
- 1Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA
- 2Department of Medicine, Harvard Medical School, Boston, MA, USA
- 3Department of Medicine, Brigham and Women’s Hospital, Boston, MA, USA
- 4Broad Institute of Harvard and MIT, Cambridge, MA, USA
- 5Ludwig Center at Dana-Farber/Harvard Cancer Center, Dana-Farber Cancer Institute, Boston, MA, USA
- 6Lowe Center for Thoracic Oncology, Dana-Farber Cancer Institute, Boston, MA, USA
Despite the ongoing “war on cancer,” cancer remains one of the major causes of human morbidity and mortality. A new paradigm of targeted therapies holds the most promise for the future, making identification of tumor-specific therapeutic targets of prime importance. ERBB2/HER2, best known for its role in breast cancer tumorigenesis, can be targeted by two types of pharmacological manipulation: antibody therapy against the extracellular receptor domain and small molecule compounds against the intracellular tyrosine kinase domain. Aberrant activation of ERBB2 by gene amplification has been shown to participate in the pathophysiology of breast, ovarian, gastric, colorectal, lung, brain, and head and neck tumors. However, the advent of next-generation sequencing technologies has enabled efficient identification of activating molecular alterations of ERBB2. In this review, we will focus on the functional role of these somatic mutations that cause ERBB2 receptor activation. We will additionally discuss the current preclinical and clinical therapeutic strategies for targeting mutationally activated ERBB2.
Introduction
Rising incidences of neoplasia worldwide are estimated to translate into 13 million cancer deaths by 2030 (World Health Organization, 2012). In order to develop more effective and less toxic targeted cancer therapies, we must utilize our knowledge of malignant cell biology and design tailored antineoplastic compounds against diverse biological targets to supplement current standard treatment modalities, such as surgical resection, chemotherapy, and radiation therapy, to eradicate this frequently fatal disease. Although encouraging response rates are achieved in a few types of cancer with these standard treatment options, the majority of patients lack sufficient therapeutic options for long-term survival, especially those with advanced disease. Hence, additional therapies are urgently needed.
Because neoplastic cells frequently show “addiction” to mutationally activated oncogenes (Weinstein, 2002; Sharma and Settleman, 2010), such oncogenes comprise the most promising group of drug targets discovered to date. In the mid 1980s, the receptor tyrosine kinase (RTK) ERBB2 (also known as HER2 – Human Epidermal Growth Factor Receptor (EGFR) 2) was identified to be an oncogenic driver (Padhy et al., 1982; Bargmann et al., 1986; Di Fiore et al., 1987; Slamon et al., 1987). ERBB2 was first targeted with the monoclonal antibody, trastuzumab, which was approved by the Food and Drug Administration (FDA) in 1998. Although the addition of trastuzumab to chemotherapy significantly prolonged survival in patients with ERBB2-overexpressing breast or gastric cancers (Piccart-Gebhart et al., 2005; Romond et al., 2005; Joensuu et al., 2006; Bang et al., 2010), these clinical benefits failed to translate in improved survival of patients with ERBB2-overexpressing non-small cell lung cancers (NSCLCs) (Gatzemeier et al., 2004; Langer et al., 2004).
Oncogenic signaling by RTKs can also be abrogated by inhibition of tyrosine kinase activity with small molecules. Imatinib mesylate demonstrated proof of principle by successfully inhibiting constitutive signaling through the BCR-ABL fusion protein in chronic myelogenous leukemia (Druker et al., 2001). Additional tyrosine kinase inhibitors (TKIs) targeting various cellular signaling pathways have entered the clinic since imatinib mesylate was approved by the FDA in 2001, including inhibitors targeting ERBB2 in breast cancer (Geyer et al., 2006) and the related RTK, EGFR, in lung adenocarcinomas (Ku et al., 2011). The emergence of sophisticated genomic methodologies like next-generation sequencing enabled high-throughput detection of known and novel oncogenic mutations, and in particular revealed the presence of activating mutations of ERBB2 in a variety of tumor types. These novel oncogenic alterations of ERBB2 potentially offer unique therapeutic opportunities to a broader range of patients than previously anticipated by analysis of ERBB2 amplification alone. However, it appears that it may be more difficult to successfully target ERBB2 mutation than ERBB2 amplification or EGFR mutation. Translation of this discovery to the clinic thus remains a major challenge.
The ERBB/HER Receptor Family
The proto-oncogene ERBB2 is a member of the ERBB/HER RTK family, additionally comprised of EGFR (EGFR/HER1/ERBB1), HER3/ERBB3, and HER4/ERBB4 (Hynes and Lane, 2005). Upon extracellular ligand binding, these four receptors mediate normal cell proliferation and cell survival via two major signaling pathways: Ras-Raf-MAPK and PI3K/Akt/mTOR. Whereas EGFR and ERBB4 have known extracellular ligands and possess active tyrosine kinase domains, no direct high-affinity ligand has been identified for ERBB2 (Carraway et al., 1994; Sliwkowski et al., 1994; Burgess et al., 2003). Furthermore, ERBB3 binds several different ligands, but has little or no tyrosine kinase activity, and is possibly able only to weakly autophosphorylate (Shi et al., 2010).
Activation of ERBB2
Signaling specificity of each ERBB receptor is transmitted through unique patterns of C-terminal autophosphorylation sites (Olayioye et al., 2000; Yarden and Sliwkowski, 2001). Further complexity is added by receptor dimerization, which can occur either between two identical (homodimerization) or two different (heterodimerization) ERBB receptors. Under resting conditions, these cell surface receptors are found as monomers folded in a so-called “closed/tethered” autoinhibited conformation to prevent dimerization (Ferguson et al., 2003). Conformational rearrangement into an “open/extended” state occurs upon ligand binding to the extracellular domain. This process exposes the dimerization arm to establish the core of the dimer interface with a homologous region of a partner molecule. The extracellular dimeric structure facilitates reciprocal transactivation of the intracellular tyrosine kinase portions of each receptor. The uniqueness of ERBB2 among its family members is not only characterized by its inability to directly bind any known EGF family ligand, but also by being permanently fixed in the active conformation. Consequently, kinase autoinhibition to prevent uncontrolled receptor activation is not mediated by the ectodomain, but by a loop connecting the αC helix and β4 sheet within the kinase domain (Fan et al., 2008).
At least in part due to its constitutively active conformation, ERBB2 is the preferred dimerization partner for other ERBB family members. Although the existence of four receptors allows several different pairings and subsequently distinct patterns of downstream pathway engagement, ERBB2 heterodimers demonstrated increased potency in conveying extracellular signals (Yarden and Sliwkowski, 2001). It comes as no surprise that the most powerful signaling heterodimer – composed of ERBB2 and ERBB3 – functions as an oncogenic unit (Holbro et al., 2003; Hsieh and Moasser, 2007; Lee-Hoeflich et al., 2008). Lack of catalytic kinase activity does not prevent ERBB3 from heterodimerizing with other ERBB molecules. In fact, the primary oncogenic signaling apparatus of ERBB2-ERBB3 is crucial for activation of the PI3K/Akt pathway (Soltoff et al., 1994). Although ERBB2 possesses no direct docking sites for PI3K, ERBB3 mediates this process with six tyrosine binding sites for the regulatory subunit of PI3K (Prigent and Gullick, 1994; Soltoff et al., 1994). Indeed, clinical data by Tokunaga et al. (2006) shows positive correlation of ERBB2-expressing breast cancers and increased activation of Akt.
Three principal mechanisms of oncogenic activation of ERBB2 have been identified to date: (i) amplification and overexpression, (ii) molecular alterations of the receptor, and (iii) inhibition of phosphatase activity (Ocana and Pandiella, 2013). Increased numbers of receptor molecules populating the cell surface increase the likelihood of dimerization and receptor tyrosine phosphorylation, even in the absence of ligand binding (Zhang et al., 2006; Endres et al., 2011). ERBB2 overexpression or amplification was initially discovered in approximately one third of human breast cancers and is associated with more aggressive tumors and poorer outcome (Slamon et al., 1987). Other human tumor types have also been reported to harbor ERBB2 amplification or overexpression, including lung cancers (Pellegrini et al., 2003; Langer et al., 2004), gastric cancers (Tanner et al., 2005; Bang et al., 2010), ovarian cancers (Tuefferd et al., 2007; Vermeij et al., 2008), prostate cancers (Minner et al., 2010), salivary gland tumors (Cornolti et al., 2007), and bladder cancers (Lae et al., 2010).
Mutational activation of ERBB2 can result from three types of somatic molecular alterations: small insertions and missense mutations in the kinase domain (Figure 1A), missense mutations in the extracellular domain (Figure 1B), or large deletions of the extracellular domain that yield the truncated form of ERBB2, p95HER2 (Figure 1C). The molecular characteristics, treatment opportunities, and potential mechanisms of resistance of these three classes will be discussed in the next sections.
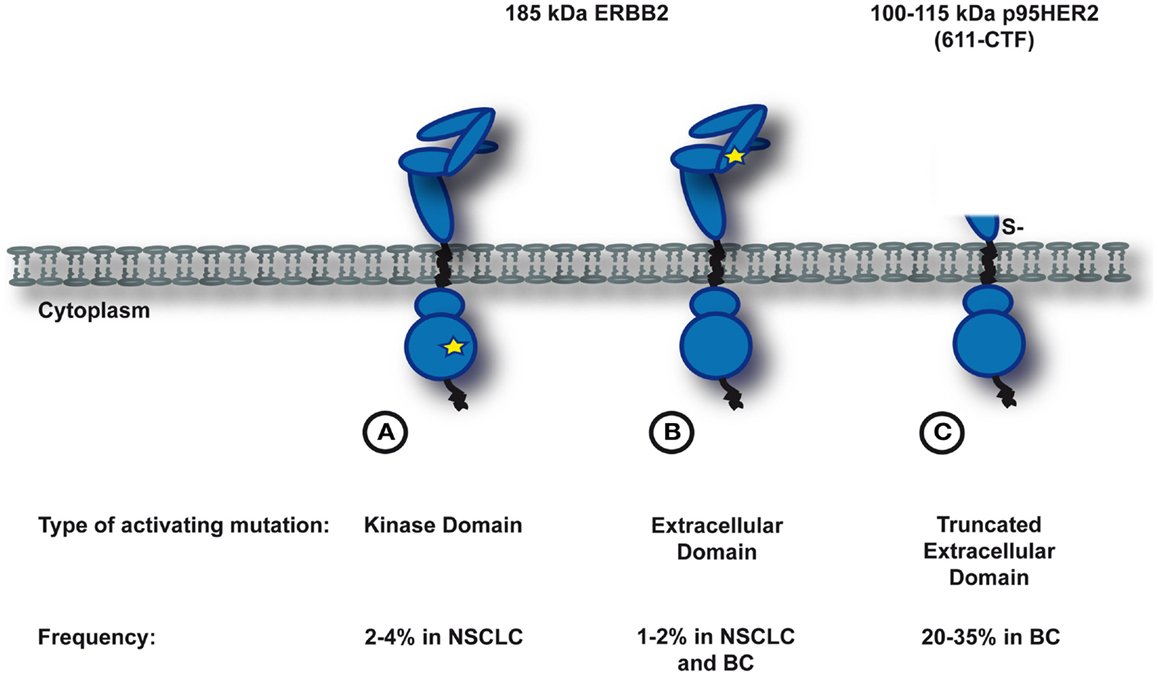
Figure 1. Somatic activating mutations in ERBB2. Depicted is a simplified schematic of three known subclasses of ERBB2-mutants. (A) and (B) Activating mutations in the full-length protein. A star indicates the position of the activating mutation (A in the kinase domain, B in the extracellular domain). (C) Large deletions of the extracellular domain that yield the trucated form of ERBB2, p95HER2.
Constitutive ERBB2 activation can also be achieved by insufficient dephosphorylation of the receptor. Although in rare cases ERBB receptors may transphosphorylate each other in the absence of ligand, overexpression, or mutations, intracellular phosphatases rapidly act as a fail-safe mechanism to dephosphorylate the receptor and terminate signaling (Ullrich and Schlessinger, 1990). Recently, studies by two different groups provided the first evidence that phosphatase activity is essential to control oncogenic ERBB2 signaling. Sun et al. (2011) demonstrated that mutational inactivation of the phosphatase PTPN12 caused activation of ERBB2 in triple negative breast cancer cell lines. Similarly, Vermeer et al. (2012) analyzed breast cancer cell lines to understand the correlation between decreased PTPN13 expression and poorer overall survival. The authors found a novel signaling complex consisting of ERBB2 and EphrinB1 which is regulated by transient association with PTPN13 and Src. In absence of the phosphatase PTPN13, activated Src associates with the ERBB2 kinase domain and phosphorylates EphrinB1, which induces Erk1/2 phosphorylation (Vermeer et al., 2012).
ERBB2 Mutations in Carcinogenesis
The clinical success of gefitinib, an inhibitor of EGFR, in a subset of lung cancers harboring activating mutations within the kinase domain of EGFR led to the investigation of analogous mutations of ERBB2. ERBB2 kinase domain mutations were found to occur in 2–4% of lung adenocarcinomas (Stephens et al., 2004; Shigematsu et al., 2005; Buttitta et al., 2006) and cause increased survival, invasiveness, and tumorigenicity in cell-based transformation assays (Wang et al., 2006). Similarly to NSCLC driven by EGFR mutation, the clinical and pathological characteristics of patients with ERBB2 mutations have been attributed to patients of the female sex, Asian ethnicity, never-smoker status, and adenocarcinoma subtype. However, a recent study of 1,478 U.S. patients with lung adenocarcinomas found no association of ERBB2 mutation with sex and race, but confirmed that mutations of the kinase domain of ERBB2 are mutually exclusive with EGFR, KRAS, and ALK mutations (Arcila et al., 2012).
The most prevalent alteration of ERBB2 involves the in-frame insertion/duplication A775_G776insYVMA within exon 20, a sequence also present in exon 20 of the EGFR gene (Stephens et al., 2004). Similar in-frame insertion mutations were also identified in ovarian cancers (Anglesio et al., 2008). These insertions induce a conformational change of the autoinhibitory αC-β4 loop, thus narrowing the ATP-binding cleft and promoting enhanced kinase activity (Gazdar et al., 2004; Fan et al., 2008). In vitro studies have shown that this ERBB2-mutant potently transphosphorylates EGFR in the absence of ERBB ligands rendering EGFR susceptible for dimerization (Wang et al., 2006). Single nucleotide missense substitutions of this region of ERBB2 have also been reported in breast cancer, gastric cancer, and colorectal cancer (Lee et al., 2006b).
Although oncogenic tyrosine kinase mutations frequently alter the ATP-binding pocket, we recently identified an alternate mechanism of ERBB2 activation resulting from extracellular domain mutations that cause reduction-sensitive covalent dimerization (Greulich et al., 2012). These missense substitutions cluster in subdomain II, a region characterized by 11 disulfide bonds (Cho et al., 2003), and impact intramolecular disulfide bond formation (Greulich et al., 2012). Mutation of cysteine residues in this region that participate in intramolecular disulfide bonds, such as S335C (Greulich, unpublished observation), or mutation of residues important to stabilization of disulfide-bonded loops, such as G309E, can both promote intermolecular disulfide bond formation, resulting in constitutively dimerized and activated ERBB2 (Greulich et al., 2012).
Reduction-sensitive dimerization is not the only mechanism by which ERBB2 extracellular domain mutations constitutively activate enzymatic activity; ERBB2 S310F and S310Y mutations, found in 1–2% of lung cancers and breast cancers, behave more similarly to the ERBB2 kinase domain mutants in that they cause elevated C-terminal tail phosphorylation without evidence of covalent dimerization. Of note, the S310F lesion was also detected in 1/316 ovarian cancers (Cancer Genome Atlas Research Network, 2011) and in a bladder cancer cell line, 5637 (Barretina et al., 2012).
Whereas activating mutations within the kinase domain of ERBB2 show close homology to their counterparts within EGFR, the extracellular domain mutations are not as closely mirrored. Oncogenic mutations affecting the ectodomain of EGFR have been identified in subdomain I, II, and IV (Lee et al., 2006a). Although the mechanism of receptor activation has not yet been characterized for these EGFR extracellular domain mutations, it is tempting to speculate that the underlying tumorigenic mechanism is caused by a less tethered conformation of the extracellular domain as most amino acid substitutions localize to interdomain interfaces (Lee et al., 2006a).
The third type of mutant ERBB2 is structurally different from the first two, as these derivatives lack substantial parts of the extracellular domain and are termed p95HER2 or HER2 carboxyl terminal fragments (CTF) (reviewed in Arribas et al., 2011). These truncated ERBB2 proteins have been predominantly found in breast cancers and cause resistance to trastuzumab (Molina et al., 2002; Scaltriti et al., 2007). Only a few cases of lung adenocarcinoma were reported to harbor these mutations (Cappuzzo et al., 2012). Two distinct mechanisms yield p95HER2 fragments: alternative mRNA translation from internal initiation codons (positions 611 and 678, respectively) and proteolytic shedding of the ectodomain of the full-length receptor (Christianson et al., 1998; Anido et al., 2006). Strikingly, in vitro studies of the membrane-anchored p95HER2 fragment known as 611-CTF (100–115 kDa) revealed more rapid and acute activation of different signaling pathways compared with the full-length receptor and the 648-CTF fragment (Pedersen et al., 2009). Additionally, this hyperactive p95HER2 fragment was shown to promote more aggressive and metastatic breast cancer progression by induction of a specific gene set (Pedersen et al., 2009). The pathological features attributed to overexpression of 611-CTF are postulated to be a result of its short extracellular domain, which contains five cysteines. Thus again, constitutive generation of activated homodimers is assumed to be maintained by intermolecular disulfide bonds (Pedersen et al., 2009).
ERBB2 as a Therapeutic Target
Two different strategies for targeting ERBB2 have successfully entered the clinic: antibodies directed against the extracellular domain of the receptor, and small molecule TKIs acting on the intracellular kinase domain.
The mechanism of action of monoclonal antibodies toward ERBB2-overexpressing cancer cells include removal of ERBB2 from the cell surface by endocytosis to diminish intracellular signaling, and induction of an immune system-mediated antitumor response. Several ERBB2-directed monoclonal antibodies have been developed, including trastuzumab and pertuzumab. Whereas trastuzumab interacts with subdomain IV of the extracellular domain (Cho et al., 2003), pertuzumab binds subdomain II, which harbors the dimerization arm and thus inhibits receptor dimerization (Franklin et al., 2004). Trastuzumab can also be conjugated to DM1, an inhibitor of tubulin polymerization derived from maytansine, to efficiently deliver DM1 to ERBB2-overexpressing cancer cells (Lewis Phillips et al., 2008). Despite promising preclinical data, clinical development of ertumaxomab, a bispecific antibody capable to bind mature T cells and ERBB2, was discontinued (Kiewe et al., 2006).
Small molecule TKIs are typically competitive inhibitors, preventing ATP from binding to its natural site within the kinase region due to the higher affinity of the TKI for the ATP-binding pocket. ERBB family TKIs fall into two categories: reversible inhibitors, like erlotinib, gefitinib, and lapatinib, that can be released from the receptor; and irreversible inhibitors, such as afatinib, neratinib, pelitinib, and dacomitinib, that covalently modify the receptor. Although the in vitro efficacy of the irreversible inhibitors was demonstrated to be superior to that of reversible inhibitors, irreversible ERBB blockade requires biosynthesis for receptor recovery, both a benefit and a drawback (Sanchez-Martin and Pandiella, 2012).
From the oncologist’s point of view, irreversible inhibition is highly desired for tumor control. However, ERBB signaling is also vital to non-malignant tissues, and inhibition of ERBB2 is associated with unwanted toxicities. For instance, trastuzumab can provoke cardiotoxicity, especially when administered in combination with anthracyclines (Slamon et al., 2001). Thus, careful evaluation is required prior to utilization of more potent irreversible inhibitors, which may result in increased toxicity. It is possible that non-competitive inhibitors could serve as a valuable alternative, particularly to combat eventual resistance to current TKIs (Ocana and Pandiella, 2013).
Despite robust preclinical and encouraging clinical data in various cancer types, a third class of antineoplastic agents active against ERBB2, HSP90 inhibitors, has still not been approved by the FDA. HSP90 is a chaperone that governs the conformational maturation and folding of ERBB2. Inhibition of HSP90 leads to ubiquitylation and proteasomal degradation of ERBB2 and its downstream signaling partners. In a Phase II study, combination treatment with trastuzumab and the HSP90 inhibitor tanespimycin (also known as 17-AAG) was demonstrated to be active in patients with breast cancer who had progressed on trastuzumab therapy (Modi et al., 2011).
Given our current knowledge of the biology of activating mutations of ERBB2, single agent antibody-based treatment strategies may be of limited clinical relevance. In particular, truncated p95HER2 fragments naturally evade antibody binding due to the absence of the extracellular domain and binding of trastuzumab to ectodomain- or kinase domain-mutated ERBB2 forms presumably fails to prevent ligand-mediated ERBB3-ERBB2 signaling (Agus et al., 2002). Our in vitro data furthermore indicates that, whereas survival of Ba/F3 cells expressing mutants of G309 and S310 was effectively inhibited upon trastuzumab treatment, other ectodomain-mutants were less responsive (Greulich et al., 2012). Further in vivo investigation will be required to determine response in a more physiological setting. Additionally, it would be of interest to evaluate whether pertuzumab is able to bind and impact survival of cancer cells expressing ectodomain-mutants. It remains unclear whether combinatorial treatment of trastuzumab and pertuzumab would be effective, given recent data obtained from ERBB2-positive metastatic breast cancer (Baselga et al., 2012).
Importantly, tissue-specific properties may hamper therapeutic success of antibody-based treatment schedules. Whereas trastuzumab has recently been approved for the treatment of metastatic gastric cancer in combination with cytotoxic agents (Bang et al., 2010), similar studies targeting overexpressed/amplified ERBB2 in NSCLC (Gatzemeier et al., 2004; Langer et al., 2004; Lara et al., 2004b; Zinner et al., 2004; Krug et al., 2005; Herbst et al., 2007) and prostate cancer (Morris et al., 2002; Lara et al., 2004a; Ziada et al., 2004) have reported modest or disappointing results. It remains to be determined whether this primary resistance to trastuzumab results from inaccessibility of the receptor. For example, Nagy et al. (2005) found that MUC4, a membrane-associated mucin, masked the extracellular domain of ERBB2. In light of this finding, evaluation of MUC4 overexpression as a possible mechanism for primary resistance to trastuzumab in NSCLC should be done. Indeed, 80–85% of NSCLCs express MUC4, and adeno- and adenosquamous-carcinomas are characterized by high levels of MUC4 expression (68 and 75%, respectively) (Kwon et al., 2007). Further analyses by Karg et al. (2006) suggest that MUC4 and ERBB2 expression are positively correlated and might be involved in the repression of apoptosis and differentiation. However, primary resistance to trastuzumab in prostate cancer may involve other mechanisms, as MUC4 expression was not detectable in malignant prostate tissue (Cozzi et al., 2005).
The identification of activating mutations within the kinase domain of ERBB2 offered an additional therapeutic possibility (Stephens et al., 2004). EGFR-TKIs were shown to be ineffective against ERBB2 mutations (Wang et al., 2006; Cappuzzo et al., 2007; Engelman et al., 2007). By contrast, several ERBB2-directed TKIs showed effective anti-proliferative properties. Despite promising preclinical data with neratinib (HKI-272; an irreversible ERBB inhibitor of EGFR and ERBB2) in the ERBB2-mutant NCI-H1781 cell line (Shimamura et al., 2006), clinical evaluation in patients with EGFR-driven lung adenocarcinomas does not support use of this inhibitor as a single agent (Wong et al., 2009; Sequist et al., 2010). However, three of four patients harboring the rare EGFR mutation G719X (X indicates substitution of glycine by either serine, cysteine, or alanine) were found to respond to neratinib (Sequist et al., 2010). Strikingly, neratinib showed promising activity in ERBB2-overexpressing breast cancers and could potentially be approved as a first-line therapy in locally advanced or metastatic ERBB2-overexpressing breast cancers (Chow et al., 2009; Limentani et al., 2009; Burstein et al., 2010; Awada et al., 2013).
Preclinical activity for afatinib (BIBW2992), a second irreversible inhibitor of EGFR and ERBB2, was demonstrated in Ba/F3 cells expressing an ERBB2-mutant with an insertional mutation at codon 776 and in transgenic lung cancer models (Li et al., 2008). De Greve et al. (2012) recently provided the first evidence of clinical benefit from treatment with afatinib. In this study, patients were initially diagnosed with lung adenocarcinoma harboring exon 20 ERBB2 mutations and had progressed under various antineoplastic regimes. Three of five such identified patients were eligible for treatment response evaluation and two patients showed rapid metabolic response within 1–2 weeks. Although single agent afatinib did not extend overall survival in patients with advanced, metastatic NSCLC after failure of other therapeutic options (Miller et al., 2012), it did prolong progression-free survival and it appears rational to investigate the synergistic effects of afatinib and paclitaxel in this patient population. Furthermore, our preclinical data utilizing an inducible mouse model of mutant ERBB2 (A775_G776insYVMA) in lung epithelium revealed that the combination of afatinib and an mTOR inhibitor (rapamycin) were effective in mediating tumor shrinkage (Perera et al., 2009). Thus, combinatorial treatment approaches are likely to positively influence clinical outcome.
Dacomitinib (PF00299804), a third irreversible pan-ERBB inhibitor, is currently under clinical investigation due to promising preclinical studies (Engelman et al., 2007; Janne et al., 2011). The rationale for irreversible TKI development to fight ERBB2-activating mutations originates from experience with reversible TKIs targeting EGFR-activating lesions. Despite their initial response, almost all of these cancers rapidly develop resistance and result in little overall survival benefit (Maemondo et al., 2010). In about half of these resistant cancers, a secondary mutation within the catalytic cleft of the kinase domain is responsible for ineffective reversible drug activity and subsequent oncogenic proliferation (details in Section Mechanisms of Resistance).
Following discovery of activating mutations within the ERBB2 extracellular domain, we analyzed the growth inhibitory effects of neratinib, afatinib, and lapatinib in Ba/F3 cells expressing the variant mutants (Greulich et al., 2012). Effective abrogation of cell survival was observed for all three inhibitors; however, the reversible inhibitor lapatinib was 5- to 10-fold less effective than neratinib and afatinib. Cells expressing the ectodomain-mutants were consistently more sensitive to these inhibitors than cells expressing the kinase domain mutant, A775_G776insYVMA.
Thus far, we focused our review on preclinical and clinical studies evaluating the existing anti-ERBB2 agents on cancers harboring activating mutations of ERBB2 as single agents with or without adjacent chemotherapy. However, the ERBB2 signaling cascade plays a pivotal role in oncogenesis and obviously affects a multitude of other key signaling nodes. Thus, combination of different ERBB2-directed agents (antibody + TKI) or with other targeted therapies (HSP90 inhibitors, MEK inhibitors, mTOR inhibitors, etc.) present valid options to combat ERBB2-driven oncogenesis.
Recent clinical data showed a significant overall survival benefit of patients with heavily pretreated metastatic ERBB2-positive breast cancer upon dual ERBB2 blockade through trastuzumab and lapatinib (Blackwell et al., 2012). Further studies are warranted to confirm the superiority of this cytotoxic agent-free regiment in earlier clinical settings. Another interesting treatment approach of synergistic efficacy was presented by Garcia-Garcia et al. (2012). The authors analyzed five different cell lines resistant to trastuzumab and lapatinib. The combination treatment of lapatinib and INK-128, an mTOR inhibitor, induced increased apoptosis in both in vitro and in vivo experiments (Garcia-Garcia et al., 2012). Along the same line, a Phase I study of neratinib and temsirolimus, an mTOR inhibitor, demonstrated encouraging antitumor activity in patients with ERBB2-overexpressing NSCLCs and breast cancers (Gandhi et al., 2011).
Although activating mutations of ERBB2 were identified in various tumor types and several potential therapeutic options are at hand, specific screening for these lesions has not been translated into clinical routine yet.
Mechanisms of Resistance
Despite the plethora of ERBB2 targeted compounds, we currently lack a sound understanding why tumor shrinkage is short-lived and only a relatively small percentage of patients benefit from these therapies. Major mechanisms of primary and acquired resistance to anti-ERBB therapeutics include (reviewed in Tortora, 2011): (1) alteration of the extracellular domain, including missense substitutions to impede epitope recognition, masking of epitopes, or expression of ectodomain-truncated ERBB2 fragments; (2) second-site mutations in the RTK domain; (3) overexpression of alternative ERBB ligands or receptors to counteract for receptor inhibition; (4) alternative signaling from other receptors such as the insulin-like growth factor-1 receptor (IGF1R) or MET; (5) aberrant signaling caused by downregulation (p27) or loss (PTEN) of downstream controllers; and (6) aberrant activation of secondary downstream growth and survival pathways, such as Ras-Raf-MAPK, PI3K/Akt/mTOR.
Retrospective studies on tumors expressing truncated p95HER2 fragments revealed that these tend to be resistant to any current therapeutic antibody approach as the required epitopes are missing (Scaltriti et al., 2007; Sperinde et al., 2010). However, two independent groups recently generated monoclonal antibodies that specifically recognize 611-CTF (Parra-Palau et al., 2010; Sperinde et al., 2010). Conceivably, this novel diagnostic tool is of valuable clinical relevance because it allows discrimination of which patients will benefit from antibody-based therapies and which will be resistant. As the studies performed by Scaltriti et al. (2010) convincingly demonstrated that treatment with lapatinib effectively inhibits p95HER2, improved treatment stratification is available for patients harboring this particular activating ERBB2 mutation. Regardless, caution is still warranted since experiments by Xia et al. (2011) revealed that chronic lapatinib treatment is capable of inducing nuclear expression of truncated ERBB2, thereby escaping further therapeutic effectiveness. Additionally, formation of nuclear lapatinib-induced p95HER2 was blocked upon proteasome inhibition (Figure 2) (Xia et al., 2011). It remains to be tested whether this phenomenon is also relevant if: (1) full-length ERBB2 is targeted, or (2) any of the irreversible TKIs employs a similar strategy to evade antitumor control.
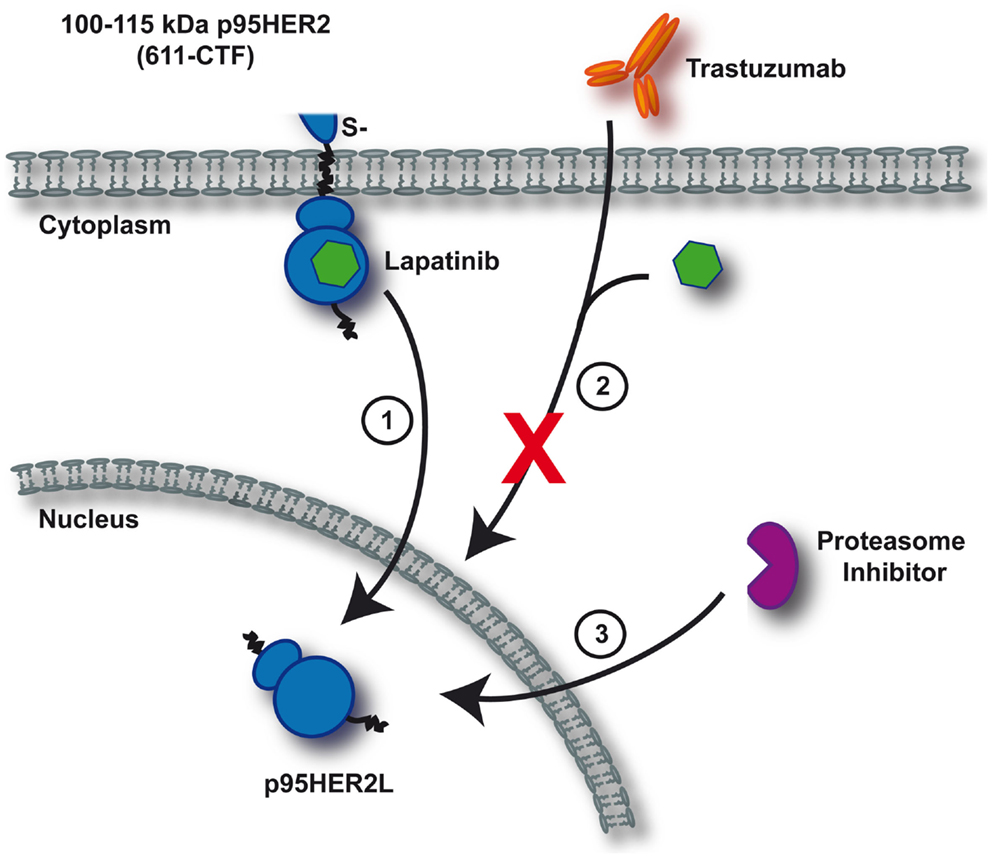
Figure 2. Mechanism of resistance upon continuous lapatinib treatment. Depicted is a simplified schematic of lapatinib-induced resistance toward current anti-ERBB2 therapeutics as identified by Xia et al. (2011). Continuous inhibition of 611-CTF with lapatinib induces nuclear p95HER2L expression (1). Trastuzumab and lapatinib are ineffective in targeting nuclear p95HER2L, thereby failing to control oncogenic proliferation (2). As formation of p95HER2L potentially depends on proteasomal processing, proteasome inhibition effectively prevents p95HER2L emergence (3).
Analogous to a commonly observed event during TKI treatment of EGFR-driven lung adenocarcinomas, lapatinib application was shown to induce secondary mutations within the ERRB2 kinase domain consequently leading to TKI resistance. In vitro analyses identified three point mutations, L755S, L755P, and T798M to confer resistance to lapatinib (Kancha et al., 2011). Threonine 798 is the ERBB2 “gatekeeper” residue that is located at the periphery of the nucleotide-binding site of ERBB2 kinase (Aertgeerts et al., 2011), and regulates access to a deep hydrophobic pocket in the active site (Schindler et al., 2000). This event is analogous to replacement of threonine 790 with methionine (T790M) in erlotinib-resistant lung adenocarcinoma. The gatekeeper mutation enhances the affinity of the oncogenic form of the receptor for ATP, allowing continued proliferation in the presence of the drug (Yun et al., 2008). The potential of irreversible EGFR/ERBB2 inhibitors to overcome drug resistance due to gatekeeper mutations was recently demonstrated in vitro and in vivo (Kobayashi et al., 2005; Engelman et al., 2007; Minami et al., 2007; Li et al., 2008; Zhou et al., 2009).
Concluding Remarks
During the past decades, the ERBB2 signaling cascade gained significant importance in the oncogenesis of many tumor types. The discovery of primary activating mutations and the emergence of acquired secondary mutations represent sophisticated challenges for effective treatment approaches. Our next steps in evaluating potential ERBB2-directed therapeutics clearly rely on: adequate diagnostic properties for specific patient selection and identification of tissue-specific mechanisms of resistance to initiate well-designed clinical trials of combinational treatment strategies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to our colleagues for the omission of many important contributions to the field, and their references, due to space limitations. We thank Dr. Camilla Laulund Christensen for assistance in illustrating the figures and the members of the Wong laboratory for helpful discussions. This work was supported by the Deutsche Forschungsgemeinschaft (HE 6897/1-1 to Grit S. Herter-Sprie), the National Cancer Institute (CA122794, CA166480, CA163896, R01CA140594, U01CA141576 to Kwok-Kin Wong) and funding from Uniting Against Lung Cancer and the Doctors Cancer Foundation (to Heidi Greulich).
References
Aertgeerts, K., Skene, R., Yano, J., Sang, B. C., Zou, H., Snell, G., et al. (2011). Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 286, 18756–18765.
Agus, D. B., Akita, R. W., Fox, W. D., Lewis, G. D., Higgins, B., Pisacane, P. I., et al. (2002). Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2, 127–137.
Anglesio, M. S., Arnold, J. M., George, J., Tinker, A. V., Tothill, R., Waddell, N., et al. (2008). Mutation of ERBB2 provides a novel alternative mechanism for the ubiquitous activation of RAS-MAPK in ovarian serous low malignant potential tumors. Mol. Cancer Res. 6, 1678–1690.
Anido, J., Scaltriti, M., Bech Serra, J. J., Santiago Josefat, B., Todo, F. R., Baselga, J., et al. (2006). Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 25, 3234–3244.
Arcila, M. E., Chaft, J. E., Nafa, K., Roy-Chowdhuri, S., Lau, C., Zaidinski, M., et al. (2012). Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin. Cancer Res. 18, 4910–4918.
Arribas, J., Baselga, J., Pedersen, K., and Parra-Palau, J. L. (2011). p95HER2 and breast cancer. Cancer Res. 71, 1515–1519.
Awada, A., Dirix, L., Manso Sanchez, L., Xu, B., Luu, T., Dieras, V., et al. (2013). Safety and efficacy of neratinib (HKI-272) plus vinorelbine in the treatment of patients with ErbB2-positive metastatic breast cancer pretreated with anti-HER2 therapy. Ann. Oncol. 24, 109–116.
Bang, Y. J., Van Cutsem, E., Feyereislova, A., Chung, H. C., Shen, L., Sawaki, A., et al. (2010). Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376, 687–697.
Bargmann, C. I., Hung, M. C., and Weinberg, R. A. (1986). Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell 45, 649–657.
Barretina, J., Caponigro, G., Stransky, N., Venkatesan, K., Margolin, A. A., Kim, S., et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607.
Baselga, J., Cortes, J., Kim, S. B., Im, S. A., Hegg, R., Im, Y. H., et al. (2012). Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 366, 109–119.
Blackwell, K. L., Burstein, H. J., Storniolo, A. M., Rugo, H. S., Sledge, G., Aktan, G., et al. (2012). Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 Study. J. Clin. Oncol. 30, 2585–2592.
Burgess, A. W., Cho, H. S., Eigenbrot, C., Ferguson, K. M., Garrett, T. P., Leahy, D. J., et al. (2003). An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 12, 541–552.
Burstein, H. J., Sun, Y., Dirix, L. Y., Jiang, Z., Paridaens, R., Tan, A. R., et al. (2010). Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J. Clin. Oncol. 28, 1301–1307.
Buttitta, F., Barassi, F., Fresu, G., Felicioni, L., Chella, A., Paolizzi, D., et al. (2006). Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int. J. Cancer 119, 2586–2591.
Cancer Genome Atlas Research Network. (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615.
Cappuzzo, F., Cho, Y. G., Sacconi, A., Ali, G., Siclari, O., Incarbone, M., et al. (2012). p95HER2 truncated form in resected non-small cell lung cancer. J. Thorac. Oncol. 7, 520–527.
Cappuzzo, F., Ligorio, C., Janne, P. A., Toschi, L., Rossi, E., Trisolini, R., et al. (2007). Prospective study of gefitinib in epidermal growth factor receptor fluorescence in situ hybridization-positive/phospho-Akt-positive or never smoker patients with advanced non-small-cell lung cancer: the ONCOBELL trial. J. Clin. Oncol. 25, 2248–2255.
Carraway, K. L. III, Sliwkowski, M. X., Akita, R., Platko, J. V., Guy, P. M., Nuijens, A., et al. (1994). The erbB3 gene product is a receptor for heregulin. J. Biol. Chem. 269, 14303–14306.
Cho, H. S., Mason, K., Ramyar, K. X., Stanley, A. M., Gabelli, S. B., Denney, D. W. Jr., et al. (2003). Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760.
Chow, L., Jiang, Z., Epstein, R., Bondarenko, I., Awada, A., Coughlin, C., et al. (2009). Safety and efficacy of neratinib (HKI-272) in combination with paclitaxel in patients with solid tumors. J. Clin. Oncol. 27, 15s, abstr. 3557.
Christianson, T. A., Doherty, J. K., Lin, Y. J., Ramsey, E. E., Holmes, R., Keenan, E. J., et al. (1998). NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res. 58, 5123–5129.
Cornolti, G., Ungari, M., Morassi, M. L., Facchetti, F., Rossi, E., Lombardi, D., et al. (2007). Amplification and overexpression of HER2/neu gene and HER2/neu protein in salivary duct carcinoma of the parotid gland. Arch. Otolaryngol. Head Neck Surg. 133, 1031–1036.
Cozzi, P. J., Wang, J., Delprado, W., Perkins, A. C., Allen, B. J., Russell, P. J., et al. (2005). MUC1, MUC2, MUC4, MUC5AC and MUC6 expression in the progression of prostate cancer. Clin. Exp. Metastasis 22, 565–573.
De Greve, J., Teugels, E., Geers, C., Decoster, L., Galdermans, D., De Mey, J., et al. (2012). Clinical activity of afatinib (BIBW 2992) in patients with lung adenocarcinoma with mutations in the kinase domain of HER2/neu. Lung Cancer 76, 123–127.
Di Fiore, P. P., Pierce, J. H., Kraus, M. H., Segatto, O., King, C. R., and Aaronson, S. A. (1987). erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science 237, 178–182.
Druker, B. J., Sawyers, C. L., Kantarjian, H., Resta, D. J., Reese, S. F., Ford, J. M., et al. (2001). Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 344, 1038–1042.
Endres, N. F., Engel, K., Das, R., Kovacs, E., and Kuriyan, J. (2011). Regulation of the catalytic activity of the EGF receptor. Curr. Opin. Struct. Biol. 21, 777–784.
Engelman, J. A., Zejnullahu, K., Gale, C. M., Lifshits, E., Gonzales, A. J., Shimamura, T., et al. (2007). PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 67, 11924–11932.
Fan, Y. X., Wong, L., Ding, J., Spiridonov, N. A., Johnson, R. C., and Johnson, G. R. (2008). Mutational activation of ErbB2 reveals a new protein kinase autoinhibition mechanism. J. Biol. Chem. 283, 1588–1596.
Ferguson, K. M., Berger, M. B., Mendrola, J. M., Cho, H. S., Leahy, D. J., and Lemmon, M. A. (2003). EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 11, 507–517.
Franklin, M. C., Carey, K. D., Vajdos, F. F., Leahy, D. J., De Vos, A. M., and Sliwkowski, M. X. (2004). Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317–328.
Gandhi, L., Bahleda, R., Cleary, J. M., Hollebecque, A., Kwak, E. L., Pandya, S., et al. (2011). Two-dimensional phase I study of neratinib (NER) combined with temsirolimus (TEM) in patients (Pts) with solid tumors. J. Clin. Oncol. 29(Suppl.), abstr. 3027.
Garcia-Garcia, C., Ibrahim, Y. H., Serra, V., Calvo, M. T., Guzman, M., Grueso, J., et al. (2012). Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin. Cancer Res. 18, 2603–2612.
Gatzemeier, U., Groth, G., Butts, C., Van Zandwijk, N., Shepherd, F., Ardizzoni, A., et al. (2004). Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Ann. Oncol. 15, 19–27.
Gazdar, A. F., Shigematsu, H., Herz, J., and Minna, J. D. (2004). Mutations and addiction to EGFR: the Achilles “heal” of lung cancers? Trends Mol. Med. 10, 481–486.
Geyer, C. E., Forster, J., Lindquist, D., Chan, S., Romieu, C. G., Pienkowski, T., et al. (2006). Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 355, 2733–2743.
Greulich, H., Kaplan, B., Mertins, P., Chen, T. H., Tanaka, K. E., Yun, C. H., et al. (2012). Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc. Natl. Acad. Sci. U.S.A. 109, 14476–14481.
Herbst, R. S., Davies, A. M., Natale, R. B., Dang, T. P., Schiller, J. H., Garland, L. L., et al. (2007). Efficacy and safety of single-agent pertuzumab, a human epidermal receptor dimerization inhibitor, in patients with non small cell lung cancer. Clin. Cancer Res. 13, 6175–6181.
Holbro, T., Beerli, R. R., Maurer, F., Koziczak, M., Barbas, C. F. III, and Hynes, N. E. (2003). The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 100, 8933–8938.
Hsieh, A. C., and Moasser, M. M. (2007). Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br. J. Cancer 97, 453–457.
Hynes, N. E., and Lane, H. A. (2005). ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 5, 341–354.
Janne, P. A., Boss, D. S., Camidge, D. R., Britten, C. D., Engelman, J. A., Garon, E. B., et al. (2011). Phase I dose-escalation study of the pan-HER inhibitor, PF299804, in patients with advanced malignant solid tumors. Clin. Cancer Res. 17, 1131–1139.
Joensuu, H., Kellokumpu-Lehtinen, P. L., Bono, P., Alanko, T., Kataja, V., Asola, R., et al. (2006). Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N. Engl. J. Med. 354, 809–820.
Kancha, R. K., Von Bubnoff, N., Bartosch, N., Peschel, C., Engh, R. A., and Duyster, J. (2011). Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS ONE 6:e26760. doi:10.1371/journal.pone.0026760
Karg, A., Dinc, Z. A., Basok, O., and Ucvet, A. (2006). MUC4 expression and its relation to ErbB2 expression, apoptosis, proliferation, differentiation, and tumor stage in non-small cell lung cancer (NSCLC). Pathol. Res. Pract. 202, 577–583.
Kiewe, P., Hasmuller, S., Kahlert, S., Heinrigs, M., Rack, B., Marme, A., et al. (2006). Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clin. Cancer Res. 12, 3085–3091.
Kobayashi, S., Ji, H., Yuza, Y., Meyerson, M., Wong, K. K., Tenen, D. G., et al. (2005). An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 65, 7096–7101.
Krug, L. M., Miller, V. A., Patel, J., Crapanzano, J., Azzoli, C. G., Gomez, J., et al. (2005). Randomized phase II study of weekly docetaxel plus trastuzumab versus weekly paclitaxel plus trastuzumab in patients with previously untreated advanced nonsmall cell lung carcinoma. Cancer 104, 2149–2155.
Ku, G. Y., Haaland, B. A., and De Lima Lopes, G. Jr. (2011). Gefitinib vs. chemotherapy as first-line therapy in advanced non-small cell lung cancer: meta-analysis of phase III trials. Lung Cancer 74, 469–473.
Kwon, K. Y., Ro, J. Y., Singhal, N., Killen, D. E., Sienko, A., Allen, T. C., et al. (2007). MUC4 expression in non-small cell lung carcinomas: relationship to tumor histology and patient survival. Arch. Pathol. Lab. Med. 131, 593–598.
Lae, M., Couturier, J., Oudard, S., Radvanyi, F., Beuzeboc, P., and Vieillefond, A. (2010). Assessing HER2 gene amplification as a potential target for therapy in invasive urothelial bladder cancer with a standardized methodology: results in 1005 patients. Ann. Oncol. 21, 815–819.
Langer, C. J., Stephenson, P., Thor, A., Vangel, M., and Johnson, D. H. (2004). Trastuzumab in the treatment of advanced non-small-cell lung cancer: is there a role? Focus on Eastern Cooperative Oncology Group Study 2598. J. Clin. Oncol. 22, 1180–1187.
Lara, P. N. Jr., Chee, K. G., Longmate, J., Ruel, C., Meyers, F. J., Gray, C. R., et al. (2004a). Trastuzumab plus docetaxel in HER-2/neu-positive prostate carcinoma: final results from the California Cancer Consortium screening and phase II trial. Cancer 100, 2125–2131.
Lara, P. N. Jr., Laptalo, L., Longmate, J., Lau, D. H., Gandour-Edwards, R., Gumerlock, P. H., et al. (2004b). Trastuzumab plus docetaxel in HER2/neu-positive non-small-cell lung cancer: a California Cancer Consortium screening and phase II trial. Clin. Lung Cancer 5, 231–236.
Lee, J. C., Vivanco, I., Beroukhim, R., Huang, J. H., Feng, W. L., Debiasi, R. M., et al. (2006a). Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 3:e485. doi:10.1371/journal.pmed.0030485
Lee, J. W., Soung, Y. H., Seo, S. H., Kim, S. Y., Park, C. H., Wang, Y. P., et al. (2006b). Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin. Cancer Res. 12, 57–61.
Lee-Hoeflich, S. T., Crocker, L., Yao, E., Pham, T., Munroe, X., Hoeflich, K. P., et al. (2008). A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 68, 5878–5887.
Lewis Phillips, G. D., Li, G., Dugger, D. L., Crocker, L. M., Parsons, K. L., Mai, E., et al. (2008). Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 68, 9280–9290.
Li, D., Ambrogio, L., Shimamura, T., Kubo, S., Takahashi, M., Chirieac, L. R., et al. (2008). BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27, 4702–4711.
Limentani, S. A., Awada, A., Dirix, L., Beck, J., Dieras, V., Binlich, F., et al. (2009). Safety and efficacy of neratinib (HKI-272) in combination with vinorelbine in patients with solid tumors. J. Clin. Oncol. 27(Suppl.), abstr. e14554.
Maemondo, M., Inoue, A., Kobayashi, K., Sugawara, S., Oizumi, S., Isobe, H., et al. (2010). Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388.
Miller, V. A., Hirsh, V., Cadranel, J., Chen, Y. M., Park, K., Kim, S. W., et al. (2012). Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 13, 528–538.
Minami, Y., Shimamura, T., Shah, K., Laframboise, T., Glatt, K. A., Liniker, E., et al. (2007). The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272. Oncogene 26, 5023–5027.
Minner, S., Jessen, B., Stiedenroth, L., Burandt, E., Kollermann, J., Mirlacher, M., et al. (2010). Low level HER2 overexpression is associated with rapid tumor cell proliferation and poor prognosis in prostate cancer. Clin. Cancer Res. 16, 1553–1560.
Modi, S., Stopeck, A., Linden, H., Solit, D., Chandarlapaty, S., Rosen, N., et al. (2011). HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 17, 5132–5139.
Molina, M. A., Saez, R., Ramsey, E. E., Garcia-Barchino, M. J., Rojo, F., Evans, A. J., et al. (2002). NH(2)-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin. Cancer Res. 8, 347–353.
Morris, M. J., Reuter, V. E., Kelly, W. K., Slovin, S. F., Kenneson, K., Verbel, D., et al. (2002). HER-2 profiling and targeting in prostate carcinoma. Cancer 94, 980–986.
Nagy, P., Friedlander, E., Tanner, M., Kapanen, A. I., Carraway, K. L., Isola, J., et al. (2005). Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 65, 473–482.
Ocana, A., and Pandiella, A. (2013). Targeting HER receptors in cancer. Curr. Pharm. Des. 19, 808–817.
Olayioye, M. A., Neve, R. M., Lane, H. A., and Hynes, N. E. (2000). The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 19, 3159–3167.
Padhy, L. C., Shih, C., Cowing, D., Finkelstein, R., and Weinberg, R. A. (1982). Identification of a phosphoprotein specifically induced by the transforming DNA of rat neuroblastomas. Cell 28, 865–871.
Parra-Palau, J. L., Pedersen, K., Peg, V., Scaltriti, M., Angelini, P. D., Escorihuela, M., et al. (2010). A major role of p95/611-CTF, a carboxy-terminal fragment of HER2, in the down-modulation of the estrogen receptor in HER2-positive breast cancers. Cancer Res. 70, 8537–8546.
Pedersen, K., Angelini, P. D., Laos, S., Bach-Faig, A., Cunningham, M. P., Ferrer-Ramon, C., et al. (2009). A naturally occurring HER2 carboxy-terminal fragment promotes mammary tumor growth and metastasis. Mol. Cell. Biol. 29, 3319–3331.
Pellegrini, C., Falleni, M., Marchetti, A., Cassani, B., Miozzo, M., Buttitta, F., et al. (2003). HER-2/Neu alterations in non-small cell lung cancer: a comprehensive evaluation by real time reverse transcription-PCR, fluorescence in situ hybridization, and immunohistochemistry. Clin. Cancer Res. 9, 3645–3652.
Perera, S. A., Li, D., Shimamura, T., Raso, M. G., Ji, H., Chen, L., et al. (2009). HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc. Natl. Acad. Sci. U.S.A. 106, 474–479.
Piccart-Gebhart, M. J., Procter, M., Leyland-Jones, B., Goldhirsch, A., Untch, M., Smith, I., et al. (2005). Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 353, 1659–1672.
Prigent, S. A., and Gullick, W. J. (1994). Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 13, 2831–2841.
Romond, E. H., Perez, E. A., Bryant, J., Suman, V. J., Geyer, C. E. Jr., Davidson, N. E., et al. (2005). Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 353, 1673–1684.
Sanchez-Martin, M., and Pandiella, A. (2012). Differential action of small molecule HER kinase inhibitors on receptor heterodimerization: therapeutic implications. Int. J. Cancer 131, 244–252.
Scaltriti, M., Chandarlapaty, S., Prudkin, L., Aura, C., Jimenez, J., Angelini, P. D., et al. (2010). Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin. Cancer Res. 16, 2688–2695.
Scaltriti, M., Rojo, F., Ocana, A., Anido, J., Guzman, M., Cortes, J., et al. (2007). Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 99, 628–638.
Schindler, T., Bornmann, W., Pellicena, P., Miller, W. T., Clarkson, B., and Kuriyan, J. (2000). Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 289, 1938–1942.
Sequist, L. V., Besse, B., Lynch, T. J., Miller, V. A., Wong, K. K., Gitlitz, B., et al. (2010). Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 28, 3076–3083.
Sharma, S. V., and Settleman, J. (2010). Exploiting the balance between life and death: targeted cancer therapy and “oncogenic shock”. Biochem. Pharmacol. 80, 666–673.
Shi, F., Telesco, S. E., Liu, Y., Radhakrishnan, R., and Lemmon, M. A. (2010). ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc. Natl. Acad. Sci. U.S.A. 107, 7692–7697.
Shigematsu, H., Takahashi, T., Nomura, M., Majmudar, K., Suzuki, M., Lee, H., et al. (2005). Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 65, 1642–1646.
Shimamura, T., Ji, H., Minami, Y., Thomas, R. K., Lowell, A. M., Shah, K., et al. (2006). Non-small-cell lung cancer and Ba/F3 transformed cells harboring the ERBB2 G776insV_G/C mutation are sensitive to the dual-specific epidermal growth factor receptor and ERBB2 inhibitor HKI-272. Cancer Res. 66, 6487–6491.
Slamon, D. J., Clark, G. M., Wong, S. G., Levin, W. J., Ullrich, A., and McGuire, W. L. (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182.
Slamon, D. J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., et al. (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792.
Sliwkowski, M. X., Schaefer, G., Akita, R. W., Lofgren, J. A., Fitzpatrick, V. D., Nuijens, A., et al. (1994). Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J. Biol. Chem. 269, 14661–14665.
Soltoff, S. P., Carraway, K. L. III, Prigent, S. A., Gullick, W. G., and Cantley, L. C. (1994). ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol. Cell. Biol. 14, 3550–3558.
Sperinde, J., Jin, X., Banerjee, J., Penuel, E., Saha, A., Diedrich, G., et al. (2010). Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clin. Cancer Res. 16, 4226–4235.
Stephens, P., Hunter, C., Bignell, G., Edkins, S., Davies, H., Teague, J., et al. (2004). Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431, 525–526.
Sun, T., Aceto, N., Meerbrey, K. L., Kessler, J. D., Zhou, C., Migliaccio, I., et al. (2011). Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 144, 703–718.
Tanner, M., Hollmen, M., Junttila, T. T., Kapanen, A. I., Tommola, S., Soini, Y., et al. (2005). Amplification of HER-2 in gastric carcinoma: association with Topoisomerase IIalpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann. Oncol. 16, 273–278.
Tokunaga, E., Kimura, Y., Oki, E., Ueda, N., Futatsugi, M., Mashino, K., et al. (2006). Akt is frequently activated in HER2/neu-positive breast cancers and associated with poor prognosis among hormone-treated patients. Int. J. Cancer 118, 284–289.
Tortora, G. (2011). Mechanisms of resistance to HER2 target therapy. J. Natl. Cancer Inst. Monographs 2011, 95–98.
Tuefferd, M., Couturier, J., Penault-Llorca, F., Vincent-Salomon, A., Broet, P., Guastalla, J. P., et al. (2007). HER2 status in ovarian carcinomas: a multicenter GINECO study of 320 patients. PLoS ONE 2:e1138. doi:10.1371/journal.pone.0001138
Ullrich, A., and Schlessinger, J. (1990). Signal transduction by receptors with tyrosine kinase activity. Cell 61, 203–212.
Vermeer, P. D., Bell, M., Lee, K., Vermeer, D. W., Wieking, B. G., Bilal, E., et al. (2012). ErbB2, EphrinB1, Src kinase and PTPN13 signaling complex regulates MAP kinase signaling in human cancers. PLoS ONE 7:e30447. doi:10.1371/journal.pone.0030447
Vermeij, J., Teugels, E., Bourgain, C., Xiangming, J., In’t Veld, P., Ghislain, V., et al. (2008). Genomic activation of the EGFR and HER2-neu genes in a significant proportion of invasive epithelial ovarian cancers. BMC Cancer 8:3. doi:10.1186/1471-2407-8-3
Wang, S. E., Narasanna, A., Perez-Torres, M., Xiang, B., Wu, F. Y., Yang, S., et al. (2006). HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 10, 25–38.
Weinstein, I. B. (2002). Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science 297, 63–64.
Wong, K. K., Fracasso, P. M., Bukowski, R. M., Lynch, T. J., Munster, P. N., Shapiro, G. I., et al. (2009). A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin. Cancer Res. 15, 2552–2558.
World Health Organization. (2012). Cancer. WHO. Available at: http://www.who.int/mediacentre/factsheets/fs297/en/ [accessed March 9, 2013].
Xia, W., Liu, Z., Zong, R., Liu, L., Zhao, S., Bacus, S. S., et al. (2011). Truncated ErbB2 expressed in tumor cell nuclei contributes to acquired therapeutic resistance to ErbB2 kinase inhibitors. Mol. Cancer Ther. 10, 1367–1374.
Yarden, Y., and Sliwkowski, M. X. (2001). Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137.
Yun, C. H., Mengwasser, K. E., Toms, A. V., Woo, M. S., Greulich, H., Wong, K. K., et al. (2008). The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U.S.A. 105, 2070–2075.
Zhang, X., Gureasko, J., Shen, K., Cole, P. A., and Kuriyan, J. (2006). An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149.
Zhou, W., Ercan, D., Chen, L., Yun, C. H., Li, D., Capelletti, M., et al. (2009). Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074.
Ziada, A., Barqawi, A., Glode, L. M., Varella-Garcia, M., Crighton, F., Majeski, S., et al. (2004). The use of trastuzumab in the treatment of hormone refractory prostate cancer; phase II trial. Prostate 60, 332–337.
Zinner, R. G., Glisson, B. S., Fossella, F. V., Pisters, K. M., Kies, M. S., Lee, P. M., et al. (2004). Trastuzumab in combination with cisplatin and gemcitabine in patients with Her2-overexpressing, untreated, advanced non-small cell lung cancer: report of a phase II trial and findings regarding optimal identification of patients with Her2-overexpressing disease. Lung Cancer 44, 99–110.
Keywords: ERBB2/HER2, activating somatic mutation, reversible and irreversible tyrosine kinase inhibitors, targeted therapies, resistance, lung cancer, breast cancer
Citation: Herter-Sprie GS, Greulich H and Wong K-K (2013) Activating mutations in ERBB2 and their impact on diagnostics and treatment. Front. Oncol. 3:86. doi: 10.3389/fonc.2013.00086
Received: 10 January 2013; Paper pending published: 12 February 2013;
Accepted: 31 March 2013; Published online: 23 April 2013.
Edited by:
Elda Tagliabue, Fondazione IRCCS Istituto Nazionale dei Tumori, ItalyReviewed by:
A. Emre Sayan, University of Southampton, UKAmedeo Columbano, University of Cagliari, Italy
Copyright: © 2013 Herter-Sprie, Greulich and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Kwok-Kin Wong, Lowe Center for Thoracic Oncology, Belfer Institute for Applied Cancer Science, Dana-Farber Cancer Institute, 450 Brookline Avenue, HIM 243, Boston, MA 02215-5450, USA. e-mail: kwong1@partners.org