 German A. Pihan
German A. Pihan- Department of Pathology and Laboratory Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA
The unique ability of centrosomes to nucleate and organize microtubules makes them unrivaled conductors of important interphase processes, such as intracellular payload traffic, cell polarity, cell locomotion, and organization of the immunologic synapse. But it is in mitosis that centrosomes loom large, for they orchestrate, with clockmaker’s precision, the assembly and functioning of the mitotic spindle, ensuring the equal partitioning of the replicated genome into daughter cells. Centrosome dysfunction is inextricably linked to aneuploidy and chromosome instability, both hallmarks of cancer cells. Several aspects of centrosome function in normal and cancer cells have been molecularly characterized during the last two decades, greatly enhancing our mechanistic understanding of this tiny organelle. Whether centrosome defects alone can cause cancer, remains unanswered. Until recently, the aggregate of the evidence had suggested that centrosome dysfunction, by deregulating the fidelity of chromosome segregation, promotes and accelerates the characteristic Darwinian evolution of the cancer genome enabled by increased mutational load and/or decreased DNA repair. Very recent experimental work has shown that missegregated chromosomes resulting from centrosome dysfunction may experience extensive DNA damage, suggesting additional dimensions to the role of centrosomes in cancer. Centrosome dysfunction is particularly prevalent in tumors in which the genome has undergone extensive structural rearrangements and chromosome domain reshuffling. Ongoing gene reshuffling reprograms the genome for continuous growth, survival, and evasion of the immune system. Manipulation of molecular networks controlling centrosome function may soon become a viable target for specific therapeutic intervention in cancer, particularly since normal cells, which lack centrosome alterations, may be spared the toxicity of such therapies.
Cancer is an evolutionary multistep process arising in single cells resulting from accumulation of non-lethal mutations that increase, decrease, deregulate, or interfere with the function of critical genes, leading to autonomous growth and loss of homeostasis. Cancer cells fail to execute programed cell death when required, fail to exit the cell cycle when prompted, or to differentiate in response to appropriate external or internal regulatory signals. This dynamic “renegade” behavior (1), which was elegantly codified by Hanahan and Weinberg in a series of publications addressing the Hallmarks of cancer cells (2, 3), lay at the core of cancer biology and dispels the notion of sporadic cancer as a simple, oligogenic somatic genetic disease. Cancer is indeed a family of complex evolutionary somatic genetic disorders resulting from dynamic and ongoing reprograming of the genome, the nature of which will continue to challenge our ingenuity for years to come.
The First Cancer Cell Hallmark
An abnormal complement of chromosomes, i.e., aneuploidy, is arguably the first identified hallmark of cancer cells. Beginning in 1890 Leo Hansemann, in a series of beautifully illustrated observations (Figure 1), documented the frequent presence of asymmetric and multipolar mitoses in carcinoma tissue (4, 5). Though uncertain of the significance of his findings, Hansemann was aware that daughter cells resulting from asymmetric mitoses received abnormal amounts of “chromatin.” Perhaps in part prompted by these findings, Theodore Boveri, who was aware of Leo Hansemann’s work and publically acknowledged his findings, formulated his now famous theory of cancer development (6). Half a century later, Torbjörn Caspersson, who pioneered cytological microspectrofluorimetric analysis of nuclei acids (7), would resoundingly confirm Hansemann and Boveri’s predictions. Caspersson was the first to observe that cancer cells, unlike normal cells, which always contained a constant amount of DNA, almost always exhibited greater, but highly variable quantities of nuclear DNA (8). It can thus be stated that an abnormal chromosome complement, possibly resulting from abnormal centrosome function, was the first hallmark of cancer ever identified. Since these pioneering observations, the nearly universal occurrence of abnormal chromosomes in cancer, in a bewildering combination of numerical and structural abnormalities, has been widely documented. A number of data repositories, such as The Cancer Genome Anatomy Project (9, 10), which includes Mitelman’s cancer cytogenetic collection currently containing 62,601 cancer karyotypes (11), provide a plethora of data and an overview of the spectrum and extent of large scale genome changes in cancer.
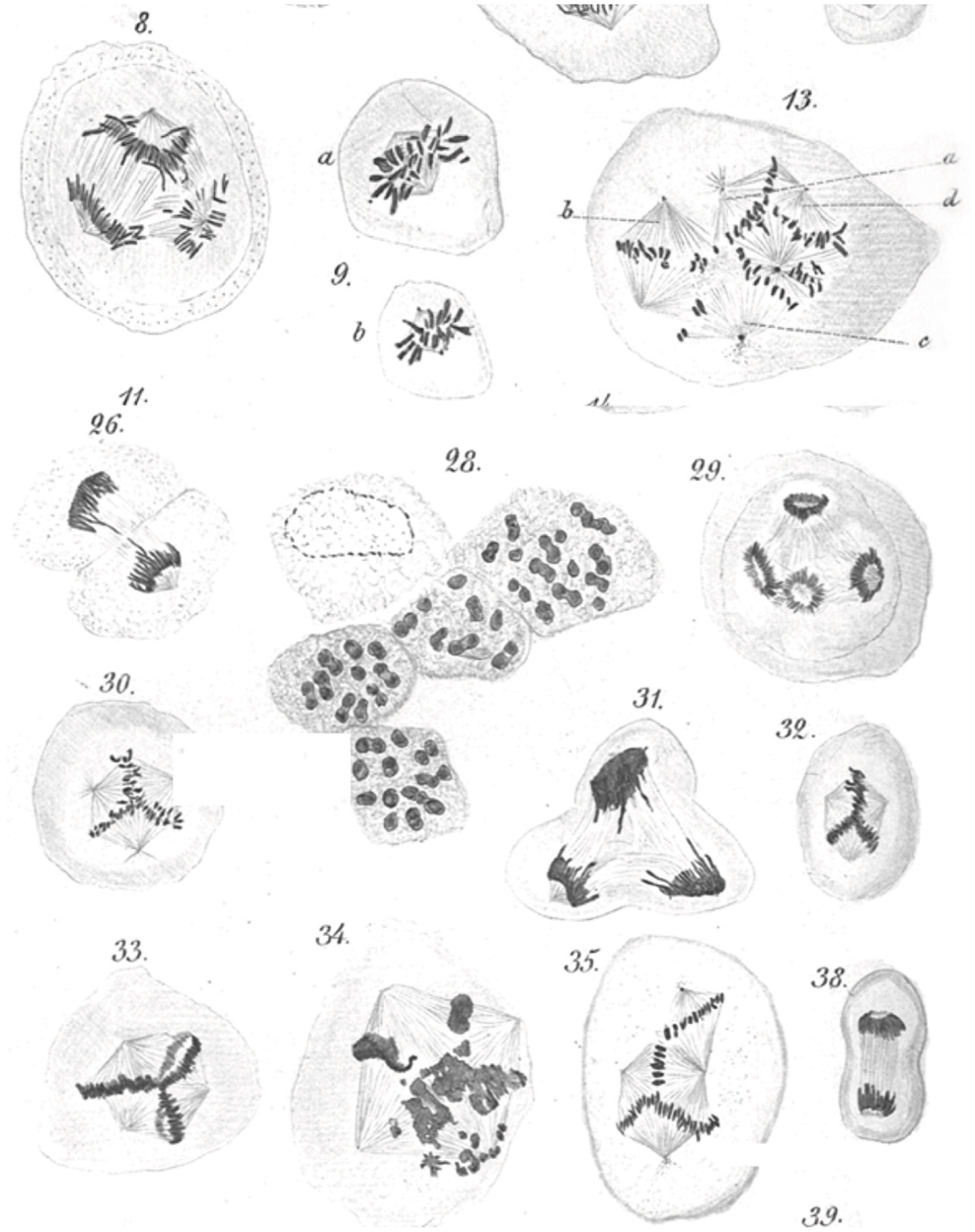
Figure 1. Leo Hansemann’s drawings of abnormal mitoses in cancer tissue. Abnormal metaphases (13, 30, 32, 33, 35), anaphases (8, 29), and telophases (31) are exquisitely represented, with many of the drawings implying supernumerary centrosomes, which could not be directly visualized in these preparations. Apparent mitotic catastrophes are represented as well (28, 34) (reproduced with permission from Virchows Archive). Hansemann took care of placing cancer tissue samples in warm fixative immediately after surgical resection to avoid anoxia-induced changes and in the process beautifully preserved spindle microtubules.
Contemporary View on Centrosomes Structure and Function
The centrosome is a multifunctional structurally complex macromolecular machine composed of hundreds of proteins (12–16) (Figure 2). It is the primary microtubule-organizing center (MTOC) in metazoans (17–26) controlling several interphase and mitotic microtubule-dependent processes (17–19, 22–25, 27).
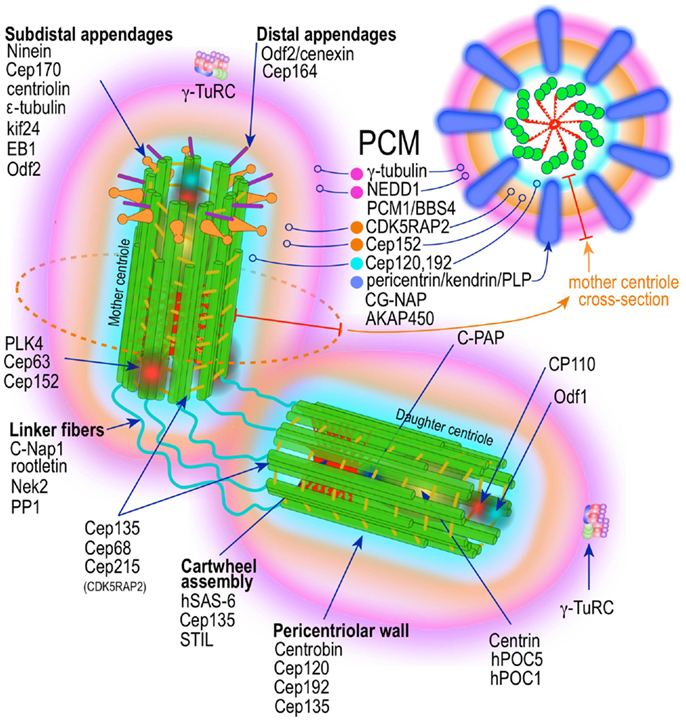
Figure 2. Prototypic vertebrate centrosome. Salient architectural features of the post-mitotic centrosome (after centriole disengagement, but before procentriole nucleation) include fully developed mother and daughter centrioles and electron dense but highly structured pericentriolar material (PCM).
In interphase, the centrosome establishes and controls the microtubule network that serves as the highway for fast intracellular payload traffic of protein and organelles (26). It contributes to the development and maintenance of cell polarity (28), cell adhesion (29), cell locomotion (30), and the organization of the immunological synapse (31). In differentiated cells the maternal centriole guides the establishment of the primary cilium (32). However, it is during mitosis that centrosomes play their most visible role. Centrosomes organize and fine-tune the microtubule arrays that form the mitotic spindle – the elegant and precisely choreographed supramolecular machine that ensures the segregation of exactly one full diploid set of chromosomes to each daughter cell during mitosis (21, 33). Additionally, astral microtubules arising in centrosomes control mitotic spindle position and orientation (34–36), thereby specifying the cell division plane, which ultimately control cell fate in some stem cell niches, such as the developing central nervous system (34–36). The centrosome, specifically its maternal centriole, is critical for the development of the primary and secondary cilium in differentiated cells.
The centrosome possesses microtubule-independent functions as well. The most important of these is its cell cycle regulatory activity. Several proteins that localize to centrosomes sequentially control a number of cell cycle processes. Centrosomes license post-mitotic G1 cells for entry into the next S phase (37). In addition, centrosomes may control the timing of S-phase and mitotic entry. Cyclins A and E have centrosome localization sequences (CLS) which are critical for Cdk2-dependent S-phase entry. Interference with cycle A/E binding to the centrosome prevents entry into S phase (38). Centrosomal localization of Cyclin E-Cdk2 is required for initiation of DNA synthesis (39), whereas Cyclin A-Cdk2 by binding MCM5 and Orc1 – two proteins involved in DNA replication – prevents centrosome overduplication in S phase (40). Centrosomes may also control the initial activation of cyclin B1-Cdk1 in the cytoplasm, which is critical for mitotic entry. Enzymatically active cyclin B1-Cdk1 appears first at the centrosome at the onset of mitosis (41, 42). Cyclin B-Cdk1 is maintained inactive via phosphorylation of ATP binding pocket amino acids Thr14 and Tyr15 by Wee kinase. Mitotic onset is triggered by a massive wave of Cyclin B-Cdk1 activation that starts at the centrosome by Cdc25B phosphatase initially dephosphorylating Cdk1. This initially activated centrosomal pool of Cyclin B-Cdk1 at the end of prophase, phosphorylates Cdc25B and Wee1, activating the first and inhibiting the second, and triggering a rapidly expanding wave of Cyclin B-Cdk1 activation with its epicenter at the centrosome. The importance of preventing activation of the centrosomal pool of Cyclin B-Cdk1 is highlighted by it being closely guarded by the checkpoint kinase Chk-1, which localizes to the centrosome in interphase, but not mitosis, and inhibits Cdk1 activation (43). Inhibition of centrosomal Chk-1 results in premature separation of centrosomes and activation of Cyclin B-Cdk1 leading to premature entry into mitosis (44). Moreover, Aurora kinase A – which localized to the centrosome and increases through S and G2 phases – appears to be the trigger for nuclear envelope breakdown (NEB) in mitosis (45). Cyclin F, the only F-box cyclin, accumulates in the centrosome in G2 and is involved in ubiquitinylation of CP110 targeting it for proteolytic degradation. Degradation of CP110 ensures restriction of centrosome replication to once in each cell cycle. Cyclin F also targets the ribonucleotide reductase RRM2, leading to downregulation of dNTP pools at the end of S phase (46). Proteolytic cleavage of CP110 and RRM2 are important to prevent genomic instability. Finally, in some cells the maternal centrosome, late during cytokinesis, moves close to the central spindle and appears to license abscission by releasing central spindle microtubules (47). This function appears to enhance the fidelity of cytokinesis (48). Considered together, these findings reveal that the centrosome is an integral component and relay station for signals that control events throughout the cell cycle, functions that if disrupted may lead to abnormal DNA replication and cell division.
Centrosome Integrity Checkpoint
In the course of experiments investigating the function of several centrosome components, an important aspect of centrosome self-regulation was revealed. Depletion of centrosomal AKAP450 and pericentrin, by targeting their common centrosome binding domains induces post-mitotic cell cycle arrest. Moreover, targeted depletion of 14 out of 15 centrosome components by siRNA, prolonged cytokinesis and triggered G1 arrest in telomerized (immortal), non-transformed human cells (49, 50). Cell cycle arrest occurred from within G1, was independent of mitosis and cytokinesis, and dependent on p38-induced phosphorylation of p53 at Ser-33. Ser-33 phosphorylated p53 localizes to the centrosome from which it translocates into the nucleus upregulating p21 and inducing S1 arrest via inhibition of Cdk2-CyclinA/E activity (50). This seemingly universal G1 arrest response of non-transformed cells to centrosome “injury” constitutes a de facto “centrosome integrity checkpoint” that precludes normal cells from reentering the cell cycle once centrosome integrity has been compromised, thus avoiding chromosome instability (CIN) and genomic instability (50).
Centrosome Structure
Centrioles
A mature centrosome is comprised of two centrioles in orthogonal configuration, surrounded by an amorphous electron-dense protein-rich cloud, termed pericentriolar material (PCM) (51–53) (Figure 2). Each centriole is a cylindrical structure made up of nine parallel microtubule triplets, their long axes running the length of the cylinder. Each microtubule triplet is composed of an internal A, middle B, and external C microtubule. Unlike A and B, the C microtubule runs only two-thirds the length of the centriole and is absent from its distal portion. In the proximal centriole, the A microtubule of one triplet is connected to the C microtubule of the previous triplet by an A-C linker, which becomes an A-B linker beyond the end of the C microtubule toward the distal end of the centriole barrel. The microtubule triplets are heavily acetylated (54) and polyglutamylated (55) and thus very stable, exchanging little if any with the soluble pool of αβ tubulin heterodimers. The ninefold radial symmetry of centrioles is apparently determined and maintained by the proximal-end cartwheel assembly (Figure 2). Comprised of nine spokes radiating from a central hub, the cartwheel assembly is composed almost entirely of a single protein, Sas-6. The cartwheel assembly is repeated six times along the interior of the proximal end of the centriole. Although nearly identical, the older, “mother” centriole, additionally exhibits distal and subdistal appendages, which are essential for centrosome tethering to the plasma membrane and initiation of ciliogenesis (56). In G1 phase of the cell cycle, disengaged mother and daughter centrioles are tethered to each other by intercentriolar fibers. Attesting to the complexity of this organelle, many structural and regulatory proteins have specific locations within the centriole (Figure 2).
Pericentriolar Material
While centrioles are essential to restrict centrosome duplication to once in every cell cycle (57, 58), they are largely dispensable for microtubule nucleation. It is instead the pericentriolar amorphous cloud of proteins, the PCM, the structure responsible for most of the centrosome microtubule-organizing activity, including microtubule nucleation and control of microtubule number, polarity, distribution, and flux (21) (Figure 2). Until both centrioles are fully mature at the end of G2, most of the PCM is associated with the mother centriole (58, 59). This ensures that the developing daughter centriole in S phase remains “inactive.” Of the hundreds of proteins that localize to the pericentriolar cloud, γ-tubulin plays a central role by providing a template for the initiation of polymerization of α- and β-tubulin heterodimers into growing microtubules. γ-tubulin (TUBG1, TUBG2) performs this task in complex with other proteins collectively known as gamma tubulin complex proteins (GCP2–6 or TUBGCPs) (60–62). GCPs exhibit conserved “grip domains” at both N- and C-terminal ends (63). Two copies of γ-tubulin (GCP1) together with GPC2 and GPC3 form a tetramer termed γ-tubulin small complex (γ-TuSC) (64, 65). Several γ-TuSC, together with GCPs 4–6, assemble into a higher-order complex with a toroidal shape known as γ-tubulin ring complex (γTuRC) (62, 65–69) (Figure 2). The γTuRC toroid is stabilized on one of its sides by a complex of GCPs 4–6 proteins (70). Two other core components of the γTuRC complex, which do not contain Grip domains, GCP-WD (GCP7, NEDD1) and GCP8 (MOZART2), are non-essential for γTuRC assembly (71–74). However, GCP-WD is essential for γTuRC attachment to the PCM. The C-terminus of GCP-WD binds γ-tubulin in the γTuRC and the N-terminus WD domains form the blades of a β-propeller structure that binds to the PCM (72, 73, 75–77). GCP8 (MOZART2) appears to play a role in the recruitment of γTuRC to the PCM during interphase (74). An additional γTuRC core component is GCP9 (MOZART1) (74, 78). In human cells MOZART1 is required for recruitment of γTuRC to mitotic centrosomes (78). While Grip-GCPs and γ-tubulin are considered structural components of the γTuRC (72, 73, 75–77), some GCPs may have regulatory functions as well. γTuRC is targeted to MTOCs with the help of several centrosomal proteins, including AKAP450 (CG-NAP, AKAP9) (79), pericentrin (PCNT) (80), and CDK5RAP2 (Cep215) (81), and in human cells is dependent on an intact mature γTuRC (82). However, attachment of γTuRC to the centrosome occurs via GCP-DW (NEDD1) and this attachment factor is recruited to the centrosome independently of the γTuRC (72, 73). A number of non-stoichiometric regulatory molecules preferentially associate with the γTuRC in mitosis (72, 73, 78). These include Plk1, and seven of the eight Augmin/HAUS complex subunits (78) (Figure 2). The Augmin complex was initially defined in Drosophila as an eight-subunit centrosome protein complex of 340 kDa that localizes γTuRC to the mitotic spindle where it is indispensable for nucleating microtubules in a centrosome-independent manner, increasing microtubule density within the spindle, and stabilizing kinetochore microtubules (83). Humans possess a similar protein complex termed HAUS1–8 (78, 83, 84). Just as Augmin, the HAUS complex resides in the centrosome PCM and moves to spindle microtubules during mitosis, where it is important to increase the density of kinetochore and polar microtubules (83). In the absence of HAUS, mitotic human cells have reduced spindle tension (insufficient number of microtubules) and are unable to extinguish the mitotic spindle assembly checkpoint, leading to stalled mitosis and microtubule-dependent centrosome fragmentation (84). HAUS1–8 complexes also have a role in cytokinesis (83). The function of the individual HAUS units is currently poorly understood. It is thought that HAUS8 directly contact microtubules, while HAUS6 binds γTuRC via NEDD1 (83).
Recently, superresolution microscopy has begun to provide insights into the organization of the PCM (85–87). With the exception of an early study using fluorescence microscopy with image deconvolution that suggested the PCM is organized in a lattice-like structure around the centriole (88), it has been generally believed that the PCM is largely unstructured. Using SIM and STORM, two forms of superresolution fluorescence microscopy, Mennella et al (87) have demonstrated that the PCM in Drosophila S2 cells, contrary to the prevailing view, is highly organized into two or three main structural domains (Figure 2). One layer juxtaposed to the centriole wall and a second, matrix-like layer, located further away. Some coil–coil proteins in the juxtacentriolar layer, such as pericentrin-like protein (PLP), have their carboxy termini located near the centriole wall from which they extend, in clusters with quasi ninefold symmetry, centrifugally into the matrix layer. RNA interference experiments indicate that the juxtacentriolar layer is fundamentally required for organization of the external matrix layer. Remarkably, many of the proteins of the PCM have distinctive and strictly defined distribution volumes around the maternal centriole. With Sas-6 located at the center of the centriole and exhibiting the smallest distribution volume, followed by Sas-4, known to localize to the centriole wall, followed by PLP and asterless (ASL) in the inner region and SPD-2, γ-tubulin, and CNN in the outer matrix-like region further away from the centriole (87). Until mitosis PLP distributes around the mother centriole only. Beginning in mitosis PLP also distributes to the daughter centriole, a process that is completed by the end of telophase. In mammalian cells, pericentrin/kendrin and CG-NAP, two PACT (pericentrin-AKAP450 centrosome targeting) proteins, have a distribution similar to Drosophila PLP, which is consistent with PCNT/Kendrin being the ortholog of PLP. PCNT associates with CEP192 (SPD-2) and CDK5Rap2 in a functional complex in the inner layer of the PCM (89–93) (Figure 2). It is apparent that this layered organization of the PCM subserves two different needs, the inner layer as an organizer of the PCM and the outer layer with most of the microtubule nucleation functionality by providing docking sites for the γTuRC (87) (Figure 2). A similar study using mammalian cells revealed essentially equivalent organization of the PCM (86). Again PCNT distribution and orientation was consistent with its role in organizing the external PCM toroid responsible for microtubule nucleation (86). γ-tubulin and NEDD1 distributed to a region estimated to be at the center of the partially extended PCNT (Figure 2). Nevertheless, the distributions of components of the PCM seem to be even more complex that these initial studies revealed. A large survey of centrosome/centriole protein localization in interphase U2OS cells using 3D-SIM revealed that γ-tubulin and NEDD/GCP-WD, in addition to their distribution around the mother centriole and to a lesser extent around the daughter centriole, are also present, in a dot-like pattern, inside the mother centriole, as the gravitational center of the pericentriolar ring (94). Remarkably, this is consistent with previous immuno-EM data demonstrating these two proteins also residing inside the centriole (95, 96). Examination of the distribution, orthogonal to the mother centriole long axis, of an additional 18 proteins revealed distinctive pattern consisting of either toroid-like or dot-like structures. Four proteins (centrin, Sas-6, STIL, and Plk-4) distributed in compact dots. All other proteins (94) revealed toroid structures of varying sizes. Only one other protein, CPAP showed both toroid and central dot-like localization. Of the toroid proteins Cep135 and CP110 formed rings of nearly identical size, but distributed to the opposite ends (proximal and distal, respectively) of the centriole, in agreement with previous immuno-EM data (97, 98). All other proteins formed toroids of larger diameter, which were separated into three arbitrary groups. The inner group is composed by Cep192, NEDD1, Cep152-C, Cap350; the intermediate by Cep152-N, Cep215, γ-tubulin; and the outer layer by CPAP and pericentrin. Cep164 localized to the distal appendages, whereas Cep170 and ninein to subdistal appendages. These data indicate the γTuRC distributes between the inner and intermediate layers of the PCM in interphase U2OS cells. By using amino- or carboxy-terminal specific antibodies, and other domain specific antibodies, it was possible to demonstrate that some of these protein have not only specific localization, but also defined orientations within the PCM (94). The proteins with the largest distribution volumes were ninein (99) and Cep170 (100), which localized to the centriole subdistal appendages, whereas the distal appendage protein Cep164 (101) had slightly lower distribution volume. The location of these proteins is concordant with their previous localization by electron microscopy (52). CP110 and Centrin localizes to the distal end of centrioles (97, 98).
These studies combined indicate that the PCM, in spite of its amorphous appearance under the regular fluorescence or electron microscope, is highly organized (Figure 2). In cells in interphase it is distributed onto roughly two pericentriolar cylinders. An inner layer closely apposed to centriolar microtubules, composed of Sas-4, Spd-2, and Polo kinase, and an outer layer composed of PACT proteins (dPLP, PCNT), Asterless (Asl), and Plk-4 kinase. As cells transit S phase and G2 and in preparation for mitosis other proteins, such as γ-tubulin and centrosomin are recruited to the external layer. Upon mitotic entry the PCM experiences a drastic expansion in which PCNT, Cep192, and Cep215 form large extended networks. γ-tubulin, however, does not co-localize with this network suggesting additional PCM components tethering γTuRC to the PCM networks (94) (vide supra).
Centriole-PCM Interactions
It has become apparent that there is crosstalk between centrioles and the PCM. While centrioles appear to control protein recruitment to the PCM, the PCM may in turn regulate and contribute components to nascent centrioles. Centrosomes experience dramatic changes during the cell cycle. They double in size from S phase to mitosis, primarily through the accrual of PCM. Yet until recently little was know about the factors that control this behavior. Since the work of Bobinec et al. (102), it has been known that the PCM is under the control of centrioles. Centriole tubulins are constitutively highly glutamylated, which render them highly stable (55). Injection of anti-glutamylated α-tubulin antibodies leads to disassembly of centrioles, and remarkably, dissipation of the PCM (102). This process is reversible as cells spontaneously reassemble centrioles de novo, which is followed by recruitment of PCM. One centriole component in particular, Sas-4 (human CPAP), which is a component of the proximal centriole and of the complex that initiates template-dependent procentriole growth (103, 104), plays a critical role in PCM recruitment (90, 105). Defects in Sas-4 lead to abnormal centrioles and defects in PCM recruitment. Centrosomal CPAP continuously exchanges with a cytoplasmic CPAP pool (106), which is highest in G2 when recruitment of proteins to the PCM is maximal. Overexpression of CPAP results in overly long centrioles and defective PCM leading to abnormal cell division (98, 107). Expression via their own promoters of mutant forms of Sas-4 lacking the conserved PN2–3 region or defective in tubulin binding, impair (90) or promote (105), respectively, PCM recruiting, without affecting centriole duplication. These separation-of-function mutations highlight how different regions of Sas-4 critically control centriologenesis and PCM recruiting through independent domains.
The Centrosome Cycle
Centrosome replication bears remarkable similarities to DNA replication. Both are semiconservative and controlled by successive waves of cyclin E-Cdk2 and cyclin A-Cdk2 activity (19, 108–112) (Figure 3). Both occur during discrete phases of the cell cycle and both rely on licensing mechanisms for tight control and prevention of re-replication during a single cell cycle (113–117). There is general agreement that centriole disengagement – the disorientation and physical separation of centrioles at their proximal ends at the end of mitosis – is a critical early step in licensing centrosomes for replication in S phase. Lack of disengagement blocks centrosome duplication in S phase (118). Centriole disengagement, which occurs late in mitosis, is blocked by non-degradable forms of securin (119) or cyclin B1 (120), both of which block the proteolytic activity of separase, directly implicating proteolysis in this process (119, 120). Indeed, recent evidence suggests a mechanism analogous to the one operating on sister chromatid separation at anaphase, in which cohesin – the tripartite ring-like protein complex composed of Scc1, Smc1, and Smc3 – at centromeres is cleaved by separase-mediated proteolysis of Scc1. Presumably, cohesin complexes localized at the junction of engaged centrioles are proteolyzed in an analogous manner. Ectopic expression of separase, or depletion of Sgo1 – a protein that protects cohesin from separase – leads to unscheduled chromosome separation and centriole disengagement (121, 122) supporting this proposal. Nevertheless, disengagement occurs much later than sister chromatid splitting raising questions about cohesin as the exclusive target. Moreover, recent experimental work in Drosophila indicates that cohesin cleavage may be insufficient for centriole disengagement (123), suggesting that additional targets may exist. Two recent papers provide evidence that the relevant second substrate may be pericentrin B (PCNTB)/kendrin (124, 125). Both studies demonstrated that PCNTB is cleaved by separase at a consensus site (R2231) releasing a rapidly degraded C-terminal fragment and retaining at the centrosome the N-terminal fragment until late in mitosis. Importantly, release of the N-terminal fragment coincided with an abrupt decrease of PCNTB at the centrosome and with centriole disengagement at the end of mitosis (125). Expression of a separase cleavage-resistant form of PCNTB blocked centriole disengagement and replication, proving in vivo proof of its physiological relevance (124, 125). Nevertheless, the critical licensing molecular event(s) that renders a disengaged centriole competent for replication has not been elucidated.
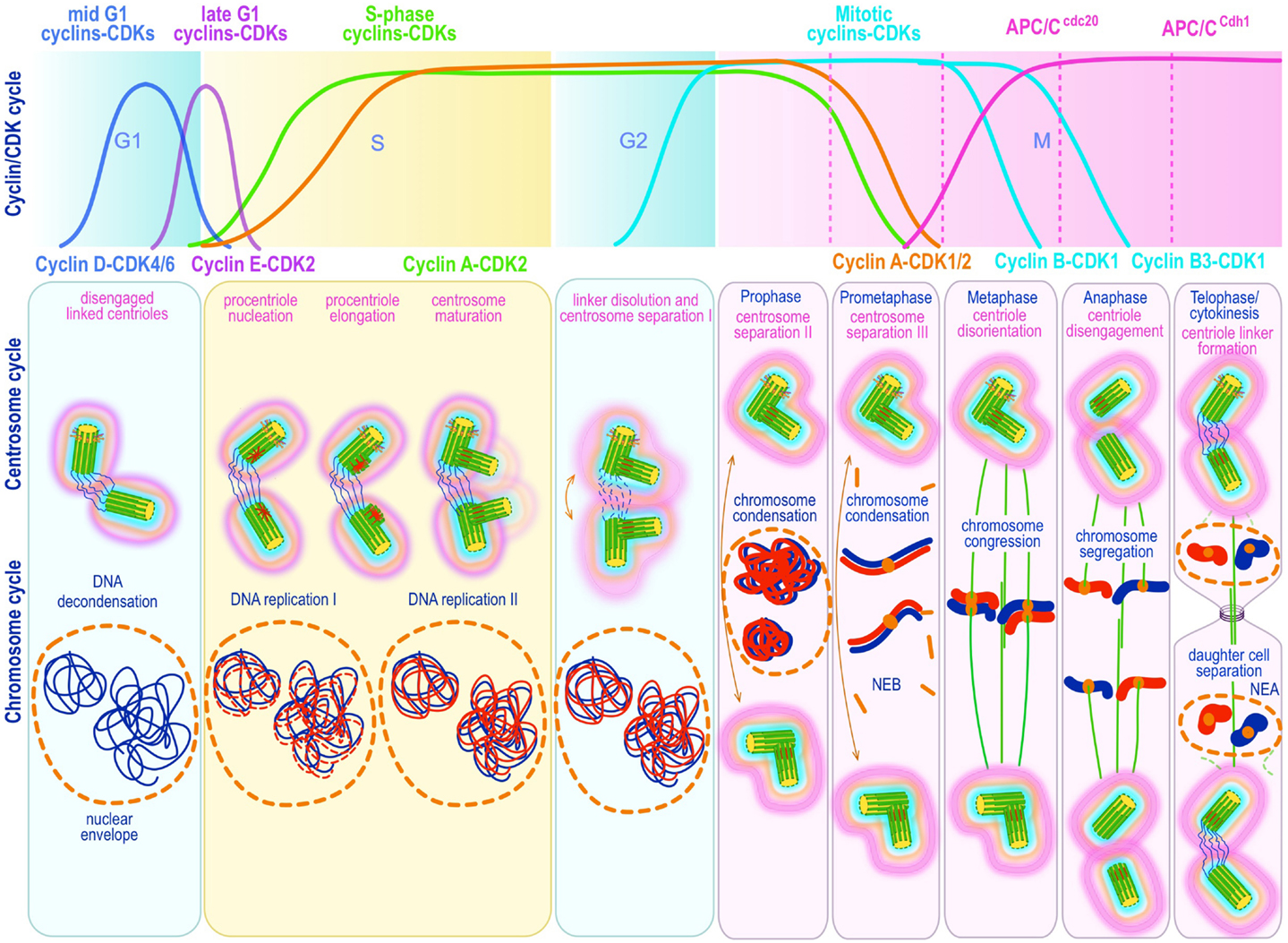
Figure 3. Canonical template-dependent centrosome duplication cycle. Normal centrosome duplication begins with centriole disengagement at the end of mitosis (anaphase-telophase). During S phase under influence of cyclin E/A-Cdk2 a procentriole is nucleated on each parental centriole, which elongate until in late S or G2 capping proteins suppress further growth. At the G2/M transition, the fibers that connect parental centrioles are dissolved while centrosomes begin to separate and mature further (acquire a full complement of PCM proteins). During prophase centrosomes continue to migrate apart until they reach opposite sides of the condensing chromosomes at metaphase and organize a bipolar spindle in preparation for chromosome segregation. The nuclear (DNA/chromosome) and CDK cycles are shown for correlation and comparison.
Raising levels of cyclin A/E-Cdk2 are necessary to trigger centriole duplication (109–112), yet the initial centrosomal molecular targets of cyclin-Cdk2 that initiate/license centrosome duplication have not been defined with absolute certainty. Several candidate proteins have ben postulated to fulfill the licensing factor role. Among these, nucleophosmin (NPM1) (126–131), Mps1 (132–139), and Polo-like kinases (Plk1, 2, and 4) (118, 127, 140) figure most prominently.
The first postulated critical target of Cdk2 in triggering centrosome duplication was nucleophosmin (NPM1/B23), which is a multifunctional chaperone protein – a large fraction of which localizes to the nucleolus. NPM1 rapidly shuttles between nucleus and cytoplasm (141) and associates with the centrosome. Inactivation of NPM1 leads to unrestricted centrosome replication (142) indicating that one of its functions is to restrain centrosome duplication. Early work by Fukasawa and collaborators showed that NPM1 is one of the most conspicuous targets of Cdk2 in unduplicated centrosomes and that phosphorylation of NPM1 on T199 by Cdk2-cyclin E leads to its dissociation from centrosomes (129, 130). Careful immunofluorescence with an antibody specific to centrosomal NPM1 showed it to localize between the centrioles in the unduplicated centrosome, to dissociate from centrosomes upon its phosphorylation, and to re-associates with the centrosome during mitosis (129, 143, 144). Based on these observations a centrosome duplication licensing role for NPM1 has been proposed (129). Phosphorylation of T199 and S4, have both been postulated as critical events for NPM1 downstream functions. Both Cdk2 and Plk1 phosphorylate T199 creating high-affinity docking site for the kinase ROCK-II, which becomes “hyperactivated” upon binding NPM1 (145). Activated ROCK-II, reportedly bypasses the Cdk2 requirement for centrosome replication (145). But NPM1 is also phosphorylated at S4 by Plk1 during mitosis (146) and by Plk-2 near the G1/S transition (147, 148). Plk1 S4 phosphorylation in coordination with separase contributes to centriole disengagement (118), while Plk-2 S4 phosphorylation has been proposed to be the trigger for centrosome replication (127), based on the fact that NPM1 S4A – a non-phosphorylatable form of NPM1 – blocks centrosome duplication, while phosphomimetic mutants have the opposite effect, i.e., centrosome overreplication (127). Clearly, further experimentation will be required to precisely map the role of NPM1 in centrosome duplication.
Mps1 (Esk) (132, 133, 136, 149) overexpression in S-arrested mouse cells leads to centrosome overduplication, while a kinase-dead form blocks centrosome duplication (133). Similarly, overexpression of a dominant negative form of Msp1 in human cells blocks centrosome duplication, while active Mps1 overexpression accelerates centrosome duplication (132). Notably, siRNA Mps1 knock-down blocks centrosome duplication and in addition cause pleiotropic defects resulting in severe mitotic abnormalities, attesting to Mps1 regulatory function in many mitotic processes, in particular on the spindle assembly checkpoint (132). Mps1 normally is under negative regulation by proteosomal destruction. Preventing the degradation of Mps1 by transient exposure to proteasome inhibition is sufficient to cause centrosome reduplication in human cells (136). Phosphorylation of Mps1 T468 appears to be the critical target of Cdk2 leading to proteasome resistance and Mps1 accumulation, since phosphomimetic mutations of T468 prevent Mps1 degradation and lead to Cdk2 independent centrosome duplication (136). This process seems to be more nuanced in human cells. Early studies showed that hMps1 was only detectable at kinetochores but not at centrosomes, and that neither overexpression of hMps1, kinase-dead hMps1, or siRNA knock-down of endogenous hMps1 in human cells revealed a centrosome phenotype, casting doubts into an hMps1 role in centrosome duplication (138). Nevertheless, GFP-hMps1T468A, a non-phosphorylatable form of hMps1, accumulates in the cytoplasm but is continuously removed from centrosomes in a proteasome-dependent manner (136), suggesting that it is the centrosomal pool of hMps1 the one relevant for the hMps1 centrosome phenotype. In contrast hMps1T468D and hMps1T468E, mutants that mimic T468 phosphorylation, and hMps1delta12/13 that lacks an Mps1 degradation signal (MDS), readily cause centrosome reduplication, even in the absence of cyclin A-Cdk activity. Accordingly, failure of wild type hMps1 overexpression to cause centrosome re-duplication appears to be the consequence of its efficient proteasome-dependent removal from the centrosome (137). The exquisite control of centrosomal hMps1 appears to depend largely on its MDS signal. Yet the MDS signal bears no resemblance to known targeting motifs for SCF or APC/C type E3-ubiquiting ligases. Emerging evidence suggest that hMps1 degradation may be controlled by ornithine decarboxylase antizyme (OAZ) (135). OAZ target substrates for ubiquitin-independent proteasome-mediated degradation (150). It has been demonstrated that OAZ localizes to the centrosome and that its activity suppresses centrosome re-duplication, while reducing OAZ at the centrosome leads to centrosome re-duplication (151). Indeed OAZ binds hMpsi1 through its MDS motif and leads to its degradation through proteasome-dependent proteolysis. Yet one additional mechanisms control the level of hMps1. A single N-terminal D-box makes hMps1 a target for APC/C-dependent degradation during mitosis, an activity that is controlled by Cdc20 and Cdh1 (152). It is possible that this second hMps1 degradation mechanism is used at the end of mitosis to rapidly reset hMps1 to low levels before reaching the G1/S transition, where the finer regulation effected by OAZ takes over (137).
Polo-like kinases have also been considered key factors in centrosome replication, possibly playing a licensing role as well. It has long being known that in addition to separase, Plk1 activity is required for centriole disengagement at the end of mitosis (118). Moreover, the recently described phosphorylation of NPM1 S4 has been proposed as a centriole duplication trigger (127). Additionally, new procentrioles require a Plk1 dependent modification, which can only occur through mitotic passage, when Plk1 activity is high. This prevents the growth of “granddaughter” centrioles, i.e., procentrioles growing from daughter centriole walls within the same cell cycle (153, 154). Plk-4 has also received considerable attention as a potential centrosome replication licensing factor (155–157) [reviewed in (140, 158)]. Plk-4 activity peaks only transiently during mitosis and is kept at very low levels during interphase (156, 159) by autoregulatory self-destruction. Plk-4 homdimerizes and autophosphorylates in trans, which triggers rapid SCFlimb E3 ubiquitination and proteasome directed proteolysis (158–162) enforcing low activity levels through most of the cell cycle. Plk-4 peak activity in mitosis is due to its interaction with Twins – the regulatory subunit of protein phosphatase 2A (PP2A), which in complex with PP2A dephosphorylates Plk-4, briefly spearing it from proteolysis (156). Plk-4/Sak activity is required in the earliest steps of procentriole formation (97, 155, 157, 163), and is recruited to the centrosome together with Cep152 (homolog of D. melanogaster Asterless) through interactions with Cep192 (164) (homolog of C. elegans Spd-2). The complex localizes to the outer surface of the proximal end of the mother centriole, precisely the site from which the procentriole will sprout (97). Thus, Plk-4 satisfies most criteria for a licensing factor for centriole duplication (116): Plk-4 levels peak only once during the cell cycle in mitosis; Plk-4 is kept low during all other stages of the cell cycle by autoregulatory phosphorylation-triggered proteolysis; increasing levels of Plk-4 experimentally leads to centriole reduplication; and, decreasing Plk-4 or suppressing its activity prevents centriole duplication (165).
Centrosome replication begins at the G1/S transition with nucleation of a procentriole at the base of the parental centrioles (108, 166–169) [reviewed in Ref. (170–172)]. Of these Plk-4 (97), Cep152 (97), and SAS-6 (172) initiates the process, with Plk-4 participating as the dominant kinase and regulator of the early steps of centriole duplication. By superresolution microscopy, Plk-4 initially localizes to a single spot within the toroid defined by N-terminal Cep152 at the proximal end of the mother centriole, but outside the one defined by C-terminal Cep152, which is consistent with the known interaction between Plk-4 and the N-terminal Cep152 (173–175). Interestingly, Plk-4 is detectable in G1 cells at the spot on the mother centriole from which later Sas-6 will initiate cartwheel assembly formation. This observation suggest that Plk-4 determines the site of initiation of procentriole formation.
SAS-4 may be important in recruiting Cep152, a PCM protein that participates in nucleation of procentrioles (164, 176). The interaction of Cep152 with Sas-4/CPAP may provide the initial scaffold for procentriole formation (147, 174). Phosphorylation of the F-box SCFFbxw5 E3-ubiquitin ligase by Plk-4 appears to stabilize SAS-6, a natural substrate of SCFFbxw5, initiating procentriole growth (177) (Figure 3). The first structure to appear at the site of procentriole formation, even before microtubules nucleation is apparent, is the cartwheel assembly. Mutations in cartwheel constituents such as SAS-6 – the central component of the cartwheel driving the establishment and maintenance of the ninefold symmetry of the centriole (178, 179) – lead to absence or severely defective centrioles. In human cells the first protein known to localize to the procentriole is indeed HsSAS-6 (180). SAS-6 molecules have conserved amino-terminal domains, followed by coil–coil domains capped by poorly conserved carboxy-terminal domains. SAS-6 homodimerizes in parallel via the coil-coil domains resulting in a rod-like structure in which the conserved globular amino-terminal domains are located next to each other at one end of the rod-like dimer. Interactions of one of these domains with a similar domain in a second homodimer leads to the progressive assembly of the cartwheel, with the globular amino-terminal domains constituting the central hub, and the coil–coil homodimers the centrifugally radiating spokes of the cartwheel assembly (181, 182) (Figure 2). The self-assembly of the cartwheel starting from homodimers of SAS-6 is a remarkable effective biological organizing principle that satisfies the need of ninefold symmetry in the simplest possible manner (182–184). SAS-6 is known to interact with SAS-5/Ana2/STIL, but the nature and consequences of the interaction are not well defined. It has ben proposed that SAS-5/Ana2/STIL interacts with SAS-6 through a STAN motif, stabilizing the procentriole cartwheel structure (185–188). Overexpressed SAS-6 and Ana2 in flies co-assemble into long cartwheel structures closely resembling the natural structure (188). Two safety mechanisms are put in place to avoid overreplicating the cartwheel structure. During S and G2 phases the SCFFbxw5 E3-ubiquitin ligase is inhibited by Plk-4 mediated phosphorylation. As the cell cycle progresses Plk-4 autophosphorylates, triggering its own proteolysis, and relieving the inhibition of SCFFbxw5, which then ubiquitinylates Sas-6 triggering its degradation (177). A second safety check is active in mitosis where Sas-6 is ubiquitinylated by APCCcdh1, which targets it for proteolysis ensuring low levels of Sas-6 throughout mitosis (180). This tandem safety mechanism prevents re-initiation of centriole duplication once a daughter centriole has emerged.
Superresolution microscopy tracing the localization in S/G2 cells of Sas-6, Cep135 and STIL, three proteins involved in template-dependent procentriole formation, shows that Sas-6 and STIL co-localize precisely (189–191), whereas Cep135 localizes away from Sas-6/STIL in a position similar to C-Nap1, a marker of the proximal end of mother centriole (170). In late G2 and M phase cells, however, Cep135 staining could be seen to extend into the area occupied by Sas-6/STIL indicating that Cep135 progressively associates with the proximal end of growing daughter centrioles. Both Sas-6 and STIL are degraded upon exit from mitosis (180, 189–191) and are no longer detectable in the centrioles from early G1 cells. Unresolved still are the mechanism that propagate the cartwheel at the proximal end of the centriole, which could occur by deposition of cartwheel structures in layers or by a helical mechanism resembling the bristles of a bottle brush, the mechanisms that limits the growth of the cartwheel, and the interacting proteins that regulate the self-assembly and precisely control the angle of interacting SAS-6 homodimers to ensure ninefold symmetry. The initial structure is stabilized further by SAS-4/CPAP, which also plays an important role in recruiting microtubules to the perimeter of the growing cartwheel structure (90, 103, 104, 107).
Microtubules are next added to the cartwheel in an orderly fashion with nucleation of the A microtubule initiated at a cone-like structure attached to the distal carboxy-terminal end of the radiating SAS-6 spokes and proceeding unidirectionally to the distal end of the nascent centriole (192). The cone-like structure may contain γ-tubulin and its interacting partner NEDD1, because depletion of these components prevents centriole growth (72). The B and C microtubules apparently require δ-tubulin and ϵ-tubulin (193, 194) and appear to polymerize bi-directionally. Additional proteins participate in the regulation and addition of microtubules to the emerging centriole. Amongst these CPAP and STIL form a complex with SAS-6 and are likely to contribute to microtubule addition (190). CPAP localization to the procentriole is dependent on phosphorylation by Plk-2 (195). Procentrioles, securely attached to their mothers, grow by elongation through G1, S, and G2 phases (Figure 3). Centriole elongation and final length are specifically controlled, and are characteristic of a species and cell type. There appears to be distinct proximal and distal elongation steps that are independently regulated. Distal growth may be regulated at least partially by the centrin-binding protein hPOC5 (proteome of centriole 5) since its depletion prevent distal but not proximal elongation (196). Conversely, CPAP, CP110, and POC1 may control proximal elongation. Overexpression of CPAP (98, 107, 197) or POC1 (198), or depletion of CP110 (98) leads to unusually long daughter centrioles. Conversely, depletion of POC1 (198) or overexpression of CP110, prevent procentriole elongation and leads to centriole overreplication (98). siRNA depletion of Cyclin F, which interacts with CP110, leads to CP110 overexpression and centrosome overreplication (46). Cyclin F is part of the ubiquitin ligase SCFcyclinF, which targets substrates for proteasome degradation. A recently described deubiquitinating enzyme named USP33 (199) specifically interacts with CP110 bound to centrioles in late S and G2/M cells, protecting it from SCFcyclinF. Depletion of USP33 leads to decreased levels of CP110 (199) and centriole elongation. Remarkably, no other centrosome SCFcyclinF substrate is also a substrate of USP33, attesting to the importance of fine-tuning the levels of CP110 in centriole replication (199). The process is completed with the distal binding of the capping proteins CP110 and Cep97, which prevent further growth of the procentriole, determining its final length (97, 98, 200, 201) (Figure 2).
Initially devoid of PCM, the daughter centriole rapidly matures, i.e., acquires PCM and the increased ability to nucleate microtubules, by the end of G2 and in mitosis, through the concerted actions of the kinases Aurora-A and Plk1 which reach high levels at the centrosome at the end of G2 (202–204). In a manner highly reminiscent of its priming function in DNA replication (205, 206), Plk1 induced centrosome maturation is essential for priming procentriole nucleation in the next cell cycle (118, 153, 154).
At the G2/M transition in a process termed centrosome disjunction, centrosomes begin to separate by dissolving the linker protein fibers connecting the proximal ends of the two parental centrioles, which were established previously around the time of centriole disengagement in late M or early G1 (207) (Figure 3). Dissolution of the linker fibers is dependent on Nek2 activity. Nek2 is a NIMA-related kinase that accumulates at the centrosome through S and G2 phases. At the G2/M transition it triggers dissolution of the intercentriolar linker. The importance of Nek2 in this process is illustrated by changing Nek2 levels at the centrosome. Overexpression of Nek2 leads to premature separation of centrosomes (170) while knock down with siRNA inhibits centrosome separation (208). Two main protein components of the linker fibers, C-Nap1 and rootletin, are phosphorylated by Nek2A (170, 209), promoting their migration onto the fibers. While rootletin appears to distribute uniformly throughout the fibers the Nek2 and C-Nap1 localize mostly to the proximal ends of the mother centriole (170) suggesting that they are docking sites for rootletin (209) (Figures 2 and 3). The levels of rootletin itself may control the length of the fibers. Overexpressing this protein lengthens the fibers (210, 211), while depleting it results in premature centrosome separation (212). Two additional putative linker proteins, Cep68 and Cep215 (CDK5RAP2), have been described, the former being a bona fide substrate of Nek2A and the latter possibly of Plk1 (213). By high-resolution fluorescence/deconvolution microscopy and immune-EM, Cep68 is seen to form fibers that attach to the proximal end of mother centrioles, while Cep215 instead, tightly surrounds the mother centriole (213). siRNA knockdown of either induces premature centrosome separation (213) implicating them in centrosome cohesion. However, overexpression of Cep68 does not induce fiber formation by itself but is readily recruited to rootletin fibers (213) indicating that Cep68 cooperates with rootletin and C-Nap1 in centrosome cohesion. Cep215 neither distributes to the linker, nor interacts with rootletin or C-Nap1, indicating that it does not represent a bona fide linker protein. Instead, its centrosome cohesion promoting function may be related to its interaction with pericentrin (213). Since Nek2 activation promotes centrosome separation by evicting linker components, it needs to be tightly regulated to prevent premature separation of centrosomes. Nek2 becomes activated by homodimerization through its coil–coil motifs, which facilitates autophosphorylation of the catalytic domain (214, 215). Further insight into Nek2 regulation has come from the discovery of KVHF motifs in its non-catalytical C-terminal domain. KVHF motifs are consensus sequences for the binding of Protein phosphatase 1 (PP1). PP1a dephosphorylates the catalytic domain of Nek2 and inactivates it. Notably PP1 itself can be inactivated by Nek2 mediated phosphorylation (214, 215). This on-off bi-stable switch is characteristic of many of the regulators that control abrupt mitotic transitions. The balance of this metastable switch appears to be tipped by the Inhibitor-2 protein (216), which binds and inhibits PP1 leading to runaway auto activation of Nek2.
Upstream regulators of the cell cycle directly control timely activation of Nek2. The main function of Aurora-A in centrosome separation is activation of Plk1 kinase by phosphorylating T120 in the T loop (217–219). Activated Plk1 phosphorylates and binds Mst2 kinase, which can now bind Nek2A and PP1γ (215, 220). Unlike PP1α, PP1γ antagonizes Nek2, not by direct dephosphorylation, but by dephosphorylating C-Nap1, its binding partner. The level of Plk1 phosphorylated Mst2 ultimately determines the dissociation of the Mst2-Nek2A-PPIγ complex (221) with increasing phosphorylated Mst2 leading to a reduction in PP1γ on the complex. Thus, phosphorylation of Mst2 by Plk1 leads to a reduction in the levels of PP1γ in the Mst2-Nek2A-PP1γ complex resulting in increased Nek2 dependent phosphorylation of C-Nap1 and dissolution of centrosome linker fibers (221). This Hippo dependent increase in Nek2 activity is counteracted by pericentrin and HEF1, the latter a focal adhesion scaffold protein. While pericentrin is an inhibitor of Nek2 kinase activity, HEF1 inhibits accumulation of Nek2 at the centrosome (222). siRNA knockdown of pericentrin causes premature separation of centrosomes in interphase. It has been proposed that pericentrin changes the structural conformation of Nek2 catalytic domain into an inhibitory conformation (223). Pericentrin and Cep125 localization to the centrosome also depends on Plk1 activity suggesting an additional level of regulation, either indirectly through Nek2A or by direct phosphorylation of some proteins involved in centrosome cohesion (213, 224).
After dissolution of the centriole linker, motor proteins bind anti-parallel astral microtubules and exert their sliding forces by walking toward MT plus ends (Figure 3). The kinesin Eg5 is the principal force generator for centrosome separation at this stage (225–229). Eg5 is a homotetrameric plus-end directed motor belonging to the kinesin-5 subfamily (230, 231). Knockdown of Eg5, by siRNA or chemical inhibition with monastrol, arrest cells in prometaphase with monopolar spindles (232, 233). Mainly cytosolic during interphase, Eg5 rapidly accumulates at spindle poles in prophase (234). There is evidence that Plk1 phosphorylation of Eg5 targets it to the spindle poles (235–237). Inhibition of Plk1 prevents accumulation of Eg5 at the centrosome, but does not change the overall level of cytoplasmic Plk1 (236). Cdk1 phosphorylates Eg5 at T927 (238). Plk1 can substitute for Cdk1. However, Plk1 phosphorylated Eg5 triggers slow and erratic centrosome separation, while Cdk1 triggers fast movement. The difference in centrosome behavior under these conditions has been attributed to differential modulation of microtubule dynamics by Cdk1 and Plk1 (237). Plk1 induced accumulation of Eg5 at the centrosome is microtubule dependent and is abolished in the presence of nocodazole, a standard MT depolymerizing agent (236). The effect of Plk1 on Eg5 may indeed be associated with the ability of Plk1 to increase the capacity of centrosomes to nucleate MT (204). Other members of the NIMA kinase family also participate in Eg5 regulation. There is experimental evidence that suggest phosphorylation of S1033 in Eg5 by Nek6 is the critical event that targets Eg5 to the centrosome after Plk1 activation (235). Tellingly, mutations in Eg5 that prevent S1033 phosphorylation abolish Eg5 localization to the centrosome (235). Moreover, differential regulation of Eg5 targeting to the centrosome, before and after NEB, appears to exist (239). Indeed, recent experimental evidence seems to validate this proposal (240). It has long been known that there are two pathways to mitotic spindle assembly. A prophase pathway occurring entirely before NEB, and a back-up pathway that occur in prometaphase, after NEB (241–246), reviewed in Ref. (225, 228). Importantly, the back-up prometaphase pathway is mechanistically more complicated and more likely to lead to chromosome segregation errors than the prophase pathway (247). Moreover, the two pathways are temporally, spatially and genetically distinct (240). These differences may be important in that the probability of a mitosis generating abnormally attached chromosomes depends on avoiding kinetochore MT from the two spindle poles contacting the same kinetochore. In the prometaphase pathway centrosomes are incompletely separated when astral microtubules can first contact kinetochores after NEB, increasing the chances of chromosomes with merothelic attachments to the spindle poles. This has indeed been demonstrated in PtK1 cells (247). In conclusion, it appears that whether a cell uses the prophase or the prometaphase centrosome separation and spindle assembly pathways, may have important consequences in the form of increased CIN in the latter. Incompletely separated centrosomes represent a mitotic liability that may translate into CIN. Nevertheless, the potential contribution of abnormal centrosome separation to CIN in cancer has not been directly studied.
A recent detailed in vivo imaging study in Eg5-mEGFP/mCherry-α-tubulin in HeLa cells in which the kinetics of centrosome separation could be precisely followed and cells indexed as either prophase or prometaphase centrosome separation pathway users, revealed interesting results (240). Cells using the “back-up” prometaphase pathway had longer lags, and lower velocity and a shorter centrosome translocation times. Of 1,388 mitotic events studied, half used the prophase centrosome separation pathway, and the other half used the prometaphase pathway (240). Whereas 0.7% of cell utilizing the prophase pathway had lagging chromosomes at metaphase (a surrogate of CIN), as many a 2.3% cells using the prometaphase pathway exhibited the same phenotype (240). It would be of great interest to repeat this experiments in immortalized non-transformed cells, since HeLa were derived from a uterine cervix carcinoma and exhibits measurable CIN under standard growth conditions [see Ref. (248) and references therein] and also to study this phenomenon in additional cancer cells. During metaphase centrosomes complete maturation achieving maximal microtubule-organizing capacity, and together with kinetochores, assemble the mitotic spindle. Late in mitosis (anaphase, telophase) daughter centrioles disengage from their mothers reinitiating he centrosome replication once again.
Abnormal Centrosomes in Cancer
Theodore Boveri, who co-discovered the centrosome, was the first to propose that centrosomes may induce CIN, which could lead to cancer (6). Boveri’s cancer development theory was singularly influenced by observations made by his colleague Leo Hansemann. Hansemann had observed abnormal mitoses in cancer tissue and had compiled beautiful renderings of their salient features (4, 5) (vide supra), which to Boveri’s trained embryologist eye, immediately suggested the work of abnormal centrosomes. Boveri theorized that a peculiar “combination of genetic determinants” may give rise to cancer and the transmission of that peculiar combination of genetic determinants (i.e., genes) may occasionally arise in daughter cells of abnormal multipolar mitoses. Boveri reasoned that the degree of centrosome abnormalities he inferred from Hansemann’s drawings, were probably detrimental as a whole, as he had directly observed in doubly fertilized sea-urchin zygotes (249). He theorized that only cells exhibiting lesser degrees of centrosome malfunction could propagate the cancer phenotype, which he ascribed directly to the genetic material (6).
Abnormal centrosomes in cancer were initially detected in some of the most common human cancers including breast, colon, lung, and brain cancer (250, 251). These findings were quickly confirmed and expanded by others (252–263). A century after Boveri and Hansemann original observations, we can affirm that centrosome defects are indeed pervasive in cancer. Excepting leukemias and some low-grade lymphomas, most carcinomas and sarcomas, and a subset of high-grade lymphoma [for a survey of clinical cancer types, see Ref. (264)] exhibit abnormal centrosomes. Furthermore, it is now well established that centrosome abnormalities in cancer correlate closely with and are an important cause of CIN (251) [for a review, see Ref. (265–268)], and that centrosome abnormalities and CIN frequently co-occur in carcinoma in situ (269, 270). These observations place centrosome abnormalities at the earliest stages of cancer development and argue against a purely secondary role or late effect. Nevertheless, despite their common occurrence, and perhaps due to the heterogeneity of centrosome abnormalities in cancer, it has been difficult to determine whether centrosomes abnormalities are caused by primary intrinsic centrosome defects, or are the consequence of dysfunction of other cellular processes that lead to the accumulation of normally replicated centrosomes, such as for instance in cases of cell division failure (271, 272). In consequence a considerable volume of ongoing research is being devoted to elucidating in detail the molecular pathways involved in centrosome dysfunction in cancer, their impact on the cancer genome, and the prospects of utilizing centrosome defects as biomarkers (205, 273, 274) and targets for cancer specific therapy (275–279).
The Spectrum of Centrosome Abnormalities in Cancer
Centrosome phenotypes in cancer are heterogeneous with both numerical and structural abnormalities documented. Nevertheless, attempts at classification of centrosome abnormalities in cancer have met with limited success, primarily because of the difficulty inherent in carrying out comprehensive surveys at the ultrastructural level using EM, which until recently has been the only technology capable of visualizing centrioles and PCM with sufficient detail (280–283). The emergence of superresolution microscopy promises to drastically change the status quo. Structured illumination microscopy is already providing images of whole centrosomes with unprecedented resolution and is poised to contribute greatly to our understanding of centrosome phenotypes in cancer in the immediate future (85) (vide supra).
The most evident and widely documented centrosome cancer phenotype is supernumerary centrosomes (250, 251) (Figure 4). In principle, supernumerary centrosomes may result from at least three separate mechanisms: template-mediated over-replication of pre-existing centrosomes within one cell cycle (hereby termed the over-replication pathway), a phenotype that has been variably referred to as centrosome amplification (254, 263, 284–286) or hyperamplification (252, 255, 287, 288), de novo formation during interphase (de novo pathway) (289, 290) (Figure 4) or from accumulation of normally (or abnormally) replicated centrosomes due to failed cell division after replication of centrosomes and chromosomes has occurred (accumulation pathway) (291, 292) (Figure 4) [reviewed in Ref. (293, 294)]. While in the latter the normal numerical relationship of one centrosome per diploid chromosome set in G1 phase is maintained, in the former two pathways it is halved or worse. This difference profoundly affects the chances of daughter cell survival in cells carrying multipolar mitoses to completion, since daughter cell viability is predicated on receiving at least a full haploid set of chromosomes. This in fact is one of the seminal experimental observations made by Boveri in dispermic see urchin eggs, which allowed him to infer that chromosomes are not interchangeable and therefore must carry different genetic determinants (249).
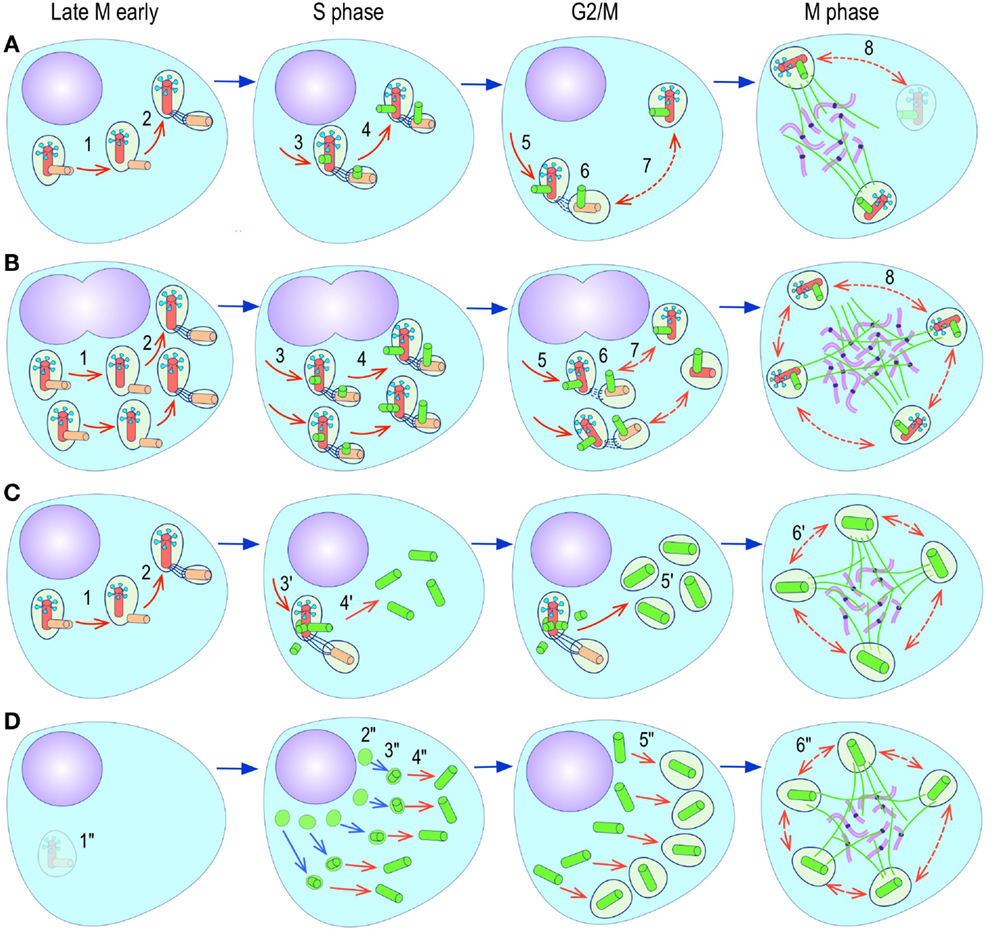
Figure 4. Pathways to supernumerary centrosomes in cancer. Canonical template-dependent centrosome replication pathway (A). Normal centrosome duplication proceeds sequentially in the following steps: centriole disengagement (1), linker fiber development (2), procentriole nucleation (3), centriole elongation (4), linker dissolution (5), centrosome maturation (6) and separation, before (7) and after (8) NEB. There are at least three pathways to supernumerary centrosomes in cancer (B–D). Centrosome accumulation pathway due to polyploidization events (B). Events such as cytokinesis failure, mitotic slippage (mitotic failure before cytokinesis), etc., with or without normal DNA replication, result in accumulation of normally replicated centrosomes, which execute all stages of replication as in the canonical template-dependent pathway A. Centrosome over-replication pathway (C). Some cancer cells, particularly if arrested in S or G2 phases, such as during DNA replication stress induced by hypoxia, chemotherapy or radiation therapy, undergo multiple rounds of templated centriole duplication (3′), which subsequently elongate (4′) and mature (5′) leading to functional centrosomes capable of enacting multipolar mitoses (6′). De novo centriole formation pathway (D). Under similar conditions certain cancer cells, even when containing resident centrosome, build new centrioles de novo, via centriole satellites (2′′). Once synthesized such centrioles (3′′) can elongate (4′′), mature by acquiring normal mitotic PCM (5′′), and become competent at mitosis (6′′) usually enacting multipolar spindles. In subsequent cell divisions, de novo centrosomes are thought to replicate via the canonical template-dependent pathway.
It is likely that only one of these three centrosome amplification pathways operates in an individual tumor. Nevertheless, there is precedent for pathway cooperation. Using a marker for the mother centriole, Duensing et al. were able to determine that the papillomavirus oncoprotein HPV16-E7 leads to over-replication of centrosomes in G2, while HPV16-E6 leads to both increased centrosomes and increased ploidy (295), by a mechanism that apparently involves cytokinesis failure.
In vivo Centrosome Biogenesis in Cancer and Non-Cancer Cells
In a series of seminal observations that rekindled interest in centrosome biology in cancer, Fukasawa et al were the first to note that p53 null mouse embryo fibroblasts (MEFs) in culture frequently acquire supernumerary centrosomes (285). The supernumerary centrosome phenotype of p53 null MEFs was exacerbated by forced overexpressing of Cyclin E, a cyclin known to promote centrosome replication (288). Furthermore in an assay that measures the entrainment of centrosome replication to the DNA replication cycle by blocking cells at the G1-S boundary with a DNA replication inhibitor, the authors could demonstrate that in p53 wild type cells, only one round of centrosome replication occurred, while in p53 null MEFs, and to a lesser extent in Waf1 null MEFs, centrosome continued to replicate several times (296) (centrosome over-replication pathway). Furthermore, restoring p53 to p53−/− MEFs restored normal centrosome replication control. These experiments clearly demonstrated that p53 played a major role in centrosome homeostasis, and that prevention of centrosome over replication was clearly dependent on an intact p53-Waf1 axis (296). These experiments however did not offer a glimpse as to the actual pathway leading to supernumerary centrosomes in continuously proliferating p53 deficient MEFs. More direct evidence for a plausible mechanisms of supernumerary centrosomes in cycling p53 null cells was obtained by overexpressing a number of mitotic kinases (Aurora-A, polo-like kinase 1 (PLK1), Aurora-B, or Aurora-C kinases) in a p53 wild or null context. High level of Aurora-A or Plk1 lead to supernumerary centrosomes via defects in cell division resulting in tetraploidization and centrosome accumulation (292, 297) (Figure 4). These results suggested that the supernumerary centrosome phenotype of p53 null cells documented by Fukasawa et al (285) in normally dividing MEFs was most likely due to polyploidization and accumulation of centrosomes rather than over replication (292). However, it is important to make clear from the outset that cell division failure, as a cause of centrosome amplification is only a viable tumorigenic mechanism insofar as it may be intermittent and stochastic, occurring only in a minority of cell divisions. Sustained division failure leads to exponential chromosome and centrosome accumulation with giant cell formation, which is detrimental to tumor growth. Accumulation of centrosomes and subsequent multipolar spindles is an attractive explanation for supernumerary centrosomes in p53 null MEFs, because it is consistent with one of the known checkpoint functions of p53, which is to impose a G1 arrest in response to tetraploidization (298–300). Absence of this p53 checkpoint explained why p53 null MEFs with extra centrosomes continued to divide and enact multipolar spindles. Moreover, these observations fit in well with the transient tetraploid state know to occur in many cancers before aneuploidy ensues (301).
Although tetraploidy and centrosome accumulation may be a prevalent pathway to supernumerary centrosomes in cancer, there is precedent for centrosome over-replication in especial situations. For instance, overexpression of PLK-4 in Hela cells arrested in S phase with aphidicolin leads to over replication of centrosomes, with multiple procentrioles attached to the mother centriole (97, 157). A similar phenotype can be induced by overexpression of other core components of the template-mediated centriole replication pathway, such as HsSAS-6 (106, 189–191, 302). However, even non-cancer cells may respond differentially to perturbations in the centrosome replication pathway depending on tissue of origin or differentiation. For instance, overexpression of DSas-6 in Drosophila leads, within on cell cycle, to a fraction of centrosome undergoing template-dependent over replication in syncytial embryo and somatic brain cells, de novo assembly of multiple centrosomes in eggs, and no abnormality in spermatocytes (303). These studies reinforce the concept that normal cells use the de novo assembly pathway only in the absence of resident centrosomes such as it exist in eggs. The need for tight control of the core proteins involved in centriole duplication and their regulatory kinases is again exemplified by PLK-4. While high levels of PLK-4 leads to multiple procentrioles (97, 157), insufficient levels of PLK-4 is associated with abnormal centrosomes with reduced microtubule nucleation capacity, abnormal spindles, and CIN (304, 305).
Most cancer cells, but not normal untransformed cells, experience centrosome over replication upon prolonged interphase arrest. This phenotype is very relevant in oncology since cancer chemotherapy, which often includes DNA replication inhibitors, could potentially cause centrosome amplification, furthering genomic instability. Competence for centrosome over replication in cancer cells arrested in S phase appears to be conferred, in addition to Plk-4 which triggers procentriole formation, by procentriole maturation normally induced by active Plk1pT210 at the S to G2 transition (153). Prolonging interphase, particularly G2 phase leads to procentriole maturation and disengagement allowing for a second procentriole to form, which in a background of persistently high Plk1pT210, matures and disengage perpetuating the over-replication cycle (153). Nevertheless, Plk1 is dispensable for centriole formation and appears to participate only in centrosome maturation/disengagement, thus coordinating the cell cycle with procentriole maturation (153). Cells arrested in S-phase by depletion of a mitotic inhibitor (early mitotic inhibitor 1, Emi1) assembled procentrioles, which do not grow further unless Plk1pT210 is available. Plk1pT210 peaks at the beginning of G2 fulfilling its role as centrosome maturation factor. Plk1pT210 does not localize to the centrosome until late S early G2 since it is not present in HU S-phase arrested cells (217) but is found at the centrosome in G2 arrested cells, all of which is consistent with Plk1pT210 being critical in the centrosome cycle in G2 (153).
De novo centrosome formation (Figure 4) is known to normally occur only in lower eukaryotes (306), in eggs (307), and in parthenogenetic embryos (308). Although it has never been documented in normal vertebrate cells, other than in Chinese hamster ovary (CHO) cells, it may occur in cancer cells under special circumstances. In CHO cells arrested in S phase by hydroxyurea, destruction of centrosomes by laser microsurgery leads to the appearance of new PCM clouds 5–8 h after ablation (289). The clouds of PCM contain γ-tubulin, pericentrin (components of the PCM), and ninein (centriole). By 24 h clouds appeared more compact and exhibited a central denser area where a centriole was located. As many as 14 centrosomes per cell developed in cells arrested in S-phase for 24 h. Of note, PCM accumulation and accretion into denser clouds was not dependent on microtubules. Only the development of centrioles was, since nocodazole pre-treatment abolished centriole formation but not PCM accumulation (289). Most centrioles had normal EM structure but some were aberrant partially open centriolar cylinders, distorted/bent walls, and different cylinder lengths. These structures are very similar to those observed during centrosome reassembly after loading cytoplasm with anti-polyglutamylated tubulin antibody a maneuver that leads to centrosome dispersal (102). In general all newly formed centrosome clustered together near the nuclear envelope and only rarely were they dispersed or away from the nuclear envelope. Interestingly, ninein distribution volume within the PCM clouds was restricted to a single dot located next to the single area of greatest γ-tubulin density, a location that suggest its normal presence in the distal end of the centriole. Nevertheless, there were no obvious appendages on the neocentrioles when examined by electron microscopy. These structures are able to nucleate microtubules and are thus functional. When cells with neocentrosomes were allowed to reenter mitosis by HU washout and exposure to caffeine, which induced rapid entry into mitosis, the majority of the cells assembled multipolar spindles, attempted multifurrow cytokinesis, which failed to complete, resulting in single daughter cells. Only two cells with multipolar spindles resulted in two (one) or three (one) daughter cells. Importantly, de novo centrosome formation in non-transformed mammalian cells does not occur in the presence of a single pre-existing normal centrosome. Taken all these data together it is reasonable to conclude that the template-dependent centriole duplication pathway is dominant, and the de novo centrosome formation pathway is only enacted if no centriole template is available to the cell. For instance, CHO cells with intact centrosome subjected to the identical S-phase block replicate centrosomes every 20 h and only using the template-dependent mechanism (309). One can conclude that de novo centrosome formation is a default back-up mechanism for cells that no longer have functional centrosomes. How the presence of centrioles suppresses the de novo pathway is currently unknown.
The significance of the de novo centrosome assembly pathway in cancer has been difficult to ascertain. For once, removal of the centrosome from normal, untransformed vertebrate cells leads to cell cycle arrest in G1 without centrosome neoformation (48, 310). Selective ablation of a single centrosome suppresses de novo centrosome formation in the daughter cell receiving the non-ablated centrosome indicating that an active pathway exist to suppress de novo centrosome formation. However, transformed cells such as HeLa cells lack such a checkpoint. Removal of resident centrosomes by laser ablation or micromechanical manipulation in HeLa cells does not result in G1 arrest, instead cells progress through mitosis and into S phase assembling centrosomes de novo (290). De novo centrosome assembly begins at the G1/S transition as faint centrin dots, which become recognizable centrosomes before mitosis. Remarkably, such centrosomes are immature, i.e., do not nucleate full arrays of microtubules, until the next cell cycle suggesting that cell cycle progression is necessary for completion of centrosome maturation. After the second mitosis, neocentrosomes coalesced into a single focus, evinced prominent PCM, and were associated with the main microtubule array focus indicating that they are by now fully competent centrosomes (290). Interestingly, de novo assembly of centrosome does not occur in cells arrested in G1 phase after centrosome ablation, but it does in cells arrested in S phase, indicating that the de novo pathway is only turned on in S phase, the cell cycle phase where normal, i.e., template-dependent centriole duplication occurs. Whether a mother or daughter centriole can suppress the de novo pathway was tested by specifically ablating the mother centriole within a centrosome at the mitotic spindle pole. The de novo pathway remained inactive in the daughter cell that received the centrosome containing the ablated mother centriole, indicating that even an immature centriole is sufficient to maintain the de novo pathway fully suppressed. During the next mitosis the daughter containing the centrosome with the ablated mother centriole enacted bipolar spindles. Since only one of the spindle poles had a centriole-containing centrosome, one of the daughters of this cell received a normal diplosomal centrosome, while the sister receiving no centrosome, promptly proceeded to assemble centrosomes de novo upon reaching S phase (290). It is important to note that HeLa cells, as many other cancer cells, lack active p53 dependent checkpoint, and that this may enable progression through G1 in the absence of centrosomes. The de novo centrosome assembly pathway may therefore be not only an important back-up mechanisms in cancer cells to regenerate lost centrosomes, but also an intrinsically destabilizing process that leads to multipolar spindles and its attendant complications, if accidentally activated.
A detailed analysis of centrosome biogenesis in HU-arrested CHO (which are p53+/−), which lack a p53 dependent G1 checkpoint (293), has added new layers of complexity and indicated that the overreplication and de novo pathways may not fundamentally differ after all (311). Time-lapse imaging of centrin1-GFP expressing CHO cells revealed that perinuclear centrin spots appeared soon after HU arrest. These spot are quite similar to those appearing shortly after laser ablation of the centrosome, which are known to represent the earliest step in de novo centriole formation (290). Further characterization revealed that such structures correspond indeed to centriolar satellites, which are known to participate in centriologenesis. Such precursors could be traced to their formation in the nucleolus, export to the cytoplasm, coalescence round the native centrosome, development of centrioles, and acquisition of functional PCM (311). These observations suggest that the phenomenon of centrosome amplification commences through a dynein/dinactin-mediated buildup of PCM material in preparation for centrosome construction. Centriolar satellites are pericentriolar protein-rich, electron dense ∼100 nm quasi-spherical bodies that appear to represent assembly factories for centrosome or ciliary components (312–314). Payload traffic to and from centriole satellites is microtubule- and dynein/dynactin-dependent (311, 313–316).
A similar process has been reported by Kramer and collaborators using a lung cancer cell line carrying a centrin-2-Dendra2 transgene in which the fluorescent protein tag is photoconvertible from green to red, permitting to distinguish unambiguously between pre-existing centrioles (photoconverted red) from newly developed centrioles (green) (317). Using this system, again numerous centrin containing centriole satellites formed after gamma-irradiation or bleomycin exposure, before centrosome amplification became evident (317). In fact, all manners of DNA damage induced the appearance of green Centrin-2 dots, i.e., newly formed centrioles, with no instance in which splitting of red signals (i.e., pre-existing centrosomes) occurred, even after X-ray irradiation, where “centrosome splitting” was first described (317). Of note, the newly formed centrin dots were mobile and loosely associated with the pre-existing centrosome (red dots). When interrogated by immunofluorescence, the newly formed green dots were negative for canonical centrosome components such as pericentrin, γ-tubulin, C-Nap1, rootletin, SAS-6, and STIL, but positive for known components of centriolar satellites such as PCM-1, BBS-4, and CEP290. The resemblance of centrin dots to normal centriolar satellites extended to their ultrastructural appearance when examined by electron microscopy using nanogold-conjugated antibodies as tracers, indicating they represent excessive production of centriolar satellites. The appearance of centrin dots preceded the appearance of newly formed centrosomes. As expected for a centriolar satellite driven process, chemical inhibition of dynein or interference of dynein/dynacting function by overexpression of dynamitin, suppressed centrosome amplification induced by DNA damage or X-ray irradiation. Moreover, chemical inhibition of Chk-1 with UCN-01 led to dose-dependent reduction of centrin dots in A549 and U2OS cells. Similar results were obtained by siRNA knock-down of Chk-1 (317).
Unless resident centrosomes are non-functional (inactivated) in transformed/cancer cells arrested in S or G2 phase, the above two studies suggest that the de novo pathway is active in cancer cells despite the presence of resident centrosomes. Indeed, some observations suggest that not all centrosomes may be functional in cancer cells, as documented by free centrosomes not associated to spindle poles in some cancer cell lines (318). Moreover, it is formally possible that the Cent2 green dots observed in Loffler et al. study (317) are templated by resident centrioles. Recent observations provide an intriguing possible explanation to this puzzle (319). Treatment of cells with Cdk1 inhibitors (RO-3306, roscovitine) or Cdk1 knockdown with siRNA, results in G2 arrest, premature centriole disengagement and chromosome endoreduplication. Under these conditions premature centriole disengagement is dependent on both separase and Plk1 activation. It is well established that centriole disengagement and displacement is a pre-requisite for growth of new procentrioles and that a mitotic Plk1 activity is required to render a new centriole competent for procentriole nucleation, thus preventing the growth of “granddaughter” procentrioles/centrioles. Under these conditions separase activation obeys to destruction of securin by activated APC/C, mediated by Plk1 induced loss of early mitotic inhibitor 1 (Emi1) (320), which normally inhibits the APC/C. Subsequent inactivation of APC/C upon prolonged arrest triggers reaccumulation of cyclin A (and to some extent cyclin E) and increased cyclin A-Cdk2 activity, promoting centriole reduplication and DNA endoreduplication (319). Similar observations were made in CHO and U2OS HU-arrested cells suggesting that this response, which includes oscillation of APC/C activity leading to first centriole disengagement (high APC/C activity) and subsequent replication (low APC/C) may be universal to prolonged S or G2 arrests (319).
Although numerical abnormalities of centrosomes are the most common centrosome “cancer phenotype,” qualitative changes frequently co-occur. Qualitative changes are far less well characterized, and little is know about their impact on the fidelity of chromosome segregation during mitosis. They include abnormally shaped centrioles, excess or deficits in PCM, and acentriolar MTOCs (250, 251, 257, 270, 318, 321–325). With the recent advances in microscopy it is formally possible to study this phenomena in vivo by multiplexed high-resolution fluorescence microscopy. Such studies will provide a wealth of new information, rapidly identifying the critical molecular events, which implicitly are targets for tumor specific targeted therapies.
Mitosis with Too Many Centrosomes: Spindle Multipolarity and Rectification Mechanisms
Regardless of the mechanism of origin, supernumerary centrosomes pose the same initial challenge to dividing cells: once two or more functionally mature centrosomes are present at the G2 phase of the cell cycle, the potential for multipolar spindles, and chromosome missegregation on the next mitosis is very real. However, the outcome of multipolar mitoses differs significantly depending on a number of additional factors (Figure 5). One key factor is the ploidy of the dividing cell, which influences the success rate of multipolar mitoses (see above). Additional factors include the ability of cancer cells with multipolar mitoses to circumvent the mitotic spindle assembly checkpoint (326); the competence of cell death execution pathways leading to mitotic (327) or post-mitotic cell death (328) of cells that cannot self-correct defects to satisfy the mitotic assembly checkpoint; the ability of the cell to exit mitosis without experiencing anaphase and cytokinesis – a process that has been termed “mitotic slippage”; and more importantly, the ability of the cell to reconfigure the multipolar spindle into a bipolar spindle before entering anaphase (328, 329) (Figure 5).
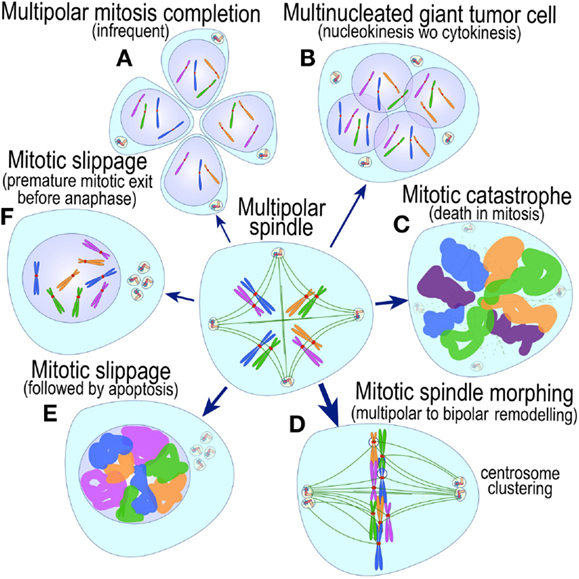
Figure 5. Possible outcomes of multipolar mitoses in cancer. A subset of cells with multipolar spindles carry mitosis to completion, resulting in highly aneuploidy cells, some with abnormal centrosome number (A). Others fail cytokinesis resulting in giant multinucleated polyploid cells, often with supernumerary centrosomes (B). Some cells exit mitosis in a process termed “mitotic slippage” and become polyploid cells with supernumerary centrosomes (F), or apoptose in the subsequent G1 phase (E). Yet others undergo mitotic catastrophe (death in mitosis) (C). Finally, most cells with multipolar mitosis, after significant delay, reconfigure their multipolar spindles into bipolar spindles resulting in (mostly) normal or abnormal (merothelic, synthelic chromosome) chromosome segregation (D). The thickness of the arrows in the figure intends to provide an estimate of the frequency of these events in cancer cells.
Mitoses with multipolar spindles are inherently inefficient, exhibiting a high rate of intra-mitotic (mitotic catastrophe) (327, 330, 331), post-mitotic cell death (328), or senescence (298), hindering tumor growth and acting as tumor suppressors rather than tumor promoters (332–334) (Figure 5). Multipolar cells that do undergo multipolar anaphase, but do not complete mitosis, and still survive, are likely to become giant multinucleated tumor cells (Figure 5). A subset of giant tumor cells is commonly present in cytologically high-grade tumors, but appear to either not divide at all or divide only sparingly. Rarely, cell with multipolar spindles may divide asymmetrically to produce viable daughter cells, a phenomenon that has been documented but does not appear to be prevalent, since the chances of a daughter cell receiving a full haploid chromosome complement is low. Nevertheless, the importance of such rare events should not be underestimated insofar as they may be critical in generating cells with properties significantly different from the main tumor population, which underscores, at least partly, the typical punctual evolution of the tumor genome (335–339).
Since a majority of multipolar mitosis outcomes are detrimental to cell growth (Figure 5) how might a tumor with supernumerary centrosomes prosper? How do cells with multipolar spindles solve the mitotic conundrum? Recent long-term in vivo observations of mitoses with multipolar spindles have revealed important clues. It turns out that a large proportion of cells with multipolar spindles utilize a spindle correction mechanism active in normal cells (340, 341) (Figure 6). After initially deploying multipolar spindles, cells delay metaphase until the extra spindle poles coalesce to form bipolar spindles, promoting “normal” bipolar mitoses (332, 341, 342). Multiple centrosomes per spindle poles were first observed by a number of investigators in mouse neuroblastoma cells (321, 323, 325), but their significance remained enigmatic until Bill Brinkley inferred their potential importance and predicted what indeed has been born out by recent experimental evidence (329). Centrosome coalescence in normal cells and some cancer cell lines is highly efficient (332, 343, 344) with cells rarely exhibiting multipolar mitoses even in the presence of greatly increased number of centrosomes (257). But not all cancers retain intact the centrosome clustering mechanisms and in some transformed cell lines (332, 341, 343, 344) and cancer tissue, multipolar anaphases may still occur. The extent to which multipolar mitoses contribute to CIN has not yet been determined. What mediates the metaphase delay needed to reconfigure a multipolar into bipolar spindle is at present poorly understood. The mitotic spindle assembly checkpoint (345) does not appear to monitor the number of spindle poles (346). However, it is possible that unattached or misattached kinetochores in multipolar spindles fail to generate the tensile forces that are key to extinguish the spindle assembly checkpoint (347, 348), delaying anaphase onset. Time spent in metaphase however is important since reducing it experimentally reduces centrosome clustering and leads to multipolar anaphases (340, 345, 349). The forces that contribute to centrosome clustering appear to be threefold: inter-centrosomal interactions, interactions of anti-parallel polar microtubules, and forces exerted by astral microtubules. Nevertheless, the molecular mechanisms coordinating these processes in cancer cells are poorly understood.
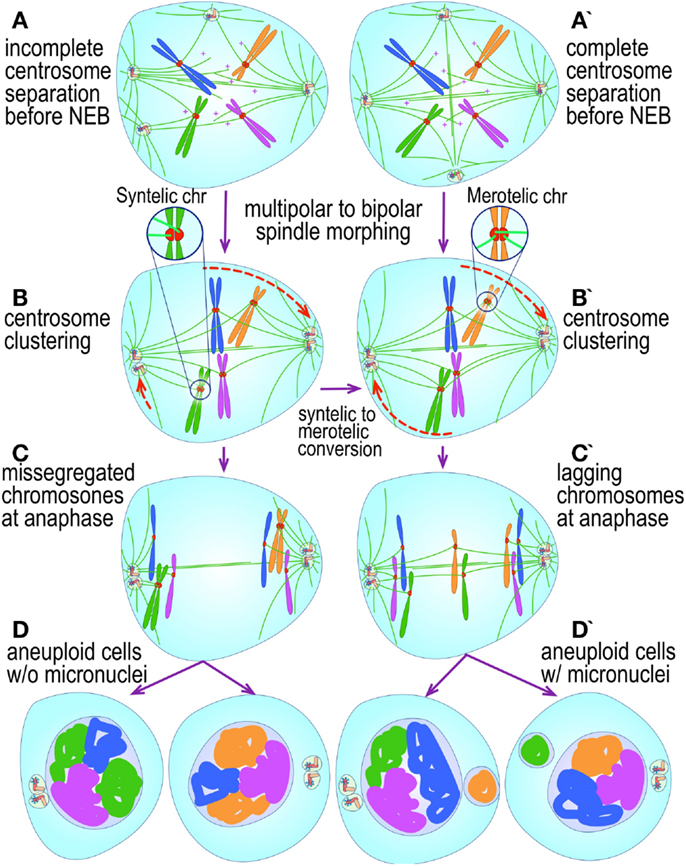
Figure 6. Multipolar to bipolar spindle reconfiguration in cancer cells. Cancer cells with supernumerary centrosomes enact multipolar spindles (A,A’), but after some delay, reconfigure their spindles to bipolar structures with centrosomes clustered at the poles (B,B’). Abnormal chromosome attachments acquired during the multipolar stage, such has synthelic (B) or merothelic (B’), may then lead to monopolar segregation (C) or lagging (C’) chromosomes, which are usually incorporated into micronuclei, resulting in aneuploid cells with supernumerary centrosomes without (D) or with micronuclei(D’).
The correction mechanism itself seems to be dependent on kinetochore microtubule dynamics and interaction of polar microtubule bundles. The minus-end-directed microtubule motors nuclear mitotic apparatus protein (NuMA) and dynein have been implicated in this process (341). Two recent screens have implicated a number of other proteins in the process suggesting mechanistic possibilities. The first, a screen in non-transformed cells (Drosophila S2 cells) revealed three main classes of proteins: components of the mitotic spindle assembly checkpoint, regulators of cortical acto-myosin contractility, and microtubule associated proteins (MAPs) (340). Also discovered in the screen was an essential role in centrosome clustering for the non-essential microtubule minus-end-directed motor non-claret disjunctional (NCD), a kinesin-14 family member (vertebrate homolog HSET). Notably, HSET is not only critical for the formation of bipolar spindles in the absence of centrosomes – through the incorporation and clustering of MTOCs at spindle poles (318), but is also critical for the clustering of canonical centrosomes in cancer cell lines, but not in non-transformed RPE1 cells (318). A second genome-wide screen for proteins that participate in centrosome clustering, this time in tumor cells, identified proteins involved in kinetochore microtubule attachment, sister chromatid cohesion, members of the augmin complex microtubule formation pathway, and chromosome passenger complexes (CPC: aurora-B, INCENP, survivin, and borealin) (350). The study suggested that kinetochore and spindle components generate the forces necessary to maintain centrosome clustering at the poles.
The Fate of Bipolar Mitoses with Clustered Centrosomes at Spindle Poles
Nevertheless, not all is well in cells that manage to convert multipolar to bipolar spindles. Multipolarity, even if transient (342), may lead to maloriented kinetochores, permitting microtubules from two or more poles to contact and bind the same kinetochore, leading to multipolar chromosome attachment (Figure 6). Upon resolution of multipolarity, the resulting bipolar spindle will contain merothelic or synthelic chromosomes. Merothelic attachments lead to lagging chromosomes at anaphase (332), resulting in anaphase bridges that interfere and delay cytokinesis. If the bridge is resolved and cytokinesis completed, the lagging chromosome becomes a micronucleus in one of the daughter cells. Alternatively, if the bridge is not resolved, the outcome is cytokinesis failure and polyploidy (Figure 6). Monothelic or synthelic attachments, which may also originate in multipolar spindles lead to monopolar segregation (both chromatids to the same daughter). Remarkably, cells with intact spindle assembly checkpoint may still correct some of these misattachments – whether merothelic, synthelic or monothelic – into perfectly amphitelic (bipolar) orientated chromosomes before anaphase onset, resulting in normal chromosome segregation (340–342).
Not all Forms of Aneuploidy are Caused by Centrosome Abnormalities
Two forms of cancer aneuploidy are readily distinguishable in clinical cancer karyotypes: stable aneuploidy and unstable or dynamic aneuploidy. In the former, all cells in a cancer growth share gains and/or losses of the same normal or structurally abnormal chromosome(s), whereas in the latter, cancer cells have more extensive gains and losses of chromosomes, only some of which are shared by most of the cells in the tumor, while others are shared only by subsets of cells. Whereas the former is thought to results from rare and transient mitotic chromosome missegregation events in a founder cancer cell, the latter is due to frequent and continuous mitotic chromosome missegregation and is a symptom of an intrinsically defective chromosome segregation machinery (278). Unstable aneuploidy, which is also known as CIN (251, 351), is the more common of the two, and is pervasive in carcinoma, some forms of sarcoma, and a subset of hematopoietic and lymphoid cancers (251, 352) [reviewed in Ref. (16, 353)]. CIN is multifactorial and may result not only from centrosome dysfunction (250, 251), but also from defects in kinetochore microtubule attachment and dynamics (354, 355), spindle assembly checkpoint (356, 357), chromosome replication/condensation/cohesion (358), cytokinesis failure (291, 359, 360), or dysfunction of checkpoints that coordinate the DNA replication, and centrosome cycles (361).
Centrosome Abnormalities Provide a Potential Mechanistic Link between Numerical and Structural Chromosome Abnormalities
In CIN, numerical (nCIN) and structural (sCIN) chromosome abnormalities nearly always co-exist [reviewed in Ref. (362–365)]. nCIN includes a spectrum of gain and losses of chromosomes fragments from kilobases to megabases, whole arms or even entire chromosomes. sCIN include translocations, inversions, end-fusions, and a number of more complex rearrangements. With the exception of break-fusion-bridge (BFB) cycles (366) (Figure 7), until recently it was thought that nCIN and sCIN, despite their frequent coexistence, were largely mechanistically unrelated. Several recent whole-genome sequencing studies of cancer tissue have uncovered new mechanistic links between nCIN, sCIN, and centrosomes (Figure 7). Grouped under the term chromoanagenesis (chromosome rebirth) (367), the first class of sCIN consists of a handful of apparently random chromosome loci per genome with highly complex structural (and copy number) sequence alterations including sequence duplications, deletions, scrambling, and polarity reversals, as if the segment had been broken in hundreds of fragments and rejoined more or less randomly (Figure 7). This phenomenon has been termed chromothripsis (chromosome shattering) (368). Chromothripsis has been postulated to occur as a single, punctual, massive event, rather than sequentially, as is seen with BFB cycles. Two possible causative mechanisms have been thus far delineated, both of which are enabled by chromosome missegregation events triggered by abnormal centrosome function in mitosis. Chromothripsis involves premature mitotic entry of a chromosome contained in a micronucleus that resulted from a chromosome missegregation event in the previous mitosis (368, 369). Mitotic entry, before completion of DNA replication in the micronucleus, leads to failure of micronucleus envelope breakdown, mitotic transit with random segregation to daughter cells, and random reassembly of the incompletely replicated chromosome fragments within the micronucleus during the subsequent interphase (368, 369). Chromothripsis [reviewed in (362, 363, 370)] appears to be common in carcinoma and neural tumors (369, 371) and is the mechanism most likely to operate in micronuclei. Micronuclei in cancer are frequent and may have many origins, one of the most important of which appears to be merotelic chromosome orientation at metaphase. A merothelic chromosome is one in which one of its two kinetochores is simultaneously attached by microtubules to the two spindle poles. Merothelic chromosomes often lag at the center of the dividing cell at anaphase and are usually not incorporated within the two groups of chromosomes at the poles in telophase before reassembly of the nuclear envelope at the end of mitosis, becoming independent “micronuclei.” Notably, a transient state of mitotic spindle multipolarity in cells with supernumerary centrosomes is thought to be the most important cause of merothelic chromosome attachment and micronuclei formation (355) (vide infra), again implicating centrosome dysfunction in the cause of sCIN.
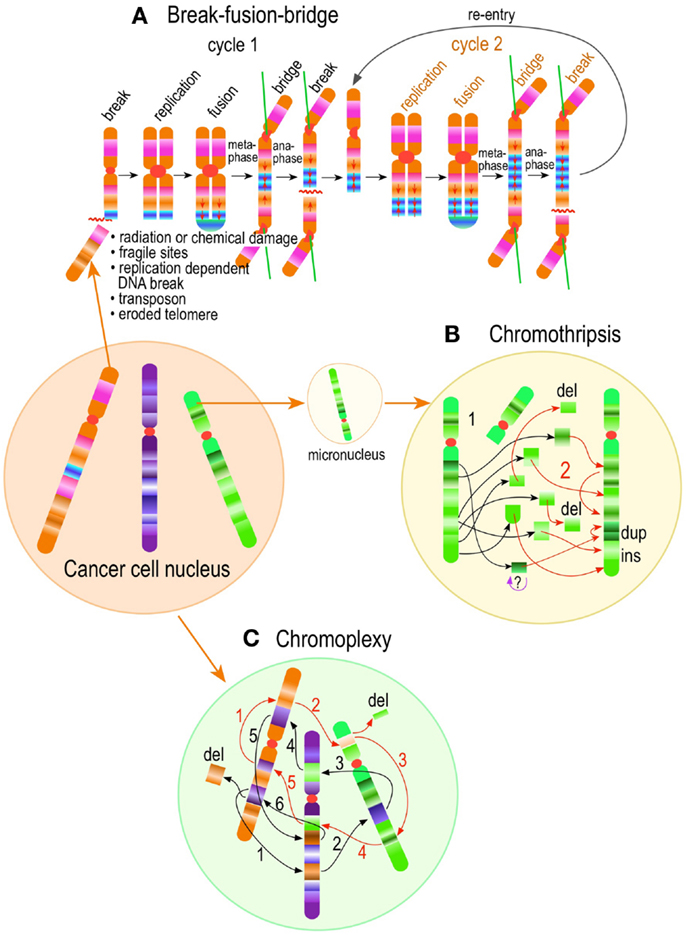
Figure 7. Chromoanagenesis in cancer. Complex structural chromosome rearrangements in cancer, involve stepwise or punctual chromosome restructuring. It includes stepwise classic intra or inter-chromosomal break-fusion-bridge cycles (A); chromothripsis (B), which are punctual localized highly complex chromosome fragmentation (1) and rejoining (2) events, and chromoplexy (C), in which complex linked translocation events involving multiple chromosomes presumably occur simultaneously. 1 through 6 (black typeface) and 1 through 5 (orange typeface) represent two such linked “chained” events.
A second class of distinct “genome level” structural abnormality in cancer gleaned from whole-genome sequences has been recently delineated in prostate cancer (335). Termed chromoplexy (chromosome restructuring), in this phenomenon numerous inter- and intra-chromosome translocations and deletion of genetic material arise in a highly interdependent manner (335) (Figure 7). These “chained rearrangements,” numbering from 3 to over 40, involved up to 6 chromosomes simultaneously, exhibited precise joints or large deletions at the joints, and occurred in the majority of prostate cancers studied. Moreover, greater than 60% of the tumors contained more than one chained rearrangements (335, 339). Statistical analyses indicated these rearrangements were unlikely to occur independently, suggesting again a single, punctual, massive genome-scrambling event (339). The cause(s) and mechanism(s) of chromoplexy, and whether centrosomes participate in its pathogenesis, are currently unknown.
Summary
A significant body of evidence implicating centrosome dysfunction on CIN has accumulated in the past 20 years. Centrosome defects are ubiquitous in cancer and are associated with dynamic CIN due to chromosome missegregation during mitosis. Surprisingly, centrosome dysfunction also participates in promoting structural CIN by a number of mechanisms, principally by initiating micronucleus formation through merothelic chromosome attachments, chromosome breakage at centromeres, and DNA damage on miss attached chromosome. The combination of structural and numerical chromosome abnormalities triggered by centrosome dysfunction, ultimately leads to gene reshuffling and reprograming of the cancer genome. Reprograming results primarily from four related events associated with gene reshuffling: deregulation of gene expression resulting from gene repositioning, which changes both the regulatory element landscape and chromatin regulatory domains; gene dosage changes resulting from numerical changes of part or whole chromosomes; gene mutations (gene fusion, inversions, indels, etc.) resulting from chromosome domain repositioning; and changes in the feasibility and probability of oncogene gain or tumor suppressor gene loss by dissociating genes from neighboring genes that normally exert opposite selection pressure. Importantly, cancer cells have developed mechanisms to cope with altered centrosome function, primarily by reconfiguring multipolar spindles into bipolar spindles prior to anaphase. The intricate molecular defects of centrosome dysfunction in cancer provide a unique opportunity for hyper-targeted therapies, which not only interfere with a specific molecule also present in normal cells, but also with a process specifically deranged in cancer, on principle avoiding harm to normal cells. Technical advances in microscopy, fluorescent protein technology and high throughput screening will permit a more rigorous examination of centrosome defects and their functional consequences in short-term cultures of human cancer samples and immortalized “non-transformed” human cells. Only through this exercise will we fully understand the magnitude and the critical differences in centrosome biology between normal and cancer tissue permitting us to develop smart therapies to combat “the emperor of all maladies.”
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
4. Hansemann L. Ueber asymmetrische zelltheilung in epithelkrebsen und deren biologische bedeutung. Arch Pathol Anat Physiol Klin Med (1890) 119:299–326.
7. Caspersson T. Über den chemischen Aufbau der Strukturen des Zellkerns. Scand Arch Physiol (1936) 73:3–151.
8. Caspersson T, Santesson L. Studies on protein metabolism in the cells of epithelial tumors. Acta Radiol (1942) 46(Suppl):1–105.
9. Atlas of Genetics and Cytogenetics in Oncology and Haematology Available from: http://atlasgeneticsoncology.org
10. Cancer Genome Anatomy Project, Available from: http://cgap.nci.nih.gov/cgap.html
11. Mitelman F, Johansson B, Mertens F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. (2013). Available at: http://cgap.nci.nih.gov/Chromosomes/Mitelman
12. Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature (2003) 426:570–4. doi:10.1038/nature02166
13. Müller H, Schmidt D, Dreher F, Herwig R, Ploubidou A, Lange BM. Gene ontology analysis of the centrosome proteomes of Drosophila and human. Commun Integr Biol (2011) 4:308–11. doi:10.4161/cib.4.3.14806
14. Müller H, Schmidt D, Steinbrink S, Mirgorodskaya E, Lehmann V, Habermann K, et al. Proteomic and functional analysis of the mitotic Drosophila centrosome. EMBO J (2010) 29:3344–57. doi:10.1038/emboj.2010.210
15. Nogales-Cadenas R, Abascal F, Diez-Perez J, Carazo JM, Pascual-Montano A. CentrosomeDB: a human centrosomal proteins database. Nucleic Acids Res (2009) 37:D175–80. doi:10.1093/nar/gkn815
16. Ren J, Liu Z, Gao X, Jin C, Ye M, Zou H, et al. MiCroKit 3.0: an integrated database of midbody, centrosome and kinetochore. Nucleic Acids Res (2010) 38:D155–60. doi:10.1093/nar/gkp784
17. Azimzadeh J, Bornens M. Structure and duplication of the centrosome. J Cell Sci (2007) 120:2139–42. doi:10.1242/jcs.005231
18. Bornens M. Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol (2002) 14:25–34. doi:10.1016/S0955-0674(01)00290-3
19. Bornens M. The centrosome in cells and organisms. Science (2012) 335:422–6. doi:10.1126/science.1209037
20. Doxsey S, McCollum D, Theurkauf W. Centrosomes in cellular regulation. Annu Rev Cell Dev Biol (2005) 21:411–34. doi:10.1146/annurev.cellbio.21.122303.120418
21. Luders J, Stearns T. Microtubule-organizing centres: a re-evaluation. Nat Rev Mol Cell Biol (2007) 8(161-167):10.1038/nrm2100. doi:10.1038/nrm2100
22. Mahen R, Venkitaraman AR. Pattern formation in centrosome assembly. Curr Opin Cell Biol (2012) 24:14–23. doi:10.1016/j.ceb.2011.12.012
23. Meraldi P, Nigg EA. The centrosome cycle. FEBS Lett (2002) 521:9–13. doi:10.1016/S0014-5793(02)02865-X
24. Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell (2009) 139:663–78. doi:10.1016/j.cell.2009.10.036
25. Stearns T, Kirschner M. In vitro reconstitution of centrosome assembly and function: the central role of gamma-tubulin. Cell (1994) 76:623–37. doi:10.1016/0092-8674(94)90503-7
26. Stearns T, Winey M. The cell center at 100. Cell (1997) 91:303–9. doi:10.1016/S0092-8674(00)80414-6
27. Bettencourt-Dias M, Glover DM. Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol (2007) 8:451–63. doi:10.1038/nrm2180
28. Musch A. Microtubule organization and function in epithelial cells. Traffic (2004) 5:1–9. doi:10.1111/j.1600-0854.2003.00149.x
29. Scliwa M, Honer B. Microtubules, centrosomes and intermediate filaments in directed cell movement. Trends Cell Biol (1993) 3:377–80. doi:10.1016/0962-8924(93)90086-G
30. Wakida NM, Botvinick EL, Lin J, Berns MW. An intact centrosome is required for the maintenance of polarization during directional cell migration. PLoS One (2010) 5:e15462. doi:10.1371/journal.pone.0015462
31. Dustin ML. Modular design of immunological synapses and kinapses. Cold Spring Harb Perspect Biol (2009) 1:a002873. doi:10.1101/cshperspect.a002873
32. Hoyer-Fender S. Centriole maturation and transformation to basal body. Semin Cell Dev Biol (2010) 21:142–7. doi:10.1016/j.semcdb.2009.07.002
33. O’Connell CB, Khodjakov AL. Cooperative mechanisms of mitotic spindle formation. J Cell Sci (2007) 120:1717–22. doi:10.1242/jcs.03442
34. Rebollo E, Sampaio P, Januschke J, Llamazares S, Varmark H, González C. Functionally unequal centrosomes drive spindle orientation in asymmetrically dividing Drosophila neural stem cells. Dev Cell (2007) 12:467–74. doi:10.1016/j.devcel.2007.01.021
35. Rusan NM, Peifer M. A role for a novel centrosome cycle in asymmetric cell division. J Cell Biol (2007) 177:13–20. doi:10.1083/jcb.200612140
36. Yamashita YM, Mahowald AP, Perlin JR, Fuller MT. Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science (2007) 315:518–21. doi:10.1126/science.1134910
37. Hinchcliffe EH, Miller FJ, Cham M, Khodjakov A, Sluder G. Requirement of a centrosomal activity for cell cycle progression through G1 into S phase. Science (2001) 291:1547–50. doi:10.1126/science.1056866
38. Matsumoto Y, Maller JL. A centrosomal localization signal in cyclin E required for Cdk2-independent S phase entry. Science (2004) 306:885–8. doi:10.1126/science.1103544
39. Ferguson RL, Maller JL. Centrosomal localization of cyclin E-Cdk2 is required for initiation of DNA synthesis. Curr Biol (2010) 20:856–60. doi:10.1016/j.cub.2010.03.028
40. Ferguson RL, Pascreau G, Maller JL. The cyclin A centrosomal localization sequence recruits MCM5 and Orc1 to regulate centrosome reduplication. J Cell Sci (2010) 123:2743–9. doi:10.1242/jcs.073098
41. Gavet O, Pines J. Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J Cell Biol (2010) 189:247–59. doi:10.1083/jcb.200909144
42. Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol (2003) 5:143–8. doi:10.1038/ncb918
43. Kramer A, Lukas J, Bartek J. Checking out the centrosome. Cell Cycle (2004) 3:1390–3. doi:10.4161/cc.3.11.1252
44. Krämer A, Mailand N, Lukas C, Syljuåsen RG, Wilkinson CJ, Nigg EA, et al. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol (2004) 6:884–91. doi:10.1038/ncb1165
45. Portier N, Audhya A, Maddox PS, Green RA, Dammermann A, Desai A, et al. A microtubule-independent role for centrosomes and Aurora A in nuclear envelope breakdown. Dev Cell (2007) 12:515–29. doi:10.1016/j.devcel.2007.01.019
46. D’Angiolella V, Esencay M, Pagano M. A cyclin without cyclin-dependent kinases: cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol (2013) 23:135–40. doi:10.1016/j.tcb.2012.10.011
47. Piel M, Nordberg J, Euteneuer U, Bornens M. Centrosome-dependent exit of cytokinesis in animal cells. Science (2001) 291:1550–3. doi:10.1126/science.1057330
48. Khodjakov A, Rieder CL. Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progression. J Cell Biol (2001) 153:237–42. doi:10.1083/jcb.153.1.237
49. Keryer G, Witczak O, Delouvée A, Kemmner WA, Rouillard D, Tasken K, et al. Dissociating the centrosomal matrix protein AKAP450 from centrioles impairs centriole duplication and cell cycle progression. Mol Biol Cell (2003) 14:2436–46. doi:10.1091/mbc.E02-09-0614
50. Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat Cell Biol (2007) 9:160–70. doi:10.1038/ncb1529
51. Bailly E, Pines J, Hunter T, Bornens M. Cytoplasmic accumulation of cyclin B1 in human cells: association with a detergent-resistant compartment and with the centrosome. J Cell Sci (1992) 101(Pt 3):529–45.
52. Paintrand M, Moudjou M, Delacroix H, Bornens M. Centrosome organization and centriole architecture: their sensitivity to divalent cations. J Struct Biol (1992) 108:107–28. doi:10.1016/1047-8477(92)90011-X
53. Paoletti A, Bornens M. Organisation and functional regulation of the centrosome in animal cells. Prog Cell Cycle Res (1997) 3:285–99. doi:10.1007/978-1-4615-5371-7_23
54. Piperno G, Fuller MT. Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. J Cell Biol (1985) 101:2085–94. doi:10.1083/jcb.101.6.2085
55. Eddé B, Rossier J, Le Caer JP, Desbruyères E, Gros F, Denoulet P. Posttranslational glutamylation of alpha-tubulin. Science (1990) 247:83–5. doi:10.1126/science.1967194
56. Tanos BE, Yang HJ, Soni R, Wang WJ, MacAluso FP, Asara JM, et al. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev (2013) 27:163–8. doi:10.1101/gad.207043.112
57. Gould RR, Borisy GG. The pericentriolar material in Chinese hamster ovary cells nucleates microtubule formation. J Cell Biol (1977) 73:601–15. doi:10.1083/jcb.73.3.601
58. Piel M, Meyer P, Khodjakov A, Rieder CL, Bornens M. The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J Cell Biol (2000) 149:317–30. doi:10.1083/jcb.149.2.317
59. Wang P, Pinson X, Archambault V. PP2A-twins is antagonized by greatwall and collaborates with polo for cell cycle progression and centrosome attachment to nuclei in Drosophila embryos. PLoS Genet (2011) 7:e1002227. doi:10.1371/journal.pgen.1002227
60. Gunawardane RN, Lizarraga SB, Wiese C, Wilde A, Zheng Y. Gamma-Tubulin complexes and their role in microtubule nucleation. Curr Top Dev Biol (2000) 49:55–73. doi:10.1016/S0070-2153(99)49004-0
61. Gunawardane RN, Martin OC, Cao K, Zhang L, Dej K, Iwamatsu A, et al. Characterization and reconstitution of Drosophila gamma-tubulin ring complex subunits. J Cell Biol (2000) 151:1513–24. doi:10.1083/jcb.151.7.1513
62. Murphy SM, Preble AM, Patel UK, O’Connell KL, Dias DP, Moritz M, et al. GCP5 and GCP6: two new members of the human gamma-tubulin complex. Mol Biol Cell (2001) 12:3340–52. doi:10.1091/mbc.12.11.3340
63. Guillet V, Knibiehler M, Gregory-Pauron L, Remy MH, Chemin C, Raynaud-Messina B, et al. Crystal structure of gamma-tubulin complex protein GCP4 provides insight into microtubule nucleation. Nat Struct Mol Biol (2011) 18:915–9. doi:10.1038/nsmb.2083
64. Kollman JM, Polka JK, Zelter A, Davis TN, Agard DA. Microtubule nucleating gamma-TuSC assembles structures with 13-fold microtubule-like symmetry. Nature (2010) 466:879–82. doi:10.1038/nature09207
65. Oegema K, Wiese C, Martin OC, Milligan RA, Iwamatsu A, Mitchison TJ, et al. Characterization of two related Drosophila gamma-tubulin complexes that differ in their ability to nucleate microtubules. J Cell Biol (1999) 144:721–33. doi:10.1083/jcb.144.4.721
66. Moritz M, Braunfeld MB, Sedat JW, Alberts B, Agard DA. Microtubule nucleation by gamma-tubulin-containing rings in the centrosome. Nature (1995) 378:638–40. doi:10.1038/378638a0
67. Moritz M, Zheng Y, Alberts BM, Oegema K. Recruitment of the gamma-tubulin ring complex to Drosophila salt-stripped centrosome scaffolds. J Cell Biol (1998) 142:775–86. doi:10.1083/jcb.142.3.775
68. Murphy SM, Urbani L, Stearns T. The mammalian gamma-tubulin complex contains homologues of the yeast spindle pole body components spc97p and spc98p. J Cell Biol (1998) 141:663–74. doi:10.1083/jcb.141.3.663
69. Zheng Y, Wong ML, Alberts B, Mitchison T. Nucleation of microtubule assembly by a gamma-tubulin-containing ring complex. Nature (1995) 378:578–83. doi:10.1038/378578a0
70. Moritz M, Braunfeld MB, Guenebaut V, Heuser J, Agard DA. Structure of the gamma-tubulin ring complex: a template for microtubule nucleation. Nat Cell Biol (2000) 2:365–70. doi:10.1038/35014058
71. Gunawardane RN, Martin OC, Zheng Y. Characterization of a new gammaTuRC subunit with WD repeats. Mol Biol Cell (2003) 14:1017–26. doi:10.1091/mbc.E02-01-0034
72. Haren L, Remy MH, Bazin I, Callebaut I, Wright M, Merdes A. NEDD1-dependent recruitment of the gamma-tubulin ring complex to the centrosome is necessary for centriole duplication and spindle assembly. J Cell Biol (2006) 172:505–15. doi:10.1083/jcb.200510028
73. Luders J, Patel UK, Stearns T. GCP-WD is a gamma-tubulin targeting factor required for centrosomal and chromatin-mediated microtubule nucleation. Nat Cell Biol (2006) 8:137–47. doi:10.1038/ncb1349
74. Teixidó-Travesa N, Villén J, Lacasa C, Bertran MT, Archinti M, Gygi SP, et al. The gammaTuRC revisited: a comparative analysis of interphase and mitotic human gammaTuRC redefines the set of core components and identifies the novel subunit GCP8. Mol Biol Cell (2010) 21:3963–72. doi:10.1091/mbc.E10-05-0408
75. Ma W, Baumann C, Viveiros MM. NEDD1 is crucial for meiotic spindle stability and accurate chromosome segregation in mammalian oocytes. Dev Biol (2010) 339:439–50. doi:10.1016/j.ydbio.2010.01.009
76. Manning JA, Shalini S, Risk JM, Day CL, Kumar S. A direct interaction with NEDD1 regulates gamma-tubulin recruitment to the centrosome. PLoS One (2010) 5:e9618. doi:10.1371/journal.pone.0009618
77. Zeng CJ, Lee YR, Liu B. The WD40 repeat protein NEDD1 functions in microtubule organization during cell division in Arabidopsis thaliana. Plant Cell (2009) 21:1129–40. doi:10.1105/tpc.109.065953
78. Hutchins JR, Toyoda Y, Hegemann B, Poser I, Hériché JK, Sykora MM, et al. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science (2010) 328:593–9. doi:10.1126/science.1181348
79. Takahashi M, Yamagiwa A, Nishimura T, Mukai H, Ono Y. Centrosomal proteins CG-NAP and kendrin provide microtubule nucleation sites by anchoring gamma-tubulin ring complex. Mol Biol Cell (2002) 13:3235–45. doi:10.1091/mbc.E02-02-0112
80. Zimmerman WC, Sillibourne J, Rosa J, Doxsey SJ. Mitosis-specific anchoring of gamma tubulin complexes by pericentrin controls spindle organization and mitotic entry. Mol Biol Cell (2004) 15:3642–57. doi:10.1091/mbc.E03-11-0796
81. Fong KW, Choi YK, Rattner JB, Qi RZ. CDK5RAP2 is a pericentriolar protein that functions in centrosomal attachment of the gamma-tubulin ring complex. Mol Biol Cell (2008) 19:115–25. doi:10.1091/mbc.E07-04-0371
82. Izumi N, Fumoto K, Izumi S, Kikuchi A. GSK-3beta regulates proper mitotic spindle formation in cooperation with a component of the gamma-tubulin ring complex, GCP5. J Biol Chem (2008) 283:12981–91. doi:10.1074/jbc.M710282200
83. Uehara R, Nozawa RS, Tomioka A, Petry S, Vale RD, Obuse C, et al. The augmin complex plays a critical role in spindle microtubule generation for mitotic progression and cytokinesis in human cells. Proc Natl Acad Sci U S A (2009) 106:6998–7003. doi:10.1073/pnas.0901587106
84. Lawo S, Bashkurov M, Mullin M, Ferreria MG, Kittler R, Habermann B, et al. HAUS, the 8-subunit human Augmin complex, regulates centrosome and spindle integrity. Curr Biol (2009) 19:816–26. doi:10.1016/j.cub.2009.04.033
85. Fu J, Glover DM. Structured illumination of the interface between centriole and peri-centriolar material. Open Biol (2012) 2:120104. doi:10.1098/rsob.120104
86. Lawo S, Hasegan M, Gupta GD, Pelletier L. Subdiffraction imaging of centrosomes reveals higher-order organizational features of pericentriolar material. Nat Cell Biol (2012) 14:1148–58. doi:10.1038/ncb2591
87. Mennella V, Keszthelyi B, McDonald KL, Chhun B, Kan F, Rogers GC, et al. Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nat Cell Biol (2012) 14:1159–68. doi:10.1038/ncb2597
88. Dictenberg JB, Zimmerman W, Sparks CA, Young A, Vidair C, Zheng Y, et al. Pericentrin and gamma-tubulin form a protein complex and are organized into a novel lattice at the centrosome. J Cell Biol (1998) 141:163–74. doi:10.1083/jcb.141.1.163
89. Conduit PT, Brunk K, Dobbelaere J, Dix CI, Lucas EP, Raff JW. Centrioles regulate centrosome size by controlling the rate of Cnn incorporation into the PCM. Curr Biol (2010) 20:2178–86. doi:10.1016/j.cub.2010.11.011
90. Gopalakrishnan J, Mennella V, Blachon S, Zhai B, Smith AH, Megraw TL, et al. Sas-4 provides a scaffold for cytoplasmic complexes and tethers them in a centrosome. Nat Commun (2011) 2:359. doi:10.1038/ncomms1367
91. O’Connell KF, Maxwell KN, White JG. The spd-2 gene is required for polarization of the anteroposterior axis and formation of the sperm asters in the Caenorhabditis elegans zygote. Dev Biol (2000) 222:55–70. doi:10.1006/dbio.2000.9714
92. Pelletier L, Ozlü N, Hannak E, Cowan C, Habermann B, Ruer M, et al. The Caenorhabditis elegans centrosomal protein SPD-2 is required for both pericentriolar material recruitment and centriole duplication. Curr Biol (2004) 14:863–73. doi:10.1016/j.cub.2004.04.012
93. Zhu F, Lawo S, Bird A, Pinchev D, Ralph A, Richter C, et al. The mammalian SPD-2 ortholog Cep192 regulates centrosome biogenesis. Curr Biol (2008) 18:136–41. doi:10.1016/j.cub.2007.12.055
94. Sonnen KF, Schermelleh L, Leonhardt H, Nigg EA. 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open (2012) 1:965–76. doi:10.1242/bio.20122337
95. Bahtz R, Seidler J, Arnold M, Haselmann-Weiss U, Antony C, Lehmann WD, et al. GCP6 is a substrate of Plk4 and required for centriole duplication. J Cell Sci (2012) 125:486–96. doi:10.1242/jcs.093930
96. Fuller SD, Gowen BE, Reinsch S, Sawyer A, Buendia B, Wepf R, et al. The core of the mammalian centriole contains gamma-tubulin. Curr Biol (1995) 5:1384–93. doi:10.1016/S0960-9822(95)00276-4
97. Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA. Plk4-induced centriole biogenesis in human cells. Dev Cell (2007) 13:190–202. doi:10.1016/j.devcel.2007.07.002
98. Schmidt TI, Kleylein-Sohn J, Westendorf J, Le Clech M, Lavoie SB, Stierhof YD, et al. Control of centriole length by CPAP and CP110. Curr Biol (2009) 19:1005–11. doi:10.1016/j.cub.2009.05.016
99. Mogensen MM, Malik A, Piel M, Bouckson-Castaing V, Bornens M. Microtubule minus-end anchorage at centrosomal and non-centrosomal sites: the role of ninein. J Cell Sci (2000) 113(Pt 17):3013–23.
100. Guarguaglini G, Duncan PI, Stierhof YD, Holmström T, Duensing S, Nigg EA. The forkhead-associated domain protein Cep170 interacts with Polo-like kinase 1 and serves as a marker for mature centrioles. Mol Biol Cell (2005) 16:1095–107. doi:10.1091/mbc.E04-10-0939
101. Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, et al. Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol (2007) 179:321–30. doi:10.1083/jcb.200707181
102. Bobinnec Y, Khodjakov A, Mir LM, Rieder CL, Eddé B, Bornens M. Centriole disassembly in vivo and its effect on centrosome structure and function in vertebrate cells. J Cell Biol (1998) 143:1575–89. doi:10.1083/jcb.143.6.1575
103. Kirkham M, Muller-Reichert T, Oegema K, Grill S, Hyman AA. SAS-4 is a C. elegans centriolar protein that controls centrosome size. Cell (2003) 112:575–87. doi:10.1016/S0092-8674(03)00117-X
104. Leidel S, Gonczy P. SAS-4 is essential for centrosome duplication in C elegans and is recruited to daughter centrioles once per cell cycle. Dev Cell (2003) 4:431–9. doi:10.1016/S1534-5807(03)00062-5
105. Gopalakrishnan J, Chim YC, Ha A, Basiri ML, Lerit DA, Rusan NM, et al. Tubulin nucleotide status controls Sas-4-dependent pericentriolar material recruitment. Nat Cell Biol (2012) 14:865–73. doi:10.1038/ncb2527
106. Kitagawa D, Kohlmaier G, Keller D, Strnad P, Balestra FR, Flückiger I, et al. Spindle positioning in human cells relies on proper centriole formation and on the microcephaly proteins CPAP and STIL. J Cell Sci (2011) 124:3884–93. doi:10.1242/jcs.089888
107. Kohlmaier G, Loncarek J, Meng X, McEwen BF, Mogensen MM, Spektor A, et al. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr Biol (2009) 19:1012–8. doi:10.1016/j.cub.2009.05.018
108. Bettencourt-Dias M, Glover DM. SnapShot: centriole biogenesis. Cell (2009) 136(188-188):e181. doi:10.1016/j.cell.2008.12.035
109. Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G. Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science (1999) 283:851–4. doi:10.1126/science.283.5403.851
110. Lacey KR, Jackson PK, Stearns T. Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci U S A (1999) 96:2817–22. doi:10.1073/pnas.96.6.2817
111. Matsumoto Y, Hayashi K, Nishida E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr Biol (1999) 9:429–32. doi:10.1016/S0960-9822(99)80191-2
112. Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol (1999) 1:88–93. doi:10.1038/10054
113. Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol (2005) 6:476–86. doi:10.1038/nrm1663
114. DePamphilis ML, Blow JJ, Ghosh S, Saha T, Noguchi K, Vassilev A. Regulating the licensing of DNA replication origins in metazoa. Curr Opin Cell Biol (2006) 18:231–9. doi:10.1016/j.ceb.2006.04.001
115. Tsou MF, Stearns T. Mechanism limiting centrosome duplication to once per cell cycle. Nature (2006) 442:947–51. doi:10.1038/nature04985
116. Tsou MF, Stearns T. Controlling centrosome number: licenses and blocks. Curr Opin Cell Biol (2006) 18:74–8. doi:10.1016/j.ceb.2005.12.008
117. Wong C, Stearns T. Centrosome number is controlled by a centrosome-intrinsic block to reduplication. Nat Cell Biol (2003) 5:539–44. doi:10.1038/ncb993
118. Tsou MF, Wang WJ, George KA, Uryu K, Stearns T, Jallepalli PV. Polo kinase and separase regulate the mitotic licensing of centriole duplication in human cells. Dev Cell (2009) 17:344–54. doi:10.1016/j.devcel.2009.07.015
119. Stemmann O, Zou H, Gerber SA, Gygi SP, Kirschner MW. Dual inhibition of sister chromatid separation at metaphase. Cell (2001) 107:715–26. doi:10.1016/S0092-8674(01)00603-1
120. Zou H, McGarry TJ, Bernal T, Kirschner MW. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science (1999) 285:418–22. doi:10.1126/science.285.5426.418
121. Nakamura A, Arai H, Fujita N. Centrosomal Aki1 and cohesin function in separase-regulated centriole disengagement. J Cell Biol (2009) 187:607–14. doi:10.1083/jcb.200906019
122. Schockel L, Mockel M, Mayer B, Boos D, Stemmann O. Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat Cell Biol (2011) 13:966–72. doi:10.1038/ncb2280
123. Oliveira RA, Nasmyth K. Cohesin cleavage is insufficient for centriole disengagement in Drosophila. Curr Biol (2013) 23:R601–3. doi:10.1016/j.cub.2013.04.003
124. Lee K, Rhee K. Separase-dependent cleavage of pericentrin B is necessary and sufficient for centriole disengagement during mitosis. Cell Cycle (2012) 11:2476–85. doi:10.4161/cc.20878
125. Matsuo K, Ohsumi K, Iwabuchi M, Kawamata T, Ono Y, Takahashi M. Kendrin is a novel substrate for separase involved in the licensing of centriole duplication. Curr Biol (2012) 22:915–21. doi:10.1016/j.cub.2012.03.048
126. Cha H, Hancock C, Dangi S, Maiguel D, Carrier F, Shapiro P. Phosphorylation regulates nucleophosmin targeting to the centrosome during mitosis as detected by cross-reactive phosphorylation-specific MKK1/MKK2 antibodies. Biochem J (2004) 378:857–65. doi:10.1042/BJ20031173
127. Krause A, Hoffmann I. Polo-like kinase 2-dependent phosphorylation of NPM/B23 on serine 4 triggers centriole duplication. PLoS One (2010) 5:e9849. doi:10.1371/journal.pone.0009849
128. Ma Z, Kanai M, Kawamura K, Kaibuchi K, Ye K, Fukasawa K. Interaction between ROCK II and nucleophosmin/B23 in the regulation of centrosome duplication ñø. Mol Cell Biol (2006) 26:9016–34. doi:10.1128/MCB.01383-06
129. Okuda M. The role of nucleophosmin in centrosome duplication. Oncogene (2002) 21:6170–4. doi:10.1038/sj.onc.1205708
130. Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell (2000) 103:127–40. doi:10.1016/S0092-8674(00)00093-3
131. Reboutier D, Troadec MB, Cremet JY, Fukasawa K, Prigent C. Nucleophosmin/B23 activates Aurora A at the centrosome through phosphorylation of serine 89. J Cell Biol (2012) 197:19–26. doi:10.1083/jcb.201107134
132. Fisk HA, Mattison CP, Winey M. Human Mps1 protein kinase is required for centrosome duplication and normal mitotic progression. Proc Natl Acad Sci U S A (2003) 100:14875–80. doi:10.1073/pnas.2434156100
133. Fisk HA, Winey M. The mouse Mps1p-like kinase regulates centrosome duplication. Cell (2001) 106:95–104. doi:10.1016/S0092-8674(01)00411-1
134. Kasbek C, Yang CH, Fisk HA. Mps1 as a link between centrosomes and genomic instability. Environ Mol Mutagen (2009) 50:654–65. doi:10.1002/em.20476
135. Kasbek C, Yang CH, Fisk HA. Antizyme restrains centrosome amplification by regulating the accumulation of Mps1 at centrosomes. Mol Biol Cell (2010) 21:3878–89. doi:10.1091/mbc.E10-04-0281
136. Kasbek C, Yang CH, Yusof AM, Chapman HM, Winey M, Fisk HA. Preventing the degradation of mps1 at centrosomes is sufficient to cause centrosome reduplication in human cells. Mol Biol Cell (2007) 18:4457–69. doi:10.1091/mbc.E07-03-0283
137. Pike AN, Fisk HA. Centriole assembly and the role of Mps1: defensible or dispensable? Cell Div (2011) 6:9. doi:10.1186/1747-1028-6-9
138. Stucke VM, Sillje HH, Arnaud L, Nigg EA. Human Mps1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. EMBO J (2002) 21:1723–32. doi:10.1093/emboj/21.7.1723
139. Yang CH, Kasbek C, Majumder S, Yusof AM, Fisk HA. Mps1 phosphorylation sites regulate the function of centrin 2 in centriole assembly. Mol Biol Cell (2010) 21:4361–72. doi:10.1091/mbc.E10-04-0298
140. Brownlee CW, Rogers GC. Show me your license, please: deregulation of centriole duplication mechanisms that promote amplification. Cell Mol Life Sci (2013) 70:1021–34. doi:10.1007/s00018-012-1102-6
141. Wang W, Budhu A, Forgues M, Wang XW. Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat Cell Biol (2005) 7:823–30. doi:10.1038/ncb1282
142. Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, et al. Role of nucleophosmin in embryonic development and tumorigenesis. Nature (2005) 437:147–53. doi:10.1038/nature03915
143. Shinmura K, Tarapore P, Tokuyama Y, George KR, Fukasawa K. Characterization of centrosomal association of nucleophosmin/B23 linked to Crm1 activity. FEBS Lett (2005) 579:6621–34. doi:10.1016/j.febslet.2005.10.057
144. Zatsepina OV, Rousselet A, Chan PK, Olson MO, Jordan EG, Bornens M. The nucleolar phosphoprotein B23 redistributes in part to the spindle poles during mitosis. J Cell Sci (1999) 112(Pt 4):455–66.
145. Hanashiro K, Brancaccio M, Fukasawa K. Activated ROCK II by-passes the requirement of the CDK2 activity for centrosome duplication and amplification. Oncogene (2011) 30:2188–97. doi:10.1038/onc.2010.607
146. Zhang H, Shi X, Paddon H, Hampong M, Dai W, Pelech S. B23/nucleophosmin serine 4 phosphorylation mediates mitotic functions of polo-like kinase 1. J Biol Chem (2004) 279:35726–34. doi:10.1074/jbc.M403264200
147. Cizmecioglu O, Warnke S, Arnold M, Duensing S, Hoffmann I. Plk2 regulated centriole duplication is dependent on its localization to the centrioles and a functional polo-box domain. Cell Cycle (2008) 7:3548–55. doi:10.4161/cc.7.22.7071
148. Warnke S, Kemmler S, Hames RS, Tsai HL, Hoffmann-Rohrer U, Fry AM, et al. Polo-like kinase-2 is required for centriole duplication in mammalian cells. Curr Biol (2004) 14:1200–7. doi:10.1016/j.cub.2004.06.059
149. Fisk HA, Winey M. Spindle regulation: Mps1 flies into new areas. Curr Biol (2004) 14:R1058–60. doi:10.1016/j.cub.2004.11.047
150. Zhang M, Pickart CM, Coffino P. Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate. EMBO J (2003) 22:1488–96. doi:10.1093/emboj/cdg158
151. Mangold U, Hayakawa H, Coughlin M, Munger K, Zetter BR. Antizyme, a mediator of ubiquitin-independent proteasomal degradation and its inhibitor localize to centrosomes and modulate centriole amplification. Oncogene (2008) 27:604–13. doi:10.1038/sj.onc.1210685
152. Cui Y, Cheng X, Zhang C, Zhang Y, Li S, Wang C, et al. Degradation of the human mitotic checkpoint kinase Mps1 is cell cycle-regulated by APC-cCdc20 and APC-cCdh1 ubiquitin ligases. J Biol Chem (2010) 285:32988–98. doi:10.1074/jbc.M110.140905
153. Loncarek J, Hergert P, Khodjakov A. Centriole reduplication during prolonged interphase requires procentriole maturation governed by Plk1. Curr Biol (2010) 20:1277–82. doi:10.1016/j.cub.2010.05.050
154. Wang WJ, Soni RK, Uryu K, Tsou MF. The conversion of centrioles to centrosomes: essential coupling of duplication with segregation. J Cell Biol (2011) 193:727–39. doi:10.1083/jcb.201101109
155. Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, Riparbelli M, Lehmann L, Gatt MK, et al. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol (2005) 15:2199–207. doi:10.1016/j.cub.2005.11.042
156. Brownlee CW, Klebba JE, Buster DW, Rogers GC. The Protein Phosphatase 2A regulatory subunit Twins stabilizes Plk4 to induce centriole amplification. J Cell Biol (2011) 195:231–43. doi:10.1083/jcb.201107086
157. Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol (2005) 7:1140–6. doi:10.1038/ncb1320
158. Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW. Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol (2010) 188:191–8. doi:10.1083/jcb.200911102
159. Rogers GC, Rusan NM, Roberts DM, Peifer M, Rogers SL. The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J Cell Biol (2009) 184:225–39. doi:10.1083/jcb.200808049
160. Cunha-Ferreira I, Rodrigues-Martins A, Bento I, Riparbelli M, Zhang W, Laue E, et al. The SCF/Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Curr Biol (2009) 19:43–9. doi:10.1016/j.cub.2008.11.037
161. Guderian G, Westendorf J, Uldschmid A, Nigg EA. Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J Cell Sci (2010) 123:2163–9. doi:10.1242/jcs.068502
162. Sillibourne JE, Tack F, Vloemans N, Boeckx A, Thambirajah S, Bonnet P, et al. Autophosphorylation of polo-like kinase 4 and its role in centriole duplication. Mol Biol Cell (2010) 21:547–61. doi:10.1091/mbc.E09-06-0505
163. Delattre M, Canard C, Gonczy P. Sequential protein recruitment in C. elegans centriole formation. Curr Biol (2006) 16:1844–9. doi:10.1016/j.cub.2006.07.059
164. Sonnen KF, Gabryjonczyk AM, Anselm E, Stierhof YD, Nigg EA. Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J Cell Sci (2013) 126:3223–33. doi:10.1242/jcs.129502
165. Holland AJ, Fachinetti D, Zhu Q, Bauer M, Verma IM, Nigg EA, et al. The autoregulated instability of Polo-like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev (2012) 26:2684–9. doi:10.1101/gad.207027.112
166. Avidor-Reiss T, Gopalakrishnan J. Building a centriole. Curr Opin Cell Biol (2013) 25:72–7. doi:10.1016/j.ceb.2012.10.016
167. Brito DA, Gouveia SM, Bettencourt-Dias M. Deconstructing the centriole: structure and number control. Curr Opin Cell Biol (2012) 24:4–13. doi:10.1016/j.ceb.2012.01.003
168. Sluder G, Khodjakov A. Centriole duplication: analogue control in a digital age. Cell Biol Int (2010) 34:1239–45. doi:10.1042/CBI20100612
169. Strnad P, Gonczy P. Mechanisms of procentriole formation. Trends Cell Biol (2008) 18:389–96. doi:10.1016/j.tcb.2008.06.004
170. Fry AM, Mayor T, Meraldi P, Stierhof YD, Tanaka K, Nigg EA. C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J Cell Biol (1998) 141:1563–74. doi:10.1083/jcb.141.7.1563
171. Gonczy P. Towards a molecular architecture of centriole assembly. Nat Rev Mol Cell Biol (2012) 13:425–35. doi:10.1038/nrm3373
172. Pelletier L, O’Toole E, Schwager A, Hyman AA, Muller-Reichert T. Centriole assembly in Caenorhabditis elegans. Nature (2006) 444:619–23. doi:10.1038/nature05318
173. Cizmecioglu O, Arnold M, Bahtz R, Settele F, Ehret L, Haselmann-Weiss U, et al. Cep152 acts as a scaffold for recruitment of Plk4 and CPAP to the centrosome. J Cell Biol (2010) 191:731–9. doi:10.1083/jcb.201007107
174. Dzhindzhev NS, Yu QD, Weiskopf K, Tzolovsky G, Cunha-Ferreira I, Riparbelli M, et al. Asterless is a scaffold for the onset of centriole assembly. Nature (2010) 467:714–8. doi:10.1038/nature09445
175. Hatch EM, Kulukian A, Holland AJ, Cleveland DW, Stearns T. Cep152 interacts with Plk4 and is required for centriole duplication. J Cell Biol (2010) 191:721–9. doi:10.1083/jcb.201006049
176. Blachon S, Gopalakrishnan J, Omori Y, Polyanovsky A, Church A, Nicastro D, et al. Drosophila asterless and vertebrate Cep152 Are orthologs essential for centriole duplication. Genetics (2008) 180:2081–94. doi:10.1534/genetics.108.095141
177. Puklowski A, Homsi Y, Keller D, May M, Chauhan S, Kossatz U, et al. The SCF-FBXW5 E3-ubiquitin ligase is regulated by PLK4 and targets HsSAS-6 to control centrosome duplication. Nat Cell Biol (2011) 13:1004–9. doi:10.1038/ncb2282
178. Culver BP, Meehl JB, Giddings TH Jr., Winey M. The two SAS-6 homologs in Tetrahymena thermophila have distinct functions in basal body assembly. Mol Biol Cell (2009) 20:1865–77. doi:10.1091/mbc.E08-08-0838
179. Nakazawa Y, Hiraki M, Kamiya R, Hirono M. SAS-6 is a cartwheel protein that establishes the 9-fold symmetry of the centriole. Curr Biol (2007) 17:2169–74. doi:10.1016/j.cub.2007.11.046
180. Strnad P, Leidel S, Vinogradova T, Euteneuer U, Khodjakov A, Gönczy P. Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev Cell (2007) 13:203–13. doi:10.1016/j.devcel.2007.07.004
181. Kitagawa D, Vakonakis I, Olieric N, Hilbert M, Keller D, Olieric V, et al. Structural basis of the 9-fold symmetry of centrioles. Cell (2011) 144:364–75. doi:10.1016/j.cell.2011.01.008
182. van Breugel M, Hirono M, Andreeva A, Yanagisawa HA, Yamaguchi S, Nakazawa Y, et al. Structures of SAS-6 suggest its organization in centrioles. Science (2011) 331:1196–9. doi:10.1126/science.1199325
183. Gopalakrishnan J, Guichard P, Smith AH, Schwarz H, Agard DA, Marco S, et al. Self-assembling SAS-6 multimer is a core centriole building block. J Biol Chem (2010) 285:8759–70. doi:10.1074/jbc.M109.092627
184. Kitagawa D, Busso C, Fluckiger I, Gonczy P. Phosphorylation of SAS-6 by ZYG-1 is critical for centriole formation in C. elegans embryos. Dev Cell (2009) 17:900–7. doi:10.1016/j.devcel.2009.11.002
185. Delattre M, Leidel S, Wani K, Baumer K, Bamat J, Schnabel H, et al. Centriolar SAS-5 is required for centrosome duplication in C. elegans. Nat Cell Biol (2004) 6:656–64. doi:10.1038/ncb1146
186. Hiraki M, Nakazawa Y, Kamiya R, Hirono M. Bld10p constitutes the cartwheel-spoke tip and stabilizes the 9-fold symmetry of the centriole. Curr Biol (2007) 17:1778–83. doi:10.1016/j.cub.2007.09.021
187. Stevens NR, Dobbelaere J, Brunk K, Franz A, Raff JW. Drosophila Ana2 is a conserved centriole duplication factor. J Cell Biol (2010) 188:313–23. doi:10.1083/jcb.200910016
188. Stevens NR, Roque H, Raff JW. DSas-6 and Ana2 coassemble into tubules to promote centriole duplication and engagement. Dev Cell (2010) 19:913–9. doi:10.1016/j.devcel.2010.11.010
189. Arquint C, Sonnen KF, Stierhof YD, Nigg EA. Cell-cycle-regulated expression of STIL controls centriole number in human cells. J Cell Sci (2012) 125:1342–52. doi:10.1242/jcs.099887
190. Tang CJ, Lin SY, Hsu WB, Lin YN, Wu CT, Lin YC, et al. The human microcephaly protein STIL interacts with CPAP and is required for procentriole formation. EMBO J (2011) 30:4790–804. doi:10.1038/emboj.2011.378
191. Vulprecht J, Vulprecht J, David A, Tibelius A, Castiel A, Konotop G, et al. STIL is required for centriole duplication in human cells. J Cell Sci (2012) 125:1353–62. doi:10.1242/jcs.104109
192. Guichard P, Chretien D, Marco S, Tassin AM. Procentriole assembly revealed by cryo-electron tomography. EMBO J (2010) 29:1565–72. doi:10.1038/emboj.2010.45
193. Dupuis-Williams P, Fleury-Aubusson A, de Loubresse NG, Geoffroy H, Vayssié L, Galvani A, et al. Functional role of epsilon-tubulin in the assembly of the centriolar microtubule scaffold. J Cell Biol (2002) 158:1183–93. doi:10.1083/jcb.200205028
194. Dutcher SK, Morrissette NS, Preble AM, Rackley C, Stanga J. Epsilon-tubulin is an essential component of the centriole. Mol Biol Cell (2002) 13:3859–69. doi:10.1091/mbc.E02-04-0205
195. Chang J, Cizmecioglu O, Hoffmann I, Rhee K. PLK2 phosphorylation is critical for CPAP function in procentriole formation during the centrosome cycle. EMBO J (2010) 29:2395–406. doi:10.1038/emboj.2010.118
196. Azimzadeh J, Hergert P, Delouvée A, Euteneuer U, Formstecher E, Khodjakov A, et al. hPOC5 is a centrin-binding protein required for assembly of full-length centrioles. J Cell Biol (2009) 185:101–14. doi:10.1083/jcb.200808082
197. Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK. CPAP is a cell-cycle regulated protein that controls centriole length. Nat Cell Biol (2009) 11:825–31. doi:10.1038/ncb1889
198. Keller LC, Geimer S, Romijn E, Yates J3rd, Zamora I, Marshall WF. Molecular architecture of the centriole proteome: the conserved WD40 domain protein POC1 is required for centriole duplication and length control. Mol Biol Cell (2009) 20:1150–66. doi:10.1091/mbc.E08-06-0619
199. Li J, D’Angiolella V, Seeley ES, Kim S, Kobayashi T, Fu W, et al. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature (2013) 495:255–9. doi:10.1038/nature11941
200. Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell (2002) 3:339–50. doi:10.1016/S1534-5807(02)00258-7
201. Spektor A, Tsang WY, Khoo D, Dynlacht BD. Cep97 and CP110 suppress a cilia assembly program. Cell (2007) 130:678–90. doi:10.1016/j.cell.2007.06.027
202. Berdnik D, Knoblich JA. Drosophila Aurora-A is required for centrosome maturation and actin-dependent asymmetric protein localization during mitosis. Curr Biol (2002) 12:640–7. doi:10.1016/S0960-9822(02)00766-2
203. Hannak E, Kirkham M, Hyman AA, Oegema K. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J Cell Biol (2001) 155:1109–16. doi:10.1083/jcb.200108051
204. Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol (1996) 135:1701–13. doi:10.1083/jcb.135.6.1701
205. Song D, Zhukov TA, Markov O, Qian W, Tockman MS. A new method for lung cancer prognosis via centrosome image feature analysis. Anal Quant Cytol Histol (2012) 34:180–8.
206. Wu ZQ, Liu X. Role for Plk1 phosphorylation of Hbo1 in regulation of replication licensing. Proc Natl Acad Sci U S A (2008) 105:1919–24. doi:10.1073/pnas.0712063105
207. Bornens M, Paintrand M, Berges J, Marty MC, Karsenti E. Structural and chemical characterization of isolated centrosomes. Cell Motil Cytoskeleton (1987) 8:238–49. doi:10.1002/cm.970080305
208. Fletcher L, Cerniglia GJ, Yen TJ, Muschel RJ. Live cell imaging reveals distinct roles in cell cycle regulation for Nek2A and Nek2B. Biochim Biophys Acta (2005) 1744:89–92. doi:10.1016/j.bbamcr.2005.01.007
209. Bahe S, Stierhof YD, Wilkinson CJ, Leiss F, Nigg EA. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J Cell Biol (2005) 171:27–33. doi:10.1083/jcb.200504107
210. Yang J, Adamian M, Li T. Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol Biol Cell (2006) 17:1033–40. doi:10.1091/mbc.E05-10-0943
211. Yang J, Li T. Focus on molecules: rootletin. Exp Eye Res (2006) 83:1–2. doi:10.1016/j.exer.2005.10.013
212. Faragher AJ, Fry AM. Nek2A kinase stimulates centrosome disjunction and is required for formation of bipolar mitotic spindles. Mol Biol Cell (2003) 14:2876–89. doi:10.1091/mbc.E03-02-0108
213. Graser S, Stierhof YD, Nigg EA. Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J Cell Sci (2007) 120:4321–31. doi:10.1242/jcs.020248
214. Fry AM, Arnaud L, Nigg EA. Activity of the human centrosomal kinase, Nek2, depends on an unusual leucine zipper dimerization motif. J Biol Chem (1999) 274:16304–10. doi:10.1074/jbc.274.23.16304
215. Mi J, Guo C, Brautigan DL, Larner JM. Protein phosphatase-1alpha regulates centrosome splitting through Nek2. Cancer Res (2007) 67:1082–9. doi:10.1158/0008-5472.CAN-06-3071
216. Eto M, Elliott E, Prickett TD, Brautigan DL. Inhibitor-2 regulates protein phosphatase-1 complexed with NimA-related kinase to induce centrosome separation. J Biol Chem (2002) 277:44013–20. doi:10.1074/jbc.M208035200
217. Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by Aurora A to promote checkpoint recovery. Nature (2008) 455:119–23. doi:10.1038/nature07185
218. Seki A, Coppinger JA, Du H, Jang CY, Yates JR 3rd, Fang G. Plk1- and beta-TrCP-dependent degradation of Bora controls mitotic progression. J Cell Biol (2008) 181:65–78. doi:10.1083/jcb.200712027
219. Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora A cooperatively activate the kinase Plk1 and control mitotic entry. Science (2008) 320:1655–8. doi:10.1126/science.1157425
220. Helps NR, Luo X, Barker HM, Cohen PT. NIMA-related kinase 2 (Nek2), a cell-cycle-regulated protein kinase localized to centrosomes, is complexed to protein phosphatase 1. Biochem J (2000) 349:509–18. doi:10.1042/0264-6021:3490509
221. Mardin BR, Lange C, Baxter JE, Hardy T, Scholz SR, Fry AM, et al. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat Cell Biol (2010) 12:1166–76. doi:10.1038/ncb2120
222. Pugacheva EN, Golemis EA. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat Cell Biol (2005) 7:937–46. doi:10.1038/ncb1309
223. Matsuo K, Nishimura T, Hayakawa A, Ono Y, Takahashi M. Involvement of a centrosomal protein kendrin in the maintenance of centrosome cohesion by modulating Nek2A kinase activity. Biochem Biophys Res Commun (2010) 398:217–23. doi:10.1016/j.bbrc.2010.06.063
224. Haren L, Stearns T, Luders J. Plk1-dependent recruitment of gamma-tubulin complexes to mitotic centrosomes involves multiple PCM components. PLoS One (2009) 4:e5976. doi:10.1371/journal.pone.0005976
225. Rosenblatt J. Spindle assembly: asters part their separate ways. Nat Cell Biol (2005) 7:219–22. doi:10.1038/ncb0305-219
226. Sharp DJ, Yu KR, Sisson JC, Sullivan W, Scholey JM. Antagonistic microtubule-sliding motors position mitotic centrosomes in Drosophila early embryos. Nat Cell Biol (1999) 1:51–4. doi:10.1038/9025
227. Tanenbaum ME, Macurek L, Galjart N, Medema RH. Dynein, Lis1 and CLIP-170 counteract Eg5-dependent centrosome separation during bipolar spindle assembly. EMBO J (2008) 27:3235–45. doi:10.1038/emboj.2008.242
228. Tanenbaum ME, Medema RH. Mechanisms of centrosome separation and bipolar spindle assembly. Dev Cell (2010) 19:797–806. doi:10.1016/j.devcel.2010.11.011
229. Woodcock SA, Rushton HJ, Castañeda-Saucedo E, Myant K, White GR, Blyth K, et al. Tiam1-Rac signaling counteracts Eg5 during bipolar spindle assembly to facilitate chromosome congression. Curr Biol (2010) 20:669–75. doi:10.1016/j.cub.2010.02.033
230. Cole DG, Saxton WM, Sheehan KB, Scholey JMA. “Slow” homotetrameric kinesin-related motor protein purified from Drosophila embryos. J Biol Chem (1994) 269:22913–6.
231. Sawin KE, Mitchison TJ, Wordeman LG. Evidence for kinesin-related proteins in the mitotic apparatus using peptide antibodies. J Cell Sci (1992) 101(Pt 2):303–13.
232. Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol (2000) 150:975–88. doi:10.1083/jcb.150.5.975
233. Whitehead CM, Rattner JB. Expanding the role of HsEg5 within the mitotic and post-mitotic phases of the cell cycle. J Cell Sci (1998) 111(Pt 17):2551–61.
234. Sawin KE, Mitchison TJ. Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc Natl Acad Sci U S A (1995) 92:4289–93. doi:10.1073/pnas.92.10.4289
235. Bertran MT, Sdelci S, Regué L, Avruch J, Caelles C, Roig J. Nek9 is a Plk1-activated kinase that controls early centrosome separation through Nek6/7 and Eg5. EMBO J (2011) 30:2634–47. doi:10.1038/emboj.2011.179
236. Mardin BR, Agircan FG, Lange C, Schiebel E. Plk1 controls the Nek2A-PP1gamma antagonism in centrosome disjunction. Curr Biol (2011) 21:1145–51. doi:10.1016/j.cub.2011.05.047
237. Smith E, Hégarat N, Vesely C, Roseboom I, Larch C, Streicher H, et al. Differential control of Eg5-dependent centrosome separation by Plk1 and Cdk1. EMBO J (2011) 30:2233–45. doi:10.1038/emboj.2011.120
238. Blangy A, Lane HA, d’Hérin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell (1995) 83:1159–69. doi:10.1016/0092-8674(95)90142-6
239. Mardin BR, Schiebel E. Breaking the ties that bind: new advances in centrosome biology. J Cell Biol (2012) 197:11–8. doi:10.1083/jcb.201108006
240. Kaseda K, McAinsh AD, Cross RA. Dual pathway spindle assembly increases both the speed and the fidelity of mitosis. Biol Open (2012) 1:12–8. doi:10.1242/bio.2011012
241. Aubin JE, Osborn M, Weber K. Variations in the distribution and migration of centriole duplexes in mitotic PtK2 cells studied by immunofluorescence microscopy. J Cell Sci (1980) 43:177–94.
242. Rattner JB, Berns MW. Centriole behavior in early mitosis of rat kangaroo cells (PTK2). Chromosoma (1976) 54:387–95. doi:10.1007/BF00292817
243. Rosenblatt J, Cramer LP, Baum B, McGee KM. Myosin II-dependent cortical movement is required for centrosome separation and positioning during mitotic spindle assembly. Cell (2004) 117:361–72. doi:10.1016/S0092-8674(04)00341-1
244. Toso A, Winter JR, Garrod AJ, Amaro AC, Meraldi P, McAinsh AD. Kinetochore-generated pushing forces separate centrosomes during bipolar spindle assembly. J Cell Biol (2009) 184:365–72. doi:10.1083/jcb.200809055
245. Waters JC, Cole RW, Rieder CL. The force-producing mechanism for centrosome separation during spindle formation in vertebrates is intrinsic to each aster. J Cell Biol (1993) 122:361–72. doi:10.1083/jcb.122.2.361
246. Whitehead CM, Winkfein RJ, Rattner JB. The relationship of HsEg5 and the actin cytoskeleton to centrosome separation. Cell Motil Cytoskeleton (1996) 35:298–308. doi:10.1002/(SICI)1097-0169(1996)35:4<298::AID-CM3>3.0.CO;2-3
247. Silkworth WT, Nardi IK, Paul R, Mogilner A, Cimini D. Timing of centrosome separation is important for accurate chromosome segregation. Mol Biol Cell (2012) 23:401–11. doi:10.1091/mbc.E11-02-0095
248. Landry JJM, Pyl PT, Rausch T, Zichner T, Tekkedil MM, Stütz AM, et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 (2013) 3(8):1213–24. doi:10.1534/g3.113.005777
249. Boveri T. Ergebnisse Uber die Konstitution der Chromatischen Substanz des Zellkerns. Jena: Gustav Fischer (1904).
250. Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci U S A (1998) 95:2950–5. doi:10.1073/pnas.95.6.2950
251. Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P, et al. Centrosome defects and genetic instability in malignant tumors. Cancer Res (1998) 58:3974–85.
252. Carroll PE, Okuda M, Horn HF, Biddinger P, Stambrook PJ, Gleich LL, et al. Centrosome hyperamplification in human cancer: chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene (1999) 18:1935–44. doi:10.1038/sj.onc.1202515
253. Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci U S A (2000) 97:10002–7. doi:10.1073/pnas.170093297
254. Ghadimi BM, Sackett DL, Difilippantonio MJ, Schröck E, Neumann T, Jauho A, et al. Centrosome amplification and instability occurs exclusively in aneuploid, but not in diploid colorectal cancer cell lines, and correlates with numerical chromosomal aberrations. Genes Chromosomes Cancer (2000) 27:183–90. doi:10.1002/(SICI)1098-2264(200002)27:2<183::AID-GCC10>3.0.CO;2-P
255. Gustafson LM, Gleich LL, Fukasawa K, Chadwell J, Miller MA, Stambrook PJ, et al. Centrosome hyperamplification in head and neck squamous cell carcinoma: a potential phenotypic marker of tumor aggressiveness. Laryngoscope (2000) 110:1798–801. doi:10.1097/00005537-200011000-00004
256. Kuo KK, Sato N, Mizumoto K, Maehara N, Yonemasu H, Ker CG, et al. Centrosome abnormalities in human carcinomas of the gallbladder and intrahepatic and extrahepatic bile ducts. Hepatology (2000) 31:59–64. doi:10.1002/hep.510310112
257. Lingle WL, Salisbury JL. Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am J Pathol (1999) 155:1941–51. doi:10.1016/S0002-9440(10)65513-7
258. Neben K, Giesecke C, Schweizer S, Ho AD, Kramer A. Centrosome aberrations in acute myeloid leukemia are correlated with cytogenetic risk profile. Blood (2003) 101:289–91. doi:10.1182/blood-2002-04-1188
259. Roshani L, Fujioka K, Auer G, Kjellman M, Lagercrantz S, Larsson C. Aberrations of centrosomes in adrenocortical tumors. Int J Oncol (2002) 20:1161–5.
260. Sato N, Mizumoto K, Nakamura M, Nakamura K, Kusumoto M, Niiyama H, et al. Centrosome abnormalities in pancreatic ductal carcinoma. Clin Cancer Res (1999) 5:963–70.
261. Sato N, Mizumoto K, Nakamura M, Tanaka M. Radiation-induced centrosome overduplication and multiple mitotic spindles in human tumor cells. Exp Cell Res (2000) 255:321–6. doi:10.1006/excr.1999.4797
262. Skyldberg B, Fujioka K, Hellström AC, Sylvén L, Moberger B, Auer G. Human papillomavirus infection, centrosome aberration, and genetic stability in cervical lesions. Mod Pathol (2001) 14:279–84. doi:10.1038/modpathol.3880303
263. Weber RG, Bridger JM, Benner A, Weisenberger D, Ehemann V, Reifenberger G, et al. Centrosome amplification as a possible mechanism for numerical chromosome aberrations in cerebral primitive neuroectodermal tumors with TP53 mutations. Cytogenet Cell Genet (1998) 83:266–9. doi:10.1159/000015168
264. Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci (2011) 7:1122–44. doi:10.7150/ijbs.7.1122
265. Chandhok NS, Pellman D. A little CIN may cost a lot: revisiting aneuploidy and cancer. Curr Opin Genet Dev (2009) 19:74–81. doi:10.1016/j.gde.2008.12.004
266. Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol (2009) 10:478–87. doi:10.1038/nrm2718
267. Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet (2008) 24:457–66. doi:10.1016/j.tig.2008.07.002
268. Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol (2010) 20:R285–95. doi:10.1016/j.cub.2010.01.034
269. Lingle WL, Barrett SL, Negron VC, D’Assoro AB, Boeneman K, Liu W, et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A (2002) 99:1978–83. doi:10.1073/pnas.032479999
270. Pihan GA, Wallace J, Zhou Y, Doxsey SJ. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res (2003) 63:1398–404.
271. Nigg EA. Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer (2002) 2:815–25. doi:10.1038/nrc924
272. Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol (2004) 5:45–54. doi:10.1038/nrm1276
273. Magnani I, Novielli C, Fontana L, Tabano S, Rovina D, Moroni RF, et al. Differential signature of the centrosomal MARK4 isoforms in glioma. Anal Cell Pathol (Amst) (2011) 34:319–38. doi:10.3233/ACP-2011-0031
274. Miyachika Y, Yamamoto Y, Matsumoto H, Nishijima J, Kawai Y, Nagao K, et al. Centrosome amplification in bladder washing cytology specimens is a useful prognostic biomarker for non-muscle invasive bladder cancer. Cancer Genet (2013) 206:12–8. doi:10.1016/j.cancergen.2012.11.004
275. Bakhoum SF, Compton DA. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest (2012) 122:1138–43. doi:10.1172/JCI59954
276. Galimberti F, Thompson SL, Ravi S, Compton DA, Dmitrovsky E. Anaphase catastrophe is a target for cancer therapy. Clin Cancer Res (2011) 17:1218–22. doi:10.1158/1078-0432.CCR-10-1178
277. Korzeniewski N, Hohenfellner M, Duensing S. The centrosome as potential target for cancer therapy and prevention. Expert Opin Ther Targets (2013) 17:43–52. doi:10.1517/14728222.2013.731396
278. Pihan GA, Doxsey SJ. The mitotic machinery as a source of genetic instability in cancer. Semin Cancer Biol (1999) 9:289–302. doi:10.1006/scbi.1999.0131
279. Yamamoto Y, Misumi T, Eguchi S, Chochi Y, Kitahara S, Nakao M, et al. Centrosome amplification as a putative prognostic biomarker for the classification of urothelial carcinomas. Hum Pathol (2011) 42:1923–30. doi:10.1016/j.humpath.2011.02.013
280. Chretien D, Buendia B, Fuller SD, Karsenti E. Reconstruction of the centrosome cycle from cryoelectron micrographs. J Struct Biol (1997) 120:117–33. doi:10.1006/jsbi.1997.3928
281. de Harven E. Early observations of centrioles and mitotic spindle fibers by transmission electron microscopy. Biol Cell (1994) 80:107–9. doi:10.1016/0248-4900(94)90028-0
282. Rieder CL, Borisy GG. The centrosome cycle in PtK2 cells: asymmetric distribution and structural changes in the pericentriolar material. Biol Cell (1982) 44:117–32.
283. Rieder CL, Faruki S, Khodjakov A. The centrosome in vertebrates: more than a microtubule-organizing center. Trends Cell Biol (2001) 11:413–9. doi:10.1016/S0962-8924(01)02085-2
284. D’Assoro AB, Lingle WL, Salisbury JL. Centrosome amplification and the development of cancer. Oncogene (2002) 21:6146–53. doi:10.1038/sj.onc.1205772
285. Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science (1996) 271:1744–7. doi:10.1126/science.271.5256.1744
286. Salisbury JL, D’Assoro AB, Lingle WL. Centrosome amplification and the origin of chromosomal instability in breast cancer. J Mammary Gland Biol Neoplasia (2004) 9:275–83. doi:10.1023/B:JOMG.0000048774.27697.30
287. Chiba S, Okuda M, Mussman JG, Fukasawa K. Genomic convergence and suppression of centrosome hyperamplification in primary p53-/- cells in prolonged culture. Exp Cell Res (2000) 258:310–21. doi:10.1006/excr.2000.4916
288. Mussman JG, Horn HF, Carroll PE, Okuda M, Tarapore P, Donehower LA, et al. Synergistic induction of centrosome hyperamplification by loss of p53 and cyclin E overexpression. Oncogene (2000) 19:1635–46. doi:10.1038/sj.onc.1203460
289. Khodjakov A, Rieder CL, Sluder G, Cassels G, Sibon O, Wang CL. De novo formation of centrosomes in vertebrate cells arrested during S phase. J Cell Biol (2002) 158:1171–81. doi:10.1083/jcb.200205102
290. La Terra S, English CN, Hergert P, McEwen BF, Sluder G, Khodjakov A. The de novo centriole assembly pathway in HeLa cells: cell cycle progression and centriole assembly/maturation. J Cell Biol (2005) 168:713–22. doi:10.1083/jcb.200411126
291. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature (2005) 437:1043–7. doi:10.1038/nature04217
292. Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J (2002) 21:483–92. doi:10.1093/emboj/21.4.483
293. Hu T, Miller CM, Ridder GM, Aardema MJ. Characterization of p53 in Chinese hamster cell lines CHO-K1, CHO-WBL, and CHL: implications for genotoxicity testing. Mutat Res (1999) 426:51–62. doi:10.1016/S0027-5107(99)00077-9
294. Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev (2007) 17:157–62. doi:10.1016/j.gde.2007.02.011
295. Duensing S, Duensing A, Crum CP, Munger K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res (2001) 61:2356–60.
296. Tarapore P, Horn HF, Tokuyama Y, Fukasawa K. Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene (2001) 20:3173–84. doi:10.1038/sj.onc.1204424
297. Dutertre S, Hamard-Peron E, Cremet JY, Thomas Y, Prigent C. The absence of p53 aggravates polyploidy and centrosome number abnormality induced by Aurora-C overexpression. Cell Cycle (2005) 4:1783–7. doi:10.4161/cc.4.12.2172
298. Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell (2001) 12:1315–28. doi:10.1091/mbc.12.5.1315
299. Khan SH, Wahl GM. p53 and pRb prevent rereplication in response to microtubule inhibitors by mediating a reversible G1 arrest. Cancer Res (1998) 58:396–401.
300. Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol (1998) 18:1055–64.
301. Shackney SE, Smith CA, Miller BW, Burholt DR, Murtha K, Giles HR, et al. Model for the genetic evolution of human solid tumors. Cancer Res (1989) 49:3344–54.
302. Leidel S, Delattre M, Cerutti L, Baumer K, Gonczy P. SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat Cell Biol (2005) 7:115–25. doi:10.1038/ncb1220
303. Peel N, Stevens NR, Basto R, Raff JW. Overexpressing centriole-replication proteins in vivo induces centriole overduplication and de novo formation. Curr Biol (2007) 17:834–43. doi:10.1016/j.cub.2007.04.036
304. Ko MA, Rosario CO, Hudson JW, Kulkarni S, Pollett A, Dennis JW, et al. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat Genet (2005) 37:883–8. doi:10.1038/ng1605
305. Pellegrino R, Calvisi DF, Ladu S, Ehemann V, Staniscia T, Evert M, et al. Oncogenic and tumor suppressive roles of polo-like kinases in human hepatocellular carcinoma. Hepatology (2010) 51:857–68. doi:10.1002/hep.23467
306. Marshall WF, Vucica Y, Rosenbaum JL. Kinetics and regulation of de novo centriole assembly. Implications for the mechanism of centriole duplication. Curr Biol (2001) 11:308–17. doi:10.1016/S0960-9822(01)00094-X
307. Palazzo RE, Vaisberg E, Cole RW, Rieder CL. Centriole duplication in lysates of Spisula solidissima oocytes. Science (1992) 256:219–21. doi:10.1126/science.1566068
308. Kallenbach RJ, Mazia D. Origin and maturation of centrioles in association with the nuclear envelope in hypertonic-stressed sea urchin eggs. Eur J Cell Biol (1982) 28:68–76.
309. Balczon R, West K. The identification of mammalian centrosomal antigens using human autoimmune anticentrosome antisera. Cell Motil Cytoskeleton (1991) 20:121–35. doi:10.1002/cm.970200205
310. Maniotis A, Schliwa M. Microsurgical removal of centrosomes blocks cell reproduction and centriole generation in BSC-1 cells. Cell (1991) 67:495–504. doi:10.1016/0092-8674(91)90524-3
311. Prosser SL, Straatman KR, Fry AM. Molecular dissection of the centrosome overduplication pathway in S-phase-arrested cells. Mol Cell Biol (2009) 29:1760–73. doi:10.1128/MCB.01124-08
312. Barenz F, Mayilo D, Gruss OJ. Centriolar satellites: busy orbits around the centrosome. Eur J Cell Biol (2011) 90:983–9. doi:10.1016/j.ejcb.2011.07.007
313. Kubo A, Sasaki H, Yuba-Kubo A, Tsukita S, Shiina N. Centriolar satellites: molecular characterization, ATP-dependent movement toward centrioles and possible involvement in ciliogenesis. J Cell Biol (1999) 147:969–80. doi:10.1083/jcb.147.5.969
314. Lopes CA, Prosser SL, Romio L, Hirst RA, O’Callaghan C, Woolf AS, et al. Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J Cell Sci (2011) 124:600–12. doi:10.1242/jcs.077156
315. Dammermann A, Merdes A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J Cell Biol (2002) 159:255–66. doi:10.1083/jcb.200204023
316. Hames RS, Crookes RE, Straatman KR, Merdes A, Hayes MJ, Faragher AJ, et al. Dynamic recruitment of Nek2 kinase to the centrosome involves microtubules, PCM-1, and localized proteasomal degradation. Mol Biol Cell (2005) 16:1711–24. doi:10.1091/mbc.E04-08-0688
317. Loffler H, Fechter A, Liu FY, Poppelreuther S, Kramer A. DNA damage-induced centrosome amplification occurs via excessive formation of centriolar satellites. Oncogene (2013) 32:2963–72. doi:10.1038/onc.2012.310
318. Kleylein-Sohn J, Pöllinger B, Ohmer M, Hofmann F, Nigg EA, Hemmings BA, et al. Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET. J Cell Sci (2012) 125:5391–402. doi:10.1242/jcs.107474
319. Prosser SL, Samant MD, Baxter JE, Morrison CG, Fry AM. Oscillation of APC/C activity during cell cycle arrest promotes centrosome amplification. J Cell Sci (2012) 125:5353–68. doi:10.1242/jcs.106096
320. Ma HT, Tsang YH, Marxer M, Poon RY. Cyclin A2-cyclin-dependent kinase 2 cooperates with the PLK1-SCFbeta-TrCP1-EMI1-anaphase-promoting complex/cyclosome axis to promote genome reduplication in the absence of mitosis. Mol Cell Biol (2009) 29:6500–14. doi:10.1128/MCB.00669-09
321. Brinkley BR, Cox SM, Pepper DA, Wible L, Brenner SL, Pardue RL. Tubulin assembly sites and the organization of cytoplasmic microtubules in cultured mammalian cells. J Cell Biol (1981) 90:554–62. doi:10.1083/jcb.90.3.554
322. Pihan GA, Purohit A, Wallace J, Malhotra R, Liotta L, Doxsey SJ. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res (2001) 61:2212–9.
323. Ring D, Hubble R, Kirschner M. Mitosis in a cell with multiple centrioles. J Cell Biol (1982) 94:549–56. doi:10.1083/jcb.94.3.549
324. Salisbury JL, Whitehead CM, Lingle WL, Barrett SL. Centrosomes and cancer. Biol Cell (1999) 91:451–60. doi:10.1111/j.1768-322X.1999.tb01100.x
325. Sharp GA, Osborn M, Weber K. Ultrastructure of multiple microtubule initiation sites in mouse neuroblastoma cells. J Cell Sci (1981) 47:1–24.
326. Gisselsson D, Håkanson U, Stoller P, Marti D, Jin Y, Rosengren AH, et al. When the genome plays dice: circumvention of the spindle assembly checkpoint and near-random chromosome segregation in multipolar cancer cell mitoses. PLoS One (2008) 3:e1871. doi:10.1371/journal.pone.0001871
327. Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol (2011) 12:385–92. doi:10.1038/nrm3115
328. Varmark H, Sparks CA, Nordberg JJ, Koppetsch BS, Theurkauf WE. DNA damage-induced cell death is enhanced by progression through mitosis. Cell Cycle (2009) 8:2951–63. doi:10.4161/cc.8.18.9539
329. Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol (2001) 11:18–21. doi:10.1016/S0962-8924(00)01872-9
330. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene (2004) 23:2825–37. doi:10.1038/sj.onc.1207528
331. Vakifahmetoglu H, Olsson M, Tamm C, Heidari N, Orrenius S, Zhivotovsky B. DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ (2008) 15:555–66. doi:10.1038/sj.cdd.4402286
332. Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature (2009) 460:278–82. doi:10.1038/nature08136
333. Weaver BA, Cleveland DW. Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res (2007) 67:10103–5. doi:10.1158/0008-5472.CAN-07-2266
334. Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell (2007) 11:25–36. doi:10.1016/j.ccr.2006.12.003
335. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell (2013) 153:666–77. doi:10.1016/j.cell.2013.03.021
336. Heng HH, Stevens JB, Bremer SW, Liu G, Abdallah BY, Ye CJ. Evolutionary mechanisms and diversity in cancer. Adv Cancer Res (2011) 112:217–53. doi:10.1016/B978-0-12-387688-1.00008-9
337. Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, Layer RM, et al. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res (2013) 23:762–76. doi:10.1101/gr.143677.112
338. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature (2011) 472:90–4. doi:10.1038/nature09807
339. Shen MM. Chromoplexy: a new category of complex rearrangements in the cancer genome. Cancer Cell (2013) 23:567–9. doi:10.1016/j.ccr.2013.04.025
340. Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev (2008) 22:2189–203. doi:10.1101/gad.1700908
341. Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. Spindle multipolarity is prevented by centrosomal clustering. Science (2005) 307:127–9. doi:10.1126/science.1104905
342. Silkworth WT, Nardi IK, Scholl LM, Cimini D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One (2009) 4:e6564. doi:10.1371/journal.pone.0006564
343. Krzywicka-Racka A, Sluder G. Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J Cell Biol (2011) 194:199–207. doi:10.1083/jcb.201101073
344. Uetake Y, Sluder G. Cell cycle progression after cleavage failure: mammalian somatic cells do not possess a “tetraploidy checkpoint”. J Cell Biol (2004) 165:609–15. doi:10.1083/jcb.200403014
345. Musacchio A. Spindle assembly checkpoint: the third decade. Philos Trans R Soc Lond B Biol Sci (2011) 366:3595–604. doi:10.1098/rstb.2011.0072
346. Sluder G, Thompson EA, Miller FJ, Hayes J, Rieder CL. The checkpoint control for anaphase onset does not monitor excess numbers of spindle poles or bipolar spindle symmetry. J Cell Sci (1997) 110(Pt 4):421–9.
347. Nicklas RB, Ward SC, Gorbsky GJ. Kinetochore chemistry is sensitive to tension and may link mitotic forces to a cell cycle checkpoint. J Cell Biol (1995) 130:929–39. doi:10.1083/jcb.130.4.929
348. Tan AL, Rida PC, Surana U. Essential tension and constructive destruction: the spindle checkpoint and its regulatory links with mitotic exit. Biochem J (2005) 386:1–13. doi:10.1042/BJ20041415
349. Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, et al. Centrosome amplification can initiate tumorigenesis in flies. Cell (2008) 133:1032–42. doi:10.1016/j.cell.2008.05.039
350. Leber B, Maier B, Fuchs F, Chi J, Riffel P, Anderhub S, et al. Proteins required for centrosome clustering in cancer cells. Sci Transl Med (2010) 2:33ra38. doi:10.1126/scitranslmed.3000915
351. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature (1997) 386:623–7. doi:10.1038/386623a0
353. McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep (2012) 13:528–38. doi:10.1038/embor.2012.61
354. Bakhoum SF, Genovese G, Compton DA. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol (2009) 19:1937–42. doi:10.1016/j.cub.2009.09.055
355. Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol (2001) 153:517–27. doi:10.1083/jcb.153.3.517
356. Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, et al. Mutations of mitotic checkpoint genes in human cancers. Nature (1998) 392:300–3. doi:10.1038/32688
357. Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol (2007) 8:379–93. doi:10.1038/nrm2163
358. Yao Y, Dai W. Shugoshins function as a guardian for chromosomal stability in nuclear division. Cell Cycle (2012) 11:2631–42. doi:10.4161/cc.20633
359. Li J, Wang J, Jiao H, Liao J, Xu X. Cytokinesis and cancer: polo loves ROCK‘n’ Rho(A). J Genet Genomics (2010) 37:159–72. doi:10.1016/S1673-8527(09)60034-5
360. Sagona AP, Stenmark H. Cytokinesis and cancer. FEBS Lett (2010) 584:2652–61. doi:10.1016/j.febslet.2010.03.044
361. Rieder CL. Mitosis in vertebrates: the G2/M and M/A transitions and their associated checkpoints. Chromosome Res (2011) 19:291–306. doi:10.1007/s10577-010-9178-z
362. Forment JV, Kaidi A, Jackson SP. Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer (2012) 12:663–70. doi:10.1038/nrc3352
363. Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell (2013) 152:1226–36. doi:10.1016/j.cell.2013.02.023
364. Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet (2012) 13:189–203. doi:10.1038/nrg3123
365. Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep (2012) 13:501–14. doi:10.1038/embor.2012.55
366. Gisselsson D. Chromosome instability in cancer: how, when, and why? Adv Cancer Res (2003) 87:1–29. doi:10.1016/S0065-230X(03)87293-7
367. Holland AJ, Cleveland DW. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med (2012) 18:1630–8. doi:10.1038/nm.2988
368. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell (2011) 144:27–40. doi:10.1016/j.cell.2010.11.055
369. Kloosterman WP, Hoogstraat M, Paling O, Tavakoli-Yaraki M, Renkens I, Vermaat JS, et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol (2011) 12:R103. doi:10.1186/gb-2011-12-10-r103
370. Tubio JM, Estivill X. Cancer: when catastrophe strikes a cell. Nature (2011) 470:476–7. doi:10.1038/470476a
Keywords: aneuploidy, centrosome, centriole, chromoanagenesis, chromothripsis, micronuclei, mitosis, CIN
Citation: Pihan GA (2013) Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front. Oncol. 3:277. doi: 10.3389/fonc.2013.00277
Received: 29 August 2013; Accepted: 28 October 2013;
Published online: 12 November 2013.
Edited by:
Samuel F. Bakhoum, Geisel School of Medicine at Dartmouth, USAReviewed by:
Valerio Donato, New York University Medical Center, USADaniela Cimini, Virginia Tech, USA
Copyright: © 2013 Pihan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: German A. Pihan, Department of Pathology and Laboratory Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, YA-309, 330 Brookline Avenue, Boston, MA 02215, USA e-mail: gpihan@bidmc.harvard.edu