 Sue Haupt
Sue Haupt Dinesh Raghu
Dinesh Raghu Ygal Haupt
Ygal Haupt- 1Tumour Suppression Laboratory, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
- 2Department of Pathology, The University of Melbourne, Parkville, VIC, Australia
- 3Sir Peter MacCallum Department of Oncology, The University of Melbourne, Parkville, VIC, Australia
- 4Department of Biochemistry and Molecular Biology, Monash University, Clayton, VIC, Australia
The tumor suppressor p53 normally acts as a brake to halt damaged cells from perpetrating their genetic errors into future generations. If p53 is disrupted by mutation, it may not only lose these corrective powers, but counterproductively acquire new capacities that drive cancer. A newly emerging manner in which mutant p53 executes its cancer promoting functions is by harnessing key proteins, which normally partner with its wild type, tumor-inhibiting counterpart. In association with the subverted activities of these protein partners, mutant p53 is empowered to act across multiple fundamental cellular pathways (regulating cell division and metabolism) and corrupt them to become cancer promoting.
Introduction
Reliance on the tumor suppressive capacity of p53 is profoundly emphasized by its near universal malfunction in all cancers. P53 is the most altered gene in cancer. More than 50% of human cancers are afflicted with a p53 mutation. Severe consequences of p53 mutation include the failure to protect against cancer stimuli, compounded by the acquisition of new cancer promoting, “neomorphic” properties, referred to as “Gain of function” (GOF), covered by other reviews in this series [reviewed in Ref. (1)].
A particularly sinister GOF constitutes the subversion by mutant p53, of molecular partners of wild type (wt) p53, and this strategy forms the focus of this review. Specifically, mutant p53 conscripts proteins that normally partner with wt p53. This new association divests them of their anticancer activities and in place, they are corrupted to act as promoters of tumorigenesis [e.g., Ref. (2)]. A number of fundamental cellular functions that are normally tumor suppressive under the directive of wt p53 become severely derailed under the influence of mutant p53 to promote cancer. Mutant p53 deregulates normally tightly controlled fundamental processes (including control of the mitotic cell cycle, glycolysis, nucleic acid, and lipid synthesis) to promote deregulated, proliferative cancer cell growth (Figure 1). Identifying the nature and the regulation of this mutant p53, GOF predicts therapeutic avenues for reining-in the impact of mutant p53 and fighting cancer.
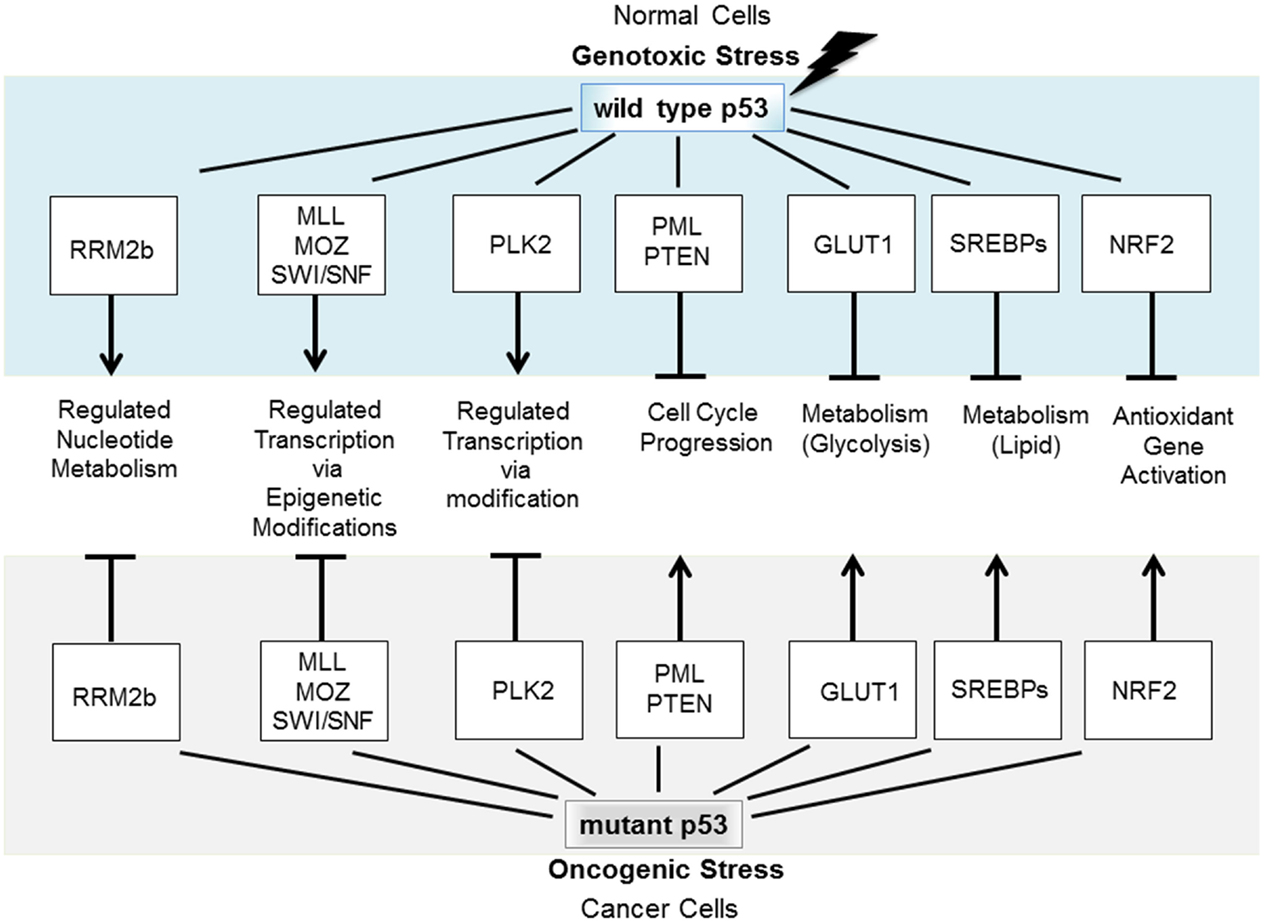
Figure 1. Wt p53 is induced to accumulate in response to stress to regulate fundamental cellular processes that protect against tumorigenesis. If p53 becomes mutated, it not only loses these tumor-protecting capacities but also may gain new functions through coercion of partner molecular partners normally engaged by wt p53.
Subversion of Cell Cycle Regulation
Promyelocytic Leukemia
Proper cell cycle regulation is vital for normal cell function. Equally critical is the capacity to sense DNA damage and to interrupt the cycle to instigate repair or eliminate cells with irreparable damage, as appropriate. Wt p53 is a key dictator of cellular fate in response to DNA damage resulting from cellular stresses. Partnership with the tumor suppressor promyelocytic leukemia (PML) protein facilitates p53 stress responses. Specifically, wt p53 stabilization and activation in response to stress is promoted by PML, through temporal co-recruitment of post-translational modifiers of p53 [kinases: CK1 (3), CK2 (4), HIPK2 (5); acetylases: CBP/p300 (6); MOZ (7)], to functional service depots, known as “PML nuclear bodies” (PML-NBs). PML-NBs facilitate the addition of post-translational modifications to p53, which relieve it from its normally labile state. Stabilized wt p53 accumulates, halts cell cycle progression, and initiates molecular responses to either repair DNA or direct the execution of incurable cells. PML in turn is a direct target of wt p53 transcriptional activation, which defines a positive regulatory loop (8). Further, PML-NBs associate with sites of active transcription and appear to facilitate gene expression (9). PML loss alone does not cause cancer [at least in mice (10)]; however, interference with its function may promote cancer, as consistent with its discovery in acute PML, where PML is fused with RAR-alpha to generate the oncogenic PML–RAR-alpha (11).
Significantly, mutant p53 enslavement of PML defines a paradigm for mutant p53 disruption of tumor suppressive partners of wt p53. We identified that when p53 is mutated in cancer cells, its association with PML is constitutive, unlike the transient association with its wt p53 counterpart in response to stress. Importantly, PML facilitates mutant p53 to aberrantly transcribe targets in the context of hijacked transcription factor NF-Y [(2), building on foundational NF-Y studies (12)].
More explicitly, wt p53 is a transcription factor that regulates its target genes (to control DNA repair, growth, and metabolic cascades), through direct engagement of its responsive elements. In stark contrast, mutant p53 is unable to directly engage these specific elements, but rather anchors onto other transcription factors and interferes with their transcription [including NF-Y (12)]. One transcriptional target of mutant p53 in association with NF-Y and PML is CDC25C, which triggers entry into mitosis (counteracting wt p53 activated growth arrest). Consistently, mutant p53 cancer cells may become growth dependent on PML, to the point where PML depletion leads to growth inhibition (2). Paradoxically, the capacity of PML to promote wt p53 as a tumor suppressor in healthy cells redefines PML as “oncogenic” when associated with mutant p53 in cancer cells [review in Ref. (13)].
At a higher level, cell cycle control is coordinated by the Circadian clock (14), and wt p53 defines a unique point of convergence between these two fundamental vital cellular regulatory systems. The Circadian clock is subject both to wt p53 (15) and PML (16) regulation and in turn regulates important cell cycle genes, including p21, independently of p53 (17) (Figure 2). While disruption of the diurnal periods of ~24 h appears insufficient alone to cause cancer, new findings suggest that it can exacerbate cancer progression [reviewed in Ref. (14)].
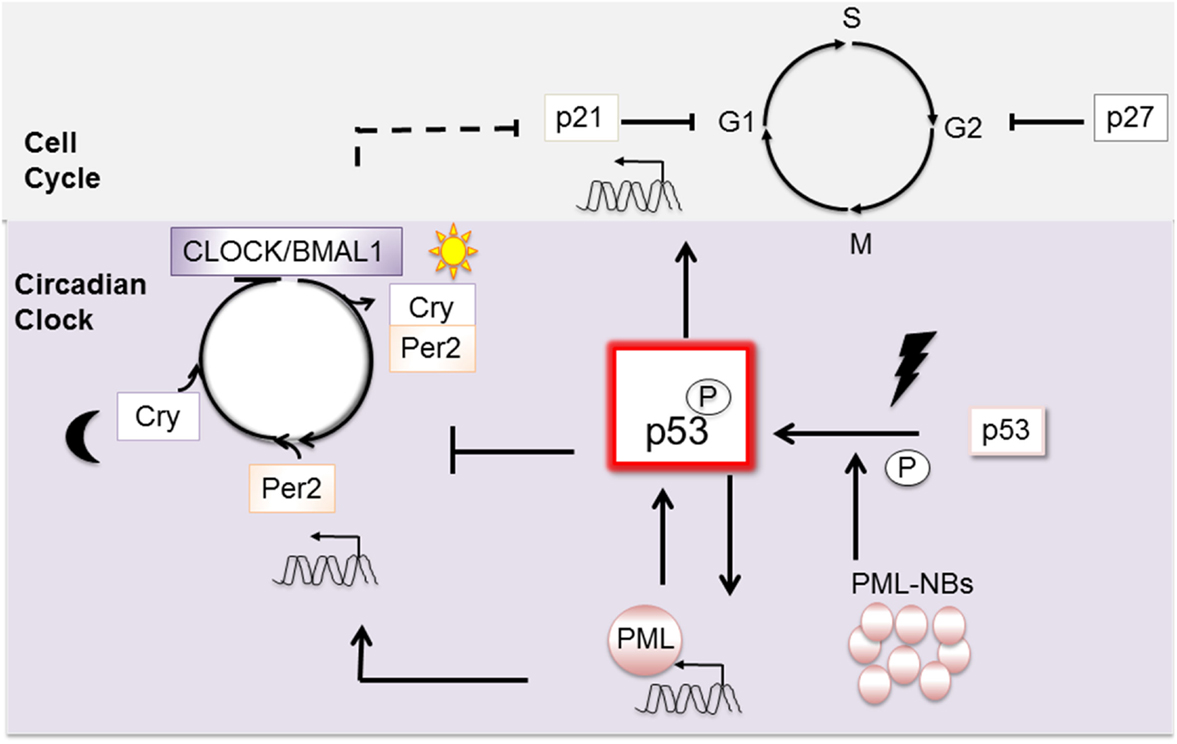
Figure 2. Wt p53 is a pivotal point of connection between the mitotic cell cycle and the circadian clock. P53 activation is promoted by its transcriptional target PML. Once activated, wt p53 intervenes in the cell cycle through expression of its target gene, the checkpoint inhibitor p21. Upon stimulation, wt p53 can also intervene to affect the circadian clock. In contrast, when p53 is mutated, its interaction with PML becomes constitutive. Cancer is exacerbated when Per is mutated on a background of p53 mutation.
At a molecular level, the clock is comprised of at least nine interplaying proteins, and we will discuss only those pertinent to this review. The clock is positively activated in a cyclic fashion through the combined activities of the two transcription factors: CLOCK and BMAL1 (Figure 2). As heterodimers, they engage E-Box motifs in the promoters of their target genes and induce transcription. Important transcriptional target genes, Per and Cry, and their protein products relocate to the nucleus and negatively regulate CLOCK and BMAL1: forming a negative feedback loop. To restart the cycle, a stimulus such as light (or pertinently to our discussion DNA damage) must prompt elimination of Per and Cry, which is mediated through proteolysis [reviewed in Ref. (18)].
Wt p53 controls the clock through negative regulation of Per2 expression (Figure 2). Mechanistically, wt p53 competes for a promoter region of Per2 normally occupied by activating CLOCK/BMAL1 (15). In normal healthy cells, p53 levels oscillate temporally and Per2 levels inversely correspond. In cells undergoing stress, wt p53 accumulation inhibits Per2 transcription. On a background of mutant p53, cancer is exacerbated by mutation of either the clock regulatory gene Per2 (19), or PML loss (20). The capacity of PML to function as an upstream regulator of Per2 is consistent with a common regulatory pathway (16). In sum, interplaying regulatory loops between p53, PML, the circadian clock, and the cell cycle are emerging, and their disruption has been linked to cancer in mouse models (19, 20). Links to human cancers are also emerging, with the possibility of sleep hormone therapies being trialed [i.e., melatonin (21)].
Phosphatase and Tensin homolog
Phosphatase and Tensin homolog (PTEN) is also a vital cell cycle regulator that has achieved its reputation as a tumor suppressor in the context of wild type (wt) p53. Pten curbs cell cycle progression and cell survival by suppressing PI3K–AKT/PKB cell survival pathway (22). PTEN functions as a tumor suppressor by stabilizing p53 protein in an Mdm2-dependent and/or -independent mechanism. (23). PTEN also increases the transcriptional activity of wt p53 through physical interaction (24). Reciprocally, wt p53 increases the transcription of PTEN by binding to the promoter of PTEN (25) and forming a feedback loop. These mutual relationships between PTEN and p53 promote tumor suppression.
In the context of mutant p53, in a diametrically opposing function, Pten promotes tumor growth (24). PTEN, in a comparable manner to PML, becomes oncogenic in cells expressing mutant p53 (26). PTEN stabilizes mutant p53 protein by inhibiting Mdm2-mediated degradation, which results in the inhibition of cell death and also in enhancement of cell proliferation (24). Additionally, PTEN increases the transcriptional activity of the mutant p53/acetylase CBP/NF-Y complex. This complex activates the transcription of c-Myc and Bcl-XL, which promotes cell survival and proliferation (26).
Polo-Like kinase-2
Polo-like kinase-2 (PLK2) is also a wt p53 target that contributes to cell cycle control. PLK2 is transcriptionally induced by wt p53 in response to the stress of DNA damage (27). PLK2 in a wt p53 setting is tumor suppressive, as engagement of p53 response elements in the promoter of PLK2 induces cell cycle arrest at the G2 checkpoint. In contrast, in a mutant p53 context, PLK2 functions as an oncogene. Distinct, indirect interaction between mutant p53 and PLK2, mediated through the conscription of the transcription factor NF-Y (to the CCAAT box promoter sequences), increases cell proliferation. A reinforcing feed back loop is created by PLK2 in turn phosphorylating mutant p53 on a site not phosphorylated on wt p53. Phosphorylated mutant p53 interacts more efficiently with p300 and promotes transcriptional activities of cell cycle activators (28). This feedback loop involving PLK2 defines a prototype cycle of reinforcement of mutant p53 GOF (29).
Diversion of Fundamental Cellular Pathways
Rapid cell proliferation inherent in cancer growth is utterly dependent on the ready supply of “molecular building blocks.” Recent studies have identified that fundamental metabolic processes normally regulated by wt p53 are extensively disrupted by mutant p53 to facilitate the supply of these necessities.
Nucleotide Metabolism
RRM2b
Proper repair of DNA damage is orchestrated by wt p53, which not only temporally halts cell cycle progression to allow repair, but also actively facilitates the supply of constituents for the repair. Specifically, in response to DNA damage, wt p53 transcriptionally activates the small subunit of the ribonucleotide reductase (RRM2b) in a temporary manner, to facilitate the catalytic conversion of ribonucleoside diphosphates to deoxyribonucleoside diphosphates, which is an essential step for DNA synthesis. In contrast, when p53 is mutated, it constitutively upregulates RRM2b expression. Importantly, the mechanism of transcriptional activation of RRM2b is dependent on the status of p53: where wt p53 engages its REs in the intronic region and in contrast mutant p53 localizes to the promoter. Further, it has emerged that mutant p53 transcriptionally drives additional nucleotide metabolic genes, both in the salvage and new synthesis pathways, through co-recruitment with the transcription factor ETS2, to its target gene promoters. ETS2 engagement by mutant p53 is a recurring theme, as we discuss below for epigenetic regulation. Overall, mutant p53 upregulates nucleotide biosynthesis, which contributes to meeting the voracious demands of rapidly proliferating and invading cancers (30).
Glucose Metabolism
Glucose Transporter 1
Regulated glucose metabolism is vital for maintaining healthy, normal cell homeostasis, in contrast to the voracious consumption of glucose that feeds cancer cell proliferation and is inherent in the “Warburg effect.” Proper glucose regulation is then an important tumor suppressive capacity of wt p53. Wt p53 regulates glucose metabolism by restricting cellular glucose at three levels through (31): (1) suppression of the expression of glucose transporter 1 (GLUT1) and 4 (32); (2) transcriptional regulation of target genes, which inhibit glycolysis [TIGAR (33)] and gluconeogenesis in the liver (34); and (3) direct binding and inhibition of the rate-limiting enzyme (glucose-6-phophate dehydrogenase) in an alternative anabolic pathway [the pentose phosphate pathway (35)].
Profoundly, when p53 is mutated, not only are these points of regulating glucose metabolism lost but further glucose uptake is accentuated through a novel GOF. This disastrous mutant p53 GOF is the shunting of the glucose transporter, Glut1, to the cell membrane surface where it stokes glucose uptake by cancer cells (36). Elevated glucose levels feed into metabolic anabolism to provide the increased demand for the molecular building blocks required to support rapid cancer cell proliferation, inherent in the Warburg effect. Reciprocally, glucose maintains mutant p53 stability and promotes cancer cell growth (37), generating a positive regulatory loop.
Reliance on a mutant p53-dependent enhanced supply of glucose to foster cell proliferation defines a unique point of vulnerability in cancer cells. This appetite for glucose identifies a potential therapy target which is currently being extensively investigated [i.e., ketogenic diets (38) and repurposing of the widely used diabetic metformin (39)].
Lipid Metabolism
Sterol Regulatory Element-Binding Proteins
A controlled supply of lipids is vital for regulated cell division and maintenance. Nearly every enzyme in the fatty acid and cholesterol synthesis are subject to regulation by the transcription factor of sterol regulatory element-binding proteins [SREBPs (40)]. Specifically, SREBP-1 dictates expression of lipogenic enzymes including fatty acid synthase, while SREBP-2 regulates cholesterol synthesis [reviewed in Ref. (41)]. In response to stress, consistent with halting cell division, wt p53 restrains lipid accumulation by inhibiting expression of the transcription factor SREBP-1, and in turn triglyceride synthesis, and lipogenic genes (41). In contrast, mutant p53 engages the SREBPs (both SREBP-1 and -2) directly. Mutant p53 is recruited to SREBP target gene promoters (although co-recruitment remains to be directly demonstrated). Mutant p53 appears to upregulate transcription of key enzymes in the sterol pathway and fatty acid biosynthesis pathway. Mutant p53 correlates with increased expression of enzymes in both the mevalonate synthesis (cholesterol) pathway and fatty acid synthesis pathways. Mutant p53 upregulation of these vital pathways is consistent with meeting increased demand for membrane lipids in rapidly proliferating cancer cells (42).
Antioxidant Pathways
Nuclear Factor Erythroid-Related Factor-2
Reactive oxygen intermediates perform important cellular functions including signaling; however, they are seriously damaging to normal cells if not properly contained and are linked to cancer [review in Ref (43)]. A master redox regulator is the transcription factor, nuclear factor erythroid-related factor-2 (NRF2) (44). P53 acts as a stress-rheostat controller of Nrf2 levels. Specifically, in response to mild stress, p53 transcriptionally activates the vital cell cycle inhibitor, p21, which binds to Nrf2 and consequently relieves it from its normal restraint (45). Relocation of NRF2 from the cytoplasm to the nucleus permits it to regulate multiple antioxidant targets, where some ~200 genes have been reported (44). These include the NADH-quinone oxidoreductase1 (NQO1), which also has differential function in a wt (46) versus mutant p53 context (47) (but will not be further elaborated here). When stress insults are severe, however, p53 inhibits Nrf2 (45). This exquisite level of control is consistent with p53 instigating repair in response to mild stress insults while intervening to prevent remedial action in those that are irrevocably damaged.
A novel GOF of mutant p53 is its capacity to reduce Nrf2 protein levels (without impacting its mRNA), in response to oxidative stress. The consequence is low levels of Nrf2 target detoxifying genes and elevated levels of reactive oxygen species (ROS). Remarkably, in contrast to growth inhibition imposed on wt p53 cells subject to oxidative stress, those with mutant p53 tolerate elevated ROS, survive, and proliferate (48).
Interference with Transcriptional Regulation
When p53 is mutated, a radical shift in transcriptional activity occurs, which is conducive to cancer promotion. An altered repertoire of transcription factor engagement is emerging for mutant p53. While mutant p53 is not able to directly engage wt p53 response elements, it may instead directly bind its wt counterpart and impose a dominant negative effect over wt p53 functions, including depriving it of capacity to regulate transcription. Mutant p53 may also engage transcription factors that wt p53 does not, including the family members p63 and p73 and disrupt their functions. More specifically, the presence of arginine at codon 72 dictates the capacity of mutant p53 to sequester p73, where mutants with proline are incapable of this inactivation (49, 50).
Beyond this negative regulation of wt p53 and its family members, mutant p53 may hijack transcription factor partners and disrupt their normal transcriptional activity (as mentioned above). Mutant p53 has been reported to engage NF-Y, NF-kappa B, SP1, E2F1, ETS1, ETS2, and SREBP. The outcome may be altered target engagement, or a change in the rate of transcription relative to a wt p53 context. These features of mutant p53 have been comprehensively reviewed recently (51), so we will concentrate on new findings.
SWI/SNF
At a higher level, mutant p53 disruption of chromatin regulation is also now emerging. In order for wt p53 to access specific DNA responsive elements in the regulatory regions (upstream promoters or introns) of its target genes, it must coordinate with numerous chromatin regulators to expose appropriate regulatory elements and associated DNA to be transcribed (52). Wt p53 exercises this activity in the context of components of the ATP-dependent nucleosomal remodeler SWI/SNF complex (53, 54). Mutant p53 has now also been identified to engage the SWI/SNF complex. However, in contrast to wt p53, mutant p53 is unable to directly engage wt p53 DNA response elements but rather localizes to distinct gene promoters through alternative transcription factors (as mentioned above). Through this co-recruitment, the SWI/SNF complex is predicted to facilitate more than 40% of all the genes transcribed by mutant p53 [where the primary example of altered regulation is the vascular endothelial growth factor receptor 2; VEGF2, which is vital for neoangiogenesis associated with oncogenesis (55)].
MLLs/MOZ
Mutant p53 can also alter transcriptional machinery, through distinct interactions from its wt counterpart. Wt p53 is able to physically interact through its core domain with the RNA polymerase II (POL II, large subunit) and limit target gene expression (56). In contrast, through engagement of the transcription factor ETS2 [as first demonstrated in Ref. (57)], mutant p53 is able to redirect POLII to transcribe the histone methyl transferases MLL1 and MLL2 and also acetyltransferase MOZ (58). This emphasizes the insidious capacity of mutant p53 to overpower fundamental transcriptional processes to support elevated proliferation. The newly emerging application of small molecule compounds to target chromatin regulators predicts application for cancers dependent on mutant p53. Specifically, cell growth inhibition of mutant p53 cancer has been demonstrated with prototoype inhibitors (58).
Conclusion
Corruption of the normal interactions between wt p53 and its molecular partners appears to lie at the heart of significant tumor promoting mutant p53 GOFs. Intriguingly, p53 mutations, which eliminate its function (e.g., deletion mutations), are rare, in contrast to the frequent activating missense mutations. To an extent, which appears unequaled by any other gene, mutation of p53 confers an exceptionally wide range of fundamental new properties that promote deregulated cell growth. These findings provide new insights directing innovative and rational approaches to therapeutically targeting cancers with mutant p53, which have proven particularly resistant to treatment. The polarized functions of these key p53 partners, would also caution that p53 status be an important criteria to consider prior to adoption of therapies directed toward these targets.
Author Contributions
SH wrote the paper and prepared the figures. DR contributed to writing the paper and to preparing a figure. YH contributed to discussion and editing the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work in the author lab is supported by NHMRC project grants (1049179 and 1063389), NHMRC Fellowship to YH (9628426), and by a grant from CCV (1085154).
References
1. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol (2010) 2(2):a001107. doi:10.1101/cshperspect.a001107
2. Haupt S, di Agostino S, Mizrahi I, Alsheich-Bartok O, Voorhoeve M, Damalas A, et al. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res (2009) 69(11):4818–26. doi:10.1158/0008-5472.CAN-08-4010
3. Alsheich-Bartok O, Haupt S, Alkalay-Snir I, Saito S, Appella E, Haupt Y. PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene (2008) 27(26):3653–61. doi:10.1038/sj.onc.1211036
4. Louria-Hayon I, Grossman T, Sionov RV, Alsheich O, Pandolfi PP, Haupt Y. The promyelocytic leukemia protein protects p53 from Mdm2-mediated inhibition and degradation. J Biol Chem (2003) 278(35):33134–41. doi:10.1074/jbc.M301264200
5. Moller A, Sirma H, Hofmann TG, Rueffer S, Klimczak E, Droge W, et al. PML is required for homeodomain-interacting protein kinase 2 (HIPK2)-mediated p53 phosphorylation and cell cycle arrest but is dispensable for the formation of HIPK domains. Cancer Res (2003) 63(15):4310–4.
6. Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature (2000) 406(6792):207–10. doi:10.1038/35018127
7. Rokudai S, Laptenko O, Arnal SM, Taya Y, Kitabayashi I, Prives C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc Natl Acad Sci U S A (2013) 110(10):3895–900. doi:10.1073/pnas.1300490110
8. de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, et al. PML is a direct p53 target that modulates p53 effector functions. Mol Cell (2004) 13(4):523–35. doi:10.1016/S1097-2765(04)00062-0
9. Kiesslich A, von Mikecz A, Hemmerich P. Cell cycle-dependent association of PML bodies with sites of active transcription in nuclei of mammalian cells. J Struct Biol (2002) 140(1–3):167–79. doi:10.1016/S1047-8477(02)00571-3
10. Bernardi R, Pandolfi PP. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene (2003) 22(56):9048–57. doi:10.1038/sj.onc.1207106
11. Pandolfi PP, Alcalay M, Fagioli M, Zangrilli D, Mencarelli A, Diverio D, et al. Genomic variability and alternative splicing generate multiple PML/RAR alpha transcripts that encode aberrant PML proteins and PML/RAR alpha isoforms in acute promyelocytic leukaemia. EMBO J (1992) 11(4):1397–407.
12. Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell (2006) 10(3):191–202. doi:10.1016/j.ccr.2006.08.013
13. Di Agostino S, Strano S, Blandino G. Gender, mutant p53 and PML: a growing “affaire” in tumor suppression and oncogenesis. Cell Cycle (2013) 12(12):1824–5. doi:10.4161/cc.25174
14. Tan XM, Ye H, Yang K, Chen D, Wang QQ, Tang H, et al. Circadian variations of clock gene Per2 and cell cycle genes in different stages of carcinogenesis in golden hamster buccal mucosa. Sci Rep (2015) 5:9997. doi:10.1038/srep09997
15. Miki T, Matsumoto T, Zhao Z, Lee CC. p53 regulates period2 expression and the circadian clock. Nat Commun (2013) 4:2444. doi:10.1038/ncomms3444
16. Miki T, Xu Z, Chen-Goodspeed M, Liu M, Van Oort-Jansen A, Rea MA, et al. PML regulates PER2 nuclear localization and circadian function. EMBO J (2012) 31(6):1427–39. doi:10.1038/emboj.2012.1
17. Grechez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem (2008) 283(8):4535–42. doi:10.1074/jbc.M705576200
18. Sahar S, Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nat Rev Cancer (2009) 9(12):886–96. doi:10.1038/nrc2747
19. Gu X, Xing L, Shi G, Liu Z, Wang X, Qu Z, et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ (2012) 19(3):397–405. doi:10.1038/cdd.2011.103
20. Haupt S, Mitchell C, Corneille V, Shortt J, Fox S, Pandolfi PP, et al. Loss of PML cooperates with mutant p53 to drive more aggressive cancers in a gender-dependent manner. Cell Cycle (2013) 12(11):1722–31. doi:10.4161/cc.24805
21. Jung-Hynes B, Huang W, Reiter RJ, Ahmad N. Melatonin resynchronizes dysregulated circadian rhythm circuitry in human prostate cancer cells. J Pineal Res (2010) 49(1):60–8. doi:10.1111/j.1600-079X.2010.00767.x
22. Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene (2008) 27(41):5443–53. doi:10.1038/onc.2008.241
23. Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell (2003) 3(2):117–30. doi:10.1016/S1535-6108(03)00021-7
24. Li Y, Guessous F, Kwon S, Kumar M, Ibidapo O, Fuller L, et al. PTEN has tumor-promoting properties in the setting of gain-of-function p53 mutations. Cancer Res (2008) 68(6):1723–31. doi:10.1158/0008-5472.CAN-07-1963
25. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of PTEN transcription by p53. Mol Cell (2001) 8(2):317–25. doi:10.1016/S1097-2765(01)00323-9
26. Huang X, Zhang Y, Tang Y, Butler N, Kim J, Guessous F, et al. A novel PTEN/mutant p53/c-Myc/Bcl-XL axis mediates context-dependent oncogenic effects of PTEN with implications for cancer prognosis and therapy. Neoplasia (2013) 15(8):952–65. doi:10.1593/neo.13376
27. Burns TF, Fei P, Scata KA, Dicker DT, El-Deiry WS. Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol Cell Biol (2003) 23(16):5556–71. doi:10.1128/MCB.23.16.5556-5571.2003
28. Valenti F, Fausti F, Biagioni F, Shay T, Fontemaggi G, Domany E, et al. Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle (2011) 10(24):4330–40. doi:10.4161/cc.10.24.18682
29. Haupt S, Haupt Y. Mutant p53 subverts PLK2 function in a novel, reinforced loop of corruption. Cell Cycle (2012) 11(2):217–8. doi:10.4161/cc.11.2.18977
30. Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun (2015) 6:7389. doi:10.1038/ncomms8389
31. Gottlieb E, Vousden KH. p53 regulation of metabolic pathways. Cold Spring Harb Perspect Biol (2010) 2(4):a001040. doi:10.1101/cshperspect.a001040
32. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res (2004) 64(7):2627–33. doi:10.1158/0008-5472.CAN-03-0846
33. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell (2006) 126(1):107–20. doi:10.1016/j.cell.2006.05.036
34. Wang YC, Lee HS, Chen SK, Yang SC, Chen CY. Analysis of K-ras gene mutations in lung carcinomas: correlation with gender, histological subtypes, and clinical outcome. J Cancer Res Clin Oncol (1998) 124(9):517–22. doi:10.1007/s004320050208
35. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol (2011) 13(3):310–6. doi:10.1038/ncb2172
36. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun (2013) 4:2935. doi:10.1038/ncomms3935
37. Rodriguez OC, Choudhury S, Kolukula V, Vietsch EE, Catania J, Preet A, et al. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle (2012) 11(23):4436–46. doi:10.4161/cc.22778
38. Paoli A, Rubini A, Volek JS, Grimaldi KA. Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur J Clin Nutr (2013) 67(8):789–96. doi:10.1038/ejcn.2013.116
39. Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med (2015) 66:17–29. doi:10.1146/annurev-med-062613-093128
40. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest (2002) 109(9):1125–31. doi:10.1172/JCI15593
41. Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, et al. p53 activation in adipocytes of obese mice. J Biol Chem (2003) 278(28):25395–400. doi:10.1074/jbc.M302364200
42. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell (2012) 148(1–2):244–58. doi:10.1016/j.cell.2011.12.017
43. Costa A, Scholer-Dahirel A, Mechta-Grigoriou F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol (2014) 25:23–32. doi:10.1016/j.semcancer.2013.12.007
44. Camp ND, James RG, Dawson DW, Yan F, Davison JM, Houck SA, et al. Wilms tumor gene on X chromosome (WTX) inhibits degradation of NRF2 protein through competitive binding to KEAP1 protein. J Biol Chem (2012) 287(9):6539–50. doi:10.1074/jbc.M111.316471
45. Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell (2009) 34(6):663–73. doi:10.1016/j.molcel.2009.04.029
46. Tsvetkov P, Reuven N, Shaul Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ (2010) 17(1):103–8. doi:10.1038/cdd.2009.67
47. Asher G, Lotem J, Tsvetkov P, Reiss V, Sachs L, Shaul Y. P53 hot-spot mutants are resistant to ubiquitin-independent degradation by increased binding to NAD(P)H:quinone oxidoreductase 1. Proc Natl Acad Sci U S A (2003) 100(25):15065–70. doi:10.1073/pnas.2436329100
48. Kalo E, Kogan-Sakin I, Solomon H, Bar-Nathan E, Shay M, Shetzer Y, et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J Cell Sci (2012) 125(22):5578–86. doi:10.1242/jcs.106815
49. Marin MC, Jost CA, Brooks LA, Irwin MS, O’Nions J, Tidy JA, et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet (2000) 25(1):47–54. doi:10.1038/75586
50. Bergamaschi D, Gasco M, Hiller L, Sullivan A, Syed N, Trigiante G, et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell (2003) 3(4):387–402. doi:10.1016/S1535-6108(03)00079-5
51. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol (2013) 15(1):2–8. doi:10.1038/ncb2641
52. Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ (2006) 13(6):951–61. doi:10.1038/sj.cdd.4401916
53. Lee D, Kim JW, Seo T, Hwang SG, Choi EJ, Choe J. SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. J Biol Chem (2002) 277(25):22330–7. doi:10.1074/jbc.M111987200
54. Xu Y, Zhang J, Chen X. The activity of p53 is differentially regulated by Brm- and Brg1-containing SWI/SNF chromatin remodeling complexes. J Biol Chem (2007) 282(52):37429–35. doi:10.1074/jbc.M706039200
55. Pfister NT, Fomin V, Regunath K, Zhou JY, Zhou W, Silwal-Pandit L, et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev (2015) 29(12):1298–315. doi:10.1101/gad.263202.115
56. Kim S, Balakrishnan SK, Gross DS. p53 interacts with RNA polymerase II through its core domain and impairs Pol II processivity in vivo. PLoS One (2011) 6(8):e22183. doi:10.1371/journal.pone.0022183
57. Do PM, Varanasi L, Fan S, Li C, Kubacka I, Newman V, et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev (2012) 26(8):830–45. doi:10.1101/gad.181685.111
Keywords: p53 mutations, gain of function, metabolism, cell cycle, transcriptional regulation
Citation: Haupt S, Raghu D and Haupt Y (2016) Mutant p53 Drives Cancer by Subverting Multiple Tumor Suppression Pathways. Front. Oncol. 6:12. doi: 10.3389/fonc.2016.00012
Received: 18 November 2015; Accepted: 12 January 2016;
Published: 27 January 2016
Edited by:
Carlotta Giorgi, University of Ferrara, ItalyReviewed by:
Mariusz R. Wieckowski, Polish Academy of Sciences, PolandGeert Bultynck, KU Leuven, Belgium
Copyright: © 2016 Haupt, Raghu and Haupt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sue Haupt, sue.haupt@petermac.org