 Ashley R. P. Hinson1 Rosanne Jones2 Lisa E. S. Crose1 Brian C. Belyea3 Frederic G. Barr4 Corinne M. Linardic1,5*
Ashley R. P. Hinson1 Rosanne Jones2 Lisa E. S. Crose1 Brian C. Belyea3 Frederic G. Barr4 Corinne M. Linardic1,5*
- 1Department of Pediatrics, Duke University Medical Center, Durham, NC, USA
- 2Duke University School of Medicine, Durham, NC, USA
- 3Department of Pediatrics, University of Virginia School of Medicine, Charlottesville, VA, USA
- 4Laboratory of Pathology, National Cancer Institute, Bethesda, MD, USA
- 5Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC, USA
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma of childhood and adolescence. Despite intergroup clinical trials conducted in Europe and North America, outcomes for high risk patients with this disease have not significantly improved in the last several decades, and survival of metastatic or relapsed disease remains extremely poor. Accrual into new clinical trials is slow and difficult, so in vitro cell-line research and in vivo xenograft models present an attractive alternative for preclinical research for this cancer type. Currently, 30 commonly used human RMS cell lines exist, with differing origins, karyotypes, histologies, and methods of validation. Selecting an appropriate cell line for RMS research has important implications for outcomes. There are also potential pitfalls in using certain cell lines including contamination with murine stromal cells, cross-contamination between cell lines, discordance between the cell line and its associated original tumor, imposter cell lines, and nomenclature errors that result in the circulation of two or more presumed unique cell lines that are actually from the same origin. These pitfalls can be avoided by testing for species-specific isoenzymes, microarray analysis, assays for subtype-specific fusion products, and short tandem repeat analysis.
Introduction
Rhabdomyosarcoma (RMS) is a malignancy that arises from skeletal muscle precursors (1). It is the most common type of soft tissue sarcoma in children and adolescents less than 20 years old, with an incidence of 4.5 cases per million children/adolescents per year (2). There are two major subtypes of RMS, embryonal and alveolar, which differ markedly in their outcomes. Embryonal RMS usually presents in children less than 10 years old and has a 5-year survival of close to 75%. Alveolar RMS, on the other hand, occurs at about the same incidence throughout childhood and adolescence and is associated with a poorer prognosis with a 5-year survival rate of less than 50%. Patients with refractory or relapsed RMS have an even worse prognosis, with survival ranging from 10 to 30% (3).
Despite numerous clinical trials, outcomes for high risk RMS have not improved significantly in the last 30 years. Given the relatively small population of children diagnosed with this disease annually, and the number and timing of new clinical trials, testing new potential treatment agents is limited. In vitro cell-line research and in vivo xenograft models aid in the basic and preclinical effort to identify potential new treatments. If shown promising, these agents can then be taken into human clinical trials, and compared to standard of care agents. The Pediatric Preclinical Testing Panel (PPTP) is an initiative formed by the National Cancer Institute, working to further characterize and validate available cell lines in multiple kinds of pediatric cancer, including RMS so that preclinical evaluations of new chemotherapeutic agents can be tested (4). Currently, there are 18 embryonal and 12 distinct alveolar human RMS cell lines described in the literature that have been used in more than one study by more than one research group. They differ in their origins, histologies, karyotypes, and methods of validation. They are described below and summarized in Table 1. There are also 16 human RMS cell lines that have been described and used by single research groups (5–17); these are listed in Table 2. [Of note, during revisions of this article an independent list of human and murine RMS cell lines was published (18).] The current article aims to summarize the published RMS cell lines, aid scientists in deciding which lines may be applicable to their research projects, and highlight important historical information and limitations for specific cell lines.
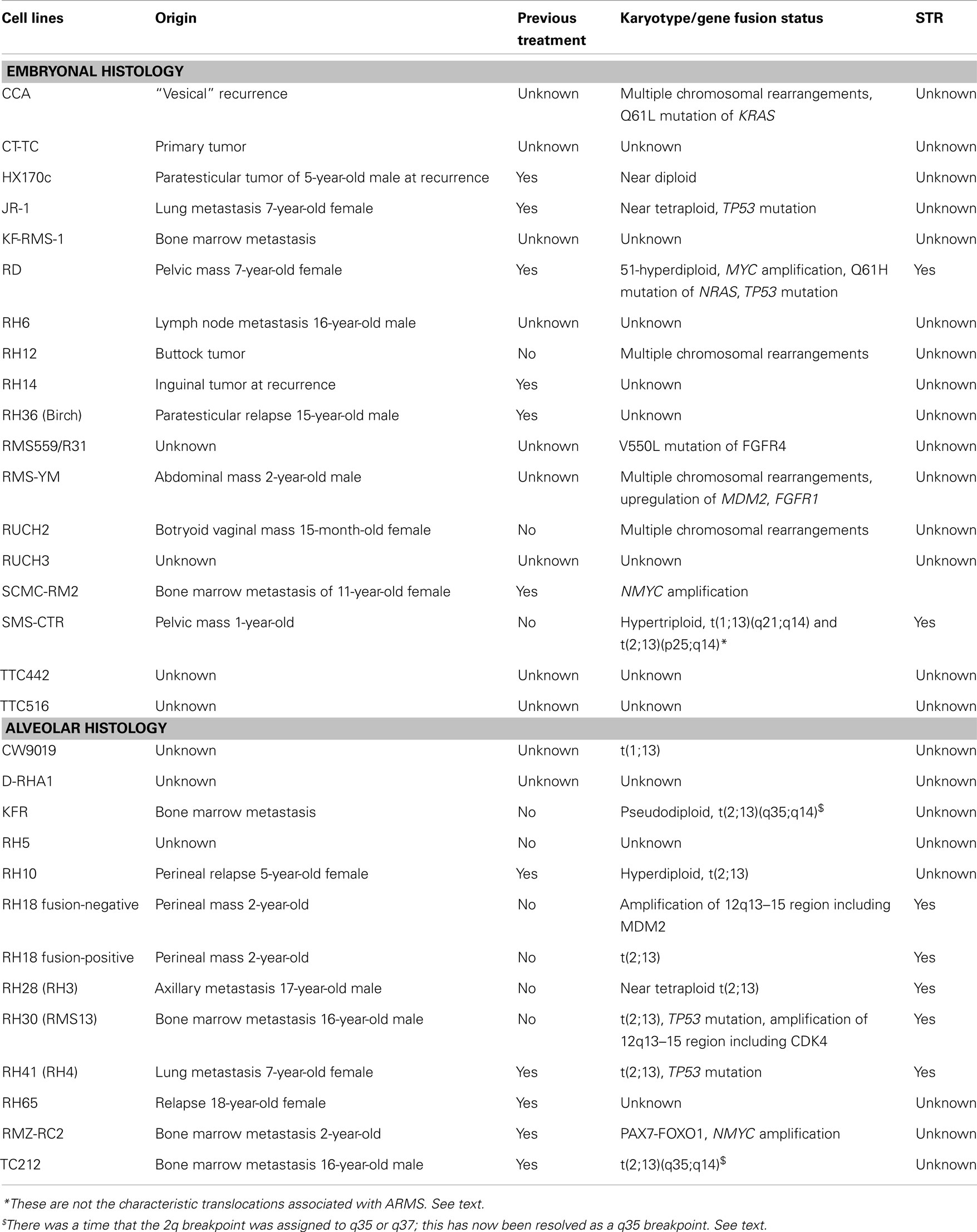
Table 1. Human RMS cell lines reported and used by multiple research groups.
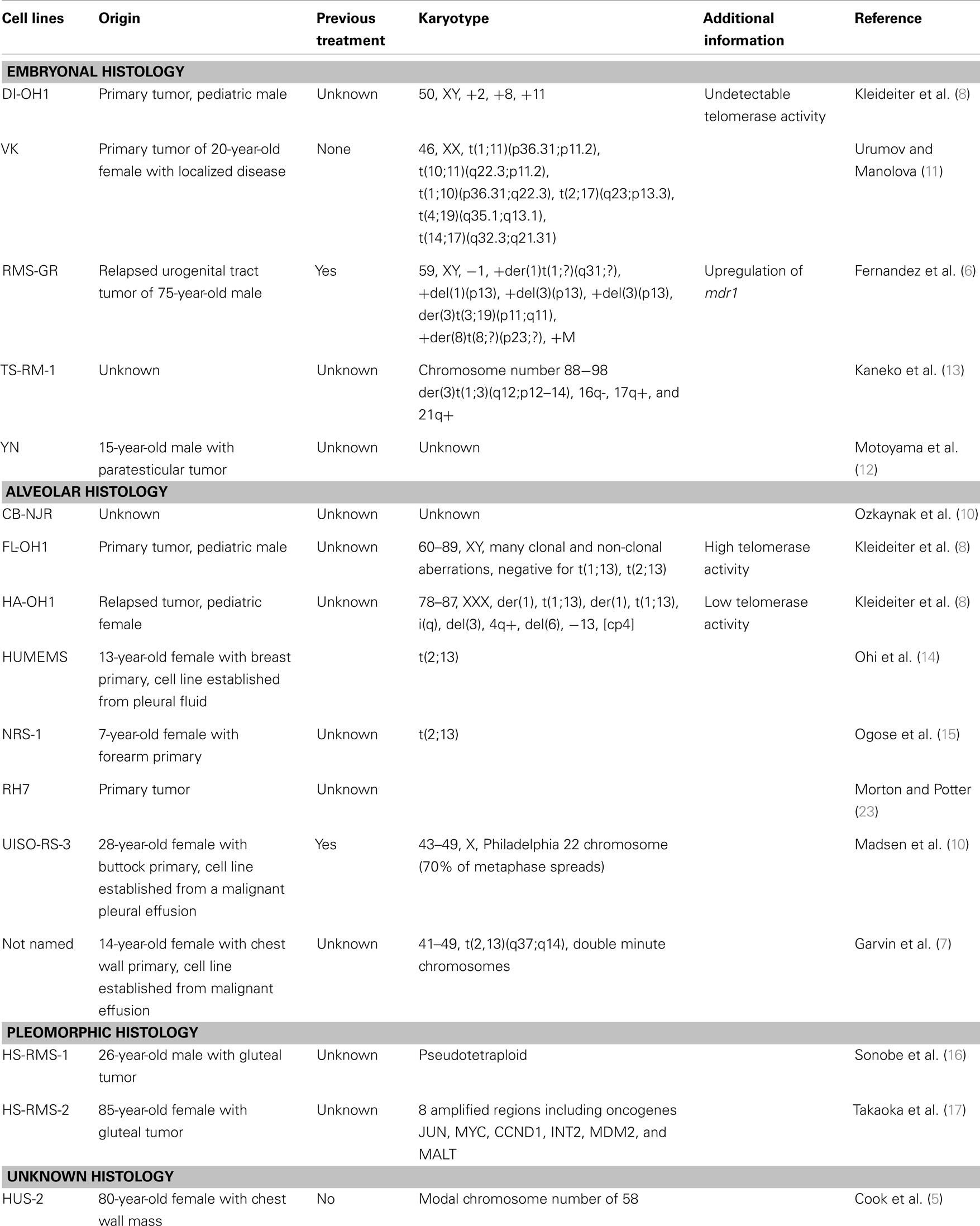
Table 2. Additional human RMS cell lines reported and used by a single research group.
Embryonal RMS Cell Lines
CCA
CCA was derived from the biopsy of a “vesical” recurrence of embryonal RMS in an 8-year-old Caucasian male (19). Multiple chromosomal rearrangements were identified upon karyotype analysis, with additional defects on chromosomes 1, 4, 6, 8, 9, 10, 11, 12, and 13 (20). CCA cells express vimentin and desmin. These cells can be used to generate xenografts in nude mice subcutaneously or intramuscularly, and form lung metastases when injected intravenously after pretreatment of the mice with cyclophosphamide. CCA cells harbor a Q61L mutation in KRAS (21). CCA has been grown in modified Dulbecco’s medium (DMEM) with 10% fetal bovine serum (FBS) (22). As with cell culture in general, it is up to the investigator whether prophylactic antibiotics penicillin and streptomycin are to be included during routine culture.
CT-TC
This cell line was derived from a primary tumor with an embryonal histology and expresses MyoD, myogenin, and desmin (at very low levels). It was originally developed by Dr. Hajime Hosoi and can be grown in DMEM with 10% FBS (23).
HX170c
HX170c was established from a paratesticular tumor of a 5-year-old Caucasian male. The patient had been previously treated with vincristine, adriamycin, cyclophosphamide, and radiotherapy. The tumor specimen was designated RMS based on the presence of desmin intermediate filaments, and assigned embryonal histology. HX170c was established simultaneously as a cell line in culture and xenograft directly from the biopsy of a local recurrence 2 months prior to the patient’s death. At early passages, HX170c was cultured in vitro on a lethally irradiated layer of mouse fibroblast 3T3 cells; the cell line was later tested and found to contain only human cells. While the tumor biopsy was positive for desmin staining, the HX170c cell line was almost completely negative for this marker when cultured in vitro, except when grown to almost complete confluence. Interestingly, HX170c subcutaneous mouse xenografts regained desmin positivity and also appeared histologically similar to the initial tumor biopsy. HX170c cells are near diploid (50 ± 6) (24).
JR-1
JR-1 was derived from a lung metastasis of a 7-year-old female with a primary uterine mass, previously treated with vincristine, cyclophosphamide, dactinomycin, and radiation therapy (25). It has a poorly differentiated embryonal histology, based on histological appearance of the biopsy specimen. This cell line was grown in RPMI-1640 medium supplemented with FBS. Chromosome analysis revealed a near tetraploid number of chromosomes, ranging from 44 to100 per cell, with 80% showing chromosomal alterations, the most common of which was 13q−. JR-1 cells have been shown to have mutation of the TP53 gene (26). Antibody staining showed similarities between the cell line and the original tumor, as both stained positive for desmin, vimentin, glycolipid, ganglioside Gq, thy-1 and Gp44; and negative for GFAP, cytokeratin, neurofilament RT97, and myoglobin.
KF-RMS-1
KF-RMS-1 has an embryonal histology (based on the histologic appearance of its tumor source) and is derived from a bone marrow metastasis. It is grown in DMEM with 10% FBS (22).
RD
RD was derived directly from biopsy specimens of a 7-year-old female with a pelvic RMS previously treated with cyclophosphamide and radiation and found to have refractory disease (27). It has an embryonal histology based on histologic appearance of the tumor biopsies and the cultured cells, and 51-hyperdiploid chromosomes. RD has been shown to have amplification of the MYC oncogene (28), Q61H mutation of NRAS (29), and homozygous mutation of TP53 (30, 31). Houghton et al. studied tumor size following injections of various chemotherapy agents and found that RD demonstrated growth inhibition to vincristine and cyclophosphamide, but no other agents tested (32). RD was also used to test tolfenamic acid, and showed decreased tumor size, decreased cell migration, and decreased expression of Sp specificity transcription factors after treatment (33). As mentioned in the Section “Discussion,” the cell line TE671, which was originally thought to be a medulloblastoma line, was later shown through cytogenetic analysis and DNA fingerprinting to likely be a subclone of RD cells (34). RD cells are one of the most commonly used cell lines in RMS research, can be obtained from ATCC, and are grown in Eagle’s medium with 10% FBS (35).
RH6
The RH6 cell line was established in the laboratory of Dr. Edwin Douglass at St. Jude’s Children’s Research Hospital (SJCRH) in Memphis, TN directly from a 16-year-old male with an infra-renal lymph node (personal communication, Susan Ragsdale, SJCRH). Of note, it has no relation to the Rh6 xenograft listed in (36). When evaluated using cDNA microarray analysis of the cell line versus original tumor, the RH6 cell line did not cluster within the RMS family of tumors and therefore, is not optimal for testing of this cancer type (37). More recently, other studies do mention that this cell line expresses myogenic markers (38).
Of historical and nomenclature import, all of the “RH” cell lines (sometimes denoted “SJRH”) were generated at SJCRH and can be obtained from a representative of that institution, or from Dr. Peter Houghton at Nationwide Children’s Hospital, Columbus, OH, USA who has maintained these lines. However, during the years in which human RMS cell lines were first being established, two separate laboratories were deriving and naming the cell lines independently. Cell lines established in the laboratory of Dr. Peter Houghton were generated by directly transplanting tumor into mice to establish xenografts, and originally labeled “Rh.” Cell lines established in the laboratory of Dr. Edwin Douglass (and subsequently Dr. David Shapiro) were generated directly from patient samples and labeled “RH.” The upper or lower case designations for these cell line names have since been interchanged in the literature and are no longer preserved.
RH12
RH12 was derived from a xenograft created from a buttock tumor in a previously untreated patient. The original tumor and corresponding xenograft have an embryonal histology based on their spindled appearances (36). Chromosome analysis showed the addition of multiple chromosomes including 2, 3, 7, 8, 9, 10, 11, 12, 13, 18, 21, and dup (1)(q11q31), +del(2)(q23) (39). In a study whereby tumor growth was measured against various chemotherapy agents, RH12 showed volume regression with vincristine, DTIC, cyclophosphamide, doxorubicin, cis-DDP, and busulfan. No growth inhibition was seen with the use of dactinomycin, mitomycin-C, or bleomycin (32). RH12 xenografts were also used to test the effect of vincristine plus topotecan on tumor growth; the combination showed improvement over topotecan alone but was no better than vincristine alone, which had progression of disease in only 1/7 mice tested (40).
RH14
The RH14 xenograft was derived from an inguinal tumor in a patient previously treated with vincristine, dactinomycin, and cyclophosphamide, with complete response and no evidence of disease for 2.5 years, then recurrence. It had an embryonal histology based on histologic appearance of the original tumor and the xenograft, both of which showed differentiation and cross striations (36). In evaluations of tumor size injections of various chemotherapy agents, RH14 had complete regression with the use of vincristine and significant regression after cis-DDP, DTIC, and mitomycin-C. No growth inhibition was seen with dactinomycin or doxorubicin (32). It is important to note that RH14 was only established as a xenograft, and to our knowledge no cell line is available.
RH36 (Also Referred to as Birch)
RH36 has an embryonal histology (based on the histologic appearance of the original tumor) and was derived from a paratesticular relapse in a 15-year-old male (41). The PPTP evaluated RH36 and found tumor xenograft inhibition in response to cyclophosphamide (41). RH36 has also been referred to as Birch in the literature (42) and is grown in RPMI-1640 with 10–20% FBS (43).
RMS559/R31
RMS559 cells were established in the laboratory of Dr. Jonathan A. Fletcher, Department of Pathology, Brigham and Women’s Hospital, Boston, MA, USA (44, 45). They can be grown in DMEM with 10% FBS. This cell line was identified to have a V550L FGFR4 mutation by direct sequencing (46).
RMS-YM
RMS-YM was derived from a relapsed abdominal mass of 2-year-old Japanese boy, at second relapse (47). The tumor was believed to originate from the urachus and was given an embryonal classification based on histology. The patient was treated with chemotherapy and radiotherapy, but died 3 years later due to multi-organ metastasis. RMS-YM is capable of forming subcutaneous xenografts in nude mice that appear histologically similar to the primary tumor. These cells stain positive for desmin and myoglobin. The karyotype of RMS-YM is 55, XY, −1, −2, −7, +8, −9, −10, −11, −16, −20, −22, +17 mars. Analysis of the MYCN and RAS genes showed no amplification or rearrangement, nor mutations of the 12th, 13th, or 61st codons of KRAS, HRAS, or NRAS. TP53 pathway gene analysis showed no mutation of the TP53 gene, but did show upregulation of MDM2 (31). RMS-YM also shows overexpression of FGFR1, located in 8p11.2 and is grown in RPMI-1640 with 10% FBS (28).
RUCH2
RUCH2 was derived from a 15-month-old female with a botryoid RMS of the vagina (48). It was originally established as a xenograft, then grown in cell culture. Karyotype at passage three revealed 55, X, –X, +7, +inv(7)(q11q32), +8, +8, +12, +13, +13, +19, +20, +21/55, idem, del(20)(q13) [cp40], almost identical to the primary tumor. It can be grown in DMEM supplemented with 10% FBS.
RUCH3
RUCH3 was derived from a patient with relapsed embryonal RMS. This cell line is first mentioned in the original reference for RUCH-2 cell line, and was found to express PAX3 but not MYF3 or MYF5 (48). It can be grown in DMEM supplemented with 10% FBS.
SCMC-RM2 (And Subclone SCMC-RM2-1)
SCMC-RM2 was derived from the bone marrow of an 11-year-old female with relapsed, metastatic embryonal RMS. This patient had a primary tumor in her right abdominal wall with extensive marrow involvement. The tumor was classified as embryonal based on the presence of cytoplasmic longitudinal fibers and cross striations, as well as cell appearance using electron microscopy. She achieved remission with standard chemotherapy but relapsed and died 11 months later after diagnosis. Tumor cells from her bone marrow at relapse were obtained and cultured as SCMC-RM2. Subsequently, at passage 20, a parent cell line was cloned in soft agar and SCMC-RM2-1 was isolated. Both cell lines stain positive for muscle markers including desmin, vimentin, myoglobin, and actin. Interestingly, molecular and cytogenetic evaluation revealed amplification of the MYCN gene with overexpression of MYCN mRNA, which was also found in the primary tumor (49). SCMC-RM2 and its subclone can be grown in RPMI-1640 with 10% FBS (50).
SMS-CTR
SMS-CTR was derived from an untreated 1-year-old male with a pelvic mass near the prostate. The original biopsy specimen is histologically consistent with a poorly differentiated embryonal RMS. This cell line was established by Dr. Patrick Reynolds in 1979. Cytogenetics were performed on this line in the 10th passage and showed hypertriploid chromosomes, with translocations involving t(1;13)(q21;q14) and t(2;13)(p25;q14) (51). It is important to point out that these are not the characteristic translocations associated with alveolar RMS. Though FOXO1 does localize to the 13q14 region, Northern blot studies of this line with a FOXO1 probe failed to show any evidence of a novel-sized transcript, thus suggesting that FOXO1 is not affected by these translocations (52).
TTC442
TTC442, also occasionally called A91 in the literature, has an embryonal histology and is grown in RPMI-1640 with 10–20% FBS. This cell line was originally obtained from the laboratory of Dr. Tim Triche of Children’s Hospital Los Angeles (CHLA) (43, 53).
TTC516
TTC516, also known as A995 in the literature, has an embryonal histology and is grown in RPMI-1640 with 10–20% FBS. This cell line was originally obtained from the laboratory of Dr. Tim Triche of CHLA (43, 53).
Alveolar RMS Cell Lines
CW9019
CW9019 has an alveolar histology and was developed by Dr. Jaclyn Biegel at the University of Pennsylvania, Philadelphia, PA, USA. It can now be obtained from Dr. Frederic Barr at the National Cancer Institute, Bethesda, MD, USA (personal communication, Dr. Frederic Barr). CW9019 has a stable reciprocal translocation of t(1;13), expresses the resulting PAX7-FOXO1 fusion protein and is grown in DMEM with 10% FBS (28). Unlike the typical PAX7-FOXO1-containing tumor, this line does not show amplification of the PAX7-FOXO1 fusion gene (54).
D-RHA1
D-RHA1 has an alveolar histology and was originally described in (43), having been obtained from Dr. W. Benedict of M.D. Anderson Cancer Center. It is grown in RPMI-1640 with 10–20% FBS.
KFR
KFR was derived from a bone marrow metastasis of a 13-year-old female and designated RMS based on positive staining for vimentin and desmin. Alveolar histology was assigned based on morphology and upon identification of the t(2;13)(q37;q14) translocation, expressing the resulting PAX3-FOXO1 (formerly known as PAX3-FKHR) fusion protein (55). Although there was a time that the 2q breakpoint was assigned to q35 or q37, this has now been resolved as a q35 breakpoint, so that the translocation would now more correctly be stated as t(2;13)(q35;q14). KFR is grown in modified DMEM with 10% FBS (22).
RH5
RH5 has an alveolar histology and has been cited in a study of the role of PDGFR-A (56).
RH10
RH10 was derived from a 15-year-old female with a perineal tumor at relapse; she had previously received extensive chemotherapy with vincristine, cyclophosphamide, dactinomycin, and doxorubicin. RH10 was first established as a xenograft. The resulting cell line is hyperdiploid and cytogenetic analysis revealed t(2;13)(q35;q14) (57). Although the tumor from which RH10 was originally derived was described as having poorly differentiated embryonal histology, in another publication from the SJCRH group at the same time, this tumor was diagnosed as mixed alveolar/embryonal or monomorphous round cell. With the presence of the t(2;13) translocation, it should not be called embryonal RMS, but either mixed or alveolar RMS (39). After injections of various chemotherapy agents, complete tumor regression was seen after administration of L-PAM, but only transient growth inhibition, if any, to the other agents tested (58). RH10 has also been evaluated by the PPTP and found to have intermediate response activity to vincristine and high response to cyclophosphamide (41).
RH18 (Fusion-Negative and Fusion-Positive Variants)
The RH18 cell line was generated from a xenograft derived from a tumor from a previously untreated 2-year-old patient with a perineal RMS assigned as embryonal histology. Interestingly, there was a small amount of alveolar histology in the tumor (although the xenograft from which the cell line is derived was entirely embryonal) (36). At the time, such a mixed presentation often received a final diagnosis of embryonal RMS, though today the mixed tumors are considered an intermediate subset in which a fraction have the PAX-FOXO1 fusion.
RH18 cell-line chromosome number ranges from 70 to 80 per cell, and cytogenetic abnormalities include del(1)(p22), der(3)t(3;?)(p14;?), del(6)(q24), and der(22)t(1;22)(q31;q11) (57). In the original cytogenetic characterization of this cell line, a t(2;13) translocation was identified in a later passage of a melphalan-resistant population (39). This sub-line also had cytogenetic markers in common with an earlier passage, suggesting that a translocation-bearing clone was selected (or arose) during culture. This finding is consistent with the presence of two available cell lines called RH18 – one fusion-negative (59) and one fusion-positive (60). Indeed an experiment comparing the parent xenograft to tumor selected for resistance after a large single dose of vincristine did show near tetraploidy (with numerous structural abnormalities) in the parent tumor, with all normal diploid cells noted in the resistant xenograft (61). This supports the idea that the original tumor, from which the xenograft was directly derived, had multiple stem lines.
The RH18 cell line also harbors amplification of the 12q13–15 region (including MDM2) (62), although unpublished data shows that the amplicon is present in the fusion-negative, but not fusion-positive, version of RH18 (personal communication, Dr. Frederic Barr). Clearly, Short Tandem Repeat (STR) analysis (see Section “Potential Pitfalls”) of the RH18 variants would be helpful to prove their relationship to the original xenograft, and users of these cell lines are well-served to determine which variant they are employing in their experiments.
When tumor volumes were measured following injections of various chemotherapy agents, RH18 showed growth inhibition with vincristine, cyclophosphamide, dactinomycin, doxorubicin, L-PAM, DTIC, cis-DDP, and mitomycin-C. No tumor growth inhibition was seen with bleomycin or busulfan (32). When evaluated by the PPTP, RH18 showed intermediate response activity to vincristine and high response to cyclophosphamide (41).
RH28 (Derived from the Same Patient as RH3)
The RH28 cell line was derived from a xenograft of tumor tissue obtained from an axillary node metastasis in a previously untreated 17-year-old male. The tumor and subsequent cell line have a poorly differentiated alveolar RMS histology. RH28 cells are near tetraploid and harbor a t(2;13)(q35;q14) translocation (57). Evaluation of RH28 proliferation in vitro shows that between passages 59–63, doubling time progressively increases until growth stops completely, and the cells appear to senesce. Further, these cells are multinucleated and show morphologies comparable to normal myoblasts (63). RH28 had complete volume regression with vincristine and L-PAM and significant growth inhibition with cyclophosphamide, doxorubicin, and dactinomycin (58). When tested by the PPTP, RH28 showed high response to both vincristine and cyclophosphamide (41).
There exists a sub-line of RH28, denoted RH28/LPAM, which was generated from RH28 cells in xenografts exposed to increasing doses of melphalan. RH28/LPAM cells are available for use and do not differentiate or stop proliferating with time (61). There are also available cells designated RH3, which have the same DNA breakpoint and pattern of DNA polymorphisms as RH28 cells (64). Indeed the RH3 cell line was developed by the Douglass laboratory from the same patient from which the RH28 xenograft was developed. The RH3 cell line has no relation to the Rh3 xenograft described in (36).
RH30 (Derived from the Same Patient as RMS13) and RH30R
The original Rh30 xenograft was established from an overnight culture of bone marrow obtained from a 16-year-old male with untreated metastatic alveolar RMS. The xenograft tumor was wild-type for TP53. The RH30 cell line was derived from the same patient bone marrow sample maintained in culture in the Douglass laboratory. The RH30 cell line expresses the t(2;13) translocation (39) and chromosomal analysis showed near-triploid chromosomes, between 51 and 87 (65). This line also has amplification of the 12q13–q15 region involving CDK4 and surrounding loci (66–68), and a heterozygous mutation of the TP53 tumor suppressor (30, 31, 65). An independent cell line, RH30R, was derived from a xenograft, made from this same patient, at the time of relapse (41). The relapse xenograft and the cell line from the xenograft both have the same TP53 mutation as the original RH30 cell line. RH30 and RH30R are cultured in RPMI-1640 with 10% FBS.
When the RH30 xenograft was tested with various chemotherapy agents, it showed complete regression using vincristine and L-PAM, significant growth inhibition after cyclophosphamide, but no growth inhibition with dactinomycin or doxorubicin (58). The PPTP evaluated the RH30 cell line and found it to have high response activity to vincristine and cyclophosphamide (41). A third experiment used the RH30 cell line to test the combination of topotecan plus vincristine against tumor growth. The combination showed better tumor control than topotecan alone but was no better than vincristine alone, where 0/5 mice tested had tumor progression (40). The RH30 cell line was also used to investigate the anti-tumor effects of tolfenamic acid. After treatment, RH30 cells showed decreased growth, decreased cell migration, and decreased expression of the PAX3-FOXO1 fusion protein (33).
An alveolar RMS cell-line labeled RMS13 (39, 69) is thought by some investigators to be related to RH30, perhaps derived from the same patient tumor as RH30, but this is not clear. Other studies have presumed RMS13 and RH30 to be independent entities (70). Finally, another cell line has been distributed with the name of RH30 (personal communication, Dr. Frederic Barr). Of interest, this other line still has the 12q amplicon but does not have the PAX3-FOXO1 fusion (unpublished data, Dr. Frederic Barr). STR analysis would help clarify the identity of both the RMS13 and fusion-negative RH30 lines.
RH41 (Derived from the Same Patient as RH4)
RH41 was established by the Houghton laboratory from a xenograft of a lung metastasis from a previously treated 7-year-old female patient with alveolar RMS. RH41 was originally designated as alveolar by histology, and more recently has been shown to harbor the t(2;13) translocation (71). RH41 also harbors mutation of the TP53 gene (26). RH41 was tested by the PPTP and showed intermediate response activity to both vincristine and cyclophosphamide (41).
The cell line designated RH4 was developed from the same patient by another laboratory at SJCRH (personal communication, Dr. Peter Houghton), in which the tumor specimen was placed directly into tissue culture. These cells designated RH4 possess the t(2;13)(q35;q14) translocation (72) and have amplified, overexpression of EYA2 (28). RH4 cells are cultured in RPMI supplemented with 10% FBS.
RH65
RH65 was derived from 18-year-old female with a relapsed RMS; it has an alveolar histology based on the original tumor (41).
RMZ-RC2
RMZ-RC2 was established as a subclone of the RMZ cell line, which was derived from a bone marrow metastasis of a 2-year-old male with alveolar RMS; the child had been treated prior with chemotherapy and radiation (73). RMZ-RC2 can be grown in DMEM with 10% FBS. Chromosomal analysis showed near tetraploid range chromosomes with numerous monosomies and double minutes (65). Both the PAX7-FOXO1 fusion gene and MYCN were found amplified (65, 72). RMZ-RC2 cells treated in culture with 2–5 Gy of radiation show dose-sensitive increases in the expression of myosin and the percentage of multinucleated cells, both markers of differentiation (74).
TC212
TC212 has an alveolar histology (based on histology of the tumor specimens) and was derived from the bone marrow metastasis of a 16-year-old male who relapsed 5 months after multi-agent chemotherapy and radiation (51). Chromosomal analysis revealed multiple translocations, inversions, and deletions including what was thought to be t(2;13)(q37;q14). Although the 2q breakpoint was assigned to q35 or q37, this has now been resolved as a q35 breakpoint, so the translocation in TC212 should correctly be described as t(2;13)(q35;q14). TC212 cells are grown in RPMI-1640 supplemented with 10% FBS.
RMS Cell Line Imposters
A204
A204 was previously (but no longer) listed in the ATCC catalog as an RMS cell line. However in 1998, A204 cells were found to lack expression of myogenin, MyoD, or desmin, suggesting that either they were not of RMS origin or that with passaging in culture there was down-regulation of myogenic genes (23). These observations were especially compelling, since a prior immunohistochemical study found that most RMS tumor samples (but not normal adult muscle including skeletal muscle) expressed MyoD (75). In 2002, A204 cells were found to have a SMARCB1 (SNF5) mutation, consistent with its classification as a rhabdoid line (76).
A673
A673 was also previously (but no longer) listed in the ATCC catalog as an RMS cell line. However, like A204 cells, in 1998 these cells were found to not express myogenic markers (23). In 2003, A673 cells were found to express the EWS-FLI1 fusion gene, indicating the cell line is derived from a Ewing sarcoma (77).
RH1
The RH1 cell line was derived from a patient tumor specimen grown directly into tissue culture. The tumor was of the hand, described in a pathology report to be consistent with alveolar RMS by histologic criteria (personal communication, Dr. Peter Houghton). RH1 has no relation to the Rh1 xenograft described in (36). In 1998, RH1 was noted to lack expression of the myogenic markers myogenin, MyoD, and desmin (23). In 2008, using gene expression profiling the PPTP determined RH1 to cluster with tumors in the Ewing sarcoma family (4). RH1 was later verified to express the EWS-FLI1 fusion characteristic of the Ewing family of tumors (78). In retrospect, it is not clear whether RH1 was contaminated by a Ewing cell line or mislabeled, however the PPTP has excluded RH1 from future RMS studies (4).
Culture of RMS Cell Lines as Rhabdospheres versus Adherent Cells
While all above mentioned studies propagated RMS cell lines as adherent cells or xenografts, Walter et al. recently described a method of culturing rhabdospheres in suspension (79). In this method, spheres are grown by placing human embryonal RMS cell lines into SC serum free medium with neurobasal medium and 10 ng/ml EGF, 20 ng/ml b-FGF, and 2× B27. In this study, RD cells were injected as adherent cells or spheres into mice, whereby tumor growth was measured over weeks. Sphere-derived tumors grew sooner, 40 days after injection, as opposed to 80 days after injection with the adherent cells. Sphere-derived tumors also required fewer cells for injection than adherent cells, indicating that rhabdospheres are more tumorigenic than adherent cell cultures. When stem cell genes associated with the stem cell phenotype were assessed using real-time PCR, sphere cultures had significantly higher expression of these genes than adherent cells, consistent with the premise of stem cell enrichment. Further, when treated with retinoic acid or DMSO, sphere cultures showed greater differentiation toward myocytes, neuronal cells or adipocytes, again indicating higher cancer stem cell potential in lines grown as rhabdospheres versus adherent cells. These data are compelling and may lead to changes in how RMS cell lines are grown for use in preclinical experimentation.
Potential Pitfalls
Several potential problems arise when using cell lines derived from human tumors in vitro or transplanted into mice as xenografts. One possibility is that tumors can be contaminated by murine stromal cells, thereby potentially affecting the outcome of the study. To combat this problem, analysis for species-specific LDH isoenzymes, as well as evaluating for acrocentric mouse chromosomes, can be performed. For example, Hazelton et al. cultured rhabdomyoblasts from the cell lines RH10, RH18, and RH28 and found human specific isoenzymes of LDH present in the lines, without evidence of murine LDH. In this same study, metaphase spreads were analyzed for acrocentric mouse chromosomes, and none of the three cell lines evaluated showed murine cells (57).
A second problem is that there may be discordance between the original tumor and the resulting cell lines or xenografts. To evaluate for this, one can perform gene expression microarray analysis. In 2008, the PPTP published a report using microarray analysis to analyze gene expression of the RMS cell lines RH10, RH18, RH28, RH30, RH41, RH65, and RD and compared expression profiles to a similar number of RMS specimens representing both alveolar and embryonal RMS. After exclusion of immunosurveillance genes (such as macrophage or B-cell markers), the cell lines resembled the other human RMS specimens (4). A similar analysis was performed using cDNA microarray analysis of the RMS cell lines RH1, RH6, RH10, RH12, RH14, RH18, RH28, RH30, RH36, and RH41. These analyses were compared to cDNA of primary tumors and all clustered appropriately by tumor type except RH1 and RH6 (37). Further cDNA microarray analysis done by Khan et al. showed comparisons between alveolar cell lines RMS13, RH3, RH4, RH5, RH18, and RH28 (69). Missiaglia et al. performed cDNA microarray analysis and gene expression profiles, and compared these profiles to primary tumors on the RMS cell lines RD, RMS-YM, RUCH2, RUCH3, RH4, RH18, RH30, RH41, and CW9019 and showed high concordance as well (28). A recent study describes an approach to develop xenografts directly from patient tumors, then test them for genomic similarities (80). In this study, after surgical resection from human subjects, tumors were either implanted into immune-compromised mice as xenografts or analyzed for histology, mutational analysis and gene expression profiles. Xenografts were later surgically resected and implanted into a second set of mice. Xenograft specimens from first and second generation implantations underwent similar analysis as the original patient tumor sample. This approach serves as a good model for evaluating and validating human tumor xenografts, which may help produce preclinical data more closely mirroring later clinical outcomes
A third problem is the situation in which two cell lines with different names are apparently from the same tumor, such as RH36 and Birch, RH30 and RMS13. This situation can represent two different nomenclatures or possibly contamination of one cell line with the other. In addition, although RH3 and RH28 appear genetically identical by fusion breakpoint and STR analysis (64, 81), it is not clear whether the two cell lines were derived from the same tumor from the same patient on the same date. In either case, the two lines may share much of their genetics but may also have diverged biologically due to continued growth as separate cultures. There are also instances where two distinct lines have the same name, but have different genetics. For example, the “RH18” cell line can refer to the version not bearing the fusion gene (59), or the version bearing the fusion gene (60), which may have emerged from the original cell culture.
A fourth problem is unintentional cross-contamination between cell lines. It is estimated that 15–35% of all cell lines are of a different parental origin than assumed, and this results in the misidentification of cell lines and ultimately the misinterpretation of scientific data (82–84). For example, the cell line TE671, which was widely used to study medulloblastoma, was thought to be a misidentified RMS cell line (RD). This statement stemmed from similar phenotypic observations and an activating point mutation in NRAS gene which was found both in TE671 and RD (34). This hypothesis was confirmed with a cytogenetic study of these cell lines (85). In addition, three esophageal adenocarcinoma cell lines were recently found to derive from other tumors (86).
Because of the problems described above, and no historical standard method to validate cell lines, the method of STR profiling was suggested as an international reference standard for cell line authentication (87). STRs (or microsatellite repeats in DNA) consist of 2–6 base pair repeats that differ from individual to individual (84). In this method, commercially available primers are used to amplify a number of polymorphic STR loci, resulting in a cell-line specific “fingerprint” that is recorded as a numerical code in a reference database. The simplicity of the code, reproducibility, and low cost of this method are the strengths supporting its use as a standard of cell line authentication (87). Indeed, several scientific journals including those managed by the American Association for Cancer Research are requiring that articles submitted for peer review contain documentation of cell line authentication (see http://www.aacrjournals.org/site/AuthServCtr/cell_line_auth.xhtml). On the other hand, it is important to acknowledge the limitations of STR in cell line authentication. For example, in a study of four human leukemia cell lines grown under variable culture conditions, STR profiles changed in two of the four cell lines due to subclone outgrowth (84). Thus, STR profiling is an important tool for cell-line validation, but has limitations, especially in tumor cell lines subject to experimental conditions that could alter cellular genetics. Other features including myogenic markers, enzyme isotypes, and cytogenetic analysis can further assist in validation of RMS cells when differences are found in STR loci (57, 84). Table 3 summarizes the validation methods that have been employed for human RMS cell lines. The cell lines with known and published STRs are listed in Table 4.
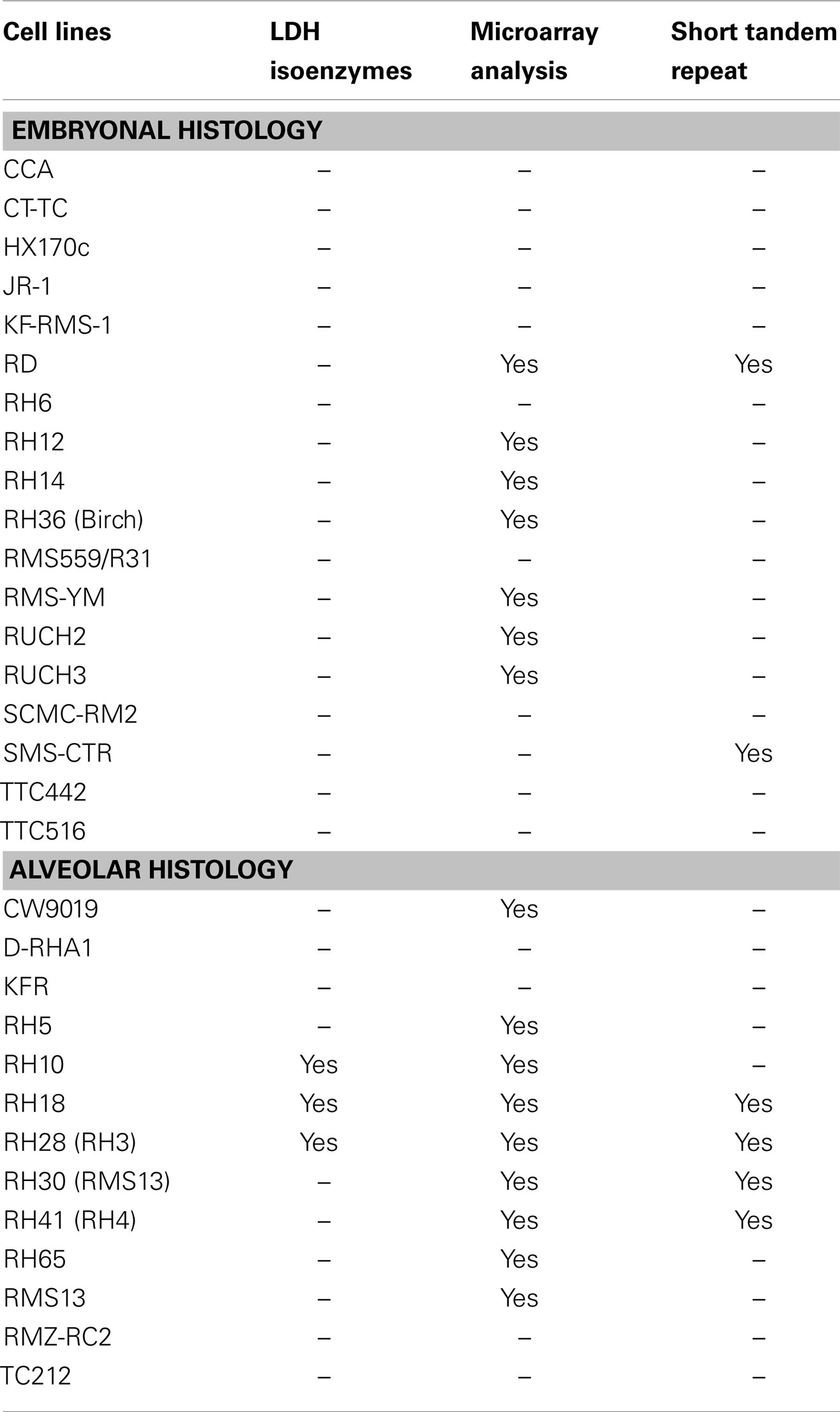
Table 3. RMS cell lines: validation methods performed to date.
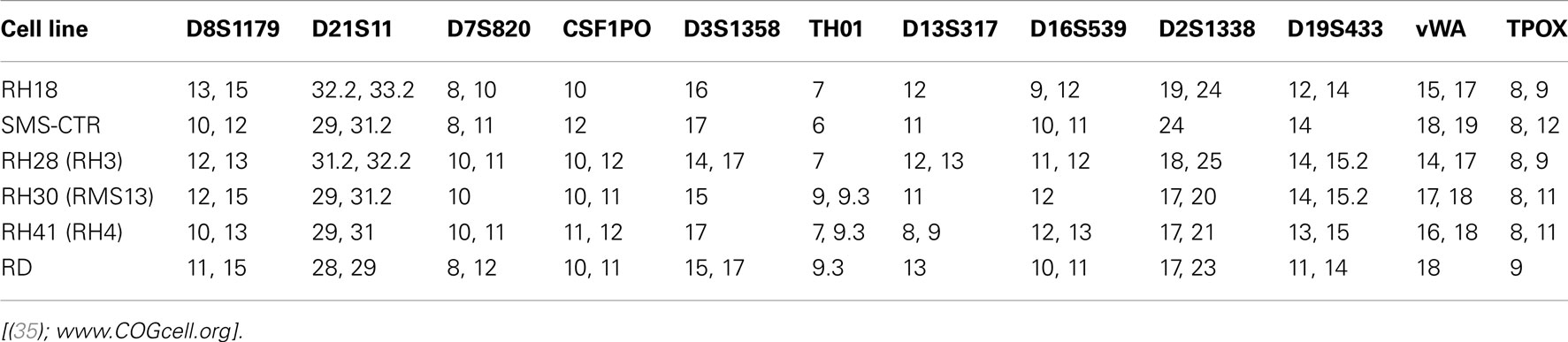
Table 4. Available RMS cell line short tandem repeats.
Discussion
Numerous RMS cell lines are available for use in basic research and preclinical testing. These cell lines differ markedly in their histologies, origins, karyotypes, degree of treatment prior to development, and the levels to which these lines have each been validated and compared to their tumors of origin. This information is of critical importance when choosing a cell line for study. For example, evaluation of results of studies performed to date on the available cell lines shows a trend that lines derived from tumors previously treated with extensive chemotherapy and/or radiation are then less susceptible (or only transiently so) to standard agents. These previously treated cell lines would therefore be optimal for preclinical testing of new agents proposed for use in relapsed or refractory disease. These further progressed tumors may also be easier to establish in culture than primary tumors. It is necessary to ensure that cell lines used in preclinical trials closely resemble the tumors from which they were derived. While some data is currently available that validates several lines, this work is ongoing and will surely continue to develop as the use of cell lines and RMS xenografts increase in pediatric oncology research.
When examining RMS cell lines as a whole, there is a surprising lack of cell lines derived from primary, untreated RMS tumors. Indeed, the majority of both embryonal and alveolar RMS cell lines are established from tumor relapses or distant metastases. Cells that arise from relapses or metastases (in addition to any adaptations as the result of radiation and/or chemotherapy given to the patient) have genetic changes distinct from the primary tumor, and may be easier to establish in culture than cells from primary tumors. Therefore, applicability of these lines in preclinical studies examining RMS primary tumor biology is questionable. This gap in our understanding of RMS reveals a significant challenge to researchers that routinely use RMS cell lines for preclinical research. How can we make significant strides to identify therapeutic targets when the reagents available do not accurately represent the biology of the primary tumor? Has RMS research been hindered by an excess of false negative hits in pharmacologic and genetic screens due to these cell-line adaptations?
In summary, it will be important for RMS scientists, as a community, to critically evaluate the cell lines we use, and identify approaches to generate improved reagents for future research. Ideally, human cell lines would be established from primary, untreated RMS tumor biopsies. Then, any cell lines generated from subsequent tumor relapse or metastasis could be paired with the primary cell line as a “matched set.” To our knowledge there are no matched set cell lines publicly available to RMS researchers. When available, such a resource would support the study of the genetic and biologic changes in the primary tumor, as well as the changes leading to recurrence or metastasis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Charles Keller (Oregon Health and Science University, Portland, OR, USA) for sharing a list of RMS cell lines, an updated version of which is recently published (18) and Peter Houghton (Nationwide Children’s, Columbus, OH, USA) for valuable insight and comments. We anticipate that readers of this article will have further insight into the origins of available human RMS cell lines, and look forward to incorporating this information in a future update. This work was supported by a Pfizer Oncology Clinical Fellowship Award (Brian C. Belyea), The Hartwell Foundation (Lisa E. S. Crose), the Intramural Research Program of the NCI (Frederic G. Barr), and the Liddy Shriver Sarcoma Initiative and NCI Award R01-CA122706 (Corinne M. Linardic).
References
1. Saab R, Spunt SL, Skapek SX. Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr Top Dev Biol (2011) 94:197–234. doi:10.1016/B978-0-12-380916-2.00007-3
2. Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer (2009) 115(18):4218–26. doi:10.1002/cncr.24465
3. Pappo AS, Anderson JR, Crist WM, Wharam MD, Breitfeld PP, Hawkins D, et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol (1999) 17(11):3487–93.
4. Neale G, Su X, Morton CL, Phelps D, Gorlick R, Lock RB, et al. Molecular characterization of the pediatric preclinical testing panel. Clin Cancer Res (2008) 14(14):4572–83. doi:10.1158/1078-0432.CCR-07-5090
5. Cook B, O’Sullivan F, Leung J, Morse P, Graham B, Chapman AL. Transformation of human embryo cells with the use of cell-free extracts of a human rhabdomyosarcoma cell line (HUS-2): brief communication. J Natl Cancer Inst (1978) 60(5):979–84.
6. Fernandez JE, Prados J, Melquizo C, Arena N, Malavasi F, Alvarez L, et al. Characterization of a new human embryonal rhabdomyosarcoma cell line, RMS-GR. Jpn J Cancer Res (1998) 89(5):525–32. doi:10.1111/j.1349-7006.1998.tb03293.x
7. Garvin AJ, Stanley WS, Bennett DD, Sullivan JL, Sens DA. The in vitro growth, heterotransplantation, and differentiation of a human rhabdomyosarcoma cell line. Am J Pathol (1986) 125(1):208–17.
8. Kleideiter E, Schwab M, Friedrich U, Koscielniak E, Schafer BW, Klotz U. Telomerase activity in cell lines of pediatric soft tissue sarcomas. Pediatr Res (2003) 54(5):718–23. doi:10.1203/01.PDR.0000086901.29111.2A
9. Madsen WE, Walker MJ, Shaughnessy EA, Brown JM, Das Gupta TK. Characterization of malignant mesenchymal cell line (UISO-RS-3) derived from a human rhabdomyosarcoma and inhibition by pharmacologic doses of estrogen. In vitro Cell Dev Biol (1990) 26(10):971–7. doi:10.1007/BF02624471
10. Ozkaynak MF, Nolta J, Parkman R. In vitro purging of human rhabdomyosarcoma cells using 4-hydroperoxycyclophosphamide. Cancer Res (1990) 50(5):1455–8.
11. Urumov IJ, Manolova Y. Cytogenetic analysis of an embryonal rhabdomyosarcoma cell line. Cancer Genet Cytogenet (1992) 61(2):214–5. doi:10.1016/0165-4608(92)90092-M
12. Motoyama T, Watanabe H, Yamamoto T. Embryonal rhabdomyosarcoma in nude mice and in vitro. Acta Pathol Jpn (1986) 36(10):1495–505.
13. Kaneko S, Kaneko M, Ohkawa H. [Establishment and characterization of a childhood embryonal rhabdomyosarcoma cell line with non-random chromosome alterations]. Hum Cell (1995) 8(1):35–41.
14. Ohi S. Characterization, anticancer drug susceptibility and atRA-induced growth inhibition of a novel cell line (HUMEMS) established from pleural effusion of alveolar rhabdomyosarcoma of breast tissue. Hum Cell (2007) 20(2):39–51. doi:10.1111/j.1749-0774.2007.00029.x
15. Ogose A, Motoyama T, Hotta T, Watanabe H. In vitro differentiation and proliferation in a newly established human rhabdomyosarcoma cell line. Virchows Arch (1995) 426(4):385–91. doi:10.1007/BF00191348
16. Sonobe H, Takeuchi T, Taguchi T, Shimizu K, Furihata M, Ohtsuki Y. A new human pleomorphic rhabdomyosarcoma cell-line, HS-RMS-1, exhibiting MyoD1 and myogenin. Int J Oncol (2000) 17(1):119–25.
17. Takaoka E, Sonobe H, Akimaru K, Sakamoto S, Shuin T, Daibata M, et al. Multiple sites of highly amplified DNA sequences detected by molecular cytogenetic analysis in HS-RMS-2, a new pleomorphic rhabdomyosarcoma cell line. Am J Cancer Res (2012) 2(2):141–52.
18. Sokolowski E, Turina CB, Kikuchi K, Langenau DM, Keller C. Proof-of-concept rare cancers in drug development: the case for rhabdomyosarcoma. Oncogene (2013). [Epub ahead of print]. doi:10.1038/onc.2013.129
19. DeGiovanni C, Nanni P, Nicoletti G, Ceccarelli C, Scotlandi K, Landuzzi L, et al. Metastatic ability and differentiative properties of a new cell line of human embryonal rhabdomyosarcoma (CCA). Anticancer Res (1989) 9(6):1943–9.
20. Magnani I, Faustinella F, Nanni P, Nicoletti G, Larizza L. Karyotypic characterization of a new human embryonal rhabdomyosarcoma cell line. Cancer Genet Cytogenet (1991) 54(1):83–9. doi:10.1016/0165-4608(91)90034-R
21. Martinelli S, McDowell HP, Vigne SD, Kokai G, Uccini S, Tartaglia M, et al. RAS signaling dysregulation in human embryonal rhabdomyosarcoma. Genes Chromosomes Cancer (2009) 48(11):975–82. doi:10.1002/gcc.20702
22. Cinatl J Jr, Cinatl J, Michaelis M, Kabickova H, Kotchetkov R, Vogel JU, et al. Potent oncolytic activity of multimutated herpes simplex virus G207 in combination with vincristine against human rhabdomyosarcoma. Cancer Res (2003) 63(7):1508–14.
23. Morton CL, Potter PM. Rhabdomyosarcoma-specific expression of the herpes simplex virus thymidine kinase gene confers sensitivity to ganciclovir. J Pharmacol Exp Ther (1998) 286(2):1066–73.
24. Kelland LR, Bingle L, Edwards S, Steel GG. High intrinsic radiosensitivity of a newly established and characterised human embryonal rhabdomyosarcoma cell line. Br J Cancer (1989) 59(2):160–4. doi:10.1038/bjc.1989.34
25. Clayton J, Pincott JR, van den Berghe JA, Kemshead JT. Comparative studies between a new human rhabdomyosarcoma cell line, JR-1 and its tumour of origin. Br J Cancer (1986) 54(1):83–90. doi:10.1038/bjc.1986.155
26. Taylor AC, Shu L, Danks MK, Poquette CA, Shetty S, Thayer MJ, et al. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Med Pediatr Oncol (2000) 35(2):96–103. doi:10.1002/1096-911X(200008)35:2<96::AID-MPO2>3.0.CO;2-Z
27. McAllister RM, Melnyk J, Finkelstein JZ, Adams EC Jr, Gardner MB. Cultivation in vitro of cells derived from a human rhabdomyosarcoma. Cancer (1969) 24(3):520–6. doi:10.1002/1097-0142(196909)24:3<520::AID-CNCR2820240313>3.0.CO;2-M
28. Missiaglia E, Selfe J, Hamdi M, Williamson D, Schaaf G, Fang C, et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: an approach to identify candidate genes involved in tumor development. Genes Chromosomes Cancer (2009) 48(6):455–67. doi:10.1002/gcc.20655
29. Schaaf G, Hamdi M, Zwijnenburg D, Lakeman A, Geerts D, Versteeg R, et al. Silencing of SPRY1 triggers complete regression of rhabdomyosarcoma tumors carrying a mutated RAS gene. Cancer Res (2010) 70(2):762–71. doi:10.1158/0008-5472.CAN-09-2532
30. Felix CA, Kappel CC, Mitsudomi T, Nau MM, Tsokos M, Crouch GD, et al. Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res (1992) 52(8):2243–7.
31. Miyachi M, Kakazu N, Yagyu S, Katsumi Y, Tsubai-Shimizu S, Kikuchi K, et al. Restoration of p53 pathway by nutlin-3 induces cell cycle arrest and apoptosis in human rhabdomyosarcoma cells. Clin Cancer Res (2009) 15(12):4077–84. doi:10.1158/1078-0432.CCR-08-2955
32. Houghton JA, Houghton PJ, Green AA. Chemotherapy of childhood rhabdomyosarcomas growing as xenografts in immune-deprived mice. Cancer Res (1982) 42(2):535–9.
33. Chadalapaka G, Jutooru I, Sreevalsan S, Pathi S, Kim K, Chen C, et al. Inhibition of rhabdomyosarcoma cell and tumor growth by targeting specificity protein (Sp) transcription factors. Int J Cancer (2013) 132(4):795–806. doi:10.1002/ijc.27730
34. Stratton MR, Darling J, Pilkington GJ, Lantos PL, Reeves BR, Cooper CS. Characterization of the human cell line TE671. Carcinogenesis (1989) 10(5):899–905. doi:10.1093/carcin/10.5.899
35. Kang MH, Smith MA, Morton CL, Keshelava N, Houghton PJ, Reynolds CP. National Cancer Institute pediatric preclinical testing program: model description for in vitro cytotoxicity testing. Pediatr Blood Cancer (2011) 56(2):239–49. doi:10.1002/pbc.22801
36. Houghton JA, Houghton PJ, Webber BL. Growth and characterization of childhood rhabdomyosarcomas as xenografts. J Natl Cancer Inst (1982) 68(3):437–43.
37. Whiteford CC, Bilke S, Greer BT, Chen Q, Braunschweig TA, Cenacchi N, et al. Credentialing preclinical pediatric xenograft models using gene expression and tissue microarray analysis. Cancer Res (2007) 67(1):32–40. doi:10.1158/0008-5472.CAN-06-0610
38. Marshall AD, Lagutina I, Grosveld GC. PAX3-FOXO1 induces cannabinoid receptor 1 to enhance cell invasion and metastasis. Cancer Res (2011) 71(24):7471–80. doi:10.1158/0008-5472.CAN-11-0924
39. Douglass EC, Valentine M, Etcubanas E, Parham D, Webber BL, Houghton PJ, et al. A specific chromosomal abnormality in rhabdomyosarcoma. Cytogenet Cell Genet (1987) 45(3-4):148–55. doi:10.1159/000132446
40. Thompson J, George EO, Poquette CA, Cheshire PJ, Richmond LB, de Graaf SS, et al. Synergy of topotecan in combination with vincristine for treatment of pediatric solid tumor xenografts. Clin Cancer Res (1999) 5(11):3617–31.
41. Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer (2007) 49(7):928–40. doi:10.1002/pbc.21078
42. Roberts I, Gordon A, Wang R, Pritchard-Jones K, Shipley J, Coleman N. Molecular cytogenetic analysis consistently identifies translocations involving chromosomes 1, 2 and 15 in five embryonal rhabdomyosarcoma cell lines and a PAX-FOXO1A fusion gene negative alveolar rhabdomyosarcoma cell line. Cytogenet Cell Genet (2001) 95(3–4):134–42. doi:10.1159/000059335
43. Decristofaro MF, Betz BL, Wang W, Weissman BE. Alteration of hSNF5/INI1/BAF47 detected in rhabdoid cell lines and primary rhabdomyosarcomas but not Wilms’ tumors. Oncogene (1999) 18(52):7559–65. doi:10.1038/sj.onc.1203168
44. Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest (2002) 110(7):923–32. doi:10.1172/JCI0215634
45. Ito H, Duxbury M, Benoit E, Farivar RS, Gardner-Thorpe J, Zinner MJ, et al. Fibronectin-induced COX-2 mediates MMP-2 expression and invasiveness of rhabdomyosarcoma. Biochem Biophys Res Commun (2004) 318(2):594–600. doi:10.1016/j.bbrc.2004.04.070
46. Shukla N, Ameur N, Yilmaz I, Nafa K, Lau CY, Marchetti A, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res (2012) 18(3):748–57. doi:10.1158/1078-0432.CCR-11-2056
47. Kubo K, Naoe T, Utsumi KR, Ishiguro Y, Ueda K, Shiku H, et al. Cytogenetic and cellular characteristics of a human embryonal rhabdomyosarcoma cell line, RMS-YM. Br J Cancer (1991) 63(6):879–84. doi:10.1038/bjc.1991.193
48. Scholl FA, Betts DR, Niggli FK, Schafer BW. Molecular features of a human rhabdomyosarcoma cell line with spontaneous metastatic progression. Br J Cancer (2000) 82(6):1239–45.
49. Mitani K, Kurosawa H, Suzuki A, Hayashi Y, Hanada R, Yamamoto K, et al. Amplification of N-myc in a rhabdomyosarcoma. Jpn J Cancer Res (1986) 77(11):1062–5.
50. Hayashi Y, Sugimoto T, Horii Y, Hosoi H, Inazawa J, Kemshead JT, et al. Characterization of an embryonal rhabdomyosarcoma cell line showing amplification and over-expression of the N-myc oncogene. Int J Cancer (1990) 45(4):705–11. doi:10.1002/ijc.2910450423
51. Whang-Peng J, Knutsen T, Theil K, Horowitz ME, Triche T. Cytogenetic studies in subgroups of rhabdomyosarcoma. Genes Chromosomes Cancer (1992) 5(4):299–310. doi:10.1002/gcc.2870050405
52. Barr FG, Galili N, Holick J, Biegel JA, Rovera G, Emanuel BS. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet (1993) 3(2):113–7. doi:10.1038/ng0293-113
53. Hu K, Lee C, Qiu D, Fotovati A, Davies A, Abu-Ali S, et al. Small interfering RNA library screen of human kinases and phosphatases identifies polo-like kinase 1 as a promising new target for the treatment of pediatric rhabdomyosarcomas. Mol Cancer Ther (2009) 8(11):3024–35. doi:10.1158/1535-7163.MCT-09-0365
54. Barr FG, Nauta LE, Davis RJ, Schafer BW, Nycum LM, Biegel JA. In vivo amplification of the PAX3-FKHR and PAX7-FKHR fusion genes in alveolar rhabdomyosarcoma. Hum Mol Genet (1996) 5(1):15–21. doi:10.1093/hmg/5.1.15
55. Engel R, Ritterbach J, Schwabe D, Lampert F. Chromosome translocation (2;13)(q37;q14) in a disseminated alveolar rhabdomyosarcoma. Eur J Pediatr (1988) 148(1):69–71. doi:10.1007/BF00441819
56. Taniguchi E, Nishijo K, McCleish AT, Michalek JE, Grayson MH, Infante AJ, et al. PDGFR-A is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene (2008) 27(51):6550–60. doi:10.1038/onc.2008.255
57. Hazelton BJ, Houghton JA, Parham DM, Douglass EC, Torrance PM, Holt H, et al. Characterization of cell lines derived from xenografts of childhood rhabdomyosarcoma. Cancer Res (1987) 47(16):4501–7.
58. Houghton JA, Cook RL, Lutz PJ, Houghton PJ. Childhood rhabdomyosarcoma xenografts: responses to DNA-interacting agents and agents used in current clinical therapy. Eur J Cancer Clin Oncol (1984) 20(7):955–60. doi:10.1016/0277-5379(84)90170-6
59. Linardic CM, Naini S, Herndon JE, Kesserwan C, Qualman SJ, Counter CM. The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res (2007) 67(14):6691–9. doi:10.1158/0008-5472.CAN-06-3210
60. Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ III, Emanuel BS, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet (1993) 5(3):230–5. doi:10.1038/ng1193-230
61. Horton JK, Houghton PJ, Houghton JA. Reciprocal cross-resistance in human rhabdomyosarcomas selected in vivo for primary resistance to vincristine and L-phenylalanine mustard. Cancer Res (1987) 47(23):6288–93.
62. Fiddler TA, Smith L, Tapscott SJ, Thayer MJ. Amplification of MDM2 inhibits MyoD-mediated myogenesis. Mol Cell Biol (1996) 16(9):5048–57.
63. Shapiro DN, Houghton PJ, Hazelton BJ, Germain GS, Murti KG, Rahman A, et al. Morphological and molecular characterization of spontaneous myogenic differentiation in a human rhabdomyosarcoma cell line. Cancer Res (1990) 50(18):6002–9.
64. Barr FG, Nauta LE, Hollows JC. Structural analysis of PAX3 genomic rearrangements in alveolar rhabdomyosarcoma. Cancer Genet Cytogenet (1998) 102(1):32–9. doi:10.1016/S0165-4608(97)00287-2
65. Rodriguez-Perales S, Martinez-Ramirez A, de Andres SA, Valle L, Urioste M, Benitez J, et al. Molecular cytogenetic characterization of rhabdomyosarcoma cell lines. Cancer Genet Cytogenet (2004) 148(1):35–43. doi:10.1016/S0165-4608(03)00216-4
66. Khatib ZA, Matsushime H, Valentine M, Shapiro DN, Sherr CJ, Look AT. Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas. Cancer Res (1993) 53(22):5535–41.
67. Weber-Hall S, Anderson J, McManus A, Abe S, Nojima T, Pinkerton R, et al. Gains, losses, and amplification of genomic material in rhabdomyosarcoma analyzed by comparative genomic hybridization. Cancer Res (1996) 56(14):3220–4.
68. Berner JM, Forus A, Elkahloun A, Meltzer PS, Fodstad O, Myklebost O. Separate amplified regions encompassing CDK4 and MDM2 in human sarcomas. Genes Chromosomes Cancer (1996) 17(4):254–9. doi:10.1002/(SICI)1098-2264(199612)17:4<254::AID-GCC7>3.0.CO;2-2
69. Khan J, Simon R, Bittner M, Chen Y, Leighton SB, Pohida T, et al. Gene expression profiling of alveolar rhabdomyosarcoma with cDNA microarrays. Cancer Res (1998) 58(22):5009–13.
70. Rodeberg DA, Erskine C, Celis E. In vitro induction of immune responses to shared tumor-associated antigens in rhabdomyosarcoma. J Pediatr Surg (2007) 42(8):1396–402. doi:10.1016/j.jpedsurg.2007.03.041
71. Missiaglia E, Shepherd CJ, Patel S, Thway K, Pierron G, Pritchard-Jones K, et al. MicroRNA-206 expression levels correlate with clinical behaviour of rhabdomyosarcomas. Br J Cancer (2010) 102(12):1769–77. doi:10.1038/sj.bjc.6605684
72. Frascella E, Lenzini E, Schafer BW, Brecevic L, Dorigo E, Toffolatti L, et al. Concomitant amplification and expression of PAX7-FKHR and MYCN in a human rhabdomyosarcoma cell line carrying a cryptic t(1;13)(p36;q14). Cancer Genet Cytogenet (2000) 121(2):139–45. doi:10.1016/S0165-4608(00)00258-2
73. Nanni P, Schiaffino S, De Giovanni C, Nicoletti G, Prodi G, Del Re B, et al. RMZ: a new cell line from a human alveolar rhabdomyosarcoma. In vitro expression of embryonic myosin. Br J Cancer (1986) 54(6):1009–14. doi:10.1038/bjc.1986.273
74. Nicoletti G, De Giovanni C, Landuzzi L, Simone G, Rocchi P, Nanni P, et al. Induction of myogenic differentiation in human rhabdomyosarcoma cells by ionising radiation, N,N-dimethylformamide and their combination. Br J Cancer (1992) 65(4):519–22. doi:10.1038/bjc.1992.107
75. Dias P, Parham DM, Shapiro DN, Tapscott SJ, Houghton PJ. Monoclonal antibodies to the myogenic regulatory protein MyoD1: epitope mapping and diagnostic utility. Cancer Res (1992) 52(23):6431–9.
76. Betz BL, Strobeck MW, Reisman DN, Knudsen ES, Weissman BE. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene (2002) 21(34):5193–203. doi:10.1038/sj.onc.1205706
77. Martinez-Ramirez A, Rodriguez-Perales S, Melendez B, Martinez-Delgado B, Urioste M, Cigudosa JC, et al. Characterization of the A673 cell line (Ewing tumor) by molecular cytogenetic techniques. Cancer Genet Cytogenet (2003) 141(2):138–42. doi:10.1016/S0165-4608(02)00670-2
78. Smith MA, Morton CL, Phelps D, Girtman K, Neale G, Houghton PJ. SK-NEP-1 and Rh1 are Ewing family tumor lines. Pediatr Blood Cancer (2008) 50(3):703–6. doi:10.1002/pbc.21099
79. Walter D, Satheesha S, Albrecht P, Bornhauser BC, D’Alessandro V, Oesch SM, et al. CD133 positive embryonal rhabdomyosarcoma stem-like cell population is enriched in rhabdospheres. PLoS ONE (2011) 6(5):e19506. doi:10.1371/journal.pone.0019506
80. Monsma DJ, Monks NR, Cherba DM, Dylewski D, Eugster E, Jahn H, et al. Genomic characterization of explant tumorgraft models derived from fresh patient tumor tissue. J Transl Med (2012) 10:125. doi:10.1186/1479-5876-10-125
81. Crose LE, Etheridge KT, Chen C, Belyea B, Talbot LJ, Bentley RC, et al. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin Cancer Res (2012) 18(14):3780–90. doi:10.1158/1078-0432.CCR-10-3063
82. Drexler HG, Dirks WG, Matsuo Y, MacLeod RA. False leukemia-lymphoma cell lines: an update on over 500 cell lines. Leukemia (2003) 17(2):416–26. doi:10.1038/sj.leu.2402799
83. Dirks WG, Drexler HG. Authentication of cancer cell lines by DNA fingerprinting. Methods Mol Med (2004) 88:43–55.
84. Parson W, Kirchebner R, Muhlmann R, Renner K, Kofler A, Schmidt S, et al. Cancer cell line identification by short tandem repeat profiling: power and limitations. FASEB J (2005) 19(3):434–6.
85. Chen TR, Dorotinsky C, Macy M, Hay R. Cell identity resolved. Nature (1989) 340(6229):106. doi:10.1038/340106b0
86. Boonstra JJ, van Marion R, Beer DG, Lin L, Chaves P, Ribeiro C, et al. Verification and unmasking of widely used human esophageal adenocarcinoma cell lines. J Natl Cancer Inst (2010) 102(4):271–4. doi:10.1093/jnci/djp499
Keywords: alveolar, embryonal, human cell line, rhabdomyosarcoma, xenograft
Citation: Hinson ARP, Jones R, Crose LES, Belyea BC, Barr FG and Linardic CM (2013) Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: utility and pitfalls. Front. Oncol. 3:183. doi: 10.3389/fonc.2013.00183
Received: 31 March 2013; Paper pending published: 21 April 2013;
Accepted: 27 June 2013; Published online: 17 July 2013.
Edited by:
Stephen Lessnick, University of Utah, USAReviewed by:
Rimas J. Orentas, National Institutes of Health, USAPeter J. Houghton, Nationwide Children’s Hospital, USA
Copyright: © 2013 Hinson, Jones, Crose, Belyea, Barr and Linardic. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Corinne M. Linardic, Departments of Pediatrics, Pharmacology and Cancer Biology, Duke University Medical Center, Box 102382, Durham, NC 27710, USA e-mail: linar001@mc.duke.edu