 Grzegorz J. Korpanty
Grzegorz J. Korpanty Donna M. Graham
Donna M. Graham Mark D. Vincent
Mark D. Vincent Natasha B. Leighl
Natasha B. Leighl- 1Division of Medical Oncology and Hematology, Princess Margaret Cancer Centre, University of Toronto, Toronto, ON, Canada
- 2London Regional Cancer Program, Department of Medical Oncology, London Health Sciences Centre, London, ON, Canada
Lung cancer remains the most lethal malignancy in the world. Despite improvements in surgical treatment, systemic therapy, and radiotherapy, the 5-year survival rate for all patients diagnosed with lung cancer remains between 15 and 20%. Newer therapeutic strategies rely on specific molecular alterations, or biomarkers, that provide opportunities for a personalized approach to specific patient populations. Classification of lung cancer is becoming increasingly focused on these biomarkers, which renders the term “non-small cell lung” cancer less clinically useful. Non-small cell lung cancer is now recognized as a complex malignancy and its molecular and genomic diversity allows for patient-centered treatment options. Here, we review advances in targeted treatment of lung adenocarcinoma with respect to five clinically relevant biomarkers – EGFR, ALK, MET, ROS-1, and KRAS.
Introduction
Lung cancer remains one of the most commonly diagnosed malignancy worldwide and the leading cause of cancer-related death (1). Until the last decade, non-small cell lung cancer (NSCLC) was considered a single disease, and systemic treatment of metastatic NSCLC was limited to platinum-based chemotherapy doublets resulting in approximately 20% response rates and median survival of 8 months (2). Only recently, we have realized that recognition of histological subtypes of NSCLC is clinically relevant when choosing systemic, platinum-based chemotherapy (3). In recent years, the oncology community has seen a paradigm shift in the molecular diagnosis and treatment of lung cancer thanks to identification of sensitizing mutations within the epidermal growth factor receptor gene (EGFR) to EGFR tyrosine kinase inhibitors (EGFR-TKIs) (erlotinib, gefitinib, and afatinib), and anaplastic lymphoma kinase gene (ALK) rearrangements (i.e., EML4-ALK) to ALK inhibitors (crizotinib and ceritinib) (4).
These breakthrough discoveries provide the unique opportunity for molecularly selected lung cancer patients to receive targeted, personalized treatment options that translate into clinically meaningful benefit (4). Molecular testing of NSCLC is now widely recommended by oncology societies because it provides personalized treatment options and better outcomes for patients with metastatic disease (5, 6).
To improve outcome, molecular profiling of lung cancer tumors should be available to all NSCLC patients in order to make targeted therapy available to patients with actionable/“druggable” driver mutations (7–9). Currently, we can offer these treatments routinely to patients with EGFR-mutated and ALK-rearranged NSCLC, the vast majority of whom have adenocarcinoma histology.
This review summarizes the most recent data on efficacy, risks, and benefits of novel biologic therapies in NSCLC focusing on EGFR, ALK, MET, ROS-1, and KRAS (Table 1).
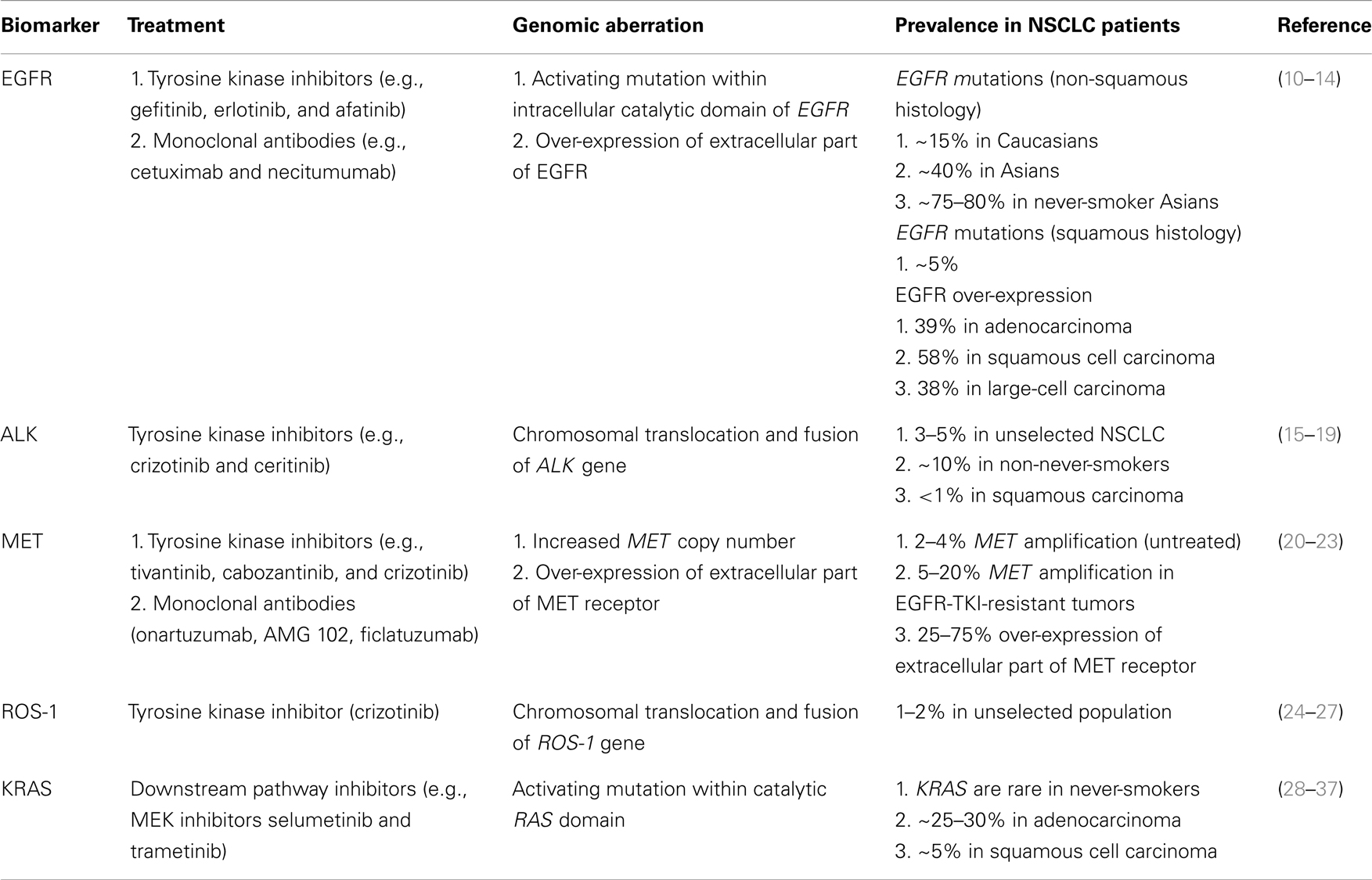
Table 1. Clinically relevant biomarkers in NSCLC.
EGFR
The epidermal growth factor receptor family (ERBB family) comprises four tyrosine kinase receptors: HER-1 (EGFR), HER-2/neu (ERBB2), HER-3 (ERBB3), and HER-4 (ERBB4) (38, 39). Following ligand-binding, EGFR receptors homo- and hetero-dimerize and promote autophosphorylation of the intracellular tyrosine kinase domain and initiate molecular cascade of events involved in growth, cell proliferation, differentiation, and survival (10, 11, 40). Small-molecule receptor tyrosine kinase inhibitors (TKIs) bind to the intracellular catalytic domain of the tyrosine kinase and inhibit receptor autophosphorylation and activation of downstream signaling pathways by competing with adenosine triphosphate (ATP) (41). Gefitinib and erlotinib are the most extensively studied reversible EGFR-TKIs in patients with metastatic NSCLC (42, 43). The majority of unselected NSCLC patients will not respond to treatment with EGFR-TKIs. Patients of Asian ethnicity, females, never-smokers, or those with adenocarcinoma histology, were initially identified as a population with the most substantial clinical benefit from EGFR-TKIs (12, 44–53). The marker of sensitivity to EGFR-TKIs was unknown until 2004 when activating mutations in exon 18, 19, and 21 of the EGFR gene were discovered (54–56). The majority of mutations are either point mutations leading to amino acid substitutions (exon 18 and 21) or in-frame deletions (exon 19) clustered around the ATP-binding pocket of the intracellular tyrosine kinase domain (13). A kinetic analysis of the intracellular domains of mutant EGFR has shown that the mutant receptor compared with a wild-type shows reduced affinity for ATP in the presence of EGFR-TKI (57).
The Iressa Pan-Asia Study (IPASS) was the first phase III randomized trial that demonstrated superior outcome with first-line EGFR-TKI treatment in patients with EGFR-mutant NSCLC when compared with platinum-based chemotherapy in a retrospective subgroup analysis (58). Other trials have employed a similar approach to the IPASS study and reported similar results (59, 60).
Four randomized phase III trials prospectively compared the efficacy of first generation EGFR-TKIs against standard platinum-based chemotherapy in patients with EGFR mutation-positive NSCLC (61–67). In all four trials, EGFR-mutated NSCLC patients treated with TKIs (erlotinib or gefitinib) had significantly better ORR, PFS, and quality of life (QOL) when compared with patients treated with platinum-based chemotherapy (58, 61, 63, 65, 67–70). Despite significant PFS benefit of EGFR-TKIs in EGFR-mutant NSCLC patients, none of the trials showed statistically significant survival improvement, which is likely related to a high rate of patient crossover to EGFR-TKI from first-line chemotherapy upon progression or development of acquired resistance.
Afatinib is a second-generation EGFR-TKI that irreversibly blocks EGFR and Her-2 (71, 72). LUX-Lung 3 was a phase III clinical trial of afatinib compared to cisplatin-pemetrexed chemotherapy in treatment-naïve patients with EGFR-mutant advanced lung adenocarcinoma (73). Both median PFS and ORR were significantly better in patients treated with afatinib compared with chemotherapy. A pooled, retrospective subgroup analysis of LUX-Lung 3 and LUX-Lung 6 trial at 2014 ASCO annual meeting demonstrated better OS for patients with EGFR exon 19 deletion vs. EGFR L858R exon 21 insertion mutations (HR = 0.59; CI 0.45–0.77; p < 0.001 vs. HR = 1.25; CI 0.92–1.71; p = 0.16) (74). First-line treatment of EGFR mutation-positive NSCLC with EGFR-TKIs (gefitinib, erlotinib, and afatinib) is now recommended worldwide (5, 9). AZD9291 and CO-1686 are irreversible selective EGFR inhibitors, which demonstrate significant activity in patients with acquired resistance to first-generation EGFR-TKI, and are currently under development. One of the most common mechanisms of resistance to EGFR-TKIs is the development of T790M mutation (~50% of patients), which prevents binding of reversible EGFR-TKI to the EGFR kinase domain while preserving its catalytic activity (75). In patients with tumors harboring T790M mutation, AZD9291 and CO-1686 show promising 64 and 58% ORR, respectively (76, 77).
ALK
The EML4-ALK fusion gene is a product of inversion within the short arm of chromosome 2, where ALK (anaplastic large-cell lymphoma kinase) joins EML4 (echinoderm microtubule-associated protein-like 4) to form a fusion gene (15). The product of EML4-ALK fusion is a chimeric protein with constitutive ALK activity and is detected in 3–6% of unselected NSCLC and especially among never-smokers or light ex-smokers who have adenocarcinoma histology (16–19). ALK rearrangements are nearly almost mutually exclusive with EGFR or KRAS mutations, although some rare exceptions exist (78). ALK-positive NSCLC represents a distinct molecular subtype that can be targeted with ALK-specific treatments (15, 24). Crizotinib is an oral small-molecule TKI that targets ALK, MET, and ROS1 tyrosine kinases (79–82). Crizotinib received accelerated US Food and Drug Administration (FDA) approval for treatment of ALK-positive NSCLC based on an objective response rate of 60% and median PFS of 8–10 months in single-arm studies (16, 79, 83, 84).
A first-line phase III study (PROFILE 1014) assessed efficacy of crizotinib vs. cisplatin/carboplatin-pemetrexed chemotherapy in patients with ALK-positive NSCLC. Recently presented data at the 2014 ASCO Annual Meeting demonstrated significantly better median PFS and ORR when compared with patients who received chemotherapy – 10.9 vs. 7.0 months and 74 vs. 45%, respectively (85). No survival benefit was demonstrated at the time of data cut-off and may never be, since patients who progressed on chemotherapy were allowed to crossover to crizotinib. The PROFILE 1007 phase III study investigated the efficacy of crizotinib vs. standard of care second-line chemotherapy (pemetrexed or docetaxel) in previously treated ALK-positive NSCLC (86). Patients treated with crizotinib demonstrated significantly improved median PFS when compared with chemotherapy – 7.0 vs. 3.0 months. No overall survival benefit was noted likely due to a high rate of patient crossover to the crizotinib arm from chemotherapy. Patients treated with single-agent pemetrexed had higher ORR when compared with docetaxel (29 vs. 13%).
After clinical recognition of acquired resistance to crizotinib, multiple second-generation ALK inhibitors (LKD378, AP26113, and TSR-011) entered early phase clinical trials for patients with ALK-positive solid tumors, including NSCLC (87, 88). Recently published results of a phase I clinical trial of ceritinib (LDK378) in patients with ALK-rearranged NSCLC demonstrated a ORR of 58% in all patients and 56% in crizotinib-resistant patients (88). Median PFS in crizotinib-naïve patients was 10.4 and 6.9 months in the crizotinib-pretreated population. Ceritinib received accelerated FDA approval in April 2014 and confirmatory trials with ceritinib in this group of patients are ongoing (http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm395299).
MET
MET is a proto-oncogene that encodes for the heterodimeric transmembrane MET tyrosine receptor kinase. Its only known ligand – hepatocyte growth factor (HGF) (89). Binding of HGF to the MET receptor activates the tyrosine kinase and downstream signaling pathways including PI3K/AKT, Ras-Rac/Rho, mitogen-activated protein kinase (MAPK), and phospholipase C (PLC) involved in cell motility and invasion (20, 21, 89). The MET receptor is expressed in approximately 40–50% of NSCLC tumors; high levels of receptor expression, as well as high MET gene copy number are independent prognostic factors of poor outcome in patients with resected NSCLC (22, 23). MET amplification is recognized as one of the potential molecular mechanisms of acquired resistance in EGFR-mutated NSCLC to EGFR-TKIs (90, 91).
Pre-clinical studies showed promising results of combined blockade of EGFR and MET signaling pathways in NSCLC (92). MET inhibitors can be divided into mAbs targeting HGF or the MET receptor (AMG 102, ficlatuzumab, and onartuzumab) or MET TKIs (tivantinib, cabozantinib, foretinib, and crizotinib) (93).
A phase II randomized study compared onartuzumab plus erlotinib vs. erlotinib alone in second- and third-line treatment. Onartuzumab, in combination with erlotinib, significantly improved PFS and OS in patients with increased MET gene copy (≥5) assessed by FISH (MET-FISH positive) as well as in patients with over-expression of MET receptor as assessed by immunohistochemistry (MET-IHC positive) regardless of gene amplification status (94). Unfortunately, a confirmatory phase III MET-Lung trial that randomized MET-IHC-positive NSCLC patients to combination onartuzumab/erlotinib vs. erlotinib alone was stopped prematurely due to lack of clinically meaningful efficacy in the combination arm (95).
Tivantinib was investigated in combination with erlotinib (EGFR-TKI) in patients with previously treated NSCLC in both phase II and phase III trials (96, 97). In the phase II trial, an exploratory subgroup analysis showed that MET-IHC-positive patients with non-squamous histology harboring KRAS mutations had better PFS and OS with tivantinib and erlotinib treatment when compared with erlotinib and placebo. MARQUEE, a phase III, double-blind trial randomized 1048 patients with metastatic pre-treated non-squamous NSCLC to tivantinib plus erlotinib vs. tivantinib plus placebo (98). While median PFS and ORR significantly favored tivantinib plus erlotinib (3.6 vs. 1.9 months; 10.3 vs. 6.5%, respectively), MARQUEE did not reach its primary endpoint of improved overall survival (http://eccamsterdam2013.ecco-org.eu/Scientific-Programme/Abstract-search.aspx?abstractid=6904). A subgroup analysis of patients with 2+-positive MET immunostaining demonstrated better OS, PFS, and ORR when compared to patients who had lower levels of tumoral MET expression. A further retrospective molecular subset analysis is underway to identify other potential biomarkers (MET copy number, KRAS, and EGFR mutations) that may help to select a target population for MET-directed treatments.
Crizotinib, which inhibits both ALK and MET, demonstrated promising results in a small pilot study (N = 13) of patients with MET-amplified NSCLC (99).
ROS-1
ROS-1 is an orphan receptor tyrosine kinase that is phylogenetically related to ALK (100–103). ROS-1 chromosomal rearrangements with CD74, EZR, SLC24A2, and FIG genes define a new genomic driver in 1–2.5% of NSCLC patients (25, 26). Clinical characteristics of NSCLC patients with ROS-1 rearrangements are similar to patients with ALK-rearranged NSCLC – more commonly seen in patients of Asian ethnicity, young age (median age 49.8 years), female sex, never-smokers, and adenocarcinoma histology (25). ROS-1 rearrangements appear mutually exclusive of other known oncogenic drivers like EGFR, KRAS, HER-2, ALK, RET, and MET aberrations (27, 104). Pre-clinical data showed activity of ALK inhibitors (i.e., crizotinib and TAE684) in ROS-1-rearranged NSCLC cell lines given the high degree of homology between ALK and ROS-1 tyrosine kinase domains (25). This led investigators to assess the benefit of crizotinib in this unique patient subset. Efficacy has been demonstrated with an overall response rate of 56% and 6-month PFS of 71% in 25 evaluable patients (105). There are a number of currently ongoing phase I and II studies investigating activity of crizotinib, dual ALK/ROS1 inhibitor PF-06463922, and ceritinib in ROS-1-rearranged NSCLC.
Since ROS-1-rearranged NSCLC is rare and detection of ROS1 fusions by a break-apart FISH assay is expensive and labor intensive, diagnostic algorithms and simpler screening methods (e.g., by immunohistochemistry) are needed to identify patients with ROS-1-rearranged NSCLC (104, 106). At this moment, patients without driver mutations like EGFR, KRAS, HER-2, ALK, and RET rearrangements and MET amplifications should be screened for ROS-1 fusions (preferentially never-smokers) since they can be offered targeted treatment with crizotinib.
KRAS
The RAS oncogene family, HRAS, KRAS, and NRAS, encodes intracellular transducer proteins (small GTPases) that are involved in transmitting signals from extracellular growth factor receptors like EGFR to the cell (107, 108). As G proteins, they are located on the intracellular side of the plasma membrane, bind guanine nucleotides, and have GTP-ase activity (109). In the resting state, RAS proteins are bound to GDP and are inactive. Upon exchange of GDP to GTP, the RAS-GTP complex activates multiple downstream pathways (MAPK, STAT, and PI3K) that regulate cell proliferation, motility, and apoptosis (110). After a short period, the signaling configuration of RAS is halted by intrinsic GTP-ase activity. Activating RAS mutations prevent GTP hydrolysis to GDP, thus the RAS protein is rendered constitutively active with uncontrolled activation of downstream signaling pathways (111).
KRAS mutations are present in approximately 30% of lung adenocarcinomas and less commonly in squamous NSCLC (~5%) (28). They are found more frequently in Caucasians with lung cancer than in the Asian population and in current- or ex-smokers when compared with never-smokers (29, 110). Most KRAS mutations in NSCLC are single amino acid substitutions in codon 12 (80%) and to a lesser extent in codons 13 and 61 (30). In current- or ex-smokers, KRAS mutations are usually transversions (pyrimidine nucleotide is exchanged for purine or vice versa; e.g., G → T or G → C) and transitions in never-smokers (purine nucleotide is exchanged for another purine or pyrimidine for another pyrimidine; e.g., G → A or C → T) (29). KRAS mutations are nearly always mutually exclusive with EGFR and BRAF mutations although rare co-existence of EGFR and KRAS mutations has been observed (12, 31–33). KRAS mutations co-exist with PIK3CA mutations in approximately 19% of PIK3CA-mutant NSCLC (32).
It has been postulated for over 20 years that KRAS-mutant NSCLC may be associated with poor outcome. However, multiple studies have shown conflicting results due to heterogeneity among the studies, including tumor type, stage, treatment, and study end points (28, 34). A meta-analysis of 28 studies published in 2005 demonstrated that KRAS mutation was a significant prognostic marker when polymerase chain reaction sequencing was used as a detection method (35). Recently published results of a LACE-Bio pooled retrospective analysis reported no prognostic or predictive (in regard to benefit from adjuvant chemotherapy) effect of KRAS mutations in patients with resected NSCLC (36). A subset analysis of patients with NSCLC with KRAS codon 13 mutations suggests that adjuvant chemotherapy may have a deleterious effect in this subgroup, but needs to be further validated (HR – 5.78; 95% CI, 2.06–16.2) (36). In the absence of prospective, large, randomized clinical trials, KRAS mutation status in NSCLC cannot be used as a prognostic nor predictive biomarker for treatment with exception of negative predictive value of KRAS mutations and response to EGFR-TKI (37, 112).
Direct inhibition of KRAS has been unsuccessful so far due to its molecular and functional complexity (113). The activation of the RAS-RAF-MEK-ERK signaling pathway as a consequence of KRAS mutations renders it an attractive target for small-molecule inhibition in KRAS-mutated NSCLC. Given the critical location in this signaling pathway, MEK has been recognized as an important target, downstream from KRAS, for anti-cancer therapy (114).
The efficacy of treatment with a combination of the orally available potent MEK inhibitor selumetinib plus docetaxel chemotherapy has been demonstrated in the treatment of patients with advanced KRAS-mutant NSCLC (115). Median PFS was 5.3 months in the selumetinib group and 2.1 months in the placebo group (p = 0.014), with a 37% ORR in the selumetinib/docetaxel arm and no response in the docetaxel alone arm (p < 0.0001).
Trametinib is another orally available MEK inhibitor that has been combined with docetaxel or pemetrexed in phase I/Ib trial in patients stratified by KRAS mutation status (116). While no difference in response rate was seen between the pemetrexed-treated groups, these response rates compare favorably with historical data for second-line chemotherapy treatment and support the absence of any negative interaction between these agents (117). Given these promising findings, ongoing studies are investigating the optimal combination of MEK inhibition and chemotherapeutic agents (www.clinicaltrials.gov).
Early studies have also suggested that the subgroup of KRAS-mutant NSCLC patients may benefit from targeting the PI3K-AKT-mTOR signaling pathway, downstream from KRAS. The mTOR inhibitor, ridaforolimus, has been investigated in patients with disease progression following chemotherapy with randomization to continued therapy or placebo after 8 weeks of treatment. Improved PFS was seen in the ongoing therapy group (4 vs. 2 months) with a trend toward survival benefit (18 vs. 5 months; p = 0.09) (118).
KRAS mutations in NSCLC, despite being the most common, remain the most intriguing and elusive of therapeutic targets. At present, targeted treatment is not available for KRAS-mutated NSCLC outside clinical trials. However, novel agents targeting downstream effector signaling pathways are under clinical development (119).
Conclusion
In the addition to the emergence of histological subtypes as key factors in the treatment decision-making process for patients with advanced NSCLC, identification of certain genomic abnormalities and protein expression signatures that drive progression and metastasis of lung cancer have led to a completely new approach to treatment of NSCLC patients (120). For the first time, we recognize NSCLC as a heterogenous entity and are able to use the differences within tumors to tailor treatment with clear improvements in outcome for patients.
Biomarker-driven treatment has proven to be a major breakthrough in the modern management of lung cancer. New therapeutic modalities target specific genomic aberrations resulting in deregulation of select signaling pathways that are crucial for proliferation and metastasis of lung cancer.
There are a number of clinically and therapeutically relevant molecular changes within the lung cancer genome that can be now effectively targeted with systemic therapy in specific subgroups of patients (14). Ongoing research involving genomic efforts to elucidate further molecular subsets of NSCLC with ongoing development of biomarker-guided targeted therapies hopefully will continue to expand the therapeutic options for NSCLC patients.
Unfortunately, the number of patients for whom targeted therapy is suitable is still very small (Figure 1). The access to tumor tissue for biomarker assessment and de novo molecular and genomic tumor heterogeneity (that may be further increased during the biomarker-driven therapy) remain a serious challenge. Ongoing research in detection of cell-free circulating tumor DNA (cfDNA) and circulating tumor cells (CTCs) may become clinically relevant alternatives for tumor biopsy that will provide measurements of the total tumor burden as well as identify mutations arising during therapy that may be responsible for development of acquired resistance (121). Genomic screening of NSCLC tumors will continue to facilitate identification of molecular mechanisms of acquired resistance to targeted therapies. Ongoing translational and clinical research will facilitate a greater understanding of genomic alterations within lung cancer, with the aim of increasing benefit to wider population of lung cancer patients.
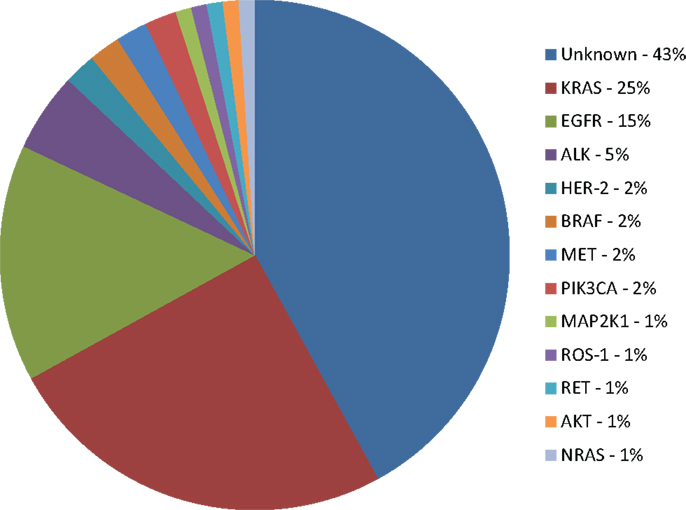
Figure 1. Molecular subsets of lung adenocarcinoma. KRAS: v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; EGFR: epidermal growth factor receptor; ALK: anaplastic lymphoma kinase fusion; HER-2: human epidermal growth factor receptor 2; BRAF: v-raf murine sarcoma viral oncogene homolog B1; PIK3CA: phosphoinositide-3-kinase, catalytic, α polypeptide; MAP2K1: mitogen-activated protein kinase kinase 1; RET: rearranged during transfection; AKT1: v-akt murine thymoma viral oncogene homolog 1; NRAS: neuroblastoma RAS viral (v-ras) oncogene homolog (4, 9, 14, 18, 24, 31, 104).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63(1):11–30. doi: 10.3322/caac.21166
2. Azzoli CG, Temin S, Aliff T, Baker S Jr, Brahmer J, Johnson DH, et al. 2011 focused update of 2009 American society of clinical oncology clinical practice guideline update on chemotherapy for stage IV non-small-cell lung cancer. J Clin Oncol (2010) 29(28):3825–31. doi:10.1200/JCO.2010.34.2774
3. Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol (2008) 26(21):3543–51. doi:10.1200/JCO.2007.15.0375
4. Mok TS. Personalized medicine in lung cancer: what we need to know. Nat Rev Clin Oncol (2011) 8(11):661–8. doi:10.1038/nrclinonc.2011.126
5. Keedy VL, Temin S, Somerfield MR, Beasley MB, Johnson DH, McShane LM, et al. American society of clinical oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol (2011) 29(15):2121–7. doi:10.1200/JCO.2010.31.8923
6. Pirker R, Herth FJ, Kerr KM, Filipits M, Taron M, Gandara D, et al. Consensus for EGFR mutation testing in non-small cell lung cancer: results from a European workshop. J Thorac Oncol (2010) 5(10):1706–13. doi:10.1097/JTO.0b013e3181f1c8de
7. Gandara DR, Li T, Lara PN Jr, Mack PC, Kelly K, Miyamoto S, et al. Algorithm for codevelopment of new drug-predictive biomarker combinations: accounting for inter- and intrapatient tumor heterogeneity. Clin Lung Cancer (2012) 13(5):321–5. doi:10.1016/j.cllc.2012.05.004
8. Haber DA, Gray NS, Baselga J. The evolving war on cancer. Cell (2011) 145(1):19–24. doi:10.1016/j.cell.2011.03.026
9. Peters S, Adjei AA, Gridelli C, Reck M, Kerr K, Felip E. Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2012) 23(Suppl 7):vii56–64. doi:10.1093/annonc/mds226
10. Scagliotti GV, Selvaggi G, Novello S, Hirsch FR. The biology of epidermal growth factor receptor in lung cancer. Clin Cancer Res (2004) 10(12 Pt 2):4227s–32s. doi:10.1158/1078-0432.CCR-040007
11. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer (2007) 7(3):169–81. doi:10.1038/nrc2088
12. Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst (2005) 97(5):339–46. doi:10.1093/jnci/dji055
13. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res (2004) 64(24):8919–23. doi:10.1158/0008-5472.CAN-04-2818
14. Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol (2011) 12(2):175–80. doi:10.1016/S1470-2045(10)70087-5
15. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature (2007) 448(7153):561–6. doi:10.1038/nature05945
16. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med (2010) 363(18):1693–703. doi:10.1056/NEJMoa1006448
17. Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol (2009) 27(26):4232–5. doi:10.1200/JCO.2009.23.6661
18. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol (2009) 27(26):4247–53. doi:10.1200/JCO.2009.22.6993
19. Inamura K, Takeuchi K, Togashi Y, Hatano S, Ninomiya H, Motoi N, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol (2009) 22(4):508–15. doi:10.1038/modpathol.2009.2
20. Cipriani NA, Abidoye OO, Vokes E, Salgia R. MET as a target for treatment of chest tumors. Lung Cancer (2009) 63(2):169–79. doi:10.1016/j.lungcan.2008.06.011
21. Sadiq AA, Geynisman DM, Salgia R. Inhibition of MET receptor tyrosine kinase and its ligand hepatocyte growth factor. J Thorac Oncol (2011) 6(11 Suppl 4):S1810–1. doi:10.1097/01.JTO.0000407568.45147.43
22. Park S, Choi YL, Sung CO, An J, Seo J, Ahn MJ, et al. High MET copy number and MET overexpression: poor outcome in non-small cell lung cancer patients. Histol Histopathol (2012) 27(2):197–207.
23. Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol (2009) 27(10):1667–74. doi:10.1200/JCO.2008.19.1635
24. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell (2007) 131(6):1190–203. doi:10.1016/j.cell.2007.11.025
25. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol (2012) 30(8):863–70. doi:10.1200/JCO.2011.35.6345
26. Kim HR, Lim SM, Kim HJ, Hwang SK, Park JK, Shin E, et al. The frequency and impact of ROS1 rearrangement on clinical outcomes in never smokers with lung adenocarcinoma. Ann Oncol (2012) 24(9):2364–70. doi:10.1093/annonc/mdt220
27. Yoshida A, Kohno T, Tsuta K, Wakai S, Arai Y, Shimada Y, et al. ROS1-rearranged lung cancer: a clinicopathologic and molecular study of 15 surgical cases. Am J Surg Pathol (2013) 37(4):554–62. doi:10.1097/PAS.0b013e3182758fe6
28. Slebos RJ, Kibbelaar RE, Dalesio O, Kooistra A, Stam J, Meijer CJ, et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N Engl J Med (1990) 323(9):561–5.
29. Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res (2008) 14(18):5731–4. doi:10.1158/1078-0432.CCR-08-0646
30. Prior IA, Lewis PD, Mattos C. A comprehensive survey of RAS mutations in cancer. Cancer Res (2012) 72(10):2457–67. doi:10.1158/0008-5472.CAN-11-2612
31. Schmid K, Oehl N, Wrba F, Pirker R, Pirker C, Filipits M. EGFR/KRAS/BRAF mutations in primary lung adenocarcinomas and corresponding locoregional lymph node metastases. Clin Cancer Res (2009) 15(14):4554–60. doi:10.1158/1078-0432.CCR-09-0089
32. Wang L, Hu H, Pan Y, Wang R, Li Y, Shen L, et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS One (2014) 9(2):e88291. doi:10.1371/journal.pone.0088291
33. Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol (2005) 23(25):5900–9. doi:10.1200/JCO.2005.02.857
34. Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med (1987) 317(15):929–35. doi:10.1056/NEJM198710083171504
35. Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer (2005) 92(1):131–9. doi:10.1038/sj.bjc.6602258
36. Shepherd FA, Domerg C, Hainaut P, Janne PA, Pignon JP, Graziano S, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol (2013) 31(17):2173–81. doi:10.1200/JCO.2012.48.1390
37. Martin P, Leighl NB, Tsao MS, Shepherd FA. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol (2013) 8(5):530–42. doi:10.1097/JTO.0b013e318283d958
38. Bazley LA, Gullick WJ. The epidermal growth factor receptor family. Endocr Relat Cancer (2005) 12(Suppl 1):S17–27. doi:10.1677/erc.1.01032
39. Hsieh AC, Moasser MM. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br J Cancer (2007) 97(4):453–7. doi:10.1038/sj.bjc.6603910
40. Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol (2006) 33(4):369–85. doi:10.1053/j.seminoncol.2006.04.003
41. Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer (2004) 4(5):361–70. doi:10.1038/nrc1360
42. Giaccone G. The role of gefitinib in lung cancer treatment. Clin Cancer Res (2004) 10(12 t 2):4233s–7s. doi:10.1158/1078-0432.CCR-040005
43. Perez-Soler R. The role of erlotinib (Tarceva, OSI 774) in the treatment of non-small cell lung cancer. Clin Cancer Res (2004) 10(12 Pt 2):4238s–40s. doi:10.1158/1078-0432.CCR-040017
44. Miller VA, Kris MG, Shah N, Patel J, Azzoli C, Gomez J, et al. Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non-small-cell lung cancer. J Clin Oncol (2004) 22(6):1103–9. doi:10.1200/JCO.2004.08.158
45. Pao W, Zakowski M, Cordon-Cardo C, Ben-Porat L, Kris MG, Miller VA. Molecular characteristics of non-small cell lung cancer (NSCLC) patients sensitive to gefitinib. ASCO Meet Abstr (2004) 22(14 Suppl):7025.
46. Kris MG, Sandler A, Miller V, Cespon M, Zakowski M, Pizzo B, et al. Cigarette smoking history predicts sensitivity to erlotinib: results of a phase II trial in patients with bronchioloalveolar carcinoma (BAC). ASCO Meet Abstr (2004) 22(14 Suppl):7062.
47. Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (the IDEAL 1 trial) [corrected]. J Clin Oncol (2003) 21(12):2237–46. doi:10.1200/JCO.2003.10.038
48. Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA (2003) 290(16):2149–58. doi:10.1001/jama.290.16.2149
49. Perez-Soler R. Phase II clinical trial data with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib (OSI-774) in non-small-cell lung cancer. Clin Lung Cancer (2004) 6(Suppl 1):S20–3. doi:10.3816/CLC.2004.s.010
50. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med (2005) 353(2):123–32. doi:10.1056/NEJMoa050753
51. Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (iressa survival evaluation in lung cancer). Lancet (2005) 366(9496):1527–37. doi:10.1016/S0140-6736(05)67625-8
52. Herbst RS, Prager D, Hermann R, Fehrenbacher L, Johnson BE, Sandler A, et al. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol (2005) 23(25):5892–9. doi:10.1200/JCO.2005.02.840
53. Lee DH, Han JY, Lee HG, Lee JJ, Lee EK, Kim HY, et al. Gefitinib as a first-line therapy of advanced or metastatic adenocarcinoma of the lung in never-smokers. Clin Cancer Res (2005) 11(8):3032–7. doi:10.1158/1078-0432.CCR-04-2149
54. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med (2004) 350(21):2129–39. doi:10.1056/NEJMoa040938
55. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science (2004) 304(5676):1497–500. doi:10.1126/science.1099314
56. Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A (2004) 101(36):13306–11. doi:10.1073/pnas.0405220101
57. Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res (2006) 66(16):8163–71. doi:10.1158/0008-5472.CAN-06-0453
58. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) 361(10):947–57. doi:10.1056/NEJMoa0810699
59. Janne PA, Wang X, Socinski MA, Crawford J, Stinchcombe TE, Gu L, et al. Randomized phase II trial of erlotinib alone or with carboplatin and paclitaxel in patients who were never or light former smokers with advanced lung adenocarcinoma: CALGB 30406 trial. J Clin Oncol (2012) 30(17):2063–9. doi:10.1200/JCO.2011.40.1315
60. Han JY, Park K, Kim SW, Lee DH, Kim HY, Kim HT, et al. First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol (2012) 30(10):1122–8. doi:10.1200/JCO.2011.36.8456
61. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol (2010) 11(2):121–8. doi:10.1016/S1470-2045(09)70364-X
62. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Seto T, et al. Updated overall survival results of WJTOG 3405, a randomized phase III trial comparing gefitinib (G) with cisplatin plus docetaxel (CD) as the first-line treatment for patients with non-small cell lung cancer harboring mutations of the epidermal growth factor receptor (EGFR). ASCO Meet Abstr (2012) 30(15 Suppl):7521.
63. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med (2010) 362(25):2380–8. doi:10.1056/NEJMoa0909530
64. Inoue A, Kobayashi K, Maemondo M, Sugawara S, Oizumi S, Isobe H, et al. Final overall survival results of NEJ002, a phase III trial comparing gefitinib to carboplatin (CBDCA) plus paclitaxel (TXL) as the first-line treatment for advanced non-small cell lung cancer (NSCLC) with EGFR mutations. ASCO Meet Abstr (2011) 29(15 Suppl):7519. doi:10.1093/annonc/mds214
65. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol (2011) 12(8):735–42. doi:10.1016/S1470-2045(11)70184-X
66. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol (2012) 13(3):239–46. doi:10.1016/S1470-2045(11)70393-X
67. Rosell R, Gervais R, Vergnenegre A, Massuti B, Felip E, Cardenal F, et al. Erlotinib versus chemotherapy (CT) in advanced non-small cell lung cancer (NSCLC) patients (p) with epidermal growth factor receptor (EGFR) mutations: interim results of the European erlotinib versus chemotherapy (EURTAC) phase III randomized trial. ASCO Meet Abstr (2011) 29(15 Suppl):7503.
68. Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Janne PA, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol (2008) 26(15):2442–9. doi:10.1200/JCO.2007.14.8494
69. Inoue A, Suzuki T, Fukuhara T, Maemondo M, Kimura Y, Morikawa N, et al. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol (2006) 24(21):3340–6. doi:10.1200/JCO.2005.05.4692
70. Asahina H, Yamazaki K, Kinoshita I, Sukoh N, Harada M, Yokouchi H, et al. A phase II trial of gefitinib as first-line therapy for advanced non-small cell lung cancer with epidermal growth factor receptor mutations. Br J Cancer (2006) 95(8):998–1004. doi:10.1038/sj.bjc.6603393
71. Minkovsky N, Berezov A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr Opin Investig Drugs (2008) 9(12):1336–46.
72. Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene (2008) 27(34):4702–11. doi:10.1038/onc.2008.109
73. Yang JC-H, Schuler MH, Yamamoto N, O’Byrne KJ, Hirsh V, Mok T, et al. LUX-Lung 3: a randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. ASCO Meet Abstr (2012) 30(18 Suppl):LBA7500.
74. Yang JC-H, Sequist LV, Schuler MH, Mok T, Yamamoto N, O’Byrne KJ, et al. Overall survival (OS) in patients (pts) with advanced non-small cell lung cancer (NSCLC) harboring common (Del19/L858R) epidermal growth factor receptor mutations (EGFR mut): pooled analysis of two large open-label phase III studies (LUX-Lung 3 [LL3] and LUX-Lung 6 [LL6]) comparing afatinib with chemotherapy (CT). ASCO Meet Abstr (2014) 32(15 Suppl):8004.
75. Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, et al. The T790M "gatekeeper" mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther (2008) 7(4):874–9. doi:10.1158/1535-7163.MCT-07-2387
76. Janne PA, Ramalingam SS, Yang JC-H, Ahn M-J, Kim D-W, Kim S-W, et al. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients (pts) with EGFR inhibitor-resistant non-small cell lung cancer (NSCLC). ASCO Meet Abstr (2014) 32(15 Suppl):8009.
77. Sequist LV, Soria J-C, Gadgeel SM, Wakelee HA, Camidge DR, Varga A, et al. First-in-human evaluation of CO-1686, an irreversible, highly selective tyrosine kinase inhibitor of mutations of EGFR (activating and T790M). ASCO Meet Abstr (2014) 32(15 Suppl):8010.
78. Camidge DR, Kono SA, Flacco A, Tan AC, Doebele RC, Zhou Q, et al. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res (2010) 16(22):5581–90. doi:10.1158/1078-0432.CCR-10-0851
79. Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol (2012) 13(10):1011–9. doi:10.1016/S1470-2045(12)70344-3
80. Ou SH, Bazhenova L, Camidge DR, Solomon BJ, Herman J, Kain T, et al. Rapid and dramatic radiographic and clinical response to an ALK inhibitor (crizotinib, PF02341066) in an ALK translocation-positive patient with non-small cell lung cancer. J Thorac Oncol (2010) 5(12):2044–6. doi:10.1097/JTO.0b013e318200f9ff
81. Ou SH, Kwak EL, Siwak-Tapp C, Dy J, Bergethon K, Clark JW, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol (2011) 6(5):942–6. doi:10.1097/JTO.0b013e31821528d3
82. Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol (2011) 12(11):1004–12. doi:10.1016/S1470-2045(11)70232-7
83. Kim D-W, Ahn M-J, Shi Y, De Pas TM, Yang P-C, Riely GJ, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). ASCO Meet Abstr (2011) 30(15 Suppl):7533. doi:10.1007/s40265-013-0142-z
84. Crino L, Kim D, Riely GJ, Janne PA, Blackhall FH, Camidge DR, et al. Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): profile 1005. ASCO Meet Abstr (2011) 29(15 Suppl):7514.
85. Mok T, Kim D-W, Wu Y-L, Solomon BJ, Nakagawa K, Mekhail T, et al. First-line crizotinib versus pemetrexed-cisplatin or pemetrexed-carboplatin in patients (pts) with advanced ALK-positive non-squamous non-small cell lung cancer (NSCLC): results of a phase III study (PROFILE 1014). ASCO Meet Abstr (2014) 32(15 Suppl):8002.
86. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med (2013) 368(25):2385–94. doi:10.1056/NEJMoa1214886
87. Camidge DR, Bazhenova L, Salgia R, Weiss GJ, Langer CJ, Shaw AT, et al. First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: updated results. ASCO Meet Abstr (2013) 31(15 Suppl):8031.
88. Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med (2014) 370(13):1189–97. doi:10.1056/NEJMoa1311107
89. Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev (2003) 22(4):309–25. doi:10.1023/A:1023768811842
90. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (2007) 316(5827):1039–43. doi:10.1126/science.1141478
91. Xu L, Kikuchi E, Xu C, Ebi H, Ercan D, Cheng KA, et al. Combined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and MET. Cancer Res (2012) 72(13):3302–11. doi:10.1158/0008-5472.CAN-11-3720
92. Robinson KW, Sandler AB. The role of MET receptor tyrosine kinase in non-small cell lung cancer and clinical development of targeted anti-MET agents. Oncologist (2013) 18(2):115–22. doi:10.1634/theoncologist.2012-0262
93. Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol (2013) 31(8):1089–96. doi:10.1200/JCO.2012.43.9422
94. Spigel DR, Ervin TJ, Ramlau R, Daniel DB, Goldschmidt JH, Blumenschein GR, et al. Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. ASCO Meet Abstr (2011) 29(15 Suppl):7505.
95. Spigel DR, Edelman MJ, O’Byrne K, Paz-Ares L, Shames DS, Yu W, et al. Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV NSCLC: results from the pivotal phase III randomized, multicenter, placebo-controlled METLung (OAM4971g) global trial. ASCO Meet Abstr (2014) 32(15 Suppl):8000.
96. Sequist LV, von Pawel J, Garmey EG, Akerley WL, Brugger W, Ferrari D, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol (2011) 29(24):3307–15. doi:10.1200/JCO.2010.34.0570
97. Munshi N, Jeay S, Li Y, Chen CR, France DS, Ashwell MA, et al. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther (2010) 9(6):1544–53. doi:10.1158/1535-7163.MCT-09-1173
98. Scagliotti GV, Novello S, Schiller JH, Hirsh V, Sequist LV, Soria JC, et al. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer (2012) 13(5):391–5. doi:10.1016/j.cllc.2012.01.003
99. Camidge DR, Ou S-HI, Shapiro G, Otterson GA, Villaruz LC, Villalona-Calero MA, et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). ASCO Meet Abstr (2014) 32(15 Suppl):8001.
100. Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta (2009) 1795(1):37–52. doi:10.1016/j.bbcan.2008.07.006
101. Birchmeier C, Sharma S, Wigler M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc Natl Acad Sci U S A (1987) 84(24):9270–4. doi:10.1073/pnas.84.24.9270
102. Gu TL, Deng X, Huang F, Tucker M, Crosby K, Rimkunas V, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One (2011) 6(1):e15640. doi:10.1371/journal.pone.0015640
103. Birch AH, Arcand SL, Oros KK, Rahimi K, Watters AK, Provencher D, et al. Chromosome 3 anomalies investigated by genome wide SNP analysis of benign, low malignant potential and low grade ovarian serous tumours. PLoS One (2011) 6(12):e28250. doi:10.1371/journal.pone.0028250
104. Go H, Kim DW, Kim D, Keam B, Kim TM, Lee SH, et al. Clinicopathologic analysis of ROS1-rearranged non-small-cell lung cancer and proposal of a diagnostic algorithm. J Thorac Oncol (2013) 8(11):1445–50. doi:10.1097/JTO.0b013e3182a4dd6e
105. Shaw AT, Camidge DR, Engelman JA, Solomon BJ, Kwak EL, Clark JW, et al. Clinical activity of crizotinib in advanced non-small cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. ASCO Meet Abstr (2012) 30(15 Suppl):7508. doi:10.1097/CCO.0b013e32835d8175
106. Mescam-Mancini L, Lantuejoul S, Moro-Sibilot D, Rouquette I, Souquet PJ, Audigier-Valette C, et al. On the relevance of a testing algorithm for the detection of ROS1-rearranged lung adenocarcinomas. Lung Cancer (2014) 83(2):168–73. doi:10.1016/j.lungcan.2013.11.019
107. Rodenhuis S, Slebos RJ. The ras oncogenes in human lung cancer. Am Rev Respir Dis (1990) 142(6 Pt 2):S27–30. doi:10.1164/ajrccm/142.6_Pt_2.S27
109. Harvey JJ. An unidentified virus which causes the rapid production of tumours in mice. Nature (1964) 204:1104–5. doi:10.1038/2041104b0
110. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene (2007) 26(22):3291–310. doi:10.1038/sj.onc.1210422
111. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol (2008) 9(7):517–31. doi:10.1038/nrm2438
112. Raponi M, Winkler H, Dracopoli NC. KRAS mutations predict response to EGFR inhibitors. Curr Opin Pharmacol (2008) 8(4):413–8. doi:10.1016/j.coph.2008.06.006
113. Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol (2011) 13(1):39–51. doi:10.1038/nrm3255
114. Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene (1999) 18(3):813–22. doi:10.1038/sj.onc.1202367
115. Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol (2013) 14(1):38–47. doi:10.1016/S1470-2045(12)70489-8
116. Kelly K, Mazieres J, Leighl NB, Barlesi F, Zalcman G, Gordon MS, et al. Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with pemetrexed for KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): a phase I/Ib trial. ASCO Meet Abstr (2013) 31(15 Suppl):8027.
117. Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol (2004) 22(9):1589–97. doi:10.1200/JCO.2004.08.163
118. Riely GJ, Brahmer JR, Planchard D, Crino L, Doebele RC, Mas Lopez LA, et al. A randomized discontinuation phase II trial of ridaforolimus in non-small cell lung cancer (NSCLC) patients with KRAS mutations. ASCO Meet Abstr (2012) 30(15 Suppl):7531.
119. Roberts PJ, Stinchcombe TE. KRAS mutation: should we test for it, and does it matter? J Clin Oncol (2013) 31(8):1112–21. doi:10.1200/JCO.2012.43.0454
120. Langer CJ, Besse B, Gualberto A, Brambilla E, Soria JC. The evolving role of histology in the management of advanced non-small-cell lung cancer. J Clin Oncol (2010) 28(36):5311–20. doi:10.1200/JCO.2010.28.8126
Keywords: EGFR, ALK, Met, ROS-1, KRAS, lung adenocarcinoma, biomarkers
Citation: Korpanty GJ, Graham DM, Vincent MD and Leighl NB (2014) Biomarkers that currently affect clinical practice in lung cancer: EGFR, ALK, MET, ROS-1, and KRAS. Front. Oncol. 4:204. doi: 10.3389/fonc.2014.00204
Received: 26 May 2014; Accepted: 16 July 2014;
Published online: 11 August 2014.
Edited by:
Barbara Melosky, British Columbia Cancer Agency, CanadaReviewed by:
Sacha I. Rothschild, University Hospital Basel, SwitzerlandRicardo Martinez, Eli Lilly, USA
Copyright: © 2014 Korpanty, Graham, Vincent and Leighl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natasha B. Leighl, Division of Medical Oncology, Princess Margaret Cancer Centre, 5-105 610 University Avenue, Toronto, ON M5G 2M9, Canada e-mail: natasha.leighl@uhn.ca