
- 1CIC bioGUNE, Bizkaia Technology Park, Derio, Spain
- 2Ikerbasque, Basque Foundation for Science, Bilbao, Spain
- 3Biochemistry and Molecular Biology Department, University of the Basque Country (UPV/EHU), Bilbao, Spain
Since the discovery of Promyelocytic leukemia (PML), this protein has been associated with the pathogenesis of several hematopoietic malignancies and solid tumors. PML was first identified as part of a fusion oncoprotein, PML-RARα, responsible for the development of acute promyelocytic leukemia (APL) (1–4). The PML-RARα fusion protein not only alters PML function but also represses transcriptional activity mediated by RAR-RXR, thereby disrupting retinoid signaling, inhibiting myeloid differentiation and enhancing the survival and proliferation of early myeloid progenitors (5). Loss of PML in cancers from multiple origins underlines its tumor-suppressive role beyond leukemia (6).
Since PML seemed to be a key regulator underlying leukemia and other cancers, these initial findings motivated a series of studies aimed at ascertaining its regulatory cues and functions. It is now well established that PML is the building block of the PML-nuclear bodies (PML-NBs). PML functions as a protein scaffold and interaction partner for a growing number of factors that shuttle in and out of these structures in a highly regulated process (7–9).
Expanding Cellular Functions of PML
Promyelocytic leukemia protein or PML exerts its anti-cancer role by modulating a number of pathways relevant to cancer biology. PML-NBs increase in number and size in response to DNA damage (10). The nuclear bodies co-localize with sites of single-stranded DNA recruitment and DNA repair. In turn, a number of DNA repair (e.g., MRE11, ATR, BLM, RAD) proteins dynamically localize to PML-NBs (11, 12). Furthermore, PML is an important regulator of both p53-dependent and p53-independent apoptotic pathways (13–15), accomplished by the activation of p53 or Fas, by the phosphorylation and activation of the checkpoint kinase CHK2 (16), or regulating mitochondrial-associated membrane (MAM) function (15). One of the PML anti-cancer and anti-proliferative functions is mediated by activation of the tumor suppressor p21, via transcriptional regulation by p53. Yang et al. demonstrated that big MAP kinase 1 (BMK1) interacts with PML suppressing p21 activation (17). BMK1 associates with PML and disrupts the interaction between PML and MDM2 (the major E3 ubiquitin ligase for p53), which leads to p53 stabilization. This effect induces an increase in tumor cell apoptosis in vitro and tumor regression in vivo (18). Additionally, PML suppresses neo-angiogenesis through the negative regulation of mTORC1 complex (19, 20).
Beyond the nucleus and the nuclear bodies, and perhaps the least studied aspect of PML, is the cytosolic localization and function of PML. Surprisingly, cytoplasmic PML mutants, with aberrant nuclear localization signal, function as a dominant negative, oncogenic forms of the tumor suppressor (21, 22). The Salomoni group (23, 24) reported that mutations in PML that re-localize the protein to the cytoplasm induced the recruitment and mis-localization of PML wildtype nuclear forms to this compartment, therefore reducing the number of PML-NBs. Cytoplasmic PML mutants inhibited p53 transcriptional, growth suppressive, and apoptotic functions (25). These data suggest that cytoplasmic expression of PML affects cell survival through inhibition of nuclear PML.
In recent years, a growing body of work has revealed that PML may provide a selective advantage for tumor cells in certain settings (Figure 1), thus presenting PML as a therapeutic target. Is it possible that PML, in specific contexts (e.g., origin of tumor cell, microenvironments, or metabolic states) can provide a selective pro-survival benefit? In contrast to its usual perception as a classical tumor suppressor, below we will review the latest reports unveiling a potentially more sinister role for PML in cancer. To fully appreciate this novel role, it is important to first mention how the PML gene and protein are regulated.
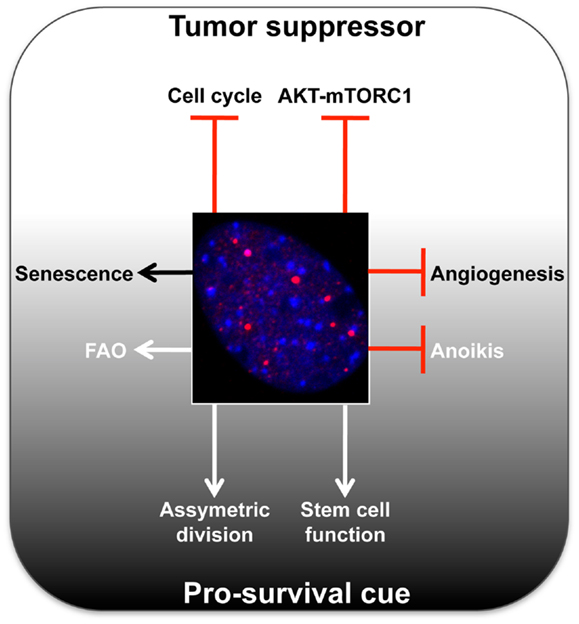
Figure 1. Summary of main tumor-suppressive and pro-survival functions of PML. A representative micrograph of PML immunofluorescence is shown in red, DAPI in blue.
The Complex Regulation of PML
Promyelocytic leukemia is subject to extensive regulation at the transcriptional, post-transcriptional, and post-translational level. At the transcriptional level PML is induced by type I and II interferons (IFN), which cause an increase in both the size and number of PML-NBs (26). This is mediated through binding of IFN-stimulated transcription factors, known as signal transducers and activators of transcription (STATs) (27) and IFN-regulatory factors, such as IRF3 (28) and IRF8 (29), important mediators of myeloid cell differentiation. PML can also be transcriptionally and post-transcriptionally up-regulated by oncogenic Ras (30–34). At the post-transcriptional level, the PML gene can undergo alternative splicing which results in the production of >10 processed mRNAs and many resultant PML protein isoforms (35, 36). The different post-translational modifications have been recently comprehensively reviewed (37), and include phosphorylation, SUMOylation, and ubiquitylation. A more recent example is PML acetylation, which may play a role in apoptotic pathways (38). These modifications regulate the ability of PML to interact with various partners and confer stress- and signal-dependent regulation of PML or its binding proteins (37).
Role of PML Beyond Tumor Suppression
An important study by the Pandolfi group (6) examined PML expression in a wide array of human cancers and revealed a surprising discovery: while PML protein expression was reduced or absent in numerous cancers (prostate, colon, breast, lung, lymphomas, CNS, germ cell tumors), PML mRNA was expressed in all tumors, rarely mutated and was not subject to loss of heterozygosity. Therefore, it was concluded that despite the presence of a functional gene, the PML protein is post-translationally degraded through proteasome-dependent mechanisms and its loss was generally associated with both tumor grade and progression.
In further studies on the role of PML, Ito et al. described that PML was highly expressed in cells from chronic myeloid leukemia (CML) patients, and in contrary to what had been described in solid tumors, loss of PML was predictive of favorable outcomes. Thus, PML expression was selected for and not against in CML (39). This unexpected finding was explained by a novel role of the PML protein in CML. PML was shown to be indispensable for quiescent leukemia-initiating cell (LIC) function; loss of PML resulted in both LIC and hematopoietic stem cell (HSC) depletion. It was also confirmed that treatment with As2O3 (arsenic trioxide or ATO), a drug that down-regulates PML through proteasomal-dependent degradation and that is currently used for the treatment of APL, was able to mimic the genetic loss of PML in mice (39). This finding pointed to a promising therapeutic application for this drug, specifically that destabilization of PML could eradicate LICs and provide a strong benefit for CML patients. The future development of additional, more selective PML-targeting drugs that promote its proteasomal degradation may be extremely helpful in the treatment of CML. These drugs may also find applications in other solid tumors where high levels of PML play a pathogenic role, as we will next discuss.
After this initial finding that PML may have a pro-survival role in CML, Ito et al. demonstrated that PML exerts its essential role in HSC maintenance through the regulation of fatty acid oxidation (FAO) (40, 41). HSCs remain in a quiescent state until environmental insults prompt them to enter the cell cycle, thus dividing and giving rise to multi-potent progenitors. Interestingly, FAO is essential to maintain this balance, under control of the peroxisome-proliferator activated receptor delta (PPARD). Moreover, PML exerts this essential role in HSC maintenance by acting upstream of PPAR signaling and FAO (41). Mechanistically, the PML-FAO PPARD pathway controls HSC asymmetric division. Loss of PML or PPARD, as well as mitochondrial FAO inhibition, resulted in symmetric commitment of HSC daughter cells, and concomitant failure to produce progenitor cells. Conversely, the pharmacological activation of PPAR increased asymmetric division and ensured the long-term self-renewal potential of HSCs.
From these studies, it seems clear that PML has two faces. PML can act as a classical tumor suppressor in many cancers, but in some cases can facilitate cancer cell survival. While studying PML expression in breast cancer biopsies, Carracedo et al. found that although PML protein expression was frequently low or undetectable in the majority of samples, a subset of breast cancer biopsies exhibited PML levels in tumor cells that were significantly higher than those observed in the normal epithelium (40). There was a significant correlation between PML protein and mRNA expression only in tumor (not stroma) cells. More importantly, high PML mRNA and protein expression was significantly associated with triple-negative breast cancer tumor subtype, high tumor grade, early tumor recurrence, and poor prognosis. This study further demonstrated that PML provides a selective advantage in response to metabolic stress triggered by conditions of loss of attachment in breast cancer cells. This was through regulation by PML of the same PPAR-FAO pathway, which stemmed from the PML-induced deacetylation and activation of the transcriptional cofactor PGC1A. PML expression in breast tumors was associated with a signature of activated PPAR signaling that controls FAO. This regulation is relevant to sustain ATP levels and potentially reduced NADP (42), when breast epithelial cells lose contact with the extracellular matrix. On the basis of these findings it is tempting to speculate that targeting both PML and FAO in triple-negative breast cancer tumors with combinations of ATO and other targeted therapies may present a novel approach to treating this tumor subtype.
Taken together, it makes sense that tumor cells would not select for the genomic loss of PML, as is seen with some classical tumor suppressor genes, since it would be irreversible and would prevent cells from utilizing the PML-mediated pro-survival or pro-self renewal pathways when challenged. Instead, transcriptional or post-translational regulation of PML expression and localization allows cancer cells to tune PML expression on the basis of the cellular context.
Two-Faced Tumor Regulators: A Selective Club
Promyelocytic leukemia is not the only protein that has been described to have a dual role as a tumor suppressor and a pro-survival protein. TGFβ also exhibits a well-documented dual activity in cancer. The TGFβ signaling pathway negatively regulates cell growth, death, and immortalization (43). Thus, mutations or deletions in the TGFβ gene can lead to the onset of several tumors. TGFβ signaling also plays an important role as a positive regulator to modulate processes such as cell invasion, immune regulation, and microenvironment remodeling that can promote cancer progression, invasion, and tumor metastasis (43–45).
Notch is another example of dual activity in cancer regulation. It is well described that Notch activates signaling pathways that regulate cell division, growth, migration, differentiation, or death (46, 47). Similar to TGFβ, Notch activity is required for the physiological development of organisms and for the maintenance of adult tissues. However, it has been demonstrated that the deregulation of Notch signaling pathway or its pathological activation can induce certain types of tumors such as leukemia, breast, colon, skin, lung, or renal carcinomas (48–50).
Other examples of genes with dual activity in tumor biology are Toll-like receptors (TLRs) (51, 52), CD44 (53), sirtuins (54), or E-cadherin (55). In general, the activation of these proteins can promote pro-tumorigenic signaling and trigger the metastatic cascade, inducing proliferation, invasion, or apoptosis resistance. However, in different scenarios or signaling through specific networks, these proteins can have opposite effects on tumors, favoring tumor-suppressive responses.
From these and other examples, it seems clear that critical genes and their downstream pathways can also be involved in the transformation of normal healthy cells into cancerous, potentially malignant forms in multiple ways depending on the cell subpopulation, and the microenvironmental milieu. This idea is under intensive research and may play a decisive role in the search for novel therapeutic treatments aimed at specific cancer types and subtypes, a key concept in personalized medicine.
Future Therapy: Combinatorial Approaches
These insights into the dual role of PML in tumorigenesis could lead to new therapeutic interventions. Twenty-five years of basic and clinical research have allowed most patients with APL to be definitively cured under ATO treatment combined with retinoic acid. ATO targets PML through oxidation-triggered disulfide bond formation and direct binding. This results in PML and PML-RARα SUMOylation, and its subsequent ubiquitylation and proteasome-mediated degradation (56, 57). As we have mentioned, in CML, a related but distinct cancer type, the PML protein is indispensable for quiescent LIC maintenance (39) through the regulation of FAO by PPAR signaling (41). Moreover, this is not the only case where PML plays a role as a pro-survival protein rather than a tumor suppressor. PML is highly expressed in a subset of breast cancers with worse prognosis and shorter time to recurrence (40). Therefore, the use of PML-targeting drugs that activate proteasomal degradation could be of remarkable interest in the treatment of CML and breast cancer. On the other hand, enhancing levels or activity of RNF4, an E3 ubiquitin ligase which is essential for ATO-induced PML degradation (56, 58), could also enhance the down-regulation of PML. With our new knowledge into the underlying mechanism of PML function, the pharmacological targeting of FAO, or the use of PPAR inhibitors in combination with low doses of ATO might exert a synergistic effect on triple-negative breast cancer tumors and possibly other solid tumors or leukemias.
In conclusion, PML has been revealed as friend and foe in cancer. More detailed studies are warranted in order to categorize diverse tumor types for these opposing activities of PML, and ultimately to explore the therapeutic potential of PML-targeting compounds, alone or in combination with drugs that target PML-linked pathways.
Acknowledgments
Apologies to those whose related publications were not cited due to space limitations. We thank the Carracedo lab for insightful discussions. The work is supported by the Ramón y Cajal award (Arkaitz Carracedo), the Basque Department of Industry, Tourism and Trade (Etortek-Arkaitz Carracedo and James D. Sutherland), health (2012111086-Arkaitz Carracedo) and education (PI2012-03-Arkaitz Carracedo; PI2012/42-James D. Sutherland), Marie Curie (277043), Spanish MICINN (BFU2011/25986-James D. Sutherland) Movember, and ISCIII (PI10/01484-Arkaitz Carracedo). Natalia Martin-Martin is supported by the Spanish Association Against Cancer (AECC).
References
1. Kakizuka A, Miller WH Jr, Umesono K, Warrell RP Jr, Frankel SR, Murty VV, et al. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell (1991) 66:663–74. doi:10.1016/0092-8674(91)90112-C
2. Goddard AD, Borrow J, Freemont PS, Solomon E. Characterization of a zinc finger gene disrupted by the t(15;17) in acute promyelocytic leukemia. Science (1991) 254:1371–4. doi:10.1126/science.1720570
3. Pandolfi PP, Grignani F, Alcalay M, Mencarelli A, Biondi A, LoCoco F, et al. Structure and origin of the acute promyelocytic leukemia myl/RAR alpha cDNA and characterization of its retinoid-binding and transactivation properties. Oncogene (1991) 6:1285–92.
4. de Thé H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell (1991) 66:675–84. doi:10.1016/0092-8674(91)90113-D
5. Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer (2001) 1:181–93. doi:10.1038/35106036
6. Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ, et al. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst (2004) 96:269–79. doi:10.1093/jnci/djh043
7. Lang M, Jegou T, Chung I, Richter K, Münch S, Udvarhelyi A, et al. Three-dimensional organization of promyelocytic leukemia nuclear bodies. J Cell Sci (2010) 123:392–400. doi:10.1242/jcs.053496
8. Shen TH, Lin H-K, Scaglioni PP, Yung TM, Pandolfi PP. The mechanisms of PML-nuclear body formation. Mol Cell (2006) 24:331–9. doi:10.1016/j.molcel.2006.09.013
9. Zhong S, Hu P, Ye TZ, Stan R, Ellis NA, Pandolfi PP. A role for PML and the nuclear body in genomic stability. Oncogene (1999) 18:7941–7. doi:10.1038/sj.onc.1203367
10. Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol (2007) 8:1006–16. doi:10.1038/nrm2277
11. Bøe SO, Haave M, Jul-Larsen A, Grudic A, Bjerkvig R, Lønning PE. Promyelocytic leukemia nuclear bodies are predetermined processing sites for damaged DNA. J Cell Sci (2006) 119:3284–95. doi:10.1242/jcs.03068
12. Dellaire G, Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays (2004) 26:963–77. doi:10.1002/bies.20089
13. Takahashi Y, Lallemand-Breitenbach V, Zhu J, de Thé H. PML nuclear bodies and apoptosis. Oncogene (2004) 23:2819–24. doi:10.1038/sj.onc.1207533
14. Bernardi R, Pandolfi PP. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene (2003) 22:9048–57. doi:10.1038/sj.onc.1207106
15. Giorgi C, Ito K, Lin H-K, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science (2010) 330:1247–51. doi:10.1126/science.1189157
16. Yang S, Jeong JH, Brown AL, Lee CH, Pandolfi PP, Chung JH, et al. Promyelocytic leukemia activates Chk2 by mediating Chk2 autophosphorylation. J Biol Chem (2006) 281:26645–54. doi:10.1074/jbc.M601356200
17. Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, et al. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell (2010) 18:258–67. doi:10.1016/j.ccr.2010.08.008
18. Yang Q, Liao L, Deng X, Chen R, Gray NS, Yates JR III, et al. BMK1 is involved in the regulation of p53 through disrupting the PML-MDM2 interaction. Oncogene (2013) 32:3156–64. doi:10.1038/onc.2012.332
19. Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, et al. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature (2006) 442:779–85. doi:10.1038/nature05029
20. Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature (2006) 441:523–7. doi:10.1038/nature04809
21. Gurrieri C, Nafa K, Merghoub T, Bernardi R, Capodieci P, Biondi A, et al. Mutations of the PML tumor suppressor gene in acute promyelocytic leukemia. Blood (2004) 103:2358–62. doi:10.1182/blood-2003-07-2200
22. Zheng P, Guo Y, Niu Q, Levy DE, Dyck JA, Lu S, et al. Proto-oncogene PML controls genes devoted to MHC class I antigen presentation. Nature (1998) 396:373–6. doi:10.1038/24628
23. Salomoni P, Bellodi C. New insights into the cytoplasmic function of PML. Histol Histopathol (2007) 22:937–46.
24. Bellodi C, Kindle K, Bernassola F, Dinsdale D, Cossarizza A, Melino G, et al. Cytoplasmic function of mutant promyelocytic leukemia (PML) and PML-retinoic acid receptor-alpha. J Biol Chem (2006) 281:14465–73. doi:10.1074/jbc.M600457200
25. Bellodi C, Kindle K, Bernassola F, Cossarizza A, Dinsdale D, Melino G, et al. A cytoplasmic PML mutant inhibits p53 function. Cell Cycle (2006) 5:2688–92. doi:10.4161/cc.5.22.3504
26. Lavau C, Marchio A, Fagioli M, Jansen J, Falini B, Lebon P, et al. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene (1995) 11:871–6.
27. Stadler M, Chelbi-Alix MK, Koken MH, Venturini L, Lee C, Saïb A, et al. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene (1995) 11:2565–73.
28. Kim TK, Lee JS, Oh SY, Jin X, Choi YJ, Lee TH, et al. Direct transcriptional activation of promyelocytic leukemia protein by IFN regulatory factor 3 induces the p53-dependent growth inhibition of cancer cells. Cancer Res (2007) 67:11133–40. doi:10.1158/0008-5472.CAN-07-1342
29. Dror N, Rave-Harel N, Burchert A, Azriel A, Tamura T, Tailor P, et al. Interferon regulatory factor-8 is indispensable for the expression of promyelocytic leukemia and the formation of nuclear bodies in myeloid cells. J Biol Chem (2007) 282:5633–40. doi:10.1074/jbc.M607825200
30. Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev (2000) 14:2015–27.
31. de StanchinaE, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, et al. PML is a direct p53 target that modulates p53 effector functions. Mol Cell (2004) 13:523–35. doi:10.1016/S1097-2765(04)00062-0
32. Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature (2000) 406:207–10. doi:10.1038/35018127
33. Jiang WQ, Ringertz N. Altered distribution of the promyelocytic leukemia-associated protein is associated with cellular senescence. Cell Growth Differ (1997) 8:513–22.
34. Scaglioni PP, Rabellino A, Yung TM, Bernardi R, Choi S, Konstantinidou G, et al. Translation-dependent mechanisms lead to PML upregulation and mediate oncogenic K-RAS-induced cellular senescence. EMBO Mol Med (2012) 4:594–602. doi:10.1002/emmm.201200233
35. Yoshida H, Ichikawa H, Tagata Y, Katsumoto T, Ohnishi K, Akao Y, et al. PML-retinoic acid receptor alpha inhibits PML IV enhancement of PU.1-induced C/EBPepsilon expression in myeloid differentiation. Mol Cell Biol (2007) 27:5819–34. doi:10.1128/MCB.02422-06
36. Jensen K, Shiels C, Freemont PS. PML protein isoforms and the RBCC/TRIM motif. Oncogene (2001) 20:7223–33. doi:10.1038/sj.onc.1204765
37. Cheng X, Kao H-Y. Post-translational modifications of PML: consequences and implications. Front Oncol (2012) 2:210. doi:10.3389/fonc.2012.00210
38. Hayakawa F, Abe A, Kitabayashi I, Pandolfi PP, Naoe T. Acetylation of PML is involved in histone deacetylase inhibitor-mediated apoptosis. J Biol Chem (2008) 283:24420–5. doi:10.1074/jbc.M802217200
39. Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature (2008) 453:1072–8. doi:10.1038/nature07016
40. Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC, Laurent G, et al. A metabolic prosurvival role for PML in breast cancer. J Clin Invest (2012) 122:3088–100. doi:10.1172/JCI62129
41. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med (2012) 18:1350–8. doi:10.1038/nm.2882
42. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer (2013) 13:227–32. doi:10.1038/nrc3483
44. Muraoka-Cook RS, Dumont N, Arteaga CL. Dual role of transforming growth factor beta in mammary tumorigenesis and metastatic progression. Clin Cancer Res (2005) 11:937s–43.
45. Meulmeester E, Ten Dijke P. The dynamic roles of TGF-β in cancer. J Pathol (2011) 223:205–18. doi:10.1002/path.2785
46. Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet (2012) 13:654–66. doi:10.1038/nrg3272
47. Becam I, Fiuza U-M, Arias AM, Milán M. A role of receptor Notch in ligand cis-inhibition in Drosophila. Curr Biol (2010) 20:554–60. doi:10.1016/j.cub.2010.01.058
48. Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene (2008) 27:5124–31. doi:10.1038/onc.2008.226
49. Shih I-M, Wang T-L. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res (2007) 67:1879–82. doi:10.1158/0008-5472.CAN-06-3958
50. Avila JL, Kissil JL. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med (2013) 19:320–7. doi:10.1016/j.molmed.2013.03.003
51. Yu L, Wang L, Chen S. Dual character of Toll-like receptor signaling: pro-tumorigenic effects and anti-tumor functions. Biochim Biophys Acta (2013) 1835:144–54. doi:10.1016/j.bbcan.2012.10.006
52. Huang B, Zhao J, Unkeless JC, Feng ZH, Xiong H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene (2008) 27:218–24. doi:10.1038/sj.onc.1210904
53. Louderbough JMV, Schroeder JA. Understanding the dual nature of CD44 in breast cancer progression. Mol Cancer Res (2011) 9:1573–86. doi:10.1158/1541-7786.MCR-11-0156
54. Bosch-Presegué L, Vaquero A. The dual role of sirtuins in cancer. Genes Cancer (2011) 2:648–62. doi:10.1177/1947601911417862
55. Rodriguez FJ, Lewis-Tuffin LJ, Anastasiadis PZ. E-cadherin’s dark side: possible role in tumor progression. Biochim Biophys Acta (2012) 1826:23–31. doi:10.1016/j.bbcan.2012.03.002
56. Lallemand-Breitenbach V, Jeanne M, Benhenda S, Nasr R, Lei M, Peres L, et al. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat Cell Biol (2008) 10:547–55. doi:10.1038/ncb1717
57. Lallemand-Breitenbach V, Zhu J, Chen Z, de Thé H. Curing APL through PML/RARA degradation by As2O3. Trends Mol Med (2012) 18:36–42. doi:10.1016/j.molmed.2011.10.001
Citation: Martin-Martin N, Sutherland JD and Carracedo A (2013) PML: not all about tumor suppression. Front. Oncol. 3:200. doi: 10.3389/fonc.2013.00200
Received: 22 March 2013; Accepted: 19 July 2013;
Published online: 05 August 2013.
Edited by:
Pier Paolo Pandolfi, Beth Israel Deaconess Medical Center, USACopyright: © 2013 Martin-Martin, Sutherland and Carracedo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: acarracedo@cicbiogune.es