 Camillo Porta
Camillo Porta Chiara Paglino1
Chiara Paglino1 Alessandra Mosca
Alessandra Mosca- 1Medical Oncology, Fondazione I.R.C.C.S. Policlinico San Matteo University Hospital Foundation, Pavia, Italy
- 2Medical Oncology, Maggiore della Carità Hospital, University of Eastern Piedmont “A. Avogadro”, Novara, Italy
The phosphatidylinositol-3-kinase (PI3K)/Akt and the mammalian target of rapamycin (mTOR) signaling pathways are two pathways crucial to many aspects of cell growth and survival, in physiological as well as in pathological conditions (e.g., cancer). Indeed, they are so interconnected that, in a certain sense, they could be regarded as a single, unique pathway. In this paper, after a general overview of the biological significance and the main components of these pathways, we address the present status of the development of specific PI3K, Akt, and mTOR inhibitors, from already registered medicines to novel compounds that are just leaving the laboratory bench.
Introduction
The phosphatidylinositol-3-kinase (PI3K)/Akt and the mammalian target of rapamycin (mTOR) signaling pathways are both crucial to many aspects of cell growth and survival, in physiological as well as in pathological conditions. They are so interconnected that, in a certain sense, they could be regarded as a single, unique pathway (Figure 1) that, in turn, heavily interacts also with many other pathways, including that of the hypoxia inducible factors (HIFs).
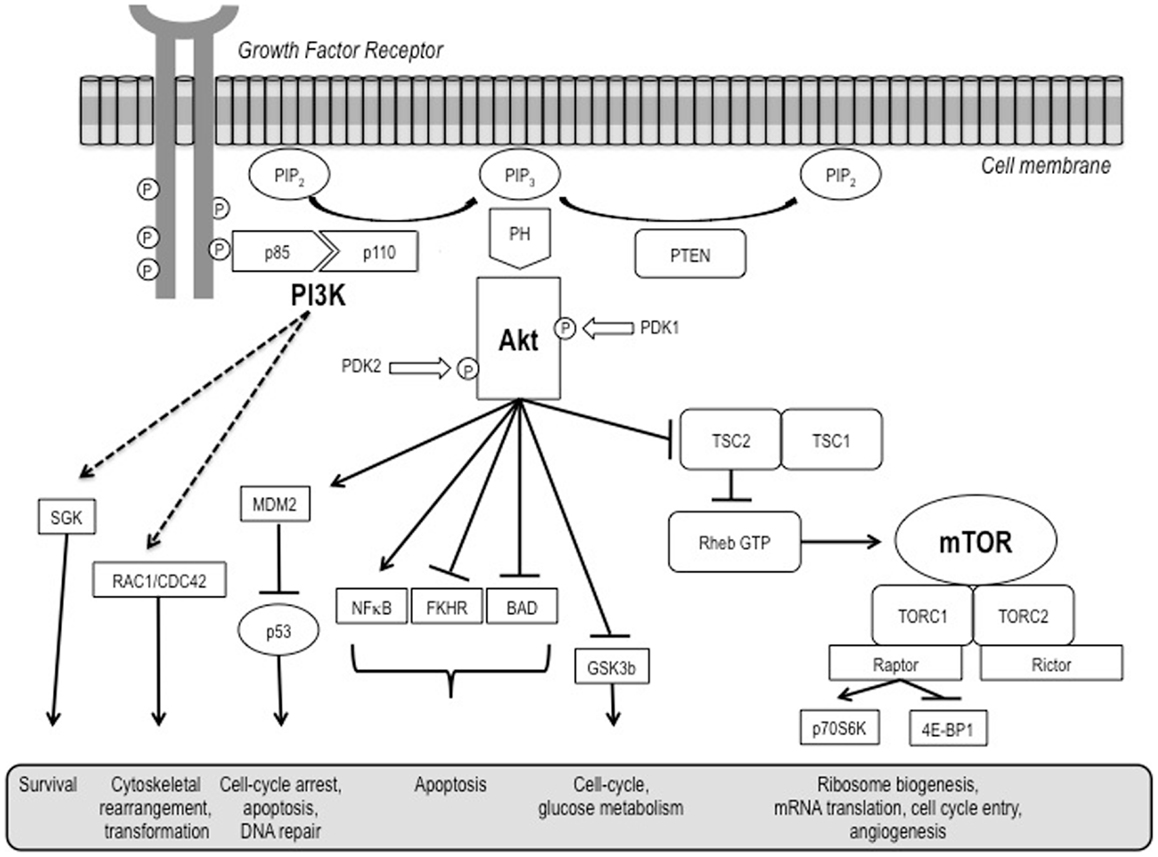
Figure 1. A schematic representation of the PI3K/Akt/mTOR pathway.
The PI3K/Akt pathway is a key regulator of survival during cellular stress (1). Since tumors exist in an intrinsically stressful environment (characterized by limited nutrient and oxygen supply, as well as by low pH), the role of this pathway in cancer appears to be crucial.
Mammalian target of rapamycin is a serine/threonine kinase ubiquitously expressed in mammalian cells (2). It picks up and integrates signals initiated by nutrient intake, growth factors, and other cellular stimuli to regulate downstream signaling and protein synthesis. Through its downstream effectors, 4EBP1 and P70S6 kinase (S6K), it is involved in the initiation of ribosomal translation of mRNA into proteins necessary for cell growth, cell cycle progression, and cell metabolism.
Somatic mutations and/or gains and losses of key genes are among a number of genetic alterations affecting these pathways in a number of different solid and hematological tumors [including big killers such as breast and colon cancer, as well as relatively less frequent neoplasms such as neuroendocrine tumors (NETs), kidney cancer, and some lymphomas]. The activation of the PI3K/Akt/mTOR pathway results in a profound disturbance of control of cell growth and survival, which ultimately leads to a competitive growth advantage, metastatic competence, angiogenesis, and therapy resistance.
Thus, this complex pathway has been taken into consideration as one of the most attractive targets for the development of anticancer agents (3, 4).
PI3K Structure and Functions
Phosphatidyl-inositol-3-kinases (PI3Ks) constitute a lipid kinase family characterized by the capability to phosphorylate inositol ring 3′-OH group in inositol phospholipids (5). Class I PI3Ks are heterodimers composed of a catalytic (CAT) subunit (i.e., p110) and an adaptor/regulatory subunit (i.e., p85).
This class is further divided into two subclasses: subclass IA (PI3Kα, β, and δ), which is activated by receptors with protein tyrosine kinase activity, and subclass IB (PI3Kγ), which is activated by receptors coupled with G proteins (5).
Activation of growth factor receptor protein tyrosine kinases results in autophosphorylation on tyrosine residues. PI3K is then recruited to the membrane by directly binding to phosphotyrosine consensus residues of growth factor receptors or adaptors through one of the two SH2 domains in the adaptor subunit. This leads to allosteric activation of the CAT subunit. In a few seconds, PI3K activation leads to the production of the second messenger phosphatidylinositol-3,4,5-triphosphate (PI3,4,5-P3) from the substrate phosphatidylinositol-4,4-bisphosphate (PI-4,5-P2). PI3,4,5-P3 then recruits a subset of signaling proteins with pleckstrin homology (PH) domains to the membrane, including protein serine/threonine kinase-3′-phosphoinositide-dependent kinase 1 (PDK1) and Akt/protein kinase B (PKB) (5, 6). Akt/PKB, on its own, regulates several cell processes involved in cell survival and cell cycle progression.
As far as cell survival is concerned, Akt/PKB can inactivate pro-apoptotic factors such as Bad and Procaspase-9, as well as the Forkhead family of transcription factors that induce the expression of other pro-apoptotic factors, such as Fas-ligand (FasL) (7, 8). Akt/PKB activation has been related to an increased resistance of prostate cancer cells to apoptosis mediated by tumor necrosis factor (TNF)-related apoptosis inducing ligand (TRAIL)/APO-2L (9). Finally, Akt/PKB also activates the IκB kinase (IKK), a positive regulator of the survival factor NFκB. Notably, a strong biological link between the NFκB and the PI3K/Akt pathways in the modulation of anti-apoptotic effects in lymphoma cells exposed to the irreversible inhibitor of the activation of NFκB and the phosphorylation of IκBα BAY11-7085 has been also shown (10).
As for cell cycle progression and cell growth, several targets of Akt are involved in protein synthesis, glycogen metabolism, and cell cycle regulation (6), including the same mTOR, glycogen synthase kinase-3 (GSK3), insulin receptor substrate-1 (IRS-1), the cyclin-dependent kinase inhibitors p21CIP1/WAF1 and p27KIP1, and possibly also Raf-1, a member of the MAPK pathway. With regard to GSK3, Akt/PKB triggers a network that positively regulates G1/S cell cycle progression through inactivation of GSK3-β, leading to increased cyclin D1, and inhibition of the Forkhead family transcription factors and the tumor suppressor tuberin (TSC2), ultimately resulting in the reduction in p27Kip1 (11).
Akt Structure and Functions
Akt kinases belong to the AGC kinase family, related to AMP/GMP kinases and protein kinase C. They consist of three conserved domains, an N-terminal PH domain, a central kinase CAT domain, and a C-terminal extension (EXT) containing a regulatory hydrophobic motif (HM) (12). Among the Akt isoforms, the PH domains are ~80% identical and ~30% identical to PH domains in pleckstrin and other proteins. The linker (LINK) region connecting the PH domain to the CAT domain is poorly conserved among the Akt isoforms (17–46% identical) and has no significant homology to any other human protein (12). The consensus CAT domain is ~90% identical among the Akt isoforms and is closely related the PKC, PKA, SGK, and S6 subfamilies of the AGC kinase family (12). The C-terminal EXT is ~70% identical among the Akt isoforms and is most closely related to the PKC family (12).
mTOR Structure and Functions
Mammalian target of rapamycin is a key protein evolutionarily conserved from yeast to man and is essential for life. Indeed, embryonic mutations in mTOR proved to be lethal.
In normal cells, mTOR activity is controlled by positive and negative upstream regulators (13). Positive regulators include growth factors and their receptors, such as insulin-like growth factor-1 (IGF-1) and its cognate receptor IFGR-1, members of the human epidermal growth factor receptor (HER) family and associated ligands, and vascular endothelial growth factor receptors (VEGFRs) and their ligands, which transmit signals to mTOR through the PI3K-Akt. Negative regulators of mTOR activity include phosphatase and tensin homolog (PTEN) that inhibits signaling through the PI3K-Akt pathway, and tuberous sclerosis complex (TSC) 1 (hamartin) and TSC2 (tuberin). Phosphorylation of TSC2 by Akt releases its inhibitory effect on mTOR and up-regulates mTOR activity. Another negative regulator, LKB1, is in an energy-sensing pathway upstream of TSC (14).
Mammalian target of rapamycin activity is carried out by two distinct complexes: mTORC1 and mTORC2.
The mTORC1 complex is made up of mTOR, Raptor, mLST8, and PRAS40. It is extremely sensitive to rapamycin and thus represents the target of first-generation mTOR inhibitors. It also activates S6K and inactivates 4EBP1, leading to protein translation and cell growth (13).
The mTORC2 complex is composed of mTOR, Rictor, Sin1, and mLST8. It is less sensitive to rapamycin and its role in normal cell function and oncogenesis has not been well clarified. However, it is known to activate Akt, thereby promoting cell proliferation and survival. The canonical pathway of mTOR activation depends on mitogen-driven signaling through PI3K/Akt, although alternative non-Akt dependent activation through the Ras/MEK/ERK pathway is now recognized (15).
Altogether, mTOR activation leads to increased synthesis of multiple proteins. These include several that have been implicated in the pathogenesis of multiple tumors, e.g., cyclin D1, which allows progression of cells through the cell cycle (16), and HIF, which drive the expression of pro-angiogenic growth factors such as VEGF (17).
PTEN as a Regulator of the PI3K/Akt/mTOR Pathway
The PTEN deleted on chromosome 10 (PTEN) is a key molecule downstream of the PI3K/Akt pathway. This phosphatase, endowed with dual activity on both lipids and proteins, acts as a tumor suppressor by inhibiting cell growth and enhancing cellular sensitivity to apoptosis and anoikis, i.e., an epithelial cell-peculiar type of apoptosis triggered by alterations in integrin–extracellular matrix interactions (18).
Phosphatase and tensin homolog is frequently mutated in several advanced human cancers. In addition, PTEN mutations in germ cell lines result in the rare hereditary syndrome known as Cowden’s disease, which is associated with a higher risk of different cancers, including breast, thyroid, and endometrial cancers (19).
The main lipid substrate of PTEN is PI3,4,5-P3, and indeed PTEN acts as a negative regulator of PI3K/Akt signaling. Thus, loss of PTEN activity leads to permanent PI3K/Akt pathway activation.
Development of mTOR Inhibitors as Anticancer Agents
Rapamycin (sirolimus), an antifungal agent with immunosuppressive properties, was first isolated in 1975 from the soil of the island of Rapa Nui or Easter Island (20). Already back in the 1980s, when tested against a panel of human cancer cell lines, rapamycin showed a broad anticancer activity (21). However, clinical development of rapamycin as an anticancer agent was hampered by unfavorable pharmacokinetic properties (22).
The relatively recent development of rapamycin analogs endowed with a more favorable pharmacokinetic profile, i.e., temsirolimus, everolimus, and ridaforolimus (a.k.a. deforolimus), opened up the present era of mTOR inhibitors as anticancer agents.
All these agents have similar structure (Figure 2) and mechanism of action, but different pharmacokinetic properties. Indeed, all these drugs are small molecule inhibitors that function intracellularly, forming a complex with the FK506 binding protein-12 (FKBP-12) that is then recognized by mTOR. The resulting complex prevents mTOR activity, leading to inhibition of cell cycle progression, survival, and angiogenesis. Notably, all these inhibitors are similar to the parental compound rapamycin in that they affect only mTORC1, and not mTORC2 (22).
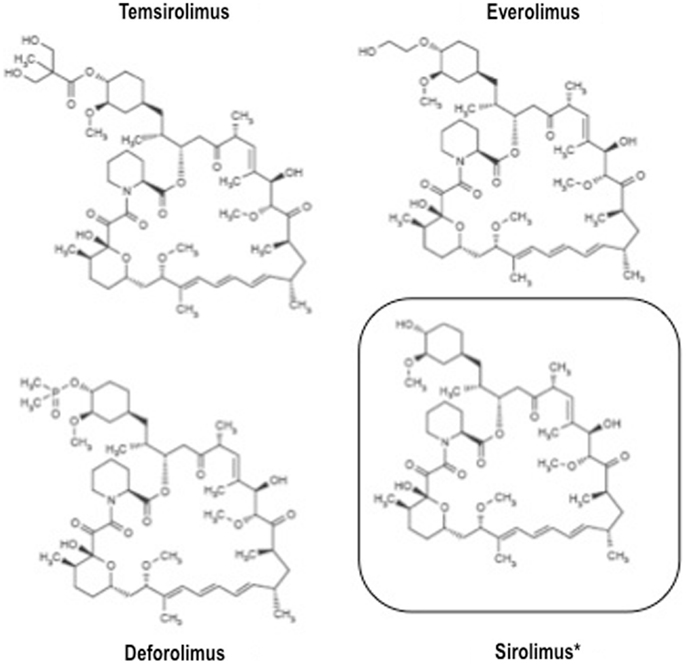
Figure 2. Structure of clinically available mTOR inhibitors.
Temsirolimus: Phase III Trials
Temsirolimus is a pro-drug whose primary active metabolite is rapamycin. Temsirolimus is administered intravenously on a once-weekly schedule (23). It has been approved for the treatment of patients with advanced renal cell carcinoma (RCC) with poor prognostic features, and of mantle cell lymphoma (MCL) patients.
Renal Cell Carcinoma
Temsirolimus registration in RCC was obtained on the basis of the positive results of a randomized, controlled, phase III trial of temsirolimus, interferon-α, or a combination of the two (24). In this study, 626 patients with previously untreated, poor-prognosis, metastatic RCC were randomized to receive 25 mg of intravenous (i.v.) temsirolimus weekly, 3 MU of interferon-α (with an increase to 18 MU) subcutaneous (s.c.) three times weekly, or a combination-therapy with 15 mg of temsirolimus weekly plus 6 MU of interferon-α three times weekly. Overall survival (OS) was the primary endpoint of the trial. Patients who received temsirolimus alone had longer OS and progression-free survival (PFS) than patients who received interferon-α alone, while there was no significant difference in OS between the combination-therapy group and the interferon group. Indeed, median OS in the temsirolimus group, the interferon-α group, and the combination-therapy group were 10.9, 7.3, and 8.4 months, respectively (24).
Mantle Cell Lymphoma
As far as MCL is concerned, the pivotal registration trial was a phase III trial evaluating two dose regimens of temsirolimus in comparison with single-agent therapy in relapsed or refractory disease (investigator’s choice) (25). Sixty-two patients with relapsed or refractory MCL were randomly assigned to receive one of two temsirolimus regimens: 175 mg weekly for 3 weeks followed by either 75 mg (175/75 mg) or 25 mg (175/25 mg) weekly, or investigator’s choice therapy from approved options. PFS was the primary endpoint of the study. Median PFS was 4.8, 3.4, and 1.9 months for the temsirolimus 175/75, 175/25 mg, and investigator’s choice groups, respectively (25). Patients treated with temsirolimus 175/75 mg had significantly longer PFS than those treated with investigator’s choice therapy [p = 0.0009; hazard ratio (HR) = 0.44]. Furthermore, objective response rate (ORR) was significantly higher in the 175/75-mg group (22%) compared with the investigator’s choice group (2%; p = 0.0019), while there was no statistical difference in OS (25).
Ridaforolimus: Phase III Trial
Ridaforolimus is not a pro-drug, but like temsirolimus, it was originally administered intravenously on an intermittent schedule, while an oral formulation has also been subsequently developed (26, 27).
Maintenance Treatment for Adult Soft Tissue and Bone Sarcomas
Recently, a large randomized, placebo-controlled, phase III trial was carried out aiming to evaluate ridaforolimus activity as a maintenance treatment in advanced sarcomas (28). In this study, 711 patients with metastatic soft tissue or bone sarcomas who achieved an objective response or at least a stable disease after standard chemotherapy were randomly assigned to receive ridaforolimus 40 mg or placebo once per day, per oral administration (o.s.) for 5 days every week. The primary endpoint was PFS. Overall, ridaforolimus treatment led to a modest, although statistically significant, improvement in PFS compared with placebo (17.7 vs. 14.6 weeks; HR: 0.72; 95% CI: 0.61–0.85; p = 0.001) (28).
Everolimus: Phase III Trials
Everolimus is another orally available mTOR inhibitor that is usually administered on a continuous daily schedule (even though a weekly schedule has been also tested, especially for combination regimens) (29).
Renal Cell Carcinoma
Everolimus has recently been approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of advanced RCC after failure of treatment with Sunitinib and/or Sorafenib, following the presentation of the results of the RECORD-1 trial.
RECORD-1 was a phase III double-blind, randomized, placebo-controlled trial aimed at evaluating the activity of everolimus in patients whose disease had progressed under treatment with one or two VEGFR tyrosine kinase inhibitors (TKIs) (30). A total of 416 patients were enrolled and stratified according to the number of previous treatments [Sorafenib or Sunitinib (1 TKI) vs. Sorafenib as well as Sunitinib (2 TKIs)] and prognostic risk group. Patients were then randomized in the ratio of 2 to 1 to receive everolimus (given at the standard dose of 10 mg daily, per o.s.) plus best supportive care (BSC), or to placebo plus BSC. After the second interim analysis, the study was terminated since the pre-specified efficacy endpoint had been met (30). Indeed, at the final trial analysis, everolimus proved able to significantly improve PFS when compared to placebo: 4.9 vs. 1.9 months, respectively (HR: 0.33; 95% CI: 0.25–0.43; p < 0.001) (31). Furthermore, everolimus significantly increased median PFS in each risk group regardless of whether patients had received one or two prior TKIs (32), had stopped prior therapy for intolerance (33), or of patient age (34).
Neuroendocrine Tumors
As most NETs are hypervascular (35) and synthesize and secrete high levels of VEGF-A (36, 37), targeted (such as everolimus and sunitinib) and untargeted (such as somatostatin analogs, interferon-α, and thalidomide) therapies, with certain or possible anti-angiogenic properties, have been tested in metastatic NET.
Everolimus, in association with octreotide LAR, first demonstrated a promising antitumor activity in a phase II trial with 30 low- to intermediate-grade NET (carcinoids) patients, showing 17% of partial remission and 80% of stable disease, added to a median PFS of 15.7 months (38).
The open-label, phase II trial RADIANT-1 enrolled 160 advanced, low- or intermediate-grade pancreatic NET (pNET) patients, with progressive (according to RECIST criteria) disease during or after cytotoxic chemotherapy (39). One hundred and fifteen patients were assigned to everolimus 10 mg/day o.s., and 45 patients were submitted to everolimus 10 mg/day o.s. + octreotide LAR 30 mg/28 days intramuscular (i.m). The response rates were 9.6% in the everolimus arm and 4.4% in the everolimus + octreotide LAR group. Median PFS by central radiology review were 9.7 months for patients receiving everolimus and 16.7 months for those receiving the combination (39). Furthermore, high baseline levels of chromogranin A and neuron-specific enolase circulating neuroendocrine markers were associated with shorter median PFS and OS (40).
The favorable results of these previous phase II trials were then confirmed in two international, multicenter, randomized, placebo-controlled, phase III studies (RADIANT-2 and RADIANT-3).
In the RADIANT-2 study (41), 429 patients with advanced progressive midgut NETs were randomized to receive everolimus 10 mg/day plus octreotide LAR 30 mg/month or octreotide LAR plus placebo. A clinically significant improvement in PFS was recorded in the everolimus arm compared with octreotide LAR/placebo arm (16.4 vs. 11.3 months, respectively), even though the pre-defined threshold for statistical significance was not reached, according to central radiological reading (41). A subsequent multivariate analysis and the local radiological reading sustained the efficacy of everolimus. Furthermore, a subgroup analysis underlined some potential primary tumor sites in particular that could benefit, such as bronchial/lung NETs or colonic NETs (42). Nevertheless, the precise therapeutic activity of everolimus in advanced progressive midgut NETs remained to be defined (43).
In RADIANT-3 (44), the largest clinical trial conducted in pNET patients, 410 patients with advanced pNET and progressive disease were randomly assigned to treatment with oral everolimus 10 mg/day or placebo. Octreotide LAR was administered at the discretion of the investigator. Everolimus was associated with an improvement in median PFS compared with placebo (11.0 vs. 4.6 months, respectively; p < 0.0001), and with an overall tumor response rate of 5% (44). The most common drug-related toxicities were G1–2 stomatitis or aphthous ulceration (44). Furthermore, everolimus therapy correlated with a reduction in VEGF pathway markers, such as soluble VEGF receptor 2 and placental growth factor, suggesting an anti-angiogenic activity of everolimus in pNET patients (45).
Even though everolimus evidently inhibited tumor growth and delayed time-to-progression, the percentages of progression events (i.e., appearance of new metastasis as the only cause of progression, appearance of new metastasis concurrent with progression of preexisting metastases, lesion growth at baseline without new metastases appearing) in the two arms (everolimus, placebo) were similar, suggesting that everolimus delayed tumor progression without affecting the pattern of progression in advanced pNET patients (46).
Following the RADIANT-3, in 2011, everolimus was approved for the treatment of progressive pNETs, but its efficacy in other NETs remains uncertain. Given that RADIANT-2, including 51% of small intestinal carcinoids, failed to achieve its primary endpoint, a placebo-controlled trial with everolimus as monotherapy in progressive gastro-intestinal and lung carcinoids (RADIANT-4) is now ongoing. In the meantime, NCCN guidelines recommend everolimus among several therapeutic options in clinically significant progressive NETs, and by ENETS guidelines in non-pNETs with progressive disease after all other medical treatments (43).
Breast Cancer
Recently, the combination of everolimus with the aromatase inhibitor Exemestane has been evaluated in a randomized, phase III trial, since a large amount of evidence supported the hypothesis that aberrant signaling through the mTOR pathway is associated with resistance to endocrine therapies (47).
In the BOLERO-2 phase III trial (48), 724 patients with hormone-receptor-positive advanced breast cancer who recurred or progressed while receiving treatment with a non-steroidal aromatase inhibitor in the adjuvant or metastatic setting, were randomized two to one to receive everolimus and exemestane or exemestane and placebo. PFS was the primary endpoint of the study. A pre-planned interim analysis was performed by an independent data and safety monitoring committee after 359 PFS events were observed. At the time of this analysis, median PFS assessed by the Investigators PFS was 6.9 months with everolimus plus Exemestane and 2.8 months with Placebo plus Exemestane (HR: 0.43; 95% CI: 0.35–0.54; p < 0.001), while centrally assessed PFS was 10.6 and 4.1 months, respectively, again in favor of the everolimus-containing combination (HR: 0.36; 95% CI: 0.27–0.47; p < 0.001) (48).
Other Indications
Everolimus also has an established role in the treatment of two rare conditions: renal angiomyolipomas associated with TSC or lymphangioleiomyomatosis, as well as TSC-related subependymal giant cell astrocytoma (SEGA), both characterized by constitutive activation of the mTOR pathway (49).
In the EXIST-2 double-blind, placebo-controlled, phase III trial (50), 118 patients with at least one angiomyolipoma 3 cm or larger in its longest diameter and a definite diagnosis of TSC or sporadic lymphangioleiomyomatosis were randomly assigned 2:1 to either everolimus 10 mg/day or Placebo. The primary efficacy endpoint of the study was the proportion of patients with confirmed angiomyolipoma response of an at least 50% reduction in total volume of target angiomyolipomas relative to baseline. Response rate (as defined above) was 42% [33 of 79 (95% CI: 31–53%)] for everolimus and 0% [0 of 39 (95% CI: 0–9%)] for placebo (p < 0.0001) (50), thus suggesting the usefulness of everolimus in this setting.
Similarly, in the EXIST-1 double-blind, placebo-controlled, phase III trial (51), 117 patients were randomized in a 2:1 ratio to everolimus 4.5 mg/m2/day (titrated to achieve blood trough concentrations of 5–15 ng/mL) or Placebo. Eligible patients had a definite diagnosis of TSC and at least one lesion with a diameter of 1 cm or greater, and either serial growth of an SEGA, a new lesion of 1 cm or greater, or new or worsening hydrocephalus. The primary endpoint of this study was the proportion of patients with confirmed response, i.e., a reduction in target volume of 50% or greater relative to baseline in SEGA. Twenty-seven (35%) patients in the everolimus group had an at least 50% reduction in SEGA volume as compared to none in the Placebo group (95% CI: 15–52; p < 0.0001) (51).
Taken together, these two studies suggest the possibility that everolimus might represent a disease-modifying treatment also for other aspects of tuberous sclerosis.
The Safety Profile of mTOR Inhibitors
Adverse events observed in patients treated with mTOR inhibitors are fairly constant, irrespective of each specific indication. They include cutaneous and mucosal events (i.e., stomatitis and skin rash), pulmonary dysfunction (non-infectious pneumonitis), metabolic abnormalities (elevated blood levels of glucose, cholesterol, and triglycerides), as well immune-related events (i.e., increased incidence of infections) (52). As far as the risk of infections is concerned, we should not forget that mTOR inhibitors were first developed as immune suppressive agents and are still widely used as such in the transplantation setting.
Metabolic and immune-related adverse events are clearly on-target effects of mTOR inhibition, while cutaneous and mucosal effects may have a less direct association with mTOR inhibition, although inhibition of mTOR-mediated growth and tissue repair and/or immune dysregulation have been proposed to be a factor in mucosal epithelia with high turnover (53, 54).
In general, the incidences of key class-effect adverse events in the three largest phase III trials of everolimus (i.e., RECORD-1 in RCC, RADIANT-3 in pNET, and BOLERO-2 in hormone receptor-positive, HER2-negative, advanced breast cancer) were comparable (30, 44, 48), as summarized in Table 1.
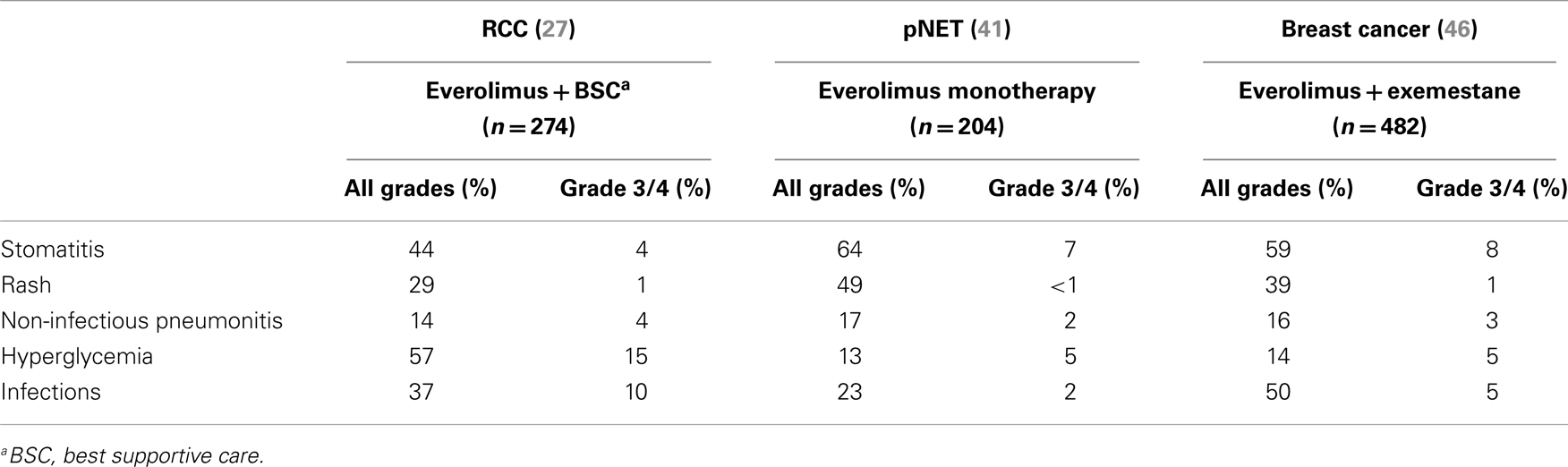
Table 1. Incidence of the main adverse events (all grades and grade 3/4) reported in the three largest phase III studies of everolimus in advanced solid tumors (RCC, pNET, and breast cancer).
The Development of PI3K and Akt Inhibitors as Anticancer Agents
In contrast to the three mTOR inhibitors discussed above, PI3K and Akt inhibitors are still at an early development phase, and so far no compound has reached the bedside. Despite this, three generations of compounds targeting PI3K have already been developed over time.
PI3K and Dual PI3K/mTOR Inhibitors
The first-generation of PI3K inhibitors included compounds like Wortmannin and LY294002, which were able to bind all class I PI3Ks, thus being called “pan-inhibitors.” These compounds have been widely used in pre-clinical models to better characterize this complex pathway. However, due to very poor pharmacokinetic properties, they were never fully developed as anticancer drugs for clinical use (55).
More recently, compounds with better pharmacokinetic properties have been developed and are currently being evaluated within clinical trials in several malignancies (55), including genitourinary cancers (56) and others. These second-generation inhibitors are characterized by greater and isoform-specific selective activity (55).
The third generation of compounds comprises the so-called “dual PI3K/mTOR inhibitors.” These were developed after consideration that the CAT sites of PI3K and mTOR share a high degree of sequence homology. The potential advantage of these novel compounds (an advantage which still has to be confirmed in vivo) is that they inhibit not only all PI3K class I isoforms, but also mTORC1 and (more notably) mTORC2. In theory, this combined activity would lead to the strongest inhibition of the whole PI3K/Akt/mTOR pathway (57).
A list of PI3K inhibitors in pre-clinical and clinical development is reported in Table 2.
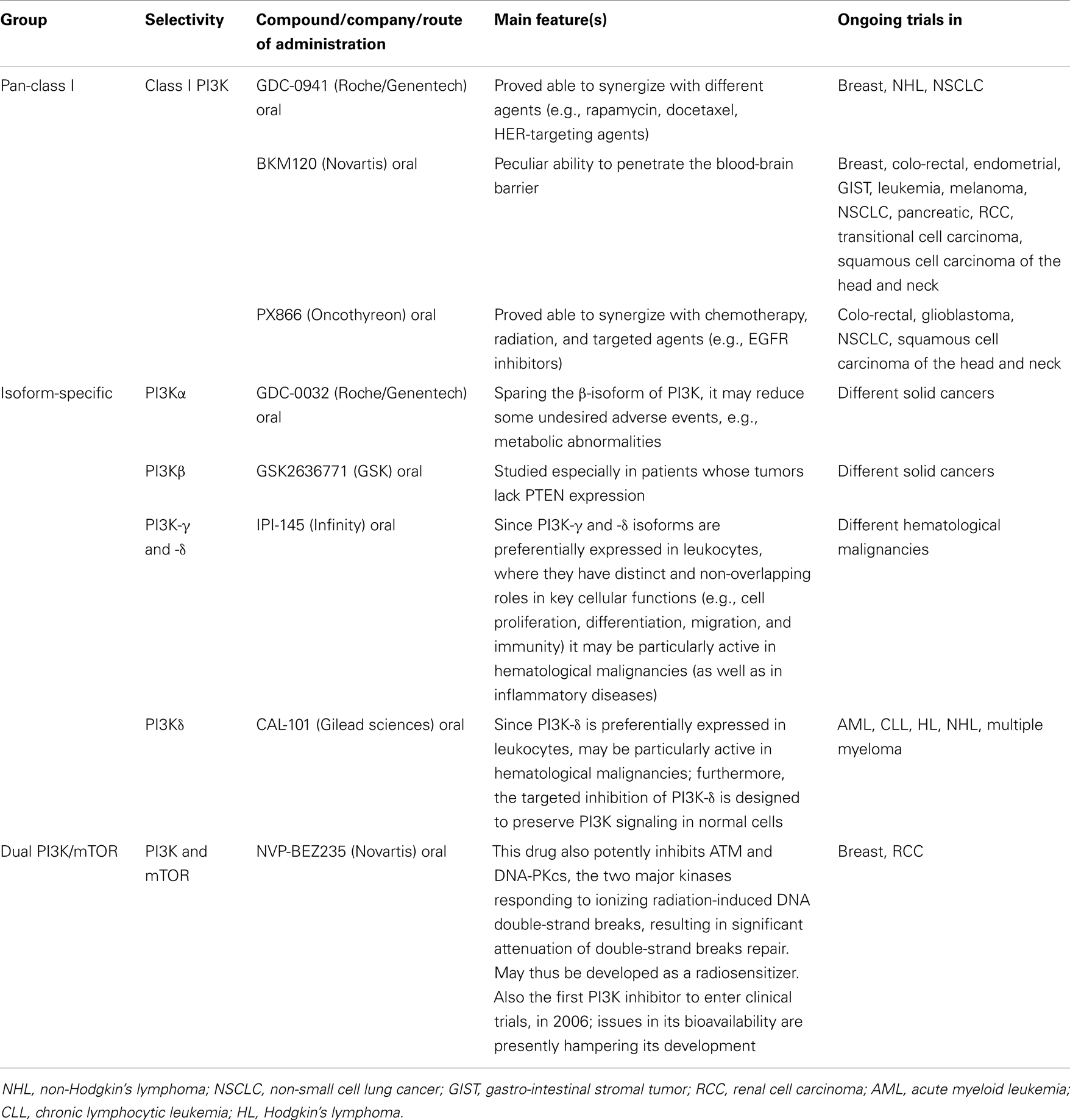
Table 2. Phosphatidylinositol-3-kinase and “dual PI3K and mTOR” inhibitors in development [modified from ref. (53)].
Akt Inhibitors
So far, compounds that target Akt ATP binding site, its PH domain, LINK, and the protein substrate sites have been developed (12, 58). Compared to PI3K, and especially mTOR inhibitors, fewer Akt-targeting agents have entered clinical development (58), even though one of them, Miltefosine, has already completed a phase III trial (59). A list of Akt inhibitors under pre-clinical and clinical development is given in Table 3.

Table 3. Akt inhibitors in development.
Mechanisms of Resistance to PI3K/Akt/mTOR Inhibitors
Despite all the successes (achieved with mTOR inhibitors) and expectations (related to novel anti-PI3K and Akt drugs), none of the above drugs is currently able to cure a single patient with cancer.
As with all antineoplastic agents, this is mainly due to the development of resistance. The underlying molecular basis of resistance, either intrinsic or acquired, remains largely unknown and has not been well characterized. So far, multiple mechanisms of resistance to targeted agents have been proposed, including secondary target mutations, activation of alternative, parallel, signaling pathways, and amplification of downstream alterations within the same pathway (60).
Resistance to mTOR inhibitors has been at least partially clarified. Indeed, it is often linked to different negative feedback loops. In one loop, mTORC1 inhibition leads to upregulation of receptor tyrosine kinases (RTKs or substrates) such as platelet-derived growth factor receptors (PDGFRs) and insulin receptor substrate-1 (IRS-1), resulting in increased PI3K-dependent Akt phosphorylation at Ser473. In another loop, mTORC1 inhibition leads to PI3K-Ras activation, which leads to an increase in MAPK signaling (61–63).
Furthermore, aberrant activation of MYC may contribute to acquired resistance to PI3K/Akt/mTOR-targeted therapy. Indeed, targeting this pathway may cause MYC activation through PDK1-dependent MYC phosphorylation and MYC amplification, which is parallel to PIK3CA-dependent Akt and MAPK activation, thus attenuating the therapeutic effect of PI3K/Akt/mTOR inhibitors (64–66).
Finally, a recent report analyzed SGK (serum- and glucocorticoid-regulated kinase) levels and the relative sensitivity of a panel of breast cancer cells toward two distinct Akt inhibitors (67). This study showed a number of Akt-inhibitor-resistant lines displaying markedly elevated SGK1 that also exhibited significant phosphorylation of the SGK1 substrate NDRG1 [neuroblastoma-derived Myc (N-Myc) downstream-regulated gene 1]. In contrast, most Akt-inhibitor-sensitive cell lines displayed low or undetectable levels of SGK1. Intriguingly, despite low SGK1 levels, several Akt-inhibitor-sensitive cell lines showed marked NDRG1 phosphorylation that, unlike resistant cells, were suppressed by Akt inhibitors. Furthermore, SGK1 knockdown markedly reduced proliferation of Akt-inhibitor-resistant cells, but not Akt-sensitive cells (67). Taken together, these results clearly suggest that SGK1 levels, as well as responses of NDRG1 phosphorylation to Akt inhibitor administration, could help us predict the sensitivity or resistance of tumor cells to Akt-targeting drugs.
Autophagy may represent another mechanism of resistance from Akt/mTOR targeting. Indeed, autophagy induction proved able to protect MCL cells from Akt/mTOR inhibition. Furthermore, selective triple knockdown of the autophagy genes ATG7, ATG5 and ATG3, and pre-treatment with the autophagy inhibitor hydroxychloroquine, efficiently overcame the resistance to Akt/mTOR inhibitors in this model, leading to the activation of the mitochondrial apoptotic pathway (68, 69). Taken together, these results suggest that counteracting autophagy may represent an attractive strategy for sensitizing lymphoma cells to everolimus-based therapy. Furthermore, autophagy facilitates cancer cell resistance also to cytotoxic chemotherapy and radiation treatment (70).
Conclusion
The PI3K/Akt/mTOR pathway represents a good example of the concept of redundancy in biological systems, particularly in cancer cells. Indeed, cancer responds to chronic treatment with drugs targeting a single pathway by adapting its signaling circuitry, taking advantage of pathway redundancy and routes of feedback and crosstalk to maintain their function and thus escape from drug-induced growth inhibition and death (71–73).
That is why, despite recent successes (achieved in completely different diseases such as kidney and breast cancer, pNETs, and other malignancies), tumors ultimately evade inhibition of this pathway.
Novel agents targeting PI3K/Akt/mTOR promise further improvement of the results achieved so far through higher selectivity and potency, as well as to combinability with other therapeutic strategies. However, only translational research, addressing this variegated and complex network of highly integrated signaling pathways and mechanisms of resistance to their inhibition, will be able to help us take another step forward.
Author Contribution
All the authors equally contributed to the preparation of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev (1999) 13:2905–27. doi:10.1101/gad.13.22.2905
2. Shaw RJ, Cantley LC. Ras, PI3K, and mTOR signalling controls tumor cell growth. Nature (2006) 441:424–30. doi:10.1038/nature04869
3. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/Akt pathway for cancer drug discovery. Nat Rev Drug Discov (2005) 4:988–1004. doi:10.1038/nrd1902
4. Chen YL, Law PY, Loh HH. Inhibition of PI3K/Akt signaling: an emerging paradigm for targeted cancer therapy. Curr Med Chem Anticancer Agents (2005) 5:575–89. doi:10.2174/156801105774574649
5. Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem (1998) 67:481–507. doi:10.1146/annurev.biochem.67.1.481
6. Fresno-Vara JA, Casado E, De Castro J, Cejas P, Belda-Iniesta C, Gonzales-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev (2004) 30:193–204. doi:10.1016/j.ctrv.2003.07.007
7. Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev (2000) 14:1027–47.
8. Testa JR, Bellacosa A. Akt plays a central role in tumorigenesis. Proc Natl Acad Sci U S A (2001) 98:10983–5. doi:10.1073/pnas.211430998
9. Chen X, Thakkar H, Tyan F, Gim S, Robinson H, Lee C, et al. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene (2001) 20:6073–7. doi:10.1038/sj.onc.1204736
10. Hussain AR, Ahmed SO, Ahmed M, Khan OS, Al Abdulmohsen S, Platanias LC, et al. Cross-talk between NFkB and the PI3-kinase/Akt pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis. PLoS One (2012) 7:e39945. doi:10.1371/journal.pone.0039945
11. Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle (2003) 2:339–45. doi:10.4161/cc.2.4.433
12. Kumar CC, Madison V. Akt crystal structure and Akt-specific inhibitors. Oncogene (2005) 24:7493–501. doi:10.1038/sj.onc.1209087
13. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev (2004) 18:1926–45. doi:10.1101/gad.1212704
14. Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell (2004) 6:91–9. doi:10.1016/j.ccr.2004.06.007
15. Memmott RM, Dennis PA. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal (2009) 21:656–64. doi:10.1016/j.cellsig.2009.01.004
16. Grewe M, Gansauge F, Schmid RM, Adler G, Seufferlein T. Regulation of cell growth and cyclin D1 expression by the constitutively active FRAP-p70s6K pathway in human pancreatic cancer cells. Cancer Res (1999) 59:3581–7.
17. Abraham RT. mTOR as a positive regulator of tumor cell responses to hypoxia. Curr Top Microbiol Immunol (2004) 279:299–319.
18. Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene (1999) 18:7034–45. doi:10.1038/sj.onc.1203183
19. Simpson L, Parson R. PTEN: life as a tumor suppressor. Exp Cell Res (2001) 264:29–41. doi:10.1006/excr.2000.5130
20. Seghal SN, Baker H, Vézina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo) (1975) 28:727–32. doi:10.7164/antibiotics.28.727
21. Douros J, Suffnes M. New antitumor substances of natural origin. Cancer Treat Rev (1981) 8:63–87. doi:10.1016/S0305-7372(81)80006-0
22. Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov (2006) 5:671–88. doi:10.1038/nrd2062
23. Hidalgo M, Buckner JC, Erlichman C, Pollack MS, Boni JP, Dukart G, et al. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res (2006) 12:5755–63. doi:10.1158/1078-0432.CCR-06-0118
24. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med (2007) 356:2271–81. doi:10.1056/NEJMoa066838
25. Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol (2009) 27:3822–9. doi:10.1200/JCO.2008.20.7977
26. Mita MM, Mita AC, Chu QS, Rowinsky EK, Fetterly GJ, Goldston M, et al. Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J Clin Oncol (2008) 26:361–7. doi:10.1200/JCO.2007.12.0345
27. Mita MM, Poplin E, Britten CD, Tap WD, Rubin EH, Scott BB, et al. Phase I/IIa trial of the mammalian target of rapamycin inhibitor ridaforolimus (AP23573; MK-8669) administered orally in patients with refractory or advanced malignancies and sarcoma. Ann Oncol (2013) 24:1104–11. doi:10.1093/annonc/mds602
28. Demetri GD, Chawla SP, Ray-Coquard I, Le Cesne A, Staddon AP, Milhem MM, et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol (2013) 31:2485–92. doi:10.1200/JCO.2012.45.5766
29. O’Donnell A, Faivre S, Burris HA III, Rea D, Papadimitrakopoulou V, Shand N, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol (2008) 26:1588–95. doi:10.1200/JCO.2007.14.0988
30. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet (2008) 372:449–56. doi:10.1016/S0140-6736(08)61039-9
31. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer (2010) 116:4256–65. doi:10.1002/cncr.25219
32. Calvo E, Escudier B, Motzer RJ, Oudard S, Hutson TE, Porta C, et al. Everolimus in metastatic renal cell carcinoma: subgroup analysis of patients with 1 or 2 previous vascular endothelial growth factor receptor-tyrosine kinase inhibitor therapies enrolled in the phase III RECORD-1 study. Eur J Cancer (2012) 48:333–9. doi:10.1016/j.ejca.2011.11.027
33. Bracarda S, Hutson TE, Porta C, Figlin RA, Calvo E, Grünwald V, et al. Everolimus in metastatic renal cell carcinoma patients intolerant to previous VEGFr-TKI therapy: a RECORD-1 subgroup analysis. Br J Cancer (2012) 106:1475–80. doi:10.1038/bjc.2012.89
34. Porta C, Calvo E, Climent MA, Vaishampayan U, Osanto S, Ravaud A, et al. Efficacy and safety of everolimus in elderly patients with metastatic renal cell carcinoma: an exploratory analysis of the outcomes of elderly patients in the RECORD-1 trial. Eur Urol (2012) 61:826–33. doi:10.1016/j.eururo.2011.12.057
35. Scoazec JY. Angiogenesis in neuroendocrine tumors: therapeutic applications. Neuroendocrinology (2013) 97:45–56. doi:10.1159/000338371
36. Terris B, Scoazec JY, Rubbia L, Bregeaud L, Pepper MS, Ruszniewski P, et al. Expression of vascular endothelial growth factor in digestive neuroendocrine tumours. Histopathology (1998) 32:133–8. doi:10.1046/j.1365-2559.1998.00321.x
37. Villaume K, Blanc M, Gouysse G, Walter T, Couderc C, Nejjari M, et al. VEGF secretion by neuroendocrine tumor cells is inhibited by octreotide and by inhibitors of the PI3K/Akt/mTOR pathway. Neuroendocrinology (2010) 91:268–78. doi:10.1159/000289569
38. Yao JC, Phan AT, Chang DZ, Wolff RA, Hess K, Gupta S, et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol (2008) 26:4311–8. doi:10.1200/JCO.2008.16.7858
39. Yao JC, Lombard-Bohas C, Baudin E, Kvols LK, Rougier P, Ruszniewski P, et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol (2010) 28:69–76. doi:10.1200/JCO.2009.24.2669
40. Yao JC, Pavel M, Phan AT, Kulke MH, Hoosen S, St Peter J, et al. Chromogranin A and neuron-specific enolase as prognostic markers in patients with advanced pNET treated with everolimus. J Clin Endocrinol Metab (2011) 96:3741–9. doi:10.1210/jc.2011-0666
41. Pavel ME, Hainsworth JD, Baudin E, Peeters M, Hoersch D, Winkler RE, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomized, placebo-controlled, phase 3 study. Lancet (2011) 378:2005–12. doi:10.1016/S0140-6736(11)61742-X
42. Yao JC, Hainsworth JD, Wolin EM, Pavel ME, Baudin E, Gross D, et al. Multivariate analysis including biomarkers in the phase III RADIANT-2 study of octreotide LAR plus everolimus (E+O) or placebo (P+O) among patients with advanced neuroendocrine tumors (NET). J Clin Oncol (2012) 30(Suppl 4):A157.
43. Pavel M, Kidd M, Modlin I. Systemic therapeutic options for carcinoid. Semin Oncol (2013) 40:84–99. doi:10.1053/j.seminoncol.2012.11.003
44. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med (2011) 364:514–23. doi:10.1056/NEJMoa1009290
45. Yao JC, Tsuchihashi Z, Panneerselvam A, Winkler RE, Bugarini R, Pavel M. Effect of everolimus treatment on markers angiogenesis in patients with advanced pancreatic neuroendocrine tumours (pNET) results from the phase III RADIANT-3 study. Eur J Cancer (2011) 47(Suppl 1):S463. doi:10.1158/0008-5472.CAN-12-3923
46. Yao JC, Phan AT, Jehl V, Shah G, Meric-Bernstam F. Everolimus in advanced pancreatic neuroendocrine tumors: the clinical experience. Cancer Res (2013) 73:1449–53. doi:10.1158/0008-5472.CAN-12-3923
47. Fedele P, Calvani N, Marino A, Orlando L, Schiavone P, Quaranta A, et al. Targeted agents to reverse resistance to endocrine therapy in metastatic breast cancer: where are we now and where are we going? Crit Rev Oncol Hematol (2012) 84:243–51. doi:10.1016/j.critrevonc.2012.03.004
48. Baselga J, Campone M, Piccart M, Burris HA III, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med (2012) 366:520–9. doi:10.1056/NEJMoa1109653
49. Astrinidis A, Henske EP. Tuberous sclerosis complex: linking growth and energy signaling pathways with human disease. Oncogene (2005) 24:7475–81. doi:10.1038/sj.onc.1209090
50. Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet (2013) 381:817–24. doi:10.1016/S0140-6736(12)61767-X
51. Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (2013) 381:125–32. doi:10.1016/S0140-6736(12)61134-9
52. Porta C, Osanto S, Ravaud A, Climent MA, Vaishampayan U, White DA, et al. Management of adverse events associated with the use of everolimus in patients with advanced renal cell carcinoma. Eur J Cancer (2011) 47:1287–98. doi:10.1016/j.ejca.2011.02.014
53. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149:274–93. doi:10.1016/j.cell.2012.03.017
54. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol (2012) 12:325–38. doi:10.1038/nri3198
55. Martini M, Ciraolo E, Gulluni F, Hirsch E. Targeting PI3K in cancer: any good news? Front Oncol (2013) 3:108. doi:10.3389/fonc.2013.00108
56. Porta C, Figlin RA. Phosphatidylinositol-3-kinase/Akt signaling pathway and kidney cancer, and the therapeutic potential of phosphatidylinositol-3-kinase/ Akt inhibitors. J Urol (2009) 182:2569–77. doi:10.1016/j.juro.2009.08.085
57. Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, et al. Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget (2012) 3:371–94.
58. Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs (2010) 19:1355–66. doi:10.1517/13543784.2010.520701
59. Leonard R, Hardy J, van Tienhoven G, Houston S, Simmonds P, David M, et al. Randomized, double-blind, placebo-controlled, multicenter trial of 6% miltefosine solution, a topical chemotherapy in cutaneous metastases from breast cancer. J Clin Oncol (2001) 19:4150–9.
60. Tan J, Yu Q. Molecular mechanisms of tumor resistance to PI3K-mTOR targeted therapy. Chin J Cancer (2013) 32:376–9. doi:10.5732/cjc.012.10287
61. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell (2006) 22:159–68. doi:10.1016/j.molcel.2006.03.029
62. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K dependent feedback loop in human cancer. J Clin Invest (2008) 118:3065–74. doi:10.1172/JCI34739
63. O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res (2006) 66:1500–8. doi:10.1158/0008-5472.CAN-05-2925
64. Tan J, Lee PL, Li Z, Jiang X, Lim YC, Hooi SC, et al. B55beta-associated PP2A complex controls PDK1-directed myc signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell (2010) 18:459–71. doi:10.1016/j.ccr.2010.10.021
65. Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci U S A (2011) 108:E699–708. doi:10.1073/pnas.1108237108
67. Sommer EM, Dry H, Cross D, Guichard S, Davies BR, Alessi DR. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem J (2013) 452:499–508. doi:10.1042/BJ20130342
68. Rosich L, Xargay-Torrent S, López-Guerra M, Campo E, Colomer D, Roué G. Counteracting autophagy overcomes resistance to everolimus in mantle cell lymphoma. Clin Cancer Res (2012) 18:5278–89. doi:10.1158/1078-0432.CCR-12-0351
69. Rosich L, Colomer D, Roue G. Autophagy controls everolimus (RAD001) activity in mantle cell lymphoma. Autophagy (2013) 9:115–7. doi:10.4161/auto.22483
70. Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophys Acta (2010) 1806:220–9. doi:10.1016/j.bbcan.2010.07.003
71. Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev (2012) 26:641–50. doi:10.1101/gad.186965.112
72. Ribatti D. Novel angiogenesis inhibitors: addressing the issue of redundancy in the angiogenic signaling pathway. Cancer Treat Rev (2011) 37:344–52. doi:10.1016/j.ctrv.2011.02.002
Keywords: PI3K, Akt, mTOR, inhibitors, temsirolimus, everolimus, ridaforolimus, novel agents
Citation: Porta C, Paglino C and Mosca A (2014) Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 4:64. doi: 10.3389/fonc.2014.00064
Received: 02 September 2013; Accepted: 17 March 2014;
Published online: 14 April 2014.
Edited by:
Alexandre Arcaro, University of Bern, SwitzerlandReviewed by:
Robert Friis, University of Bern, SwitzerlandRosa Bernardi, Fondazione Centro San Raffaele del Monte Tabor, Italy
Patrizia Agostinis, Catholic University of Leuven, Belgium
Copyright: © 2014 Porta, Paglino and Mosca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Camillo Porta, Medical Oncology, Fondazione I.R.C.C.S. Policlinico San Matteo, Piazzale C. Golgi 19, Pavia 27100, Italy e-mail: c.porta@smatteo.pv.it