 Shubhankar Suman
Shubhankar Suman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 08 October 2015
Sec. Radiation Oncology
Volume 5 - 2015 | https://doi.org/10.3389/fonc.2015.00218
This article is part of the Research TopicCharged Particles in OncologyView all 60 articles
Although it is known that accumulation of oncogenic β-catenin is critical for intestinal tumorigenesis, the underlying mechanisms have not yet been fully explored. Post-translational β-catenin level is regulated via the adenomatous polyposis coli (APC)-dependent as well as the APC-independent ubiquitin–proteasome pathway (UPP). Employing an APC-mutant mouse model (APCMin/+) the present study aimed to investigate the status of RXRα, an APC-independent factor involved in targeting β-catenin to UPP for degradation, in tumor-bearing and tumor-free areas of intestine after exposure to energetic 56Fe ions. APCMin/+ mice were exposed to energetic 56Fe ions (4 or 1.6 Gy) and intestinal tumor samples and tumor-free normal intestinal samples were collected 100–110 days after exposure. The status of TCF4, β-catenin, cyclin D1, and RXRα was examined using immunohistochemistry and immunoblots. We observed increased accumulation of the transcription factor TCF4 and its co-activator β-catenin as well as their downstream oncogenic target protein cyclin-D1 in 56Fe ion-induced intestinal tumors. Further, decreased expression of RXRα in tumors as well as in adjacent normal epithelium was indicative of perturbations in β-catenin proteasomal-targeting machinery. This indicates that decreased UPP targeting of β-catenin due to downregulation of RXRα can contribute to further accumulation of β-catenin and to 56Fe-induced tumorigenesis.
Heavy ion charged particle (HZE) radiation, such as 56Fe ions, is prevalent in deep space, and is a major concern for astronauts’ health (1). Recently, using APCMin/+ mice, a well-accepted mouse model for human colorectal cancer (CRC), we found increased risk of CRC development accompanied by increased nuclear accumulation of oncogenic β-catenin and activation of its downstream signaling after exposure to 56Fe ions (2–5). However, the mechanisms behind the accumulation of oncogenic β-catenin are not yet fully understood.
Cellular levels of free β-catenin are tightly regulated via the ubiquitin–proteasome pathway (UPP). Targeting of β-catenin to the proteasome and its subsequent degradation involves two adenomatous polyposis coli (APC)-dependent (i.e., APC/GSK3β/AXIN and APC/Siah1) and one APC-independent (RXRα-mediated) mechanisms (6). In gastrointestinal (GI) tumors, genes involved in APC-dependent (APC, Siah1, and Axin) targeting of β-catenin are often mutated (7–11), and similarly in APCMin/+ mice, tumor formation is mostly driven through inactivation of the wild type APC allele (12). Thus, APC-dependent proteasomal targeting of β-catenin is eventually disabled in these tumors. In the absence of proteasomal targeting, β-catenin accumulates and interacts with T-cell factor transcription factors (TCF4) in the nucleus leading to activation of oncogenic signaling pathways (13). In view of the known perturbations in APC-dependent proteasomal targeting of β-catenin early in the GI tumorigenesis process, only the APC-independent (RXRα-dependent) pathway would remain to control its accumulation. However, the status of the APC-independent proteasomal targeting of the β-catenin in heavy ion radiation-induced intestinal tumors has not been explored. In this study, using the APCMin/+ intestinal tumor mouse model (14), we demonstrated downregulation of RXRα expression, which may complement the disabled APC-dependent proteasomal degradation pathway to increase β-catenin accumulation in 56Fe-induced tumors. Downregulation of RXRα observed in this study could potentially play a crucial role in heavy ion radiation-induced increased risk of intestinal tumorigenesis and would warrant further investigation.
Male APCMin/+ mice (The Jackson Laboratory, Bar Harbor, ME, USA) were bred with female C57BL/6J mice at the Georgetown University (GU)’s animal facility. Genotyping using tail DNA samples were done using reverse-transcription polymerase chain reaction (RT-PCR) to identify heterozygous offspring as per the Jackson Laboratory protocol. The mouse colony was maintained on standard certified rodent diet and filtered water in a humidity and temperature-controlled room with 12 h dark/light cycle. All experimental procedures were performed in compliance with the protocols approved by the Institutional Animal Care and Use Committee (IACUC) at GU and Brookhaven National Laboratory (BNL). Both the facilities are Association for Assessment and Accreditation of Laboratory and Animal Care International (AAALACI) accredited facilities and we followed The Guide for the Care and Use of Laboratory Animals.
APCMin/+ female mice (6–8-weeks old) were placed in well-ventilated transparent plastic boxes (1 mouse/box) allowing easy movement and irradiated with 4 or 1.6 Gy whole body 56Fe radiation (energy: 1000 MeV/n; LET: 148 keV/μm; dose rate: 1 Gy/min) at the NASA Space Radiation Laboratory (NSRL) at BNL. These two doses were used in our previously published tumorigenesis experiments and samples collected during that study were used for molecular analysis in this study. For 56Fe exposure, both control and treatment groups were shipped to BNL for irradiation and brought back to GU after irradiation in a temperature-controlled vehicle for a same day delivery to minimize stress to the animals. Age-matched 56Fe-irradiated and control mice were euthanized by CO2 asphyxiation between 100 and 110 days after radiation exposure. The small intestinal tract was surgically removed, washed with phosphate-buffered saline (PBS), and cut open longitudinally at room temperature. A dissecting scope (Leica MZ6, Buffalo Grove, IL, USA) was used to visualize and dissect tumors, which were then flash frozen in liquid nitrogen and stored at −80°C for further use. Also, intestinal samples (~3 cm) with tumor-bearing and surrounding tumor-free area were fixed overnight in 10% buffered formalin, embedded in paraffin, and 4 μm-thick sections were obtained for immunohistochemistry staining.
Intestinal sections (n = 5 mice per group) were used for immunohistochemistry with a protocol described earlier (3). Briefly, immunostaining for active-β-catenin (Cat#05-665, Millipore, Billerica, MA, USA; dilution: 1:100), TCF4 (Cat#05-511, Millipore; dilution: 1:100), RXRα (Cat#sc-553, Santa Cruz Biotechnology, Dallas, TX, USA; dilution: 1:40), and cyclin D1 (Cat#04-1151; Millipore; dilution: 1:150) were performed by soaking slides in antigen retrieval citrate buffer (pH 6.0; Dako, Carpinteria, CA, USA) and heating at 100°C for 15 min in a microwave oven. Further, endogenous peroxidase activity was quenched using 3% hydrogen peroxide in methanol followed by incubation in blocking buffer (5% bovine serum albumin in PBS) for 30 min. After blocking sections were incubated overnight at 4°C with the respective primary antibody. Signal detection and color development was done using SuperPicture 3rd Gen IHC detection kit (Cat#87-9673; Invitrogen, Carlsbad, CA, USA). Sections were counterstained using hematoxylin and images were acquired using bright field microscopy at a magnification of 20×. At least 10 randomly chosen images from the tumor-bearing as well as from the tumor-free areas were acquired from each mouse and a representative image from each group is shown in the results. Images were analyzed using color deconvolution and image-based tool for counting nuclei (ITCN) plug-ins of ImageJ v1.45 software (National Institutes of Health, Bethesda, MD, USA). Quantification data were statistically analyzed using two-tailed paired Student’s t-test and difference between control and irradiated group was considered significant when p-value was <0.05. Error bars represent mean ± SEM.
Frozen intestinal tumor samples (n = 5 mice per group) were pooled and used for immunoblot analysis of RXRα level with a protocol described previously (3). Briefly, samples were homogenized in ice-cold lysis buffer, centrifuged, and supernatant collected. Protein was estimated in supernatant and equal amount of protein was used for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Protein was transferred to PVDF membrane, incubated with RXRα antibody, and protein detected using horseradish peroxidase (HRP) conjugated secondary antibody and enhanced chemiluminescence (ECL) detection system (Cat# 34080, Thermo Fisher Scientific, Rockford, IL, USA) and representative images shown in the results.
Intestinal tumors stained for β-catenin showed increased level in 4 Gy 56Fe-irradiated samples relative to control tumors from sham-irradiated mice (Figures 1A,B) and this is consistent with our previous results after 1.6 Gy 56Fe irradiation (3). Higher levels were also observed for TCF4 in 56Fe-irradiated intestinal tumors relative to controls (Figures 1C,D). Transcription factor TCF4 along with the transcriptional co-activator β-catenin are involved in transcribing pro-proliferative factors, such as cyclin D1, and increased cyclin D1 was observed in the current study (Figures 1E,F) as well.
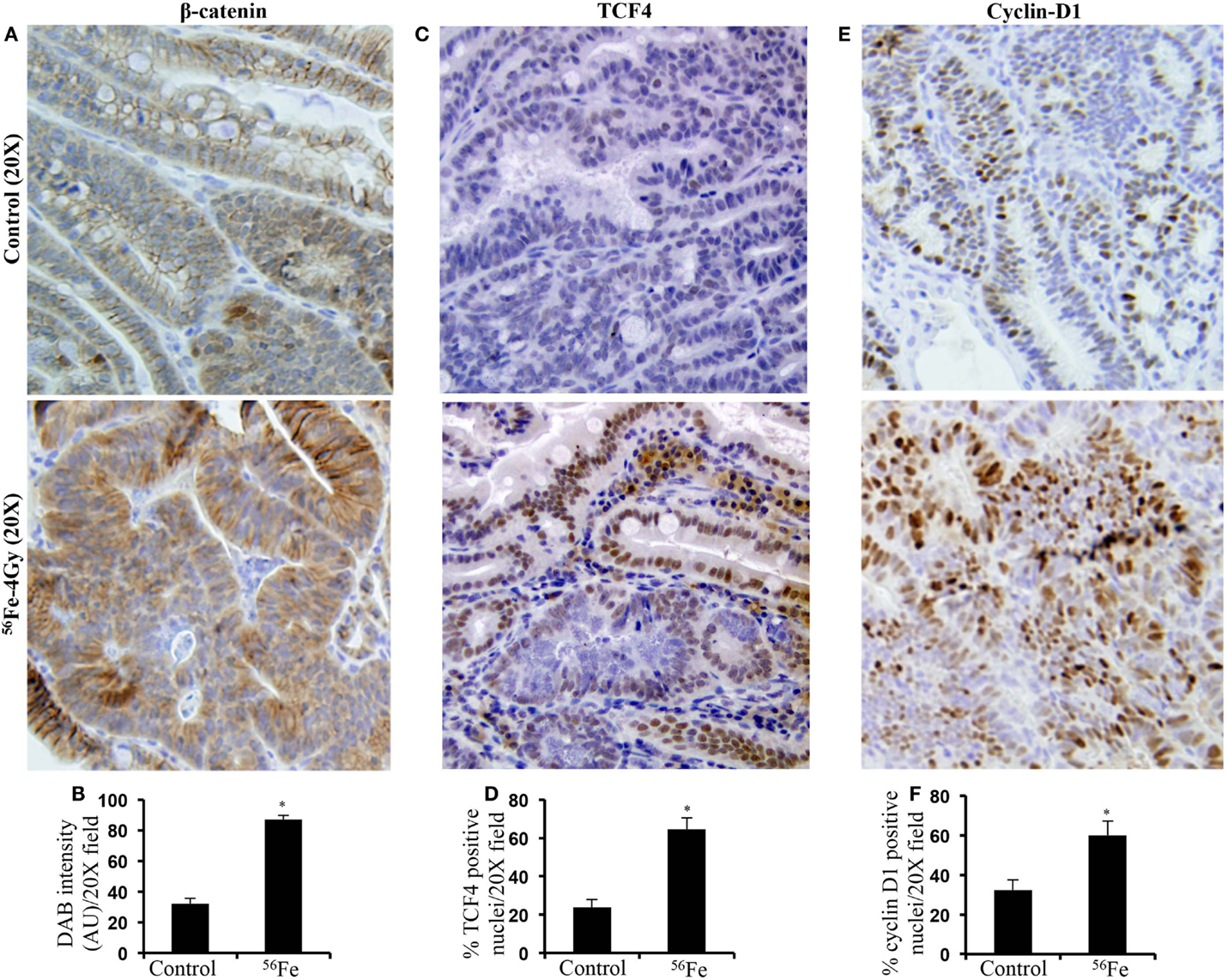
Figure 1. Accumulation and activation of oncogenic β-catenin signaling in 56Fe-induced intestinal tumors compared to spontaneous tumors from sham-irradiated mice (control). (A) Immunohistochemical detection of active-β-catenin in 56Fe-induced intestinal tumors. (B) Quantification of β-catenin expression in intestinal tumors. (C) Immunohistochemical detection of β-catenin transcriptional regulator TCF4 in 56Fe-induced intestinal tumors. (D) Quantification of TCF4 positive nuclei in intestinal tumors. (E) Immunohistochemical detection of β-catenin/TCF4 oncogenic target cyclin-D1 in 56Fe-induced intestinal tumors. (F) Quantification of cyclin-D1 positive nuclei in intestinal tumors. Error bars represent mean ± SEM and p < 0.05 was considered significant. AU – Arbitrary Unit.
Immunohistochemistry in tumor samples demonstrated that expression of RXRα was reduced after 4 Gy (Figure 2A). Quantification and statistical analysis of stained sections from five mice showed that RXRα was significantly lowered in 56Fe-irradiated tumors relative to sham-irradiated tumors (Figure 2B). Intestinal tumors from 1.6 Gy 56Fe-irradiated mice also showed decreased RXRα staining (Figure 2C) and quantification and statistical analysis showed that the staining in irradiated samples were significantly lower compared to controls (Figure 2D). However, quantification did not show significant difference in RXRα staining between two radiation doses. Immunoblots of 4 Gy (Figure 2E) and 1.6 Gy (Figure 2F) intestinal tumor samples also showed decreased RXRα. We also performed immunohistochemistry for RXRα on tumor-free intestinal sections from APCMin/+ mice exposed to either 1.6 or 4 Gy 56Fe ions. Staining of tumor-free intestinal section showed lower expression of RXRα after 4 Gy 56Fe relative to corresponding controls (Figure 3A). Decreased RXRα after 4 Gy 56Fe was statistically significant compared to sham-irradiated controls (Figure 3B). Conversely, we also observed downregulation of RXRα in 1.6 Gy irradiated samples compared to controls (Figure 3C) and quantification showed statistically significant difference between irradiated and sham-irradiated samples (Figure 3D).
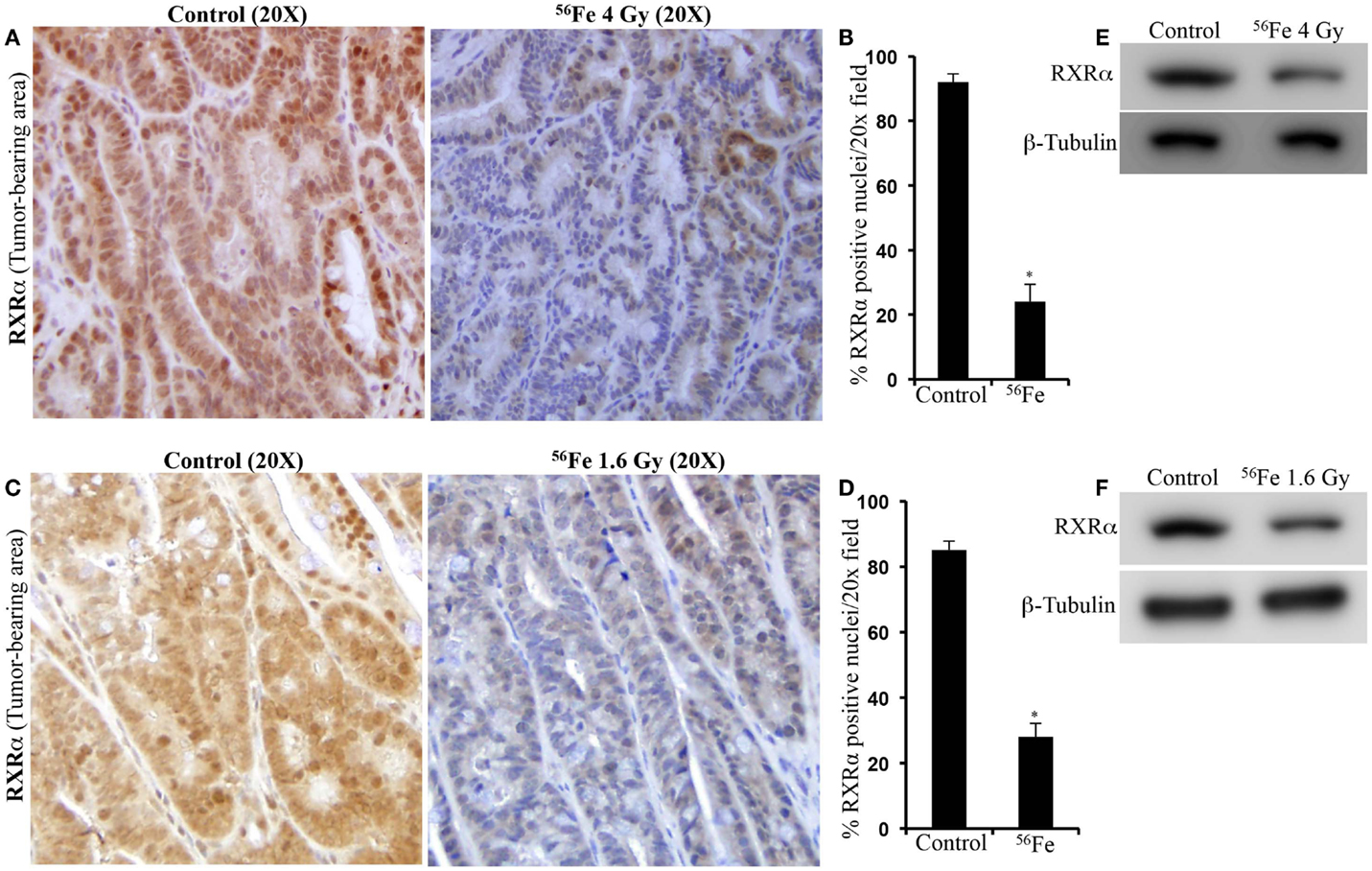
Figure 2. Lower RXRα expression in 56Fe-induced intestinal tumors. (A) Representative immunohistochemistry images showing expression of RXRα in spontaneous and 4 Gy 56Fe-induced intestinal tumors. (B) Quantification of RXRα in spontaneous and 4 Gy 56Fe-induced intestinal tumors. (C) Expression of RXRα in spontaneous and 1.6 Gy 56Fe-induced intestinal tumors. (D) Quantification of RXRα in spontaneous and 1.6 Gy 56Fe-induced intestinal tumors. (E) Immunoblots of RXRα in spontaneous and 4 Gy 56Fe-induced intestinal tumors. (F) Immunoblots of RXRα in spontaneous and 1.6 Gy 56Fe-induced intestinal tumors. Error bars represent mean ± SEM and p < 0.05 was considered significant.
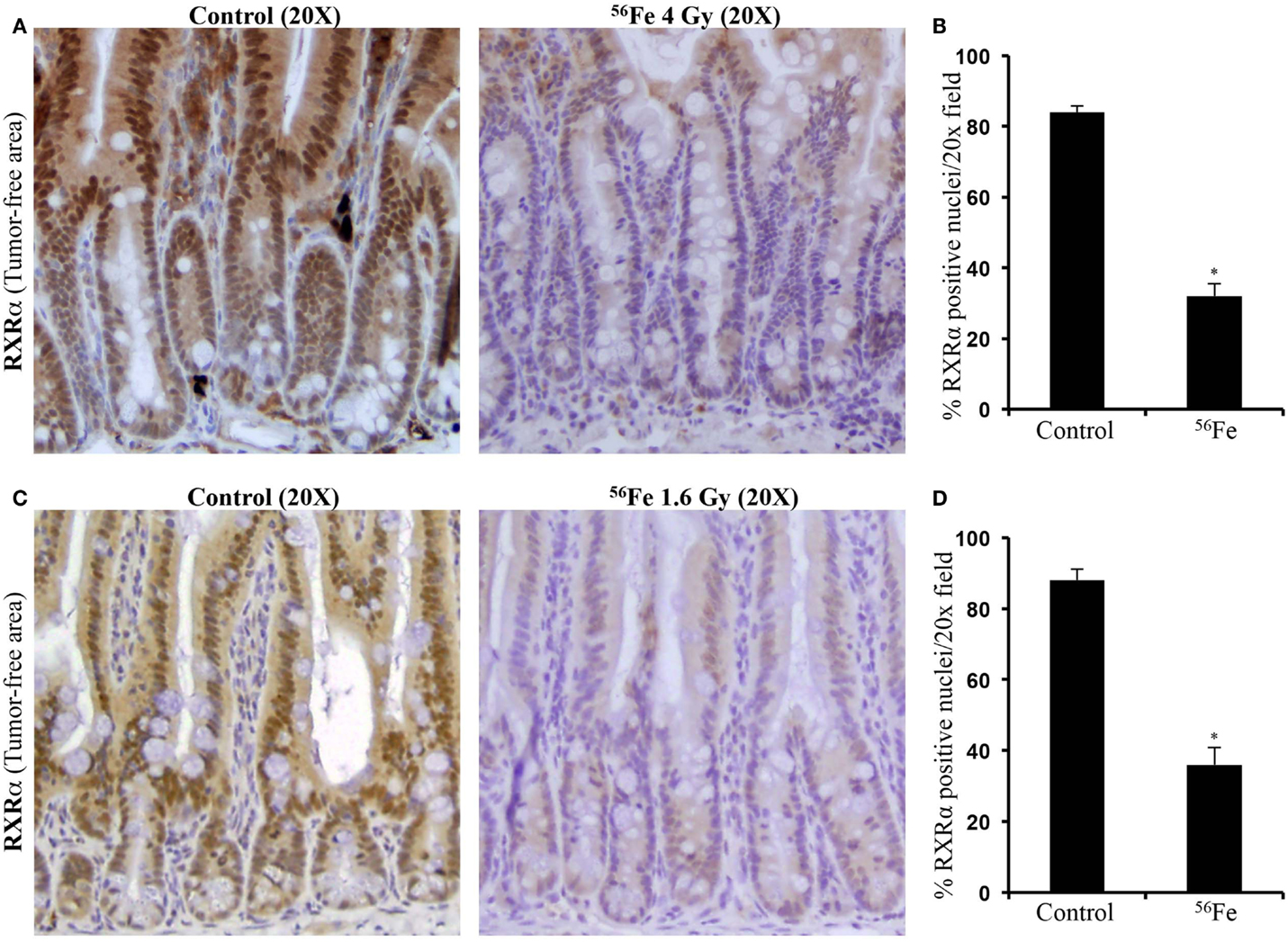
Figure 3. Downregulation of RXRα was observed in tumor-free areas of intestinal samples from APCMin/+ mice. (A) Decreased RXRα expression after 4 Gy 56Fe radiation. (B) Quantification of immunohistochemistry images showed significant decrease in RXRα after 4 Gy 56Fe. (C) Decreased RXRα expression after 1.6 Gy 56Fe radiation. (D) Quantification of immunohistochemistry images showed significant decrease in RXRα after 1.6 Gy 56Fe. Error bars represent mean ± SEM and p < 0.05 was considered significant.
The carcinogenic potential of ionizing radiation is well known and using animal models it has been established that high-LET heavy ion radiation has higher carcinogenic potential compared to low-LET radiation (15). Increased frequencies of site-specific cancer following heavy ion exposure have been reported in various rodent models with upregulation of oncogenic signaling mediated through genetic, epigenetic, and/or physiological changes (3, 15–17). Earlier studies conducted in APCMin/+ mice revealed increased tumor induction and a higher number of adenocarcinomas, which was associated with greater upregulation of β-catenin signaling after 56Fe exposure relative to γ radiation; this is indicative of perturbations in the molecular events upstream of β-catenin (3). The purpose of the current study was to develop mechanistic insight into greater tumorigenesis observed in our previous work in APCMin/+ mice after two doses of 56Fe radiation relative to γ radiation. While pathways can be investigated in the wild-type mice, they are resistant to intestinal tumorigenesis. Therefore, we used APC-mutant mice not only to quantitatively assess tumor frequency but also to understand molecular pathway alterations, which may have contributed to tumor development after radiation exposure. While we reported previously that two doses of 56Fe caused higher tumor frequency, we are yet to fully understand molecular characteristics of the tumors and tumor-adjacent normal tissues after 56Fe irradiation. To this end, the results presented in the current study explain in part potential underlying mechanisms contributing to increased tumor frequency after 56Fe irradiation. We have focused on the APC-independent mechanism of β-catenin degradation via UPP. In APC-deficient adenoma, accumulation of β-catenin complexed with nuclear TCF4 results in the increased expression of its oncogenic target genes, such as cyclin-D1 that promotes intestinal cell proliferation and polyp formation (18). In agreement with our published reports in APCMin/+ mice exposed to 1.6 Gy of 56Fe ion, the current study also observed similar activation of β-catenin at 4 Gy of 56Fe ion along with increased TCF4 and cyclin-D1. Significant loss of RXRα was evident in tumors as well as in tumor-free areas of intestine after 56Fe radiation and this could contribute to decreased proteasomal targeting of β-catenin, therefore enhancing cell survival and proliferation through β-catenin/TCF4 signaling. Notably, RXRα was downregulated in both the radiation doses tested suggesting that the effect is independent of radiation dose and that the lower dose may have a proportionately greater effect relative to the higher dose. We recognize that the mean absorbed doses of energetic 56Fe ions used in the current study are higher than the doses astronauts are expected to receive during prolonged space missions. These high doses of energetic 56Fe ions were used as a proof of principle in our initial studies for establishing the differential effects, quantitatively and qualitatively, of space compared to γ radiation.
Loss of the remaining wild type APC allele has often been implicated as the primary mechanism for increased β-catenin signaling leading to tumor development in APCMin/+ mice (12, 19, 20). In addition to APC, the β-catenin cellular level is also regulated through a direct proteasomal targeting mediated by RXRα (21) and downregulation of RXRα in human and rodent colonic tumors has been reported previously (22). Considering that protein turnover is critical for cellular homeostasis, availability of multiple independent pathways for protein degradation ensures that the potentially pro-carcinogenic β-catenin level remains within physiologic limits to limit cancer initiation and progression. Downregulation of RXRα in our model system may have played a role in 56Fe radiation-induced more aggressive tumorigenesis reported earlier (3). Apart from driving proteasomal degradation of β-catenin, RXRα also functions to suppress β-catenin-mediated upregulation of oncogenes through direct protein–protein interaction (23) in colon cancer cells. Thus, loss of RXRα expression could further stabilize β-catenin signaling in tumor cells, leading to greater cell proliferation and higher number of invasive cancers associated with 56Fe relative to γ radiation.
Nuclear receptor RXRα is known to heterodimerize with a host of other nuclear receptors, such as the vitamin D receptor (VDR) and retinoid acid receptor (RAR), and is involved through transactivation of target genes, such as p21, in regulating normal growth and development (23, 24). Consequently, loss of RXRα is expected to cause disordered cellular proliferation, and indeed, downregulation of RXRα has been widely reported in a number of cancers including CRC (21–26). Our result demonstrates for the first time that RXRα is downregulated in tumor-free areas of APCMin/+ intestine ~100 days after radiation exposure. Considering that a significant number of intestinal adenomas has also been reported to arise without the loss of heterozygosity of the APC gene and these adenomas are often polyclonal (8, 20, 27), our results supports an APC-independent mechanism of β-catenin stabilization during 56Fe-irradiated tumorigenesis. We believe that decreased RXRα expression in tumor-free areas of the intestine may be a reflection of the RXRα status in other areas of the GI tract and that this molecular event may be preceding intestinal tumorigenesis in APCMin/+ mice. Furthermore, RXRα signaling is also linked to cellular redox regulation and it has been demonstrated that RXRα activation protects cell from oxidative stress and inhibition promotes ROS production (28, 29). Downregulation of RXRα observed ~100 days post-exposure in the current study aligns with our previous studies demonstrating chronic oxidative stress even 1 year after exposure to energetic 56Fe ions (30). Although we observed persistent oxidative stress after γ radiation, it was less pronounced relative to equitoxic doses of 56Fe radiation. Additionally, intestinal tumor frequency and grade was also higher after 56Fe relative to equitoxic doses of γ radiation. Considering that γ radiation responses were consistently lower relative to 56Fe, in the current study, we have analyzed and presented 56Fe-induced alterations of an alternate pathway involved in β-catenin regulation via RXRα. Our data, previous and current, demonstrate that effects of radiation on redox balance, carcinogenesis, and related molecular pathways are dependent on radiation quality and energy deposition characteristics. However, further in depth studies will be required to dissect the link between heavy ion radiation exposure and long-term molecular alterations, such as oxidative stress and RXRα downregulation. In summary, our results show that energetic heavy ion radiation is capable of lowering RXRα in tumor as well as non-tumor intestinal epithelial cells. Due to its roles in multiple cellular processes, continuous downregulation of RXRα, we believe, will have major ramifications for intestinal cellular homeostasis with implications for carcinogenesis including colorectal carcinogenesis (Figure 4).
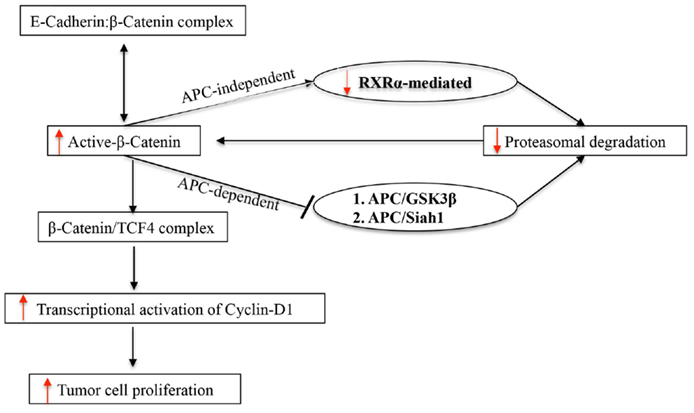
Figure 4. Schematic representation of APC-dependent and APC-independent pathways of UPP involved in β-catenin degradation in APCMin/+ mice.
Conceived and designed the experiments: SS and KD. Performed the experiments: SS and SK. Analyzed the data: SS, SK, and KD. Contributed reagents/materials/analysis tools: AF and KD. Wrote the paper: SS, AF, and KD. All authors read and approved this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study is supported in part by NASA grants NNX13AD58G and NNX09AU95G. We are very much thankful to the members of the NASA Space Radiation Laboratory (NSRL), especially to Drs. Peter Guida and Adam Rusek from Brookhaven National Laboratory for their excellent support in conducting heavy ion radiation exposures. We are also thankful to Steve Strawn and Pelagie Ake for administrative and animal facility supports. We acknowledge the Histopathology and Tissue Shared Resources at the Georgetown University supported by Award Number P30CA051008 from the National Cancer Institute.
1. Cucinotta FA, Kim MH, Chappell LJ, Huff JL. How safe is safe enough? Radiation risk for a human mission to Mars. PLoS One (2013) 8:e74988. doi:10.1371/journal.pone.0074988
2. Trani D, Datta K, Doiron K, Kallakury B, Fornace AJJ. Enhanced intestinal tumor multiplicity and grade in vivo after HZE exposure: mouse models for space radiation risk estimates. Radiat Environ Biophys (2010) 49:389–96. doi:10.1007/s00411-010-0292-2
3. Datta K, Suman S, Kallakury BV, Fornace AJJ. Heavy ion radiation exposure triggered higher intestinal tumor frequency and greater beta-catenin activation than gamma radiation in APC(Min/+) mice. PLoS One (2013) 8:e59295. doi:10.1371/journal.pone.0059295
4. Cucinotta FA. Space radiation risks for astronauts on multiple International Space Station missions. PLoS One (2014) 9:e96099. doi:10.1371/journal.pone.0096099
5. Trani D, Nelson SA, Moon BH, Swedlow JJ, Williams EM, Strawn SJ, et al. High-energy particle-induced tumorigenesis throughout the gastrointestinal tract. Radiat Res (2014) 181:162–71. doi:10.1667/RR13502.1
6. Xiao JH, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N, et al. Adenomatous polyposis coli (APC)-independent regulation of beta-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem (2003) 278:29954–62. doi:10.1074/jbc.M304761200
7. Kim CJ, Cho YG, Park CH, Jeong SW, Nam SW, Kim SY, et al. Inactivating mutations of the Siah-1 gene in gastric cancer. Oncogene (2004) 23:8591–6. doi:10.1038/sj.onc.1208113
8. Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene (2006) 25:7531–7. doi:10.1038/sj.onc.1210059
9. Russo A, Bazan V, Iacopetta B, Kerr D, Soussi T, Gebbia N. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol (2005) 23:7518–28. doi:10.1200/JCO.2005.00.471
10. Hegde MR, Roa BB. Detecting mutations in the APC gene in familial adenomatous polyposis (FAP). Curr Protoc Hum Genet (2006) Chapter 10:Unit10.8. doi:10.1002/0471142905.hg1008s50
11. Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet (2000) 24:245–50. doi:10.1038/73448
12. Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res (1994) 54:5953–8.
13. Wei W, Chua MS, Grepper S, So S. Small molecule antagonists of Tcf4/beta-catenin complex inhibit the growth of HCC cells in vitro and in vivo. Int J Cancer (2010) 126:2426–36. doi:10.1002/ijc.24810
14. Suman S, Fornace AJJ, Datta K. Animal models of colorectal cancer in chemoprevention and therapeutics development. In: Ettarh R, editor. Colorectal Cancer – From Prevention to Patient Care. Rijeka: InTech (2012). p. 277–300. doi:10.5772/28497
15. Bielefeldt-Ohmann H, Genik PC, Fallgren CM, Ullrich RL, Weil MM. Animal studies of charged particle-induced carcinogenesis. Health Phys (2012) 103:568–76. doi:10.1097/HP.0b013e318265a257
16. Peng Y, Brown N, Finnon R, Warner CL, Liu X, Genik PC, et al. Radiation leukemogenesis in mice: loss of PU.1 on chromosome 2 in CBA and C57BL/6 mice after irradiation with 1 GeV/nucleon 56Fe ions, X rays or gamma rays. Part I. Experimental observations. Radiat Res (2009) 171:474–83. doi:10.1667/RR1547.1
17. Weil MM, Bedford JS, Bielefeldt-Ohmann H, Ray FA, Genik PC, Ehrhart EJ, et al. Incidence of acute myeloid leukemia and hepatocellular carcinoma in mice irradiated with 1 GeV/nucleon (56)Fe ions. Radiat Res (2009) 172:213–9. doi:10.1667/RR1648.1
18. Barker N, Morin PJ, Clevers H. The Yin-Yang of TCF/beta-catenin signaling. Adv Cancer Res (2000) 77:1–24. doi:10.1016/S0065-230X(08)60783-6
19. Luongo C, Dove WF. Somatic genetic events linked to the Apc locus in intestinal adenomas of the Min mouse. Genes Chromosomes Cancer (1996) 17:194–8. doi:10.1002/1098-2264(199611)17:3<194::AID-GCC2870170302>3.0.CO;2-E
20. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell (2000) 103:311–20. doi:10.1016/S0092-8674(00)00122-7
21. Dillard AC, Lane MA. Retinol increases beta-catenin-RXRalpha binding leading to the increased proteasomal degradation of beta-catenin and RXRalpha. Nutr Cancer (2008) 60:97–108. doi:10.1080/01635580701586754
22. Janakiram NB, Mohammed A, Qian L, Choi CI, Steele VE, Rao CV. Chemopreventive effects of RXR-selective rexinoid bexarotene on intestinal neoplasia of Apc(Min/+) mice. Neoplasia (2012) 14:159–68. doi:10.1593/neo.111440
23. Han A, Tong C, Hu D, Bi X, Yang W. A direct protein-protein interaction is involved in the suppression of beta-catenin transcription by retinoid X receptor alpha in colorectal cancer cells. Cancer Biol Ther (2008) 7:454–9. doi:10.4161/cbt.7.3.5455
24. Shimizu M, Shirakami Y, Imai K, Takai K, Moriwaki H. Acyclic retinoid in chemoprevention of hepatocellular carcinoma: targeting phosphorylated retinoid X receptor-α for prevention of liver carcinogenesis. J Carcinog (2012) 11:11. doi:10.4103/1477-3163.100398
25. Yamazaki K, Shimizu M, Okuno M, Matsushima-Nishiwaki R, Kanemura N, Araki H, et al. Synergistic effects of RXR alpha and PPAR gamma ligands to inhibit growth in human colon cancer cells – phosphorylated RXR alpha is a critical target for colon cancer management. Gut (2007) 56:1557–63. doi:10.1136/gut.2007.129858
26. Zhang F, Meng F, Li H, Dong Y, Yang W, Han A. Suppression of retinoid X receptor alpha and aberrant β-catenin expression significantly associates with progression of colorectal carcinoma. Eur J Cancer (2011) 47:2060–7. doi:10.1016/j.ejca.2011.04.010
27. Merritt AJ, Gould KA, Dove WF. Polyclonal structure of intestinal adenomas in ApcMin/+ mice with concomitant loss of Apc+ from all tumor lineages. Proc Natl Acad Sci U S A (1997) 94:13927–31. doi:10.1073/pnas.94.25.13927
28. Shan P, Pu J, Yuan A, Shen L, Shen L, Chai D, et al. RXR agonists inhibit oxidative stress-induced apoptosis in H9c2 rat ventricular cells. Biochem Biophys Res Commun (2008) 375:628–33. doi:10.1016/j.bbrc.2008.08.074
29. Ning RB, Zhu J, Chai DJ, Xu CS, Xie H, Lin XY, et al. RXR agonists inhibit high glucose-induced upregulation of inflammation by suppressing activation of the NADPH oxidase-nuclear factor-κB pathway in human endothelial cells. Genet Mol Res (2013) 12:6692–707. doi:10.4238/2013.December.13.3
Keywords: APCMin/+, intestinal tumor, space radiation, heavy ion radiation, tumorigenesis, proteasome, β-catenin
Citation: Suman S, Kumar S, Fornace AJ Jr. and Datta K (2015) Decreased RXRα is associated with increased β-catenin/TCF4 in 56Fe-induced intestinal tumors. Front. Oncol. 5:218. doi: 10.3389/fonc.2015.00218
Received: 22 July 2015; Accepted: 23 September 2015;
Published: 08 October 2015
Edited by:
Marco Durante, GSI, GermanyReviewed by:
Kevin Prise, Queen’s University Belfast, UKCopyright: © 2015 Suman, Kumar, Fornace and Datta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kamal Datta, Department of Biochemistry and Molecular and Cellular Biology, Georgetown University, Research Building, Room E518, 3970 Reservoir Road, NW, Washington, DC 20057, USA,a2QyNTdAZ2VvcmdldG93bi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.