 Freja A. Venning
Freja A. Venning Lena Wullkopf
Lena Wullkopf Janine T. Erler
Janine T. Erler- Biotech Research and Innovation Centre (BRIC), University of Copenhagen (UCPH), Copenhagen, Denmark
Metastatic complications are responsible for more than 90% of cancer-related deaths. The progression from an isolated tumor to disseminated metastatic disease is a multistep process, with each step involving intricate cross talk between the cancer cells and their non-cellular surroundings, the extracellular matrix (ECM). Many ECM proteins are significantly deregulated during the progression of cancer, causing both biochemical and biomechanical changes that together promote the metastatic cascade. In this review, the influence of several ECM proteins on these multiple steps of cancer spread is summarized. In addition, we highlight the promising (pre-)clinical data showing benefits of targeting these ECM macromolecules to prevent cancer progression.
Introduction
Metastases cause more than 90% of cancer patient death (1). The spread of the tumor cells to secondary sites of the body is a complex process involving reciprocal interaction between tumor cells and their microenvironment (2). Metastases are the result of a series of complex processes, including the escape from the primary tumor, the invasion into adjacent tissue, hematogenous or lymphatic spread, the establishment of micrometastases, and the final outgrowth and colonization at the distant site of the body (3). Today, it is becoming widely accepted that the tumor microenvironment crucially affects cancer progression. The tumor microenvironment consists of not only the cancer cells, all non-malignant cell types such as immune cells, fibroblasts, pericytes, endothelial cells, adipocytes, and mesenchymal stem cells, but also the interstitial fluids and the extracellular matrix (ECM). This review will summarize how ECM composition and structure, at both the primary and the secondary site, are key factors for a successful metastatic spread.
The ECM is a complex meshwork of macromolecules secreted by the different cell types of a tissue, made up of both proteins and proteoglycans (PGs) with covalently attached sugar chains, glycosaminoglycans (GAGs). Besides providing the structural support of an organ, the ECM is instrumental in modulating cell functions. Beyond direct interaction with cellular signaling receptors, the network of macromolecules also functions as a reservoir for growth factors or signaling molecules, thus influencing cellular behavior indirectly (4). Together ECM signaling can be involved in proliferation, migration, invasion, the onset of angiogenesis, or the resistance to apoptotic stimuli. In addition, ECM proteins can work as an anchor and promote cellular adhesion. Moreover, fibers of ECM proteins such as collagens can build migration tracks for the tumor cells. At the same time, the ECM can function as a barrier blocking, e.g., the penetration of immune cells into the tumor, or it can create a high interstitial fluid pressure (IFP) preventing the perfusion of drugs, which facilitates chemoresistance.
In this review, we will first briefly introduce a handful of well-studied ECM components and then describe their contribution to the steps of the metastatic cascade (Figure 1). On the protein side, the focus will be on the matricellular proteins periostin and tenascin C as well as on the fibrillar collagen I. We will also look into the role played by the unique GAG hyaluronan (HA) and its often partner-in-crime versican. Second, we will discuss therapeutic approaches that either directly target the ECM components or their modification.
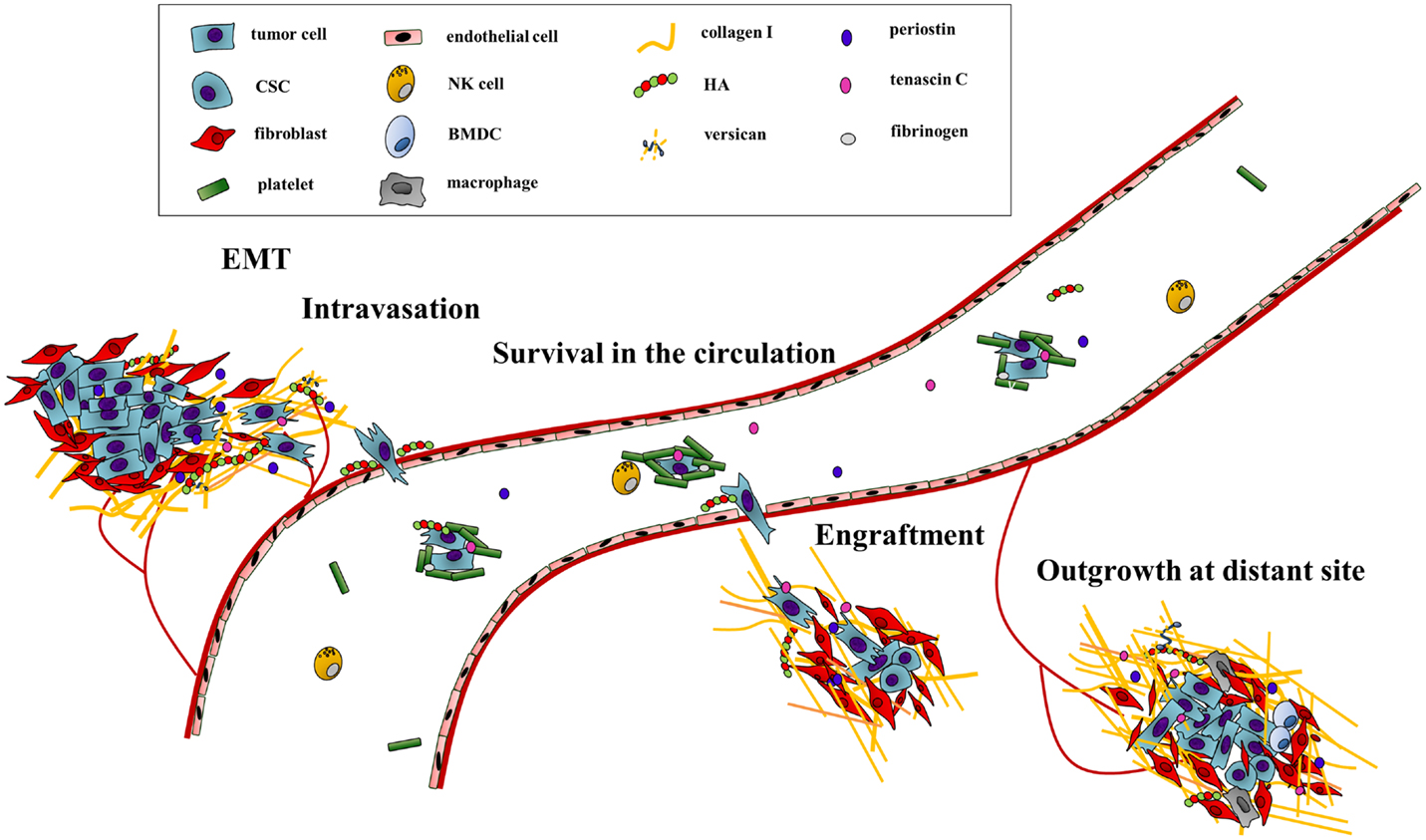
Figure 1. The ECM drives the progression of cancer cells along the metastatic cascade. The metastatic cascade is composed of multiple complex processes, which are critically influenced by ECM components. First, ECM-regulated signaling pathways increase cancer cell motility and promote the egress from the primary tumor. In addition, the stability of the endothelial cell barrier is critically regulated by HA, thus influencing intra- and extravasation efficacy of cancer cells. The survival in the circulation system is also directly and indirectly modulated by ECM components as they function as physical shields as well as attractants for platelets. Through deposition and modification of ECM components at distant sites, the initial engraftment and final colonization of cancer are enhanced. Hereby, biochemical as well as biomechanical cues of the ECM promote metastatic outgrowth.
Key Players
Periostin
Periostin, also known as osteoblast-specific factor 2 (OSF-2), is a secreted N-glycoprotein that was identified as a cell adhesion protein in a mouse osteoblast cell line (5). This matricellular protein was shown to interact with itself and other ECM proteins as collagen I, fibronectin, and tenascin C (6). Initially, periostin was associated to cancer as high expression levels were found in patient samples of common solid tumor types such as breast, colon, lung, and pancreatic cancer, as well as melanoma (7–11). The expression level correlates with tumor progression and was shown to be especially elevated in the secondary sites in 75% of the lymph node metastases of breast cancer patients (12). Besides, periostin can be found in the serum of patients with advanced metastatic disease (13–15). Therefore, periostin qualifies as a tumor marker in the clinic, especially for advanced breast cancer.
Tenascin C
The tenascin family has five members named tenascin C, R, W, X, and Y (16). The secreted glycoproteins bind to a great variety of proteins as periostin, fibronectin, integrins, and several collagens. This review will focus on the role of tenascin C in advanced tumorigenesis. Tenascin C expression is restricted to connective tissues and interestingly stem cell niches in adult tissues but is very prominent in tumor tissue (17–22). Stromal expression was particularly observed in late-stage tumors, with a particular strong staining at the zone of tumor–stroma interaction (17, 18, 23). Elevated serum levels make tenascin C suitable for clinical monitoring of the most common cancer types (24–26).
Hyaluronan
Hyaluronan is an important GAG in the ECM of many adult tissues, and an increase in HA deposition is seen in many solid cancers, particularly of the prostate, pancreas, breast, and bladder (27), often correlating to a poor prognosis (28). HA is the only GAG composed of unsulfated disaccharides (d-glucuronic acid and N-acetylglucosamine), and it is never covalently attached to a proteoglycan core protein (29). HA is synthesized by hyaluronan synthases (HAS), which produce high-molecular-weight HA (HMW-HA) above 1000 kDa (30), which can be degraded into low-molecular-weight (LMW)-HA and even smaller oligo-HA by either hyaluronidases (HYAL-2, -2, -3, and PH20) or reactive oxygen species (ROS) (31). Interest in HA’s role in cancer progression was recently rekindled by the seminal finding that the extremely large HMW-HA produced by the naked mole rat (five times larger than human HMW-HA) is essential for its remarkable resistance to cancer development (32–34).
Versican
Versican is a large chondroitin sulfate proteoglycan (CSPG) that is present around cells in most healthy tissue in low amounts and upregulated in malignancies of, e.g., the breast, colon, prostate, lung, and ovaries, among others (35). Versican binds to a number of other ECM components, notably HA, contributing to the formation of a biomechanically active pericellular matrix that affects the proliferation, adhesion, and motility of cells (36–39). Clinically, increased versican expression correlates to a decreased progression-free survival in prostate cancer (40), increased relapse in breast cancer (41), advanced disease and lymph node metastasis in adenocarcinomas of the lung (42), and is also diagnostically relevant in other cancers [reviewed in Ref. (35)].
Collagen I
Collagen I is the main fibrillar collagen in the ECM, providing tensile strength to the tissue and limiting its distensibility. Collagen I fibrils in normal tissue are made up of processed heterotrimers of two col1α1 and one col1α2, which self-assemble in the extracellular space into fibrils. The fibrils are cross-linked by enzymes of the lysyl oxidase (LOX) family (43), forming larger mechanotransductive fibers that increase the density and rigidity of the tissue (44). Increased mammographic density correlates to an increase in collagen deposition (45) and more importantly to an increased risk of developing breast cancer (46). Abnormally large collagen deposition is the most well-documented ECM alteration in many tumor types, and collagen deposition has been causally linked to an increase in mammary tumor and metastasis incidence (47).
Escaping the Primary Tumor: EMT and Intravasation
The first step of tumor dissemination is for the cancer cells to break free from the confinements of the primary tumor. They have to acquire the ability to move and invade through the basement membrane as well as the walls of vessels of the blood stream or the lymphatic system, a process called intravasation (48). In order to detach from the primary site, some tumor cells undergo epithelial–mesenchymal transition (EMT). EMT is defined by a simultaneous downregulation of epithelial proteins such as E-cadherin and an upregulation of mesenchymal proteins such as N-cadherin and vimentin leading to the loss of cell–cell contacts and an increase in cell motility (49). Many ECM proteins are associated with the induction of EMT by activating receptor-mediated signaling cascades (Figure 2) (50, 51).
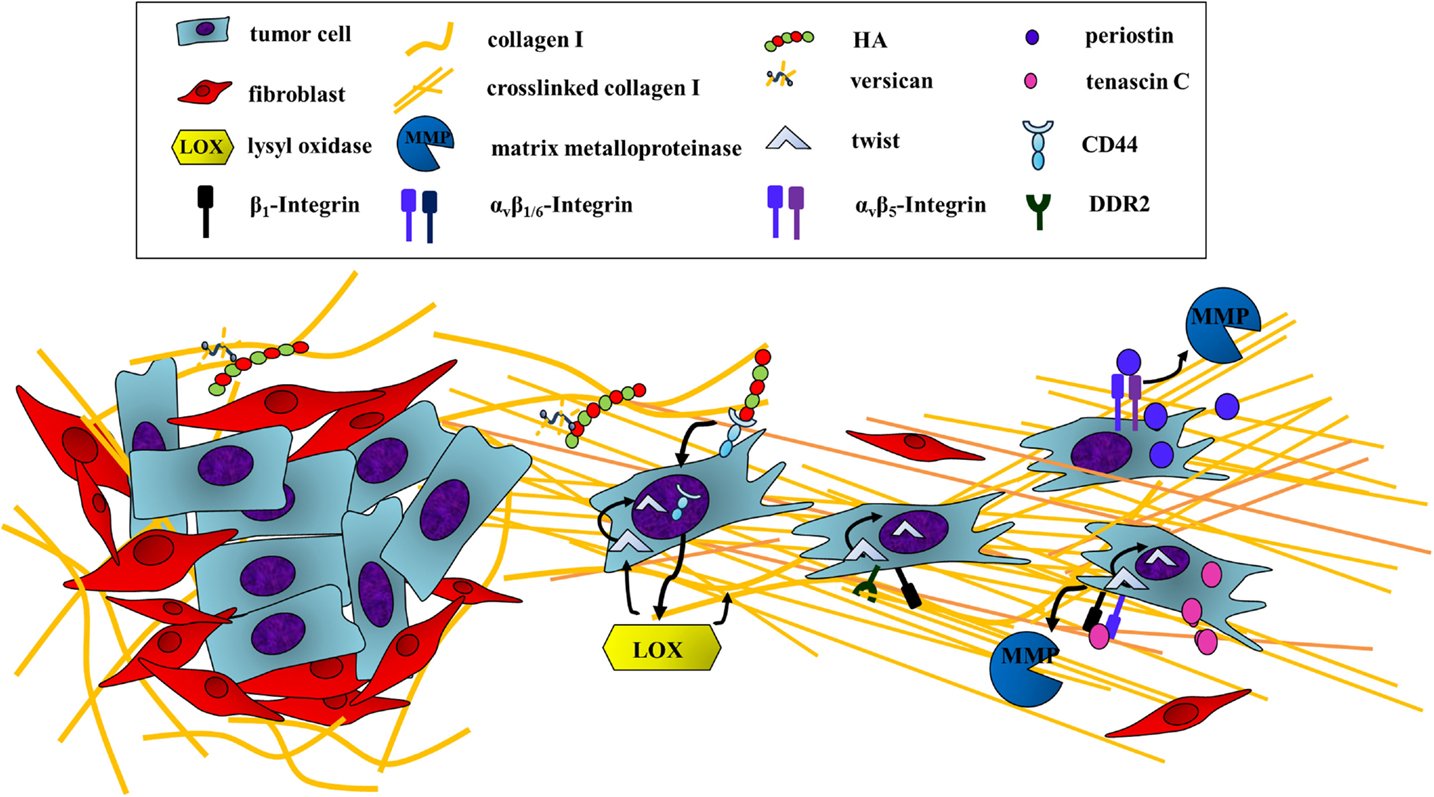
Figure 2. Escaping the primary tumor: ECM components induce cancer cell motility. In order to leave the primary tumor, cancer cells undergo epithelial-to-mesenchymal transition (EMT), which can be induced by ECM proteins and GAGs activating receptor-mediated signaling pathways. First, HA binding to CD44 on tumor cells induces EMT through the translocation of the receptor to the nucleus. CD44 is then able to induce a transcriptional upregulation of LOX. LOX promotes EMT by regulating the EMT transcription factor TWIST-1 through two mechanisms. First, LOX is able to activate the expression of TWIST-1. In addition, the activity of TWIST-1 is indirectly enhanced by LOX through association of cross-linked collagen I with integrin β1 and DDR2, which promotes the nuclear translocation of cytoplasmatic-bound TWIST-1. Besides, tenascin C also influences the transcriptional regulation of EMT indicated by the downregulation of E-cadherin and a simultaneous upregulation of vimentin as well as several MMPs. Periostin also enhances MMP expression, thus inducing EMT through binding to tumor cell αvβ5-integrin.
First, the production of the glycosaminoglycan HA has been demonstrated to induce EMT in both normal and transformed epithelial cells in vitro [reviewed in Ref. (31) and references therein]. In vivo, accumulation of HA in pancreatic and mammary tumor models is associated with loss of E-cadherin and nuclear translocation of β-catenin (52, 53), both hallmarks of EMT. However, overproduction of HA is not in itself enough to create an invasive phenotype, on its own it actually decreases cell motility and tumorigenesis (54). However, if the general turnover of HA is increased due to high levels of both HA synthases and hyaluronidases, i.e., increased levels of LMW-HA, then this leads to an increase in cell motility in vitro. This is mirrored in vivo by the appearance of spontaneous lymph node metastasis in a mouse model of pancreatic cancer with increased HA-turnover (54). One mechanism of HA-induced EMT involves binding to the cellular receptor CD44, which then translocates to the nucleus and by binding to the promoter leads to the upregulation of LOX. Next, LOX catalytic activity is, in a so far undetermined manner, necessary for the expression of the EMT transcription factor TWIST-1 (Figure 2) (55).
The formation of an HA-rich pericellular matrix is important for proliferation and motility of normal mesenchymal cells (36), a phenomenon cancer cells also utilize (37–39, 56, 57). Studies of ovarian cancer cells and leiomyosarcoma cells have showed that versican is necessary for the formation of this HA-rich pericellular matrix (38, 39). Knockdown of versican expression in ovarian cancer cells decreased their motility in vitro and more interestingly also their ability to form experimental metastases after injection into the peritoneal cavity (58).
Besides its role in general motility, HA has a particular important function in the process of intravasation. HA regulates blood vessel integrity, with HMW-HA and LMW-HA degradation products playing opposite roles. HMW-HA promotes endothelial cell barrier function through several mechanisms while LMW-HA disrupts it (59–61). Furthermore, LMW-HA is also angiogenic (62), so the production of LMW-HA fragments in the tumor microenvironment can thus compromise the tumor vessel integrity and promote angiogenesis, making it easier for cancer cells to intravasate and continue the metastatic process.
Studies of both patient material and mouse models of cancer have shown that the deposition of a collagen-rich matrix is linked to tumor progression and metastasis (47). Collagen I is indeed intricately involved in the induction and maintenance of EMT and an invasive phenotype. In vitro studies have shown that interaction between collagen I and integrin β1 leads to destabilization of the E-cadherin–beta-catenin complex and also to upregulation of N-cadherin (63, 64). Recently, it has been reported that inhibition of collagen synthesis in human MDA-MB231 breast cancer xenografts leads to a decrease in local invasion into the surrounding adipose tissue and to a decrease in metastasis to both the draining lymph nodes and lungs (65, 66). The level of circulating tumor cells was decreased in mice where collagen synthesis was inhibited, further demonstrating that the collagen content of the primary tumor is important for generating invasive cancer cells capable of intravasation (65).
Changes in the collagen matrix in tumors also provide altered biomechanical cues to tumor cells. Enzymes of the LOX family catalyze the cross-linking of collagens and elastin, increasing the tissue stiffness (43). LOX and LOX family members are frequently overexpressed in cancers (43), and their collagen cross-linking activity has been proven to promote tumor progression through increased integrin signaling (67–70). Additionally, the tissue stiffness is essential for determining the cellular response to the potent EMT inducer TGF-β, as EMT signaling is only induced in cells residing in a stiff tissue, with apoptosis being the go-to program for cells in a soft ECM (71). The mechanism behind this stiffness-regulated switch was decoded recently, showing that the transcription factor TWIST-1, which is essential for EMT, translocates to the nucleus due to stiffness-induced release from its cytoplasmic anchor G3BP2 (Figure 2) (72).
It is not only the amount and stiffness of the collagen network that is important; the orientation of collagen fibers also appears to be central to the progression of cancer. Through intravital imaging of tumors several studies have shown that the organization of collagen into straight, aligned fibers promotes cell invasion along these fibers (73). In breast cancer, the orientation of these collagen fibers in relation to the tumor is an independent prognostic indicator, with fibers aligned perpendicular to the tumor correlating to a poor disease-specific and disease-free survival (hazard ratio >3) (74). Molecular evidence for this clinical correlation was provided by Zhang et al. in a mouse model of breast cancer, showing that binding of collagen I to the cell surface receptor discoidin domain receptor 2 (DDR2), a receptor tyrosine kinase, leads to the formation of collagen fibers oriented perpendicular to the surface of the tumor, facilitating cells invading out along these (75). Furthermore, DDR2 is upregulated in cells undergoing EMT, and binding of collagen I to DDR2 further sustained the EMT transcriptional program, providing a positive feedback loop (Figure 2) (75).
Bornstein et al. introduced the term matricellular proteins to describe the family of non-structural extracellular proteins (76), among which periostin and tenascin C are especially important for the metastatic cascade. During the last decade, increasing evidence has amassed for the functional importance of periostin in the initiation of the metastatic cascade via the induction of EMT. First, overexpression of the periostin in 293T cells led to an augmented expression of vimentin correlating with a mesenchymal morphology as well as an increase in migration and invasion. These effects were dependent on αvβ5-integrin binding to periostin (Figure 2) (77). In addition, these cells also showed an increase in expression of the matrix metalloproteinase MMP-9. Proteases degrade the ECM in the cell’s surroundings, paving the way for the cells through the dense environment (78). Moreover, elevated levels of periostin mRNA and protein were revealed at the invasive front of immortalized esophageal cells in vitro and in vivo. Overexpression of periostin in these cells increased their ability to move while depriving them of the matricellular protein reduced the invasive potential significantly (79). The same was reported in oral squamous cell carcinomas, where overexpression of periostin facilitated motility and invasiveness in vitro as well as in an orthotopic mouse model (80).
A further mediator of cell motility is tenascin C. By binding to either integrin αvβ1 or αvβ6, recombinant tenascin C induced a change in the morphology of the breast cancer cell line MCF-7 to a more mesenchymal phenotype (Figure 2) (81). Besides, Tavazoi et al. were able to diminish the invasive potential of a metastatic breast cancer cell line by knocking down tenascin C expression. These cells were also deprived of the ability to form lung metastasis in vivo (82). Moreover, tenascin C induces expression of several matrix metalloproteinases (Figure 2) (83), again linking it to the onset of invasion (84). Both observations are in accordance with the striking expression of tenascin C seen at sites of epithelial–mesenchymal interactions during development (85) and at the invasive front of human breast cancer samples (86). Oskarsson et al. could even demonstrate a deposition of tenascin C at the margin of lung metastases of both mice injected with the breast cancer cell line MDA231-LM2 and breast cancer patient samples (23).
A Journey in the Blood: Resisting Mechanical Forces and Immunoescape
Once tumor cells manage to evade the constraints of the primary tumor and successfully invade the lymphatic or hematologic system, they are exposed to a hostile and deadly environment. As the transition to distant organs depends on the blood system, we will focus on the tumor cell survival in the blood circulation. Here, cancer cells have to resist shear forces and turbulence. In addition, they have to escape the immune surveillance, especially natural killer (NK) cells. The main protective mechanism for tumor cells is an interaction with platelets that work as a shield against immune cell lysis and mechanical stress (87). The most important mediator is the acute phase protein fibrinogen. Fibrinogen is mainly secreted by hepatocytes and megakaryocytes but can also derive from tumor cells (88). Although being primarily a plasma protein, fibrinogen has many roles as an ECM protein in cancer (89). Fibrinogen can bind to thrombocyte receptors inducing platelet adhesion. Both fibrinogen and especially its protease converted form fibrin can provoke αvβ3-integrin and αIIbβ3-mediated cancer-cell–fibrin(ogen)–platelet complexes (Figure 3) (90, 91). The promoting role of fibrinogen and platelets in hematogenous metastasis was confirmed by a striking decrease in melanoma cells colonizing the lung in either fibrinogen knockout or platelet deprived, protease-activated thrombin receptor 4 (PAR4) knockout animals (92–94).
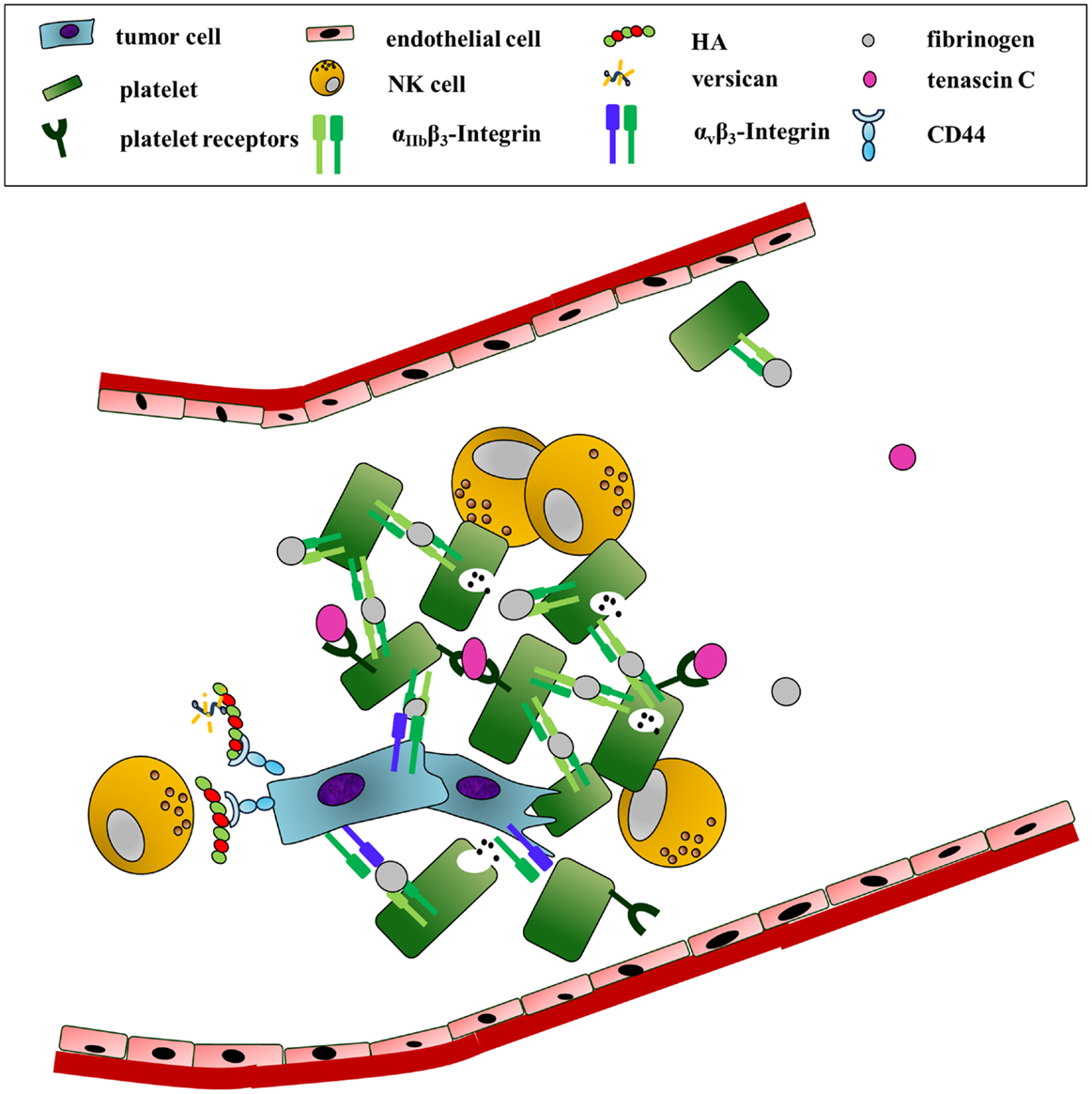
Figure 3. Survival in the blood: ECM components as physical shields and mediators of platelet recruitment. In order to survive the high shear forces and patrolling NK-cells in the blood circulation, tumor cells interact with platelets. This association is mainly initiated by fibrinogen. Fibrinogen binding to tumor-expressed αvβ3 and platelet-derived integrin αIIbβ3 induces the formation of cancer-cell–fibrinogen–platelet complexes. In addition, tenascin C association with integrin α2β1 or the glycoprotein Ib-IX complex enhances platelet adhesion and activation. An HA-rich pericellular matrix further shields the cancer cells from NK-cell attack.
However, other ECM proteins can also promote the recruitment, binding, and activation of platelets (95, 96). Platelets adhered efficiently to tenascin C in static in vitro assays as well as under dynamic flow. Tenascin C also enhanced platelet activation (97). Although not specifically shown in cancer, tenascin C therefore could indirectly support tumor survival in the blood by promoting platelet recruitment and activation (Figure 3).
An HA-rich pericellular matrix could be another type of shield the cancer cells employ to ward off NK cells. In vitro studies have shown that HA effectively keeps lymphocytes such as NK cells from getting in close contact with the cancer cells, preventing them from killing the cancer cells (Figure 3) (57, 98). Thus, the ability of the cancer cell to produce such a pericellular matrix, requiring CD44, HA, and versican (or aggrecan), could add to the chances of survival in the circulation.
Finding a New Home: The Challenge to Extravasate and Survive
Seeding of cancer cells in a secondary organ requires the extravasation from the circulation, initial adherence, and the initiation of proliferation under the unpermissive conditions of the secondary sites.
In order to leave the circulation, cancer cells need to adhere to the endothelium and push through into the tissue. Endothelial cells have a thick, pericellular matrix rich in HA, the glycocalyx, and tumor cells can interact with this through CD44 to initiate adhesion (Figure 4) (99). The importance of cancer cell interaction with HA on endothelial cells in the process of extravasation is backed by a recent in vivo study, which showed that knockdown of the HA receptor CD44 in MDA-MB-231 breast cancer cells drastically decreased the number of experimental metastases in an intracardiac dissemination model (100).
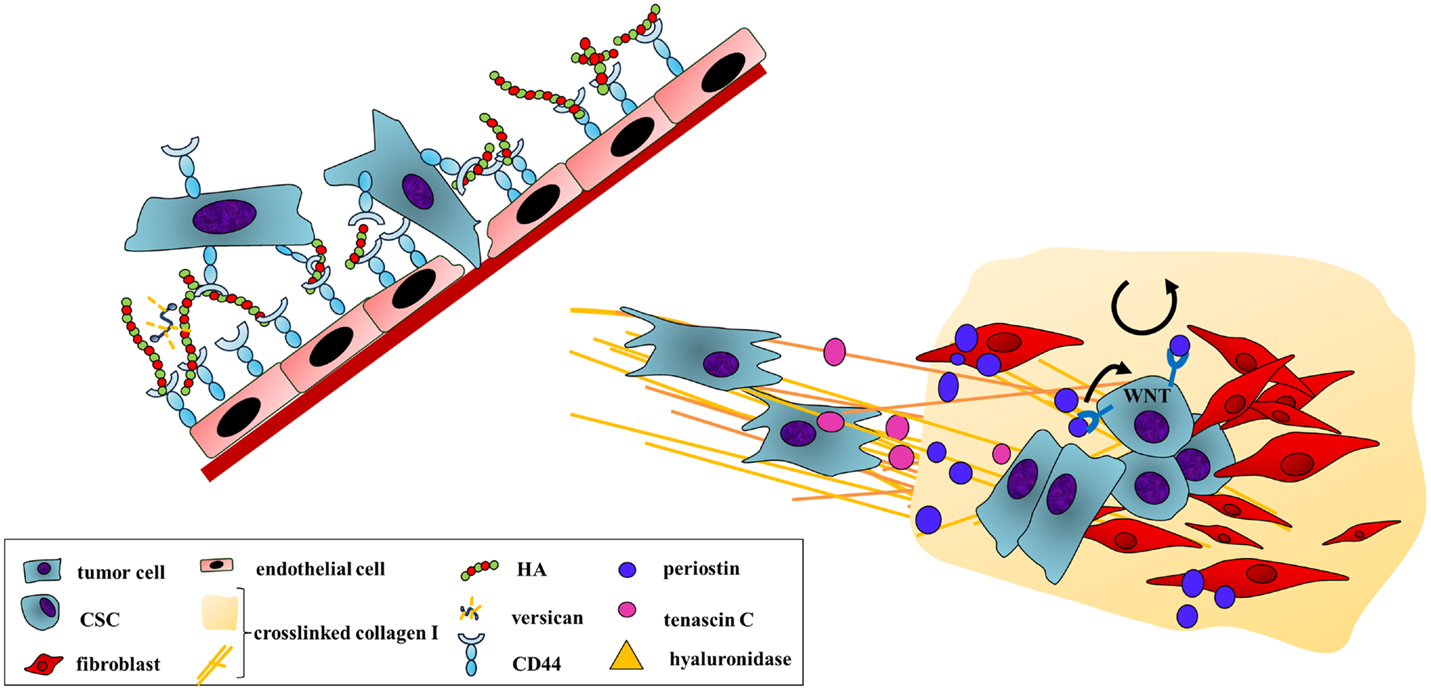
Figure 4. A distant home: tumor cell extravasation and engraftment at secondary sites are enhanced by ECM secretion and remodeling. Extravasation requires cancer cell adhesion to the vessel wall and an invasion into the foreign tissue. HA mediates an initial adherence as both endothelial and cancer cells bind to the GAG through CD44. A subsequent secretion of hyaluronidase breaks down HMW-HA. The local increase of LMW-HA disrupts the endothelial cell barrier supporting transendothelial migration of the cancer cells. After arrival at a distant site, cancer cells are exposed to a foreign environment. To allow survival under these different conditions, cancer cells secrete ECM components and ECM-remodeling enzymes to favor the engraftment at the foreign site. An example is tumor-derived LOX altering collagen cross-linking at pre-metastatic sites. Tumor cell-secreted tenascin C also enhances the establishment of micrometastases. However, tumor cells also induce stromal cells to produce cancer-promoting ECM proteins, creating a more permissive environment. Here, stromal periostin improves cancer cells adhesion by binding to αvβ5-integrin. In addition, periostin supports the self-renewal and proliferation of CSCs through the activation of the WNT signaling pathway, which may enhance outgrowth of metastases.
Cancer cells may also construct a pericellular matrix rich in HA (57), and the reciprocal interaction of this with receptors on endothelial cells also appears to be important for adhesion (101–104) and the following transmigration into tissue (103). Once cancer cells have adhered to the endothelium, it is possible that tumor cell-secreted hyaluronidases create a local increase in LMW-HA, from the breakdown of the glycocalyx, which then in turn can lead to the disruption of the endothelial barrier, opening up the door for the cancer cell.
Hirose et al. recently showed that the levels of circulating LMW-HA in the blood is an important factor for melanoma cell adhesion to endothelial cells. Increased serum levels of LMW-HA, created by blocking the HA receptor for endocytosis (HARE/Stab2) found in sinusoidal endothelium in the liver, bone marrow, and lymph nodes, prevented B16F10 melanoma cells from binding to endothelial cells and producing lung foci in tail-vein injected mice (105). Strikingly, elevated LMW-HA in serum also prevented spontaneous lung and lymph node metastasis of orthotopically implanted human MDA-MB-231 and mouse 4T1 breast cancer cells (105). Similarly, Simpson et al. have reported that prostate cancer cells bind to sinusoidal endothelial cells through HARE/Stab2 and that blocking HARE in vivo via an antibody completely prevented spontaneous lymph node metastasis in an orthotopic prostate cancer model (104). Two cooperating mechanisms may be at play here. Blocking HARE or saturating it with HA can prevent direct binding of cancer cells to HARE+ cells through their HA-rich pericellular matrix and simultaneously the elevated blood levels of HA, created by lack of HARE-mediated clearance, might also saturate CD44 on the cancer cells, preventing them from utilizing this previously demonstrated important factor for extravasation. Yet, a recent study has found that elevated plasma levels of LMW-HA correlate with lymph node metastasis in breast cancer patients (106). An explanation for this discrepancy could be the fact that serum levels of LMW-HA that prevent extravasation and metastasis in mice were more than fourfold higher than normal serum levels, and the difference in serum LMW-HA that separated metastatic vs. non-metastatic patients was less than twofold. This suggests that the modest increase in serum LMW-HA seen in metastatic breast cancer patients, while prognostically indicative, confers no protective advantage to offset the pro-metastatic benefits of increased intra-tumoral LMW-HA.
Lysyl oxidase, LOXL2, or LOXL4 expression in the primary breast tumor leads to pre-metastatic deposition of collagen I in the lungs of mice (Figure 4) (65, 107), favoring the formation of metastases in the lungs (65, 107–109). Furthermore, LOX-mediated cross-linking of collagen I increases the fibrotic response in lungs and livers of mice and helps form a favorable metastatic niche in these organs. Mice with fibrotic lungs and livers had an increased number of spontaneous 4T1 breast cancer metastases to these organs, which was abrogated by blocking LOX activity (110).
Periostin also showed a proadhesive effect on 293T cells, which was αvβ5-integrin-dependent (Figure 4) (77). This is concurrent with the identification of periostin as one of the key components of the so-called metastatic niche (111). In order to convert the new unfavorable surrounding into a more permissive environment, tumor cells secrete factors before and upon arrival at the distant site. Furthermore, they induce stromal cells to produce cancer-promoting extracellular proteins (112). Contié et al. reported a prominent expression of periostin in bone metastasis of tail vein-injected MDA-B02 breast cancer cells (15). Malanchi et al. confirmed this result in the polyomavirus middle T antigen (PyMT) mouse model for spontaneous breast cancer. Here, a deposition of periostin was revealed not only in the primary breast tumor but also even more pronounced in the lung metastasis. By combining the breast cancer model with periostin null mice, the authors could reveal that stromal periostin supports the survival and proliferation of cancer stem cells (CSCs, CD90+, and CD24+) through the activation of WNT signaling (Figure 4) (12). CSCs have been implicated in the establishment of new micrometastases that then subsequently grow out into macroscopic secondary tumors (113). The reduction in number of metastasis in periostin knockout mice to less than 10% of the control, with a lower number of CSCs and abrogated WNT signaling, affirms this concept (12).
The initial engraftment of tumor cells at secondary sites is also critically influenced by tenascin C (Figure 4). In depriving a human metastatic breast cancer cell line of tenascin C, Oskarsson et al. proved the need of tumor-secreted tenascin C for the establishment of micrometastasis. The tenascin C knockdown resulted in a 90% inhibition of lung colonization in either experimental or spontaneous lung metastases of these cells. Immunohistochemistry probing for the apoptotic marker caspase-3 revealed that tumor cell survival is dependent on tenascin C. In order to specify the effect of tumor endogenous protein production, Oskarsson et al. used an inducible knockdown model narrowing down the time frame for the dependency on tumor-derived tenascin C. Interestingly, depriving the breast cancer cells of tenascin C only affected the outgrowth of metastases when they reached a certain size (23). In pancreatic cancer, ectopic tenascin C expression in RIP-Taq2 mice significantly increased the establishment of micrometastases, whereas a tenascin C knockout reduced tumor cell engraftment in the lungs (22).
Making Yourself at Home: Macroscopic Outgrowth in an Inhospitable Environment
The low efficiency of engrafted cancer cells to establish bona fide macroscopic metastases indicates that the microenvironment of secondary sites is generally unsupportive for tumor cells. In order to convert an unfavorable surrounding of a distant site into a more permissive environment, which allows macroscopic lesions to develop, tumor cells secrete factors before and upon arrival at the distant site. Furthermore, they induce stromal cells to produce cancer-promoting extracellular proteins (112).
Collagen deposition in the metastases is very important for the progression into larger lesions. Extravasated MDA-MB-231 cells with a knockdown of prolyl-4-hydroxylases, an enzyme essential for correct collagen biosynthesis, were able to survive but formed smaller lung metastases than their wild-type counterparts (65, 66). In line with this, successful colonization of draining lymph nodes by MDA-MB-231 xenografts was accompanied by an increase in collagen deposition in the lymph nodes (114). Additionally, mice with collagen-rich fibrotic lungs and livers tended to develop larger breast cancer metastases (110).
The recruitment of bone marrow-derived cells (BMDCs) is another key step in the formation of the metastatic niche (115). LOX-mediated cross-linking of collagen IV recruits CD11b+ BMDC to the metastatic niche in the lungs (107, 109), and these CD11b+ cells further remodel the ECM into a favorable home for extravasating cancer cells, e.g., by laying down the proteoglycan versican (Figure 5) (116). Inhibiting versican production by CD11b+ BDMCs radically decreased the burden of lung metastases in a mouse model of spontaneous breast cancer, specifically preventing the progression from micrometastases to macrometastases (116). Previously, versican had been shown to promote mesenchymal to epithelial transition (MET) in fibroblasts in vitro (117). In the study by Gao et al., versican also promoted MET in vivo, changing the phenotype of the intravasated cancer cells from migratory and slowly proliferative to more adhesive and proliferative (116). Versican could also contribute to the growth of metastases by interacting with HA and forming complexes that can recruit macrophages and induce angiogenesis (53, 118), further molding the secondary site into a supportive home (Figure 5).
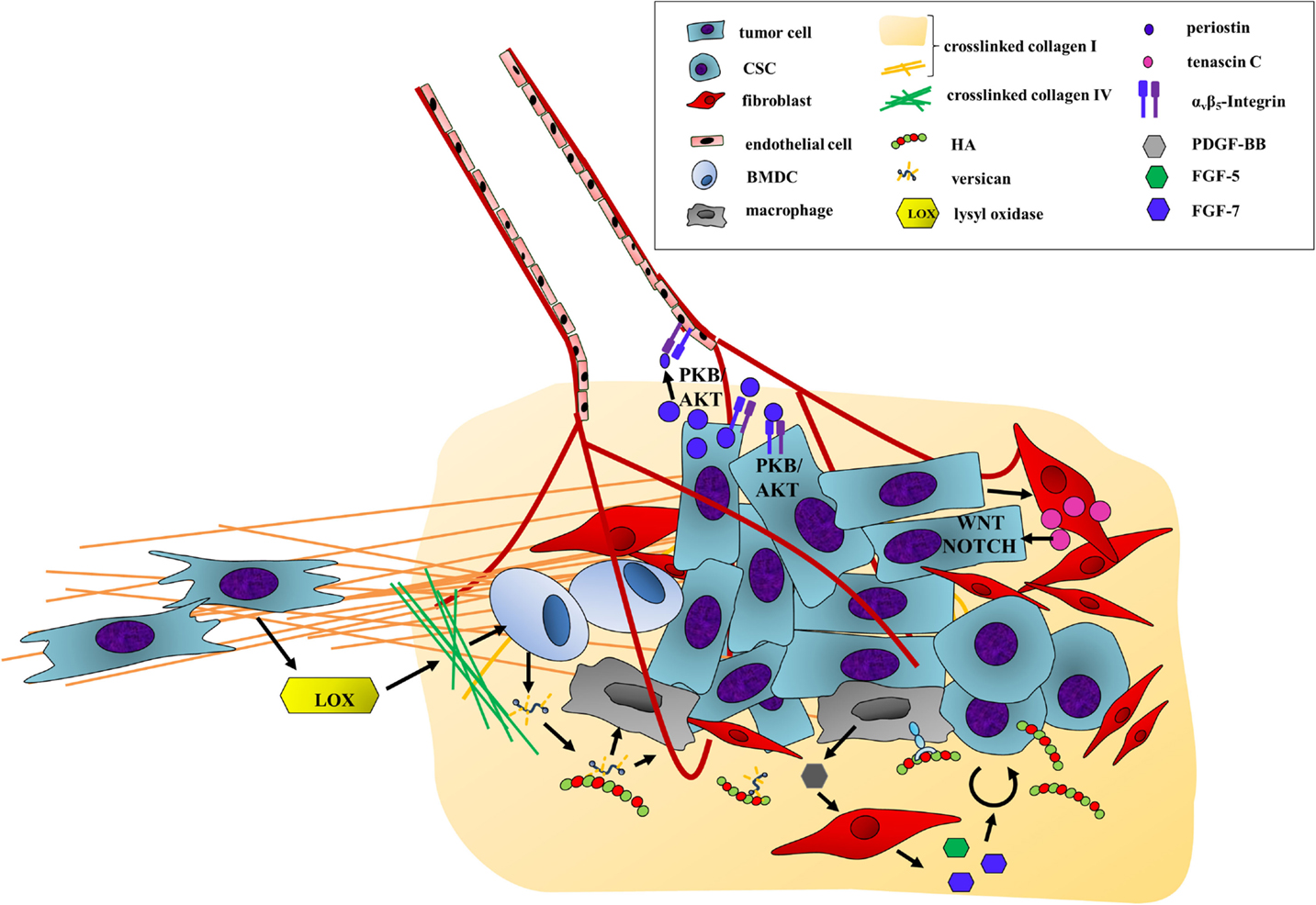
Figure 5. Final colonization: ECM deposition and remodeling facilitate the outgrowth of metastases. The final outgrowth of macrometastases is influenced by the deposition, remodeling, and signaling of ECM components. An example is the extensive secretion and alignment of collagen I fibers promoting the final colonization of cancer cells at a distant site. In addition, tumor-secreted LOX cross-links collagen IV, thereby attracting BMDCs. These cells indirectly enhance CSC survival and renewal. BMDC-secreted versican associates with HA. The complex can bind CD44 of macrophages activating their production of PDGF-BB. PDGF-BB in turn mediates other stromal cell secretion of FGF, which finally stimulates the proliferation of CSCs. In addition, versican also induces angiogenesis. An increase in angiogenic activity is also mediated by periostin. Tumor-secreted periostin binds integrin αvβ3 of endothelial and tumor cells. This activates PKB/AKT signaling, promoting cell survival enhancing angiogenesis as well as tumor cell outgrowth. Another mechanism supporting tumor cell survival is the induction of stromal tenascin C secretion. Tenascin C can activate WNT and NOTCH signaling pathways ensuring tumor cell viability under unpermissive conditions.
Cancer cell-produced HA and the accumulation of HA in experimental bone lesions of MDA-MB-231 breast cancer cells appear to be important for the growth of the lesions (119). One mechanism for HA-induced bone lesion growth was deciphered by Okuda et al., showing that CSCs (CD24−, CD44+, EpCAM+) from bone tropic MDA-MB-231-BoM depend on autologous HA synthesis to survive and renew in the metastatic bone niche. CSC-produced HA interacts with CD44 on macrophages, activating them to produce the growth factor PDGF-BB, which in turn activates other stromal cells to produce FGF7 and FGF9 that stimulate CSC proliferation and self-renewal (Figure 5) (103). This places HA at a central position in the development of a favorable metastatic niche that permits secondary tumor development.
While Malanchi et al. (12) reported an essential role of stromal periostin in the establishment of micrometastases, an earlier study revealed a role of tumor-endogenous periostin in the final step of colon cancer colonization of the liver. Colon cancer cells overexpressing periostin showed a strong increase in hepatic metastases in an experimental metastasis model. Yet, while an equal amount of micrometastases was detected in the first days after intraportal injection, the further outgrowth of these cells was dependent on tumor cell-derived periostin. Tumor cell survival was promoted as periostin binds to αvβ3-integrin, enhancing the PKB/AKT pathway at secondary sites (Figure 5). Besides, the authors linked periostin to angiogenesis as the survival-promoting signaling pathway was also activated in endothelial cells (Figure 5) (7). This is in accordance with the increased occurrence of tumor blood vessel in metastases of periostin-overexpressing 293T cells (77).
Oskarsson et al. revealed an inverse time course of the role of tumor-endogenous and stromal tenascin C. While an induced knockdown of the protein diminished metastatic engraftment in the beginning, tumor cells induced myofibroblasts to produce tenascin C when the tumors reached a certain size, compensating the loss of the matricellular protein. This was consistent with immunohistochemical staining of human breast cancer samples, revealing tumor cell tenascin C expression in early malignant stages, and a strong reaction in stromal cells in advanced cirrhotic carcinomas (17). As a mechanism, Oskarsson et al. suggested the augmentation of signaling pathways, ensuring tumor cell survival at the unpermissive site, namely WNT and NOTCH signaling (Figure 5). Although having its main function in creating a metastatic niche and inducing WNT and NOTCH signaling pathways, tenascin C depletion did not correlate with a reduction of stem cell characteristics of the breast cancer cells in this study (23). These results are supported by another study in the 4T1 orthotopic model of breast cancer, where S100A4+ stromal cells were found to promote metastatic colonization to the lungs by producing high levels of tenascin C. In addition, tail vein-injected 4T1 cells showed a significantly lower efficiency of final engraftment in the lungs in tenascin C knockout mice (120).
Rationale of Targeting the ECM
As metastases rather than the primary tumor cause the poor prognosis of most cancer patients, it is clearly important to stop the dissemination of cancer to and growth at distant sites of the body. Targeting the ECM, often secreted by stromal cells, is hereby of particular interest as these targets are less prone to rapid mutation as seen for signaling pathways in cancer cells. In addition, combinatorial therapies, integrating standard chemotherapy with ECM targeting, are an important strategy of cancer treatment in the adjuvant setting. Here, the hope is that the disruption of the dense tumor microenvironment will promote drug delivery and prevent chemoresistance. Although former studies concentrate on enzymes as LOXs or proteases like the MMPs responsible for remodeling ECM, here we focus on studies about targeting key ECM proteins.
Targeting HA
HA Deposition and Synthesis
The HA-rich pericellular matrix around cancer cells can be an obstacle for monoclonal antibody therapy, as the NK cells mediating the antibody-dependent cell-mediated cytolysis (ADCC) cannot come in close enough contact with the cancer cell to form the cytolytic synapse (98). Dissolving this protective halo in vitro through a PEGylated recombinant human hyaluronidase PH20 (PEGPH20) increased ADCC, and in vivo the combined treatment of HA-overexpressing SKOV3 ovarian cancer xenografts with PEGPH20, trastuzumab, and NK cells resulted in a drastic inhibition of tumor growth (98).
Besides sensitizing cancer cells to ADCC, hyaluronidase treatment of cancer also affects the biomechanics of HA-rich tumors. Accumulation of HA increases the colloid osmotic pressure leading to an increase in IFP within the tumor, causing multiple problems for treatment (121). An increase in IFP results in decreased transcapillary transport of solutes into the tumor, including systemic treatment. The increased IFP can also cause unstable tumor vessels to collapse, cutting off perfusion to areas of the tumor, creating hypoxia and further limiting tumor penetration of any systemic treatment. Accordingly, treating tumors with hyaluronidase lowers the IFP and increases the perfusion of tumors and the penetration of therapeutics (27, 122–124).
Although hyaluronidase treatment of cancers produces LMW-HA fragments, which can increase angiogenesis and disrupt vessel integrity, PEGPH20 treatment of experimental pancreatic cancer in combination with chemotherapy increased overall survival (27, 124) and decreased the metastatic incidence (124). It is reassuring to see that the creation of more functional vessels did not lead to an increase in metastasis, as it might have been feared to do. It is conceivable that by stripping cancer cells of their pericellular matrix, the hyaluronidase treatment is sensitizing any extravasating cancer cells to immune cell lysis, in this way counteracting the effect of restoring tumor vessel function. PEGPH20 is now in a randomized phase 2 clinical trial for metastatic pancreatic cancer and shows promising results (125). However, the clinical trial also revealed a serious side effect of the treatment, as the incidence of thromboemboli increased in the PEGPH20 treatment arm. Fortunately, this appears to be preventable by giving the patients the prophylaxis enoxoparin, and now a large, randomized, double-blinded, placebo-controlled phase 3 study is planned to start in the beginning of 2016 (125).
Instead of breaking down already produced HMW-HA, another option is to prevent its synthesis. 4-Methylumbelliferone (4-MU) is a cumarin derivative that inhibits the production of HA, most likely by depleting the cell of the UDP-glucuronic acid that is a precursor of HA (126). 4-MU is a naturally occurring compound, and it is has been approved in Europe since the 1960s to treat biliary spasms, thus repurposing it for cancer treatment is a very real possibility. Several in vivo studies have reported promising inhibition of tumor growth and metastasis formation upon treatment with 4-MU [reviewed in Ref. (127)]; however, so far no clinical trials or safety studies aimed at evaluating long-term safety have been initiated.
Using HA as a Tag to Target Drugs to Cells
Many malignancies express CD44, thus using HA as a homing missile for cancer therapeutics is a logical step. Coupling of nanoparticles loaded with the chemotherapeutic drug paclitaxel to ultrashort HA polymers leads to improved drug uptake in brain tumor lesions of breast cancer in a preclinical model of brain metastases, significantly improving the overall survival of the mice treated with the nanoconjugate (128). Another study found that liposomes coated with PEG and HMW-HA had increased cellular uptake and tumor penetration in subcutaneous MDA-MB-231 tumors, even though the overall accumulation in the tumor was the same as PEG liposomes (129). Loading HA-coated liposomes with doxorubicin had an increased therapeutic effect in multiple tumor models, including experimental B16F10 melanoma metastases in the lungs (130).
One caveat of using HA as a tag to target chemotherapy to CD44+ cancer cells is the abundant expression of the HA receptor HARE/Stab2 in the liver, thus special attention to liver toxicity is essential in such studies. One way of limiting potential liver toxicity could be to try to exhaust the endocytic HARE/Stab2 presence on the hepatocytes by increasing the levels of HA in the blood before administering the HA–drug conjugate. Such an approach might even have the added benefit of preventing circulating tumor cells from attaching to endothelial cells and seeding new organs (104, 105), and elevated blood HA levels have shown no adverse effects in mice (105).
Small Oligo-HA as Direct Treatment
Small oligo-HA (sHA) has been tested in multiple pre-clinical studies following the rationale that it can prevent the normal signaling from endogenous, often larger, HA molecules by occupying the HA binding partners/receptors. Indeed, inhibition of tumor growth in vivo in breast cancer, lung cancer, osteosarcoma, and melanoma has been reported. The addition of sHA prevents the formation of the versican–HA-rich pericellular matrix (36, 38) and inhibits the binding of ovarian cancer cells to the peritoneal wall in vivo (38). Most interestingly, the direct injections of sHA into tibial tumors of MDA-MB-231 breast cancer cells reduced the progression of already established osteolytic lesions (119). However, sHA fragments of a similar size correlate to increased lymphatic invasion and development of lymph node metastasis in colorectal cancer patients, suggesting that small sHA may promote tumor progression (131). The conflicting reports of the pro- and anti-tumorigenic effects of sHA are possibly due to a combination of differences in both size and concentration. Low concentrations of sHA stimulated the neovascularization of Matrigel plugs in vivo while very high concentrations inhibited angiogenesis (53). Thus, further studies with sHA should test a range of different sizes and concentrations to evaluate whether there is a large enough therapeutic window regarding bioavailability in the tumor that could make the treatment translational.
Targeting Periostin
One example is the monoclonal periostin-blocking antibody OC-20. Although Orecchia et al. only reported beneficial aspects of the antibody on the primary tumor in their mouse model of human melanomas (132), the monoclonal antibody could be utilized in further preclinical studies investigating its influence on metastatic spread. The combination of a periostin-blocking antibody and 5-fluorouracil enhanced the apoptotic effect of the pyrimidine analog in vitro (133).
A further approach to target periostin is the benzyl-d(U)TP-modified nucleic acid aptamer PNDA-3. PNDA-3 specifically antagonized periostin-induced adhesion and invasion of breast cancer cells. In the 4T1 orthotopic mouse model, intratumoral PNDA-3 administration significantly reduced the size of the primary tumor and, more interestingly, of metastatic foci in the lung. The aptamer abrogated AKT/PKB pathway-mediated survival at the metastatic sites (134). PNDA-3 also reduced the metastatic burden of gastric cancer cells in the liver (135). Taken together, the DNA aptamer PNDA-3 is a promising drug for targeting periostin. New approaches combining nucleic acid aptamers with nanoparticle leading to a prolonged stability and specific binding to the tumor tissue could further strengthen the beneficial effect of the treatment qualifying the drug for clinical trials.
Targeting Tenascin C
The large splice variant of tenascin C is specifically expressed in tumors and expression levels increase with grade of malignancy in many cancer types including brain, lung, and squamous cell cancer (21, 136). Therefore, targeting tenascin C is of particular interest for diagnostic and therapeutic approaches. Several (pre-)clinical studies using tenascin C-targeting agents were performed, in particular in advanced brain tumors such as glioblastoma multiforme and malignant astrocytomas.
First, a murine anti-tenascin monoclonal antibody named 81C6 was shown to bind specifically to tumor tissue (137). Neuradiab, the (131)I-labeled 81C6, showed decent benefits in a glioblastoma xenograft mouse models (138). Phase I and II studies where the antibody was injected directly into the resection cavity of glioblastoma patients revealed an improvement in the median survival of the patients (139–141); however, phase III trials were suspended owing to a delay in site initiation (clinicaltrials.gov).
To overcome the restriction of a non-recurrent and local administration of the murine IgG2, several human antibodies targeting tenascin C were generated. Silacci et al. developed a tenascin C-targeting antibody, named G11. The human monoclonal antibody stained patient-derived tumor sections with high specificity confirming the restricted expression of tenascin C to cancerous tissue. In addition, biodistribution studies in either mice bearing subcutaneous human glioma cell tumors or rats with orthotopic brain tumors, showed a highly selective tumor uptake of the antibody (136). In addition, Brack et al. developed a 125I-labeled humanized antibody targeting tenascin C, called F16. In a xenograft model, this radiolabeled human IgE antibody showed a selective targeting to human glioblastoma tissue with a clearance from the blood and other organs within 24h after intravenous injection (142). Both antibodies are promising approaches for further clinical trials, allowing a repeated treatment of cancer patients.
In addition, Kim et al. developed a tenascin C-targeting peptide binding specifically to the large splice variant of the protein either in a xenograft model of glioblastoma or in human lung and colon cancer or squamous carcinoma samples. This peptide reduced the migration of a glioblastoma cell line in vitro (143). Coupling this peptide to chemotherapeutics or radioactive isotopes could improve the clinical outcome of many cancer types.
Another way to target tenascin C is RNAi. When ATN-RNA, a double-stranded RNA complementary to human tenascin C, was applied locally to patients with advance brain tumors, the drug showed a survival benefit of 18 weeks in grade III astrocytoma and 10 weeks in glioblastoma multiforme (144). Thus, targeting tenascin C particularly for brain cancers looks very promising.
Targeting Collagen I
Collagen prolyl-4-hydroxylases (CP4H) are essential enzymes for the correct biosynthesis of collagens. CP4H catalyzes the conversion of proline to hydroxyproline, facilitating the assembly of three procollagen proteins into a procollagen triple helix. Recent studies in preclinical breast cancer models have shown that inhibition of CP4H either completely prevents or dramatically decreases spontaneous metastasis to the lungs (65, 66).
Interfering with post-translational cross-linking of collagens by LOX-family members has been proposed as a promising therapeutic avenue due to successful inhibition of metastasis in pre-clinical models (43, 110). So far, a LOXL2-targeting antibody (145) is in clinical trials for the treatment of fibrosis and cancer, but recently phase II clinical trials combining this antibody with chemotherapy (gemcitabine) in pancreatic cancer did not yield positive results (Gilead, press release). However, recent preclinical data have shown efficacy of targeting LOX in combination with gemcitabine in preclinical models of pancreatic cancer, provided treatment was administered early (146). Thus, LOXL2 inhibition combined with gemcitabine could still be effective in early-stage patients.
Concluding Remarks and Future Approaches
The ECM has crucial impact on cancer progression. In this review, we highlighted how ECM proteins influence every step of the complex cascade of tumor spread with a focus on some of the key players (Figure 1). The ECM not only provides cues favoring migration, attachment, survival, or proliferation but also physically protects the cells from the shear stress in the blood, immune cell attack, and the impact of drugs. Targeting ECM proteins is therefore of great interest in the adjuvant therapeutic setting. In particular, combination treatment with gold-standard therapeutics seems to be a promising strategy, as targeting the macromolecules facilitates the delivery and efficacy of the standard treatment and inhibits protumorigenic signaling of the ECM itself.
Extracellular matrix proteins can be targeted in many different ways. Beyond the direct targeting of the macromolecules, one can also prevent their synthesis, cross-linking, or correct post-translational processing, opening up many possible avenues of treatment. However, although targeting of ECM components holds much promise for improved cancer management, it is important to keep in mind that any treatment aimed at cancer ECM also may affect the same ECM component in healthy tissue. Indeed, one of the main reasons why the much anticipated MMP inhibitors had such a poor success rate in clinical trials was the unexpected toxicity in healthy tissue [this reason and others are reviewed in Ref. (147)]. It is thus crucial to develop and rigorously study preclinical models of ECM targeting in many cancer types, paying particular attention to any side effects in healthy tissue to overcome this translational hurdle and move from bench to bedside. Additionally, further research into the exact mechanisms of how the individual ECM macromolecules stimulate migration, adhesion, and cell survival, and how this may be different in cancerous contra normal settings, will help to tailor targeting strategies to suit cancer ECM better than healthy ECM. Another important lesson that can be learned from the initial failure of MMP inhibitors is about the timing of the treatment with regard to the cancer progression stage. The ECM components reviewed here often play a part in more than one of the steps of the metastatic cascade, but the involvement in one step might turn out to be more targetable than others. This makes it paramount for preclinical studies to carefully dissect when a given target should be hit to have an effect and, in effect, making it very important to stratify clinical trials accordingly.
Nevertheless, targeting components of the ECM in order to block cancer-driving signaling pathways and to facilitate the penetrance of standard chemotherapeutics is a promising approach to prevent the life-threatening spread of cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the Danish Council for Independent Research for the YDUN grant that supports FV (108481001) and the FNU grant that supports LW (DFF – 4002-00099), as well as the Novo Nordisk Foundation for the Hallas Møller Stipendum that supports JE. The review was cowritten by FV and LW, and edited by JE.
References
2. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer (2009) 9(4):239–52. doi:10.1038/nrc2618
3. Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell (2006) 127(4):679–95. doi:10.1016/j.cell.2006.11.001
4. Hynes RO. The extracellular matrix: not just pretty fibrils. Science (2009) 326(5957):1216–9. doi:10.1126/science.1176009
5. Takeshita S, Kikuno R, Tezuka K, Amann E. Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J (1993) 294(Pt 1):271–8. doi:10.1042/bj2940271
6. Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem (2007) 101(3):695–711. doi:10.1002/jcb.21224
7. Bao S, Ouyang G, Bai X, Huang Z, Ma C, Liu M, et al. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell (2004) 5(4):329–39. doi:10.1016/S1535-6108(04)00081-9
8. Tilman G, Mattiussi M, Brasseur F, van Baren N, Decottignies A. Human periostin gene expression in normal tissues, tumors and melanoma: evidences for periostin production by both stromal and melanoma cells. Mol Cancer (2007) 6:80. doi:10.1186/1476-4598-6-80
9. Fukushima N, Kikuchi Y, Nishiyama T, Kudo A, Fukayama M. Periostin deposition in the stroma of invasive and intraductal neoplasms of the pancreas. Mod Pathol (2008) 21(8):1044–53. doi:10.1038/modpathol.2008.77
10. Puglisi F, Puppin C, Pegolo E, Andreetta C, Pascoletti G, D’Aurizio F, et al. Expression of periostin in human breast cancer. J Clin Pathol (2008) 61(4):494–8. doi:10.1136/jcp.2007.052506
11. Takanami I, Abiko T, Koizumi S. Expression of periostin in patients with non-small cell lung cancer: correlation with angiogenesis and lymphangiogenesis. Int J Biol Markers (2008) 23(3):182–6.
12. Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Abstract SY28-02: interactions between cancer stem cells and their niche govern metastatic colonization. Cancer Res (2012) 72(8 Suppl):SY28–22. doi:10.7554/eLife.06938
13. Sasaki H, Dai M, Auclair D, Fukai I, Kiriyama M, Yamakawa Y, et al. Serum level of the periostin, a homologue of an insect cell adhesion molecule, as a prognostic marker in nonsmall cell lung carcinomas. Cancer (2001) 92(4):843–8. doi:10.1002/1097-0142(20010815)92:4<843::AID-CNCR1391>3.0.CO;2-P
14. Sasaki H, Yu CY, Dai M, Tam C, Loda M, Auclair D, et al. Elevated serum periostin levels in patients with bone metastases from breast but not lung cancer. Breast Cancer Res Treat (2003) 77(3):245–52. doi:10.1023/A:1021899904332
15. Contié S, Voorzanger-Rousselot N, Litvin J, Clézardin P, Garnero P. Increased expression and serum levels of the stromal cell-secreted protein periostin in breast cancer bone metastases. Int J Cancer (2011) 128(2):352–60. doi:10.1002/ijc.25591
16. Jones FS, Jones PL. The tenascin family of ECM glycoproteins: structure, function, and regulation during embryonic development and tissue remodeling. Dev Dyn (2000) 218(2):235–59. doi:10.1002/(SICI)1097-0177(200006)218:2<235::AID-DVDY2>3.0.CO;2-G
17. Yoshida T, Matsumoto E, Hanamura N, Kalembeyi I, Katsuta K, Ishihara A, et al. Co-expression of tenascin and fibronectin in epithelial and stromal cells of benign lesions and ductal carcinomas in the human breast. J Pathol (1997) 182(4):421–8. doi:10.1002/(SICI)1096-9896(199708)182:4<421::AID-PATH886>3.3.CO;2-L
18. Hanamura N, Yoshida T, Matsumoto E, Kawarada Y, Sakakura T. Expression of fibronectin and tenascin-C mRNA by myofibroblasts, vascular cells and epithelial cells in human colon adenomas and carcinomas. Int J Cancer (1997) 73(1):10–5. doi:10.1002/(SICI)1097-0215(19970926)73:1<10::AID-IJC2>3.0.CO;2-4
19. Mori M, Muramatsu Y, Yamada K, Shrestha P, Takai Y. Intracellular localization of tenascin in squamous cell carcinoma of oral cavity: an immunohistochemical study. Anticancer Res (1996) 16(5B):3075–9.
20. Ilmonen S, Jahkola T, Turunen JP, Muhonen T, Asko-Seljavaara S. Tenascin-C in primary malignant melanoma of the skin. Histopathology (2004) 45(4):405–11. doi:10.1111/j.1365-2559.2004.01976.x
21. Pedretti M, Soltermann A, Arni S, Weder W, Neri D, Hillinger S. Comparative immunohistochemistry of L19 and F16 in non-small cell lung cancer and mesothelioma: two human antibodies investigated in clinical trials in patients with cancer. Lung Cancer (2009) 64(1):28–33. doi:10.1016/j.lungcan.2008.07.013
22. Saupe F, Schwenzer A, Jia Y, Gasser I, Spenlé C, Langlois B, et al. Tenascin-C downregulates wnt inhibitor dickkopf-1, promoting tumorigenesis in a neuroendocrine tumor model. Cell Rep (2013) 5(2):482–92. doi:10.1016/j.celrep.2013.09.014
23. Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med (2011) 17(7):867–74. doi:10.1038/nm.2379
24. Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Sports (2009) 19(4):511–9. doi:10.1111/j.1600-0838.2009.00916.x
25. Didem T, Faruk T, Senem K, Derya D, Murat S, Murat G, et al. Clinical significance of serum tenascin-C levels in epithelial ovarian cancer. Tumour Biol (2014) 35(7):6777–82. doi:10.1007/s13277-014-1923-z
26. Pauli C, Stieber P, Schmitt UM, Andratschke M, Hoffmann K, Wollenberg B. The significance of tenascin-C serum level as tumor marker in squamous cell carcinoma of the head and neck. Anticancer Res (2002) 22(5):3093–7.
27. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut (2013) 62(1):112–20. doi:10.1136/gutjnl-2012-302529
28. Sironen RK, Tammi M, Tammi R, Auvinen PK, Anttila M, Kosma VM. Hyaluronan in human malignancies. Exp Cell Res (2011) 317(4):383–91. doi:10.1016/j.yexcr.2010.11.017
29. Gandhi NS, Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des (2008) 72(6):455–82. doi:10.1111/j.1747-0285.2008.00741.x
30. Itano N, Kimata K. Mammalian hyaluronan synthases. IUBMB Life (2002) 1(4):195–9. doi:10.1080/15216540214929
31. Heldin P, Basu K, Olofsson B, Porsch H, Kozlova I, Kahata K. Deregulation of hyaluronan synthesis, degradation and binding promotes breast cancer. J Biochem (2013) 154(5):395–408. doi:10.1093/jb/mvt085
32. Tian X, Azpurua J, Hine C, Vaidya A, Myakishev-Rempel M, Ablaeva J, et al. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature (2013) 499(7458):346–9. doi:10.1038/nature12234
33. Delaney MA, Nagy L, Kinsel MJ, Treuting PM. Spontaneous histologic lesions of the adult naked mole rat (Heterocephalus glaber): a retrospective survey of lesions in a zoo population. Vet Pathol (2013) 50(4):607–21. doi:10.1177/0300985812471543
34. Buffenstein R. Negligible senescence in the longest living rodent, the naked mole-rat: insights from a successfully aging species. J Comp Physiol B (2008) 178(4):439–45. doi:10.1007/s00360-007-0237-5
35. Ricciardelli C, Sakko AJ, Ween MP, Russell DL, Horsfall DJ. The biological role and regulation of versican levels in cancer. Cancer Metastasis Rev (2009) 28(1–2):233–45. doi:10.1007/s10555-009-9182-y
36. Evanko SP, Angello JC, Wight TN. Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol (1999) 19(4):1004–13. doi:10.1161/01.ATV.19.4.1004
37. Ricciardelli C, Russell DL, Ween MP, Mayne K, Suwiwat S, Byers S, et al. Formation of hyaluronan- and versican-rich pericellular matrix by prostate cancer cells promotes cell motility. J Biol Chem (2007) 282(14):10814–25. doi:10.1074/jbc.M606991200
38. Ween MP, Hummitzsch K, Rodgers RJ, Oehler MK, Ricciardelli C. Versican induces a pro-metastatic ovarian cancer cell behavior which can be inhibited by small hyaluronan oligosaccharides. Clin Exp Metastasis (2011) 28(2):113–25. doi:10.1007/s10585-010-9363-7
39. Keire PA, Bressler SL, Lemire JM, Edris B, Rubin BP, Rahmani M, et al. A role for versican in the development of leiomyosarcoma. J Biol Chem (2014) 289(49):34089–103. doi:10.1074/jbc.M114.607168
40. Ricciardelli C, Mayne K, Sykes PJ, Raymond WA, McCaul K, Marshall VR, et al. Elevated levels of versican but not decorin predict disease progression in early-stage prostate cancer. Clin Cancer Res (1998) 4(4):963–71.
41. Ricciardelli C, Brooks JH, Suwiwat S, Sakko AJ, Mayne K, Raymond WA, et al. Regulation of stromal versican expression by breast cancer cells and importance to relapse-free survival in patients with node-negative primary breast cancer. Clin Cancer Res (2002) 8(4):1054–60.
42. Pirinen R, Leinonen T, Böhm J, Johansson R, Ropponen K, Kumpulainen E, et al. Versican in nonsmall cell lung cancer: relation to hyaluronan, clinicopathologic factors, and prognosis. Hum Pathol (2005) 36(1):44–50. doi:10.1016/j.humpath.2004.10.010
43. Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nat Rev Cancer (2012) 12(8):540–52. doi:10.1038/nrc3319
44. Cox TR, Erler JT. The importance of LOX family members on modulating cell-ECM interactions in carcinogenesis. J Carcinog Mutagen (2013) S13:001. doi:10.4172/2157-2518.S13-001
45. Alowami S, Troup S, Al-Haddad S, Kirkpatrick I, Watson PH. Mammographic density is related to stroma and stromal proteoglycan expression. Breast Cancer Res (2003) 5(5):R129–35. doi:10.1186/bcr622
46. Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E, et al. Mammographic density and the risk and detection of breast cancer. N Engl J Med (2007) 356(3):227–36. doi:10.1056/NEJMoa062790
47. Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med (2008) 6:11. doi:10.1186/1741-7015-6-11
48. Reymond N, d’Água BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer (2013) 13(12):858–70. doi:10.1038/nrc3628
49. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119(6):1420–8. doi:10.1172/JCI39104
50. Chiodoni C, Colombo MP, Sangaletti S. Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev (2010) 29(2):295–307. doi:10.1007/s10555-010-9221-8
51. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi:10.1038/nrm3758
52. Kultti A, Zhao C, Singha NC, Zimmerman S, Osgood RJ, Symons R, et al. Accumulation of extracellular hyaluronan by hyaluronan synthase 3 promotes tumor growth and modulates the pancreatic cancer microenvironment. Biomed Res Int (2014) 2014:817613. doi:10.1155/2014/817613
53. Koyama H, Hibi T, Isogai Z, Yoneda M, Fujimori M, Amano J, et al. Hyperproduction of hyaluronan in neu-induced mammary tumor accelerates angiogenesis through stromal cell recruitment: possible involvement of versican/PG-M. Am J Pathol (2007) 170(3):1086–99. doi:10.2353/ajpath.2007.060793
54. Bharadwaj AG, Kovar JL, Loughman E, Elowsky C, Oakley GG, Simpson MA. Spontaneous metastasis of prostate cancer is promoted by excess hyaluronan synthesis and processing. Am J Pathol (2009) 174(3):1027–36. doi:10.2353/ajpath.2009.080501
55. El-Haibi CP, Bell GW, Zhang J, Collmann AY, Wood D, Scherber CM, et al. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci U S A (2012) 109(43):17460–5. doi:10.1073/pnas.1206653109
56. Hosono K, Nishida Y, Knudson W, Knudson CB, Naruse T, Suzuki Y, et al. Hyaluronan oligosaccharides inhibit tumorigenicity of osteosarcoma cell lines MG-63 and LM-8 in vitro and in vivo via perturbation of hyaluronan-rich pericellular matrix of the cells. Am J Pathol (2007) 171(1):274–86. doi:10.2353/ajpath.2007.060828
57. McBride WH, Bard JBL. Hyaluronidase-sensitive halos around adherent cells. Their role in blocking lymphocyte-mediated cytolysis. J Exp Med (1979) 149(2):507–15. doi:10.1084/jem.149.2.507
58. Desjardins M, Xie J, Gurler H, Muralidhar GG, Sacks JD, Burdette JE, et al. Versican regulates metastasis of epithelial ovarian carcinoma cells and spheroids. J Ovarian Res (2014) 7:70. doi:10.1186/1757-2215-7-70
59. Singleton PA, Dudek SM, Ma SF, Garcia JG. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J Biol Chem (2006) 281(45):34381–93. doi:10.1074/jbc.M603680200
60. Singleton PA, Mirzapoiazova T, Guo Y, Sammani S, Mambetsariev N, Lennon FE, et al. High-molecular-weight hyaluronan is a novel inhibitor of pulmonary vascular leakiness. Am J Physiol Lung Cell Mol Physiol (2010) 299(5):L639–51. doi:10.1152/ajplung.00405.2009
61. Mambetsariev N, Mirzapoiazova T, Mambetsariev B, Sammani S, Lennon FE, Garcia JG, et al. Hyaluronic acid binding protein 2 is a novel regulator of vascular integrity. Arterioscler Thromb Vasc Biol (2010) 30(3):483–90. doi:10.1161/ATVBAHA.109.200451
62. Lennon FE, Mirzapoiazova T, Mambetsariev N, Mambetsariev B, Salgia R, Singleton PA. Transactivation of the receptor-tyrosine kinase ephrin receptor A2 is required for the low molecular weight hyaluronan-mediated angiogenesis that is implicated in tumor progression. J Biol Chem (2014) 289(35):24043–58. doi:10.1074/jbc.M114.554766
63. Shintani Y, Hollingsworth MA, Wheelock MJ, Johnson KR. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH(2)-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res (2006) 66(24):11745–53. doi:10.1158/0008-5472.CAN-06-2322
64. Koenig A, Mueller C, Hasel C, Adler G, Menke A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res (2006) 66(9):4662–71. doi:10.1158/0008-5472.CAN-05-2804
65. Gilkes DM, Chaturvedi P, Bajpai S, Wong CC, Wei H, Pitcairn S, et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res (2013) 73(11):3285–96. doi:10.1158/0008-5472.CAN-12-3963
66. Xiong G, Deng L, Zhu J, Rychahou PG, Xu R. Prolyl-4-hydroxylase α subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer (2014) 14(1):1. doi:10.1186/1471-2407-14-1
67. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell (2009) 139(5):891–906. doi:10.1016/j.cell.2009.10.027
68. Baker AM, Cox TR, Bird D, Lang G, Murray GI, Sun XF, et al. The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer. J Natl Cancer Inst (2011) 103(5):407–24. doi:10.1093/jnci/djq569
69. Baker AM, Bird D, Lang G, Cox TR, Erler JT. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene (2013) 32(14):1863–8. doi:10.1038/onc.2012.202
70. Barker HE, Bird D, Lang G, Erler JT. Tumor-secreted LOXL2 activates fibroblasts through FAK signaling. Mol Cancer Res (2013) 11(11):1425–36. doi:10.1158/1541-7786.MCR-13-0033-T
71. Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell (2012) 23(5):781–91. doi:10.1091/mbc.E11-06-0537
72. Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, et al. Matrix stiffness drives epithelial–mesenchymal transition and tumour metastasis through a TWIST1–G3BP2 mechanotransduction pathway. Nat Cell Biol (2015) 17(5):678–88. doi:10.1038/ncb3157
73. Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol (2010) 22(5):697–706. doi:10.1016/j.ceb.2010.08.015
74. Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol (2011) 178(3):1221–32. doi:10.1016/j.ajpath.2010.11.076
75. Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol (2013) 15(6):677–87. doi:10.1038/ncb2743
76. Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol (2002) 14(5):608–16. doi:10.1016/S0955-0674(02)00361-7
77. Yan W, Shao R. Transduction of a mesenchyme-specific gene periostin into 293T cells induces cell invasive activity through epithelial-mesenchymal transformation. J Bioll Chem (2006) 281(28):19700–9. doi:10.1074/jbc.M601856200
78. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell (2010) 141(1):52–67. doi:10.1016/j.cell.2010.03.015
79. Michaylira CZ, Wong GS, Miller CG, Gutierrez CM, Nakagawa H, Hammond R, et al. Periostin, a cell adhesion molecule, facilitates invasion in the tumor microenvironment and annotates a novel tumor-invasive signature in esophageal cancer. Cancer Res (2010) 70(13):5281–92. doi:10.1158/0008-5472.CAN-10-0704
80. Siriwardena BS, Kudo Y, Ogawa I, Kitagawa M, Kitajima S, Hatano H, et al. Periostin is frequently overexpressed and enhances invasion and angiogenesis in oral cancer. Br J Cancer (2006) 95(10):1396–403. doi:10.1038/sj.bjc.6603431
81. Nagaharu K, Zhang X, Yoshida T, Katoh D, Hanamura N, Kozuka Y, et al. Tenascin C induces epithelial-mesenchymal transition-like change accompanied by SRC activation and focal adhesion kinase phosphorylation in human breast cancer cells. Am J Pathol (2011) 178(2):754–63. doi:10.1016/j.ajpath.2010.10.015
82. Tavazoie SF, Alarcón C, Oskarsson T, Padua D, Wang Q, Bos PD, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature (2008) 451(7175):147–52. doi:10.1038/nature06487
83. Kalembeyi I, Inada H, Nishiura R, Imanaka-Yoshida K, Sakakura T, Yoshida T. Tenascin-C upregulates matrix metalloproteinase-9 in breast cancer cells: direct and synergistic effects with transforming growth factor beta1. Int J Cancer (2003) 105(1):53–60. doi:10.1002/ijc.11037
84. Nisticò P, Bissell MJ, Radisky DC. Epithelial-mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harbor Perspect Biol (2012) 4(2):a011908. doi:10.1101/cshperspect.a011908
85. Chiquet-Ehrismann R, Mackie EJ, Pearson CA, Sakakura T. Tenascin: an extracellular matrix protein involved in tissue interactions during fetal development and oncogenesis. Cell (1986) 47(1):131–9. doi:10.1016/0092-8674(86)90374-0
86. Jahkola T, Toivonen T, Virtanen I, von Smitten K, Nordling S, von Boguslawski K, et al. Tenascin-C expression in invasion border of early breast cancer: a predictor of local and distant recurrence. Br J Cancer (1998) 78(11):1507–13. doi:10.1038/bjc.1998.714
87. Amo L, Tamayo-Orbegozo E, Maruri N, Eguizabal C, Zenarruzabeitia O, Riñón M, et al. Involvement of platelet-tumor cell interaction in immune evasion. Potential role of podocalyxin-like protein 1. Front Oncol (2014) 4:1–7. doi:10.3389/fonc.2014.00245
88. Rybarczyk BJ, Simpson-Haidaris PJ. Fibrinogen assembly, secretion, and deposition into extracellular matrix by MCF-7 human breast carcinoma cells. Cancer Res (2000) 60(7):2033–9.
89. Simpson-Haidaris PJ, Rybarczyk B. Tumors and fibrinogen. The role of fibrinogen as an extracellular matrix protein. Ann N Y Acad Sci (2001) 936:406–25. doi:10.1111/j.1749-6632.2001.tb03525.x
90. Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov (2012) 2(12):1091–9. doi:10.1158/2159-8290.CD-12-0329
91. Sharma D, Brummel-Ziedins KE, Bouchard BA, Holmes CE. Platelets in tumor progression: a host factor that offers multiple potential targets in the treatment of cancer. J Cell Physiol (2014) 229(8):1005–15. doi:10.1002/jcp.24539
92. Palumbo JS, Kombrinck KW, Drew AF, Grimes TS, Kiser JH, Degen JL, et al. Fibrinogen is an important determinant of the metastatic potential of circulating tumor cells. Blood (2000) 96(10):3302–9.
93. Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood (2004) 104(2):397–401. doi:10.1182/blood-2004-02-0434
94. Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E, et al. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res (2010) 70(11):4327–34. doi:10.1158/0008-5472.CAN-09-3312
95. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer (2011) 11(2):123–34. doi:10.1038/nrc3004
96. Menter DG, Tucker SC, Kopetz S, Sood AK, Crissman JD, Honn KV. Platelets and cancer: a casual or causal relationship: revisited. Cancer Metastasis Rev (2014) 33(1):231–69. doi:10.1007/s10555-014-9498-0
97. Schaff M, Receveur N, Bourdon C, Wurtz V, Denis CV, Orend G, et al. Novel function of tenascin-C, a matrix protein relevant to atherosclerosis, in platelet recruitment and activation under flow. Arterioscler Thromb Vasc Biol (2011) 31(1):117–24. doi:10.1161/ATVBAHA.110.206375
98. Singha NC, Nekoroski T, Zhao C, Symons R, Jiang P, Frost GI, et al. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol Cancer Ther (2014) 14(2):523–32. doi:10.1158/1535-7163.MCT-14-0580
99. Singleton PA. Hyaluronan regulation of endothelial barrier function in cancer. Adv Cancer Res (2014) 123:191–209. doi:10.1016/B978-0-12-800092-2.00007-1
100. McFarlane S, Coulter JA, Tibbits P, O’Grady A, McFarlane C, Montgomery N, et al. CD44 increases the efficiency of distant metastasis of breast cancer. Oncotarget (2015) 6(13):11465–76. doi:10.18632/oncotarget.3410
101. Simpson MA, Reiland J, Burger SR, Furcht LT, Spicer AP, Oegema TR Jr, et al. Hyaluronan synthase elevation in metastatic prostate carcinoma cells correlates with hyaluronan surface retention, a prerequisite for rapid adhesion to bone marrow endothelial cells. J Biol Chem (2001) 276(21):17949–57. doi:10.1074/jbc.M010064200
102. Draffin JE, McFarlane S, Hill A, Johnston PG, Waugh DJ. CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res (2004) 64(16):5702–11. doi:10.1158/0008-5472.CAN-04-0389
103. Okuda H, Kobayashi A, Xia B, Watabe M, Pai SK, Hirota S, et al. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res (2012) 72(2):537–47. doi:10.1158/0008-5472.CAN-11-1678
104. Simpson MA, Weigel JA, Weigel PH. Systemic blockade of the hyaluronan receptor for endocytosis prevents lymph node metastasis of prostate cancer. Int J Cancer (2012) 131(5):E836–40. doi:10.1002/ijc.27427
105. Hirose Y, Saijou E, Sugano Y, Takeshita F, Nishimura S, Nonaka H, et al. Inhibition of Stabilin-2 elevates circulating hyaluronic acid levels and prevents tumor metastasis. Proc Natl Acad Sci U S A (2012) 109(11):4263–8. doi:10.1073/pnas.1117560109
106. Wu M, Cao M, He Y, Liu Y, Yang C, Du Y, et al. A novel role of low molecular weight hyaluronan in breast cancer metastasis. FASEB J (2014) 1(4):1–9. doi:10.1096/fj.14-259978
107. Wong CC, Zhang H, Gilkes DM, Chen J, Wei H, Chaturvedi P, et al. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J Mol Med (Berl) (2012) 90(7):803–15. doi:10.1007/s00109-011-0855-y
108. Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le QT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature (2006) 440(7088):1222–6. doi:10.1038/nature04695
109. Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell (2009) 15(1):35–44. doi:10.1016/j.ccr.2008.11.012
110. Cox TR, Bird D, Baker AM, Barker HE, Ho MW, Lang G, et al. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res (2013) 73(6):1721–32. doi:10.1158/0008-5472.CAN-12-2233
111. Wong GS, Rustgi AK. Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis. Br J Cancer (2013) 108(4):755–61. doi:10.1038/bjc.2012.592
112. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer (2009) 9(4):285–93. doi:10.1038/nrc2621
113. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells – perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res (2006) 66(19):9339–44. doi:10.1158/0008-5472.CAN-06-3126
114. Rizwan A, Bulte C, Kalaichelvan A, Cheng M, Krishnamachary B, Bhujwalla ZM, et al. Metastatic breast cancer cells in lymph nodes increase nodal collagen density. Sci Rep (2015) 5:10002. doi:10.1038/srep10002
115. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438(7069):820–7. doi:10.1038/nature04186
116. Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res (2012) 72(6):1384–94. doi:10.1158/0008-5472.CAN-11-2905
117. Sheng W, Wang G, La Pierre DP, Wen J, Deng Z, Wong CK, et al. Versican mediates mesenchymal-epithelial transition. Mol Biol Cell (2006) 17(4):2009–20. doi:10.1091/mbc.E05-10-0951
118. Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res (2010) 70(18):7073–83. doi:10.1158/0008-5472.CAN-09-4687
119. Urakawa H, Nishida Y, Knudson W, Knudson CB, Arai E, Kozawa E, et al. Therapeutic potential of hyaluronan oligosaccharides for bone metastasis of breast cancer. J Orthop Res (2012) 30(4):662–72. doi:10.1002/jor.21557
120. O’Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci USA (2011) 108(38):16002–7. doi:10.1073/pnas.1109493108
121. Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer (2004) 4(10):806–13. doi:10.1038/nrc1456
122. Thompson CB, Shepard HM, O’Connor PM, Kadhim S, Jiang P, Osgood RJ, et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther (2010) 9(11):3052–64. doi:10.1158/1535-7163.MCT-10-0470
123. Brekken C, de Lange Davies C. Hyaluronidase reduces the interstitial fluid pressure in solid tumours in a non-linear concentration-dependent manner. Cancer Lett (1998) 131(1):65–70. doi:10.1016/S0304-3835(98)00202-X
124. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21(3):418–29. doi:10.1016/j.ccr.2012.01.007
125. Hingorani S, Harris WP, Hendiraf AE, Bullock AJ, Wu XU, Ping J, et al. High response rate and PFS with PEGPH20 added to nab-paclitaxel/gemcitabine in stage IV previously untreated pancreatic cancer patients with high-HA tumors: interim results of a randomized phase 2 study. ASCO Annual Meeting (2015). Availble from: http://meetinglibrary.asco.org/content/152325-156
126. Kakizaki I, Kojima K, Takagaki K, Endo M, Kannagi R, Ito M, et al. A novel mechanism for the inhibition of hyaluronan biosynthesis by 4-methylumbelliferone. J Biol Chem (2004) 279(32):33281–9. doi:10.1074/jbc.M405918200
127. Nagy N, Kuipers HF, Frymoyer AR, Ishak HD, Bollyky JB, Wight TN, et al. 4-methylumbelliferone treatment and hyaluronan inhibition as a therapeutic strategy in inflammation, autoimmunity, and cancer. Front Immunol (2015) 6:123. doi:10.3389/fimmu.2015.00123
128. Mittapalli RK, Liu X, Adkins CE, Nounou MI, Bohn KA, Terrell TB, et al. Paclitaxel-hyaluronic nanoconjugates prolong overall survival in a preclinical brain metastases of breast cancer model. Mol Cancer Ther (2013) 12(11):2389–99. doi:10.1158/1535-7163.MCT-13-0132
129. Qhattal HS, Hye T, Alali A, Liu X. Hyaluronan polymer length, grafting density, and surface poly(ethylene glycol) coating influence in vivo circulation and tumor targeting of hyaluronan-grafted liposomes. ACS Nano (2014) 8(6):5423–40. doi:10.1021/nn405839n
130. Peer D, Margalit R. Tumor-targeted hyaluronan nanoliposomes increase the antitumor activity of liposomal Doxorubicin in syngeneic and human xenograft mouse tumor models. Neoplasia (2004) 6(4):343–53. doi:10.1593/neo.03460
131. Schmaus A, Klusmeier S, Rothley M, Dimmler A, Sipos B, Faller G, et al. Accumulation of small hyaluronan oligosaccharides in tumour interstitial fluid correlates with lymphatic invasion and lymph node metastasis. Br J Cancer (2014) 111:1–9. doi:10.1038/bjc.2014.332
132. Orecchia P, Conte R, Balza E, Castellani P, Borsi L, Zardi L, et al. Identification of a novel cell binding site of periostin involved in tumour growth. Euro J Cancer (2011) 47(14):2221–9. doi:10.1016/j.ejca.2011.04.026
133. Tai IT, Dai M, Chen LB. Periostin induction in tumor cell line explants and inhibition of in vitro cell growth by anti-periostin antibodies. Carcinogenesis (2005) 26(5):908–15. doi:10.1093/carcin/bgi034
134. Lee YJ, Kim IS, Park SA, Kim Y, Lee JE, Noh DY, et al. Periostin-binding DNA aptamer inhibits breast cancer growth and metastasis. Mol Ther (2013) 21(5):1004–13. doi:10.1038/mt.2013.30
135. Liu G-X. Role of periostin and its antagonist PNDA-3 in gastric cancer metastasis. World J Gastroenterol (2015) 21(9):2605. doi:10.3748/wjg.v21.i9.2605
136. Silacci M, Brack SS, Späth N, Buck A, Hillinger S, Arni S, et al. Human monoclonal antibodies to domain C of tenascin-C selectively target solid tumors in vivo. Protein Eng Des Sel (2006) 19(10):471–8. doi:10.1093/protein/gzl033
137. Bourdon MA, Wikstrand CJ, Furthmayr H, Matthews TJ, Bigner DD. Human glioma-mesenchymal extracellular matrix antigen defined by monoclonal antibody. Cancer Res (1983) 43:2796–805.
138. Lee YS, Bullard DE, Zalutsky MR, Coleman RE, Wikstrand CJ, Friedman HS, et al. Therapeutic efficacy of antiglioma mesenchymal extracellular matrix 131I-radiolabeled murine monoclonal antibody in a human glioma xenograft model. Cancer Res (1988) 48(3):559–66.
139. Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, Herndon JE II, et al. Phase II trial of murine (131)I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol (2002) 20(5):1389–97. doi:10.1200/JCO.20.5.1389
140. Akabani G, McLendon RE, Bigner DD, Zalutsky MR. Vascular targeted endoradiotherapy of tumors using alpha-particle-emitting compounds: theoretical analysis. Int J Radiat Oncol Biol Phys (2002) 54(4):1259–75. doi:10.1016/S0360-3016(02)03794-X
141. Reardon DA, Zalutsky MR, Akabani G, Coleman RE, Friedman AH, Herndon JE II, et al. A pilot study: 131I-antitenascin monoclonal antibody 81c6 to deliver a 44-Gy resection cavity boost. Neuro Oncol (2008) 10(2):182–9. doi:10.1215/15228517-2007-053
142. Brack SS, Silacci M, Birchler M, Neri D. Tumor-targeting properties of novel antibodies specific to the large isoform of tenascin-C. Clin Cancer Res (2006) 12(10):3200–8. doi:10.1158/1078-0432.CCR-05-2804
143. Kim MY, Kim OR, Choi YS, Lee H, Park K, Lee CT, et al. Selection and characterization of tenascin C targeting peptide. Mol Cells (2012) 33(1):71–7. doi:10.1007/s10059-012-2214-4
144. Wyszko E, Rolle K, Nowak S, Zukiel R, Nowak M, Piestrzeniewicz R, et al. A multivariate analysis of patients with brain tumors treated with ATN-RNA. Acta Pol Pharm (2008) 65(6):677–84.
145. Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med (2010) 16(9):1009–17. doi:10.1038/nm.2208
146. Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med (2015) 7(8):1063–76. doi:10.15252/emmm.201404827
Keywords: extracellular matrix, collagen I, hyaluronan, periostin, tenascin C, metastasis
Citation: Venning FA, Wullkopf L and Erler JT (2015) Targeting ECM disrupts cancer progression. Front. Oncol. 5:224. doi: 10.3389/fonc.2015.00224
Received: 12 August 2015; Accepted: 30 September 2015;
Published: 20 October 2015
Edited by:
Jai Prakash, University of Twente, NetherlandsReviewed by:
Justin Lathia, Lerner Research Institute, USAKatarzyna Anna Rejniak, H. Lee Moffitt Cancer Center and Research Institute, USA
Copyright: © 2015 Venning, Wullkopf and Erler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Janine T. Erler, janine.erler@bric.ku.dk
†Freja A. Venning and Lena Wullkopf have contributed equally to this work.