinkawa, H., Kelley, P. M., and Kimberling, W. J. (2000). Prevalent connexin 26 gene (GJB2) mutations in Japanese. J. Med. Genet. 37, 41–43. doi: 10.1136/jmg.37.1.41
Ambrosi, C., Walker, A. E., Depriest, A. D., Cone, A. C., Lu, C., Badger, J., et al. (2013). Analysis of trafficking, stability and function of human connexin 26 gap junction channels with deafness-causing mutations in the fourth transmembrane helix. PLoS One 8:e70916. doi: 10.1371/journal.pone.0070916
Anselmi, F., Hernandez, V. H., Crispino, G., Seydel, A., Ortolano, S., Roper, S. D., et al. (2008). ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc. Natl. Acad. Sci. U S A 105, 18770–18775. doi: 10.1073/pnas.0800793105
Azaiez, H., Chamberlin, G. P., Fischer, S. M., Welp, C. L., Prasad, S. D., Taggart, R. T., et al. (2004). GJB2: the spectrum of deafness-causing allele variants and their phenotype. Hum. Mutat. 24, 305–311. doi: 10.1002/humu.20084
Azaiez, H., and Smith, R. J. H. (2007). In reference to temporal bone imaging in GJB2 deafness. Laryngoscope 117:1127. doi: 10.1097/MLG.0b013e3180421729
Barashkov, N. A., Teryutin, F. M., Pshennikova, V. G., Solovyev, A. V., Klarov, L. A., Solovyeva, N. A., et al. (2014). Age-Related Hearing Impairment (ARHI) associated with GJB2 single mutation IVS1+1G>A in the Yakut population isolate in Eastern Siberia. PLoS One 9:e100848. doi: 10.1371/journal.pone.0100848
Bartsch, O., Vatter, A., Zechner, U., Kohlschmidt, N., Wetzig, C., Baumgart, A., et al. (2010). GJB2 mutations and genotype-phenotype correlation in 335 patients from germany with nonsyndromic sensorineural hearing loss: evidence for additional recessive mutations not detected by current methods. Audiol. Neurootol. 15, 375–382. doi: 10.1159/000297216
Bauer, P. W., Geers, A. E., Brenner, C., Moog, J. S., and Smith, R. J. (2003). The effect of GJB2 allele variants on performance after cochlear implantation. Laryngoscope 113, 2135–2140. doi: 10.1097/00005537-200312000-00015
Beltramello, M., Piazza, V., Bukauskas, F. F., Pozzan, T., and Mammano, F. (2005). Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat. Cell. Biol. 7, 63–69. doi: 10.1038/ncb1205
Bicego, M., Beltramello, M., Melchionda, S., Carella, M., Piazza, V., Zelante, L., et al. (2006). Pathogenetic role of the deafness-related M34T mutation of Cx26. Hum. Mol. Genet. 15, 2569–2587. doi: 10.1093/hmg/ddl184
Bliznetz, E. A., Lalayants, M. R., Markova, T. G., Balanovsky, O. P., Balanovska, E. V., Skhalyakho, R. A., et al. (2017). Update of the GJB2/DFNB1 mutation spectrum in Russia: a founder Ingush mutation del(GJB2–D13S175) is the most frequent among other large deletions. J. Hum. Genet. 62, 789–795. doi: 10.1038/jhg.2017.42
Bliznetz, E. A., Makienko, O. N., Okuneva, E. G., Markova, T. G., and Polyakov, A. V. (2014). New recurrent large deletion, encompassing both GJB2 and GJB6 genes, results in isolated sensorineural hearing impairment with autosomal recessive mode of inheritance. Russ. J. Genet. 50, 415–420. doi: 10.1134/s1022795414020045
Boulay, A. C., del Castillo, F. J., Giraudet, F., Hamard, G., Giaume, C., Petit, C., et al. (2013). Hearing is normal without connexin30. J. Neurosci. 33, 430–434. doi: 10.1523/JNEUROSCI.4240-12.2013
Brobby, G. W., Müller-Myhsok, B., and Horstmann, R. D. (1998). Connexin 26 R143W mutation associated with recessive nonsyndromic sensorineural deafness in Africa. N. Engl. J. Med. 338, 548–550. doi: 10.1056/NEJM199802193380813
Brown, C. W., Levy, M. L., Flaitz, C. M., Reid, B. S., Manolidis, S., Hebert, A. A., et al. (2003). A novel GJB2 (connexin 26) mutation, F142L, in a patient with unusual mucocutaneous findings and deafness. J. Invest. Dermatol. 121, 1221–1223. doi: 10.1046/j.1523-1747.2003.12550_4.x
Bruzzone, R., Veronesi, V., Gomès, D., Bicego, M., Duval, N., Marlin, S., et al. (2003). Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 533, 79–88. doi: 10.1016/s0014-5793(02)03755-9
Burke, W. F., Warnecke, A., Schöner-Heinisch, A., Lesinski-Schiedat, A., Maier, H., and Lenarz, T. (2016). Prevalence and audiological profiles of GJB2 mutations in a large collective of hearing impaired patients. Hear. Res. 333, 77–86. doi: 10.1016/j.heares.2016.01.006
Carrasquillo, M. M., Zlotogora, J., Barges, S., and Chakravarti, A. (1997). Two different connexin 26 mutations in an inbred kindred segregating non-syndromic recessive deafness: implications for genetic studies in isolated populations. Hum. Mol. Genet. 6, 2163–2172. doi: 10.1093/hmg/6.12.2163
Ceriani, F., Pozzan, T., and Mammano, F. (2016). Critical role of ATP-induced ATP release for Ca2+ signaling in nonsensory cell networks of the developing cochlea. Proc. Natl. Acad. Sci. U S A 113, E7194–E7201. doi: 10.1073/pnas.1616061113
Chai, Y., Chen, D., Sun, L., Li, L., Chen, Y., Pang, X., et al. (2015). The homozygous p.V37I variant of GJB2 is associated with diverse hearing phenotypes. Clin. Genet. 87, 350–355. doi: 10.1111/cge.12387
Chan, D. K., and Chang, K. W. (2014). GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 124, E34–E53. doi: 10.1002/lary.24332
Chan, D. K., Schrijver, I., and Chang, K. W. (2010). Connexin-26-associated deafness: phenotypic variability and progression of hearing loss. Genet. Med. 12, 174–181. doi: 10.1097/GIM.0b013e3181d0d42b
Chan, D. K., Schrijver, I., and Chang, K. W. (2011). Diagnostic yield in the workup of congenital sensorineural hearing loss is dependent on patient ethnicity. Otol. Neurotol. 32, 81–87. doi: 10.1097/MAO.0b013e3181fc786f
Chang, Q., Tang, W., Ahmad, S., Zhou, B., and Lin, X. (2008). Gap junction mediated intercellular metabolite transfer in the cochlea is compromised in connexin30 null mice. PLoS One 3:e4088. doi: 10.1371/journal.pone.0004088
Chen, J., Zhu, Y., Liang, C., Chen, J., and Zhao, H. B. (2015). Pannexin1 channels dominate ATP release in the cochlea ensuring endocochlear potential and auditory receptor potential generation and hearing. Sci. Rep. 5:10762. doi: 10.1038/srep10762
Choi, S. Y., Lee, K. Y., Kim, H. J., Kim, H. K., Chang, Q., Park, H. J., et al. (2011). Functional evaluation of GJB2 variants in nonsyndromic hearing loss. Mol. Med. 17, 550–556. doi: 10.2119/molmed.2010.00183
Choung, Y. H., Moon, S. K., and Park, H. J. (2002). Functional study of GJB2 in hereditary hearing loss. Laryngoscope 112, 1667–1671. doi: 10.1097/00005537-200209000-00026
Cohen-Salmon, M., Ott, T., Michel, V., Hardelin, J. P., Perfettini, I., Eybalin, M., et al. (2002). Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr. Biol. 12, 1106–1111. doi: 10.1016/s0960-9822(02)00904-1
Cohen-Salmon, M., Regnault, B., Cayet, N., Caille, D., Demuth, K., Hardelin, J. P., et al. (2007). Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proc. Natl. Acad. Sci. U S A 104, 6229–6234. doi: 10.1073/pnas.0605108104
Cohn, E. S., Kelley, P. M., Fowler, T. W., Gorga, M. P., Lefkowitz, D. M., Kuehn, H. J., et al. (1999). Clinical studies of families with hearing loss attributable to mutations in the connexin 26 gene (GJB2/DFNB1). Pediatrics 103, 546–550. doi: 10.1542/peds.103.3.546
Common, J. E. A., Bitner-Glindzicz, M., O’Toole, E. A., Barnes, M. R., Jenkins, L., Forge, A., et al. (2005). Specific loss of connexin 26 expression in ductal sweat gland epithelium associated with the deletion mutation del(GJB6–D13S1830). Clin. Exp. Dermatol. 30, 688–693. doi: 10.1111/j.1365-2230.2005.01878.x
Connell, S. S., Angeli, S. I., Suarez, H., Hodges, A. V., Balkany, T. J., and Liu, X. Z. (2007). Performance after cochlear implantation in DFNB1 patients. Otolaryngol. Head Neck Surg. 137, 596–602. doi: 10.1016/j.otohns.2007.02.017
Cucci, R. A., Prasad, S., Kelley, P. M., Green, G. E., Storm, K., Willocx, S., et al. (2000). The M34T allele variant of connexin 26. Genet. Test. 4, 335–344. doi: 10.1089/109065700750065063
Cullen, R. D., Buchman, C. A., Brown, C. J., Copeland, B. J., Zdanski, C., Pillsbury, H. C., et al. (2004). Cochlear implantation for children with GJB2-related deafness. Laryngoscope 114, 1415–1419. doi: 10.1097/00005537-200408000-00019
D’Andrea, P., Veronesi, V., Bicego, M., Melchionda, S., Zelante, L., Di Iorio, E., et al. (2002). Hearing loss: frequency and functional studies of the most common connexin26 alleles. Biochem. Biophys. Res. Commun. 296, 685–691. doi: 10.1016/s0006-291x(02)00891-4
Dahl, E., Manthey, D., Chen, Y., Schwarz, H. J., Chang, Y. S., Lalley, P. A., et al. (1996). Molecular cloning and functional expression of mouse connexin-30, a gap junction gene highly expressed in adult brain and skin. J. Biol. Chem. 271, 17903–17910. doi: 10.1074/jbc.271.30.17903
Dahl, H. H., Saunders, K., Kelly, T. M., Osborn, A. H., Wilcox, S., Cone-Wesson, B., et al. (2001). Prevalence and nature of connexin 26 mutations in children with non-syndromic deafness. Med. J. Aust. 175, 191–194.
Dahl, H. H., Tobin, S. E., Poulakis, Z., Rickards, F. W., Xu, X., Gillam, L., et al. (2006). The contribution of GJB2 mutations to slight or mild hearing loss in Australian elementary school children. J. Med. Genet. 43, 850–855. doi: 10.1136/jmg.2006.042051
de Wolf, E., van de Wiel, J., Cook, J., and Dale, N. (2016). Altered CO2 sensitivity of connexin26 mutant hemichannels in vitro. Physiol. Rep. 4:13038. doi: 10.14814/phy2.13038
Decrock, E., Krysko, D. V., Vinken, M., Kaczmarek, A., Crispino, G., Bol, M., et al. (2012). Transfer of IP3 through gap junctions is critical, but not sufficient, for the spread of apoptosis. Cell Death Differ. 19, 947–957. doi: 10.1038/cdd.2011.176
del Castillo, F. J., and del Castillo, I. (2011). The DFNB1 subtype of autosomal recessive non-syndromic hearing impairment. Front. Biosci. 16, 3252–3274. doi: 10.2741/3910
del Castillo, I., Moreno-Pelayo, M. A., Del Castillo, F. J., Brownstein, Z., Marlin, S., Adina, Q., et al. (2003). Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am. J. Hum. Genet. 73, 1452–1458. doi: 10.1086/380205
del Castillo, F. J., Rodríguez-Ballesteros, M., Alvarez, A., Hutchin, T., Leonardi, E., de Oliveira, C. A., et al. (2005). A novel deletion involving the connexin-30 gene, del(GJB6–S1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J. Med. Genet. 42, 588–594. doi: 10.1136/jmg.2004.028324
del Castillo, I., Villamar, M., Moreno-Pelayo, M. A., del Castillo, F. J., Alvarez, A., Tellería, D., et al. (2002). A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 346, 243–249. doi: 10.1056/NEJMoa012052
Denoyelle, F., Lina-Granade, G., Plauchu, H., Bruzzone, R., Chaïb, H., Lévi-Acobas, F., et al. (1998). Connexin 26 gene linked to a dominant deafness. Nature 393, 319–320. doi: 10.1038/30639
Denoyelle, F., Marlin, S., Weil, D., Moatti, L., Chauvin, P., Garabédian, E. N., et al. (1999). Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet 353, 1298–1303. doi: 10.1016/s0140-6736(98)11071-1
Denoyelle, F., Weil, D., Maw, M. A., Wilcox, S. A., Lench, N. J., Allen-Powell, D. R., et al. (1997). Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum. Mol. Genet. 6, 2173–2177. doi: 10.1093/hmg/6.12.2173
Dodson, K. M., Blanton, S. H., Welch, K. O., Norris, V. W., Nuzzo, R. L., Wegelin, J. A., et al. (2011). Vestibular dysfunction in DFNB1 deafness. Am. J. Med. Genet. A 155A, 993–1000. doi: 10.1002/ajmg.a.33828
Du, Y., Huang, L., Cheng, X., Zhao, L., Ruan, Y., and Ni, T. (2016). Analysis of p.V37I compound heterozygous mutations in the GJB2 gene in Chinese infants and young children. Biosci. Trends 10, 220–226. doi: 10.5582/bst.2016.01096
Engel-Yeger, B., Zaaroura, S., Zlotogora, J., Shalev, S., Hujeirat, Y., Carrasquillo, M., et al. (2002). The effects of a connexin 26 mutation—35delG—on oto-acoustic emissions and brainstem evoked potentials: homozygotes and carriers. Hear. Res. 163, 93–100. doi: 10.1016/s0378-5955(01)00386-0
Engel-Yeger, B., Zaaroura, S., Zlotogora, J., Shalev, S., Hujeirat, Y., Carrasquillo, M., et al. (2003). Otoacoustic emissions and brainstem evoked potentials in compound carriers of connexin 26 mutations. Hear. Res. 175, 140–151. doi: 10.1016/s0378-5955(02)00719-0
Estivill, X., Fortina, P., Surrey, S., Rabionet, R., Melchionda, S., D’Agruma, L., et al. (1998). Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 351, 394–398. doi: 10.1016/s0140-6736(98)24006-2
Feldmann, D., Denoyelle, F., Chauvin, P., Garabédian, E. N., Couderc, R., Odent, S., et al. (2004a). Large deletion of the GJB6 gene in deaf patients heterozygous for the GJB2 gene mutation: genotypic and phenotypic analysis. Am. J. Med. Genet. A 127A, 263–267. doi: 10.1002/ajmg.a.20588
Feldmann, D., Denoyelle, F., Loundon, N., Weil, D., Garabedian, E. N., Couderc, R., et al. (2004b). Clinical evidence of the nonpathogenic nature of the M34T variant in the connexin 26 gene. Eur. J. Hum. Genet. 12, 279–284. doi: 10.1038/sj.ejhg.5201147
Feldmann, D., Le Marechal, C., Jonard, L., Thierry, P., Czajka, C., Couderc, R., et al. (2009). A new large deletion in the DFNB1 locus causes nonsyndromic hearing loss. Eur. J. Med. Genet. 52, 195–200. doi: 10.1016/j.ejmg.2008.11.006
Forge, A., Becker, D., Casalotti, S., Edwards, J., Marziano, N., and Nevill, G. (2003a). Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessment of connexin composition in mammals. J. Comp. Neurol. 467, 207–231. doi: 10.1002/cne.10916
Forge, A., Marziano, N. K., Casalotti, S. O., Becker, D. L., and Jagger, D. (2003b). The inner ear contains heteromeric channels composed of cx26 and cx30 and deafness-related mutations in cx26 have a dominant negative effect on cx30. Cell. Commun. Adhes. 10, 341–346. doi: 10.1080/714040450
Franzé, A., Caravelli, A., Di Leva, F., Marciano, E., Auletta, G., D’Aulos, F., et al. (2005). Audiometric evaluation of carriers of the connexin 26 mutation 35delG. Eur. Arch. Otorhinolaryngol. 262, 921–924. doi: 10.1007/s00405-005-0918-1
Frei, K., Lucas, T., Ramsebner, R., Schöfer, C., Baumgartner, W. D., Weipoltshammer, K., et al. (2004). A novel connexin 26 mutation associated with autosomal recessive sensorineural deafness. Audiol. Neurootol. 9, 47–50. doi: 10.1159/000074186
Frei, K., Szuhai, K., Lucas, T., Weipoltshammer, K., Schöfer, C., Ramsebner, R., et al. (2002). Connexin 26 mutations in cases of sensorineural deafness in eastern Austria. Eur. J. Hum. Genet. 10, 427–432. doi: 10.1038/sj.ejhg.5200826
Fukushima, K., Sugata, K., Kasai, N., Fukuda, S., Nagayasu, R., Toida, N., et al. (2002). Better speech performance in cochlear implant patients with GJB2-related deafness. Int. J. Pediatr. Otorhinolaryngol. 62, 151–157. doi: 10.1016/s0165-5876(01)00619-x
Fuse, Y., Doi, K., Hasegawa, T., Sugii, A., Hibino, H., and Kubo, T. (1999). Three novel connexin26 gene mutations in autosomal recessive non-syndromic deafness. Neuroreport 10, 1853–1857. doi: 10.1097/00001756-199906230-00010
Gandía, M., del Castillo, F. J., Rodríguez-Álvarez, F. J., Garrido, G., Villamar, M., Calderón, M., et al. (2013). A novel splice-site mutation in the GJB2 gene causing mild postlingual hearing impairment. PLoS One 8:e73566. doi: 10.1371/journal.pone.0073566
García, I. E., Maripillán, J., Jara, O., Ceriani, R., Palacios-Muñoz, A., Ramachandran, J., et al. (2015). Keratitis-ichthyosis-deafness syndrome-associated Cx26 mutants produce nonfunctional gap junctions but hyperactive hemichannels when co-expressed with wild type Cx43. J. Invest. Dermatol. 135, 1338–1347. doi: 10.1038/jid.2015.20
Gardner, P., Oitmaa, E., Messner, A., Hoefsloot, L., Metspalu, A., and Schrijver, I. (2006). Simultaneous multigene mutation detection in patients with sensorineural hearing loss through a novel diagnostic microarray: a new approach for newborn screening follow-up. Pediatrics 118, 985–994. doi: 10.1542/peds.2005-2519
Gopalarao, D., Kimberling, W. J., Jesteadt, W., Kelley, P. M., Beauchaine, K. L., and Cohn, E. S. (2008). Is hearing loss due to mutations in the Connexin 26 gene progressive? Int. J. Audiol. 47, 11–20. doi: 10.1080/14992020701602087
Green, G. E., Scott, D. A., McDonald, J. M., Teagle, H. F., Tomblin, B. J., Spencer, L. J., et al. (2002). Performance of cochlear implant recipients with GJB2-related deafness. Am. J. Med. Genet. 109, 167–170. doi: 10.1002/ajmg.10330
Green, G. E., Scott, D. A., McDonald, J. M., Woodworth, G. G., Sheffield, V. C., and Smith, R. J. H. (1999). Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA 281, 2211–2216. doi: 10.1001/jama.281.23.2211
Green, G. E., Smith, R. J., Bent, J. P., and Cohn, E. S. (2000). Genetic testing to identify deaf newborns. JAMA 284:1245. doi: 10.1001/jama.284.10.1239
Grifa, A., Wagner, C. A., D’Ambrosio, L., Melchionda, S., Bernardi, F., Lopez-Bigas, N., et al. (1999). Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat. Genet. 23, 16–18. doi: 10.1038/12612
Griffith, A. J., Chowdhry, A. A., Kurima, K., Hood, L. J., Keats, B., Berlin, C. I., et al. (2000). Autosomal recessive nonsyndromic neurosensory deafness at DFNB1 not associated with the compound-heterozygous GJB2 (connexin 26) genotype M34T/167delT. Am. J. Hum. Genet. 67, 745–749. doi: 10.1086/303045
Groh, D., Seeman, P., Jilek, M., Popelář, J., Kabelka, Z., and Syka, J. (2013). Hearing function in heterozygous carriers of a pathogenic GJB2 gene mutation. Physiol. Res. 62, 323–330.
Guilford, P., Ben Arab, S., Blanchard, S., Levilliers, J., Weissenbach, J., Belkahia, A., et al. (1994). A non-syndrome form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat. Genet. 6, 24–28. doi: 10.1038/ng0194-24
Hilgert, N., Huentelman, M. J., Thorburn, A. Q., Fransen, E., Dieltjens, N., Mueller-Malesinska, M., et al. (2009). Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene. Eur. J. Hum. Genet. 17, 517–524. doi: 10.1038/ejhg.2008.201
Hoefsloot, L. H., Roux, A. F., Bitner-Glindzicz, M., and contributors to EMQN DFNB1 best practice meeting. (2013). EMQN Best Practice guidelines for diagnostic testing of mutations causing non-syndromic hearing impairment at the DFNB1 locus. Eur. J. Hum. Genet. 21, 1325–1329. doi: 10.1038/ejhg.2013.83
Houseman, M. J., Ellis, L. A., Pagnamenta, A., Di, W. L., Rickard, S., Osborn, A. H., et al. (2001). Genetic analysis of the connexin-26 M34T variant: identification of genotype M34T/M34T segregating with mild-moderate non-syndromic sensorineural hearing loss. J. Med. Genet. 38, 20–25. doi: 10.1136/jmg.38.1.20
Huang, S., Huang, B., Wang, G., Yuan, Y., and Dai, P. (2015). The relationship between the p.V37I mutation in GJB2 and hearing phenotypes in Chinese individuals. PLoS One 10:e0129662. doi: 10.1371/journal.pone.0129662
Huckstepp, R. T. R., Eason, R., Sachdec, A., and Dale, N. (2010a). CO2-dependent opening of connexin 26 and related β connexins. J. Physiol. 588, 3921–3931. doi: 10.1113/jphysiol.2010.192096
Huckstepp, R. T. R., id Bihi, R., Eason, R., Spyer, K. M., Dicke, N., Willecke, K., et al. (2010b). Connexin hemichannel-mediated CO2-dependent release of ATP in the medulla oblongata contributes to central respiratory chemosensitivity. J. Physiol. 588, 3901–3920. doi: 10.1113/jphysiol.2010.192088
Huculak, C., Bruyere, H., Nelson, T. N., Kozak, F. K., and Langlois, S. (2006). V37I connexin 26 allele in patients with sensorineural hearing loss: evidence of its pathogenicity. Am. J. Med. Genet. 140A, 2394–2400. doi: 10.1002/ajmg.a.31486
Hwa, H. L., Ko, T. M., Hsu, C. J., Huang, C. H., Chiang, Y. L., Oong, J. L., et al. (2003). Mutation spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with prelingual deafness. Genet. Med. 5, 161–165. doi: 10.1097/01.gim.0000066796.11916.94
Johnson, S. L., Ceriani, F., Houston, O., Polishchuk, R., Polishchuk, E., Crispino, G., et al. (2017). Connexin-mediated signaling in nonsensory cells is crucial for the development of sensory inner hair cells in the mouse cochlea. J. Neurosci. 37, 258–268. doi: 10.1523/JNEUROSCI.2251-16.2016
Jun, A. I., McGuirt, W. T., Hinojosa, R., Green, G. E., Fischel-Ghodsian, N., and Smith, R. J. (2000). Temporal bone histopathology in connexin 26-related hearing loss. Laryngoscope 110, 269–275. doi: 10.1097/00005537-200002010-00016
Kasai, M., Hayashi, C., Iizuka, T., Inoshita, A., Kamiya, K., Okada, H., et al. (2010). Vestibular function of patients with profound deafness related to GJB2 mutation. Acta Otolaryngol. 130, 990–995. doi: 10.3109/00016481003596508
Kelley, P. M., Harris, D. J., Comer, B. C., Askew, J. W., Fowler, T., Smith, S. D., et al. (1998). Novel mutations in the connexin 26 Gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am. J. Hum. Genet. 62, 792–799. doi: 10.1086/301807
Kelsell, D. P., Dunlop, J., Stevens, H. P., Lench, N. J., Liang, J., Parry, G., et al. (1997). Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387, 80–83. doi: 10.1038/387080a0
Kenna, M. A., Feldman, H. A., Neault, M. W., Frangulov, A., Wu, B. L., Fligor, B., et al. (2010). Audiologic phenotype and progression in GJB2 (Connexin 26) hearing loss. Arch. Otolaryngol. Head Neck Surg. 136, 81–87. doi: 10.1001/archoto.2009.202
Kenna, M. A., Rehm, H. L., Frangulov, A., Feldman, H. A., and Robson, C. D. (2011). Temporal bone abnormalities in children with GJB2 mutations. Laryngoscope 121, 630–635. doi: 10.1002/lary.21414
Kenna, M. A., Wu, B. L., Cotanche, D. A., Korf, B. R., and Rehm, H. L. (2001). Connexin 26 studies in patients with sensorineural hearing loss. Arch. Otolaryngol. Head Neck Surg. 127, 1037–1042. doi: 10.1001/archotol.127.9.1037
Kiang, D. T., Jin, N., Tu, Z. J., and Lin, H. H. (1997). Upstream genomic sequence of the human connexin26 gene. Gene 199, 165–171. doi: 10.1016/s0378-1119(97)00365-x
Kikuchi, T., Kimura, R. S., Paul, D. L., and Adams, J. C. (1995). Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat. Embryol. 191, 101–118. doi: 10.1007/bf00186783
Kim, H. R., Oh, S. K., Lee, E. S., Choi, S. Y., Roh, S. E., Kim, S. J., et al. (2016). The pathological effects of connexin 26 variants related to hearing loss by in silico and in vitro analysis. Hum. Genet. 135, 287–298. doi: 10.1007/s00439-015-1625-7
Kim, S. Y., Park, G., Han, K. H., Kim, A., Koo, J. W., Chang, S. O., et al. (2013). Prevalence of p.V37I variant of GJB2 in mild or moderate hearing loss in a pediatric population and the interpretation of its pathogenicity. PLoS One 8:e61592. doi: 10.1371/journal.pone.0061592
Kimura, K., Wakamatsu, A., Suzuki, Y., Ota, T., Nishikawa, T., Yamashita, R., et al. (2006). Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 16, 55–65. doi: 10.1101/gr.4039406
Kokotas, H., Theodosiou, M., Korres, G., Grigoriadou, M., Ferekidou, E., Giannoulia-Karantana, A., et al. (2008). Sudden hearing loss in a family with GJB2 related progressive deafness. Int. J. Pediatr. Otorhinolaryngol. 72, 1735–1740. doi: 10.1016/j.ijporl.2008.08.006
Kudo, T., Ikeda, K., Kure, S., Matsubara, Y., Oshima, T., Watanabe, K., et al. (2000). Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am. J. Med. Genet. 90, 141–145. doi: 10.1002/(sici)1096-8628(20000117)90:2<141::aid-ajmg10>3.0.co;2-g
Kudo, T., Kure, S., Ikeda, K., Xia, A. P., Katori, Y., Suzuki, M., et al. (2003). Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum. Mol. Genet. 12, 995–1004. doi: 10.1093/hmg/ddg116
Kumar, N. M., and Gilula, N. B. (1996). The gap junction communication channel. Cell 84, 381–388. doi: 10.1016/s0092-8674(00)81282-9
Lang, F., Vallon, V., Knipper, M., and Wangemann, P. (2007). Functional significance of channels and transporters expressed in the inner ear and kidney. Am. J. Physiol. Cell Physiol. 293, C1187–C1208. doi: 10.1152/ajpcell.00024.2007
Lautermann, J., ten Cate, W. J. F., Altenhoff, P., Grümmer, R., Traub, O., Frank, H. G., et al. (1998). Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res. 294, 415–420. doi: 10.1007/s004410051192
Lee, K. H., Larson, D. A., Shott, G., Rasmussen, B., Cohen, A. P., Benton, C., et al. (2009). Audiologic and temporal bone imaging findings in patients with sensorineural hearing loss and GJB2 Mutations. Laryngoscope 119, 554–558. doi: 10.1002/lary.20162
Lerer, I., Sagi, M., Ben-Neriah, Z., Wang, T., Levi, H., and Abeliovich, D. (2001). A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: a novel founder mutation in Ashkenazi Jews. Hum. Mutat. 18:460. doi: 10.1002/humu.1222
Lim, L. H. Y., Bradshaw, J. K., Guo, Y., Pilipenko, V., Madden, C., Ingala, D., et al. (2003). Genotypic and phenotypic correlations of DFNB1-related hearing impairment in the Midwestern United States. Arch. Otolaryngol. Head Neck Surg. 129, 836–840. doi: 10.1001/archotol.129.8.836
Lindholm, D., Korhonen, L., Eriksson, O., and Kõks, S. (2017). Recent insights into the role of unfolded protein response in ER stress in health and disease. Front. Cell Dev. Biol. 5:48. doi: 10.3389/fcell.2017.00048
Liu, W., Boström, M., Kinnefors, A., and Rask-Andersen, H. (2009). Unique expression of connexins in the human cochlea. Hear. Res. 250, 55–62. doi: 10.1016/j.heares.2009.01.010
Liu, W., Edin, F., Blom, H., Magnusson, P., Schrott-Fischer, A., Glueckert, R., et al. (2016). Super-resolution structured illumination fluorescence microscopy of the lateral wall of the cochlea: the Connexin26/30 proteins are separately expressed in man. Cell Tissue Res. 365, 13–27. doi: 10.1007/s00441-016-2359-0
Liu, W., Li, H., Edin, F., Brännström, J., Glueckert, R., Schrott-Fischer, A., et al. (2017). Molecular composition and distribution of gap junctions in the sensory epithelium of the human cochlea-a super-resolution structured illumination microscopy (SR-SIM) study. Ups. J. Med. Sci. 122, 160–170. doi: 10.1080/03009734.2017.1322645
Liu, X. Z., Pandya, A., Angeli, S., Telischi, F. F., Arnos, K. S., Nance, W. E., et al. (2005). Audiological features of GJB2 (connexin 26) deafness. Ear Hear. 26, 361–369. doi: 10.1097/00003446-200506000-00011
Lykke-Andersen, S., and Jensen, T. H. (2015). Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 16, 665–677. doi: 10.1038/nrm4063
Lynn, B. D., Tress, O., May, D., Willecke, K., and Nagy, J. I. (2011). Ablation of connexin30 in transgenic mice alters expression patterns of connexin26 and connexin32 in glial cells and leptomeninges. Eur. J. Neurosci. 34, 1783–1793. doi: 10.1111/j.1460-9568.2011.07900.x
Maeda, S., Nakagawa, S., Suga, M., Yamashita, E., Oshima, A., Fujiyoshi, Y., et al. (2009). Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature 458, 597–604. doi: 10.1038/nature07869
Maestrini, E., Korge, B. P., Ocaña-Sierra, J., Calzolari, E., Cambiaghi, S., Scudder, P. M., et al. (1999). A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum. Mol. Genet. 8, 1237–1243. doi: 10.1093/hmg/8.7.1237
Mammano, F., and Bortolozzi, M. (2017). Ca2+ signaling, apoptosis and autophagy in the developing cochlea: milestones to hearing acquisition. Cell Calcium doi: 10.1016/j.ceca.2017.05.006 [Epub ahead of print].
Mani, R. S., Ganapathy, A., Jalvi, R., Srikumari Srisailapathy, C. R., Malhotra, V., Chadha, S., et al. (2009). Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur. J. Hum. Genet. 17, 502–509. doi: 10.1038/ejhg.2008.179
Marcus, D. C., Wu, T., Wangemann, P., and Kofuji, P. (2002). KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am. J. Physiol. Cell Physiol. 282, C403–C407. doi: 10.1152/ajpcell.00312.2001
Marlin, S., Feldmann, D., Blons, H., Loundon, N., Rouillon, I., Albert, S., et al. (2005). GJB2 and GJB6 mutations: genotypic and phenotypic correlations in a large cohort of hearing-impaired patients. Arch. Otolaryngol. Head Neck Surg. 131, 481–487. doi: 10.1001/archotol.131.6.481
Marlin, S., Garabédian, E. N., Roger, G., Moatti, L., Matha, N., Lewin, P., et al. (2001). Connexin 26 gene mutations in congenitally deaf children: pitfalls for genetic counseling. Arch. Otolaryngol. Head Neck Surg. 127, 927–933. doi: 10.1001/archotol.127.8.927
Martin, P. E. M., Coleman, S. L., Casalotti, S. O., Forge, A., and Evans, W. H. (1999). Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal hereditary deafness. Hum. Mol. Genet. 8, 2369–2376. doi: 10.1093/hmg/8.13.2369
Marziano, N. K., Casalotti, S. O., Portelli, A. E., Becker, D. L., and Forge, A. (2003). Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum. Mol. Genet. 12, 805–812. doi: 10.1093/hmg/ddg076
Matos, T. D., Caria, H., Simões-Teixeira, H., Aasen, T., Nickel, R., Jagger, D. J., et al. (2007). A novel hearing-loss-related mutation occurring in the GJB2 basal promoter. J. Med. Genet. 44, 721–725. doi: 10.1136/jmg.2007.050682
Matsushiro, N., Doi, K., Fuse, Y., Nagai, K., Yamamoto, K., Iwaki, T., et al. (2002). Successful cochlear implantation in prelingual profound deafness resulting from the common 233delC mutation of the GJB2 gene in the Japanese. Laryngoscope 112, 255–261. doi: 10.1097/00005537-200202000-00011
Mei, L., Chen, J., Zong, L., Zhu, Y., Liang, C., Jones, R. O., et al. (2017). A deafness mechanism of digenic Cx26 (GJB2) and Cx30 (GJB6) mutations: reduction of endocochlear potential by impairment of heterogeneous gap junctional function in the cochlear lateral wall. Neurobiol. Dis. 108, 195–203. doi: 10.1016/j.nbd.2017.08.002
Meigh, L., Greenhalgh, S. A., Rodgers, T. L., Cann, M. J., Roper, D. I., and Dale, N. (2013). CO2 directly modulates connexin 26 by formation of carbamate bridges between subunits. Elife 2:e01213. doi: 10.7554/elife.01213
Michel, V., Hardelin, J. P., and Petit, C. (2003). Molecular mechanism of a frequent genetic form of deafness. N. Engl. J. Med. 349, 716–717. doi: 10.1056/nejmc030327
Minami, S. B., Mutai, H., Nakano, A., Arimoto, Y., Taiji, H., Morimoto, N., et al. (2013). GJB2-associated hearing loss undetected by hearing screening of newborns. Gene 532, 41–45. doi: 10.1016/j.gene.2013.08.094
Mustapha, M., Salem, N., Delague, V., Chouery, E., Ghassibeh, M., Rai, M., et al. (2001). Autosomal recessive non-syndromic hearing loss in the Lebanese population: prevalence of the 30delG mutation and report of two novel mutations in the connexin 26 (GJB2) gene. J. Med. Genet. 38:E36. doi: 10.1136/jmg.38.10.e36
Norris, V. W., Arnos, K. S., Hanks, W. D., Xia, X., Nance, W. E., and Pandya, A. (2006). Does universal newborn hearing screening identify all children with GJB2 (Connexin 26) deafness? Penetrance of GJB2 deafness. Ear Hear. 27, 732–741. doi: 10.1097/01.aud.0000240492.78561.d3
Ortolano, S., Di Pasquale, G., Crispino, G., Anselmi, F., Mammano, F., and Chiorini, J. A. (2008). Coordinated control of connexin 26 and connexin 30 at the regulatory and functional level in the inner ear. Proc. Natl. Acad. Sci. U S A 105, 18776–18781. doi: 10.1073/pnas.0800831105
Orzan, E., and Murgia, A. (2007). Connexin 26 deafness is not always congenital. Int. J. Pediatr. Otorhinolaryngol. 71, 501–507. doi: 10.1016/j.ijporl.2006.12.002
Pallares-Ruiz, N., Blanchet, P., Mondain, M., Claustres, M., and Roux, A. F. (2002). A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur. J. Hum. Genet. 10, 72–76. doi: 10.1038/sj.ejhg.5200762
Parzefall, T., Lucas, T., Koenighofer, M., Ramsebner, R., Frohne, A., Czeiger, S., et al. (2017). The role of alternative GJB2 transcription in screening for neonatal sensorineural deafness in Austria. Acta Otolaryngol. 137, 356–360. doi: 10.1080/00016489.2016.1249946
Piazza, V., Ciubotaru, C. D., Gale, J. E., and Mammano, F. (2007). Purinergic signalling and intercellular Ca2+ wave propagation in the organ of Corti. Cell Calcium 41, 77–86. doi: 10.1016/j.ceca.2006.05.005
Pollak, A., Skórka, A., Mueller-Malesińska, M., Kostrzewa, G., Kisiel, B., Waligóra, J., et al. (2007). M34T and V37I mutations in GJB2 associated hearing impairment: evidence for pathogenicity and reduced penetrance. Am. J. Med. Genet. A 143A, 2534–2543. doi: 10.1002/ajmg.a.31982
Prasad, S., Cucci, R. A., Green, G. E., and Smith, R. J. H. (2000). Genetic testing for hereditary hearing loss: connexin 26 (GJB2) allele variants and two novel deafness-causing mutations (R32C and 645–648delTAGA). Hum. Mutat. 16, 502–508. doi: 10.1002/1098-1004(200012)16:6<502::aid-humu7>3.0.co;2-4
Preciado, D. A., Lim, L. H., Cohen, A. P., Madden, C., Myer, D., Ngo, C., et al. (2004). A diagnostic paradigm for childhood idiopathic sensorineural hearing loss. Otolaryngol. Head Neck Surg. 131, 804–809. doi: 10.1016/j.otohns.2004.06.707
Primignani, P., Trotta, L., Castorina, P., Lalatta, F., Sironi, F., Radaelli, C., et al. (2009). Analysis of the GJB2 and GJB6 genes in Italian patients with nonsyndromic hearing loss: frequencies, novel mutations, genotypes, and degree of hearing loss. Genet. Test. Mol. Biomarkers 13, 209–217. doi: 10.1089/gtmb.2008.0086
Propst, E. J., Blaser, S., Stockley, T. L., Harrison, R. V., Gordon, K. A., and Papsin, B. C. (2006). Temporal bone imaging in GJB2 deafness. Laryngoscope 116, 2178–2186. doi: 10.1097/01.mlg.0000244389.68568.a7
Propst, E. J., Harrison, R. V., Gordon, K. A., Papsin, B. C., Blaser, S., and Stockley, T. L. (2007). In reply to “In reference to temporal bone imaging in GJB2 deafness”. Laryngoscope 117, 1127–1129. doi: 10.1097/MLG.0b013e3180421729
Putcha, G. V., Bejjani, B. A., Bleoo, S., Booker, J. K., Carey, J. C., Carson, N., et al. (2007). A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet. Med. 9, 413–426. doi: 10.1097GIM.0b013e3180a03276
Rabionet, R., Zelante, L., López-Bigas, N., D’Agruma, L., Melchionda, S., Restagno, G., et al. (2000). Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum. Genet. 106, 40–44. doi: 10.1007/s004390051007
Richard, G., Brown, N., Ishida-Yamamoto, A., and Krol, A. (2004). Expanding the phenotypic spectrum of Cx26 disorders: bart-pumphrey syndrome is caused by a novel missense mutation in GJB2. J. Invest. Dermatol. 123, 856–863. doi: 10.1111/j.0022-202x.2004.23470.x
Richard, G., Rouan, F., Willoughby, C. E., Brown, N., Chung, P., Ryynänen, M., et al. (2002). Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am. J. Hum. Genet. 70, 1341–1348. doi: 10.1086/339986
Richard, G., White, T. W., Smith, L. E., Bailey, R. A., Compton, J. G., Paul, D. L., et al. (1998). Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum. Genet. 103, 393–399. doi: 10.1007/s004390050839
Richardson, G. P., de Monvel, J. B., and Petit, C. (2011). How the genetics of deafness illuminates auditory physiology. Annu. Rev. Physiol. 73, 311–334. doi: 10.1146/annurev-physiol-012110-142228
Rickard, S., Kelsell, D. P., Sirimana, T., Rajput, K., MacArdle, B., and Bitner-Glindzicz, M. (2001). Recurrent mutations in the deafness gene GJB2 (connexin 26) in British Asian families. J. Med. Genet. 38, 530–533. doi: 10.1136/jmg.38.8.530
Rodriguez-Paris, J., and Schrijver, I. (2009). The digenic hypothesis unraveled: the GJB6 del (GJB6–D13S1830) mutation causes allele-specific loss of GJB2 expression in cis. Biochem. Biophys. Res. Commun. 389, 354–359. doi: 10.1016/j.bbrc.2009.08.152
Rodriguez, L., Simeonato, E., Scimemi, P., Anselmi, F., Calì, B., Crispino, G., et al. (2012). Reduced phosphatidylinositol 4,5-bisphosphate synthesis impairs inner ear Ca2+ signaling and high-frequency hearing acquisition. Proc. Natl. Acad. Sci. U S A 109, 14013–14018. doi: 10.1073/pnas.1211869109
Rodriguez-Paris, J., Tamayo, M. L., Gelvez, N., and Schrijver, I. (2011). Allele-specific impairment of GJB2 expression by GJB6 deletion del(GJB6–D13S1854). PLoS One 6:e21665. doi: 10.1371/journal.pone.0021665
Rouan, F., White, T. W., Brown, N., Taylor, A. M., Lucke, T. W., Paul, D. L., et al. (2001). trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J. Cell. Sci. 114, 2105–2113.
Rubinato, E., Azaiez, H., Black-Ziegelbein, E., Morgan, A., Booth, K., Vozzi, D., et al. (2016). High frequency of GJB2 splice-site mutation c.-22–2A>C in a large cohort of Italian age related hearing loss patients and matched controls. Assoc. Res. Otolaryngol. Abs. 39:358.
Safieddine, S., El-Amraoui, A., and Petit, C. (2012). The auditory hair cell ribbon synapse: from assembly to function. Annu. Rev. Neurosci. 35, 509–528. doi: 10.1146/annurev-neuro-061010-113705
Santos-Sacchi, J., and Dallos, P. (1983). Intercellular communication in the supporting cells of the organ of Corti. Hear. Res. 9, 317–326. doi: 10.1016/0378-5955(83)90034-5
Schrijver, I., and Chang, K. W. (2006). Two patients with the V37I/235delC genotype: are radiographic cochlear anomalies part of the phenotype? Int. J. Pediatr. Otorhinolaryngol. 70, 2109–2113. doi: 10.1016/j.ijporl.2006.07.015
Scott, D. A., Kraft, M. L., Carmi, R., Ramesh, A., Elbedour, K., Yairi, Y., et al. (1998). Identification of mutations in the connexin 26 gene that cause autosomal recessive nonsyndromic hearing loss. Hum. Mutat. 11, 387–394. doi: 10.1002/(sici)1098-1004(1998)11:5<387::aid-humu6>3.0.co;2-8
Shahin, H., Walsh, T., Sobe, T., Lynch, E., King, M. C., Avraham, K. B., et al. (2002). Genetics of congenital deafness in the Palestinian population: multiple connexin 26 alleles with shared origins in the Middle East. Hum. Genet. 110, 284–289. doi: 10.1007/s00439-001-0674-2
Shearer, A. E., and Smith, R. J. (2015). Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol. Head Neck Surg. 153, 175–182. doi: 10.1177/0194599815591156
Sinnathuray, A. R., Toner, J. G., Geddis, A., Clarke-Lyttle, J., Patterson, C. C., and Hughes, A. E. (2004). Auditory perception and speech discrimination after cochlear implantation in patients with connexin 26 (GJB2) gene-related deafness. Otol. Neurotol. 25, 930–934. doi: 10.1097/00129492-200411000-00012
Snoeckx, R. L., Huygen, P. L. M., Feldmann, D., Marlin, S., Denoyelle, F., Waligora, J., et al. (2005). GJB2 mutations and degree of hearing loss: a multi-center study. Am. J. Hum. Genet. 77, 945–957. doi: 10.1086/497996
Stanghellini, I., Genovese, E., Palma, S., Ravani, A., Falcinelli, C., Guarnaccia, M. C., et al. (2014). New and rare GJB2 alleles in patients with nonsyndromic sensorineural hearing impairment: a genotype/auditory phenotype correlation. Genet. Test. Mol. Biomarkers 18, 839–844. doi: 10.1089/gtmb.2014.0185
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677. doi: 10.1007/s00439-017-1779-6
Taitelbaum-Swead, R., Brownstein, Z., Muchnik, C., Kishon-Rabin, L., Kronenberg, J., Megirov, L., et al. (2006). Connexin-associated deafness and speech perception outcome of cochlear implantation. Arch. Otolaryngol. Head Neck Surg. 132, 495–500. doi: 10.1001/archotol.132.5.495
Takeuchi, S., Ando, M., and Kakigi, A. (2000). Mechanism generating endocochlear potential: role played by intermediate cells in stria vascularis. Biophys. J. 79, 2572–2582. doi: 10.1016/s0006-3495(00)76497-6
Taniguchi, M., Matsuo, H., Shimizu, S., Nakayama, A., Suzuki, K., Hamajima, N., et al. (2015). Carrier frequency of the GJB2 mutations that cause hereditary hearing loss in the Japanese population. J. Hum. Genet. 60, 613–617. doi: 10.1038/jhg.2015.82
Tayoun, A. N., Mason-Suares, H., Frisella, A. L., Bowser, M., Duffy, E., Mahanta, L., et al. (2016). Targeted droplet-digital PCR as a tool for novel deletion discovery at the DFNB1 locus. Hum. Mutat. 37, 119–126. doi: 10.1002/humu.22912
Teubner, B., Michel, V., Pesch, J., Lautermann, J., Cohen-Salmon, M., Söhl, G., et al. (2003). Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum. Mol. Genet. 12, 13–21. doi: 10.1093/hmg/ddg001
Thönnissen, E., Rabionet, R., Arbonès, M. L., Estivill, X., Willecke, K., and Ott, T. (2002). Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum. Genet. 111, 190–197. doi: 10.1007/s00439-002-0750-2
Todt, I., Hennies, H. C., Basta, D., and Ernst, A. (2005). Vestibular dysfunction of patients with mutations of connexin 26. Neuroreport 16, 1179–1181. doi: 10.1097/00001756-200508010-00009
Tritsch, N. X., and Bergles, D. E. (2010). Developmental regulation of spontaneous activity in the mammalian cochlea. J. Neurosci. 30, 1539–1550. doi: 10.1523/JNEUROSCI.3875-09.2010
Tritsch, N. X., Rodríguez-Contreras, A., Crins, T. T., Wang, H. C., Borst, J. G., and Bergles, D. E. (2010). Calcium action potentials in hair cells pattern auditory neuron activity before hearing onset. Nat. Neurosci. 13, 1050–1052. doi: 10.1038/nn.2604
Tsukada, K., Fukuoka, H., and Usami, S. (2015). Vestibular functions of hereditary hearing loss patients with GJB2 mutations. Audiol. Neurootol. 20, 147–152. doi: 10.1159/000368292
Tsukada, K., Nishio, S., Usami, S., and The Deafness Gene Study Consortium. (2010). A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin. Genet. 78, 464–470. doi: 10.1111/j.1399-0004.2010.01407.x
Tu, Z. J., and Kiang, D. T. (1998). Mapping and characterization of the basal promoter of the human connexin26 gene. Biochim. Biophys. Acta 1443, 169–181. doi: 10.1016/s0167-4781(98)00212-7
Uyguner, O., Emiroglu, M., Üzümcü, A., Hafiz, G., Ghanbari, A., Baserer, N., et al. (2003). Frequencies of gap- and tight-junction mutations in Turkish families with autosomal-recessive non-syndromic hearing loss. Clin. Genet. 64, 65–69. doi: 10.1034/j.1399-0004.2003.00101.x
van Geel, M., van Steensel, M. A., Küster, W., Hennies, H. C., Happle, R., Steijlen, P. M., et al. (2002). HID and KID syndromes are associated with the same connexin 26 mutation. Br. J. Dermatol. 146, 938–942. doi: 10.1046/j.1365-2133.2002.04893.x
van Steensel, M. A., van Geel, M., Nahuys, M., Smitt, J. H. S., and Steijlen, P. M. (2002). A novel connexin 26 mutation in a patient diagnosed with keratitis-ichthyosis-deafness syndrome. J. Invest. Dermatol. 118, 724–727. doi: 10.1046/j.1523-1747.2002.01735.x
Verselis, V. K., Ginter, C. S., and Bargiello, T. A. (1994). Opposite voltage gating polarities of two closely related connexins. Nature 368, 348–351. doi: 10.1038/368348a0
Wang, H. L., Chang, W. T., Li, A. H., Yeh, T. H., Wu, C. Y., Chen, M. S., et al. (2003). Functional analysis of connexin-26 mutants associated with hereditary recessive deafness. J. Neurochem. 84, 735–742. doi: 10.1046/j.1471-4159.2003.01555.x
Wang, H. C., Lin, C. C., Cheung, R., Zhang-Hooks, Y., Agarwal, A., Ellis-Davies, G., et al. (2015). Spontaneous activity of cochlear hair cells triggered by fluid secretion mechanism in adjacent support cells. Cell 163, 1348–1359. doi: 10.1016/j.cell.2015.10.070
Wangemann, P., Liu, J., and Marcus, D. C. (1995). Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro. Hear. Res. 84, 19–29. doi: 10.1016/0378-5955(95)00009-s
Wattanasirichaigoon, D., Limwongse, C., Jariengprasert, C., Yenchitsomanus, P. T., Tocharoenthanaphol, C., Thongnoppakhun, W., et al. (2004). High prevalence of V37I genetic variant in the connexin-26 (GJB2) gene among non-syndromic hearing-impaired and control Thai individuals. Clin. Genet. 66, 452–460. doi: 10.1111/j.1399-0004.2004.00325.x
Wilch, E., Azaiez, H., Fisher, R. A., Elfenbein, J., Murgia, A., Birkenhäger, R., et al. (2010). A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin. Genet. 78, 267–274. doi: 10.1111/j.1399-0004.2010.01387.x
Wilch, E., Zhu, M., Burkhart, K. B., Regier, M., Elfenbein, J. L., Fisher, R. A., et al. (2006). Expression of GJB2 and GJB6 is reduced in a novel DFNB1 allele. Am. J. Hum. Genet. 79, 174–179. doi: 10.1086/505333
Wu, B. L., Lindeman, N., Lip, V., Adams, A., Amato, R. S., Cox, G., et al. (2002). Effectiveness of sequencing connexin 26 (GJB2) in cases of familial or sporadic childhood deafness referred for molecular diagnostic testing. Genet. Med. 4, 279–288. doi: 10.1097/00125817-200207000-00006
Xiao, Z., Yang, Z., Liu, X., and Xie, D. (2011). Impaired membrane targeting and aberrant cellular localization of human Cx26 mutants associated with inherited recessive hearing loss. Acta Otolaryngol. 131, 59–66. doi: 10.3109/00016489.2010.506885
Zagólski, O. (2007). Vestibular system in infants with hereditary nonsyndromic deafness. Otol. Neurotol. 28, 1053–1055. doi: 10.1097/MAO.0b013e31815145e9
Zazo-Seco, C., Wesdorp, M., Feenstra, I., Pfundt, R., Hehir-Kwa, J. Y., Lelieveld, S. H., et al. (2017). The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur. J. Hum. Genet. 25, 308–314. doi: 10.1038/ejhg.2016.182
Zelante, L., Gasparini, P., Estivill, X., Melchionda, S., D’Agruma, L., Govea, N., et al. (1997). Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 6, 1605–1609. doi: 10.1093/hmg/6.9.1605
Zhang, J., Scherer, S. S., and Yum, S. W. (2011). Dominant Cx26 mutants associated with hearing loss have dominant-negative effects on wild type Cx26. Mol. Cell. Neurosci. 47, 71–78. doi: 10.1016/j.mcn.2010.10.002
Zhao, H. B. (2017). Hypothesis of K+-recycling defect is not a primary deafness mechanism for Cx26 (GJB2) deficiency. Front. Mol. Neurosci. 10:162. doi: 10.3389/fnmol.2017.00162
Zhao, F. F., Ji, Y. B., Wang, D. Y., Lan, L., Han, M. K., Li, Q., et al. (2011). Phenotype-genotype correlation in 295 Chinese deaf subjects with biallelic causative mutations in the GJB2 gene. Genet. Test. Mol. Biomarkers 15, 619–625. doi: 10.1089/gtmb.2010.0192
Zhao, H. B., Yu, N., and Fleming, C. R. (2005). Gap junctional hemichannel-mediated ATP release and hearing controls in the inner ear. Proc. Natl. Acad. Sci. U S A 102, 18724–18729. doi: 10.1073/pnas.0506481102
Zhu, Y., Liang, C., Chen, J., Zong, L., Chen, G. D., and Zhao, H. B. (2013). Active cochlear amplification is dependent on supporting cell gap junctions. Nat. Commun. 4:1786. doi: 10.1038/ncomms2806
Zhu, Y., Zong, L., Mei, L., and Zhao, H. B. (2015). Connexin26 gap junction mediates miRNA intercellular genetic communication in the cochlea and is required for inner ear development. Sci. Rep. 5:15647. doi: 10.1038/srep15647
Zong, L., Zhu, Y., Liang, R., and Zhao, H. B. (2016). Gap junction mediated miRNA intercellular transfer and gene regulation: a novel mechanism for intercellular genetic communication. Sci. Rep. 6:19884. doi: 10.1038/srep19884
Zonta, F., Buratto, D., Cassini, C., Bortolozzi, M., and Mammano, F. (2014a). Molecular dynamics simulations highlight structural and functional alterations in deafness-related M34T mutation of connexin 26. Front. Physiol. 5:85. doi: 10.3389/fphys.2014.00085
Zonta, F., Mammano, F., Torsello, M., Fortunati, N., Orian, L., and Polimeno, A. (2014b). Role of gamma carboxylated Glu47 in connexin 26 hemichannel regulation by extracellular Ca2+: insight from a local quantum chemistry study. Biochem. Biophys. Res. Commun. 445, 10–15. doi: 10.1016/j.bbrc.2014.01.063
Keywords: hearing impairment, inner ear, DFNB1, GJB2, connexin-26, GJB6, connexin-30
Citation: del Castillo FJ and del Castillo I (2017) DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 10:428. doi: 10.3389/fnmol.2017.00428
Received: 09 October 2017; Accepted: 07 December 2017;
Published: 22 December 2017.
Edited by:
Isabel Varela-Nieto, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Veronica Fuentes, Universidad de Castilla-La Mancha, SpainRégis Nouvian, INSERM U1051 Institut des Neurosciences de Montpellier, France
Copyright © 2017 del Castillo and del Castillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ignacio del Castillo, ignacio.castillo@salud.madrid.org