
- Department of Experimental Oncology, European Institute of Oncology, Milan, Italy
The role of the promyelocytic leukemia (PML) protein has been widely tested in many different contexts, as attested by the hundreds of papers present in the literature. In most of these studies, PML is regarded as a tumor suppressor, a notion on the whole accepted by the scientific community. In this review, we examine how the concept of tumor-suppressor gene has evolved until now and then systematically assess whether this assumption for PML is supported by unambiguous experimental evidence.
Tumor Suppressor, an Evolving Concept
Despite more than 80 years of research on “characterizing properties,” the tumor-suppressor definition is still controversial. The existence of chromosomal inherited traits that negatively regulate tumor development was first postulated by Theodor Boveri (1), who suggested the existence of inhibitory chromosomes that keep cell proliferation in check and favor tumor development upon removal. Formal proof for this concept came from cell fusion experiments by Henry Harris, who showed that hybrids of normal rodent cells and cancer cells exhibited growth properties similar to normal cells (2).
Few years later, Alfred Knudson Jr. (3) proposed a genetic mechanism for hereditary retinoblastoma (RB) that has influenced cancer genetics until today. Knudson, on the basis of mathematical and statistical analyses, hypothesized that the disease was caused by two rate-limiting genetic lesions, one inherited and phenotypically silent, and one acquired somatically during life. His theory was then validated by cloning the RB gene, and it is commonly referred to as the “two hit hypothesis.” In his model, Knudson introduced a new class of cancer genes, the recessive anti-oncogenes, now known as tumor-suppressor genes (TSGs) (4–6). Since then, the two hit model has defined the experimental strategies for the identification of novel TSGs: (i) identification of genes involved in cancer-predisposition, where the first “lesion” is inherited and the second is acquired somatically; (ii) identification of genes inside genomic regions displaying loss-of-heterozygosity (LOH) in cancer cells; (iii) identification of chromosomal regions from normal cells that suppress the transformed phenotype. Those strategies are embedded in the concept of double mutation, emphasizing this aspect over gene function. For this reason many today consider two inactivating hits in cancer to be the only acceptable proof for the existence of a TSG.
In 1997, Haber and Harlow proposed a novel description of TSGs as “genes that sustain loss-of-function mutations in the development of cancer” (7), thus introducing a functional aspect in the definition. The same year, Kinzler and Vogelstein distinguished two functional classes of TSGs, namely, “gatekeepers” and “caretakers” (8). Gatekeeper genes are designated to control cell-cycle progression, e.g., adenomatous polyposis coli (APC), β-catenin (CTNNB1), and RB, whereas caretaker genes maintain genome integrity, e.g., breast cancer gene 1 (BRCA1), Bloom syndrome gene (BLM), and ataxia telangiectasia mutated gene (ATM). Caretaker mutations would lead to increased mutation rate, consequently increasing the probability that gatekeepers become inactivated. There are several studies on caretaker and gatekeeper genes in the literature and the examples that follow below have been intentionally chosen to reject the belief that two inactivating mutations are required to define a tumor suppressor.
CDKN1B/p27KIP1 is a member of the cyclin-dependent kinase inhibitor family and regulates cell-cycle progression. Two series of evidence suggest that CDKN1B behaves as a classical TSG: LOH at chromosome 12p13, encompassing the CDKN1B locus, is frequently found in breast, prostate, and ovarian cancers (9–14), and reduced CDKN1B protein is significantly associated with high-grade and high-stage disease in those tumors [reviewed in (14)]. Reverse genetic experiments in mouse models, however, suggest a different scenario: (i) Cdkn1b−/− and Cdkn1b± mice developed intestinal and lung adenomas at higher frequency than wild type mice, yet with similar latency and penetrance (15); (ii) tumors from Cdkn1b± mice did not show mutations of the second Cdkn1b allele; (iii) the CDKN1B protein was still expressed at about 50% of the levels of wild type animals. In this case, as for other genes like Dmtf1 (16, 17), Fhit (18, 19), and Apc (20), to mention a few, it is clear that the mutation of a single allele is sufficient to promote tumorigenesis, a phenomenon that is called haplo-insufficiency. Haplo-insufficient TSGs have been proposed to act by increasing the population of mutated cells available for further mutations [reviewed in (21)].
Haplo-insufficiency is not the only exception to the two hit model. For example, ATM missense mutations are more common than ATM null mutations in breast cancers (22, 23), and mice carrying a copy of a mutated Atm allele develop more tumors than mice heterozygous for a silenced Atm allele (Atm±) (24), suggesting the existence of mono-allelic, dominant negative mutations in tumor suppressors. Thus, in open contrast to the classical definition of TSGs, dominant negative and haplo-insufficient genes are able to give rise to tumors following a single mutation insult. Finally, genetic mutations are not the only mechanism of functional inactivation for TSGs. Analysis of CDKN2A (p16INK4) in primary tumors, for example, revealed few inactivating mutations, and CDKN2A CpG island hypermethylation in 20% of cases (25), suggesting that epigenetic silencing of TSGs can drive tumorigenesis as well.
Mounting evidence and exceptions to the two hit model suggest that this definition of TSGs is too restrictive and in need of revision. A TSG operative definition should probably focus more on functional aspects, clear support from reverse genetic experiments, and determination of gene/protein dosage for which the two hit model is the extrema ratio. According to this view, we would then define a TSG as a gene whose alteration is found in human tumors, causes a reduction of its function, favors tumorigenesis (as demonstrated by the introduction of the same modification in animal experimental models), and if the overexpression of its wild type form elicits anti-tumoral effects. Functional reduction can be due to a reduced level of expression and to mutational events, including dominant negative effects on the wild type allele. With this paradigm in mind we questioned whether promyelocytic leukemia (PML) fulfills all the above requirements to be considered a true TSG.
PML Expression in Human Tumors
The first criteria we have examined to assess if PML is a tumor suppressor is the presence of genetic alterations or abnormal expression in human cancers.
About 95% of human acute promyelocytic leukemia (APL) cases harbor the oncogenic fusion gene PML-RARA as a result of the translocation t(15;17), whose breakpoints are located within the PML and the retinoic acid receptor α, RARA loci on chromosome 15 and 17, respectively. In principle, PML function in these cells is impaired by two concomitant events: PML haplo-insufficiency, for the presence of a single WT PML autosome and the expression of PML-RARA, which might interfere with the function of the WT PML protein. PML is involved in the regulation of many cellular functions and some of them, such as the activation of P53 by the DNA damage checkpoint response, rely on its localization and organization in subnuclear structures called PML-nuclear bodies (PML-NBs). PML-RARA functions as a dominant negative factor in the process of PML-NB assembly, causing a microspeckled distribution of PML. In this context, treatment with retinoic acid and arsenic, two agents that induce disease-remission in APL patients, leads to PML-RARA degradation, cell differentiation, re-assembly of PML-NBs, and tumor regression. These “therapeutic” effects establish a correlation between PML-NB assembly and tumor suppression, yet formal evidence that PML-NBs per se are responsible for tumor suppression is still missing. Indeed, it cannot be excluded that APL regression is due, exclusively, to other missing PML-RARA-associated functions after its degradation. Notably, expression of X-RARA chimera, which does not interfere with PML-NB assembly (P50-RARA, GCN4-RARA), and where X represents a coiled-coil domain mediating RARA homodimerization, maintains transforming potential, and recapitulates the main biological properties of PML–RARA (26, 27). However, the expression of X-RARA fusion proteins in mice, like P50-RARA or GCN4-RARA, is not sufficient to drive tumorigenesis as PML-RARA or the CCPML-RARA protein, where the RARA is fused to the coiled-coil region of PML, do (26). Pml−/− mice expressing P50-Rara do not show increased incidence or acceleration of leukemia onset. This suggests that loss of Pml cannot complement P50-Rara in restoring the leukemogenic potential to a level comparable to that of PML-RARA, and that the PML-RARA oncogene does not simply interfere with Pml and Rara functions but has additional activities that cannot be recapitulated by separating the two components in this way (27). In contrast with this observation, Pml−/− Pml-Rara transgenic-mice show increased incidence and acceleration of leukemia onset (28). Therefore is not clear if PML-NBs really exert tumor suppressive functions or not.
Immunohistochemical analysis of PML expression in human tumors of different histologic origins shows that PML expression-levels are reduced in a considerable number of cases, as compared to the corresponding normal tissues. PML expression is absent in 49% of central nervous system (CNS) tumors (in 100% of medulloblastomas and over 90% of oligodendroglial tumors), 17% of colon adenocarcinomas, 21% of lung tumors, 27% of prostate adenocarcinomas, 31% of breast adenocarcinomas, 49% of germ cell tumors, and 68% of non-Hodgkin’s lymphomas (in 83% of diffuse large-cell lymphomas and 77% of follicular lymphomas) (29). In addition, PML low-expression correlates with bad prognosis and high-grade tumors for breast adenocarcinomas and prostate carcinomas (29). PML is phosphorylated by the extracellular signal-regulated kinase ERK2 which facilitates the recruitment of the peptidyl-prolyl cis/trans isomerase PIN1 and the following degradation of PML by the proteasome (30, 31). Given the frequent activation of ERK2 in several types of tumors, due to the sustained action of paracrine growth factors or mutations, the ERK2/PML axis may partly explain why PML expression is low in a considerable fraction of human tumors. However, mutational analysis of the PML gene in 132 samples from different primary tumors and human cell lines showed that PML is rarely mutated or subjected to LOH mutations. Moreover, the few PML mutations detected did not correlate with PML protein-loss, suggesting that mutation is not the main mechanism of PML inactivation in the tumor types analyzed (29).
Triple negative breast cancer (TNBC) is a noticeable exception to the common theme of PML loss in tumors. PML expression is strongly associated with TNBC and basal high tumor-grade breast cancers, which are among the most undifferentiated and untreatable breast cancers (32). Here, high levels of PML expression correlate with early tumor recurrence, a signature of poor prognosis, and mutations of the tumor-suppressor P53 (33). In vitro experiments performed on MCF10A cells overexpressing PML show increased survival to anoikis via the regulation of PPAR-α and fatty acid oxidation, suggesting a crucial role for PML in the regulation of cell metabolism (33).
Chronic myeloid leukemia (CML) is also an exception. Indeed, high levels of PML expression correlate with bad prognosis in CML, and its function is critical to maintain leukemic initiating cells (34). PML degradation induced by As2O3 (arsenic trioxide) treatment allows exit from quiescence and exhaustion of cancer stem cells (CSCs) in a murine model of the disease (34). This parallels the role of PML in hematopoietic stem cells (HSCs) where it is highly expressed and controls HSC self-renewal and symmetric division through its regulation of mTOR and PPAR-δ signaling (fatty acid oxidation) (34). Additional proof of the general role that PML plays in stem cell homeostasis comes from studies on the nervous system. In mice PML expression is restricted to neural progenitor cells (NPCs) in the developing neocortex where PML regulates NPC proliferation and differentiation (35).
In summary, analysis of PML expression in human tumors reveals two distinct situations: (i) reduced/loss-of expression in different tumor types, most frequently in CNS tumors, which suggests that PML may be a tumor suppressor; (ii) high expression in CML and TNBC tumors, which, on the contrary, rely on PML expression to, respectively, maintain unlimited self-renewal and survive under metabolic stressing conditions. A controversial picture emerges from all these observations, indicating that the role of PML in human malignancies is context dependent.
Pml Loss is Not a General Tumor-Promoting Event
Pml−/− mice are fertile, with birth rates in line with the expected Mendelian frequency, and without gross phenotypic differences compared to Pml± or Pml+/+ littermates (36). However, Pml−/− mice are leukopenic, show an increased susceptibility to infections, a reduction of both granulocytes and monocytes, and have an impaired capacity for terminal maturation of myeloid cells in response to retinoic acid (36). Pml−/− mouse embryonic fibroblasts (MEFs) show an increased rate of proliferation as compared to Pml± or Pml+/+ MEFs without the hallmarks of transformation (36). Notably, Pml−/− mice do not develop spontaneous tumors at higher frequency than WT syngenic mice.
Experiments of cooperation between carcinogens/oncogenes and the concomitant inactivation of Pml show a more complicated picture. Cooperation between Pml-Rara and Pml inactivation was assessed in a murine model of APL expressing the Pml-Rara under the control of the human cathepsin G promoter (28). In this case, the leukemia-free survival (LFS) of hCG-Pml-Rara±Pml −/− and hCG-Pml-Rara±Pml ± mice was significantly reduced as compared to hCG-Pml-Rara±Pml +/+ mice [mean LFS ± SD, respectively: 434.4 ± 30.6 days (p < 0.0001), 498.9 ± 31.3 days (p = 0.003), 686.4 ± 35.5 days] showing a clear acceleration of leukemia onset. Moreover, the incidence of leukemia was significantly increased in Pml absence or haplo-insufficiency (p < 0.001 for both) (28). However, these data were not confirmed in a second independent model system where WT and Pml−/− cells transduced with a retroviral vector expressing Pml-Rara were compared for their ability to generate leukemia upon transplantation into irradiated recipients. In contrast to the previous study, leukemias developed significantly faster in a WT background (p < 0.05) (37). Since the two experimental settings were different it is difficult to draw a definitive conclusion about the role of Pml in Pml-Rara – driven APL.
Pml cooperation has also been assessed in a murine model of lung tumorigenesis driven by the oncogene K-RasG12D, which induces non-small cell lung carcinomas (NSCLC) in mice (38). In this model, Pml−/− mice showed an increased tumor burden compared to Pml+/+ control animals, assessed as the number of lung carcinomas/mouse after 8 weeks of continuous expression of the K-RasG12D oncogene (p < 0.05) (39).
In a murine model of kidney tumor driven by the haplo-insufficiency of the tuberous sclerosis protein 2 (TSC2), the picture, again, is different. Tsc2 heterozygosity leads to high MTORC1 activity, to the development of kidney cysts and, after a long latency, to carcinomas (40, 41). Kidney tumor initiation, assessed by the number of cysts and small carcinomas/kidney, is not affected in Pml−/− Tsc2± mice. However, tumors from these mice were more vascularized, showed higher proliferation and a more aggressive histological profile (42) suggesting changes in tumor progression rather than tumor initiation.
Pml−/− mice, challenged with 7,12-dimethylbenz(α)anthracene (DMBA) and 12-O-tetradecanoylphorbol-13-acetate (TPA) two-stage skin carcinogenesis, showed an increased number of papillomas indicative of benign tumor initiation but only a mild increase in the frequency of tumors undergoing malignant transformation (1.8 Pml+/+ vs. 2.3% Pml−/−) (36). Finally, Pml haplo-insufficiency or complete inactivation does not affect frequency, latency, and size of breast tumors in the MMTV/neu murine model of mammary tumorigenesis (28).
Thus, deletion of Pml does not lead to transformation per se but can favor tumorigenesis in some specific instances. Indeed, far from providing a coherent and unifying picture for the role of PML in tumorigenesis, these observations suggest that PML contribution depends on the specific background in which its inactivation is achieved.
PML Functions and Tumor Suppressive Pathways
Promyelocytic leukemia is a pleiotropic protein placed at the crossroad of many regulatory pathways. DNA damage and repair, anti-viral response, metabolic adaptation, induction of apoptosis/senescence, and telomere maintenance are some of the important processes which regulate and are, in turn, regulated by PML. Aim of this section is to examine the main tumor suppressive pathways and factors that are engaged by PML.
PML, Cell Death, and Senescence
Promyelocytic leukemia modulates the activity and expression of the known tumor-suppressor P53. Indeed, Pml−/− cells express less P53 than WT controls, both under steady-state conditions and upon γ-irradiation (43, 44). PML interacts directly with the DNA binding domain of the tumor-suppressor P53, colocalizes with P53 in the PML-NBs and acts as a P53 transcriptional coactivator (45–47). It activates P53 through multiple mechanisms: increasing its acetylation and phosphorylation (44, 46, 48–50), binding and inhibiting the main negative regulator of P53, the mouse double minute 2 homolog, MDM2 (51–53), and promoting P53 deubiquitination through the herpes-virus-associated ubiquitin-specific protease, HAUSP (54, 55). In turn, activated P53 can directly induce PML expression generating a positive feedback loop (56). Biologically, the influence of PML on the P53 pathway may result in the regulation of stress-induced apoptosis or senescence on a cell type-dependent basis. All these features are coherent with a tumor suppressive role of PML exerted via P53.
Promyelocytic leukemia function in apoptosis is not limited to the regulation of P53 or to the P53-dependent apoptotic pathway. Indeed, both murine Pml−/− cells and PML siRNA interfered human cells are defective for the induction of apoptosis triggered by several factors, like type I and type II interferons (IFN), FAS, tumor necrosis factor, ceramides, ionizing radiations (57, 58), and transforming growth factor (TGF)-β, which induce apoptosis (59).
How can a single protein being involved in the regulation of apoptosis from so many different stimuli? PML has been shown to interact with the phosphatase PP2A and to modulate its activity on specific targets. Via PP2A PML can modify the sensitivity to pro-survival and pro-apoptotic cues in at least two ways: (i) recruiting PP2A phosphatase to nuclear PML-NBs, thereby de-phosphorylating and inactivating AKT and its pro-survival functions (60); (ii) recruiting PP2A to the inositol 1,4,5-trisphosphate receptor, IP3R-3, to modulate Ca2+ release and storage at the endosplasmic reticulum (ER). The physical structures where both this regulation is achieved and cytoplasmic PML is localized are called mitochondria-associated membranes (MAMs). Indeed, sensitivity to apoptosis is dependent on the ability of cells to transfer Ca2+ from the ER to the mitochondria. Ca2+ release, in turn, induces mitochondrial Ca2+ loading, with a consequent release of mitochondrial proteins involved in the apoptotic process, such as cytochrome c, the apoptosis initiating factor AIF, and the complex of second mitochondria-derived activator of caspases (SMAC) with the direct IAP binding protein with low pI (DIABLO) (61, 62). Conditions that reduce ER Ca2+ storage, and thus Ca2+ release from the ER to the mitochondria, lower the probability of Ca2+ dependent apoptosis (62). In Pml−/− MEFs, both ER steady-state (Ca2+) values and the increase of cytosolic Ca2+ concentration after treatment with apoptotic stimuli like H2O2 are significantly smaller than in Pml+/+ MEFs (63). PML appears to be essential for PP2A tethering to the ER localized receptor IP3R-3, favoring IP3R-3 dephosphorylation and Ca2+ storage. Conversely, IP3R-3 is hyper-phosphorylated in the absence of PML and this modification decreases the Ca2+ release from the ER and the overload of mitochondria increasing the apoptotic threshold (64).
The effect of PML overexpression is consistent with it having a function in the regulation of survival, yet it is variable and depends on cell-type and experimental conditions. Three kinds of responses are observed upon PML overexpression: cell death without the hallmarks of apoptosis, caspase3-dependent apoptosis, and senescence. PML overexpression leads to rapid cell death without the typical features of apoptosis in immortalized rat embryonic fibroblasts (65). Caspase-3 activity is not induced upon overexpression of PML in these cells, implying that PML may quickly trigger apoptosis with a caspases-3-independent mechanism. Caspase-independent apoptosis is not unprecedented and has been also observed after staurosporine treatment, serum deprivation, and overexpression of oncogenes like c-MYC or E1A (66). Caspase-3-dependent apoptosis is triggered via P53 stabilization and the regulation of mitochondrial Ca2+ overload as mentioned above. Finally, introduction of PML into Pml−/− cells leads to increased P53 levels, recruitment of P53 and its acetyltransferase CREB binding protein (CBP) which mediates P53 stabilization to PML-NBs, and cellular senescence (43, 44). PML is essential for the induction of V-H-Ras-induced senescence via P53 acetylation and activation (48). Interestingly, although all isoforms can bind P53 and recruit it to PML-NBs, only the PML-IV isoform activates P53 and triggers senescence even in the absence of PML-NBs (67).
Overexpression of PML-IV also induces senescence through an RB-dependent mechanism (67, 68). Intriguingly, during the induction of senescence, PML-NBs colocalize with RB, the E2F transcription factor (69), and senescence-associated heterochromatin foci (SAHF), suggesting that heterochromatin proteins (such as HIRA, HP1, and ASF1) transit via NBs before the establishment of heterochromatin foci and senescence (70, 71). PML involvement in senescence induction has recently gained central stage from studies on human ubiquitin ligase E6AP−/− B-cell lymphomas. E6AP is an E3 ubiquitin ligase shown to target PML for degradation via the proteasome (72). E6AP−/− lymphomas express elevated levels of PML and PML-NBs and show a parallel increase in markers of cellular senescence, including P21, H3K9me3, and P16 (73). Equally, the expression of PML is restored by down-regulation of E6AP in B-lymphoma cells, with concurrent induction of cellular senescence, suggesting that the overriding of PML-induced senescence is essential for B-cell lymphoma progression (73).
PML and Cell-Cycle Restriction
Promyelocytic leukemia function is also intertwined with a second tumor suppressor called P21WAF1/CIP1. P21 belongs to the cyclin-dependent kinase inhibitor family and its role in cell-cycle restriction and differentiation has been widely investigated. PML is required for the transactivation of the P21 promoter following physiological doses of all-trans-retinoic acid (ATRA) treatment. Concentrations of ATRA in the range of 10−7–10−6 M do not activate the endogenous P21 gene in Pml−/− bone-marrow cells. Since P21 up-regulation results in the terminal differentiation of hematopoietic cells (74), the lack of P21 induction in Pml−/− cells might contribute to the role of PML in controlling hematopoietic myeloid differentiation (36). Terminal differentiation is indeed a powerful tumor suppressive mechanism, which leads a cell to a post-mitotic stage. Interestingly, PML-dependent accumulation of P21 is both P53 dependent and independent. Indeed, PML knock-down in P53−/− H1299 and HCT116 cells by siRNA interference causes down-regulation of P21 expression, inhibition of the γ-irradiation-induced up-regulation of P21, and decreased half-life of the P21 protein (75). These data provide evidence for a P53-independent functional relationship between PML and P21 in γ-irradiation-induced DNA damage responses, and identify PML as a positive post-translational regulator of P21 in P53−/− tumor cells.
Two lines of evidence link PML and P21 activity in normal and cancer cells. Interestingly, P21 and PML are important determinants of HSC stemness, self-renewal, and quiescence (34, 76). Experiments suggest that PML and P21 functions are important to maintain self-renewal of CML stem cells and to sustain AML leukemogenesis, respectively (77). In this context, PML and P21 both act as oncogenes rather than tumor suppressors. Notably, stem cells may be endowed with unique checkpoint pathways, as compared to progenitors or more differentiated cells, suggesting that cell-cycle restriction might increase their self-renewal potential (78). Importantly, P21 is also involved in DNA repair and it is recruited to UV irradiation-induced DNA damage sites where it interacts with proliferating cell nuclear antigen (PCNA), regulates the interaction of repair factors with PCNA, and protects PCNA from degradation (79).
Previous studies also indicated that PML post-transcriptionally represses several genes involved in growth promotion and cell-cycle progression, including cyclins (D1, E1, A2, B1) and c-MYC (43, 80 – 82). In addition, PML can influence entry into the cell cycle by regulating the activation of the tumor-suppressor RB. Interestingly, PML binds to the phosphatase PP1, and promotes PP1-dependent dephosphorylation of RB in neural stem cells (35). In PML−/− mice, the amount of phosphorylated RB in NPCs is increased and correlates with higher proliferation and defective exit from the cell cycle without any substantial effect on apoptosis (35).
PML and DNA Repair
An important role for PML in the response to DNA damage and repair is also suggested by the dynamic localization of several players of the DNA damage detection and repair machinery, such as the Bloom syndrome protein (BLM) and others like the ataxia telangiectasia and Rad3 related protein (ATR), the cds1 homolog kinase 2 (CHK2), and the Nijmegen Breakage Syndrome 1 (NBS1), to PML-NBs [reviewed in (52)]. Indeed, Pml−/− MEFs show a greatly augmented frequency of sister chromatid exchange (SCE) which is a distinctive molecular feature of Bloom syndrome cell genomic instability, suggesting that the localization of BLM to PML-NB is important to ensure BLM proper function (83). Thus, PML expression is a barrier to genomic instability, contributing to restrain the effects of DNA damage and oncogenic mutations. On the other hand, DNA repair systems are also potentially harmful for the organism, particularly in cells with a chronic source of DNA damage like cancer cells.
PML and Viral Infection
Promyelocytic leukemia confers resistance on RNA viruses by interacting with viral proteins and inhibiting their functions or, in a P53-dependent way, by inducing apoptosis of infected cells (84). In this way, PML expression restrains the propagation of infection and reduces the chance of possible oncogenic mutations due to viral DNA integration into the cell genome. The IFN is the best characterized inducer of PML, and all IFNs (α, β, and γ) sharply enhance mRNA and protein levels of PML, leading to a marked increase in the number and size of PML-NBs (85). Most of the studies implicating PML in anti-viral defense have been performed with the isoforms PML III, IV, or VI, whereas those implicating PML in apoptosis and senescence were performed with PML-IV isoform.
PML and Angiogenesis
Promyelocytic leukemia affects angiogenesis by negatively regulating the Akt-mTOR pathway which controls the synthesis of the hypoxia inducible factor HIF-1α, the main factor regulating cell response to hypoxia (86). Therefore, PML loss not only decreases PML tumor suppressive functions but also amplifies tumor hypoxia responses, such as angiogenesis, migration, metabolic reprograming, epithelial-to-mesenchymal transition, tumor growth, and chemoresistance, and these functions collectively generate very aggressive tumors. The HIF-1α high, Kelch-like protein KLHL20 high (PML E3 ubiquitin ligase), the peptidyl-prolyl cis-trans isomerase NIMA-interacting Pin1 high and the resulting PML low-expression profile correlate with high-grade tumors (87). PML deficiency, indeed, leads to increased neoangiogenesis and elevated expression of pro-angiogenic factors such as the vascular endothelial growth factor (VEGF) in human and mouse tumors, thus supporting growth and spreading of the disease (86).
PML and Telomere Maintenance
Promyelocytic leukemia is also involved in a mechanism of telomere maintenance known as alternative lengthening of telomeres (ALT). ALT occurs in telomerase-negative cell lines and PML forms ALT associated PML bodies (APBs) (88, 89). The appearance of APBs correlates with the stabilization of telomere length in ALT cell lines, suggesting that these bodies are responsible for the preservation of telomere length in the absence of telomerase activity (88, 89). The involvement of PML as a positive regulator of telomere maintenance on one hand suggests that its function may prevent genomic instability, on the other, it may grant telomerase-negative cancer cells a way to overcome replicative senescence.
PML and Autophagy
Autophagy is a catabolic process that allows lysosomal-mediated degradation of unnecessary or dysfunctional cellular components. Autophagy functions as an adaptive mechanism, which grants cell survival under several stress conditions (e.g., starvation, hypoxia) and maintains cellular integrity by clearing of subcellular debris and regeneration of metabolic precursors (90–93). In cells growing in a nutrient-rich environment (94, 95) autophagy is negatively regulated by mTOR and AKT signaling, two factors that are often constitutively active in AML (96). In line with these observations, mice lacking the autophagy-related protein 7 (Atg7) in HSCs develop an atypical myeloproliferative disorder, reminiscent of myelodisplastic syndrome (MDS), which progresses to AML (97). PML inhibits the AKT-mTOR signaling pathway, thus suggesting an involvement in the regulation of autophagy. A study from Laane and colleagues (98) shows that impairment of autophagy by siRNA-mediated repression of the autophagy regulator BECN1 interferes with glucocorticoid dexamethasone-mediated lymphoblastic leukemia cell death. Dexamethasone treatment leads to PML up-regulation, its interaction with AKT, and PML-dependent AKT dephosphorylation, which was required for dexamethasone-dependent cytotoxicity effects on thymocytes. Conversely, autophagy was shown to contribute to ATRA mediated differentiation and PML-RARA degradation in NB4 cells, where PML-RARA is targeted to lysosomes by the ubiquitin binding adaptor protein p62/SQSTM1 (99, 100). These studies coherently associate PML activity with autophagy induction and tumor suppression in acute promyelocytic leukemia. However, the role of autophagy in tumorigenesis and tumor maintenance is still controversial.
In conclusion, PML functions are complex, and a comprehensive model which clearly summarizes how they are interlinked is still missing. Definitely, PML mediates apoptosis and senescence, two very important strategies for limiting tumor development and progression. However, some of PML activities, such as its control of cell-cycle progression via P21 and RB or its role in telomere maintenance, hide an oncogenic potential that some tumors might exploit to survive. Thus, PML functions cannot be considered univocally tumor suppressive.
Discussion
Promyelocytic leukemia has an important role in the regulation of stress response in normal cells. Its complex regulatory network allows integration of signals coming from several pathways. As a consequence, PML seems to be a key factor in cell fate decisions, i.e., self-renewal vs. differentiation, adaptation vs. apoptosis or senescence. In vitro, PML activity is necessary for the induction of apoptosis in response to many different stimuli, and overexpression of PML leads to apoptosis or senescence, as demonstrated from both in vitro experiments and in vivo observations. Despite this centrality in mediating apoptosis or cell-cycle arrest, PML inactivation is not sufficient to increase the rate of spontaneous cancers in mice. However, in murine experimental models of cancer, PML loss can cooperate with some oncogenic lesions to increase tumor initiation and progression.
Altogether, the emerging picture is that of a context-dependent contribution of PML to both tumorigenesis and tumor suppression, and it is not possible to conclude which way the balance tilts (Figure 1).
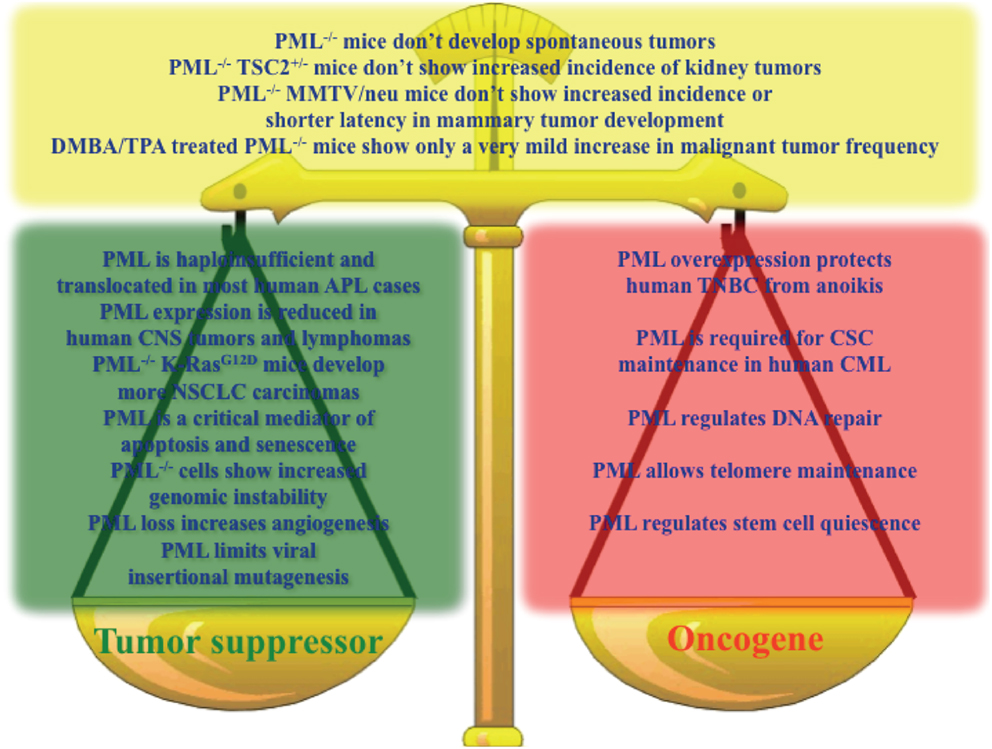
Figure 1. Key results sustaining a role for PML as a tumor suppressor (green box), oncogene (red box), or in contrast with the hypothesis that PML is a tumor suppressor (yellow box).
Non-small cell lung carcinoma tumorigenesis driven by the oncogene K-RasG12D, for example, benefits from PML loss, which leads to an increased number of lung adenocarcinomas. However, Pml−/− mice treated with the DMBA-TPA protocol, while showing increased papilloma incidence, develop few more malignant tumors, and Tsc2± and MMTV neu driven kidney and mammary tumors, respectively, do not show differences in tumor initiation. Moreover, PML expression is required to maintain self-renewal and avoid cell death by anoikis in CML and TNBC, respectively, suggesting that some tumors may even rely on PML expression to sustain the malignant phenotype. Hence, the simple notion that PML is a general tumor suppressor that serves as a strong pro-apoptotic and pro-senescence determinant is not supported by the available data. Which of the multiple functions of PML is crucial or contrasts cancer development and maintenance depends probably on the specific physiological state of each neoplasia, and it is determined by the mutations occurring in that specific cell. Consistent with this hypothesis is the observation that PML expression correlates with high frequency of mutated P53 in TNBC. It is tempting to speculate that also the reciprocal may be true, so that reduced PML expression could affect tumors where the P53 or RB pathways are not inactivated. Furthermore, PML loss of expression in human tumors, which is mostly regulated at the level of protein stability, may be the result of the constitutive activation of common growth factor cues, which impinge into the same ERK2/PIN1/KLHL20/PML axis leading to PML protein degradation.
In recent years supporting evidence for the CSC or hierarchical model has been collected, shedding light on the crucial role of CSCs in tumor maintenance. The emerging concept that CSCs are the real biological reservoir of malignancies and that it is possible to exhaust CSC self-renewal by driving CSCs out of quiescence is a powerful paradigm which has just begun to be exploited experimentally and in clinical trials. Proof of concept experiments on this issue have been performed in murine models of CML (34) and AML (77). In this regard, PML is an interesting potential target since it is highly expressed in stem cells, restrains cell-cycle progression and maintains stem cell self-renewal [reviewed in (101)].
Future experiments should probably reassess the expression level of PML more specifically in the CSC compartment of human tumors. It is indeed possible that PML expression is confined to the CSCs and absent in the more differentiated cancer progenitors. Since CSCs very often constitute only a minor fraction of the whole tumor population, PML expression may have gone unnoticed in previous analyses, underestimating the importance of PML in the maintenance of a specific tumor.
If this is the case, PML function in support of self-renewal may be more important than its tumor-suppressor activities and the applicability of well-established therapeutic agents, like As2O3, which lead to PML degradation may be much broader than expected.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We kindly thank Paola Dalton for critical reading and editing of the manuscript.
References
2. Harris H, Miller OJ, Klein G, Worst P, Tachibana T. Suppression of malignancy by cell fusion. Nature (1969) 223(5204):363–8. doi:10.1038/223363a0
3. Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A (1971) 68(4):820–3. doi:10.1073/pnas.68.4.820
4. Knudson AG. The genetics of childhood cancer. Cancer (1975) 35(Suppl 3):1022–6. doi:10.1002/1097-0142(197503)35:3<1022::AID-CNCR2820350726>3.0.CO;2-A
5. Knudson AG. Antioncogenes and human cancer. Proc Natl Acad Sci U S A (1993) 90(23):10914–21. doi:10.1073/pnas.90.23.10914
6. Murphree AL, Benedict WF. Retinoblastoma: clues to human oncogenesis. Science (1984) 223(4640):1028–33. doi:10.1126/science.6320372
7. Haber D, Harlow E. Tumour-suppressor genes: evolving definitions in the genomic age. Nat Genet (1997) 16(4):320–2. doi:10.1038/ng0897-320
8. Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature (1997) 386(6627):761, 763.
9. Hatta Y, Takeuchi S, Yokota J, Koeffler HP. Ovarian cancer has frequent loss of heterozygosity at chromosome 12p12.3-13.1 (region of TEL and Kip1 loci) and chromosome 12q23-ter: evidence for two new tumour-suppressor genes. Br J Cancer (1997) 75(9):1256–62. doi:10.1038/bjc.1997.214
10. Kibel AS, Freije D, Isaacs WB, Bova GS. Deletion mapping at 12p12-13 in metastatic prostate cancer. Genes Chromosomes Cancer (1999) 25(3):270–6. doi:10.1002/(SICI)1098-2264(199907)25:3<270::AID-GCC9>3.0.CO;2-Z
11. Pietenpol JA, Bohlander SK, Sato Y, Papadopoulos N, Liu B, Friedman C, et al. Assignment of the human p27Kip1 gene to 12p13 and its analysis in leukemias. Cancer Res (1995) 55(6):1206–10.
12. Takeuchi S, Bartram CR, Wada M, Reiter A, Hatta Y, Seriu T, et al. Allelotype analysis of childhood acute lymphoblastic leukemia. Cancer Res (1995) 55(22):5377–82.
13. Alsheich-Bartok O, Haupt S, Alkalay-Snir I, Saito S, Appella E, Haupt Y. PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene (2008) 27(26):3653–61. doi:10.1038/sj.onc.1211036
14. Tsihlias J, Kapusta L, Slingerland J. The prognostic significance of altered cyclin-dependent kinase inhibitors in human cancer. Annu Rev Med (1999) 50:401–23. doi:10.1146/annurev.med.50.1.401
15. Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature (1998) 396(6707):177–80. doi:10.1038/24179
16. Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev (1999) 13(20):2658–69. doi:10.1101/gad.13.20.2658
17. Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, Roussel MF, et al. Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev (2000) 14(14):1797–809.
18. Fong LY, Fidanza V, Zanesi N, Lock LF, Siracusa LD, Mancini R, et al. Muir-Torre-like syndrome in Fhit-deficient mice. Proc Natl Acad Sci U S A (2000) 97(9):4742–7. doi:10.1073/pnas.080063497
19. Zanesi N, Fidanza V, Fong LY, Mancini R, Druck T, Valtieri M, et al. The tumor spectrum in FHIT-deficient mice. Proc Natl Acad Sci U S A (2001) 98(18):10250–5. doi:10.1073/pnas.191345898
20. Yan H, Dobbie Z, Gruber SB, Markowitz S, Romans K, Giardiello FM, et al. Small changes in expression affect predisposition to tumorigenesis. Nat Genet (2002) 30(1):25–6. doi:10.1038/ng799
21. Quon KC, Berns A. Haplo-insufficiency? Let me count the ways. Genes Dev (2001) 15(22):2917–21. doi:10.1101/gad.949001
22. Izatt L, Greenman J, Hodgson S, Ellis D, Watts S, Scott G, et al. Identification of germline missense mutations and rare allelic variants in the ATM gene in early-onset breast cancer. Genes Chromosomes Cancer (1999) 26(4):286–94. doi:10.1002/(SICI)1098-2264(199912)26:4<286::AID-GCC2>3.3.CO;2-O
23. Teraoka SN, Malone KE, Doody DR, Suter NM, Ostrander EA, Daling JR, et al. Increased frequency of ATM mutations in breast carcinoma patients with early onset disease and positive family history. Cancer (2001) 92(3):479–87. doi:10.1002/1097-0142(20010801)92:3<479::AID-CNCR1346>3.0.CO;2-G
24. Spring K, Ahangari F, Scott SP, Waring P, Purdie DM, Chen PC, et al. Mice heterozygous for mutation in Atm, the gene involved in ataxia-telangiectasia, have heightened susceptibility to cancer. Nat Genet (2002) 32(1):185–90. doi:10.1038/ng958
25. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med (1995) 1(7):686–92. doi:10.1038/nm0795-686
26. Occhionorelli M, Santoro F, Pallavicini I, Gruszka A, Moretti S, Bossi D, et al. The self-association coiled-coil domain of PML is sufficient for the oncogenic conversion of the retinoic acid receptor (RAR) alpha. Leukemia (2011) 25(5):814–20. doi:10.1038/leu
27. Sternsdorf T, Phan VT, Maunakea ML, Ocampo CB, Sohal J, Silletto A, et al. Forced retinoic acid receptor alpha homodimers prime mice for APL-like leukemia. Cancer Cell (2006) 9(2):81–94. doi:10.1016/j.ccr.2005.12.030
28. Rego EM, Wang ZG, Peruzzi D, He LZ, Cordon-Cardo C, Pandolfi PP. Role of promyelocytic leukemia (PML) protein in tumor suppression. J Exp Med (2001) 193(4):521–9. doi:10.1084/jem.193.4.521
29. Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ, et al. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst (2004) 96(4):269–79. doi:10.1093/jnci/djh043
30. Lim JH, Liu Y, Reineke E, Kao H-Y. Mitogen-activated protein kinase extracellular signal-regulated kinase 2 phosphorylates and promotes Pin1 protein-dependent promyelocytic leukemia protein turnover. J Biol Chem (2011) 286(52):44403–11. doi:10.1074/jbc.M111.289512
31. Reineke EL, Lam M, Liu Q, Liu Y, Stanya KJ, Chang K-S, et al. Degradation of the tumor suppressor PML by Pin1 contributes to the cancer phenotype of breast cancer MDA-MB-231 cells. Mol Cell Biol (2008) 28(3):997–1006. doi:10.1128/MCB.01848-07
32. Huber KE, Carey LA, Wazer DE. Breast cancer molecular subtypes in patients with locally advanced disease: impact on prognosis, patterns of recurrence, and response to therapy. Semin Radiat Oncol (2009) 19(4):204–10. doi:10.1016/j.semradonc.2009.05.004
33. Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VCJ, Laurent G, et al. A metabolic prosurvival role for PML in breast cancer. J Clin Invest (2012) 122(9):3088–100. doi:10.1172/JCI62129
34. Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature (2008) 453(7198):1072–8. doi:10.1038/nature07016
35. Regad T, Bellodi C, Nicotera P, Salomoni P. The tumor suppressor Pml regulates cell fate in the developing neocortex. Nat Neurosci (2009) 12(2):132–40. doi:10.1038/nn.2251
36. Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, et al. Role of PML in cell growth and the retinoic acid pathway. Science (1998) 279(5356):1547–51. doi:10.1126/science.279.5356.1547
37. Insinga A, Pelicci PG, Inucci S. Leukemia-associated fusion proteins. Multiple mechanisms of action to drive cell transformation. Cell Cycle (2005) 4(1):67–9. doi:10.4161/cc.4.1.1400
38. Fisher GH, Orsulic S, Holland E, Hively WP, Li Y, Lewis BC, et al. Development of a flexible and specific gene delivery system for production of murine tumor models. Oncogene (1999) 18(38):5253–60. doi:10.1038/sj.onc.1203087
39. Scaglioni PP, Yung TM, Cai LF, Erdjument-Bromage H, Kaufman AJ, Singh B, et al. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell (2006) 126(2):269–83. doi:10.1016/j.cell.2006.05.041
40. Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda T. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res (1999) 59(6):1206–11.
41. Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2(±) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest (1999) 104(6):687–95. doi:10.1172/JCI7319
42. Bernardi R, Papa A, Egia A, Coltella N, Teruya-Feldstein J, Signoretti S, et al. Pml represses tumour progression through inhibition of mTOR. EMBO Mol Med (2011) 3(5):249–57. doi:10.1002/emmm.201100130
43. Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev (2000) 14(16):2015–27.
44. Louria-Hayon I, Grossman T, Sionov RV, Alsheich O, Pandolfi PP, Haupt Y. The promyelocytic leukemia protein protects p53 from Mdm2-mediated inhibition and degradation. J Biol Chem (2003) 278(35):33134–41. doi:10.1074/jbc.M301264200
45. Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J (2000) 19(22):6185–95. doi:10.1093/emboj/19.22.6185
46. Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell (1997) 90(4):595–606. doi:10.1016/S0092-8674(00)80521-8
47. Pearson M, Pelicci PG. PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene (2001) 20(49):7250–6. doi:10.1038/sj.onc.1204856
48. Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature (2000) 406(6792):207–10. doi:10.1038/35018127
49. Möller A, Sirma H, Hofmann TG, Rueffer S, Klimczak E, Dröge W, et al. PML is required for homeodomain-interacting protein kinase 2 (HIPK2)-mediated p53 phosphorylation and cell cycle arrest but is dispensable for the formation of HIPK domains. Cancer Res (2003) 63(15):4310–4.
50. Yang S, Jeong J-H, Brown AL, Lee C-H, Pandolfi PP, Chung JH, et al. Promyelocytic leukemia activates Chk2 by mediating Chk2 autophosphorylation. J Biol Chem (2006) 281(36):26645–54. doi:10.1074/jbc.M604391200
51. Bernardi R, Pandolfi PP. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene (2003) 22(56):9048–57. doi:10.1038/sj.onc.1207106
52. Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays (2004) 26(9):963–77. doi:10.1002/bies.20089
53. Kurki S, Latonen L, Laiho M. Cellular stress and DNA damage invoke temporally distinct Mdm2, p53 and PML complexes and damage-specific nuclear relocalization. J Cell Sci (2003) 116(Pt 19):3917–25. doi:10.1242/jcs.00714
54. Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J (1997) 16(3):566–77. doi:10.1093/emboj/16.7.1519
55. Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature (2002) 416(6881):648–53. doi:10.1038/nature737
56. de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, et al. PML is a direct p53 target that modulates p53 effector functions. Mol Cell (2004) 13(4):523–35. doi:10.1016/S1097-2765(04)00062-0
57. Crowder C, Dahle O, Davis RE, Gabrielsen OS, Rudikoff S. PML mediates IFN-alpha-induced apoptosis in myeloma by regulating TRAIL induction. Blood (2005) 105(3):1280–7. doi:10.1182/blood-2004-04-1614
58. Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, et al. PML is essential for multiple apoptotic pathways. Nat Genet (1998) 20(3):266–72. doi:10.1038/3030
59. Lin H-K, Bergmann S, Pandolfi PP. Cytoplasmic PML function in TGF-beta signalling. Nature (2004) 431(7005):205–11. doi:10.1038/nature02783
60. Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature (2006) 441(7092):523–7. doi:10.1038/nature04809
61. Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene (2008) 27(50):6407–18. doi:10.1038/onc.2008.308
62. Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J (2001) 20(11):2690–701. doi:10.1093/emboj/20.11.2690
63. Giorgi C, Ito K, Lin H-K, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science (2010) 330(6008):1247–51. doi:10.1126/science.1189157
64. Pinton P, Giorgi C, Pandolfi PP. The role of PML in the control of apoptotic cell fate: a new key player at ER-mitochondria sites. Cell Death Differ (2011) 18(9):1450–6. doi:10.1038/cdd.2011.31
65. Quignon F, De Bels F, Koken M, Feunteun J, Ameisen JC, de Thé H. PML induces a novel caspase-independent death process. Nat Genet (1998) 20(3):259–65. doi:10.1038/3068
66. Rathmell JC, Thompson CB. The central effectors of cell death in the immune system. Annu Rev Immunol (1999) 17:781–828. doi:10.1146/annurev.immunol.17.1.781
67. Bischof O, Kirsh O, Pearson M, Itahana K, Pelicci PG, Dejean A. Deconstructing PML-induced premature senescence. EMBO J (2002) 21(13):3358–69. doi:10.1093/emboj/cdf341
68. Mallette FA, Goumard S, Gaumont-Leclerc M-F, Moiseeva O, Ferbeyre G. Human fibroblasts require the Rb family of tumor suppressors, but not p53, for PML-induced senescence. Oncogene (2004) 23(1):91–9. doi:10.1038/sj.onc.1206886
69. Vernier M, Bourdeau V, Gaumont-Leclerc M-F, Moiseeva O, Bégin V, Saad F, et al. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev (2011) 25(1):41–50. doi:10.1101/gad.1975111
70. Ye X, Zerlanko B, Zhang R, Somaiah N, Lipinski M, Salomoni P, et al. Definition of pRB- and p53-dependent and -independent steps in HIRA/ASF1a-mediated formation of senescence-associated heterochromatin foci. Mol Cell Biol (2007) 27(7):2452–65. doi:10.1128/MCB.01592-06
71. Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell (2005) 8(1):19–30. doi:10.1016/j.devcel.2004.10.019
72. Louria-Hayon I, Alsheich-Bartok O, Levav-Cohen Y, Silberman I, Berger M, Grossman T, et al. E6AP promotes the degradation of the PML tumor suppressor. Cell Death Differ (2009) 16(8):1156–66. doi:10.1038/cdd.2009.31
73. Wolyniec K, Shortt J, de Stanchina E, Levav-Cohen Y, Alsheich-Bartok O, Louria-Hayon I, et al. E6AP ubiquitin ligase regulates PML-induced senescence in Myc-driven lymphomagenesis. Blood (2012) 120(4):822–32. doi:10.1182/blood-2011-10-387647
74. Liu M, Iavarone A, Freedman LP. Transcriptional activation of the human p21(WAF1/CIP1) gene by retinoic acid receptor. Correlation with retinoid induction of U937 cell differentiation. J Biol Chem (1996) 271(49):31723–8. doi:10.1074/jbc.271.49.31723
75. Cao L, Song Y, Tian B, Liu J, Liu B, Zhang J, et al. PML contributes to p53-independent p21 up-regulation in gamma-irradiation induced DNA damage responses. Chin Sci Bull (2011) 56(30):3148–54. doi:10.1007/s11434-011-4566-0
76. Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science (2000) 287(5459):1804–8. doi:10.1126/science.287.5459.1804
77. Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature (2009) 457(7225):51–6. doi:10.1038/nature07618
78. Insinga A, Cicalese A, Faretta M, Gallo B, Albano L, Ronzoni S, et al. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc Natl Acad Sci U S A (2013) 110(10):3931–6. doi:10.1073/pnas.1213394110
79. Lee JY, Kim HS, Kim JY, Sohn J. Nuclear translocation of p21(WAF1/CIP1) protein prior to its cytosolic degradation by UV enhances DNA repair and survival. Biochem Biophys Res Commun (2009) 390(4):1361–6. doi:10.1016/j.bbrc.2009.10.160
80. Cohen N, Sharma M, Kentsis A, Perez JM, Strudwick S, Borden KL. PML RING suppresses oncogenic transformation by reducing the affinity of eIF4E for mRNA. EMBO J (2001) 20(16):4547–59. doi:10.1093/emboj/20.16.4547
81. Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KLB. eIF4E is a central node of an RNA regulon that governs cellular proliferation. J Cell Biol (2006) 175(3):415–26. doi:10.1083/jcb.200607020
82. Lai HK, Borden KL. The promyelocytic leukemia (PML) protein suppresses cyclin D1 protein production by altering the nuclear cytoplasmic distribution of cyclin D1 mRNA. Oncogene (2000) 19(13):1623–34. doi:10.1038/sj.onc.1203473
83. Zhong S, Hu P, Ye TZ, Stan R, Ellis NA, Pandolfi PP. A role for PML and the nuclear body in genomic stability. Oncogene (1999) 18(56):7941–7. doi:10.1038/sj.onc.1203367
84. McNally BA, Trgovcich J, Maul GG, Liu Y, Zheng P. A role for cytoplasmic PML in cellular resistance to viral infection. PLoS ONE (2008) 3(5):e2277. doi:10.1371/journal.pone.0002277
85. Geoffroy M-C, Chelbi-Alix MK. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res (2011) 31(1):145–58. doi:10.1089/jir.2010.0111
86. Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, et al. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature (2006) 442(7104):779–85. doi:10.1038/nature05029
87. Chen RH, Lee YR, Yuan WC. The role of PML ubiquitination in human malignancies. J Biomed Sci (2012) 19:81. doi:10.1186/1423-0127-19-81
88. Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res (1999) 59(17):4175–9.
89. Grobelny JV, Godwin AK, Broccoli D. ALT-associated PML bodies are present in viable cells and are enriched in cells in the G(2)/M phase of the cell cycle. J Cell Sci (2000) 113(Pt 24):4577–85.
90. Bøe SO, Simonsen A. Autophagic degradation of an oncoprotein. Autophagy (2010) 6(7):964–5. doi:10.4161/auto.6.7.13066
91. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell (2008) 132(1):27–42. doi:10.1016/j.cell.2007.12.018
92. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature (2008) 451(7182):1069–75. doi:10.1038/nature06639
93. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev (2010) 90(4):1383–435. doi:10.1152/physrev.00030.2009
94. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet (2009) 43:67–93. doi:10.1146/annurev-genet-102808-114910
95. Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol (2004) 36(12):2445–62. doi:10.1016/j.biocel.2004.02.002
96. Yecies JL, Manning BD. mTOR links oncogenic signaling to tumor cell metabolism. J Mol Med (2011) 89(3):221–8. doi:10.1007/s00109-011-0726-6
97. Watson AS, Mortensen M, Simon AK. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle (2011) 10(11):1719–25. doi:10.4161/cc.10.11.15673
98. Laane E, Pokrovskaja Tamm K, Buentke E, Ito K, Kharaziha P, Khahariza P, et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ (2009) 16(7):1018–29. doi:10.1038/cdd.2009.46
99. Isakson P, Bjørås M, Bøe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood (2010) 116(13):2324–31. doi:10.1182/blood-2010-01-261040
100. Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y, et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy (2011) 7(4):401–11. doi:10.4161/auto.7.4.14397
Keywords: PML, tumor suppressor, cancer stem cells, cancer, cancer therapy
Citation: Mazza M and Pelicci PG (2013) Is PML a tumor suppressor? Front. Oncol. 3:174. doi: 10.3389/fonc.2013.00174
Received: 29 March 2013; Accepted: 20 June 2013;
Published online: 09 July 2013.
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Alexandre Arcaro, University of Bern, SwitzerlandMichelangelo Campanella, Royal Veterinary College and University College London, UK
Copyright: © 2013 Mazza and Pelicci. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Pier Giuseppe Pelicci, Department of Experimental Oncology, European Institute of Oncology, Via Adamello 16, 20139 Milan, Italy e-mail: piergiuseppe.pelicci@ieo.eu