
- Department of Physiology and Biochemistry, Faculty of Medicine and Surgery, University of Malta, Msida, Malta
Yeasts, such as Saccharomyces cerevisiae, have long served as useful models for the study of oxidative stress, an event associated with cell death and severe human pathologies. This review will discuss oxidative stress in yeast, in terms of sources of reactive oxygen species (ROS), their molecular targets, and the metabolic responses elicited by cellular ROS accumulation. Responses of yeast to accumulated ROS include upregulation of antioxidants mediated by complex transcriptional changes, activation of pro-survival pathways such as mitophagy, and programmed cell death (PCD) which, apart from apoptosis, includes pathways such as autophagy and necrosis, a form of cell death long considered accidental and uncoordinated. The role of ROS in yeast aging will also be discussed.
Introduction
It is a well known fact that the majority of living organisms depend on oxygen for survival. However, organisms also had to evolve a multitude of antioxidant defenses to protect their cells from oxygen’s toxic properties (Bilinski, 1991; Halliwell, 1999), which stem mainly from its propensity to produce reactive oxygen species (ROS) such as the superoxide radical hydrogen peroxide (H2O2), and the hydroxyl radical These ROS typically arise because of electron leakage from the electron transport chain onto dioxygen (O2) during aerobic respiration (Boveris et al., 1972; Turrens and Boveris, 1980; Turrens, 1997; Halliwell and Gutteridge, 2007). Exposure to heavy metals (Liang and Zhou, 2007; Xu et al., 2011), ultraviolet (UV) irradiation, herbicides, air pollutants, xenobiotics, and other exogenous factors can also induce significant generation of ROS (Halliwell and Cross, 1994; Gille and Sigler, 1995). Failure of cell antioxidant defenses to impede ROS accumulation inevitably results in oxidative stress, a condition broadly defined as an imbalance between prooxidants and antioxidants, in favor of the former. This potentially leads to a situation where important cell biomolecules suffer severe oxidative damage, thus compromising the viability of cells (Sies, 1991; Halliwell and Cross, 1994; Halliwell and Gutteridge, 2007).
In fact, accumulated ROS have been shown to inflict oxidative damage upon essential biomolecules such as nucleic acids (Yakes and Van Houten, 1997), proteins (Cabiscol et al., 2000), and lipids (Bilinski et al., 1989). Furthermore, ROS accumulation has long been found to play an important role in mediating programmed cell death (PCD) such as apoptosis, or even – at very high concentrations – necrosis, in various cell types (Pierce et al., 1991; Kasahara et al., 1997; Madeo et al., 1999; Chandra et al., 2000; Simon et al., 2000; Ludovico et al., 2001; Jeon et al., 2002; Avery, 2011). More significantly, oxidative damage and cell death induced by ROS have been linked to many serious human pathologies including diabetes (Giugliano et al., 1996; Baynes and Thorpe, 1999; Yokozawa et al., 2011) and neurodegenerative diseases such as Parkinson’s disease (Hirsch, 1993; Jenner and Olanow, 1996; Jenner, 2003; Facecchia et al., 2011), Alzheimer’s disease (Behl, 1999; Nunomura et al., 2001; Reddy et al., 2009), and amyotrophic lateral sclerosis (ALS; Andrus et al., 1998; Barber et al., 2006; Barber and Shaw, 2010). ROS have also been implicated in the aging process (Harman, 1956; Orr and Sohal, 1994; Barja, 2004; Fabrizio et al., 2004; Herker et al., 2004) and are known to play a pivotal role in the development of cancer (Ames et al., 1993, 1995; Loft and Poulson, 1996; Naka et al., 2008; Khandrika et al., 2009; Acharya et al., 2010).
Yeast cells have steadily evolved into one of the most preferred experimental models for the study of oxidative stress and its effects, in the context of PCD and aging. Yeast species such as the budding yeast Saccharomyces cerevisiae and the fission yeast Schizosaccharomyces pombe, enjoy several advantages over other experimental models. For instance, they have a short life-cycle and are easy to manipulate genetically – much like bacteria, whilst still retaining the core cellular processes that are characteristic of eukaryotes (Carmona-Gutierrez et al., 2010a). Most importantly, past studies have shown that, amongst these cellular processes, core elements of PCD, such as apoptosis, are also conserved in yeast (Madeo et al., 1997, 1999, 2002; Fröhlich and Madeo, 2001; Fahrenkrog et al., 2004; Wissing et al., 2004; Büttner et al., 2007) and that accumulation of ROS plays a key role in these pathways (Madeo et al., 1999; Jeon et al., 2002). Hence, yeast can make a significant contribution to our understanding of oxidative stress, and its consequences such as PCD and the misregulation of PCD in the context of pathologies such as cancer, neurodegenerative diseases, and aging (Carmona-Gutierrez et al., 2010a).
This review aims to present a broad overview of current knowledge of oxidative stress and its role in yeast PCD, with emphasis on S. cerevisiae. The role of oxidative stress and ROS in yeast aging will also be discussed.
Oxygen Toxicity and Oxidative Stress in Yeast
The accumulation of ROS, in yeast, generally stems from internal metabolic processes associated with respiration, but can also be triggered by environmental stress stimuli (Jamieson, 1998; Perrone et al., 2008).
Reactive Oxygen Species Generation in Yeast
Mitochondria have long been established as a major source of ROS (Longo et al., 1996) such as the superoxide radical which is generated from O2 by electron leakage originating in the mitochondrial transport chain, during respiration. (Fridovich, 1998; Cadenas and Davies, 2000). In yeast species such as S. cerevisiae, the specific sources of in the mitochondrial chain include the external NADH dehydrogenases Nde1p and Nde2p (the active sites of which face the mitochondrial intermembrane space) and complex III (Fang and Beattie, 2003).
Although superoxide can directly inactivate certain proteins such as catalases (Kono and Fridovich, 1983; Fridovich, 1989) and dehydratases (Murakami and Yoshino, 1997), it is a relatively selective and unreactive ROS (Fridovich, 1998; Halliwell and Gutteridge, 2007). Nevertheless, the prompt and efficient removal of from living cells is of great biological importance, given its tendency to generate H2O2 and subsequent, more harmful ROS (Fridovich, 1989; Gille and Sigler, 1995). In fact, studies have shown that detoxification mechanisms such as the superoxide dismutase enzymes (SODs), are essential for survival of S. cerevisiae against hyperoxia (Outten et al., 2005). Recently has also been implicated as a signaling molecule in yeast cell processes such as chronological aging (Weinberger et al., 2010; Lewinska et al., 2011).
Superoxide radical dismutation, which mainly occurs via enzymatic SOD activity, is a major source of H2O2 in vivo (McCord and Fridovich, 1969). In yeast, H2O2 is also generated by D-amino acid oxidases, peroxisomal acyl-coenzyme A oxidases (Halliwell and Cross, 1994; Herrero et al., 2008), and protein folding events in the endoplasmic reticulum (ER; Gross et al., 2006). Like H2O2 is relatively unreactive (Gille and Sigler, 1995; Halliwell and Gutteridge, 2007) but can also travel long distances and penetrate biological membranes, allowing it to exert oxidative damage in locations far from its point of origin (Saran and Bors, 1991). It is also a signaling molecule in mammals (Sundaresan et al., 1995), plants (Vergara et al., 2012), and yeast (Bienert et al., 2006; Bartosz, 2009). In fact, H2O2 signaling activates transcription factors that regulate antioxidant gene expression in yeast (Wemmie et al., 1997; Delaunay et al., 2000, 2002; Kuge et al., 2001). This is believed to take place primarily via the direct oxidation and concomitant functional alteration of redox-sensitive thiol peroxidases (Fomenko et al., 2011). Furthermore, pre-treatment with 150 μM H2O2 enhances the sensitivity of budding yeast cells to heat stress, suggesting that ROS (presumably H2O2) relay signals induced by heat stress to yeast heat shock transcription factors that initiate the heat shock response (Moraitis and Curran, 2004).
Both and H2O2 can combine to form the hydroxyl radical via the Fenton and Haber–Weiss reactions catalyzed by free metal cations such as those of iron (Fe; Haber and Weiss, 1934). Ferrous Fe2+ ions are oxidized to ferric Fe3+ ions by H2O2 to produce These Fe3+ ions can be reduced again by resulting in a reaction cycle capable of generating an infinite supply of
The radical is an exceedingly powerful oxidant which indiscriminately oxidizes cell biomolecules at a diffusion-limited rate (Fridovich, 1989, 1998). Biomolecules oxidized by can become radicals themselves, which propagate even further non-specific cell oxidative damage (Evans et al., 1998). In fact, most oxidative damage in cells is mediated by , which is far more toxic than its precursors and H2O2, the impact of which mainly lies in their propensity to form (Halliwell and Cross, 1994).
The nitric oxide radical is another important free radical species associated with oxidative damage in organisms. Upon reacting with it produces peroxynitrite (ONOO−), a strongly oxidizing reactive nitrogen species (RNS) which can generate further radicals such as (Beckman et al., 1990) and induce oxidation of proteins and nucleic acids (Radi, 2004; Poyton et al., 2009). The radical has also been linked to increased ROS generation and cell death in S. cerevisiae, where endogenous generated by synthase-like activity (Osório et al., 2007), was found to induce ROS accumulation and apoptosis in yeast cells treated with H2O2 (Almeida et al., 2007). Importantly, this was also accompanied by S-nitrosation of the glycolytic glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Almeida et al., 2007). Given that S-nitrosation, defined as the covalent attachment of to the cysteine thiol groups of target proteins, is a mechanism by which regulates varied cellular processes in organisms (Hess et al., 2005), these observations also exemplify the role of radicals as signaling molecules (Almeida et al., 2007).
Under normoxic conditions, cells can very efficiently prevent ROS accumulation and attenuate oxidative damage, using various defensive strategies and antioxidants. However, environmental conditions can undergo frequent changes, such as progressive depletion of nutrients, increases in ambient temperature, or sudden xenobiotic contamination. Regardless of their nature, such environmental changes invariably cause an element of stress to organisms, to which they must effectively adapt in order to survive. This stress is frequently associated with ROS (Avery, 2011), which either deplete functional antioxidants or induce further ROS accumulation, or both (Costa and Moradas-Ferreira, 2001).
For instance, UVA radiation (Kozmin et al., 2005) and cadmium (Brennan and Schiestl, 1996) cause oxidative damage or outright deletion of genes in yeast cells. Deletion or impairment of genes associated with antioxidant enzymes such as manganese superoxide dismutase (MnSOD) and copper-zinc superoxide dismutase (CuZnSOD) causes increased mitochondrial protein oxidation (O’Brien et al., 2004) and loss of viability of respiring stationary phase cells (Longo et al., 1996). Also, prooxidant xenobiotics, including aminotriazole (AMT) and diethyldithiocarbamate (DDC), directly inactivate catalase (Bayliak et al., 2008) and CuZnSOD (Lushchak et al., 2005), respectively, sensitizing yeast to oxidative stress (Lushchak, 2010). Redox-cycling xenobiotics, such as paraquat and menadione, stimulate superoxide production in yeast by accepting electrons from cellular reducers and transferring them to oxygen (Lushchak, 2010). Other environmental stimuli such as heat stress (Davidson et al., 1996), ethanol-induced diauxic shift (Drakulic et al., 2005), H2O2 (Madeo et al., 1999), acetic acid (Ludovico et al., 2001), cadmium (Brennan and Schiestl, 1996), and arsenic (Du et al., 2007) amongst several other agents, can all induce ROS accumulation in yeast.
Molecular Targets of ROS in Yeast
The accumulation of cellular ROS inevitably results in oxidative damage to important cell biomolecules such as proteins, DNA, and lipids. Failure to curb such damage is ultimately associated with cell death (Costa and Moradas-Ferreira, 2001). The following sections will briefly outline some of the most prominent molecular targets of ROS in yeast.
Oxidative damage of proteins
Oxidative damage of proteins adversely influences cell homeostatic functions and rapidly compromises cell viability. In fact, accumulation of oxidized proteins has been associated with aging and several ROS-related diseases such as ALS and Alzheimer’s disease. All ROS, including lipid peroxides, can oxidize proteins via several different pathways, the net result of which is usually the formation of protein carbonyls. This is also accelerated by free metal cations as a result of Fenton chemistry (Berlett and Stadtman, 1997).
Protein carbonyls are irreversible products and thus susceptible to proteolytic degradation (Wolff and Dean, 1986). Prompt removal of protein carbonyls is important, given their tendency to form large protein aggregates which cannot then be degraded by cells via normal proteolytic pathways. Failure to effectively degrade such aggregates will only enhance further carbonyl accumulation and eventually disrupt cell homeostasis altogether (Cecarini et al., 2007).
Thiol (–SH) groups of cysteine, methionine, and aromatic phenylalanine amino acid residues are particularly sensitive to oxidation by all ROS (Berlett and Stadtman, 1997). Thiol groups can be transiently oxidized to disulfide groups (S–S) and sulfenic acid groups (–SOH), or irreversibly oxidized to sulfinic (–SO2H) or sulfonic (–SO3H) acid groups (Klatt and Lamas, 2000).
Additionally, ROS can cause dysfunctional protein synthesis, such as by mistranslation of mRNA. For instance, in S. cerevisiae, oxygen-dependent chromate ion Cr (VI)-induced mistranslation of mRNA was found to cause accumulation of insoluble and toxic protein carbonyl aggregates composed of improperly synthesized, inactive proteins. This is considered the primary means of Cr (VI) toxicity in yeast (Holland et al., 2007).
Otherwise, major yeast protein targets include mitochondrial citric acid cycle proteins, such as α-ketoglutarate dehydrogenase, pyruvate dehydrogenase, and the iron-sulfur [4Fe-4S] cluster enzymes aconitase and succinate dehydrogenase, all of which are readily inactivated by H2O2 and by menadione-derived superoxide radicals (Cabiscol et al., 2000; Cecarini et al., 2007). The superoxide-mediated oxidation of labile [4Fe-4S] clusters in dehydratase enzymes, such as aconitase, also releases free Fe2+ ions that participate in Fenton chemistry, promoting further protein oxidation (Fridovich, 1998). Aconitase in yeast is also inactivated by -induced oxidative stress (Lushchak and Lushchak, 2008). Important enzymes such as GAPDH (Cabiscol et al., 2000) and CuZnSOD (Costa et al., 2002) are also easily inactivated via H2O2-induced carbonylation, which is largely mediated by metal ion-catalyzed formation of (Costa and Moradas-Ferreira, 2001).
The yeast actin cytoskeleton is another important target of ROS. The recessive loss-of-function mutation of an ASC1 sequence in yeast induces hyper-assembly of actin filaments and consequent cytoskeletal morphological defects, whilst further increasing actin’s sensitivity to oxidative stress (Haarer and Amberg, 2010). The physiological consequences of actin oxidation in yeast include accelerated aging and apoptotic cell death (Farah and Amberg, 2007). Similarly, the decline of actin dynamics and subsequent cytoskeletal stabilization in yeast, induced by the disruption of actin regulatory proteins Sla1p and End3p, leads to ROS accumulation and cell death, mediated by hyperactivation of the Ras/cAMP kinase pathway (Gourlay and Ayscough, 2006).
Oxidative damage of DNA
Nucleic acids can suffer various forms of mutational damage caused by ROS. These include single or double strand breaks, base modification, abasic sites, and protein-DNA cross-linkage (Henle and Linn, 1997; Finn et al., 2011). Oxidatively induced mutations such as these, which compromise DNA integrity and functionality, have long been implicated in the development of pathologies such as cancer (Ames et al., 1995) and in aging (Burhans and Weinberger, 2012). Oxidized DNA also interferes with the normal response to environmental oxidative stress in yeast (Salmon et al., 2004), causing ROS accumulation and PCD in both S. cerevisiae and S. pombe (Burhans et al., 2003). Oxidative DNA damage and chromosome fragmentation are in fact frequently detected in ROS-mediated killing of yeast exposed to acetic acid (Ludovico et al., 2002), H2O2 (Madeo et al., 1999), and hyperosmotic stresses (Ribeiro et al., 2006).
The ROS which plays a dominant role in oxidizing DNA is Singlet oxygen (1O2) also oxidizes guanine bases (Halliwell and Cross, 1994) into 7,8-dihydro-8-oxoguanine, a well-characterized marker of oxidative DNA damage (Jenner, 1994; Daroui et al., 2004). Other prooxidants or ROS can only oxidize DNA via these two species (Halliwell and Aruoma, 1991). This is true for stresses such as ionizing radiation (Frankenberg et al., 1993), UVA radiation (Kozmin et al., 2005), heavy metals such as cadmium (IV) (Jin et al., 2003) and prooxidants that produce H2O2 such as mitomycin and paraquat (Brennan et al., 1994) or such as menadione (Lee and Park, 1998) – these induce damages such as strand breakages, oxidation of bases, and intrachromosomal recombination in the DNA of S. cerevisiae cells, primarily via formation of
Oxidative damage of lipids
Oxidative damage of lipid molecules generally involves lipid peroxidation, an autocatalytic process initiated by the oxidation of polyunsaturated fatty acids (PUFAs) into labile lipid hydroperoxides, by radicals. Lipid hydroperoxides propagate the synthesis of further hydroperoxides and other reactive derivatives, all of which can inflict extensive oxidative damage to cell biomolecules (Halliwell and Gutteridge, 2007).
Yeast cells are incapable of synthesizing PUFAs, but will readily incorporate them into their membrane structures if cultured in PUFA-enriched growth medium, thus raising the risk of lipid peroxidation on exposure to oxidative stress (Bilinski et al., 1989). In fact, yeast cells with PUFA-enriched yeast membranes, showed hypersensitivity to cadmium toxicity, which was mediated by lipid peroxidation (Howlett and Avery, 1997). Expression of human Bax in yeast cells containing PUFA-enriched mitochondrial membranes also caused pronounced lipid peroxidation (Priault et al., 2002). Lipid peroxidation was also reported in yeast cells undergoing rapamycin-induced autophagy (Kissová et al., 2006), and oxidative stress induced by H2O2 (Reekmans et al., 2005) and menadione (Kim et al., 2011).
Pro-Survival Cellular Responses to Oxidative Stress in Yeast
Yeast cells express limited pools of antioxidants which sufficiently protect against ROS. However, these constitutive defenses cannot protect the cells from sudden oxidative insults. Therefore, yeast has had to evolve the ability to sense aggressive stress stimuli and to generate rapid adaptive responses to increased ROS accumulation. The onset of oxidative stress in yeast cells generally induces an early response, where pre-existing antioxidant defenses provide immediate protection against the initial sub-lethal accumulation of ROS. There is also early transmission of stress signals and consequent activation of transcription factors which promote the synthesis of further antioxidant defenses (Gasch et al., 2000; Temple et al., 2005). This leads to the late response involving synthesis and activation of new antioxidant defenses that scavenge ROS, repair oxidized biomolecules, and restore cellular redox balance (Costa and Moradas-Ferreira, 2001).
Yeast Transcription Factors and Their Role in Yeast Oxidative Stress Responses
The adaptive response mechanisms to oxidative stress in S. cerevisiae are regulated at the transcriptional level (Collinson and Dawes, 1992; Jamieson et al., 1994) mainly by the transcription factors Yap1p, Skn7p, Msn2p, and Msn4p (Morgan et al., 1997; Takeuchi et al., 1997; Lee et al., 1999; Gasch et al., 2000). These transcription factors collectively coordinate appropriate responses to different oxidative stressors, by repressing or upregulating the transcription of specific genes, many of which are associated with antioxidant defenses (Gasch et al., 2000; Temple et al., 2005).
Transcription factor Yap1p
The yeast activator protein 1 (AP-1), Yap1p, is a basic leucine zipper domain (b/ZIP) type transcription factor that bears great similarity to mammalian AP-1 type transcription factors such as c-Jun, in terms of sequence structure and DNA-binding affinity. It generally binds to a consensus Yap1p recognition element (YRE; 5′-TT/GAC/GTC/AA-3′) contained in the promoter sequences of several genes (Fernandes et al., 1997) and plays a key role in the cellular response of S. cerevisiae yeast cells to oxidative stress and xenobiotic insults, particularly drugs and heavy metals (Wemmie et al., 1994; Kolaczkowska and Goffeau, 1999; Gasch et al., 2000). In fact, Yap1p-deficient (Δyap1) mutants exhibit hypersensitivity and an impaired adaptive response to oxidants such as H2O2 (Stephen et al., 1995), which is accompanied by reduced activities of antioxidants such as Γ-L-glutamyl-L-cysteinylglycine or glutathione (GSH), SODs, and GSH reductase (Schnell et al., 1992).
Activation of Yap1p in response to oxidants, particularly H2O2, has been well studied (Wemmie et al., 1997; Delaunay et al., 2000, 2002; Kuge et al., 2001). Under normoxic conditions, most of the cell’s Yap1p molecules are retained in the cytosol by the nuclear export activity of chromosome regional maintenance protein 1 (Crm1p), which is bound to a nuclear export signal (NES) in the C-terminal cysteine-rich domain (CRD) of Yap1p (Kuge et al., 1998). These redox-sensitive domains are highly conserved among Yap1p and its homolog proteins, and are essential for Crm1p-mediated nuclear export and binding (Kuge et al., 1998; Kudo et al., 1999). Because of their redox-sensitive nature, the CRDs easily respond to ROS-induced oxidative signaling. However, Yap1p is not directly oxidized by ROS (Delaunay et al., 2002).
In H2O2-induced oxidative stress, Yap1p is activated by a yeast GSH peroxidase (GPX)-like protein called oxidant receptor peroxidase 1 (Gpx3p/Orp1p). The cysteine residues of this peroxidase are themselves oxidized to cysteine-sulfenic acid (–SOH) groups by H2O2. The oxidized cysteine residues of Orp1p then react with the CRDs of Yap1p, inducing the formation of an intra-molecular disulfide bond between the cysteine residues of the Yap1p C- and N-terminal CRDs (Delaunay et al., 2002). This bond masks the NES contained in the C-terminal CRD and thus inhibits Yap1p-to-Crm1p bond formation, allowing the newly oxidized Yap1p transcription factor to migrate back into the nucleus (Delaunay et al., 2000). Once it approaches nuclear DNA, Yap1p initiates the transcription of specific genes by binding to the YREs contained in their promoter sequences (Lushchak, 2010). This Orp1p-mediated activation of Yap1p is further facilitated by Yap1p-binding protein (Ybp1p), which forms an H2O2-induced complex with Yap1p (Veal et al., 2003).
In response to H2O2, Yap1p promotes the transcription of several genes encoding antioxidants or antioxidant-associated molecules, such as those involved in the thioredoxin (TRX) and GSH systems. Upregulated sequences include GSH1 (encoding Γ-glutamylcysteine synthetase), GSH2 (encoding GSH synthetase), TRX2 (encoding TRX), SOD1 (encoding CuZnSOD), SOD2 (encoding MnSOD), and several others (Schnell et al., 1992; Stephen et al., 1995; Gasch et al., 2000). Activated Yap1p further upregulates expression of Ret tyrosine kinase inhibitor RPI1 in yeast cells (Dumond et al., 2000). This represses the cAMP-dependent Ras/protein kinase A (Ras/PKA) signaling pathway which, under non-stress conditions, actively represses the activation of Yap1p and of other stress response transcription factors (Fernandes et al., 1997). The Ras/PKA pathway is then itself repressed by RPI1 expression when cells are at stationary phase or exposed to oxidative stress (Dumond et al., 2000).
Oxidized Yap1p can be reduced and rendered H2O2-sensitive again by TRX-mediated reduction of the disulfide bond (Kuge and Jones, 1994; Delaunay et al., 2000; Kuge et al., 2001). TRX is in turn reduced by TRX reductase at the expense of reduced nicotinamide adenine dinucleotide (NADPH; Holmgren, 1989). Therefore, Yap1p’s functionality is intimately associated with the redox status of the cell and with cellular NADPH content in particular (Lushchak, 2010). By promoting the synthesis of antioxidant molecules, Yap1p essentially sustains its own transcription activity until sufficiently inhibited by TRX, at which point the increase in antioxidants is likely sufficient for the cells to adapt to oxidant-induced stress (Delaunay et al., 2000; Temple et al., 2005).
Yap1p can also be activated via alternate pathways, as in the Orp1p- and Ybp1p-independent response to diamide, where the N- and C-terminal CRD cysteine residues of Yap1p do not form a disulfide between themselves, as they do in H2O2-induced activation (Wemmie et al., 1997; Delaunay et al., 2000; Kuge et al., 2001). Besides its sensitivity to ROS, Yap1p also regulates the yeast cell response to cadmium ion toxicity by upregulating transcription of yeast cadmium factor YCF1. This sequence encodes a membrane transport protein involved in conferring cadmium tolerance to cells (Wemmie et al., 1994).
At least seven other Yap proteins – Yap2p to Yap8p – have been identified in S. cerevisiae. It has been suggested that these proteins each have different, albeit slightly overlapping, physiological roles in the regulation of cellular responses to oxidative stresses and xenobiotic insults, along with Yap1p (Fernandes et al., 1997).
Similarly to budding yeast, fission yeast S. pombe (Toone et al., 1998), facultative yeast pathogen Candida albicans (Zhang et al., 2000) and lactose-metabolizing yeast species Kluyveromyces lactis (Billard et al., 1997) each possess their own Yap1p homologs, which mediate responses to oxidative stress (Toone and Jones, 1999).
Transcription factor Skn7
The Skn7 protein is a kinase-regulated transcription factor that also modulates the response to oxidative insults in budding yeast. It possesses a C-terminus receiver domain which is essential for its role in the regulation of cell wall biosynthesis, the cell cycle, and the response to osmotic shock and to oxidative stress (Brown et al., 1994; Maeda et al., 1994; Morgan et al., 1995, 1997; Krems et al., 1996). The Skn7p transcription factor also shows homology to heat shock factor protein (Hsf1p). In fact, both Skn7p and Hsf1p upregulate heat shock proteins by binding to heat shock elements (HSEs) in H2O2-treated S. cerevisiae cells exposed to heat shock (Raitt et al., 2000).
Skn7p also acts as an auxiliary transcription factor to Yap1p, in response to oxidants such as H2O2 (Lee et al., 1999) where it upregulates proteins such as TRX reductase (encoded by TRR1) and TRX (Morgan et al., 1997). Both Yap1p and Skn7p upregulate the transcription of genes encoding proteins such as SODs, catalases, TRXs, and heat shock proteins (Lee et al., 1999). Other yeast species have their own respective homolog of Skn7p, such as Prr1p in S. pombe (Ohmiya et al., 1999).
Transcription factors Msn2p and Msn4p
Another two yeast transcription factors involved in the response of S. cerevisiae cells to oxidative stress are Msn2p and Msn4p. These are proteins with zinc-finger type DNA-binding domains. In response to a variety of general stresses such as osmotic shock, hypersalinity, heat stress, oxidants, and also diauxic transition, Msn2p and Msn4p are reversibly translocated to the nucleus, where they bind to the stress response element (STRE) 5′-CCCCT-3′contained in certain DNA promoter sequences (Martínez-Pastor et al., 1996; Schmitt and McEntee, 1996; Boy-Marcotte et al., 1998). They play a considerably important role in the response to oxidative stress in yeast (Gasch et al., 2000), as shown by the increased sensitivity of Δmsn2Δmsn4 budding yeast mutants to high concentrations of H2O2 and their limited ability to adapt to low H2O2 concentrations (Hasan et al., 2002). Two antioxidant-encoding sequences are upregulated by Msn2p and Msn4p: CTT1 (encoding cytosolic catalase) and GRX1 (encoding glutaredoxin). Otherwise, they largely upregulate genes encoding proteases, heat shock proteins, metabolic enzymes, and other molecules implicated in stress recovery, restoration of metabolic homeostasis, and the repair or removal of damaged biomolecules (Hasan et al., 2002). Activation of general Msn2p/4p-mediated stress responses is negatively regulated by kinase enzyme signaling systems such as cAMP-dependent Ras/PKA activity (Hasan et al., 2002).
Yeast Antioxidants and Their Role in Yeast Oxidative Stress Responses
Following their upregulation by transcription factors and their subsequent synthesis, yeast cell antioxidant defenses remove excess ROS and restore redox balance. Prominent among these are mitochondrial MnSOD and cytosolic CuZnSOD, which scavenge and convert it to H2O2 (McCord and Fridovich, 1969; Weisiger and Fridovich, 1973a,b). The SOD-derived H2O2 can in turn be degraded to water and oxygen by the redox-sensitive heme groups of cytosolic catalase T (Ctt1p; Hartig and Ruis, 1986) and peroxisomal catalase A (Cta1p) enzymes (Cohen et al., 1988). Other enzymatic antioxidants include GPXs and TRX peroxidases/peroxiredoxins (PRX), which use electron-donating cysteine thiol groups as active sites to catalyze the reduction of hydroperoxides (Jamieson, 1998). In fact, GPXs protect against lipid peroxidation (Avery and Avery, 2001) and heavy metal toxicity (Avery et al., 2004) and help activate Yap1p transcription factors in response to H2O2 signaling (Delaunay et al., 2000; Gasch et al., 2000; Fomenko et al., 2011). PRX also respond to heavy metal toxicity (Nguyên-Nhu and Knoops, 2002) and zinc-metal deficiency (Wu et al., 2007), and reduce ROS, such as H2O2 and alkylhydroperoxides (Chae et al., 1994). PRX also reduce peroxynitrites (Wood et al., 2003).
Yeast cells also possess thiol oxidoreductase enzymes such as glutaredoxins and TRXs, which both catalyze the reduction of disulfides to thiols, using thiolated cysteine residues as an active site (Herrero et al., 2008). TRXs, the functionality of which is maintained by NADPH and the catalytic action of TRX reductase, restore functionality to oxidized PRX, maintain redox balance (Holmgren, 1989), and confer protection against hydroperoxides in yeast (Garrido and Grant, 2002). Similarly, glutaredoxins protect yeast against superoxide radicals (Luikenhuis et al., 1998) and hydroperoxides (Pujol-Carrion et al., 2006), reduce GSH-protein mixed disulfides and protein disulfides, and also play an important role in iron homeostasis (Yamaguchi-Iwai et al., 2002; Ojeda et al., 2006).
Glutathione, an abundant non-enzymatic antioxidant in yeast (Izawa et al., 1995; Zechmann et al., 2011), is required for the glutaredoxin-catalyzed reduction of disulfides and also reduces oxidized GPXs (Holmgren, 1989). Oxidized GSH (GSSG) is reduced back to GSH by GSH reductase, in a reaction that requires electrons from NADPH, the main source of which is the pentose phosphate pathway (Izawa et al., 1995; Juhnke et al., 1996). Besides being a crucial source of electrons for many antioxidants, GSH can also protect protein thiol groups from oxidation, by S-thiolation (Klatt and Lamas, 2000). Other prominent non-enzymatic yeast antioxidants include -scavenging manganese ion complexes regulated by nutrient-sensing kinase pathways (Reddi et al., 2009; Reddi and Culotta, 2011), erythroascorbates, and vitamin E (Herrero et al., 2008).
Intracellular Degradation of Oxidized Biomolecules
Despite the rapid upregulation and high efficiency of yeast antioxidant defenses, cell biomolecules can still sustain severe oxidative damage that cannot be repaired. Irreparably damaged cell components such as carbonylated proteins and oxidatively damaged mitochondria must be efficiently removed before they are allowed to form large aggregate masses that lead to cell death. Yeast has two stress response mechanisms that facilitate the removal of irreparably oxidized biomolecules: the ubiquitin-dependent proteasome system (UPS) and autophagy.
The ubiquitin-dependent proteasome system in yeast
The UPS is a highly conserved mechanism of targeted protein degradation, which facilitates the removal of small oxidized proteins from cells. The precise mechanism by which polyubiquitinated substrates are delivered to the proteasome is presently unclear, despite the identification of key UBL/UBA ubiquitin receptor proteins such as Rad23 (Schauber et al., 1998; Chen and Madura, 2002; Elsasser et al., 2004), Dsk2 (Wilkinson et al., 2001), and Ddi1 (Bertolaet et al., 2001) in yeast. A recent study by Li et al. (2010) has shed some light on this aspect, demonstrating that Rad4, a regulatory protein previously associated only with nuclear excision repair (NER) of DNA, together with Rad23, regulate a post-ubiquitination event, presumably the delivery of specific ubiquitinated substrates to the proteasome. In fact, Rad4 shares specific substrates with Rad23 and possesses a binding domain which is crucial for Rad23 to exert its degradative function. The authors suggested that Rad4 plays an accessory role in proteolysis by keeping the Rad23 protein in the required active conformation to associate with ubiquitinated substrates. On the other hand, Rad23 is believed to play an accessory role in NER, by protecting Rad4 from degradation by proteins such as the yeast deubiquitinase Ubp3 (Mao and Smerdon, 2010).
Because the UPS mainly operates in the cytosol, oxidized proteins located within cell compartments segregated by membranes, such as the ER and the mitochondria, cannot be degraded by this mechanism unless they are exported into the cytosol (Taylor and Rutter, 2011). In fact, an ER-associated degradation (ERAD) pathway facilitates the translocation of oxidized or misfolded ER proteins to the cytosol for UPS-mediated proteasomal degradation (Ye et al., 2001). An ERAD-like mechanism, called the mitochondria-associated degradation (MAD) pathway, has also been proposed whereby oxidized mitochondrial proteins are extruded through the outer mitochondrial membrane into the cytosol for UPS-mediated degradation (Chatenay-Lapointe and Shadel, 2010; Heo et al., 2010).
Autophagic pathways in yeast
Autophagy involves the intracellular degradation and recycling of long-lived biomolecules such as macromolecular proteins and organelles, by lysosomes (in mammalian cells) or vacuoles (in yeast cells; Klionsky et al., 2010). These pathways are activated by the expression of autophagy-related genes (ATG), of which there are at least 33 in yeast, along with many other ATG homologs in mammals (Reggiori and Klionsky, 2002; Goldman et al., 2010). Autophagy is indeed a highly conserved process among eukaryotes. It is involved in metazoan cell differentiation and plays a crucial pro-survival role in response to stresses such as nutrient starvation and ROS accumulation in eukaryotes such as yeast (Kissová et al., 2004, 2006, 2007; Bhatia-Kissová and Camougrand, 2010). In fact, deficiency of autophagy in yeast causes ROS accumulation after stresses such as nitrogen starvation (Suzuki et al., 2011). The protective role of autophagy against oxidative stress is also very evident in ROS-associated pathologies such as aging (Donati et al., 2001) and neurodegenerative diseases (Rubinsztein, 2006), where autophagic pathways are largely disrupted and cells suffer extensive intracellular aggregation of oxidized proteins (Terman and Brunk, 1998; Rubinsztein, 2006; Scherz-Shouval and Elazar, 2007; Rubinsztein et al., 2011).
The principal autophagic pathway in yeast and in other eukaryotes is macroautophagy. This involves the non-selective sequestration of intracellular macromolecules and portions of cytosol by a double-membraned vesicle called the autophagosome, which then delivers its enclosed material to a vacuole (or lysosome in higher eukaryotes) replete with hydrolytic enzymes that facilitate biomolecular degradation (Klionsky et al., 2010). Another non-selective autophagic pathway, called microautophagy, involves the direct sequestration and degradation of organelles and portions of cytoplasm by vacuoles/lysosomes themselves, excluding the involvement of autophagosomes. Autophagy predominantly proceeds via these two non-selective pathways, both of which take place in yeast depending on factors such as the metabolic state of the cells (Kissová et al., 2007).
Selective autophagic degradation of specific biomolecular targets can also take place, often in conjunction with non-selective autophagy (Nair and Klionsky, 2005; Kissová et al., 2007; Kraft et al., 2009). This includes selective autophagic degradation of the Golgi apparatus (crinophagy; Glaumann, 1989), ER (reticulophagy; Hamasaki et al., 2005), ribosomes (ribophagy; Kraft et al., 2008), peroxisomes (pexophagy; Sakai et al., 1998), and mitochondria (mitophagy; Lemasters et al., 1998; Lemasters, 2005; Kissová et al., 2007).
In terms of its physiological role in yeast, it is believed that selective autophagy is required for (i) selective removal of defective organelles under normal physiological conditions to maintain homeostatic cell structure integrity, and (ii) specific removal of excess or damaged organelles in response to stresses such as nutrient starvation and ROS accumulation (Goldman et al., 2010). During oxidative stress, autophagic pathways involved in the specific removal of mitochondria assume particular importance (Bhatia-Kissová and Camougrand, 2010). Mitochondria, as previously described, constitute a major production site and target of ROS, therefore the risk of accumulation of defective mitochondria in cells increases during oxidative stress. If left unabated, such accumulation will only lead to further cell oxidative damage and eventual disruption of cell homeostasis. Therefore the rapid removal of these dysfunctional organelles, a function facilitated by mitophagy, is of paramount importance (Lemasters, 2005; Scherz-Shouval and Elazar, 2007; Bhatia-Kissová and Camougrand, 2010).
Mitophagy in yeast as a response to oxidative stress
Mitochondrial degradation by autophagy in yeast was first reported by Takeshige et al. (1992), who observed sequestered mitochondria in the autophagic bodies of yeast cells subjected to nitrogen starvation, after growth on glucose (Takeshige et al., 1992). Similarly, Ashford and Porter (1962) had earlier reported an abundance of mitochondria in rat hepatocyte autophagosomes. These observations suggested that autophagy is a primary removal mechanism of mitochondria in cells. In fact, subsequent studies showed that selective autophagic degradation of mitochondria, defined by Lemasters (2005) as mitophagy, can indeed take place in yeast in response to adverse stimuli involving mitochondrial damage and ROS accumulation (Goldman et al., 2010).
Campbell and Thorsness (1998) were among the first to indicate a link between mitochondrial damage and the upregulation of specific mitochondrial degradation in yeast. S. cerevisiae Δyme1 mutants, the mitochondria of which were impaired by the absence of yeast mitochondrial ATP-dependent metalloprotease enzyme Yme1p, exhibited increased migration of mtDNA from their dysfunctional mitochondria, when grown on ethanol/glycerol medium. This was accompanied by the increased degradation and proximity of dysfunctional mitochondria to the surfaces of degradative vacuoles, although this did not conclusively demonstrate that the mitochondria were degraded by autophagy.
Priault et al. (2005) then unequivocally demonstrated the preferential removal of dysfunctional mitochondria by autophagy in yeast. It was shown that Δfmc1 yeast mutants, the mitochondria of which had an impaired mitochondrial membrane potential due to aggregation of mitochondrial ATP synthase, exhibited preferential degradation of their dysfunctional mitochondria by mitophagy under anaerobic conditions. The possibility that autophagy was induced by ATP depletion (due to ATP synthase aggregation) as opposed to mitochondrial damage was excluded, given that Δfmc1 yeast cells had similar cellular ATP levels to the wildtype cells. Further studies showed that osmotic swelling of mitochondria due to impairment of K+/H+ exchange proteins (Nowikovsky et al., 2007) and impairment of the mitochondrial membrane potential due to malfunctioning mtDNA replication machinery (Zhang et al., 2007), also initiate mitophagy.
Kissová et al. (2006) further showed that upregulation of mitophagy in wildtype yeast cells, in response to the target of rapamycin (TOR) kinase inhibitor, rapamycin, is accompanied by early ROS accumulation and mitochondrial membrane lipid peroxidation. The addition of resveratrol to rapamycin-treated cells inhibited these oxidative effects and largely impaired mitophagy, clearly suggesting that ROS-induced mitochondrial damage also plays a crucial role in activating this pathway. This is further corroborated by the fact that, unlike macroautophagy, selective mitophagy is specifically suppressed by the antioxidant N-acetylcysteine (NAC), which replenishes the cellular GSH pool (Deffieu et al., 2009; Kissová and Camougrand, 2009). This showed that mitophagy is sensitive to cellular redox imbalances induced by the depletion of GSH and that it is a discretely, independently regulated pathway.
Key genetic evidence that mitophagy is a discrete process, regulated independently of non-selective macroautophagy in yeast, was provided by Kissová et al. (2007). Nitrogen-starved wildtype yeast cells, grown under aerobic conditions, exhibited early selective mitochondrial autophagy followed by non-selective autophagic removal of mitochondria after prolonged stress. The early selective mitophagic process is dependent on an outer mitochondrial membrane protein, Uth1p. Removal of this protein, facilitated by the deletion of UTH1, resulted in the activation of only the non-selective (Uth1p-independent) autophagic degradation of mitochondria. Subsequent genetic screening has since identified that other sequences, such as ATG32 (Kanki et al., 2009a; Okamoto et al., 2009) and ATG33 (Kanki et al., 2009b) also encode proteins without which mitophagy cannot take place, further proving it to be a discretely regulated process.
Overall, these studies established that, in yeast, mitochondrial damage stimulates the specific degradation of mitochondria by mitophagy. This implies that the physiological role of mitophagy is that of a discretely controlled cellular surveillance mechanism which monitors mitochondrial functionality and specifically removes irreparably damaged mitochondria (Goldman et al., 2010). Given that such damage can be induced by ROS accumulation (Kissová et al., 2006), it is likely that mitophagy also constitutes an important defense against oxidative stress in yeast, in conjunction with the other antioxidant defenses described previously.
However, mitophagy’s hypothetic role as a mitochondrial functionality control system in yeast is uncertain, given that mitophagy-deficient ΔAtg32 mutants did not show any significant increase in ROS and did not exhibit reduced growth in comparison to wildtype cells, when grown on non-fermentable media (Kanki et al., 2009a; Okamoto et al., 2009). This might imply that mitophagy is a non-essential and redundant process in yeast, with respect to non-selective autophagy. This is surprising, given that mitophagy’s role as a mitochondrial functionality control mechanism in mammalian cells has been established (Narendra et al., 2008; Geisler et al., 2010; Matsuda et al., 2010), naturally leading to the assumption that it has the same such role in yeast. Clearly, further studies of mitophagy’s poorly understood mechanism and of its regulation are required for this issue to be resolved. Progress in this regard is already being made, given the recent discovery of two mitogen-activated protein kinases (MAPKs), Slt2p and Hog1p, which regulate mitophagy in yeast (Mao et al., 2011).
Also, the physiological role of mitophagy as a clearance mechanism of excess mitochondria has been confirmed by studies of stationary phase yeast cells grown under respiratory conditions (Kanki and Klionsky, 2008). Such clearance behavior serves to reduce the high metabolic cost of maintaining excess mitochondria, which are made redundant by the reduced growth demands of stationary phase cells, whilst concurrently lowering the risk of cellular ROS accumulation (Kanki et al., 2011).
Programmed Cell Death and Oxidative Stress in Yeast
In the event that yeast cells suffer prolonged or sudden exposure to harmful doses of ROS, it is often the case that their pro-survival response mechanisms and antioxidant defenses fail. This consequently elicits the yeast cell’s final response to oxidative stress: the initiation of PCD (Madeo et al., 1999). This is essentially an “orderly” process of cell elimination, which can proceed via different pathways, depending on the physiological state of the cells and the nature of the oxidative insult. At least two main established PCD pathways have been observed in yeast in response to oxidative stress: the first is apoptosis, which is typically induced by exposure to low doses of oxidants such as H2O2 (Madeo et al., 1999). Alternatively, yeast cells can undergo necrosis, a PCD pathway which, until recently, was conveniently dismissed as uncoordinated and accidental (Eisenberg et al., 2010; Galluzzi et al., 2011, 2012). Finally, even autophagic pathways such as mitophagy have been implicated in ROS-induced PCD in yeast (Bhatia-Kissová and Camougrand, 2010), which is intriguing given their main established role as pro-survival pathways.
Oxidative Stress and Apoptosis in Yeast
The first observation of an apoptotic phenotype in yeast cells was made by Madeo et al. (1997), who observed that mutant S. cerevisiae cells lacking the ATPase AAA cell division cycle gene CDC48 (Latterich et al., 1995) showed key morphological hallmarks of mammalian apoptosis such as phosphatidylserine externalization, chromatin condensation, and DNA fragmentation. Since then, further markers characteristic of mammalian apoptosis have been discovered in yeast, including the release of mitochondrial cytochrome c (Manon et al., 1997; Ludovico et al., 2002; Sapienza et al., 2008), and the execution of conserved proteolytic pathways associated with mammalian apoptosis, such as Cdc6 protein degradation (Blanchard et al., 2002).
Undoubtedly, the most compelling evidence of apoptosis in yeast is derived from the discovery of several orthologs of key mammalian apoptotic regulators, such as the yeast metacaspase protein Yca1p/Mca1p (Madeo et al., 2002), apoptosis inducing factor Aif1p (Wissing et al., 2004), the Htr2A/Omi serine protease Nma111p (Fahrenkrog et al., 2004), inhibitor of apoptosis protein (IAP) Bir1p (Walter et al., 2006), and the mitochondrial endonuclease G Nuclp (Büttner et al., 2007). The yeast NADH dehydrogenase Ndi1p, located in the inner mitochondrial membrane, has also been implicated in cell death (Li et al., 2006). It is in fact a homolog of AIF-like mitochondrial-associated inducer of cell death (AMID; Li et al., 2006), a protein associated with caspase-independent apoptosis in humans (Wu et al., 2002). Similarly, the yeast protein Cdc48 also has a human ortholog that negatively regulates apoptosis (Shirogane et al., 1999). Yeast also possesses a true homolog of highly conserved (Chae et al., 2003) Bax-inhibitor BI-1 proteins called Bxi1p (Cebulski et al., 2011), anti-apoptotic Bax-suppressor homologs called Sno1p and Fyv10p (Khoury et al., 2008), and also a pro-apoptotic BH3 homolog called Ybh3p (Büttner et al., 2011). Furthermore, apoptotic markers have consistently appeared in aging yeast cells (Laun et al., 2001; Herker et al., 2004). ROS, which have a central role in regulating mammalian apoptosis, also act as crucial modulators of apoptosis in yeast (Madeo et al., 1999). We have confirmed this by specific ROS probes (unpublished observations). This wealth of evidence has largely confirmed the existence of a highly conserved apoptotic pathway in yeast, which seems to have evolved early on as a response to harmful stimuli that are in large part associated with ROS and oxidative stress.
Proponents of apoptosis in yeast have also presented a strong hypothesis to account for its physiological role in an organism which, despite being unicellular, naturally exists in the large, densely packed cell populations that make up yeast colonies (Váchová and Palková, 2007). With this paradigm, apoptosis in yeast was suggested to be an altruistic clearance mechanism of old or damaged cells, designed to free up nutrients and enhance the survival chances of younger, healthier clones in aging, or stressed yeast colonies (Fabrizio et al., 2004; Herker et al., 2004; Knorre et al., 2005). It has also been suggested that apoptosis-associated ROS accumulation may raise the chances of clone mutation in a yeast population, leading to an increased number of genetically varied individual cells, which are better adapted to altered environmental conditions (Fabrizio et al., 2004).
Still, the notion of apoptosis in yeast has been met with skepticism by some researchers, a number of whom have suggested that the standard flow-cytometric techniques used to assay Yca1p activity and ROS accumulation in yeast cells are artifact-prone, implying that previous evidence of yeast apoptosis was artifactual and that yeast cell death is independent of both ROS and Yca1p (Wysocki and Kron, 2004). Counter-arguments to these claims assert that these authors exposed their yeast cells to extreme death conditions, which either induced non-apoptotic cell death or caspase-independent apoptosis (LeBrasseur, 2004), the latter of which has since been observed in yeast cells, and which accounts for the rough majority of reported apoptotic death scenarios in yeast (Madeo et al., 2009).
Some have also proposed that Yca1p, the yeast ortholog of mammalian caspases (the cysteine-protease enzymes which mediate mammalian apoptosis) is not a true caspase at all and has no caspase-like activity. The argument behind this is that, whilst metacaspases and caspases probably share a common origin given the presence of a conserved caspase fold in both proteins, this does not justify putting them into the same protein family, since evolutionary conservation of protein structure does not necessarily mean that protein function is also conserved (Vercammen et al., 2007). The differing substrate affinity of metacaspases, attributed to their preference for protein cleavage-sites preceding basic arginine residues, as opposed to the preference of caspases for cleavage-sites preceding acidic aspartate residues, helped to strengthen the argument that the physiological function of metacaspases differs from that of caspases (Vercammen et al., 2007). However, it has since been shown that, despite their proteolytic discrepancies, metacaspases and caspases do indeed share a number of common substrates known to be involved in PCD, such as Tudor staphylococcal nuclease (TSN) derived from Pinus abies (Norway Spruce Tree; Sundström et al., 2009; Carmona-Gutierrez et al., 2010b).
Whilst many of its core aspects, such as the mechanics of yeast metacaspase activity, are still poorly understood (Abdelwahid et al., 2011), the paradigm of yeast apoptosis has by now been largely accepted within the research community, in spite of the lingering doubts expressed by some.
One aspect that leaves little room for doubt is the role of ROS as a key inducer of apoptotic cell death in yeast, as shown early on by Madeo et al. (1999) who observed significant accumulation in yeast cells undergoing H2O2-induced apoptosis – a cell death scenario which is exacerbated by deletion of GSH1 and prevented by hypoxia or quenching of ROS using spin traps (Madeo et al., 1999) and antioxidants (Jeon et al., 2002). In fact, ROS accumulation and the involvement of mitochondria, where this usually takes place, are arguably the most unifying features shared by apoptotic yeast cells treated with different pro-apoptotic stimuli (Pereira et al., 2008). These include exogenous stimuli, ranging from metals such as copper (Liang and Zhou, 2007), lead (Bussche and Soares, 2011), iron (Almeida et al., 2008), and cadmium (Nargund et al., 2008) to plant toxins (Narasimhan et al., 2005), amiodarone (Pozniakovsky et al., 2005), ethanol (Kitagaki et al., 2007), and anti-tumor drugs including paclitaxel (Foland et al., 2005), arsenic (Du et al., 2007), valproate (Mitsui et al., 2005), and aspirin which commits yeast cells lacking mitochondrial MnSOD to apoptosis by disrupting the cellular redox balance (Sapienza and Balzan, 2005; Sapienza et al., 2008). There are also several ROS-associated endogenous stimuli of apoptosis in yeast, which include aging (Laun et al., 2001; Herker et al., 2004) and the disruption of metabolic pathways involved in DNA metabolism (Weinberger et al., 2005), protein modification (Hauptmann et al., 2006), and actin dynamics (Gourlay and Ayscough, 2006).
Furthermore, yeast cell death induced by ROS-associated stress stimuli can proceed either via caspase-dependent apoptosis or caspase-independent apoptosis (Madeo et al., 2009), as described below.
Oxidative stress and caspase-dependent apoptosis in yeast
Caspase-dependent apoptosis in yeast was first reported by Madeo et al. (2002), who observed that yeast cells undergoing H2O2-induced apoptotic cell death exhibited an upregulation of caspase-like proteolytic activity, attributed to yeast caspase protein 1, Yca1p, encoded by the YCA1 gene. The absence of Yca1p prevented H2O2-induced apoptosis entirely, whereas its overexpression exacerbated apoptosis, providing the first clear evidence of an apoptotic pathway in yeast which is caspase-dependent and induced by ROS.
Acetic acid-induced PCD of yeast cells, which bears typical markers of mammalian apoptosis, is also accompanied by an accumulation of ROS in yeast cell compartments such as the mitochondria (Ludovico et al., 2001, 2002) and can involve Yca1p (Madeo et al., 2002; Guaragnella et al., 2006). It has been suggested that H2O2 triggers acetic acid-induced PCD in yeast, given the early accumulation of H2O2 in acetic acid-induced cell death. This is corroborated by the observed absence of H2O2 buildup in acid-stress adapted yeast cells in which acetic acid-induced PCD is prevented (Giannattasio et al., 2005; Guaragnella et al., 2007). In fact, further studies have shown that the quenching of acetic acid-induced ROS, using antioxidants such as NAC, abrogates both cytochrome c release and Yca1p activity in wildtype yeast cells, preventing cell death altogether. However, it is worth noting that NAC does not prevent acetic acid-induced death in yeast knockout mutants deficient in Yca1p and cytochrome c, implying that acetic acid-induced PCD can also proceed along pathways that are Yca1p and ROS-independent (Guaragnella et al., 2010).
The accumulation of ROS is also implicated in yeast apoptosis induced by salt stress (NaCl; Wadskog et al., 2004), heat stress (Lee et al., 2007), and hyperosmotic stress (Silva et al., 2005) which, in all cases, cannot take place without Yca1p activity. Stress induced by disruption of iron homeostasis (Almeida et al., 2008), cadmium (Nargund et al., 2008), and arsenic (Du et al., 2007) can also result in a caspase-mediated apoptotic phenotype associated with significant accumulation of ROS. Likewise, both Yca1p activity and ROS accumulation are key requirements for apoptosis in yeast cells treated with low to moderate doses of virally encoded killer toxins (Reiter et al., 2005).
Apoptosis induced by the heterologous expression, in yeast, of Parkinson’s disease-associated protein α-synuclein, also requires the activation of Yca1p. The latter’s removal totally abolishes α-synuclein’s ability to induce ROS accumulation and apoptotic cell death. Likewise, the addition of antioxidant GSH prevented α-synuclein-induced apoptosis in yeast, further demonstrating the important mediatory role of ROS in this pathway (Flower et al., 2005).
Treatment of yeast cells with drugs such as valproic acid, a teratogenic, short chain fatty acid with anticonvulsant and anti-tumor properties (Blaheta and Cinatl, 2002), also induces a significant accumulation of ROS that in turn upregulates the activity of Yca1p and initiates apoptotic cell death (Mitsui et al., 2005). Recent studies have also shown that propolis, a product derived from plant resins, which is utilized by bees to protectively seal the apertures in their hives, can elicit caspase-dependent apoptosis associated with ROS accumulation in yeast cells (de Castro et al., 2011). This corroborates the potential use of propolis as an antifungal agent.
Additionally, mutations that induce defects in yeast cellular processes such as those associated with mRNA stability (Mazzoni et al., 2005), initiation of DNA replication (Weinberger et al., 2005), ubiquitination (Bettiga et al., 2004), and mitochondrial fragmentation (Fannjiang et al., 2004) can induce Yca1p-mediated apoptotic cell death accompanied by ROS accumulation. A marked increase in ROS is also observed during pro-apoptotic chronological aging of yeast cells, the survival of which is initially improved by deletion of YCA1 (Herker et al., 2004). However, aging Δyca1 yeast mutants later lose the ability of regrowth, as opposed to aged wildtype cells, which actually outlive their YCA1-deficient counterparts (Herker et al., 2004). This is likely because Δyca1 yeast mutants suffer greater accumulation of oxidized proteins than the wildtype cells (Khan et al., 2005), thus indicating the importance of Yca1p-dependent apoptosis as a clearance mechanism of aged or oxidatively damaged cells in a yeast colony (Herker et al., 2004; Madeo et al., 2009).
Finally, it has been argued that the caspase-like activity detected upon stimulation of apoptosis in yeast is not entirely attributable to Yca1p, this implicating the participation of other caspase-like proteases (Wilkinson and Ramsdale, 2011). One such protein is Esp1p, which plays an important pro-apoptotic role in yeast treated with ROS such as H2O2. Upon induction of apoptosis, Esp1p is released from anaphase inhibitor Pds1p, and cleaves Mcd1p (the yeast homolog of human cohesin subunit Rad21) such that the latter’s C-terminal fragment migrates from the nucleus to the mitochondria. This induces disruption of the mitochondrial membrane potential and cytochrome c release, followed by apoptosis (Yang et al., 2008). Another yeast protein associated with caspase-like activity in yeast is Kex1 protease, which mediates apoptotic cell death induced by defective N-glycosylation, acetic acid and chronological aging (Hauptmann and Lehle, 2008).
Oxidative stress and caspase-independent apoptosis in yeast
Apoptotic cell death in yeast caused by ROS-associated stress stimuli, can also take place without the need for Yca1p activity. For instance, ROS accumulation and apoptosis caused by defective N-glycosylation in Ost2p-deficient yeast cells (Ost2p being the yeast homolog of mammalian defender of apoptosis-1 DAD1) are not prevented by deletion of YCA1 (Hauptmann et al., 2006). Similarly, absence of Yca1p activity does not impede ROS accumulation, mitochondrial degradation, and apoptosis of yeast cells treated with formic acid (Du et al., 2008). Caspase-independent apoptosis triggered by release of ammonia in differentiating yeast cell colonies (Váchová and Palková, 2005), in yeast cells treated with copper (which generates ROS via Fenton/Haber–Weiss pathways; Liang and Zhou, 2007), and in yeast cells treated with ceramide (Carmona-Gutierrez et al., 2011b) also share the common feature of ROS accumulation. In fact, the latter two apoptotic scenarios are both suppressed in mtDNA-deficient rhoo (ρo) “petite” mutant cells, where mitochondrial respiration is impaired. Likewise, functional mitochondria are a key requirement of caspase-independent apoptosis of aging yeast cells caused by heterologous expression of α-synuclein, and characterized by massive ROS accumulation (Büttner et al., 2008).
The requirement of functional mitochondria in caspase-independent yeast apoptosis is no surprise given that, aside from their major role as a source and target of ROS, mitochondria are also the source of important yeast apoptotic regulators capable of operating independently of Yca1p. One such regulator is the yeast AIF homolog Aif1p (Wissing et al., 2004). This protein can translocate from the mitochondrial intermembrane space to the nucleus, in response to oxidative pro-apoptotic stimuli such as H2O2, aging, acetic acid (Wissing et al., 2004), and fungal toxins such as bostrycin (Xu et al., 2010). Like its mammalian counterpart, Aif1p operates in tandem with cyclophilin A once it is inside the nucleus, inducing chromatin condensation, DNA fragmentation, and subsequent apoptotic cell death (Candé et al., 2004; Wissing et al., 2004). Similarly, the endonuclease G yeast homolog Nuc1p translocates from the mitochondria to the nucleus, in response to oxidative stimuli such as H2O2, resulting in a caspase-independent apoptotic phenotype involving ROS accumulation (Büttner et al., 2007).
Additionally, the yeast nucleus itself contains the Yca1p-independent death regulator Nma111p, the yeast homolog of pro-apoptotic mammalian serine protease HtrA2/Omi. In response to heat stress and oxidative stimuli such as H2O2, Nma111p starts to aggregate in the nucleus and induces yeast cell apoptosis in a manner that is dependent on its serine protease activity (Fahrenkrog et al., 2004). The pro-apoptotic effects of Nma111p are suppressed by its antagonistic substrate Bir1p, the yeast homolog of mammalian IAP, as demonstrated by the enhanced apoptotic phenotype of Δbir1 yeast cells exposed to oxidative stress (Walter et al., 2006).
Finally, yeast nuclei contain another trigger of caspase-independent apoptosis, which is induced by ROS such as H2O2. This involves the epigenetic modification of the chromatin histone structure H2B, the N-terminal tail of which is deacetylated at lysine 11 by histone deacetylase (HDAC) Hos3, and subsequently phosphorylated at serine residue 10 by Ste20 kinase (Ahn et al., 2005, 2006), which also plays a key role in pheromone-induced apoptotic cell death in yeast (Severin and Hyman, 2002). The resulting chromatin modification promotes apoptosis.
Oxidative Stress and Necrosis in Yeast
Yeast can also undergo necrotic cell death in response to oxidative stress (Madeo et al., 1999). Necrosis is characterized by bioenergetic failure and morphological features such as random DNA fragmentation, an increase in cell volume or oncosis, swelling of organelles, loss of cell plasma membrane integrity, and subsequent leakage of intracellular contents (Zong and Thompson, 2006). Because of its seemingly disordered features, necrosis was long dismissed as an accidental form of cell death (Galluzzi et al., 2011) which generally occurs in response to extreme environmental stresses (Madeo et al., 1999). However, this paradigm has changed, given recent evidence that certain necrotic cell death scenarios are regulated by factors such as signaling and catabolic proteins (Baines, 2010). The fact that necrosis can be regulated by the active involvement of such proteins, provides strong evidence that it can indeed proceed as a programmed pathway referred to as “programmed necrosis” (Galluzzi et al., 2012). Furthermore, accumulating evidence suggests that, as in apoptosis, the accumulation of ROS and the presence of mitochondria are key requirements of this form of PCD, both in mammals and in yeast (Baines, 2010; Eisenberg et al., 2010).
In yeast cells, exposure to very high concentrations of pro-apoptotic stimuli including H2O2 (Madeo et al., 1999), acetic acid (Ludovico et al., 2001), and heavy metals (Liang and Zhou, 2007), generally causes accidental necrosis as a result of very severe oxidative damage to cell components. Acetic acid also causes disruption of pH homeostasis (Ludovico et al., 2001). However, considerable evidence suggests that yeast cells also possess a programmed necrotic cell death pathway similar to that of their mammalian counterparts, where many of the conditions and processes which regulate programmed necrosis in mammals have also been conserved (Galluzzi et al., 2011).
For instance, vacuolar dysfunction and subsequent cytoplasmic acidification can commit yeast cells to necrosis (Schauer et al., 2009), just like in mammalian cells (Hitomi et al., 2008). Necrosis in yeast is also regulated by yeast heat shock protein Hsp90 (Dudgeon et al., 2008), the homolog of human Hsp90p. The latter is a cytosolic chaperone of several kinases, one of which is the pro-necrotic signaling protein RIP1 kinase (Lewis et al., 2000; Vanden Berghe et al., 2003). Whilst a clear homolog of RIP1 kinase in yeast has not yet been elucidated, yeast Hsp90p has proved to be a key requirement for tunicamycin-induced necrosis of yeast cells deficient in calcineurin, a phosphatase enzyme that suppresses necrosis in yeast (Dudgeon et al., 2008). Additionally, necrotic death under these conditions is preceded by the accumulation of ROS, implying that oxidative stress is an important inducer of this death phenotype in yeast, as in mammals.
Further evidence of the involvement of ROS in programmed necrosis in yeast is the fact that yeast mitochondria have also been implicated as a key requirement of this pathway. For instance, necrosis induced by expression of proteinaceous elicitor harpin (Pss) derived from Pseudomonas syringae, was avoided altogether in S. cerevisiae rho− (ρ−) “petite” cells containing dysfunctional mitochondria (Sripriya et al., 2009). Similarly, functional mitochondria are required for both apoptotic and necrotic cell death of aging yeast cells induced by heterologous expression of α-synuclein, which is accompanied by ROS accumulation and disruption of mitochondrial membrane potential (Büttner et al., 2008). Even unsaturated free fatty acid (FFA)-induced necrosis in yeast depends on the presence of functional mitochondria and is accompanied by the accumulation of ROS (Rockenfeller et al., 2010).
Peroxisomes, which generate ROS such as H2O2, can also act as key regulators of necrosis in yeast. Deletion of PEX6, encoding a crucial component of the peroxisomal protein import machinery in S. cerevisiae cells, caused increased sensitivity to acetic acid-induced stress, accompanied by necrosis and ROS accumulation in cells approaching stationary phase growth (Jungwirth et al., 2008). Similarly, removal of peroxisomal peroxiredoxin Pmt20 induced massive necrotic cell death accompanied by pronounced ROS accumulation and lipid peroxidation in the yeast Hansenula polymorpha (Bener Aksam et al., 2008).
Another prominent regulator of necrosis in yeast is the mitochondrial yeast endonuclease G homolog Nuc1p, which also triggers apoptosis when released from the mitochondria. Deletion of its coding sequence NUC1 was found to inhibit apoptotic death of yeast cells grown on glycerol medium, whilst enhancing necrosis of aging or peroxide-treated yeast cells grown on fermentative glucose medium (Büttner et al., 2007).
Similarly, a recent study has shown that deletion of the PEP4 sequence, encoding the yeast ortholog of mammalian cathepsin D, Pep4p, upregulates necrotic and apoptotic death in chronologically aging yeast cells. Conversely, prolonged hyperexpression of Pep4p increased cell longevity by specifically inhibiting necrosis. This anti-necrotic effect involved epigenetic deacetylation of histones mediated by polyamines, the biosynthesis of which was enhanced by the expression of Pep4p (Carmona-Gutiérrez et al., 2011a). Polyamines are molecules with antioxidant properties, which suppress the oxidative and inflammatory stresses associated with aging (Løvaas and Carlin, 1991). In fact, polyamine cell content is known to decline as aging progresses in both yeast (Eisenberg et al., 2009; Carmona-Gutiérrez et al., 2011a) and mammals (Scalabrino and Ferioli, 1984; Nishimura et al., 2006).
Eisenberg et al. (2009) were the first to establish an association between age-induced decline of polyamine content and necrosis in yeast cells, reporting a large percentage of chronologically aging yeast cells that were necrotic and that depletion of natural yeast cell polyamines not only accelerated necrosis, but increased ROS accumulation and shortened cell lifespan. Conversely, addition of the polyamine spermidine actually prevented necrosis, reduced oxidative stress, and increased longevity. Intriguingly, the cytoprotective effects of spermidine were mediated by the activation of autophagy, and were in fact abolished upon deletion of crucial autophagy genes such as ATG7, further demonstrating the pro-survival role of autophagy under conditions of age-related stress. Furthermore, the study showed that spermidine-induced suppression of necrosis in aging yeast cells is mediated by epigenetic deacetylation of histones (Eisenberg et al., 2009).
In the light of all this evidence, it is clear that necrosis in yeast is indeed a highly regulated and therefore PCD pathway. Its involvement in highly physiological processes such as aging, along with its requirement of functional mitochondria and, in particular, the fact that it can be actively suppressed by mechanisms as intricate as epigenetic modulation, all strongly support this argument. Furthermore, given the near ubiquitous involvement of ROS in various yeast necrotic scenarios, it is evident that oxidative stress and ROS accumulation play an important, if not central role in programmed necrosis in yeast, just like in mammals (Eisenberg et al., 2010). At present, however, the full extent of this role remains to be elucidated.
From a physiological perspective, the role of programmed necrosis as opposed to apoptosis in yeast has been suggested to be that of a “noisy” response, which involves the release of damage-associated molecular patterns (DAMPs) that could act as warning signals for surviving cells in a colony under extreme stress (Galluzzi et al., 2011). This proposed hypothesis is corroborated by the observed release, from necrotic yeast cells, of Nhp6Ap (Eisenberg et al., 2009). This is the yeast ortholog of mammalian high-mobility group box 1 (HMGB1), which serves as a danger signal (Apetoh et al., 2007). However, whether Nhp6Ap shares the same function as HMGB1 remains to be elucidated (Galluzzi et al., 2011).
Oxidative Stress, Autophagy, and Cell Death in Yeast
Although primarily a homeostatic pro-survival response to stresses such as nutrient depletion and ROS accumulation, it has been suggested that autophagy might also act as a mediator of cell death when it occurs at high levels (Pattingre et al., 2005), given observed instances of numerous autophagic bodies present in the dying cells of various organisms (Tsujimoto and Shimizu, 2005; Kourtis and Tavernarakis, 2009). Amongst these organisms are yeast cells, where large scale autophagy induced by stimuli such as rapamycin is followed by growth arrest and a sharp decline in cell viability (Kissová et al., 2004). Also, as stated earlier, addition of rapamycin to respiring yeast cells induces selective mitophagy (Kissová et al., 2007). Furthermore, rapamycin-induced autophagy is accompanied by early ROS accumulation and early mitochondrial lipid peroxidation. Inhibition of these oxidative events by resveratrol largely impairs autophagy of cell components and delays rapamycin-induced cell death (Kissová et al., 2006). What this evidence collectively implies is that autophagic pathways, such as mitophagy, can have a pro-death role in yeast, driven by massive accumulation of ROS in the mitochondria. Furthermore, a recent study by Dziedzic and Caplan (2012) presented evidence suggesting that autophagy may accelerate cell death in S. cerevisiae under starvation conditions, given that deletion of autophagic proteins, encoded by ATG8, delayed cell death under leucine starvation.
Therefore, a complex interplay exists between autophagy and cell death, two distinct stress responses which, depending on the circumstances of the cells, can either compete against each other or cooperate (Carmona-Gutierrez et al., 2010a) in a manner which is probably regulated, at least in part, by the ROS-dependent mitophagic turnover of mitochondria. This is probably one of the few pro-death aspects of autophagy which is understood. Otherwise, the extent to which autophagy acts as a cell death response in yeast is largely unclear.
There has in fact been much heated debate as to whether autophagy is even a true cell death mechanism per se. Some argue that the very term “autophagic cell death” – a definition based solely on the morphological appearance of vacuolization in dying cells – can be misleading and often incorrectly used to refer to cases where cells actually die with autophagy but not by autophagy (Kroemer and Levine, 2008). Whilst the plausibility of cell death by autophagy has not been entirely excluded, there seems to be a consensus that in the overwhelming majority of cases, autophagy constitutes a cytoprotective pathway, the removal of which accelerates rather than prevents cell mortality (Galluzzi et al., 2012).
Oxidative Stress and Aging
The physiological process of aging in budding yeast is heavily associated with ROS accumulation and PCD (Laun et al., 2001; Fabrizio et al., 2004; Herker et al., 2004). Replicative aging is used as a model for aging in mammalian proliferating cells (particularly stem cells), and chronological aging is used as an aging model for mammalian post-mitotic cells (Kaeberlein, 2010).
Replicative aging in yeast is defined by the number of times a specific yeast mother cell can divide before it reaches senescence and dies (Mortimer and Johnston, 1959; Müller et al., 1980). Budding yeast cells replicate asymmetrically (Hartwell and Unger, 1977) in such a way that aging factors are retained by the mother cell. These include oxidized proteins (Aguilaniu et al., 2003; Erjavec and Nystrom, 2007), protein aggregates (Erjavec et al., 2007), and extrachromosomal ribosomal DNA circles (ERCs) believed to be generated by rDNA recombination (Sinclair and Guarente, 1997; Kaeberlein et al., 1999). The same mother cell can replicate several times until a certain threshold of damaged cell content is accumulated, at which time the cell dies via PCD pathways such as apoptosis (Laun et al., 2001).
One other prominent form of cell damage believed to contribute to replicative aging is the accumulation of dysfunctional mitochondria that are rich in ROS in old yeast mother cells (Laun et al., 2001). This clearly indicates that ROS promote replicative aging in yeast. In fact, several other studies have shown that genetic or environmental alterations which increase the ROS burden on yeast mother cells result in a shortening of their lifespan. Such alterations include the deletion of genes encoding SODs (Barker et al., 1999; Wawryn et al., 1999) and catalases, the increase of partial pressure of atmospheric oxygen and the modulation of GSH abundance (Nestelbacher et al., 2000).
Likewise, studies have shown that ROS originating from dysfunctional mitochondria contribute to chronological aging in yeast (Longo et al., 1996;Fabrizio et al., 2003, 2004). This aging model is defined by how long a yeast cell can survive once it has reached the post-diauxic, stationary phase of growth, when nutrients become scarce (Longo and Fabrizio, 2012). The pro-aging effects of ROS are corroborated by the observed accumulation of oxidatively damaged proteins (Reverter-Branchat et al., 2004) and ROS (Herker et al., 2004) in chronologically aged cells. Furthermore, yeast chronological lifespan can be extended by interventions that induce upregulated expression of SOD2, activation of Msn2p and Msn4p transcription factors, and activation of protein kinase Rim15p, all of which are involved in the stress response that mitigates oxidative stress (Fabrizio et al., 2001, 2003).
Further evidence of the role of oxidative stress in the aging of yeast cells lies in the manner by which, both replicative and chronological aging in yeast are regulated. Both aging types depend on the same set of nutrient-sensing kinases: Ras/cAMP-dependent PKA, Sch9, and Tor, the activities of which are, in turn, dependent on nutrient availability in the environment. Under nutrient-rich conditions, all three kinases naturally promote cell growth, cell division, and thus also aging of yeast cells, culminating in PCD. The pro-aging effect of these kinases is largely due to their suppressive influence upon the yeast stress response mechanisms, in the presence of nutrients (Fabrizio et al., 2001, 2003; Inoue and Klionsky, 2010; Longo and Fabrizio, 2012).
Conversely, when nutrients are scarce, the Sch9, Tor, and Ras/cAMP-dependent PKA pathways are downregulated and cell longevity is increased (Fabrizio and Longo, 2008). A similar longevity-inducing scenario of nutrient starvation called calorie restriction has consistently been shown to increase longevity of both replicatively (Lin et al., 2002) and chronologically aging cells (Reverter-Branchat et al., 2004). It is believed that the downregulation of Sch9, Tor, and Ras/cAMP-dependent PKA signaling is coupled with a concomitant upregulation of cytoprotective stress response regulators, which are activated downstream of the kinases. These include Msn2/4p and Gis1 transcription factors, along with Rim15 kinase (Wei et al., 2008) the collective activation of which enhances cytoprotective pathways such as autophagy (Inoue and Klionsky, 2010), a process associated with increased longevity and reduced ROS in aging yeast (Eisenberg et al., 2009).
Thus yeast cell aging is a conserved, genetically regulated process, where a cell’s ability to upregulate its cytoprotective stress response mechanisms above a certain threshold is actively suppressed by a sustained baseline activity of nutrient-sensing Sch9, Tor, and Ras/cAMP-dependent PKA kinases (Longo et al., 2005). In this way, aging cells accumulate a certain amount of oxidative damage, without upregulating the current activity of their antioxidant defenses. They consequently accumulate ROS such as superoxide radicals (Fabrizio et al., 2004) and succumb to PCD, be it apoptosis (Herker et al., 2004) or necrosis (Eisenberg et al., 2009). Thus, in a physiological context, replicatively aged cells that are no longer capable of segregating damaged cell components from their daughter cells, are eliminated before they seriously compromise the integrity of a population. Likewise, chronologically aged cells, which have suffered too much oxidative damage to be worth maintaining, are eliminated to free up nutrients and enhance the survival chances of younger, fitter cells in a yeast population, especially during starvation. In the meantime, these same fitter individuals of a starved yeast population may further increase their chances of survival by actively suppressing the aforementioned kinase pathways to enhance their cytoprotective systems, until new nutrient sources are made available (Fabrizio and Longo, 2008).
Concluding Remarks
The evidence presented in this discussion clearly illustrates the remarkable ability of yeast to detect and appropriately respond to the constant threat of oxidative stress, using a diverse array of strategies such as ROS detoxification, autophagic degradation, and even cell death (Figure 1). Importantly, these defensive pathways are largely conserved in mammals. Thus, yeast has long proved itself to be a powerful research model, the use of which has thrown light upon many obscure aspects of important human pathologies that are harder to elucidate using other, more complex eukaryotic models.
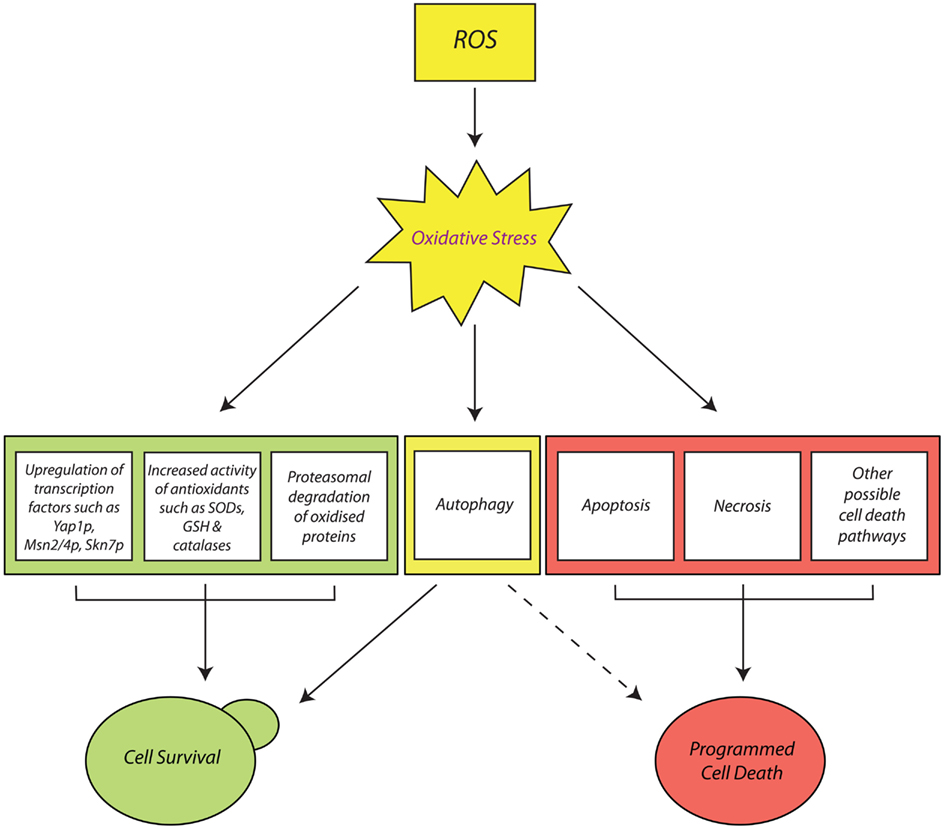
Figure 1. Cellular responses to oxidative stress in Saccharomyces cerevisiae. Oxidative stress, induced by the accumulation of reactive oxygen species (ROS) such as H2O2, and , can elicit a range of stress responses in budding yeast cells, that either result in cell survival (shown in green) or cell death (shown in red). Stress signals relayed by ROS themselves (such as H2O2) can activate transcription factors which upregulate the expression of genes encoding enzymatic (such as catalases) and non-enzymatic antioxidants (such as GSH). These response mechanisms, together with the targeted removal of small, oxidized proteins by the ubiquitin-dependent proteasome system (UPS), help ensure the survival of cells. Additionally, cells can activate cytoprotective autophagic pathways (bordered in yellow) that remove irreparably oxidized macromolecules or dysfunctional organelles, such as mitochondria. However, an abnormally high degree of autophagy might also mediate programmed cell death (PCD). Finally, exposure of cells to severe oxidative insults can elicit lethal response pathways such as apoptosis, necrosis, and possibly other forms of PCD which have yet to be discovered.
For instance, the prevalent involvement of ROS and functional mitochondria among most pro-death stimuli in yeast has served to highlight the central role of mitochondrial dysfunction and ROS accumulation in most important human pathologies such as aging (Laun et al., 2001) and Parkinson’s disease (Büttner et al., 2008). Furthermore, the conserved metabolic machinery and simplicity of yeast has allowed researchers to perform very revealing mechanistic studies of anti-tumor drugs (Balzan et al., 2004; Mitsui et al., 2005; Aouida et al., 2007) and disease-inducing toxins (Sokolov et al., 2006; Franssens et al., 2010), paving the way toward enhanced therapeutic treatment and better understanding of human diseases. The yeast model can even be used to examine important aspects of mammalian aging, such as the contributive roles of DNA damage and genome instability (Wei et al., 2011). Finally, the chance of uncovering more conserved PCD pathways in yeast cannot be excluded, especially given the increasing number of newly elucidated PCD pathways in other eukaryotes (Galluzzi et al., 2012).
A potential example of these new pathways is “linker cell death” which has so far only been observed in Caenorhabditis elegans. This cell death pathway, which proceeds independently of known death genes and caspases, is characterized by non-apoptotic markers such as uncondensed chromatin, indentations of the nuclear envelope, organelle swelling, and accumulation of membrane bound cytoplasmic structures (Abraham et al., 2007). It requires the expression of a pqn-41 sequence, encoding a polyglutamine repeat protein that bears similarity to Huntingtin (Blum et al., 2012), the cause of Huntington’s disease (Harper, 1999). The expression of pqn-41, which is itself dependent on the activity of a MAP kinase kinase called SEK-1, promotes cell death in parallel to a zinc-finger protein called LIN-29p.
In budding yeast, expression of expanded mammalian polyglutamines elicits physiological consequences similar to those of degenerating neurons in Huntington’s disease patients. These include an apoptotic phenotype and nuclear polyglutamine aggregation, both of which are caspase-dependent (Sokolov et al., 2006; Bocharova et al., 2008). It is very tempting to speculate whether yeast cells also possess a conserved pqn-41-like polyglutamine protein, the expression of which might elicit an alternative caspase-independent PCD pathway that is similar or identical to linker cell death in C. elegans. If this is the case, what impact might this have on our understanding of neurodegenerative pathologies such as Huntington’s disease? This is an intriguing question which has yet to be answered.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the authors’ laboratory is partly funded by the Malta Government Scholarship Scheme (MGSS) award, fund number ME 367/07/8, to Gianluca Farrugia and partly by Research Fund grants to Rena Balzan from the University of Malta.
References
Abdelwahid, E., Rolland, S., Teng, X., Conradt, B., Hardwick, J. M., and White, K. (2011). Mitochondrial involvement in cell death of non-mammalian eukaryotes. Biochim. Biophys. Acta 1813, 597–607.
Abraham, M. C., Lu, Y., and Shaham, S. (2007). A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev. Cell 12, 73–86.
Acharya, A., Das, I., Chandhok, D., and Saha, T. (2010). Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxid. Med. Cell. Longev. 3, 23–34.
Aguilaniu, H., Gustafsson, L., Rigoulet, M., and Nyström, T. (2003). Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753.
Ahn, S. H., Cheung, W. L., Hsu, J. Y., Diaz, R. L., Smith, M. M., and Allis, C. D. (2005). Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell 120, 25–36.
Ahn, S. H., Diaz, R. L., Grunstein, M., and Allis, C. D. (2006). Histone H2B deacetylation at lysine 11 is required for yeast apoptosis induced by phosphorylation of H2B at serine 10. Mol. Cell 24, 211–220.
Almeida, B., Büttner, S., Ohlmeier, S., Silva, A., Mesquita, A., Sampaio-Marques, B., Osório, N. S., Kollau, A., Mayer, B., Leão, C., Laranjinha, J., Rodrigues, F., Madeo, F., and Ludovico, P. (2007). NO-mediated apoptosis in yeast. J. Cell Sci. 120, 3279–3288.
Almeida, T., Marques, M., Mojzita, D., Amorim, M. A., Silva, R. D., Almeida, B., Rodrigues, P., Ludovico, P., Hohmann, S., Moradas-Ferreira, P., Côrte-Real, M., and Costa, V. (2008). Isc1p plays a key role in hydrogen peroxide resistance and chronological lifespan through modulation of iron levels and apoptosis. Mol. Biol. Cell 19, 865–876.
Ames, B. N., Gold, L. S., and Willett, W. C. (1995). The causes and prevention of cancer. Proc. Natl. Acad. Sci. U.S.A. 92, 5258–5265.
Ames, B. N., Shigenaga, M. K., and Hagen, T. M. (1993). Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. U.S.A. 90, 7915–7922.
Andrus, P. K., Fleck, T. J., Gurney, M. E., and Hall, E. D. (1998). Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 71, 2041–2048.
Aouida, M., Mekid, H., Belhadj, O., Mir, L. M., and Tounekti, O. (2007). Mitochondria-independent morphological and biochemical apoptotic alterations promoted by the anti-tumor agent bleomycin in Saccharomyces cerevisiae. Biochem. Cell Biol. 85, 49–55.
Apetoh, L., Ghiringhelli, F., Tesniere, A., Obeid, M., Ortiz, C., Criollo, A., Mignot, G., Maiuri, M. C., Ullrich, E., Saulnier, P., Yang, H., Amigorena, S., Ryffel, B., Barrat, F. J., Saftig, P., Levi, F., Lidereau, R., Nogues, C., Mira, J. P., Chompret, A., Joulin, V., Clavel-Chapelon, F., Bourhis, J., André, F., Delaloge, S., Tursz, T., Kroemer, G., and Zitvogel, L. (2007). Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059.
Ashford, T. P., and Porter, K. R. (1962). Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 12, 198–202.
Avery, A. M., and Avery, S. V. (2001). Saccharomyces cerevisiae expresses three phospholipid hydroperoxide glutathione peroxidases. J. Biol. Chem. 276, 33730–33735.
Avery, A. M., Willetts, S. A., and Avery, S. V. (2004). Genetic dissection of the phospholipid hydroperoxidase activity of yeast gpx3 reveals its functional importance. J. Biol. Chem. 279, 46652–46658.
Baines, C. P. (2010). Role of the mitochondrion in programmed necrosis. Front. Physiol. 1:156. doi:10.3389/fphys.2010.00156
Balzan, R., Sapienza, K., Galea, D. R., Vassallo, N., Frey, H., and Bannister, W. H. (2004). Aspirin commits yeast cells to apoptosis depending on carbon source. Microbiology 150, 109–115.
Barber, S. C., Mead, R. J., and Shaw, P. J. (2006). Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 1762, 1051–1067.
Barber, S. C., and Shaw, P. J. (2010). Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 48, 629–641.
Barker, M. G., Brimage, L. J., and Smart, K. A. (1999). Effect of Cu, Zn superoxide dismutase disruption mutation on replicative senescence in Saccharomyces cerevisiae. FEMS Microbiol. Lett. 177, 199–204.
Bartosz, G. (2009). Reactive oxygen species: destroyers or messengers? Biochem. Pharmacol. 77, 1303–1315.
Bayliak, M., Gospodaryov, D., Semchyshyn, H., and Lushchak, V. (2008). Inhibition of catalase by aminotriazole in vivo results in reduction of glucose-6-phosphate dehydrogenase activity in Saccharomyces cerevisiae cells. Biochemistry (Mosc.) 73, 420–426.
Baynes, J. W., and Thorpe, S. R. (1999). Role of oxidative stress in diabetes: a new perspective on an old paradigm. Diabetes 48, 1–9.
Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624.
Behl, C. (1999). Alzheimer’s disease and oxidative stress: implications for novel therapeutic approaches. Prog. Neurobiol. 57, 301–323.
Bener Aksam, E., Jungwirth, H., Kohlwein, S. D., Ring, J., Madeo, F., Veenhuis, M., and van der Klei, I. J. (2008). Absence of the peroxiredoxin Pmp20 causes peroxisomal protein leakage and necrotic cell death. Free Radic. Biol. Med. 45, 1115–1124.
Berlett, B. S., and Stadtman, E. R. (1997). Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272, 20313–20316.
Bertolaet, B. L., Clarke, D. J., Wolff, M., Watson, M. H., Henze, M., Divita, G., and Reed, S. I. (2001). UBA domains of DNA damage-inducible proteins interact with ubiquitin. Nat. Struct. Biol. 8, 417–422.
Bettiga, M., Calzari, L., Orlandi, I., Alberghina, L., and Vai, M. (2004). Involvement of the yeast metacaspase Yca1 in ubp10Delta-programmed cell death. FEMS Yeast Res. 5, 141–147.
Bhatia-Kissová, I., and Camougrand, N. (2010). Mitophagy in yeast: actors and physiological roles. FEMS Yeast Res. 10, 1023–1034.
Bienert, G. P., Schjoerring, J. K., and Jahn, T. P. (2006). Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 1758, 994–1003.
Bilinski, T., Litwinska, J., Blaszczynski, M., and Bajus, A. (1989). Superoxide dismutase deficiency and the toxicity of the products of autooxidation of polyunsaturated fatty acids in yeast. Biochem. Biophys. Acta 1001, 102–106.
Billard, P., Dumond, H., and Bolotin-Fukuhara, M. (1997). Characterization of an AP-1-like transcription factor that mediates an oxidative stress response in Kluyveromyces lactis. Mol. Gen. Genet. 257, 62–70.
Blaheta, R. A., and Cinatl, J. Jr. (2002). Anti-tumor mechanisms of valproate: a novel role for an old drug. Med. Res. Rev. 22, 492–511.
Blanchard, F., Rusiniak, M. E., Sharma, K., Sun, X., Todorov, I., Castellano, M. M., Gutierrez, C., Baumann, H., and Burhans, W. C. (2002). Targeted destruction of DNA replication protein Cdc6 by cell death pathways in mammals and yeast. Mol. Biol. Cell 13, 1536–1549.
Blum, E. S., Abraham, M. C., Yoshimura, S., Lu, Y., and Shaham, S. (2012). Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science 335, 970–973.
Bocharova, N. A., Sokolov, S. S., Knorre, D. A., Skulachev, V. P., and Severin, F. F. (2008). Unexpected link between anaphase promoting complex and the toxicity of expanded polyglutamines expressed in yeast. Cell Cycle 7, 3943–3946.
Boveris, A., Oshino, N., and Chance, B. (1972). The cellular production of hydrogen peroxide. Biochem. J. 128, 617–630.
Boy-Marcotte, E., Perrot, M., Bussereau, F., Boucherie, H., and Jacquet, M. (1998). Msn2p and Msn4p control a large number of genes induced at the diauxic transition which are repressed by cyclic AMP in Saccharomyces cerevisiae. J. Bacteriol. 180, 1044–1052.
Brennan, R. J., and Schiestl, R. H. (1996). Cadmium is an inducer of oxidative stress in yeast. Mutat. Res. 356, 171–178.
Brennan, R. J., Swoboda, B. E., and Schiestl, R. H. (1994). Oxidative mutagens induce intrachromosomal recombination in yeast. Mutat. Res. 308, 159–167.
Brown, J. L., Bussey, H., and Stewart, R. C. (1994). Yeast Skn7p functions in a eukaryotic two-component regulatory pathway. EMBO J. 13, 5186–5194.
Burhans, W. C., and Weinberger, M. (2012). DNA damage and DNA replication stress in yeast models of aging. Subcell. Biochem. 57, 187–206.
Burhans, W. C., Weinberger, M., Marchetti, M. A., Ramachandran, L., D’Urso, G., and Huberman, J. A. (2003). Apoptosis-like yeast cell death in response to DNA damage and replication defects. Mutat. Res. 532, 227–243.
Bussche, J. V., and Soares, E. V. (2011). Lead induces oxidative stress and phenotypic markers of apoptosis in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 90, 679–687.
Büttner, S., Bitto, A., Ring, J., Augsten, M., Zabrocki, P., Eisenberg, T., Jungwirth, H., Hutter, S., Carmona-Gutierrez, D., Kroemer, G., Winderickx, J., and Madeo, F. (2008). Functional mitochondria are required for alpha-synuclein toxicity in aging yeast. J. Biol. Chem. 283, 7554–7560.
Büttner, S., Eisenberg, T., Carmona-Gutierrez, D., Ruli, D., Knauer, H., Ruckenstuhl, C., Sigrist, C., Wissing, S., Kollroser, M., Fröhlich, K.-U., Sigrist, S. J., and Madeo, F. (2007). Endonuclease G regulates budding yeast life and death. Mol. Cell 25, 233–246.
Büttner, S., Ruli, D., Vögtle, F. N., Galluzzi, L., Moitzi, B., Eisenberg, T., Kepp, O., Habernig, L., Carmona-Gutierrez, D., Rockenfeller, P., Laun, P., Breitenbach, M., Khoury, C., Fröhlich, K.-U., Rechberger, G., Meisinger, C., Kroemer, G., and Madeo, F. (2011). A yeast BH3-only protein mediates the mitochondrial pathway of apoptosis. EMBO J. 30, 2779–2792.
Cabiscol, E., Piulats, E., Echave, P., Herrero, E., and Ros, J. (2000). Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J. Biol. Chem. 35, 27393–27398.
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230.
Campbell, C. L., and Thorsness, P. E. (1998). Escape of mitochondrial DNA to the nucleus in yme1 yeast is mediated by vacuolar-dependent turnover of abnormal mitochondrial compartments. J. Cell Sci. 111, 2455–2464.
Candé, C., Vahsen, N., Kouranti, I., Schmitt, E., Daugas, E., Spahr, C., Luban, J., Kroemer, R. T., Giordanetto, F., Garrido, C., Penninger, J. M., and Kroemer, G. (2004). AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene 23, 1514–1521.
Carmona-Gutiérrez, D., Bauer, M. A., Ring, J., Knauer, H., Eisenberg, T., Büttner, S., Ruckenstuhl, C., Reisenbichler, A., Magnes, C., Rechberger, G. N., Birner-Gruenberger, R., Jungwirth, H., Fröhlich, K. U., Sinner, F., Kroemer, G., and Madeo, F. (2011a). The propeptide of yeast cathepsin D inhibits programmed necrosis. Cell Death Dis. 2, e161.
Carmona-Gutierrez, D., Reisenbichler, A., Heimbucher, P., Bauer, M. A., Braun, R. J., Ruckenstuhl, C., Büttner, S., Eisenberg, T., Rockenfeller, P., Fröhlich, K. U., Kroemer, G., and Madeo, F. (2011b). Ceramide triggers metacaspase-independent mitochondrial cell death in yeast. Cell Cycle 10, 3973–3978.
Carmona-Gutierrez, D., Eisenberg, T., Büttner, S., Meisinger, C., Kroemer, G., and Madeo, F. (2010a). Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ. 17, 763–773.
Carmona-Gutierrez, D., Fröhlich, K. U., Kroemer, G., and Madeo, F. (2010b). Metacaspases are caspases. Doubt no more. Cell Death Differ. 17, 377–378.
Cebulski, J., Malouin, J., Pinches, N., Cascio, V., and Austriaco, N. (2011). Yeast Bax inhibitor, Bxi1p, is an ER-localized protein that links the unfolded protein response and programmed cell death in Saccharomyces cerevisiae. PLoS ONE 6, e20882. doi:10.1371/journal.pone.0020882
Cecarini, V., Gee, J., Fioretti, E., Amici, M., Angeletti, M., Eleuteri, A. M., and Keller, J. N. (2007). Protein oxidation and cellular homeostasis: emphasis on metabolism. Biochim. Biophys. Acta 1773, 93–104.
Chae, H. J., Ke, N., Kim, H. R., Chen, S., Godzik, A., Dickman, M., and Reed, J. C. (2003). Evolutionarily conserved cytoprotection provided by Bax inhibitor-1 homologs from animals, plants, and yeast. Gene 323, 101–113.
Chae, H. Z., Chung, S. J., and Rhee, S. G. (1994). Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 269, 27670–27678.
Chandra, J., Samali, A., and Orrenius, S. (2000). Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 29, 323–333.
Chatenay-Lapointe, M., and Shadel, G. S. (2010). Stressed-out mitochondria get MAD. Cell Metab. 12, 559–560.
Chen, L., and Madura, K. (2002). Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol. Cell. Biol. 22, 4902–4913.
Cohen, G., Rapatz, W., and Ruis, H. (1988). Sequence of the Saccharomyces cerevisiae CTA1 gene and amino acid sequence of catalase A derived from it. Eur. J. Biochem. 176, 159–163.
Collinson, L. P., and Dawes, I. W. (1992). Inducibility of the response of yeast cells to peroxide stress. J. Gen. Microbiol. 138, 329–335.
Costa, V., and Moradas-Ferreira, P. (2001). Oxidative stress and signal transduction in Saccharomyces cerevisiae: insights into ageing, apoptosis and diseases. Mol. Aspects Med. 22, 217–246.
Costa, V. M., Amorim, M. A., Quintanilha, A., and Moradas-Ferreira, P. (2002). Hydrogen peroxide-induced carbonylation of key metabolic enzymes in Saccharomyces cerevisiae: the involvement of the oxidative stress response regulators Yap1 and Skn7. Free Radic. Biol. Med. 33, 1507–1515.
Daroui, P., Desai, S. D., Li, T. K., Liu, A. A., and Liu, L. F. (2004). Hydrogen peroxide induces topoisomerase I-mediated DNA damage and cell death. J. Biol. Chem. 279, 14587–14594.
Davidson, J. F., Whyte, B., Bissinger, P. H., and Schiestl, R. H. (1996). Oxidative stress is involved in heat-induced cell death in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 93, 5116–5121.
de Castro, P. A., Savoldi, M., Bonatto, D., Barros, M. H., Goldman, M. H., Berretta, A. A., and Goldman, G. H. (2011). Molecular characterization of propolis-induced cell death in Saccharomyces cerevisiae. Eukaryot. Cell 10, 398–411.
Deffieu, M., Bhatia-Kissová, I., Salin, B., Galinier, A., Manon, S., and Camougrand, N. (2009). Glutathione participates in the regulation of mitophagy in yeast. J. Biol. Chem. 284, 14828–14837.
Delaunay, A., Isnard, A. D., and Toledano, M. B. (2000). H2O2 sensing through oxidation of the Yap1 transcription factor. EMBO J. 19, 5157–5166.
Delaunay, A., Pflieger, D., Barrault, M. B., Vinh, J., and Toledano, M. B. (2002). A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111, 471–481.
Donati, A., Cavallini, G., Paradiso, C., Vittorini, S., Pollera, M., Gori, Z., and Bergamini, E. (2001). Age-related changes in the autophagic proteolysis of rat isolated liver cells: effects of antiaging dietary restrictions. J. Gerontol. A Biol. Sci. Med. Sci. 56, 375–383.
Drakulic, T., Temple, M. D., Guido, R., Jarolim, S., Breitenbach, M., Attfield, P. V., and Dawes, I. W. (2005). Involvement of oxidative stress response genes in redox homeostasis, the level of reactive oxygen species, and ageing in Saccharomyces cerevisiae. FEMS Yeast Res. 5, 1215–1228.
Du, L., Su, Y., Sun, D., Zhu, W., Wang, J., Zhuang, X., Zhou, S., and Lu, Y. (2008). Formic acid induces Yca1p-independent apoptosis-like cell death in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 8, 531–539.
Du, L., Yu, Y., Chen, J., Liu, Y., Xia, Y., Chen, Q., and Liu, X. (2007). Arsenic induces caspase- and mitochondria-mediated apoptosis in Saccharomyces cerevisiae. FEMS Yeast Res. 7, 860–865.
Dudgeon, D. D., Zhang, N., Ositelu, O. O., Kim, H., and Cunningham, K. W. (2008). Nonapoptotic death of Saccharomyces cerevisiae cells that is stimulated by Hsp90 and inhibited by calcineurin and Cmk2 in response to endoplasmic reticulum stresses. Eukaryot. Cell 7, 2037–2051.
Dumond, H., Danielou, N., Pinto, M., and Bolotin-Fukuhara, M. (2000). A large-scale study of Yap1p-dependent genes in normal aerobic and H2O2-stress conditions: the role of Yap1p in cell proliferation control in yeast. Mol. Microbiol. 36, 830–845.
Dziedzic, S. A., and Caplan, A. B. (2012). Autophagy proteins play cytoprotective and cytocidal roles in leucine starvation-induced cell death in Saccharomyces cerevisiae. Autophagy 8, 1–8.
Eisenberg, T., Carmona-Gutierrez, D., Büttner, S., Tavernarakis, N., and Madeo, F. (2010). Necrosis in yeast. Apoptosis 15, 257–268.
Eisenberg, T., Knauer, H., Schauer, A., Büttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., Ring, J., Schroeder, S., Magnes, C., Antonacci, L., Fussi, H., Deszcz, L., Hartl, R., Schraml, E., Criollo, A., Megalou, E., Weiskopf, D., Laun, P., Heeren, G., Breitenbach, M., Grubeck-Loebenstein, B., Herker, E., Fahrenkrog, B., Fröhlich, K. U., Sinner, F., Tavernarakis, N., Minois, N., Kroemer, G., and Madeo, F. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314.
Elsasser, S., Chandler-Militello, D., Müller, B., Hanna, J., and Finley, D. (2004). Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 279, 26817–26822.
Erjavec, N., Larsson, L., Grantham, J., and Nystrom, T. (2007). Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 21, 2410–2421.
Erjavec, N., and Nystrom, T. (2007). Sir2p-dependent protein segregation gives rise to a superior reactive oxygen species management in the progeny of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 104, 10877–10881.
Evans, M. V., Turton, H. E., Grant, C. M., and Dawes, I. W. (1998). Toxicity of linoleic acid hydroperoxide to Saccharomyces cerevisiae: involvement of a respiration-related process for maximal sensitivity and adaptive response. J. Bacteriol. 180, 483–490.
Fabrizio, P., Battistella, L., Vardavas, R., Gattazzo, C., Liou, L. L., Diaspro, A., Dossen, J. W., Gralla, E. B., and Longo, V. D. (2004). Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae. J. Cell Biol. 166, 1055–1067.
Fabrizio, P., Liou, L. L., Moy, V. N., Diaspro, A., Valentine, J. S., Gralla, E. B., and Longo, V. D. (2003). SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics 163, 35–46.
Fabrizio, P., and Longo, V. D. (2008). Chronological aging-induced apoptosis in yeast. Biochim. Biophys. Acta 1783, 1280–1285.
Fabrizio, P., Pozza, F., Pletcher, S. D., Gendron, C. M., and Longo, V. D. (2001). Regulation of longevity and stress resistance by Sch9 in yeast. Science 292, 288–290.
Facecchia, K., Fochesato, L.-A., Ray, S. D., Stohs, S. J., and Pandey, S. (2011). Oxidative toxicity in neurodegenerative diseases: role of mitochondrial dysfunction and therapeutic strategies. J. Toxicol. 2011, 1–11.
Fahrenkrog, B., Sauder, U., and Aebi, U. (2004). The S. cerevisiae HtrA-like protein Nma111p is a nuclear serine protease that mediates yeast apoptosis. J. Cell Sci. 117, 115–126.
Fang, J., and Beattie, D. S. (2003). External alternative NADH dehydrogenase of Saccharomyces cerevisiae: a potential source of superoxide. Free Radic. Biol. Med. 34, 478–488.
Fannjiang, Y., Cheng, W. C., Lee, S. J., Qi, B., Pevsner, J., McCaffery, J. M., Hill, R. B., Basañez, G., and Hardwick, J. M. (2004). Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev. 18, 2785–2797.
Farah, M. E., and Amberg, D. C. (2007). Conserved actin cysteine residues are oxidative stress sensors that can regulate cell death in yeast. Mol. Biol. Cell 18, 1359–1365.
Fernandes, L., Rodrigues-Pousada, C., and Struhl, K. (1997). Yap, a novel family of eight bZIP proteins in Saccharomyces cerevisiae with distinct biological functions. Mol. Cell. Biol. 17, 6982–6993.
Finn, K., Lowndes, N. F., and Grenon, M. (2011). Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell. Mol. Life Sci. 69, 1447–1473.
Flower, T. R., Chesnokova, L. S., Froelich, C. A., Dixon, C., and Witt, S. N. (2005). Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. J. Mol. Biol. 351, 1081–1100.
Foland, T. B., Dentler, W. L., Suprenant, K. A., Gupta, M. L. Jr., and Himes, R. H. (2005). Paclitaxel-induced microtubule stabilization causes mitotic block and apoptotic-like cell death in a paclitaxel-sensitive strain of Saccharomyces cerevisiae. Yeast 22, 971–978.
Fomenko, D. E., Koc, A., Agisheva, N., Jacobsen, M., Kaya, A., Malinouski, M., Rutherford, J. C., Siu, K. L., Jin, D. Y., Winge, D. R., and Gladyshev, V. N. (2011). Thiol peroxidases mediate specific genome-wide regulation of gene expression in response to hydrogen peroxide. Proc. Natl. Acad. Sci. U.S.A. 108, 2729–2734.
Frankenberg, D., Frankenberg-Schwager, M., and Harbich, R. (1993). Mechanisms of oxygen radiosensitization in irradiated yeast. I. DNA double-strand breakage. Int. J. Radiat. Biol. 64, 511–521.
Franssens, V., Boelen, E., Anandhakumar, J., Vanhelmont, T., Büttner, S., and Winderickx, J. (2010). Yeast unfolds the road map toward alpha-synuclein-induced cell death. Cell Death Differ. 17, 746–753.
Fridovich, I. (1989). Superoxide dismutase: an adaptation to a paramagnetic gas. J. Biol. Chem. 264, 7761–7764.
Fröhlich, K.-U., and Madeo, F. (2001). Apoptosis in yeast: a new model for aging research. Exp. Gerontol. 37, 27–31.
Galluzzi, L., Vanden Berghe, T., Vanlangenakker, N., Buettner, S., Eisenberg, T., Vandenabeele, P., Madeo, F., and Kroemer, G. (2011). Programmed necrosis from molecules to health and disease. Int. Rev. Cell Mol. Biol. 289, 1–35.
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., Dawson, T. M., Dawson, V. L., El-Deiry, W. S., Fulda, S., Gottlieb, E., Green, D. R., Hengartner, M. O., Kepp, O., Knight, R. A., Kumar, S., Lipton, S. A., Lu, X., Madeo, F., Malorni, W., Mehlen, P., Nuñez, G., Peter, M. E., Piacentini, M., Rubinsztein, D. C., Shi, Y., Simon, H. U., Vandenabeele, P., White, E., Yuan, J., Zhivotovsky, B., Melino, G., and Kroemer, G. (2012). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120.
Garrido, E. O., and Grant, C. M. (2002). Role of thioredoxins in the response of Saccharomyces cerevisiae to oxidative stress induced by hydroperoxides. Mol. Microbiol. 43, 993–1003.
Gasch, A. P., Spellman, P. T., Kao, C. M., Carmel-Harel, O., Eisen, M. B., Storz, G., Botstein, D., and Brown, P. O. (2000). Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11, 4241–4257.
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., and Springer, W. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131.
Giannattasio, S., Guaragnella, N., Côrte-Real, M., Passarella, S., and Marra, E. (2005). Acid stress adaptation protects Saccharomyces cerevisiae from acetic acid-induced programmed cell death. Gene 354, 93–98.
Gille, G., and Sigler, K. (1995). Oxidative stress and living cells. Folia Microbiol. (Praha) 40, 131–152.
Giugliano, D., Ceriello, A., and Paolisso, G. (1996). Oxidative stress and diabetic vascular complications. Diabetes Care 19, 257–267.
Glaumann, H. (1989). Crinophagy as a means for degrading excess secretory proteins in rat liver. Revis. Biol. Celular. 20, 97–110.
Goldman, S. J., Taylor, R., Zhang, Y., and Jin, S. (2010). Autophagy and the degradation of mitochondria. Mitochondrion 10, 309–315.
Gourlay, C. W., and Ayscough, K. R. (2006). Actin-induced hyperactivation of the Ras signalling pathway leads to apoptosis in Saccharomyces cerevisiae. Mol. Cell. Biol. 26, 6487–6501.
Gross, E., Sevier, C. S., Heldman, N., Vitu, E., Bentzur, M., Kaiser, C. A., Thorpe, C., and Fass, D. (2006). Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. U.S.A. 103, 299–304.
Guaragnella, N., Antonacci, L., Passarella, S., Marra, E., and Giannattasio, S. (2007). Hydrogen peroxide and superoxide anion production during acetic acid-induced yeast programmed cell death. Folia Microbiol. (Praha) 52, 237–240.
Guaragnella, N., Passarella, S., Marra, E., and Giannattasio, S. (2010). Knock-out of metacaspase and/or cytochrome c results in the activation of a ROS-independent acetic acid-induced programmed cell death pathway in yeast. FEBS Lett. 584, 3655–3660.
Guaragnella, N., Pereira, C., Sousa, M. J., Antonacci, L., Passarella, S., Côrte-Real, M., Marra, E., and Giannattasio, S. (2006). YCA1 participates in the acetic acid induced yeast programmed cell death also in a manner unrelated to its caspase-like activity. FEBS Lett. 580, 6880–6884.
Haarer, B. K., and Amberg, D. C. (2010). Retraction. Old yellow enzymes protects the actin cytoskeleton from oxidative stress. Mol. Biol. Cell 21, 842.
Haber, F., and Weiss, J. (1934). The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. Lond. B Biol. Sci. 147, 332–351.
Halliwell, B. (1999). Antioxidant defence mechanisms: from the beginning to the end (of the beginning). Free Radic. Res. 31, 261–272.
Halliwell, B., and Aruoma, O. I. (1991). DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 281, 9–19.
Halliwell, B., and Cross, C. E. (1994). Oxygen-derived species: their relation to human disease and environmental stress. Environ. Health Perspect. 102, 5–12.
Halliwell, B., and Gutteridge, J. M. C. (2007). Free Radicals in Biology and Medicine, 4th Edn. Oxford: Oxford University Press.
Hamasaki, M., Noda, T., Baba, M., and Ohsumi, Y. (2005). Starvation triggers the delivery of the endoplasmic reticulum to the vacuole via autophagy in yeast. Traffic 6, 56–65.
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300.
Harper, P. S. (1999). Huntington’s disease: a clinical, genetic and molecular model for polyglutamine repeat disorders. Philos. Trans. R Soc. Lond. B Biol. Sci. 354, 957–961.
Hartig, A., and Ruis, H. (1986). Nucleotide sequence of the Saccharomyces cerevisiae CTT1 gene and deduced amino-acid sequence of yeast catalase T. Eur. J. Biochem. 160, 487–490.
Hartwell, L. H., and Unger, M. W. (1977). Unequal division in Saccharomyces cerevisiae and its implications for the control of cell division. J. Cell Biol. 75, 422–435.
Hasan, R., Leroy, C., Isnard, A. D., Labarre, J., Boy-Marcotte, E., and Toledano, M. B. (2002). The control of the yeast H2O2 response by the Msn2/4 transcription factors. Mol. Microbiol. 45, 233–241.
Hauptmann, P., and Lehle, L. (2008). Kex1 protease is involved in yeast cell death induced by defective N-glycosylation, acetic acid, and chronological aging. J. Biol. Chem. 283, 19151–19163.
Hauptmann, P., Riel, C., Kunz-Schughart, L. A., Fröhlich, K. U., Madeo, F., and Lehle, L. (2006). Defects in N-glycosylation induce apoptosis in yeast. Mol. Microbiol. 59, 765–778.
Henle, E. S., and Linn, S. (1997). Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J. Biol. Chem. 272, 19095–19098.
Heo, J. M., Livnat-Levanon, N., Taylor, E. B., Jones, K. T., Dephoure, N., Ring, J., Xie, J., Brodsky, J. L., Madeo, F., Gygi, S. P., Ashrafi, K., Glickman, M. H., and Rutter, J. (2010). A stress-responsive system for mitochondrial protein degradation. Mol. Cell 40, 465–480.
Herker, E., Jungwirth, H., Lehmann, K. A., Maldener, C., Fröhlich, K.-U., Wissing, S., Büttner, S., Fehr, M., Sigrist, S., and Madeo, F. (2004). Chronological aging leads to apoptosis in yeast. J. Cell Biol. 164, 501–507.
Herrero, E., Ros, J., Bellí, G., and Cabiscol, E. (2008). Redox control and oxidative stress in yeast cells. Biochim. Biophys. Acta 1780, 1217–1235.
Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E., and Stamler, J. S. (2005). Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166.
Hirsch, E. C. (1993). Does oxidative stress participate in nerve cell death in Parkinson’s disease? Eur. Neurol. 33, 52–59.
Hitomi, J., Christofferson, D. E., Ng, A., Yao, J., Degterev, A., Xavier, R. J., and Yuan, J. (2008). Identification of a molecular signalling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323.
Holland, S., Lodwig, E., Sideri, T., Reader, T., Clarke, I., Gkargkas, K., Hoyle, D. C., Delneri, D., Oliver, S. G., and Avery, S. V. (2007). Application of the comprehensive set of heterozygous yeast deletion mutants to elucidate the molecular basis of cellular chromium toxicity. Genome Biol. 8, R268.
Howlett, N. G., and Avery, S. V. (1997). Relationship between cadmium sensitivity and degree of plasma membrane fatty acid unsaturation in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 48, 539–545.
Inoue, Y., and Klionsky, D. J. (2010). Regulation of macroautophagy in Saccharomyces cerevisiae. Semin. Cell Dev. Biol. 21, 664–670.
Izawa, S., Inoue, Y., and Kimura, A. (1995). Oxidative stress response in yeast: effect of glutathione on adaptation to hydrogen peroxide stress in Saccharomyces cerevisiae. FEBS Lett. 368, 73–76.
Jamieson, D. J. (1998). Oxidative stress responses of the yeast Saccharomyces cerevisiae. Yeast 14, 1511–1527.
Jamieson, D. J., Rivers, S. L., and Stephen, D. W. S. (1994). Analysis of Saccharomyces cerevisiae proteins induced by peroxide and superoxide stress. Microbiology 140, 3277–3283.
Jenner, P., and Olanow, C. W. (1996). Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 47, 161–170.
Jeon, B. W., Kim, K. T., Chang, S.-I., and Kim, H. Y. (2002). Phosphoinositide 3-OH kinase/protein kinase B inhibits apoptotic cell death induced by reactive oxygen species in Saccharomyces cerevisiae. J. Biochem. 131, 693–699.
Jin, Y. H., Clark, A. B., Slebos, R. J. C., Al-Refai, H., Taylor, J. A., Kunkel, T. A., Resnick, M. A., and Gordenin, D. A. (2003). Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat. Genet. 34, 326–329.
Juhnke, H., Krems, B., Kötter, P., and Entian, K. D. (1996). Mutants that show increased sensitivity to hydrogen peroxide reveal an important role for the pentose phosphate pathway in protection of yeast against oxidative stress. Mol. Gen. Genet. 252, 456–464.
Jungwirth, H., Ring, J., Mayer, T., Schauer, A., Büttner, S., Eisenberg, T., Carmona-Gutierrez, D., Kuchler, K., and Madeo, F. (2008). Loss of peroxisome function triggers necrosis. FEBS Lett. 582, 2882–2886.
Kaeberlein, M., McVey, M., and Guarente, L. (1999). The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580.
Kanki, T., and Klionsky, D. J. (2008). Mitophagy in yeast occurs through a selective mechanism. J. Biol. Chem. 283, 32386–32393.
Kanki, T., Klionsky, D. J., and Okamoto, K. (2011). Mitochondria autophagy in yeast. Antioxid. Redox Signal. 14, 1989–2001.
Kanki, T., Wang, K., Cao, Y., Baba, M., and Klionsky, D. J. (2009a). Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 17, 98–109.
Kanki, T., Wang, K., Baba, M., Bartholomew, C. R., Lynch-Day, M. A., Du, Z., Geng, J., Mao, K., Yang, Z., Yen, W. L., and Klionsky, D. J. (2009b). A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol. Biol. Cell 20, 4730–4738.
Kasahara, Y., Iwai, K., Yachie, A., Ohta, K., Konno, A., Seki, H., Miyawaki, T., and Taniguchi, N. (1997). Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood 89, 1748–1753.
Khan, M. A., Chock, P. B., and Stadtman, E. R. (2005). Knockout of caspase-like gene, YCA1, abrogates apoptosis and elevates oxidized proteins in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 102, 17326–17331.
Khandrika, L., Kumar, B., Koul, S., Maroni, P., and Koul, H. K. (2009). Role of oxidative stress in prostrate cancer. Cancer Lett. 282, 125–136.
Khoury, C. M., Yang, Z., Li, X. Y., Vignali, M., Fields, S., and Greenwood, M. T. (2008). A TSC22-like motif defines a novel antiapoptotic protein family. FEMS Yeast Res. 8, 540–563.
Kim, I. S., Sohn, H. Y., and Jin, I. (2011). Adaptive stress response to menadione-induced oxidative stress in Saccharomyces cerevisiae KNU5377. J. Microbiol. 49, 816–823.
Kissová, I., Deffieu, M., Manon, S., and Camougrand, N. (2004). Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 279, 39068–39074.
Kissová, I., Deffieu, M., Samokhvalov, V., Velours, G., Bessoule, J. J., Manon, S., and Camougrand, N. (2006). Lipid oxidation and autophagy in yeast. Free Radic. Biol. Med. 41, 1655–1661.
Kissová, I., Salin, B., Schaeffer, J., Bhatia, S., Manon, S., and Camougrand, N. (2007). Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 3, 329–336.
Kissová, I. B., and Camougrand, N. (2009). Glutathione participates in the regulation of mitophagy in yeast. Autophagy 5, 872–873.
Kitagaki, H., Araki, Y., Funato, K., and Shimoi, H. (2007). Ethanol-induced death in yeast exhibits features of apoptosis mediated by mitochondrial fission pathway. FEBS Lett. 581, 2935–2942.
Klatt, P., and Lamas, S. (2000). Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 267, 4928–4944.
Klionsky, D. J., Codogno, P., Cuervo, A. M., Deretic, V., Elazar, Z., Fueyo-Margareto, J., Gewirtz, D. A., Kroemer, G., Levine, B., Mizushima, N., Rubinsztein, D. C., Thumm, M., and Tooze, S. A. (2010). A comprehensive glossary of autophagy-related molecules and processes. Autophagy 6, 438–448.
Knorre, D. A., Smirnova, E. A., and Severin, F. F. (2005). Natural conditions inducing programmed cell death in the yeast Saccharomyces cerevisiae. Biochemistry (Mosc.) 70, 264–266.
Kolaczkowska, A., and Goffeau, A. (1999). Regulation of pleiotropic drug resistance in yeast. Drug Resist. Updat. 2, 403–414.
Kono, Y., and Fridovich, I. (1983). Inhibition and reactivation of Mn-catalase. Implications for valence changes at the active site manganese. J. Biol. Chem. 258, 13646–13648.
Kourtis, N., and Tavernarakis, N. (2009). Autophagy and cell death in model organisms. Cell Death Differ. 16, 21–30.
Kozmin, S., Slezak, G., Reynaud-Angelin, A., Elie, C., de Rycke, Y., Boiteux, S., and Sage, E. (2005). UVA radiation is highly mutagenic in cells that are unable to repair 7,8-dihydro-8-oxoguanine in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 102, 13538–13543.
Kraft, C., Deplazes, A., Sohrmann, M., and Peter, M. (2008). Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 10, 602–610.
Kraft, C., Reggiori, F., and Peter, M. (2009). Selective types of autophagy in yeast. Biochim. Biophys. Acta 1793, 1404–1412. doi:10.1371/journal.pone.0017412
Krems, B., Charizanis, C., and Entian, K. D. (1996). The response regulator-like protein Pos9/Skn7 of Saccharomyces cerevisiae is involved in oxidative stress resistance. Curr. Genet. 29, 327–334.
Kroemer, G., and Levine, B. (2008). Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010.
Kudo, N., Taoka, H., Toda, T., Yoshida, M., and Horinouchi, S. (1999). A novel nuclear export signal sensitive to oxidative stress in the fission yeast transcription factor Pap1. J. Biol. Chem. 274, 15151–15158.
Kuge, S., Arita, M., Murayama, A., Maeta, K., Izawa, S., Inoue, Y., and Nomoto, A. (2001). Regulation of the yeast Yap1p nuclear export signal is mediated by redox signal-induced reversible disulfide bond formation. Mol. Cell. Biol. 21, 6139–6150.
Kuge, S., and Jones, N. (1994). YAP1 dependent activation of TRX2 is essential for the response of Saccharomyces cerevisiae to oxidative stress by hydroperoxides. EMBO J. 13, 655–664.
Kuge, S., Toda, T., Iizuka, N., and Nomoto, A. (1998). Crm1 (XpoI) dependent nuclear export of the budding yeast transcription factor yAP-1 is sensitive to oxidative stress. Genes Cells 3, 521–532.
Latterich, M., Fröhlich, K. U., and Schekman, R. (1995). Membrane fusion and the cell cycle: Cdc48p participates in the fusion of ER membranes. Cell 82, 885–893.
Laun, P., Pichova, A., Madeo, F., Fuchs, J., Ellinger, A., Kohlwein, S., Dawes, I., Fröhlich, K. U., and Breitenbach, M. (2001). Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis. Mol. Microbiol. 39, 1166–1173.
Lee, J., Godon, C., Lagniel, G., Spector, D., Garin, J., Labarre, J., and Toledano, M. B. (1999). Yap1 and Skn7 control two specialized oxidative stress response regulons in yeast. J. Biol. Chem. 274, 16040–16046.
Lee, S. M., and Park, J. W. (1998). A yeast mutant lacking thiol-dependent protector protein is hypersensitive to menadione. Biochim. Biophys. Acta 1382, 167–175.
Lee, Y. J., Hoe, K. L., and Maeng, P. J. (2007). Yeast cells lacking the CIT1-encoded mitochondrial citrate synthase are hypersusceptible to heat- or aging-induced apoptosis. Mol. Biol. Cell 18, 3556–3567.
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5.
Lemasters, J. J., Nieminen, A. L., Qian, T., Trost, L. C., Elmore, S. P., Nishimura, Y., Crowe, R. A., Cascio, W. E., Bradham, C. A., Brenner, D. A., and Herman, B. (1998). The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1366, 177–196.
Lewinska, A., Macierzynska, E., Grzelak, A., and Bartosz, G. (2011). A genetic analysis of nitric oxide-mediated signalling during chronological aging in the yeast. Biogerontology 12, 309–320.
Lewis, J., Devin, A., Miller, A., Lin, Y., Rodriguez, Y., Neckers, L., and Liu, Z. G. (2000). Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J. Biol. Chem. 275, 10519–10526.
Li, W., Sun, L., Liang, Q., Wang, J., Mo, W., and Zhou, B. (2006). Yeast AMID homologue Ndi1p displays respiration-restricted apoptotic activity and is involved in chronological aging. Mol. Biol. Cell 17, 1802–1811.
Li, Y., Yan, J., Kim, I., Liu, C., Huo, K., and Rao, H. (2010). Rad4 regulates protein turnover at a postubiquitylation step. Mol. Biol. Cell 21, 177–185.
Liang, Q., and Zhou, B. (2007). Copper and manganese induce yeast apoptosis via different pathways. Mol. Biol. Cell 18, 4741–4749.
Lin, S. J., Kaeberlein, M., Andalis, A. A., Sturtz, L. A., Defossez, P. A., Culotta, V. C., Fink, G. R., and Guarente, L. (2002). Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418, 344–348.
Loft, S., and Poulson, H. E. (1996). Cancer risk and oxidative damage in man. J. Mol. Med. 74, 297–312.
Longo, V. D., and Fabrizio, P. (2012). Chronological aging in Saccharomyces cerevisiae. Subcell. Biochem. 57, 101–121.
Longo, V. D., Gralla, E. B., and Valentine, J. S. (1996). Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo. J. Biol. Chem. 271, 12275–12280.
Longo, V. D., Mitteldorf, J., and Skulachev, V. P. (2005). Programmed and altruistic ageing. Nat. Rev. Genet. 6, 866–872.
Løvaas, E., and Carlin, G. (1991). Spermine: an anti-oxidant and anti-inflammatory agent. Free Radic. Biol. Med. 11, 455–461.
Ludovico, P., Rodrigues, F., Almeida, A., Silva, M. T., Barrientos, A., and Côrte-Real, M. (2002). Cytochrome c release and mitochondria involvement in programmed cell death induced by acetic acid in Saccharomyces cerevisiae. Mol. Biol. Cell 13, 2598–2606.
Ludovico, P., Sousa, M. J., Silva, M. T., Leao, C., and Côrte-Real, M. (2001). Saccharomyces cerevisiae commits to a programmed cell death process in response to acetic acid. Microbiology 147, 2409–2415.
Luikenhuis, S., Perrone, G., Dawes, I. W., and Grant, C. M. (1998). The yeast Saccharomyces cerevisiae contains two glutaredoxin genes that are required for protection against reactive oxygen species. Mol. Biol. Cell 9, 1081–1091.
Lushchak, O. V., and Lushchak, V. I. (2008). Catalase modifies yeast Saccharomyces cerevisiae response towards S-nitrosoglutathione-induced stress. Redox Rep. 13, 144–152.
Lushchak, V., Semchyshyn, H., Lushchak, O., and Mandryk, S. (2005). Diethyldithiocarbamate inhibits in vivo Cu,Zn-superoxide dismutase and perturbs free radical processes in the yeast Saccharomyces cerevisiae cells. Biochem. Biophys. Res. Commun. 338, 1739–1744.
Madeo, F., Carmona-Gutierrez, D., Ring, J., Büttner, S., Eisenberg, T., and Kroemer, G. (2009). Caspase-dependent and caspase-independent cell death pathways in yeast. Biochem. Biophys. Res. Commun. 382, 227–231.
Madeo, F., Fröhlich, E., and Fröhlich, K.-U. (1997). A yeast mutant showing diagnostic markers of early and late apoptosis. J. Cell Biol. 139, 729–734.
Madeo, F., Fröhlich, E., Ligr, M., Grey, M., Sigrist, S. J., Wolf, D. H., and Fröhlich, K.-U. (1999). Oxygen stress: a regulator of apoptosis in yeast. J. Cell. Biol. 145, 757–767.
Madeo, F., Herker, E., Maldiner, C., Wissing, S., Lachelt, S., Herlan, M., Fehr, M., Lauber, K., Sigrist, S. J., Wesselborg, S., and Fröhlich, K.-U. (2002). A caspase-related protease regulates apoptosis in yeast. Mol. Cell 9, 911–917.
Maeda, T., Wurgler-Murphy, S. M., and Saito, H. (1994). A two-component system that regulates an osmosensing MAP kinase cascade in yeast. Nature 369, 242–245.
Manon, S., Chaudhuri, B., and Guérin, M. (1997). Release of cytochrome c and decrease of cytochrome c oxidase in Bax-expressing yeast cells, and prevention of these effects by coexpression of Bcl-xL. FEBS Lett. 415, 29–32.
Mao, K., Wang, K., Zhao, M., Xu, T., and Klionsky, D. J. (2011). Two MAPK-signalling pathways are required for mitophagy in Saccharomyces cerevisiae. J. Cell Biol. 193, 755–767.
Mao, P., and Smerdon, M. J. (2010). Yeast deubiquitinase Ubp3 interacts with the 26 S proteasome to facilitate Rad4 degradation. J. Biol. Chem. 285, 37542–37550.
Martínez-Pastor, M. T., Marchler, G., Schüller, C., Marchler-Bauer, A., Ruis, H., and Estruch, F. (1996). The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J. 15, 2227–2235.
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., Sou, Y. S., Saiki, S., Kawajiri, S., Sato, F., Kimura, M., Komatsu, M., Hattori, N., and Tanaka, K. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221.
Mazzoni, C., Herker, E., Palermo, V., Jungwirth, H., Eisenberg, T., Madeo, F., and Falcone, C. (2005). Yeast caspase 1 links messenger RNA stability to apoptosis in yeast. EMBO Rep. 6, 1076–1081.
McCord, J. M., and Fridovich, I. (1969). Superoxide dismutase: an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244, 6049–6055.
Mitsui, K., Nakagawa, D., Nakamura, M., Okamoto, T., and Tsurugi, K. (2005). Valproic acid induces apoptosis dependent of Yca1p at concentrations that mildly affect the proliferation of yeast. FEBS Lett. 579, 723–727.
Moraitis, C., and Curran, B. P. (2004). Reactive oxygen species may influence the heat shock response and stress tolerance in the yeast Saccharomyces cerevisiae. Yeast 21, 313–323.
Morgan, B. A., Banks, G. R., Toone, W. M., Raitt, D., Kuge, S., and Johnston, L. H. (1997). The Skn7 response regulator controls gene expression in the oxidative stress response of the budding yeast Saccharomyces cerevisiae. EMBO J. 16, 1035–1044.
Morgan, B. A., Bouquin, N., Merrill, G. F., and Johnston, L. H. (1995). A yeast transcription factor bypassing the requirement for SBF and DSC1/MBF in budding yeast has homology to bacterial signal transduction proteins. EMBO J. 14, 5679–5689.
Mortimer, R. K., and Johnston, J. R. (1959). Life span of individual yeast cells. Nature 183, 1751–1752.
Müller, I., Zimmermann, M., Becker, D., and Flomer, M. (1980). Calendar life span versus budding life span of Saccharomyces cerevisiae. Mech. Ageing Dev. 12, 47–52.
Murakami, K., and Yoshino, M. (1997). Inactivation of aconitase in yeast exposed to oxidative stress. Biochem. Mol. Biol. Int. 41, 481–486.
Nair, U., and Klionsky, D. J. (2005). Molecular mechanisms and regulation of specific and nonspecific autophagy pathways in yeast. J. Biol. Chem. 280, 41785–41788.
Naka, K., Muraguchi, T., Hoshii, T., and Hirao, A. (2008). Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid. Redox Signal. 10, 1883–1894.
Narasimhan, M. L., Coca, M. A., Jin, J., Yamauchi, T., Ito, Y., Kadowaki, T., Kim, K. K., Pardo, J. M., Damsz, B., Hasegawa, P. M., Yun, D. J., and Bressan, R. A. (2005). Osmotin is a homolog of mammalian adiponectin and controls apoptosis in yeast through a homolog of mammalian adiponectin receptor. Mol. Cell 17, 171–180.
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803.
Nargund, A. M., Avery, S. V., and Houghton, J. E. (2008). Cadmium induces a heterogeneous and caspase-dependent apoptotic response in Saccharomyces cerevisiae. Apoptosis 13, 811–821.
Nestelbacher, R., Laun, P., Vondráková, D., Pichová, A., Schüller, C., and Breitenbach, M. (2000). The influence of oxygen toxicity on yeast mother cell-specific aging. Exp. Gerontol. 35, 63–70.
Nguyên-Nhu, N. T., and Knoops, B. (2002). Alkyl hydroperoxide reductase 1 protects Saccharomyces cerevisiae against metal ion toxicity and glutathione depletion. Toxicol. Lett. 135, 219–228.
Nishimura, K., Shiina, R., Kashiwagi, K., and Igarashi, K. (2006). Decrease in polyamines with aging and their ingestion from food and drink. J. Biochem. 139, 81–90.
Nowikovsky, K., Reipert, S., Devenish, R. J., and Schweyen, R. J. (2007). Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ. 14, 1647–1656.
Nunomura, A., Perry, G., Aliey, G., Hirai, K., Takeda, A., Balraj, E. K., Jones, P. K., Ghanbari, H., Wataya, T., Shimohama, S., Chiba, S., Atwood, C. S., Peterson, R. B., and Smith, M. A. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767.
O’Brien, K. M., Dirmeier, R., Engle, M., and Poyton, R. O. (2004). Mitochondrial protein oxidation in yeast mutants lacking manganese-(MnSOD) or copper- and zinc-containing superoxide dismutase (CuZnSOD): evidence that MnSOD and CuZnSOD have both unique and overlapping functions in protecting mitochondrial proteins from oxidative damage. J. Biol. Chem. 279, 51817–51827.
Ohmiya, R., Kato, C., Yamada, H., Aiba, H., and Mizuno, T. (1999). A fission yeast gene (prr1(+)) that encodes a response regulator implicated in oxidative stress response. J. Biochem. 125, 1061–1066.
Ojeda, L., Keller, G., Muhlenhoff, U., Rutherford, J. C., Lill, R., and Winge, D. R. (2006). Role of glutaredoxin-3 and glutaredoxin-4 in the iron regulation of the Aft1 transcriptional activator in Saccharomyces cerevisiae. J. Biol. Chem. 281, 17661–17669.
Okamoto, K., Kondo-Okamoto, N., and Ohsumi, Y. (2009). A landmark protein essential for mitophagy: Atg32 recruits the autophagic machinery to mitochondria. Autophagy 5, 1203–1205.
Orr, W. C., and Sohal, R. S. (1994). Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 263, 1128–1130.
Osório, N. S., Carvalho, A., Almeida, A. J., Padilla-Lopez, S., Leão, C., Laranjinha, J., Ludovico, P., Pearce, D. A., and Rodrigues, F. (2007). Nitric oxide signaling is disrupted in the yeast model for Batten disease. Mol. Biol. Cell 18, 2755–2767.
Outten, C. E., Falk, R. L., and Culotta, V. C. (2005). Cellular factors required for protection from hyperoxia toxicity in Saccharomyces cerevisiae. Biochem. J. 388, 93–101.
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., Packer, M., Schneider, M. D., and Levine, B. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939.
Pereira, C., Silva, R. D., Saraiva, L., Johansson, B., Sousa, M. J., and Côrte-Real, M. (2008). Mitochondria-dependent apoptosis in yeast. Biochim. Biophys. Acta 1783, 1286–1302.
Perrone, G. G., Tan, S. X., and Dawes, I. W. (2008). Reactive oxygen species and yeast apoptosis. Biochim. Biophys. Acta 1783, 1354–1368.
Pierce, G. B., Parchment, R. E., and Lewellyn, A. L. (1991). Hydrogen peroxide as a mediator of programmed cell death in the blastocyst. Differentiation 46, 181–186.
Poyton, R. O., Ball, K. A., and Castello, P. R. (2009). Mitochondrial generation of free radicals and hypoxic signalling. Trends Endocrinol. Metab. 20, 332–340.
Pozniakovsky, A. I., Knorre, D. A., Markova, O. V., Hyman, A. A., Skulachev, V. P., and Severin, F. F. (2005). Role of mitochondria in the pheromone- and amiodarone-induced programmed death of yeast. J. Cell Biol. 168, 257–269.
Priault, M., Bessoule, J. J., Grelaud-Coq, A., Camougrand, N., and Manon, S. (2002). Bax-induced cell death in yeast depends on mitochondrial lipid oxidation. Eur. J. Biochem. 269, 5440–5450.
Priault, M., Salin, B., Schaeffer, J., Vallette, F. M., di Rago, J. P., and Martinou, J. C. (2005). Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 12, 1613–1621.
Pujol-Carrion, N., Belli, G., Herrero, E., Nogues, A., and de la Torre-Ruiz, M. A. (2006). Glutaredoxins Grx3 and Grx4 regulate nuclear localisation of Aft1 and the oxidative stress response in Saccharomyces cerevisiae. J. Cell Sci. 119, 4554–4564.
Radi, R. (2004). Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. U.S.A. 101, 4003–4008.
Raitt, D. C., Johnson, A. L., Erkine, A. M., Makino, K., Morgan, B., Gross, D. S., and Johnston, L. H. (2000). The Skn7 response regulator of Saccharomyces cerevisiae interacts with Hsf1 in vivo and is required for the induction of heat shock genes by oxidative stress. Mol. Biol. Cell 11, 2335–2347.
Reddi, A. R., and Culotta, V. C. (2011). Regulation of manganese antioxidants by nutrient sensing pathways in Saccharomyces cerevisiae. Genetics 189, 1261–1270.
Reddi, A. R., Jensen, L. T., Naranuntarat, A., Rosenfeld, L., Leung, E., Shah, R., and Culotta, V. C. (2009). The overlapping roles of manganese and Cu/Zn SOD in oxidative stress protection. Free Radic. Biol. Med. 46, 154–162.
Reddy, V. P., Zhu, X., Perry, G., and Smith, M. A. (2009). Oxidative stress in diabetes and Alzheimer’s disease. J. Alzheimers Dis. 16, 763–774.
Reekmans, R., De Smet, K., Chen, C., Van Hummelen, P., and Contreras, R. (2005). Old yellow enzyme interferes with Bax-induced NADPH loss and lipid peroxidation in yeast. FEMS Yeast Res. 5, 711–725.
Reggiori, F., and Klionsky, D. J. (2002). Autophagy in the eukaryotic cell. Eukaryot. Cell 1, 11–21.
Reiter, J., Herker, E., Madeo, F., and Schmitt, M. J. (2005). Viral killer toxins induce caspase-mediated apoptosis in yeast. J. Cell Biol. 168, 353–358.
Reverter-Branchat, G., Cabiscol, E., Tamarit, J., and Ros, J. (2004). Oxidative damage to specific proteins in replicative and chronological-aged Saccharomyces cerevisiae: common targets and prevention by calorie restriction. J. Biol. Chem. 279, 31983–31989.
Ribeiro, G. F., Côrte-Real, M., and Johansson, B. (2006). Characterization of DNA damage in yeast apoptosis induced by hydrogen peroxide, acetic acid, and hyperosmotic shock. Mol. Biol. Cell 17, 4584–4591.
Rockenfeller, P., Ring, J., Muschett, V., Beranek, A., Buettner, S., Carmona-Gutierrez, D., Eisenberg, T., Khoury, C., Rechberger, G., Kohlwein, S. D., Kroemer, G., and Madeo, F. (2010). Fatty acids trigger mitochondrion-dependent necrosis. Cell Cycle 9, 2836–2842.
Rubinsztein, D. C. (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786.
Sakai, Y., Koller, A., Rangell, L. K., Keller, G. A., and Subramani, S. (1998). Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. J. Cell Biol. 141, 625–636.
Salmon, T. B., Evert, B. A., Song, B., and Doetsch, P. W. (2004). Biological consequences of oxidative stress-induced DNA damage in Saccharomyces cerevisiae. Nucleic Acids Res. 32, 3712–3723.
Sapienza, K., and Balzan, R. (2005). Metabolic aspects of aspirin-induced apoptosis in yeast. FEMS Yeast Res. 5, 1207–1213.
Sapienza, K., Bannister, W., and Balzan, R. (2008). Mitochondrial involvement in aspirin-induced apoptosis in yeast. Microbiology 154, 2740–2747.
Saran, M., and Bors, W. (1991). Direct and indirect measurements of oxygen radicals. Klin. Wochenschr. 69, 957–964.
Scalabrino, G., and Ferioli, M. E. (1984). Polyamines in mammalian ageing: an oncological problem, too? A review. Mech. Ageing Dev. 26, 149–164.
Schauber, C., Chen, L., Tongaonkar, P., Vega, I., Lambertson, D., Potts, W., and Madura, K. (1998). Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 391, 715–718.
Schauer, A., Knauer, H., Ruckenstuhl, C., Fussi, H., Durchschlag, M., Potocnik, U., and Fröhlich, K. U. (2009). Vacuolar functions determine the mode of cell death. Biochim. Biophys. Acta 1793, 540–545.
Scherz-Shouval, R., and Elazar, Z. (2007). ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 17, 422–427.
Schmitt, A. P., and McEntee, K. (1996). Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 93, 5777–5782.
Schnell, N., Krems, B., and Entian, K. D. (1992). The PAR1 (YAP1/SNQ3) gene of Saccharomyces cerevisiae, a c-jun homologue, is involved in oxygen metabolism. Curr. Genet. 21, 269–273.
Severin, F. F., and Hyman, A. A. (2002). Pheromone induces programmed cell death in S. cerevisiae. Curr. Biol. 12, 233–235.
Shirogane, T., Fukada, T., Muller, J. M., Shima, D. T., Hibi, M., and Hirano, T. (1999). Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity 11, 709–719.
Sies, H. (1991). Oxidative stress: from basic research to clinical application. Am. J. Med. 91, 31S–38S.
Silva, R. D., Sotoca, R., Johansson, B., Ludovico, P., Sansonetty, F., Silva, M. T., Peinado, J. M., and Côrte-Real, M. (2005). Hyperosmotic stress induces metacaspase- and mitochondria-dependent apoptosis in Saccharomyces cerevisiae. Mol. Microbiol. 58, 824–834.
Simon, H.-U., Haj-Yehia, A., and Levi-Schaffer, F. (2000). Role of reactive oxygen species in apoptosis induction. Apoptosis 5, 415–418.
Sinclair, D. A., and Guarente, L. (1997). Extrachromosomal rDNA circles: a cause of aging in yeast. Cell 91, 1033–1042.
Sokolov, S., Pozniakovsky, A., Bocharova, N., Knorre, D., and Severin, F. (2006). Expression of an expanded polyglutamine domain in yeast causes death with apoptotic markers. Biochim. Biophys. Acta 1757, 660–666.
Sripriya, P., Vedantam, L. V., and Podile, A. R. (2009). Involvement of mitochondria and metacaspase elevation in harpin Pss-induced cell death of Saccharomyces cerevisiae. J. Cell. Biochem. 107, 1150–1159.
Stephen, D. W., Rivers, S. L., and Jamieson, D. J. (1995). The role of the YAP1 and YAP2 genes in the regulation of the adaptive oxidative stress responses of Saccharomyces cerevisiae. Mol. Microbiol. 16, 415–423.
Sundaresan, M., Yu, Z. X., Ferrans, V. J., Irani, K., and Finkel, T. (1995). Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270, 296–299.
Sundström, J. F., Vaculova, A., Smertenko, A. P., Savenkov, E. I., Golovko, A., Minina, E., Tiwari, B. S., Rodriguez-Nieto, S., Zamyatnin, A. A. Jr., Välineva, T., Saarikettu, J., Frilander, M. J., Suarez, M. F., Zavialov, A., Ståhl, U., Hussey, P. J., Silvennoinen, O., Sundberg, E., Zhivotovsky, B., and Bozhkov, P. V. (2009). Tudor staphylococcal nuclease is an evolutionarily conserved component of the programmed cell death degradome. Nat. Cell Biol. 11, 1347–1354.
Suzuki, S. W., Onodera, J., and Ohsumi, Y. (2011). Starvation induced cell death in autophagy-defective yeast mutants is caused by mitochondria dysfunction. PLoS ONE 6, e17412.
Takeshige, K., Baba, M., Tsuboi, S., Noda, T., and Ohsumi, Y. (1992). Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 119, 301–311.
Takeuchi, T., Miyahara, K., Hirata, D., and Miyakawa, T. (1997). Mutational analysis of Yapl protein, an AP-1-like transcriptional activator of Saccharomyces cerevisiae. FEBS Lett. 416, 339–343.
Taylor, E. B., and Rutter, J. (2011). Mitochondrial quality control by the ubiquitin-proteasome system. Biochem. Soc. Trans. 39, 1509–1513.
Temple, M. D., Perrone, G. G., and Dawes, I. W. (2005). Complex cellular responses to reactive oxygen species. Trends Cell Biol. 15, 319–326.
Terman, A., and Brunk, U. T. (1998). Lipofuscin: mechanisms of formation and increase with age. APMIS 106, 265–276.
Toone, W. M., and Jones, N. (1999). AP-1 transcription factors in yeast. Curr. Opin. Genet. Dev. 9, 55–61.
Toone, W. M., Kuge, S., Samuels, M., Morgan, B. A., Toda, T., and Jones, N. (1998). Regulation of the fission yeast transcription factor Pap1 by oxidative stress: requirement for the nuclear export factor Crm1 (Exportin) and the stress-activated MAP kinase Sty1/Spc1. Genes Dev. 12, 1453–1463.
Tsujimoto, Y., and Shimizu, S. (2005). Another way to die: autophagic programmed cell death. Cell Death Differ. 12, 1528–1534.
Turrens, J. F. (1997). Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 17, 3–8.
Turrens, J. F., and Boveris, A. (1980). Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 191, 421–427.
Váchová, L., and Palková, Z. (2005). Physiological regulation of yeast cell death in multicellular colonies is triggered by ammonia. J. Cell Biol. 169, 711–717.
Váchová, L., and Palková, Z. (2007). Caspases in yeast apoptosis-like death: facts and artefacts. FEMS Yeast Res. 7, 12–21.
Vanden Berghe, T., Kalai, M., van Loo, G., Declercq, W., and Vandenabeele, P. (2003). Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J. Biol. Chem. 278, 5622–5629.
Veal, E. A., Ross, S. J., Malakasi, P., Peacock, E., and Morgan, B. A. (2003). Ybp1 is required for the hydrogen peroxide-induced oxidation of the Yap1 transcription factor. J. Biol. Chem. 278, 30896–30904.
Vercammen, D., Declercq, W., Vandenabeele, P., and Van Breusegem, F. (2007). Are metacaspases caspases? J. Cell Biol. 179, 375–380.
Vergara, R., Parada, F., Rubio, S., and Pérez, F. J. (2012). Hypoxia induces H2O2 production and activates antioxidant defence system in grapevine buds through mediation of H2O2 and ethylene. J. Exp. Bot. doi:10.1093/jxb/ers094, 1–9. [Epub ahead of print].
Wadskog, I., Maldener, C., Proksch, A., Madeo, F., and Adler, L. (2004). Yeast lacking the SRO7/SOP1-encoded tumor suppressor homologue show increased susceptibility to apoptosis-like cell death on exposure to NaCl stress. Mol. Biol. Cell 15, 1436–1444.
Walter, D., Wissing, S., Madeo, F., and Fahrenkrog, B. (2006). The inhibitor-of-apoptosis protein Bir1p protects against apoptosis in S. cerevisiae and is a substrate for the yeast homologue of Omi/HtrA2. J. Cell Sci. 119, 1843–1851.
Wawryn, J., Krzepiłko, A., Myszka, A., and Biliński, T. (1999). Deficiency in superoxide dismutases shortens life span of yeast cells. Acta Biochim. Pol. 46, 249–253.
Wei, M., Fabrizio, P., Hu, J., Ge, H., Cheng, C., Li, L., and Longo, V. D. (2008). Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 4, e13. doi:10.1371/journal.pgen.0040013
Wei, M., Madia, F., and Longo, V. D. (2011). Studying age-dependent genomic instability using the S. cerevisiae chronological lifespan model. J. Vis. Exp. 55, 3030.
Weinberger, M., Mesquita, A., Caroll, T., Marks, L., Yang, H., Zhang, Z., Ludovico, P., and Burhans, W. C. (2010). Growth signaling promotes chronological aging in budding yeast by inducing superoxide anions that inhibit quiescence. Aging (Albany N.Y.) 2, 709–726.
Weinberger, M., Ramachandran, L., Feng, L., Sharma, K., Sun, X., Marchetti, M., Huberman, J. A., and Burhans, W. C. (2005). Apoptosis in budding yeast caused by defects in initiation of DNA replication. J. Cell Sci. 118, 3543–3553.
Weisiger, R. A., and Fridovich, I. (1973a). Mitochondrial superoxide dismutase. J. Biol. Chem. 248, 4793–4796.
Wemmie, J. A., Steggerda, S. M., and Moye-Rowley, W. S. (1997). The Saccharomyces cerevisiae AP-1 protein discriminates between oxidative stress elicited by the oxidants H2O2 and diamide. J. Biol. Chem. 272, 7908–7914.
Wemmie, J. A., Szczypka, M. S., Thiele, D. J., and Moye-Rowley, W. S. (1994). Cadmium tolerance mediated by the yeast AP-1 protein requires the presence of an ATP-binding cassette transporter-encoding gene, YCF1. J. Biol. Chem. 269, 32592–32597.
Wilkinson, C. R., Seeger, M., Hartmann-Petersen, R., Stone, M., Wallace, M., Semple, C., and Gordon, C. (2001). Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nat. Cell Biol. 3, 939–943.
Wilkinson, D., and Ramsdale, M. (2011). Proteases and caspase-like activity in the yeast Saccharomyces cerevisiae. Biochem. Soc. Trans. 39, 1502–1508.
Wissing, S., Ludovico, P., Herker, E., Büttner, S., Engelhardt, S. M., Decker, T., Link, A., Proksch, A., Rodrigues, F., Côrte-Real, M., Fröhlich, K.-U., Manns, J., Candé, C., Sigrist, S. J., Kroemer, G., and Madeo, F. (2004). An AIF orthologue regulates apoptosis in yeast. J. Cell Biol. 166, 969–974.
Wolff, S. P., and Dean, R. T. (1986). Fragmentation of proteins by free radicals and its effect on their susceptibility to enzymic hydrolysis. Biochem. J. 234, 399–403.
Wood, Z. A., Schröder, E., Robin Harris, J., and Poole, L. B. (2003). Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 28, 32–40.
Wu, C. Y., Bird, A. J., Winge, D. R., and Eide, D. J. (2007). Regulation of the yeast TSA1 peroxiredoxin by ZAP1 is an adaptive response to the oxidative stress of zinc deficiency. J. Biol. Chem. 282, 2184–2195.
Wu, M., Xu, L. G., Li, X., Zhai, Z., and Shu, H. B. (2002). AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 277, 25617–25623.
Wysocki, R., and Kron, S. J. (2004). Yeast cell death during DNA damage arrest is independent of caspase or reactive oxygen species. J. Cell Biol. 166, 311–316.
Xu, B., Chen, S., Luo, Y., Chen, Z., Liu, L., Zhou, H., Chen, W., Shen, T., Han, X., Chen, L., and Huang, S. (2011). Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS ONE 6, e19052. doi:10.1371/journal.pone.0019052
Xu, C., Wang, J., Gao, Y., Lin, H., Du, L., Yang, S., Long, S., She, Z., Cai, X., Zhou, S., and Lu, Y. (2010). The anthracenedione compound bostrycin induces mitochondria-mediated apoptosis in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 10, 297–308.
Yakes, F. M., and Van Houten, B. (1997). Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 94, 516–519.
Yamaguchi-Iwai, Y., Ueta, R., Fukunaka, A., and Sasaki, R. (2002). Subcellular localization of Aft1 transcription factor responds to iron status in Saccharomyces cerevisiae. J. Biol. Chem. 277, 18914–18918.
Yang, H., Ren, Q., and Zhang, Z. (2008). Cleavage of Mcd1 by caspase-like protease Esp1 promotes apoptosis in budding yeast. Mol. Biol. Cell 19, 2127–2134.
Ye, Y., Meyer, H. H., and Rapoport, T. A. (2001). The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656.
Yokozawa, T., Cho, E. J., Park, C. H., and Kim, J. H. (2011). Protective effect of proanthocyanidin against diabetic oxidative stress. Evid. Based Complement. Alternat. Med. 2012, 1–11.
Zechmann, B., Liou, L. C., Koffler, B. E., Horvat, L., Tomašic, A., Fulgosi, H., and Zhang, Z. (2011). Subcellular distribution of glutathione and its dynamic changes under oxidative stress in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 11, 631–642.
Zhang, X., De Micheli, M., Coleman, S. T., Sanglard, D., and Moye-Rowley, W. S. (2000). Analysis of the oxidative stress regulation of the Candida albicans transcription factor, Cap1p. Mol. Microbiol. 36, 618–629.
Zhang, Y., Qi, H., Taylor, R., Xu, W., Liu, L. F., and Jin, S. (2007). The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy 3, 337–346.
Keywords: oxidative stress, yeast, apoptosis, necrosis, mitophagy, autophagy, aging
Citation: Farrugia G and Balzan R (2012) Oxidative stress and programmed cell death in yeast. Front. Oncol. 2:64. doi: 10.3389/fonc.2012.00064
Received: 03 April 2012; Accepted: 02 June 2012;
Published online: 21 June 2012.
Edited by:
Frank Madeo, Karl-Franzens-Universitat Graz, AustriaReviewed by:
Michael Breitenbach, University of Salzburg, AustriaIan Dawes, University of New South Wales, Australia
Copyright: © 2012 Farrugia and Balzan. This is an openaccess article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Rena Balzan, Department of Physiology and Biochemistry, Faculty of Medicine and Surgery, University of Malta, Msida MSD 2080, Malta. e-mail: rena.balzan@um.edu.mt