Altered balance of activity in the striatal direct and indirect pathways in mouse models of Huntington’s disease
- Intellectual and Developmental Disabilities Research Center, Department of Psychiatry and Biobehavioral Sciences, David Geffen School of Medicine, Semel Institute, University of California at Los Angeles, Los Angeles, CA, USA
Imbalance in the activity of striatal direct and indirect pathway neurons contributes to motor disturbances in several neurodegenerative diseases. In Huntington’s disease (HD), indirect pathway [dopamine (DA) D2 receptor-expressing] medium-sized spiny neurons (MSNs) are believed to show earlier vulnerability than direct pathway MSNs. We examined synaptic activity and DA modulation in MSNs forming the direct and indirect pathways in YAC128 and BACHD mouse models of HD. To visualize the two types of MSNs, we used mice expressing enhanced green fluorescent protein under the control of the promoter for the DA D1 or D2 receptor. Experiments were performed in early symptomatic (1.5 months) and symptomatic (12 months) mice. Behaviorally, early symptomatic mice showed increased stereotypies while symptomatic mice showed decreased motor activity. Electrophysiologically, at the early stage, excitatory and inhibitory transmission onto D1-YAC128 and D1-BACHD MSNs were increased, while there was no change in D2 MSNs. DA modulation of spontaneous excitatory postsynaptic currents (sEPSCs) in slices was absent in YAC128 cells at the early stage, but was restored by treating the slices with the DA depleter tetrabenazine (TBZ). In BACHD mice TBZ restored paired-pulse ratios and a D1 receptor antagonist induced a larger decrease of sEPSCs than in D1-WT cells, suggesting increased DA tone. Finally, TBZ decreased stereotypies in BACHD mice. These results indicate that by reducing DA or antagonizing D1 receptors, increases in inhibitory and excitatory transmission in early phenotypic direct pathway neurons can be normalized. In symptomatic YAC128 mice, excitatory synaptic transmission onto D1 MSNs was decreased, while inhibitory transmission was increased in D2 MSNs. These studies provide evidence for differential and complex imbalances in glutamate and GABA transmission, as well as in DA modulation, in direct and indirect pathway MSNs during HD progression.
Introduction
Huntington’s disease (HD) is caused by the mutation of a gene inducing expansion of CAG triplets on the huntingtin (htt) protein (The Huntington’s Disease Collaborative Research Group, 1993). In HD, the most obvious neuropathology is observed in the cortex and in the striatum. More specifically, striatal medium-sized spiny neurons (MSNs) expressing enkephalin and dopamine (DA) D2 receptors, forming the indirect pathway, are believed to be the most vulnerable and are preferentially lost in brains of presymptomatic and fully symptomatic patients. In contrast, MSNs expressing substance P and D1 receptors, forming the direct pathway, are believed to be relatively spared early in the progression of the disorder (Vonsattel et al., 1985; Albin et al., 1990).
The earlier vulnerability of indirect pathway MSNs in HD could induce imbalances in striatal output to other basal ganglia nuclei, which will affect the final outflow to the thalamus and contribute to the motor disturbances in the disease. Direct pathway MSNs preferentially project to the substantia nigra pars reticulata and to the internal segment of the globus pallidus (Gerfen, 1992; Bolam et al., 2000) and facilitate movement when activated (Chevalier et al., 1985a,b). In addition, direct pathway MSNs express high levels of DA D1 receptors that enhance synaptic activity when stimulated by DA. Indirect pathway neurons preferentially project to the external segment of the globus pallidus and inhibit movement when activated (Smith et al., 1998; Kravitz et al., 2010). They express D2 receptors that attenuate synaptic activity when stimulated by DA. Thus, direct and indirect pathways have opposite effects on movement and express DA receptors that have opposite effects on cell excitability.
Medium-sized spiny neurons are driven by two main glutamatergic inputs, those from cortex and thalamus. Intrastriatal inputs from GABAergic interneurons and axon collaterals from other MSNs provide feedforward and feedback inhibition, respectively. DA also exerts control of neurotransmitter release by directly activating presynaptic D2 receptors present on many terminals in the striatum or indirectly by interacting with other endogenous modulators (Malenka and Kocsis, 1988; Lovinger, 2008). In HD, glutamate and DA transmission are altered, which likely produces an imbalance in the activity of the direct and indirect pathways and contributes to the motor, cognitive, and psychiatric symptoms of HD.
Huntington’s disease is a neurodegenerative disease characterized by a progression of motor symptoms manifesting as chorea in early stages and as akinesia in later stages (Thompson et al., 1988). Thus, we hypothesized that alterations in direct and indirect pathway activity would be different at early and late stages of the disease. We examined the YAC128 (yeast artificial chromosome) mouse model of HD at an early symptomatic stage (1.5 months) and a symptomatic stage (12 months). YAC mice with 128 CAG repeats show a biphasic motor phenotype with hyperactivity at 3 months followed by hypoactivity in the open field at 12 months. Modest striatal atrophy occurs at 9 months and a small but significant decrease in striatal neurons is evident at 12 months (Slow et al., 2003). We also examined the BACHD (bacterial artificial chromosome) model at an early symptomatic stage (2 months). BACHD mice express human mutant HTT with 97 CAG repeats and exhibit progressive motor deficits starting at 2 months, followed by late-onset selective neuropathology at 12 months (Gray et al., 2008). BAC and YAC transgenics are used to permit expression of the full-length human mutant HTT, in order to reproduce more faithfully the human condition. BACHD and YAC128 mice develop a more protracted and progressive phenotype than the fragment models such as the R6/2, recapitulating the juvenile form of the disease (Mangiarini et al., 1996; Schilling et al., 1999). Full-length models of HD allow the investigation at an early stage of the disease, when symptoms start to develop while it is more difficult to separate time-dependent changes in fragment models showing rapid and aggressive progression of the disease. They also present more robust motor deficits and neuropathology than the knock-in models (Shelbourne et al., 1999; Wheeler et al., 2000). Although BAC and YAC transgenes allow the expression of large genomic sequence, they display a limited neuronal loss (10–15%) and do not recapitulate a shorter lifespan, as seen in the human condition (Tang et al., 2007).
To assess functional activity of specifically identified neurons, we crossed YAC128 or BACHD mice with mice expressing a reporter gene under the control of the promoter for either DA D1 or D2 receptors. In the double transgenic mice we examined excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs) and DA modulation in direct and indirect pathway MSNs. As tetrabenazine (TBZ) is the only drug providing significant benefit in the treatment of chorea associated with HD, we also tested the effect of this monoamine-depleting agent in vitro and in vivo. Portions of this study were published previously (André et al., 2011).
Materials and Methods
Animals
All experimental procedures were performed in accordance with the United States Public Health Service Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of California Los Angeles (UCLA). Every effort was made to minimize pain and discomfort. Experiments were conducted in two mouse models of adult-onset HD crossbred to heterozygous FVB/N mice expressing enhanced green fluorescent protein (EGFP) under the control of the promoter for dopamine receptor D1 or D2, obtained from our colony at UCLA. One mouse model carried full-length human mutant HTT, including 128 CAG repeats, on a yeast artificial chromosome (YAC128, line 53; Slow et al., 2003). YAC128 and WT littermates expressing D1- or D2-EGFP were obtained from our colonies at UCLA at 1.5 and 12 months of age. The second mouse model carried full-length human mutant HHT, including 97 CAG repeats, on a bacterial artificial chromosome (BACHD; Gray et al., 2008). BACHD and WT littermates expressing D1- or D2-EGFP at 2 months of age were also obtained from our colonies.
Cell Visualization
Enhanced green fluorescent protein-positive cells were visualized in slices using a 40× water immersion lens. The microscope (Olympus BX50WI or Zeiss Axioscope) was equipped with differential interference contrast optics and fluorescence. For infrared videomicroscopy, a halogen lamp and an infrared filter (790 nm, Ealing Optics, Holliston, MA, USA) were used. For fluorescence, ultra-violet light was filtered to obtain 480 nm excitation light. The light source consisted of a mercury lamp (100 W) and fluorescent light was detected with a video camera (QICAM-IR Fast 1394, Burnaby, BC, Canada) optimized to detect EGFP fluorescence and infrared light. Images were digitized and saved using the Q Capture Pro software (version 5, Burnaby, BC, Canada). Once a viable MSN was identified with infrared videomicroscopy, the filter was switched to fluorescence to determine if it was labeled with EGFP. The digitized infrared image was superimposed over the fluorescence image, and electrophysiological recordings proceeded only if the cell identified with infrared light showed a complete overlap with EGFP fluorescence.
Experiments in Slices
Detailed procedures have been published (Cepeda et al., 1993; André et al., 2010). Briefly, mice were deeply anesthetized with halothane and sacrificed. The brains were dissected and immediately placed in oxygenated ice-cold low-Ca2+ artificial cerebrospinal fluid (ACSF) containing (in millimolar); NaCl, 130; KCl, 3; NaH2PO4, 1.25; NaHCO3, 26; MgCl2, 5; CaCl2, 1; and glucose, 10. Coronal slices (350 μm) were cut and transferred to an incubating chamber containing ACSF (with 2 mM CaCl2 and 2 mM MgCl2) oxygenated with 95% 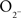 5% CO2 (pH 7.2–7.4, 290–310 mOsm, 25 ± 2°C). Slices were visualized and EGFP-expressing cells were selected for recordings. The patch pipette contained (in millimolar): Cs-methanesulfonate 130, CsCl 10, NaCl 4, MgCl2 1, MgATP 5, EGTA 5, HEPES 10, GTP 0.5, phosphocreatine 10, leupeptin 0.1 for voltage clamp recordings. Access resistances were <20 MΩ. Spontaneous postsynaptic currents were recorded in standard ACSF at room temperature. Membrane current was filtered at 1 kHz and digitized at 100–200 μs using Clampex 10.2 (gap-free mode). Cells were voltage clamped at −70 mV to determine basic membrane properties and to examine spontaneous glutamate receptor-mediated EPSCs. Bicuculline methiodide (Bic, 10 μM) was applied in order to block GABAA receptor-mediated currents. For IPSCs, voltage was held at +10 mV. For experiments examining the modulation of GABA receptor-mediated currents, AMPA and NMDA receptor blockers 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) and D-(−)-2-amino-5-phosphonopentanoic acid (APV), respectively were added. Baseline activity was recorded for 3–5 min after a 10- to 15-min baseline and another 5–10 min after addition of D1 or D2 receptor agonists (SKF81297 or quinpirole, 5–10 μM). To evoke synaptic currents, a monopolar glass stimulating electrode (patch pipette filled with ACSF, impedance 1.5–1.8 MΩ) was placed in the corpus callosum, 200–300 μm from the recorded cell.
5% CO2 (pH 7.2–7.4, 290–310 mOsm, 25 ± 2°C). Slices were visualized and EGFP-expressing cells were selected for recordings. The patch pipette contained (in millimolar): Cs-methanesulfonate 130, CsCl 10, NaCl 4, MgCl2 1, MgATP 5, EGTA 5, HEPES 10, GTP 0.5, phosphocreatine 10, leupeptin 0.1 for voltage clamp recordings. Access resistances were <20 MΩ. Spontaneous postsynaptic currents were recorded in standard ACSF at room temperature. Membrane current was filtered at 1 kHz and digitized at 100–200 μs using Clampex 10.2 (gap-free mode). Cells were voltage clamped at −70 mV to determine basic membrane properties and to examine spontaneous glutamate receptor-mediated EPSCs. Bicuculline methiodide (Bic, 10 μM) was applied in order to block GABAA receptor-mediated currents. For IPSCs, voltage was held at +10 mV. For experiments examining the modulation of GABA receptor-mediated currents, AMPA and NMDA receptor blockers 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) and D-(−)-2-amino-5-phosphonopentanoic acid (APV), respectively were added. Baseline activity was recorded for 3–5 min after a 10- to 15-min baseline and another 5–10 min after addition of D1 or D2 receptor agonists (SKF81297 or quinpirole, 5–10 μM). To evoke synaptic currents, a monopolar glass stimulating electrode (patch pipette filled with ACSF, impedance 1.5–1.8 MΩ) was placed in the corpus callosum, 200–300 μm from the recorded cell.
Test-stimuli (500 μs duration) were applied every 20 s and averaged over three consecutive trials. Stimulation intensities were set to evoke responses at 30% maximal amplitude. Paired-pulse ratios (PPRs) were used to assess probability of release, and paired-pulses were applied at intervals between 25 and 400 ms.
Drugs
Stocks of Bic (20 mM, Tocris), D1 receptor agonist SKF81297 (1 mM, Sigma), D2 receptor agonist quinpirole (10 mM, Sigma) were all dissolved in H2O. TBZ (10 mM, Tocris) was dissolved in 100% DMSO for stock solution and final concentration for electrophysiology was 0.1%. For in vivo injections, TBZ (1 mg/ml) was dissolved in a mixture of DMSO/NaCl (50/50). Procedures for determination of the appropriate TBZ dosage in vivo have been published (André et al., 2011).
Acutely Dissociated Neurons
Detailed procedures have been published (Flores-Hernandez et al., 2002). Briefly, coronal slices containing the dorsal striatum were dissected, placed in an oxygenated cell-stir chamber (Wheaton, Millville, NJ, USA) and enzymatically treated for 15–20 min with papain (0.5 mg/ml, Calbiochem) at 35°C in a N-[2-hydroxyethyl] piperazine-N-[2-ethanesulfonic acid] (HEPES)-buffered Hank’s balanced salts solution (HBSS, Sigma Chemical) supplemented with (in millimolar) 1 pyruvic acid, 0.005 glutathione, 0.1 NG-nitro-L-arginine, and 1 kynurenic acid. The tissue was rinsed with HEPES-buffered Na-isethionate solution containing (in millimolar) 140 Na isethionate, 2 KCl, 2 MgCl2, 0.1 CaCl2, 23 glucose, and 15 HEPES. Striatal slices were mechanically dissociated with fire-polished Pasteur pipettes. The cell suspension was then plated into a 35-mm Petri dish mounted on the stage of an upright fixed-stage microscope containing a HEPES-buffered salt solution. Only fluorescent cells were selected for recordings.
The internal solution consisted of (in millimolar) 175 N-methyl-D-glucamine (NMDG), 40 HEPES, 2 MgCl2, 10 ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetra-acetic acid (EGTA), 12 phosphocreatine, 2 Na2ATP, 0.2 Na2GTP, and 0.1 leupeptin (pH 7.25, 265–270 mOsm). The external solution consisted of (in millimolar) 135 NaCl, 20 CsCl, 3 BaCl2, 2 CaCl2, 10 glucose, 10 HEPES, and 0.0003 tetrodotoxin (TTX). Drugs were applied through a pressure-driven fast perfusion system using application capillaries positioned a few hundred micrometers from the cell. A DC drive system controlled by a SF-77B perfusion system (Warner Instruments, Hamden, CT, USA) synchronized by pClamp changed solutions by altering the position of the capillary array. NMDA currents (100 μM, 3 s duration every 10 s) were evoked by applying the agonist while holding the cell at −70 mV. D1 and D2 agonists were applied as previously described (André et al., 2010).
Data Analysis and Statistics
In the text, values are presented as mean ± SEM. Data analyses were performed with Origin (Microcal Software, Northampton, MA, USA) and pClamp 10.2. Group means for all measures were compared using Student’s t-tests (for two group comparisons) using SigmaStat software (SPSS, Chicago, IL, USA). Differences were considered statistically significant when p < 0.05. Spontaneous synaptic currents were analyzed offline using the automatic detection protocol within the Mini Analysis Program (Justin Lee, Synaptosoft, 1999) and subsequently checked manually for accuracy. Event analyses were done blind to genotype. The threshold amplitude for the detection of an excitatory event (5 pA) was set above the root mean square background noise level (1–2 pA at Vhold = −70 mV). For inhibitory events, the detection was set at 10 pA because the noise level was higher (2–3 pA at Vhold = −10 mV). Amplitude–frequency and cumulative inter-event interval distributions were constructed and compared among the different groups using two-way repeated measures (RM) ANOVAs to compare events at each amplitude and interval, followed by Bonferroni t-tests (multiple group comparisons) using SigmaStat software (SPSS, Chicago, IL, USA).
Results
Glutamate Transmission and GABA Transmissions in D1-YAC128 Cells
At 1.5 months, D1-YAC128 cells displayed a higher frequency of spontaneous excitatory postsynaptic currents (sEPSCs) than D1-WT cells (t38 = 2.65, p = 0.012) while at 12 months, this difference was reversed (t29 = 3.24, p = 0.003; Figures 1A,B). Cumulative inter-event interval histograms also showed a significantly increased probability of release onto D1-YAC128 cells at the early stage, while at 12 months, they showed a decreased probability of glutamate release onto D1-YAC128 cells (Figure 1C).
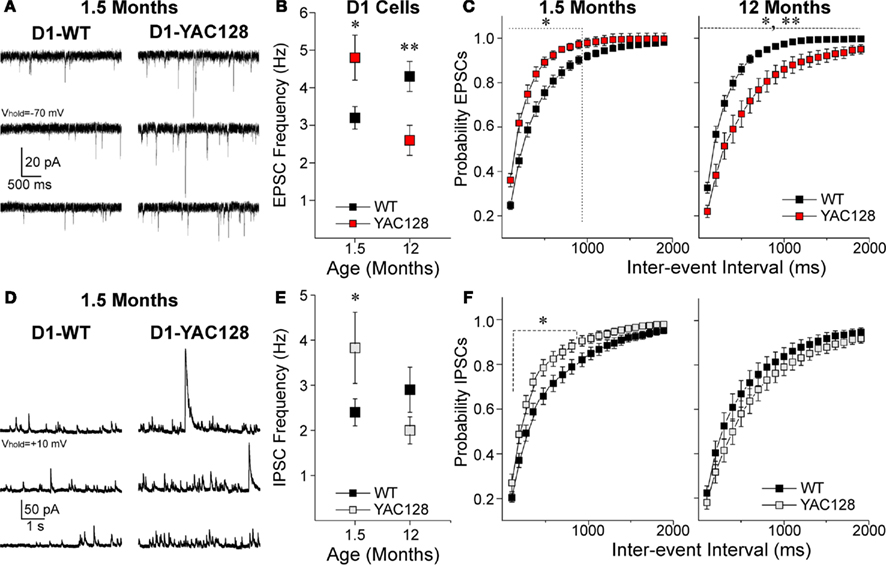
Figure 1. Alterations in excitatory and inhibitory transmission in D1-YAC128 cells. (A) Traces show sEPSCs in the presence of Bic in D1 cells from 1.5-month-old WT (left) and YAC128 (right) mice. (B) Graph shows that at 1.5 months, mean sEPSC frequency is higher in D1-YAC128 cells (n = 9) compared to D1-WT (n = 31) while at 12 months, it is lower in D1-YAC128 cells (n = 15) than in D1-WT cells (n = 16). (C) There was a leftward shift in the cumulative probability distribution of inter-event intervals for sEPSCs in D1-YAC128 cells at 1.5 months, while at 12 months there was a rightward shift D1-YAC128 cells. (D) Traces show sIPSCs in D1 cells from 1.5-month-old WT (left) and YAC128 (right) mice. (E) Graph shows that mean sIPSC frequency at 1.5 months is higher in D1-YAC128 (n = 20) than in D1-WT (n = 31) while at 12 months, it is similar in D1-WT (n = 17) and D1-YAC128 cells (n = 14). (F) There was a leftward shift in the cumulative probability distribution of inter-event intervals for sIPSCs at 1.5 months, while at 12 months the distributions of sIPSC are similar. *, **: significant difference between WT and YAC128 cells.
D1-YAC128 also displayed a higher frequency of sIPSCs at 1.5 months (t47 = 2.36, p = 0.022) while there was no difference at 12 months (t29 = 1.44, p = 0.161, Figures 1D,E). Cumulative inter-event interval histograms for IPSCs also showed a significantly increased probability of GABA release onto D1-YAC128 cells at 1.5 months but not at 12 months (Figure 1F).
Glutamate Transmission and GABA Transmission in D2-YAC128 Cells
In contrast to the D1 cells, D2-YAC128 cells did not show any significant differences in mean sEPSCs at 1.5 (t37 = 0.86, p = 0.395) or at 12 months (t31 = 1.083, p = 0.287, Figures 2A,B). Cumulative inter-event interval histograms did not show any differences at 1.5 months but at 12 months, the significant rightward shift suggests a decrease of glutamate release in late stage D2-YAC128 cells (Figure 2C).
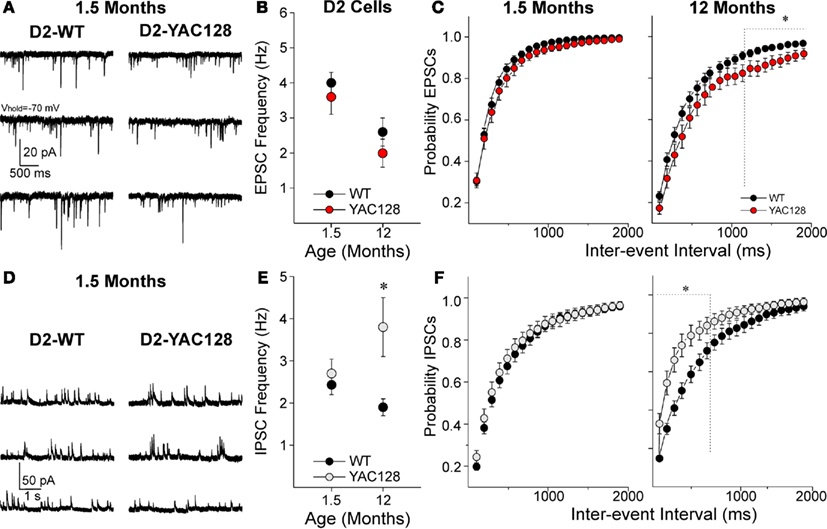
Figure 2. Alterations in excitatory and inhibitory transmission in D2-YAC128 cells. (A) Traces show sEPSCs in D2 cells from 1.5-month-old WT (left) and YAC128 (right) mice. (B) Graph shows there are no significant differences in mean EPSC frequency between D2-YAC128 (n = 25 at 1.5 and n = 13 at 12 months) and D2-WT (n = 14 at 1.5 and n = 20 at 12 months) cells at any age. (C) Cumulative probability distributions of inter-event interval histograms for sEPSCs show that D2-WT and D2-YAC128 cells display similar distributions at 1.5 months, while at 12 months D2-YAC128 cells display significantly fewer events at the longer intervals only. (D) Traces show sIPSCs in D2 cells from 1.5-month-old WT (left) and YAC128 (right) mice. (E) Graph shows that mean sIPSC frequency in D2 cells is similar in D2-WT (n = 26) and D2-YAC128 cells (n = 16) at 1.5 months while at 12 months, IPSC frequency is higher in D2-YAC128 (n = 14) than in D2-WT cells (n = 15). (F) Cumulative probability distributions of inter-event intervals for sIPSCs show that D2-YAC128 cells display more events at 12 months but not at 1.5 months. *: significant difference between WT and YAC128 cells.
In 1.5 month D2 cells, there was no difference in the mean IPSC frequency (t40 = 0.693, p = 0.492) or the inter-event interval histogram (Figures 2D–F). However, at 12 months, IPSC frequency was increased (t27 = 2.668, p = 0.013) in the D2-YAC128 cells and the cumulative inter-event interval histogram suggested increased probability of GABA release onto D2-YAC128 cells (Figures 2E,F).
Glutamate Transmission and GABA Transmission are Increased in D1 Cells in BACHD Mice at 2 Months
Similar to D1-YAC128 cells, D1-BACHD cells displayed a higher sEPSC frequency at 2 months (t18 = 2.781, p = 0.012, Figure 3A). To provide further evidence that the differences in EPSC frequencies were due to altered glutamate release, we examined responses evoked by stimulation of corticostriatal afferents (eEPSCs) and measured PPRs (André et al., 2011). At 2 months, D1-BACHD cells displayed a significant decrease in PPRs (two-way RM ANOVA F1,89 = 15.6, p = 0.001) at 25 (t = 3.8, p < 0.001) and 50 (t = 3.57, p < 0.001) ms (Figures 3B,C) suggesting an increased probability of glutamate release onto cells of the direct pathway in BACHD mice at an early stage. D2-BACHD cells did not show any difference in PPRs compared to their WT littermates (two-way RM ANOVA F1,83 = 0.19, p = 0.662, Figure 3C), again similar to that finding in D2-YAC128 cells at the early stage.
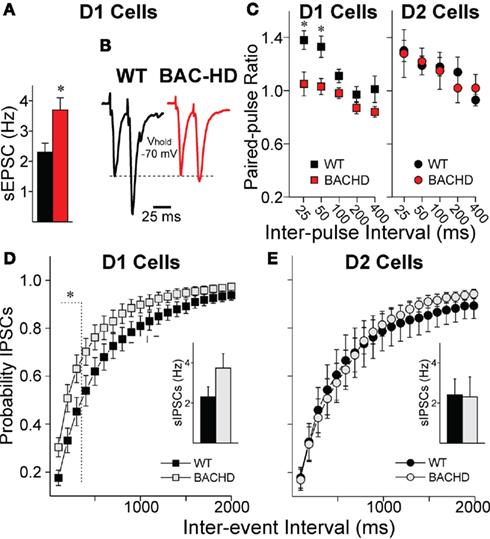
Figure 3. Glutamate and GABA transmission in BACHD mice at 2 months. (A) Mean sEPSC frequency is increased in D1-BACHD cells (n = 11) compared to D1-WT cells (n = 9). (B) Traces of two evoked glutamate responses at 25 ms interval. (C) PPRs were significantly decreased in D1-BACHD cells (n = 10) compared to D1-WT cells (n = 8) at 2 months (25, 50, and 100 ms intervals), indicating increased probability of glutamate release. In D2 cells, PPRs were similar in WT (n = 11) and BACHD mice (n = 6). (D,E) Cumulative probability distributions of inter-event intervals for sIPSCs show that D1-BACHD (n = 14) cells display more events at shorter intervals than D1-WT cells (n = 11) at 1.5 months, while D2 cells show no differences in sIPSC distribution (WT, n = 6; BACHD, n = 6). Insets in (D,E) show mean sIPSC frequencies *: significant difference between WT and BACHD.
At this early stage, we found similar increases in frequency of sIPSCs in D1-BACHD as we showed in D1-YAC128 cells with a significant leftward shift in cumulative probability histograms in the D1-BACHD cells suggesting increased probability of GABA release (Figure 3D). However, as the changes in mean sIPSC frequency did not reach significance in D1-BACHD compared to D1-WT cells (t23 = 1.736, p = 0.096, Figure 3D inset), the increase of GABA release might not be of the same amplitude in the BACHD mice as it was in the YAC128 mice, at least at the early stage. D2-BACHD cells did not show any differences in sIPSC mean frequency or in cumulative inter-event interval distribution (Figure 3E).
D1 and D2 Receptors are Still Functional in YAC128
As EGFP was visible in YAC128 and WT cells and no difference in the intensity of fluorescence was observed (Figure 4A), we hypothesized that DA D1 and D2 receptors are still expressed in the YAC128 mice. To examine whether D1 and D2 receptors are transported to the surface and functional, we tested the effects of D1 and D2 agonists on postsynaptic NMDA currents in acutely dissociated D1 and D2 MSNs at 1.5 months. It is well known that D1 receptor activation increases NMDA currents while D2 receptor activation decreases them (Cepeda et al., 1993; André et al., 2010). The D1 agonist (SKF81297, 1 μM) increased NMDA currents in D1 cells similarly in WT (paired t-test, t8 = 6.953, p < 0.001) and YAC128 (t9 = 2.888, p = 0.018) cells (Figure 4B). The D2 agonist (quinpirole, 10 μM) similarly decreased NMDA current amplitudes in WT (t4 = 3.290, p = 0.03) and YAC128 (t4 = 4.103, p = 0.015) cells (Figure 4C).
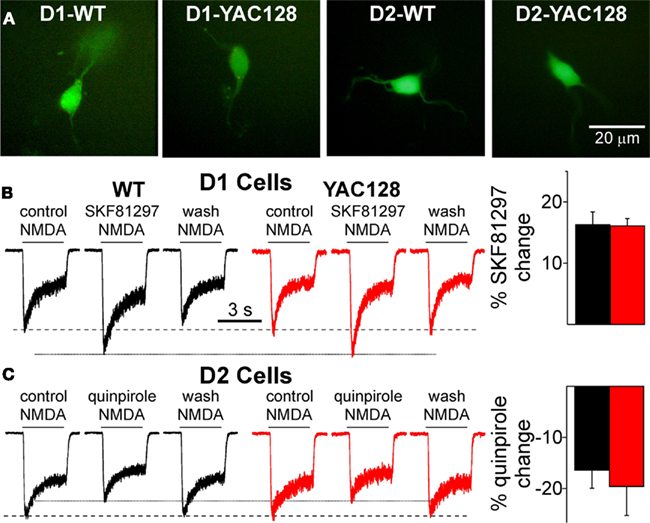
Figure 4. D1 and D2 receptor modulation in acutely dissociated MSNs is similar in WT and YAC128 cells at 1. 5 months. (A) Examples of acutely dissociated MSNs in WT and YAC128. (B) Traces of NMDA currents (100 μM) in D1 cells show the D1 agonist SKF81297 (1 μM) increases the amplitude of NMDA currents in D1-WT (n = 9) and D1-YAC128 cells (n = 10). Graph shows the percent potentiation by SKF81297 is similar in D1-WT and D1-YAC128 cells. (C) Traces of NMDA currents (100 μM) in D2 cells show the D2 agonist quinpirole (10 μM) decreases NMDA currents in D2-WT (n = 5) and D2-YAC128 cells (n = 5). Graph shows the percent attenuation by quinpirole is similar in D2-WT and D2-YAC128 cells.
Loss of D1 Modulation is Reversed by DA Blockers
We examined DA modulation in slices by testing the effect of the D1 agonist SKF81297 on sEPSC frequency at 1.5 months. While in D1-WT cells, SKF81297 increased sEPSC frequency (paired t-test: t20 = 5.867, p < 0.001), the D1 agonist did not have any effect on D1-YAC128 cells (t7 = 0.607, p = 0.563; Figures 5A,B). By contrast, at a later stage, the D1 agonist effect was similar in D1-WT and D1-YAC128 cells (André et al., 2011). This early alteration in DA neurotransmission might be induced by abnormal DA concentrations as we show that DA receptors are functional (Figure 4). Previous studies showed that elevated striatal DA concentrations decrease DA receptor function (Giros et al., 1996; Dumartin et al., 2000; Wu et al., 2007). In addition, decreasing DA concentrations with TBZ alleviates chorea in HD patients (Huntington Study Group, 2006). TBZ is a monoamine depleter, but it ultimately depletes DA stores (Pettibone et al., 1984), suggesting that decreasing DA might be beneficial for hyperkinetic symptoms. To test whether DA function could be rescued in early HD, we examined the effect of TBZ and D1 antagonists in early HD.
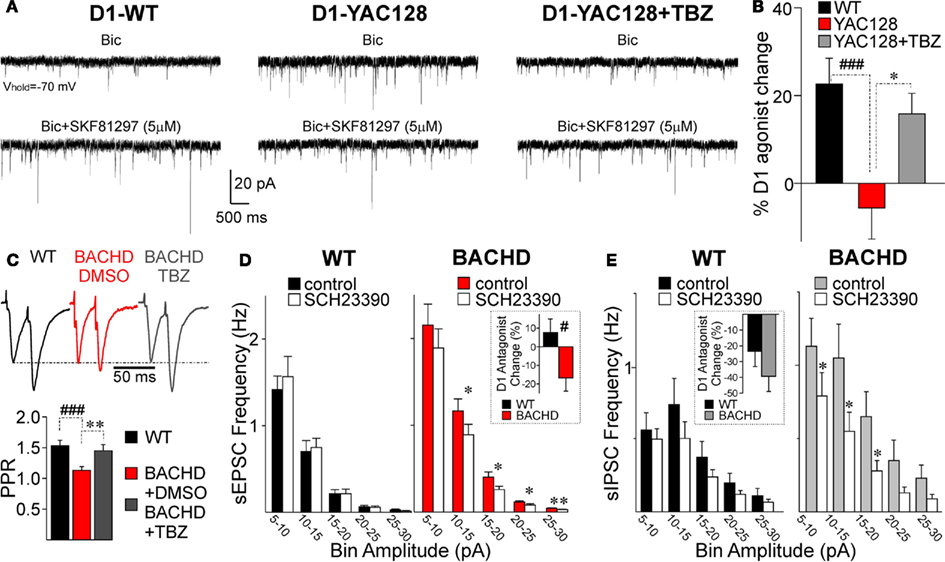
Figure 5. Alterations in DA modulation in early HD. (A) Traces of sEPSCs in a D1-WT MSN (left), a D1-YAC128 MSN (middle) and a TBZ-treated D1-YAC128 MSN before and after application of the D1 agonist SKF81297 at 1.5 months. (B) Bar graph shows that the D1 agonist increased sEPSC frequency in D1-WT cells (n = 12) while it had no effect in D1-YAC128 cells (n = 10). The D1 agonist effect on sEPSC frequency was restored by TBZ treatment in D1-YAC128 cells (n = 8). (C) Top panel shows traces of paired-pulse evoked glutamate currents in D1-BACHD cells at 2 months. Bottom panel graph shows the decrease in PPRs in DMSO-treated D1-BACHD cells (n = 17) is increased in TBZ-treated D1-BACHD cells (n = 8). (D) Amplitude–frequency histogram shows that in D1-WT cells, the D1 antagonist SCH23390 (20 μM) had no effect on sEPSC frequency. In contrast, in D1-BACHD cells (2 months), the D1 antagonist decreased sEPSC frequency. Inset shows that the percent change induced by SCH23390 is significantly different in D1-WT (n = 9) and D1-BACHD cells (n = 11). (E) Amplitude–frequency histogram shows that in D1-WT cells (n = 8), the D1 antagonist SCH23390 (10 μM) did not significantly decrease the frequency of sIPSCs. In D1-BACHD cells (n = 6), the D1 antagonist decreased the frequency of sIPSCs. Inset shows that although the percent change induced by SCH23390 tended to be higher in D1-BACHD cells, it was not significantly different from that in D1-WT cells. *p < 0.05, **p < 0.01, effect of TBZ or SCH23390 compared to control; #p < 0.05 and ###p < 0.001, difference between WT and YAC128 or BACHD cells.
In slices incubated in TBZ (10 μM) for at least 2 h, SKF81297 significantly increased sEPSC frequency in D1-YAC128 cells (t8 = 2.672, p = 0.028), and its effect was similar to that of the D1 agonist in D1-WT cells (Figures 5A,B). In D1-BACHD cells, pre-incubation in TBZ also rescued the decrease in PPRs (Figure 5C). As TBZ was dissolved in DMSO, we tested the effect of DMSO alone on PPR in D1-BACHD cells. In the BACHD cells, DMSO had no effect on PPRs, indicating that the rescue effect was induced by TBZ. PPRs from TBZ-incubated slices in D1-BACHD cells were similar to those of D1-WT cells. To examine whether alterations in glutamate and GABA transmission could be induced by abnormally elevated activation of D1 receptors, we tested the effect of the D1 receptor antagonist SCH23390 (10 μM) on sEPSC and sIPSC frequency in D1-BACHD cells. SCH23390 was added after a stable baseline of sEPSCs or sIPSCs was established (in different cells), and the change in frequency was measured after 6–10 min of incubation. In D1-WT cells, SCH23390 (10 and 20 μM) did not have a significant effect on sEPSC frequency. In contrast 20 μM SCH23390 significantly decreased sEPSC frequency in D1-BACHD cells (t10 = 2.416, p = 0.036). The D1 antagonist effect was significantly different between WT and BACHD cells (t18 = 2.858, p = 0.01; Figure 5D, inset). SCH23390 (10 μM) did not significantly decrease sIPSC frequency in D1-WT cells although there was a trend (Figure 5E). In D1-BACHD cells, SCH23390 decreased sIPSC frequency by −36.3 ± 11.5% (t5 = 3.027, p = 0.029). However, the difference between D1-WT and D1-BACHD cells was not statistically different (Figure 5E, inset).
Discussion
Visualization of direct and indirect pathway MSNs by EGFP expression allowed us to differentiate the two types of MSNs and to uncover alterations in the two pathways that differed with the progression of the disease. The use of mice expressing the reporter protein EGFP through a BAC transgene has proved to alter the expression of the D2 receptor, and induce electrophysiological hypersensitivity to D2 receptor agonists in midbrain DA neurons, as well as hyperactivity, suggesting the need for better tools to visualize D2 receptor-expressing neurons (Kramer et al., 2011). This characterization was mainly performed on homozygote BAC transgenic mice, and heterozygote mice might display a milder phenotype although the study also found some behavioral changes in heterozygote mice. In our study, we used heterozygote mice and we excluded all D2-EGFP mice for behavioral studies. Although we did not extensively compare EGFP-positive and negative MSNs, we found that the effect of the D2 agonist was decreased in HD mice, and that in early HD, most alterations were present in direct pathway MSNs, validating the use of EGFP to compare WT and HD mice.
Glutamate Transmission and GABA Transmission are Increased onto Direct Pathway Neurons at the Early Stage of HD
In both YAC128 and BACHD animal models, we observed an increase in sEPSC and in sIPSC frequency in D1 cells at an early symptomatic age. In addition, there was a decrease in PPRs for evoked glutamate responses (Table 1). Low PPRs at short intervals indicate depletion of neurotransmitter from the presynaptic terminal and an increased probability of release of the immediately releasable store (Zucker, 1989; Choi and Lovinger, 1997). There were also increased frequencies of miniature EPSCs but no change in event amplitude (André et al., 2011). These results suggest that glutamate release is increased onto direct pathway MSNs. Although we did not perform paired-pulse stimulation to measure PPRs for IPSCs in this particular study, our data on sIPSCs show that the frequency of GABA-mediated events is also higher onto direct pathway MSNs in YAC128 and BACHD mice. Furthermore, we have shown that PPRs of evoked IPSCs are decreased in MSNs in the R6/2 mouse, providing further evidence for the hypothesis of increased GABA release in HD (Rao et al., 2010). Taken together, our results suggest that both glutamate and GABA release are increased on direct pathway MSNs. Previously we demonstrated that the frequency of sIPSCs increases in HD (Cepeda et al., 2004). The present study indicates that the increase is specific to D1-expressing neurons in the early stage.
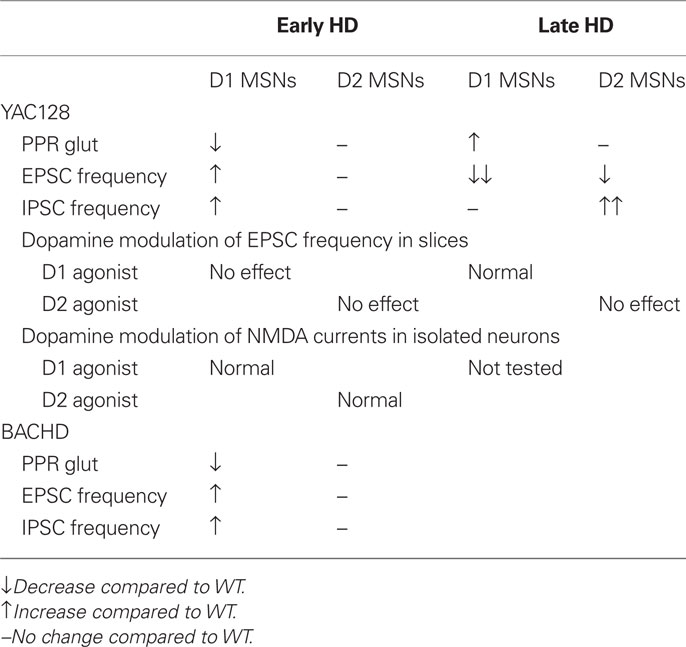
Table 1. Summary of changes in spontaneous excitatory and inhibitory currents and dopamine modulation in D1 and D2 cells in early and late stage HD in the YAC128 and BACHD models.
D1 and D2 receptors are functionally active in acutely dissociated YAC128 cells while D1 and D2 receptors were not responsive in slices. The dissociated preparation isolates neurons from synaptic inputs and from ambient DA, while in slices neurons have active synapses and are exposed to modulators. As striatal neurotransmitter release is modulated by DA receptors located on presynaptic terminals and postsynaptically on the two types of MSNs, we hypothesized that abnormal DA transmission in slices could underlie differential alterations of excitatory and inhibitory transmission onto direct and indirect pathway neurons. Our data show that blocking DA function with a D1 antagonist had larger effects in D1-BACHD cells than in WT cells and that depleting DA with TBZ decreased stereotypies in the BACHD model (André et al., 2011). These findings suggest that at an early stage, DA tone or basal function is increased and might contribute to some of the symptoms in HD by altering neuronal activity in the direct pathway. Our results are in line with observations that DA receptor antagonists and agents that decrease DA reduce chorea and motor symptoms in patients and in animal models while dopaminergic stimulation can exacerbate symptoms (Huntington Study Group, 2006; Tang et al., 2007; Mestre et al., 2009). It is unclear why we did not observe any changes in GABA or glutamate transmission in D2 cells at the early stage. As DA induces reduction of neurotransmitter release onto D2 cells via activation of pre and postsynaptic receptors (Kreitzer and Malenka, 2007; André et al., 2010), a decrease of neurotransmitter release could be expected in D2 cells at an early stage. However, in another study, an increased facilitation of evoked EPSCs was reported in 26-day-old YAC128. The authors suggested this could be interpreted as a decrease in the probability of glutamate release in indirect pathway MSNs (Milnerwood and Raymond, 2007). We could have missed those alterations onto indirect pathway MSNs since we performed our early stage recordings in slightly older mice between 45 and 60 days.
It is also unclear why changes in DA transmission would affect glutamate and GABA synaptic activity in D1 MSNs while having no effects in D2 MSNs, especially if considering that the proportion of high-affinity D2 receptors is much greater than the proportion of high-affinity D1 receptors in the striatum (Richfield et al., 1989). It is important to consider that DA influence also depends on the number of receptors within a sphere of DA spillover (Rice and Cragg, 2008). Although no study evaluated the number of high- and low-affinity DA receptors, it was reported that D1 receptors are in considerable excess of D2 receptors in the striatum (Richfield et al., 1989; Benn et al., 2007). It is therefore possible that in cases of abnormal DA transmission, or DA spillover, D1 receptors are more affected than D2 receptors. Other factors such as differential glutamate cortical and GABAergic striatal inputs formed onto D1 and D2 MSNs (Lei et al., 2004; Kreitzer and Malenka, 2006; Cepeda et al., 2008; Gittis et al., 2010) could also contribute to prominent changes in D1 MSNs in early HD.
Glutamate Transmission is Decreased onto All MSNs at a Late Stage while GABA Transmission is Increased onto Indirect Pathway MSNs
The fact that glutamate transmission is decreased onto both direct and indirect pathway MSNs is consistent with a progressive disconnection between cortex and striatum (Cepeda et al., 2003). However, this effect was more marked onto direct pathway MSNs, showing both decreased sEPSC frequency and increased PPRs, while indirect pathway MSNs showed only a subtle decrease of sEPSCs (Figure 6). In contrast to glutamate transmission, GABA transmission was increased onto indirect pathway MSNs, suggesting that unlike glutamate terminals, GABA terminals might not disconnect in the YAC128 model. In the R6/2 model, we have provided evidence that the increased GABA transmission emanates from GABAergic interneurons that may be hyperexcitable (Rao et al., 2010). The fact that increased inhibition was observed only onto indirect pathway MSNs indicates that cannabinoid signaling might be involved. Cannabinoid type 1 (CB1) receptors are located on striatal GABAergic terminals, inhibiting GABA release when activated (Szabo et al., 1998; Kofalvi et al., 2005). Endogenous ligands for the cannabinoid receptors are released by MSNs via activation of DA D2 receptors located on D2 receptor-expressing MSNs (Giuffrida et al., 1999; Patel et al., 2003). In late HD, a lack of DA or D2 receptors in indirect pathway MSNs could therefore increase GABA release onto indirect pathway MSN via a lack of endocannabinoid release. This effect will not occur in D1 cells, not expressing D2 receptors.
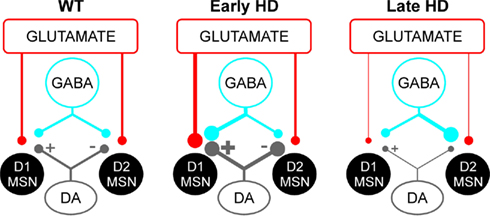
Figure 6. Representation of glutamate, GABA, and DA projections onto direct (D1) and indirect pathway (D2) MSNs in HD. In WT (left), DA released by nigrostriatal inputs activates glutamate and GABA release onto D1 MSNs while it decreases glutamate and GABA release onto D2 MSNs. In early HD (middle), increased DA transmission leads to increased release of glutamate and GABA onto D1 MSNs. There is no change onto D2 MSNs, suggesting changes in indirect pathway MSNs might occur at a different time point or involve other mechanisms. In late HD (right), D1 MSNs display decreases in glutamate transmission, presumably due to loss of corticostriatal inputs and/or decreased DA transmission. In contrast, D2 MSNs display only a small decrease in glutamate synaptic inputs while GABA synaptic transmission is increased, probably through altered D2 receptor function.
Decreased DA tone or DA function could also contribute to hyperexcitability of interneurons and increased GABA release (Dehorter et al., 2009). Our data showing a lack of effect of a D2 antagonist in D2-YAC128 cells (André et al., 2011) support the hypothesis that DA function is decreased in late HD. In this case, one would also expect an increase of sIPSCs onto direct pathway MSNs, except if direct and indirect pathway MSNs are connected by different types of interneurons as suggested by a recent study (Gittis et al., 2010). In the case of abnormally low DA function, an increase in glutamate release would also be expected since DA D2 receptors are also present on presynaptic glutamate terminals and act to decrease neurotransmitter release when activated by DA (Wang and Pickel, 2002; Bamford et al., 2004). However, sEPSC frequency was not increased in the indirect pathway MSNs, and was even slightly decreased. This might be explained by the loss of corticostriatal inputs at this late stage of the disease.
Functional Consequences of Alterations in Direct and Indirect Pathway MSNs
Increased probability of glutamate release onto D1 cells will shift the balance of direct and indirect pathway activity, disinhibiting output structures such as the colliculus and the thalamus, and increasing movement (Chevalier and Deniau, 1990). Thus, stereotypies observed in young BACHD and YAC128 mice might be induced by overactivation of D1 cells as has been observed in cases of elevated DA transmission (Usiello et al., 2000; Horvitz et al., 2001). Although hypoactivity in symptomatic HD animals has been reported, there is evidence that hyperactivity and stereotyped behaviors occur in lesion models in rats (Borlongan et al., 1997; Brouillet et al., 1999; Bolivar et al., 2004) and in early stage in transgenic mouse models carrying a short fragment or the full length of the human gene in different strains (Sanberg et al., 1989; Carter et al., 1999; Bolivar et al., 2004; Van Raamsdonk et al., 2005, 2007). Abnormal involuntary movements, dyskinesia and chorea are hallmarks of human pathology at early stages and the hyperkinetic syndrome is progressively replaced by a more hypokinetic syndrome (Thompson et al., 1988). In contrast, a decrease in the activity of direct pathway MSNs could explain the decreased locomotor activity observed in the late symptomatic YAC128 mice.
The resulting effect of increased GABA release could also alter direct and indirect pathway activity. The effect of GABAA receptor activation on striatal cell excitability depends on the cell membrane potential as the reversal potential for Cl− is ~−60 mV and the resting membrane potential for MSNs is hyperpolarized (~ −80 to −90 mV). In vivo, MSN membrane potentials oscillate between hyperpolarized “down” and more depolarized “up” states (Wilson and Kawaguchi, 1996). Therefore, activation of GABAA receptors depolarizes MSN membrane at rest while it hyperpolarizes membrane potential near the action potential threshold (Mercuri et al., 1991; Koos and Tepper, 1999). Although our results in current-clamp show there are no changes in membrane excitability in YAC128 cells at the early stage (André et al., 2011), more data are needed to determine the consequences of increased GABA on MSN activity.
Conclusion
The present findings suggest anomalies in glutamate and GABA transmission in HD might be induced by abnormal DA transmission. Early in the progression of the disease, increased glutamate release onto D1 cells and increased stereotypies are consistent with increased DA tone (Presti et al., 2004). In symptomatic animals, decreased glutamate release onto D1 cells and hypoactive behavior are consistent with decreased DA tone and/or loss of corticostriatal terminals. Our data suggest that modulation of direct pathway neurons with D1 antagonists or agents decreasing DA function in early HD stage might prove useful as a strategy to treat hyperkinetic motor symptoms. In contrast, DA agonists at more advanced stages might prove useful as a strategy to treat hypokinetic motor symptoms. In the future, studies are necessary to understand the consequences of altered GABA and glutamate transmissions onto basal ganglia output neurons in HD to help direct new therapies and understand the mechanisms underlying the phenotypic changes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health Grant NS41574, CHDI, and The Hereditary Disease Foundation.
References
Albin, R. L., Reiner, A., Anderson, K. D., Penney, J. B., and Young, A. B. (1990). Striatal and nigral neuron subpopulations in rigid Huntington’s disease: implications for the functional anatomy of chorea and rigidity-akinesia. Ann. Neurol. 27, 357–365.
André, V. M., Cepeda, C., Cummings, D. M., Jocoy, E. L., Fisher, Y. E., Yang, X. W., and Levine, M. S. (2010). Dopamine modulation of excitatory currents in the striatum is dictated by the expression of D1 or D2 receptors and modified by endocannabinoids. Eur. J. Neurosci. 31, 14–28.
André, V. M., Cepeda, C., Fisher, Y. E., Huynh, M., Bardakjian, N., Singh, S., Yang, X. W., and Levine, M. S. (2011). Differential electrophysiological changes in striatal output neurons in Huntington’s disease. J. Neurosci. 31, 1170–1182.
Bamford, N. S., Robinson, S., Palmiter, R. D., Joyce, J. A., Moore, C., and Meshul, C. K. (2004). Dopamine modulates release from corticostriatal terminals. J. Neurosci. 24, 9541–9552.
Benn, C. L., Slow, E. J., Farrell, L. A., Graham, R., Deng, Y., Hayden, M. R., and Cha, J. H. (2007). Glutamate receptor abnormalities in the YAC128 transgenic mouse model of Huntington’s disease. Neuroscience 147, 354–372.
Bolam, J. P., Hanley, J. J., Booth, P. A., and Bevan, M. D. (2000). Synaptic organisation of the basal ganglia. J. Anat. 196(Pt 4), 527–542.
Bolivar, V. J., Manley, K., and Messer, A. (2004). Early exploratory behavior abnormalities in R6/1 Huntington’s disease transgenic mice. Brain Res. 1005, 29–35.
Borlongan, C. V., Koutouzis, T. K., Freeman, T. B., Hauser, R. A., Cahill, D. W., and Sanberg, P. R. (1997). Hyperactivity and hypoactivity in a rat model of Huntington’s disease: the systemic 3-nitropropionic acid model. Brain Res. Brain Res. Protoc. 1, 253–257.
Brouillet, E., Conde, F., Beal, M. F., and Hantraye, P. (1999). Replicating Huntington’s disease phenotype in experimental animals. Prog. Neurobiol. 59, 427–468.
Carter, R. J., Lione, L. A., Humby, T., Mangiarini, L., Mahal, A., Bates, G. P., Dunnett, S. B., and Morton, A. J. (1999). Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J. Neurosci. 19, 3248–3257.
Cepeda, C., André, V. M., Yamazaki, I., Wu, N., Kleiman-Weiner, M., and Levine, M. S. (2008). Differential electrophysiological properties of dopamine D1 and D2 receptor-containing striatal medium-sized spiny neurons. Eur. J. Neurosci. 27, 671–682.
Cepeda, C., Buchwald, N. A., and Levine, M. S. (1993). Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc. Natl. Acad. Sci. U.S.A. 90, 9576–9580.
Cepeda, C., Hurst, R. S., Calvert, C. R., Hernández-Echeagaray, E., Nguyen, O. K., Jocoy, E., Christian, L. J., Ariano, M. A., and Levine, M. S. (2003). Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J. Neurosci. 23, 961–969.
Cepeda, C., Starling, A. J., Wu, N., Nguyen, O. K., Uzgil, B., Soda, T., André, V. M., Ariano, M. A., and Levine, M. S. (2004). Increased GABAergic function in mouse models of Huntington’s disease: reversal by BDNF. J. Neurosci. Res. 78, 855–867.
Chevalier, G., and Deniau, J. M. (1990). Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci. 13, 277–280.
Chevalier, G., Vacher, S., Deniau, J. M., and Desban, M. (1985a). Disinhibition as a basic process in the expression of striatal functions. I. The striato-nigral influence on tecto-spinal/tecto-diencephalic neurons. Brain Res. 334, 215–226.
Chevalier, G., Vacher, S., Deniau, J. M., and Desban, M. (1985b). Disinhibition as a basic process in the expression of striatal functions. I. The striato-nigral influence on tecto-spinal/tecto-diencephalic neurons. Brain Res. 334, 215–226.
Choi, S., and Lovinger, D. M. (1997). Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc. Natl. Acad. Sci. U.S.A. 94, 2665–2670.
Dehorter, N., Guigoni, C., Lopez, C., Hirsch, J., Eusebio, A., Ben-Ari, Y., and Hammond, C. (2009). Dopamine-deprived striatal GABAergic interneurons burst and generate repetitive gigantic IPSCs in medium spiny neurons. J. Neurosci. 29, 7776–7787.
Dumartin, B., Jaber, M., Gonon, F., Caron, M. G., Giros, B., and Bloch, B. (2000). Dopamine tone regulates D1 receptor trafficking and delivery in striatal neurons in dopamine transporter-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 97, 1879–1884.
Flores-Hernandez, J., Cepeda, C., Hernandez-Echeagaray, E., Calvert, C. R., Jokel, E. S., Fienberg, A. A., Greengard, P., and Levine, M. S. (2002). Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J. Neurophysiol. 88, 3010–3020.
Gerfen, C. R. (1992). The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 15, 133–139.
Giros, B., Jaber, M., Jones, S. R., Wightman, R. M., and Caron, M. G. (1996). Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379, 606–612.
Gittis, A. H., Nelson, A. B., Thwin, M. T., Palop, J. J., and Kreitzer, A. C. (2010). Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J. Neurosci. 30, 2223–2234.
Giuffrida, A., Parsons, L. H., Kerr, T. M., Rodriguez de Fonseca, F., Navarro, M., and Piomelli, D. (1999). Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat. Neurosci. 2, 358–363.
Gray, M., Shirasaki, D. I., Cepeda, C., Andre, V. M., Wilburn, B., Lu, X. H., Tao, J., Yamazaki, I., Li, S. H., Sun, Y. E., Li, X. J., Levine, M. S., and Yang, X. W. (2008). Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 28, 6182–6195.
Horvitz, J. C., Williams, G., and Joy, R. (2001). Time-dependent actions of D2 family agonist quinpirole on spontaneous behavior in the rat: dissociation between sniffing and locomotion. Psychopharmacology (Berl.) 154, 350–355.
Huntington Study Group. (2006). Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology 66, 366–372.
Kofalvi, A., Rodrigues, R. J., Ledent, C., Mackie, K., Vizi, E. S., Cunha, R. A., and Sperlagh, B. (2005). Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: a combined immunochemical and pharmacological analysis. J. Neurosci. 25, 2874–2884.
Koos, T., and Tepper, J. M. (1999). Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat. Neurosci. 2, 467–472.
Kramer, P. F., Christensen, C. H., Hazelwood, L. A., Dobi, A., Bock, R., Sibley, D. R., Mateo, Y., and Alvarez, V. A. (2011). Dopamine D2 receptor overexpression alters behavior and physiology in Drd2-EGFP mice. J. Neurosci. 31, 126–132.
Kravitz, A. V., Freeze, B. S., Parker, P. R., Kay, K., Thwin, M. T., Deisseroth, K., and Kreitzer, A. C. (2010). Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626.
Kreitzer, A. C., and Malenka, R. C. (2006). Distinct function and plasticity at striatal direct and indirect pathway synapses. Abstr. Soc. Neurosci. 732.20.
Kreitzer, A. C., and Malenka, R. C. (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647.
Lei, W., Jiao, Y., Del Mar, N., and Reiner, A. (2004). Evidence for differential cortical input to direct pathway versus indirect pathway striatal projection neurons in rats. J. Neurosci. 24, 8289–8299.
Lovinger, D. M. (2008). “Presynaptic modulation by endocannabinoids,” in Handbook of Experimental Pharmacology, eds T. C. Sudhoh and K. Starke (Berlin: Springer-Verlag) 435–477.
Malenka, R. C., and Kocsis, J. D. (1988). Presynaptic actions of carbachol and adenosine on corticostriatal synaptic transmission studied in vitro. J. Neurosci. 8, 3750–3756.
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S. W., and Bates, G. P. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506.
Mercuri, N. B., Calabresi, P., Stefani, A., Stratta, F., and Bernardi, G. (1991). GABA depolarizes neurons in the rat striatum: an in vivo study. Synapse 8, 38–40.
Mestre, T., Ferreira, J., Coelho, M. M., Rosa, M., and Sampaio, C. (2009). Therapeutic interventions for symptomatic treatment in Huntington’s disease. Cochrane Database Syst. Rev. 3, CD006456.
Milnerwood, A. J., and Raymond, L. A. (2007). Corticostriatal synaptic function in mouse models of Huntington’s disease: early effects of huntingtin repeat length and protein load. J. Physiol. (Lond.) 585, 817–831.
Patel, S., Rademacher, D. J., and Hillard, C. J. (2003). Differential regulation of the endocannabinoids anandamide and 2-arachidonylglycerol within the limbic forebrain by dopamine receptor activity. J. Pharmacol. Exp. Ther. 306, 880–888.
Pettibone, D. J., Totaro, J. A., and Pflueger, A. B. (1984). Tetrabenazine-induced depletion of brain monoamines: characterization and interaction with selected antidepressants. Eur. J. Pharmacol. 102, 425–430.
Presti, M. F., Gibney, B. C., and Lewis, M. H. (2004). Effects of intrastriatal administration of selective dopaminergic ligands on spontaneous stereotypy in mice. Physiol. Behav. 80, 433–439.
Rao, S. R., André, V. M., Cepeda, C., and Levine, M. S. (2010). Altered inhibitory inputs onto striatal medium-sized spiny neurons in the R6/2 mouse model of Huntington’s disease. Abstr. Soc. Neurosci. 861.16.
Rice, M. E., and Cragg, S. J. (2008). Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain Res. Rev. 58, 303–313.
Richfield, E. K., Penney, J. B., and Young, A. B. (1989). Anatomical and affinity state comparisons between dopamine D1 and D2 receptors in the rat central nervous system. Neuroscience 30, 767–777.
Sanberg, P. R., Calderon, S. F., Giordano, M., Tew, J. M., and Norman, A. B. (1989). The quinolinic acid model of Huntington’s disease: locomotor abnormalities. Exp. Neurol. 105, 45–53.
Schilling, G., Becher, M. W., Sharp, A. H., Jinnah, H. A., Duan, K., Kotzuk, J. A., Slunt, H. H., Ratovitski, T., Cooper, J. K., Jenkins, N. A., Copeland, N. G., Price, D. L., Ross, C. A., and Borchelt, D. R. (1999). Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 8, 397–407.
Shelbourne, P. F., Killeen, N., Hevner, R. F., Johnston, H. M., Tecott, L., Lewandoski, M., Ennis, M., Ramirez, L., Li, Z., Iannicola, C., Littman, D. R., and Myers, R. M. (1999). A Huntington’s disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum. Mol. Genet. 8, 763–774.
Slow, E. J., van Raamsdonk, J., Rogers, D., Coleman, S. H., Graham, R. K., Deng, Y., Oh, R., Bissada, N., Hossain, S. M., Yang, Y. Z., Li, X. J., Simpson, E. M., Gutekunst, C. A., Leavitt, B. R., and Hayden, M. R. (2003). Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 12, 1555–1567.
Smith, Y., Bevan, M. D., Shink, E., and Bolam, J. P. (1998). Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 86, 353–387.
Szabo, B., Dorner, L., Pfreundtner, C., Norenberg, W., and Starke, K. (1998). Inhibition of GABAergic inhibitory postsynaptic currents by cannabinoids in rat corpus striatum. Neuroscience 85, 395–403.
Tang, T. S., Chen, X., Liu, J., and Bezprozvanny, I. (2007). Dopaminergic signaling and striatal neurodegeneration in Huntington’s disease. J. Neurosci. 27, 7899–7910.
The Huntington’s Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983.
Thompson, P. D., Berardelli, A., Rothwell, J. C., Day, B. L., Dick, J. P., Benecke, R., and Marsden, C. D. (1988). The coexistence of bradykinesia and chorea in Huntington’s disease and its implications for theories of basal ganglia control of movement. Brain 111(Pt 2), 223–244.
Usiello, A., Baik, J. H., Rouge-Pont, F., Picetti, R., Dierich, A., LeMeur, M., Piazza, P. V., and Borrelli, E. (2000). Distinct functions of the two isoforms of dopamine D2 receptors. Nature 408, 199–203.
Van Raamsdonk, J. M., Metzler, M., Slow, E., Pearson, J., Schwab, C., Carroll, J., Graham, R. K., Leavitt, B. R., and Hayden, M. R. (2007). Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol. Dis. 26, 189–200.
Van Raamsdonk, J. M., Pearson, J., Rogers, D. A., Bissada, N., Vogl, A. W., Hayden, M. R., and Leavitt, B. R. (2005). Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 14, 1379–1392.
Vonsattel, J. P., Myers, R. H., Stevens, T. J., Ferrante, R. J., Bird, E. D., and Richardson, E. P. Jr. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577.
Wang, H., and Pickel, V. M. (2002). Dopamine D2 receptors are present in prefrontal cortical afferents and their targets in patches of the rat caudate-putamen nucleus. J. Comp. Neurol. 442, 392–404.
Wheeler, V. C., White, J. K., Gutekunst, C. A., Vrbanac, V., Weaver, M., Li, X. J., Li, S. H., Yi, H., Vonsattel, J. P., Gusella, J. F., Hersch, S., Auerbach, W., Joyner, A. L., and MacDonald, M. E. (2000). Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet. 9, 503–513.
Wilson, C. J., and Kawaguchi, Y. (1996). The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J. Neurosci. 16, 2397–2410.
Wu, N., Cepeda, C., Zhuang, X., and Levine, M. S. (2007). Altered corticostriatal neurotransmission and modulation in dopamine transporter knock-down mice. J. Neurophysiol. 98, 423–432.
Keywords: dopamine, electrophysiology, synaptic activity, glutamate, GABA, postsynaptic currents, Huntington’s disease
Citation: André VM, Fisher YE and Levine MS (2011) Altered balance of activity in the striatal direct and indirect pathways in mouse models of Huntington’s disease. Front. Syst. Neurosci. 5:46. doi: 10.3389/fnsys.2011.00046
Received: 25 February 2011; Paper pending published: 20 April 2011;
Accepted: 03 June 2011; Published online: 16 June 2011.
Edited by:
Charles J. Wilson, University of Texas at San Antonio, USACopyright: © 2011 André, Fisher and Levine. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Véronique M. André, David Geffen School of Medicine, Semel Institute for Neuroscience and Human Behavior, University of California at Los Angeles, 760 Westwood Plaza, NPI 58-258A, Los Angeles, CA 90095, USA. e-mail: vandre@mednet.ucla.edu