 Menglin Cheng1
Menglin Cheng1 Zaver M. Bhujwalla
Zaver M. Bhujwalla Kristine Glunde
Kristine Glunde- 1Division of Cancer Imaging Research, Russell H. Morgan Department of Radiology and Radiological Science, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 2Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
All cancers tested so far display abnormal choline and ethanolamine phospholipid metabolism, which has been detected with numerous magnetic resonance spectroscopy (MRS) approaches in cells, animal models of cancer, as well as the tumors of cancer patients. Since the discovery of this metabolic hallmark of cancer, many studies have been performed to elucidate the molecular origins of deregulated choline metabolism, to identify targets for cancer treatment, and to develop MRS approaches that detect choline and ethanolamine compounds for clinical use in diagnosis and treatment monitoring. Several enzymes in choline, and recently also ethanolamine, phospholipid metabolism have been identified, and their evaluation has shown that they are involved in carcinogenesis and tumor progression. Several already established enzymes as well as a number of emerging enzymes in phospholipid metabolism can be used as treatment targets for anticancer therapy, either alone or in combination with other chemotherapeutic approaches. This review summarizes the current knowledge of established and relatively novel targets in phospholipid metabolism of cancer, covering choline kinase α, phosphatidylcholine-specific phospholipase D1, phosphatidylcholine-specific phospholipase C, sphingomyelinases, choline transporters, glycerophosphodiesterases, phosphatidylethanolamine N-methyltransferase, and ethanolamine kinase. These enzymes are discussed in terms of their roles in oncogenic transformation, tumor progression, and crucial cancer cell properties such as fast proliferation, migration, and invasion. Their potential as treatment targets are evaluated based on the current literature.
Introduction
Phospholipids, which form the bilayer structures of all cellular membranes, are an essential component of all cells. The phospholipid content was shown to increase with cell transformation and tumor progression (1–4). For example, lipid analyses of samples from breast cancer tissue displayed an increase in phospholipid content as compared to non-cancerous adjacent healthy breast tissue (5). Concentrations of the two major phospholipid components phosphatidylcholine (PtdCho) and phosphatidylethanolamine (PtdEtn) increased with increasing breast cancer tumor grade (6), indicating that the phospholipid synthesis rate increases with oncogenesis and tumor progression as compared to normal tissue.
Metabolic intermediates of PtdCho metabolism were consistently observed to display abnormal levels with magnetic resonance spectroscopy (MRS) (7). An increased total choline (tCho) signal, which consists of signals from phosphocholine (PC), glycerophosphocholine (GPC), and free choline (Cho), and whose chemical structures are shown in Figure 1, has been detected by 1H MRS in all cancers tested so far (7). High-resolution 1H MRS, which can resolve the signals from the individual components of the tCho signal, confirmed the increase of PC in breast (8–12), prostate (13, 14), ovarian (15), endometrial (16), cervical (15), and brain cancers (17–19). In some cancer cell types such as isolated breast and ovarian cancer cells, a relative decrease of GPC was observed as well, which led to the suggestion to use a higher ratio of PC/GPC as a marker of tumor progression (20, 21). PC was also reported as a useful imaging biomarker of tumor response to several targeted treatments of cancers, enabling 1H MRS to be used for monitoring changes in PC as a non-invasive marker of pharmacodynamic treatment response (22–26). A PC decrease and GPC increase was reported after docetaxel treatment in breast cancer cell lines and xenograft models (27).
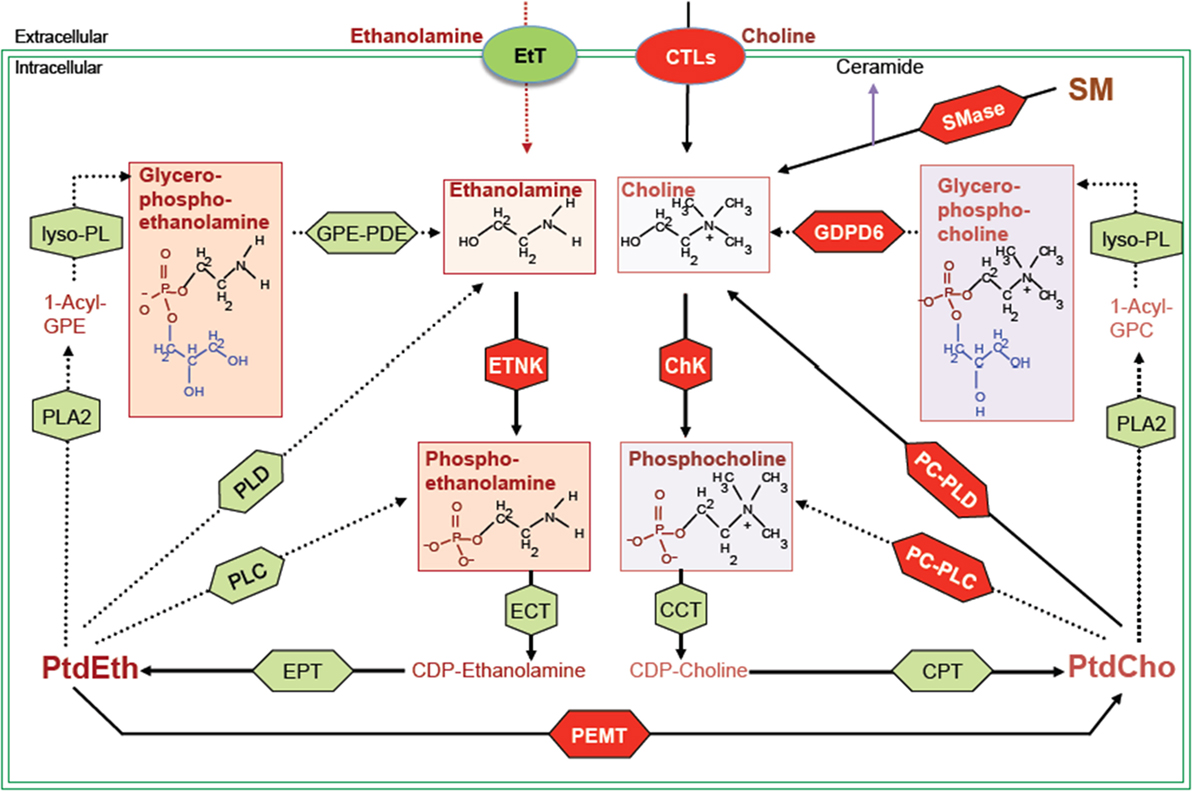
Figure 1. Biochemical network of ethanolamine and choline phospholipid metabolism showing crosstalk between these two metabolic cycles. MR detectable metabolites are drawn in boxes. Red shapes show enzymes discussed in this review, and green shapes show other enzymes responsible for the indicated reactions, but not discussed in this review. Metabolite abbreviations: 1-acyl-GPE, 1-acyl-glycerophosphoethanolamine; 1-acyl-GPC, 1-acyl-glycerophosphocholine; PtdCho, phosphatidylcholine; PtdEth, phosphatidylethanolamine; SM, sphingomyelin. Enzyme abbreviations: CCT, phosphocholine cytidylyltransferase; ChK, choline kinase; CPT, diacylglycerol cholinephosphotransferase; CTL, choline transporter-like protein; EtT, ethanolamine transporter; ECT, phosphoethanolamine cytidylyltransferase; EPT, diacylglycerol ethanolaminephosphotransferase; ETNK, ethanolamine kinase; GPE-PDE, glycerophosphoethanolamine phosphodiesterase; GDPD6, glycerophosphodiester phosphodiesterase domain containing 6; lyso-PL, lysophospholipase; PEMT, phosphatidylethanolamine N-methyltransferase; PLA2, phospholipase A2; PLC, phospholipase C; PLD, phospholipase D.
Magnetic resonance spectroscopy detection of tCho allows for non-invasive and longitudinal monitoring of deep-seated tumors, and it is currently being evaluated as non-invasive biomarker for cancer diagnosis, monitoring of treatment response, and anticancer drug development. In clinical studies, in vivo MRS detection of the tCho signal was proposed as a marker of breast cancer malignancy (8, 28–31). The tCho signal has been used to monitor neoadjuvant chemotherapy of breast tumors in patients (32), and decreased tCho was associated with the pathology-detected tumor response to chemotherapy (10, 12, 33). One limitation of using choline-based 1H MRS is the difficulty of resolving the signals of PC and GPC and free choline in the in vivo 1H MRS setting.
Phosphorus MRS (31P MRS) is also able to detect phospholipid metabolites and displays two sets of phospholipid metabolite peaks: phosphomonoesters (PMEs), which are mainly composed of PC and PE (phosphoethanolamine), and diphosphodiesters, with GPC and GPE (glycerophosphoethonolamine) as the main components as shown in Figure 2. Early 31P MRS cancer studies, which were not able to separate the individual components of these two sets of 31P peaks very well, detected that PMEs were increased in human breast tumors (34–37), neuroblastomas (38), prostate cancers (39), and hepatic lymphomas (40) as compared to corresponding normal tissues. Following chemotherapy or radiotherapy, a PME reduction was observed in human neuroblastoma (38), breast tumors (35, 36), and hepatic lymphoma (40). Recent developments of innovative 31P MRS approaches that are based on 1H to 31P polarization transfer enable the quantification of in vivo levels of PE, PC, GPE, and GPC on small animal and clinical MR scanners (41–44). PE and GPE are the metabolic intermediates of PtdEtn, the second most abundant phospholipid. PE was found to consistently increase in tumors similar to PC (see Figure 2) (45), although its role in cancers is much less explored than PC. The potential of PE and GPE to be monitored is currently being explored along with PC and GPC (41, 44). The changes that occur in tCho, PMEs, and PDEs with oncogenesis and in response to therapy are the result of complex molecular pathways and are therefore not always consistent (8, 37, 46, 47), making it necessary to improve the resolution of clinical in vivo MRS applications and to investigate the molecular mechanism underlying these metabolic changes.
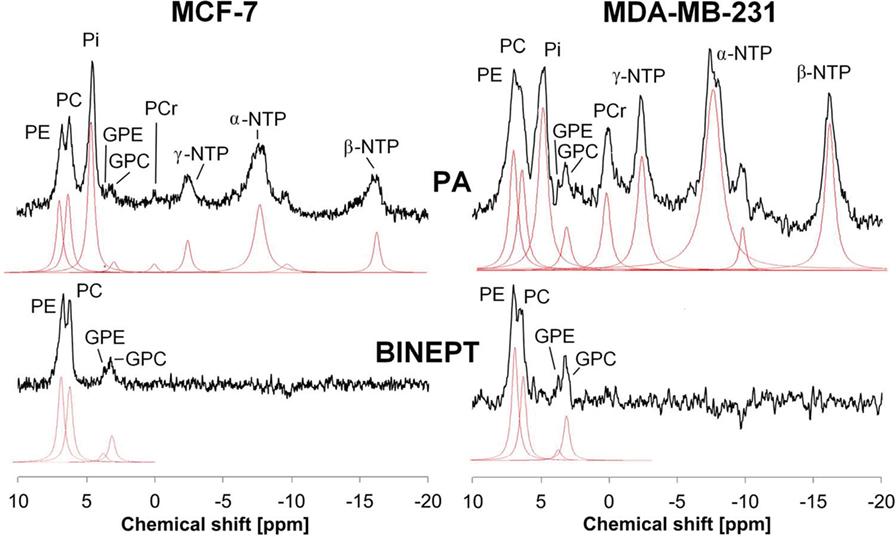
Figure 2. Example of in vivo pulse-acquire (PA, top) and BINEPT (bottom) 31P MR spectra of a representative MCF-7 (left) and MDA-MB-231 (right) tumor. Lorentzian lines as fitted by the software jMRUI (http://www.jmrui.eu/) are shown below each MR spectrum. All phosphorylated metabolites are visible in the PA spectrum, whereas the BINEPT spectrum only contains signals from phospholipid metabolites with H-P-coupling such as phosphoethanolamine (PE), phosphocholine (PC), glycerophosphoethanolamine (GPE), and glycerophosphocholine (GPC). Note the broad, uneven baseline in the 0–5 ppm region of the PA spectra, where signals from mobile membrane phospholipids are resonating. The signal of β-nucleoside triphosphate (NTP) is formed by β-NTP only. The signal labeled α-NTP is an overlapping signal from α-NTP, α-nucleoside diphosphate (α-NDP), nicotinamide adenine diphosphate, and diphosphodiesters. The signal labeled γ-NTP is an overlapping signal from γ-NTP and β-NDP. Typically, β-NTP is the smallest peak of the three NTP signals; however, here, γ-NTP overlaps with a broad baseline signal that makes it appear smaller than β-NTP. Adapted from Wijnen et al. (44).
Choline uptake and retention can also be imaged using positron emission tomography (PET), mainly with the tracers [11C]-choline, [18F]-fluoromethylcholine, and [18F]-fluoroethylcholine. The use of [11C]-choline PET was approved for clinical use in prostate cancer by the federal drug administration of the United States in 2012 (48). Choline PET/computed tomography (CT) is used in the clinic as well and has the advantages of providing improved local disease evaluation and staging of prostate cancer as compared to conventional [18F]-FDG PET, giving additional information on nodal staging and suspected metastasis in prostate cancer patients (49).
While choline-based imaging has been explored extensively, discoveries of genes and signaling pathways leading to the changes in choline-containing metabolites are still emerging. The function and regulation of the two enzymes choline kinase α (ChKα) and phospholipase D (PLD) has been more widely explored in cancer, but research into other important enzymes in choline metabolism is still at an early stage. These enzymes may provide new targets for cancer therapy. In this review, we will provide a brief update on the established targets in choline phospholipid metabolism and discuss some new anticancer targets in the choline and ethanolamine phospholipid metabolic pathways as highlighted in red in Figure 1.
Choline Kinase α
Choline kinase is the enzyme that phosphorylates free choline and produces PC. There are two genes that encode this enzyme: ChKα and choline kinase beta (ChKβ) (50–52). ChKα is well established as an oncogene that promotes tumor initiation and progression (7, 53, 54). Its overexpression and elevated enzyme activity is one of the most established factors that contribute to increased PC and tCho in various tumor tissues including breast (55), ovarian (21), colorectal (56), prostate (56), lung (56, 57), endometrial (16), and pancreatic (58) cancer.
In addition to catalyzing the formation of PC in the Kennedy pathway of PtdCho synthesis, ChKα also has other functions in regulating cell signaling pathways. For example, the non-receptor tyrosine kinase c-Src was shown to phosphorylate ChKα, which in turn forms a protein complex with epidermal growth factor receptor to regulate breast cancer cell proliferation and tumorigenesis (59). Downregulation of ChKα decreased the phosphorylation of the mitogen-activated protein kinase (MAPK) signaling pathway protein ERK1/2 to p-ERK1/2 on T202/Y204 (60), and the PI3K/AKT signaling pathway protein AKT to p-AKT on S473 (61). ChKα inhibition induced exacerbated endoplasmic reticulum (ER) stress and triggered apoptosis via the CHOP pathway in cancer cells, but not in the non-tumorigenic mammary epithelial cell line MCF-10A (62). CHOP is the major pro-apoptotic transcription factor that is induced by ER stress. Inhibition of ChKα in cancer cells also resulted in an intracellular increase in ceramides, leading to apoptotic cell death, suggesting that ChKα activity disruption may be a highly specific and selective cytotoxic antitumoral strategy. ChKα knockout mice, but not knockout mice with a loss of ChKβ, are embryonically lethal, implying that ChKα is essential for PtdCho biosynthesis (7, 63, 64).
Several small molecule inhibitors of ChKα- and siRNA-based nanoparticles have been developed to test their potential for MRS-monitored anticancer treatment. Lacal and colleagues have tested several groups of compounds that inhibit ChKα activity, some of which displayed significant levels of antiproliferative activity and led to a reduction of tumor growth (65–67). One of these inhibitors, TCD-717 (68), is currently being tested in a phase I clinical trial (https://clinicaltrials.gov/ct2/show/NCT01215864). Bhujwalla and colleagues have developed siRNA and shRNA strategies (Figure 3) (69, 70), and siRNA-based nanoparticles against ChKα (71) for anticancer therapy. Both methods of therapy can be monitored by detecting ChKα activity with MRS (22, 69, 70, 72). siRNA silencing of ChKα demonstrated synergistic effects with 5-fluorouracil (5-FU) in the treatment of breast cancer cells (73), and ChKα inhibitors showed similar effects in preclinical studies of treating colorectal cancer (74). Overall, ChKα is a well-established antitumor target, with multiple inhibitors and siRNA-based agents against ChKα under development and in initial clinical testing phases. It is possible that ChKα may enter clinical application as stand-alone treatment or in combination with other first-line chemotherapeutic drugs such as 5-FU. MRS monitoring of tumor tCho or PME levels in patients undergoing treatment with ChKα-targeted drugs may facilitate non-invasive assessment of tumor response for more precise treatment monitoring and dynamic adjustment of the treatment plan.
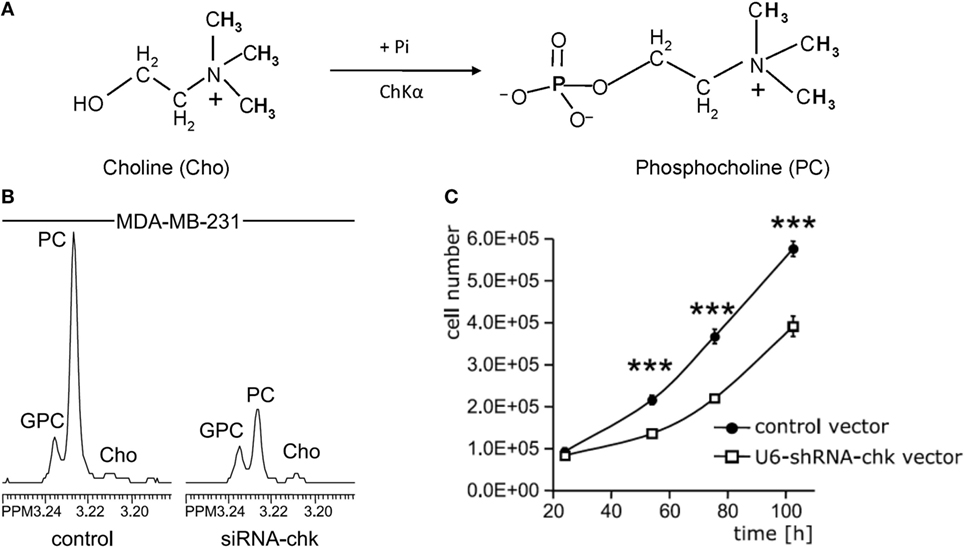
Figure 3. Choline kinase α (ChKα) as target for anticancer treatment. (A) ChKα converts free choline to phosphocholine (PC). (B) Knockdown of ChKα resulted in a dramatic reduction of cellular PC as evident by the decreased PC signal in high-resolution 1H MR spectrum obtained from water-soluble metabolites of MDA-MB-231 cells transfected with anti-ChKα siRNA compared to the spectrum from non-target siRNA control. (C) Stable knockdown of ChKα with a pSHAG-U6-shRNA vector significantly decreased MDA-MB-231 cell growth rate. Abbreviations: GPC, glycerophosphocholine. ***represents P < 0.001, average proliferation rates were calculated from five independent experiments. Adapted from Glunde et al. (69).
PtdCho-Specific Phospholipase D (PLD)
The phospholipase D enzyme hydrolyzes the most abundant phospholipid PtdCho and thereby produces phosphatidic acid (PA) and free choline (75). PA is a second messenger that directly binds to the mammalian target of rapamycin (mTOR) (76). PA can also be converted to other secondary messengers such as diacylglycerol (DAG) by lipid phosphatidate phosphatase, or to lysophosphatidic acid (LPA) by phospholipase A2 (75). There are two isoforms of PLD in the human genome: phospholipase D1 (PLD1) and PLD2, which share 50% amino acid identity and display a similar protein structure (77, 78). PLD1, PLD2, and PA have been reported to interact with several protein partners, forming a molecular network that mediates various cellular functions (79). These cellular functions include vesicle trafficking (endocytosis and exocytosis), trans-Golgi network vesicle formation, cytoskeleton organization, cell migration, cell morphogenesis, autophagy (80), and the regulation of growth, proliferation, survival, and apoptosis (81).
Elevated PtdCho-specific PLD activity and/or increased expression of PLD1 or PLD2 have been observed in many different types of cancer and in transformed cells. A single-nucleotide polymorphism without protein change in PLD1 was reported to increase the risk of small cell lung cancer (82). A polymorphism in PLD2 is associated with the prevalence of colorectal cancer (83). Overexpression of either PLD1 or PLD2 in murine fibroblasts alters their cell growth, confers anchorage-independent growth ability in soft agar, and sarcoma formation in nude mice (84). Genetically enforced overexpression of PLD1 or PLD2 in a human breast cancer xenograft model led to primary tumor initiation and increased metastasis (85). PLD activity was shown to confer rapamycin resistance in human breast cancer cells (86). PLD activity was also demonstrated to couple survival and migration signals in human cancer cells to stress response (87). PLD1 and ChKα are interactive, such that ChKα silencing increases PLD1 expression and PLD1 silencing increases ChKα expression (88). The contributions of PLD to cancer have been summarized by Bruntz et al. as sustaining proliferative signaling, transducing MAPK signaling, regulating mTOR, evading growth suppression, resisting apoptosis- or autophagy-mediated cell death, activating invasion and metastasis, inducing angiogenesis, and deregulating cellular energetics (81).
Targeting PLD for anticancer therapy has been extensively explored. siRNA silencing or genetic ablation of either PLD1 or PLD2 were demonstrated to reduce cell proliferation, cell migration, and/or xenograft growth (85, 89). In ApcMin/+ and azoxymethane/dextran sodium sulfate (AOM/DSS) mouse models, spontaneous and colitis-associated intestinal tumorigenesis was reported to be disrupted by genetic or pharmacological targeting of PLD1, but not PLD2 (90). This was the case because PLD1 inactivation suppressed the self-renewal capacity of colon cancer-initiating cells, which in turn decreased the expression of β-catenin (90). The combination of PLD1 and autophagy inhibition was shown to synergize in inducing tumor cell apoptosis and tumor regression, providing a potential rational target for anticancer therapy (91). In earlier studies, short-chain primary alcohols were used as PLD inhibitor, but concerns were raised about insufficient suppression of PA production and off-target effects (92). Several PLD-specific inhibitors have been screened and evaluated in preclinical studies (81, 92, 93). 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a dual inhibitor of PLD1 and PLD2 (93), has shown anticancer potential. Delivering FIPI with an osmotic pump to a xenograft model of breast cancer in mice was able to inhibit primary tumor growth and reduce metastatic growth of nodules in the axillary lymph nodes and lungs (85). Tumors grew more slowly in FIPI-treated mice and showed a significant decrease in microvessel density as compared to non-treated control mice (94). Later studies have explored the anticancer properties of isoform-selective inhibitors of PLD1 or PLD2 (95). The PLD1 selective inhibitor VU0155069 was shown to attenuate intestinal tumorigenesis in the ApcMin/+ and AOM/DSS mouse models (90). Both PLD1 and PLD2 selective inhibitors were found to enhance the radiosensitivity of breast cancer cells (96).
The generation of PLD1-deficient transgenic mice provided the opportunity of studying the role of PLD1 in the tumor microenvironment. Tumor xenografts growing in Pld1−/−, but not in Pld2−/−, mice were demonstrated to have limited primary tumor growth and reduced lung metastasis as shown in Figure 4 (94). Further examination showed that vascular endothelial cells of PLD1-deficient mice displayed reduced activities in signaling pathways that affect vascular endothelial growth factor as a downstream target (94). These signaling pathways were mediated by reduced phosphorylation of Akt, the MAPK proteins ERK1/2, and P38, leading to decreased integrin-dependent cell adhesion to and migration on extracellular matrices, as well as reduced tumor angiogenesis (Figure 4B) (94). Partly mediated by impaired activation of αIIbβ3 integrin in platelets, tumor cell–platelet interactions decreased in Pld1−/− mice, resulting in reduced seeding of tumor cells into the lung parenchyma (Figure 4C) (94). Small molecule inhibitors of PLD1 displayed similar effects on tumors as the genetic ablation of PLD1 in the host mice (94).
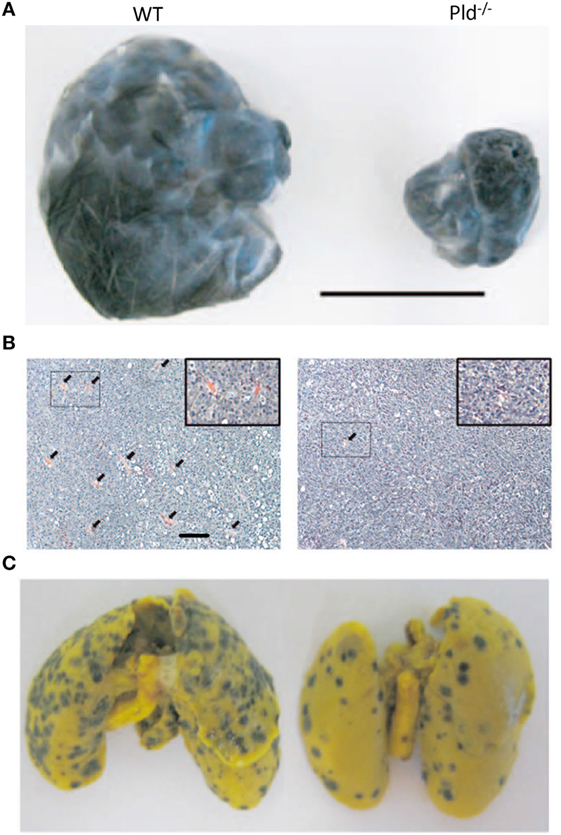
Figure 4. Phospholipase D1 activity in the tumor microenvironment is important for tumor growth and metastasis. (A) Representative picture showing that melanoma xenografts grown in Pld−/− mice have limited growth volume compared to that growing in wild-type (WT) mice. Scale bar, 1 cm. (B) Melanoma xenografts growing in Pld−/− mice have fewer blood vessels as compared to those grown in wild-type mice, as evident from H&E stains of the respective tumor sections. Arrowheads in panel (B) indicate blood vessels. Scale bar, 50 µm. (C) For melanoma xenografts, lung metastasis has been significantly lower in lungs of Pld−/− mice than in lungs of wild-type mice. Adapted from Chen et al. (94).
Both Pld1−/− and Pld2−/− mice developed overtly normal (80, 97, 98), indicating that PLD1 inhibitors may have low toxicity. Although the existing small molecule inhibitors against PLD1 only impair cell migration and under-perform in killing cancer cells or reducing the proliferation rate of cancer cells as a stand-alone treatment, they may synergize well with other therapy approaches in combatting cancers, due to PLD1’s multiple roles in important cell functions. Moreover, PLD1’s role in the tumor microenvironment, which confers the ability to disrupt tumor angiogenesis in some studies (94), makes it an attractive target. Although no PLD1 inhibitors have entered into clinical trials for anticancer therapy yet, promising results emerging from recent preclinical studies may pave the way for PLD1 inhibitors into clinical studies in the near future.
Phosphatidylcholine-Specific Phospholipase C (PtdCho-PLC)
Phosphatidylcholine-specific phospholipase C cleaves the PC moiety from PtdCho, thereby generating and releasing DAG and PC into cytosol. DAG is an important second messenger that activates protein kinase C and induces mitogenic signal transduction. Considering the large abundance of PtdCho, it is possible that PtdCho-PLC hydrolysis of phospholipids can produce a more sustained DAG elevation than phosphatidylinositol-specific PLC cleavage of phosphatidylinositol (99). The sustained mitogenic signal produced by PtdCho-PLC generated DAG may lead to cell transformation. Indeed, NIH3T3 fibroblasts stably transfected with bacterial PtdCho-PLC displayed chronically elevated levels of DAG and PC, and a transformed phenotype that was characterized by anchorage-independent growth in soft agar, formation of transformed foci in tissue culture, and loss of contact inhibition (100). Increased PtdCho-PLC enzyme activity was observed in ovarian cancer cells (101, 102), breast cancer cells (103), and squamous cell carcinoma cells (104) as compared to the corresponding non-malignant, immortalized cells. PtdCho-PLC selectively accumulated in raft domains in the plasma membrane of HER2-overexpressing breast cancer cells, in which it colocalized with HER2 (105).
Since the genes encoding PtdCho-PLC have not yet been cloned from mammalian genomes, it is currently not possible to genetically ablate PtdCho-PLC activity, and mechanistic PtdCho-PLC studies have to rely on small molecule inhibitors of PtdCho-PLC, such as tricyclodecan-9-yl-potassium xanthate (D609). D609 competitively inhibits PtdCho-PLC activity and was shown to confer antiviral and antitumor effects (106). Inhibition of PtdCho-PLC with D609 reduced the cellular PC content and blocked cell proliferation in human ovarian cancer cells (102). D609 treatment of human MDA-MB-231 breast cancer cells led to a loss of mesenchymal traits, such as decreased Vimentin and N-Cadherin expression, and a changed cell morphology more similar to mature breast epithelial cells, as well as growth arrest and reduced migration and invasion potential (103). PtdCho-PLC inhibition with D609 also resulted in enhanced HER2 internalization and lysosomal degradation, strong retardation of HER2 re-expression on the membrane, and reduced HER2 content in HER2-overexpressing breast cancer cells, suggesting that PtdCho-PLC inhibitors may be potential candidates for the treatment of HER2-amplified or trastuzumab-resistant breast cancers (105). D609 treatment of the two squamous cell carcinoma cell lines A431 and CaSki, which display different stemness potential, led to reduced cell proliferation, decreased sphere-forming efficiency, and down-modulated mRNA levels of stemness-related markers (104). Although the mammalian PtdCho-PLC genes have not yet been identified, it is evident that PtdCho-PLC activity is important in cell transformation and cancer progression, and that inhibiting PtdCho-PLC activity may have potential as an anticancer strategy. Efforts should focus on cloning mammalian PtdCho-PLC gene(s) and identifying compounds that inhibit PtdCho-PLC and are suitable for clinical application.
Sphingomyelinases (SMAs)
Sphingomyelin is a sphingosine-based phospholipid that exists in cell membranes with a typical mole percentage of around 10–20 (107). Sphingomyelin typically contains a PC or sometimes a PE headgroup similar to glycerol-based phospholipids. SMAs are enzymes that act analogous to phospholipase C, release PC or PE from sphingomyelin, and thereby generate ceramide (108). According to the pH for their optimum enzyme function, they are classified as acidic, neutral, and alkaline SMAs (109).
The importance of acid sphingomyelinase (ASM) function in tumor irradiation has been well established with genetic studies (110). Niemann–Pick patients have an inherited deficiency of ASM activity, which results in their lymphoblasts failing to respond to ionizing radiation with ceramide generation and apoptosis (110). ASM knockout mice also displayed failure of radiation-induced ceramide generation and apoptosis in vivo (110). MCA/129 fibrosarcomas and B16F1 melanomas grown in ASM-deficient mice grow much faster than in control mice and showed reduced endothelial apoptosis upon irradiation (111). When these cells were grown in severe combined immunodeficient mice with ASM-deficiency, the xenografts acquired resistance to single-dose radiotherapy (112), implying that ASM-mediated, radiotherapy-induced apoptosis occurs both in cancer cells and in the tumor microenvironment. Production of ceramide after tumor irradiation or treatment with chemotherapeutic agents, including commonly used daunorubicin, doxorubicin, cisplatin, paclitaxel, gemicitabine, among others, is a well-known treatment response (113). The production of ceramide through hydrolysis of sphingomyelin by ASM following chemotherapy is an important mechanism of cellular ceramide increase which leads to tumor cell apoptosis, as siRNA knockdown of ASM prevents cancer cell death induced by cisplatin (114) or gemcitabine (115, 116).
The molecular mechanism by which ASM promotes cell apoptosis is mainly through its ceramide-producing ability. Moreover, the formation of ceramide-enriched lipid domains can trap and cluster specific membrane receptors such as CD95 and intracellular signaling molecules, and thus, facilitate cellular signal transduction, which leads to apoptosis, autophagic responses, and cell cycle arrest (114, 117, 118). To improve the therapeutic efficacy of ceramide-producing chemotherapy, further increasing the ceramide generation in cancer cells through activation of SMAs may be a worthwhile strategy to enhance anticancer treatment. However, it is generally hard to activate protein function. On the contrary, most rational drug design strategies inhibit rather than activate protein function. Based on this reasoning, ASM does not seem to be a suitable anticancer target at first sight. However, on the other hand, ASM-deficient human patients suffering from Niemann–Pick disease have cells with lysosomal storage disorders and significantly decreased lysosomal stability (119), suggesting that ASM is essential for maintaining normal lysosomal integrity. To escape from apoptosis, tumor cells, especially therapy resistant cancer cells, probably decrease their ASM activity to a critically low level, making them more sensitive than normal cells to ASM inhibition that can lead to lysosomal destabilization (120). Siramesine and several clinically relevant ASM inhibitors were found to trigger cancer-selective lysosomal cell death, displaying great anticancer potential as evident by reducing tumor growth in vivo, and reverting multidrug resistance (Figure 5) (121). So the strength of escaping from apoptosis by decreasing ASM levels becomes a weakness of cancers, through which cancer-specific killing agents can be developed especially for malignant and multidrug-resistant cancers (122).
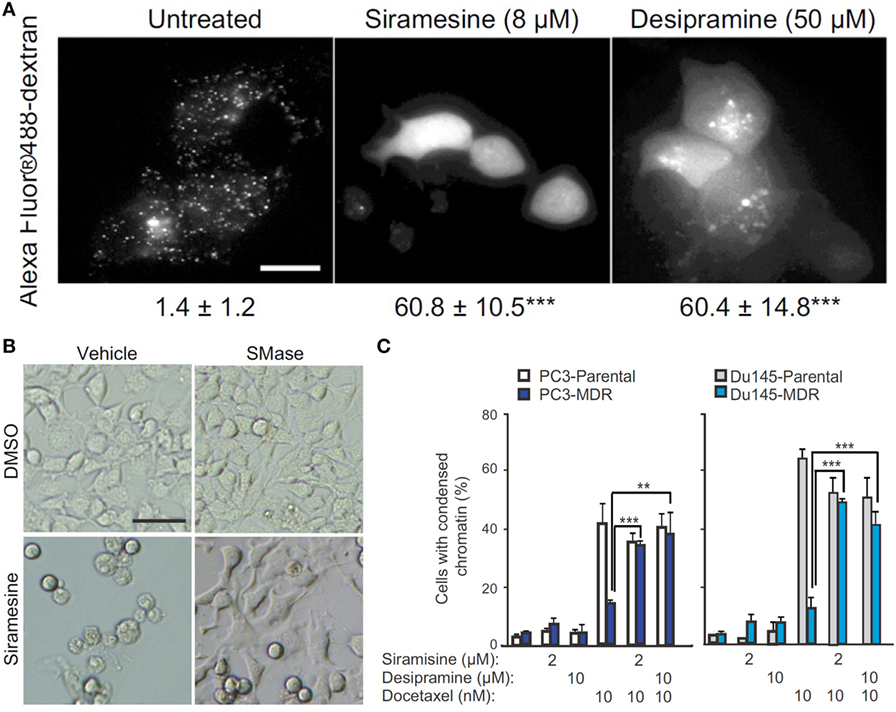
Figure 5. Targeting acid sphingomyelinases (ASM) is an effective strategy to overcome drug-resistant cancer through lysosome destabilization-mediated cell kill. (A) Siramesine and Desipramine, two cationic amphiphilic drugs and inhibitors of ASM, cause lysosomal membrane permeabilization in MCF-7 cells. The numbers given underneath each image are the respective percentage of cells with lysosomal membrane permeabilization as indicated by cytosolic Alexa Fluor 488-dextran staining. Scale bar, 20 µm. (B) Cell death of transformed NIH 3T3-c-srcY527F cells caused by siramesine can be rescued by adding 75 µM ASM from Bacillus cereus prior to starting drug treatment. Scale bar, 50 µm. (C) ASM inhibition reverts drug resistance of cancer cells. Siramesine and Desipramine treatment of multidrug-resistant (MDR) PC3-MDR or Du145-MDR greatly increased the percentage of cells undergoing apoptotic cell death as evident by condensed chromatin when co-treated with docetaxel. **represents P < 0.01, ***represents P < 0.001. Adapted from Petersen Nikolaj et al. (121).
There are several genes in the mammalian genome encoding neutral sphingomyelinases (N-SMase): the nSMase1 gene most likely encodes a lyso-platelet activating factor with phospholipase C activity (123), the nSMase2 and nSMase3 genes encode bona fide SMAs (124), and the newly identified MA-nSMase is a mitochondria-associated protein (125). Similar to ASM, N-SMase activity also promotes apoptosis through production of ceramide (124). The antitumoral chemotherapeutic reagents daunorubicine and arabinoside-C activate N-SMase in leukemic cells, resulting in ceramide release, which activates Jun-N-terminal kinases and Lyn protein tyrosine kinase to mediate cell apoptosis (126–128). Doxorubicin also induces nSMase2 mRNA and protein in a dose-dependent manner, and nSMase2 knockdown can prevent doxorubicin-induced growth arrest (129).
Alkaline sphingomyelinase activity is mostly found in the gastrointestinal tract and in human bile (130), and it is associated with colon cancer. Early studies showed that dietary sphingomyelin intake inhibited the formation of colon cancer that was induced with the carcinogen 1,2-dimethylhydrazine in mice (131). Both, human colorectal carcinoma and familial adenomatous polyposis, display a marked reduction in alkaline sphingomyelinase activity (130, 132, 133). Knockout of the alkaline sphingomyelinase encoding gene NPP7 in mice significantly enhanced colonic tumorigenesis as compared to wild-type mice, after both groups of mice had been treated with a carcinogenic formulation of azoxymethane and dextran sulfate sodium (134). Loss of function mutated NPP7 gene copies were identified in the colon cancer cell line HT-29, the liver cancer cell line HepG2, and in liver tumor tissues (135, 136).
In summary, the family of SMAs generally acts in an anticancer role through ceramide release leading to apoptosis. Radiation therapy, chemotherapeutic agents, and some targeted therapy inhibitors exerted their tumor killing ability by activating sphingomyelinase enzyme activity. The functioning of ASM in lysosomal integration has lead to the concept of inhibiting ASM to destabilize lysosomes and kill cancer cells (120, 122). The cationic amphiphilic drug siramesine, initially developed as an anti-depression agent which failed for having no effect, may be alternatively developed as an anticancer drug, and may be particularly successful when combined with microtubule-destabilizing antimitotic drugs (137). To date, the effects of sphingomyelinase activation or inhibition on the choline metabolite profile and tCho levels have not yet been studied.
Choline Transporters
The accelerated synthesis of PtdCho to satisfy the need of fast proliferation of cancer cells requires a large quantity of choline as precursor. Cancer cells typically take up free choline from their environment, which in some cases is a rate-limiting step in forming PC and in the Kennedy pathway (138). Free choline, which is an organic cation, cannot cross cell membranes freely, and hence requires active transporters for its import into cells. The human genome has four groups of proteins with choline transport ability: choline transporter 1 (CHT1/SLC5A7), choline transporter-like proteins (CTLs), organic cation transporters (OCTs), and organic cation/carnitine transporters.
A microarray survey of CTL1 revealed that it was expressed in cancers of the central nervous system, ovary, breast, prostate, and leukemia, while highly expressed in melanoma, renal, and colon cancer (139). Functional expression analysis showed that the expression of the CTL family was different across different cancer cell lines, and that cancer cells expressed at least one set of choline transporters to guarantee the import of this essential nutrient. Both the expression of choline transporter genes and the rate of choline uptake in breast cancer cells were shown to be much higher than in the non-malignant human mammary epithelial cell (HMEC) line MCF-10A (140). CHT1 mRNA was only slightly expressed in the neuroblastoma cell lines SH-SY5Y and LA-N-2 (140), while CTL1 was the major choline transporter in these cancer cells (141). CHT1 and OCT2 were upregulated in breast cancer cell lines as compared to HMEC (142), while another study showed that CTL1 and CTL2 were the two genes overexpressed in the breast cancer cell line MCF-7 as compared to the non-malignant mammary epithelial cell line MCF-10A, and CTL2 specifically was detected in human breast cancers by immunohistochemical staining (140). CTL1 was highly overexpressed in human pulmonary adenocarcinoma tissues as compared to matched normal tissues (143). CTL1 was also the major choline transporter that accounts for the enhanced choline uptake in lung adenocarcinoma cell lines, the human HT-29 colon carcinoma cell line (144), leukemia cells (140), and human neuroblastoma cells (141).
The requirement for an increased supply of choline implies that cancer cells may be vulnerable to choline transporter inhibition. Organic cation drugs, which likely share the same transporters with choline, might inhibit choline uptake, and thereby reduce cancer cell viability. The choline analog hemicholinium-3 (HC-3) and tetrahexylammonium chloride were reported to inhibit choline uptake and reduce cell proliferation when treating the human colon carcinoma cell line HT-29 (144). The Na+/H+ exchanger inhibitor dimethylamiloride and various organic cations such as quinine, quinidine, desipramine, imipramine, clomipramine, diphenhydramine, and fluvoxamine, among others were shown to inhibit choline uptake and cell viability in the small cell lung carcinoma cell line NCI-H69 (145). In the same study, HC-3 and CTL1 siRNA were shown to inhibit choline uptake and cell viability as well, along with increasing caspase-3/7 activity (145). CTL1 inhibitors significantly block choline uptake in the lung adenocarcinoma cell lines A549, H1299, and SPC-A-1, and their ability to block uptake was closely associated with their efficacy in reducing cell proliferation (143). HC-3 also significantly inhibited choline uptake in both HL-60 and Jurkat leukemia cells, thereby leading to increased caspase-3/7 activity and decreased cell viability (140).
The high-affinity choline transporter CHT1 was thought to be unique to cholinergic neurons, in which choline is used to synthesize the neurotransmitter acetylcholine (ACh) (146). However, there is evidence that non-neuronal cholinergic systems exist in lung cancers (147, 148), colon adenocarcinoma (149), and leukemia (140). In these cancers, ACh acts as an autocrine or local paracrine growth factor that stimulates tumor growth, with muscarinic cholinergic receptor subtype M3 or nicotinic cholinergic receptors functioning as its receptors (147, 149, 150), which is indicated in Figure 6. However, the free choline transported into some cells for this non-neuronal ACh synthesis is not dependent on CHT1 transport (150–152). Knockdown of CTL4, but not other genes in the CTL family, in both lung and colon cancer cells significantly decreased ACh secretion and cell growth, suggesting that CTL4 is a reasonable target for certain types of cancer therapy without affecting neuronal ACh synthesis (150, 153). CTL1 was reported to be associated with ACh production in human neuroblastoma cells (141), but negatively associated with ACh secretion from non-small cell lung carcinoma (154). CTL1 inhibition to reduce ACh production may be an effective way of killing cancer cells in certain types of cancer.
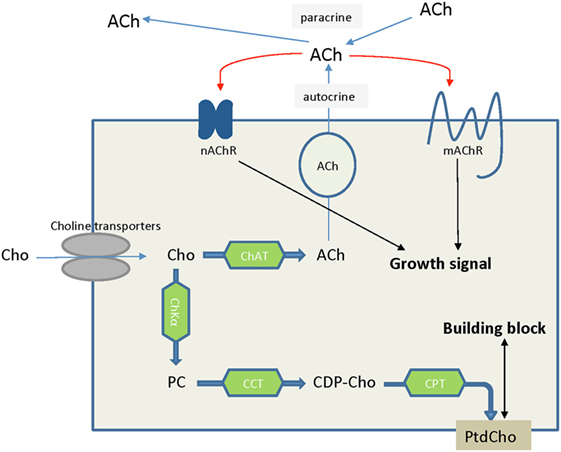
Figure 6. Choline is an essential nutrient transported into cells by choline transporters. In some cancer types such as lung cancers, choline that is taken up into cells is converted to acetylcholine, serving as an autocrine or paracrine growth factor which stimulates cancer cell growth. Alternatively, choline is converted to phosphocholine and ultimately phosphatidylcholine, serving as building block to satisfy the increased proliferation rate of cancer cells and tumor growth. Abbreviations: ACh, acetylcholine; CCT, phosphocholine cytidylyltransferase; CDP-Cho, cytidine 5′-diphosphocholine; ChAT, choline acetyltransferase; ChKα, choline kinase α; Cho, choline; CPT, diacylglycerol cholinephosphotransferase; nAChR, nicotinic acetycholine receptor; mAChR, muscarinic acetycholine receptor; PC, phosphocholine; PtdCho, phosphatidylcholine.
Choline transporters are crucial for cells to import extracellular choline. Inhibition of choline transport would likely lead to choline deficiency and ceramide accumulation, which in turn would trigger apoptosis via p53-independent pathway (140, 155, 156). Overall, choline transporters, and in particular CTL1 and CTL4, are interesting targets for anticancer drug development because their inhibition has a great potential to reduce proliferation and induce apoptosis or block the tumor promoting ACh-M3 muscarinic cholinergic receptor system. As CHT1 is necessary for neuronal ACh biogenesis, as evident by the lethal effect of CHT1 knockout in mice (157) and the severe toxicity of its inhibitor hemicholinium-3 (158), developing inhibitors specific to CTLs with low affinity to CHT1 is a must in targeted drug development to avoid possible toxic side effects.
Glycerophosphodiesterases
Glycerophosphodiester phosphodiesterases (GDPDs) or glycerophosphodiesterases (GDEs) are enzymes that use glycerophosphodiesters as substrates and break them down into glycerol 3-phosphate and alcohols (see Figure 7A). Although bacterial GDPDs can hydrolyze various glycerophosphodiesters, the mammalian genes display substrate preference, and the substrates are not limited to glycerophosphodiesters (159). The human genome encodes seven members of the GDE family, and three of them, as well as their close homologs in mice, were reported to hydrolyze glycerophosphodiesters (159). GDE1 displays substrate preference toward glycerophosphoinositol (GPI), while glycerophosphoserine (GPS) and glycerophosphoglycerate (GPG) are direct GDE1 substrates as well, and GPE is able to block the glycerophosphoinositol phosphodiesterase activity of GDE1, but not GPC (160). The two GDPDs: GDPD5 (or GDE2) and GDPD6 (or GDE5, GPCPD1) were reported to display glycerophosphocholine phosphodiesterase (GPC-PDE) activity (161, 162).
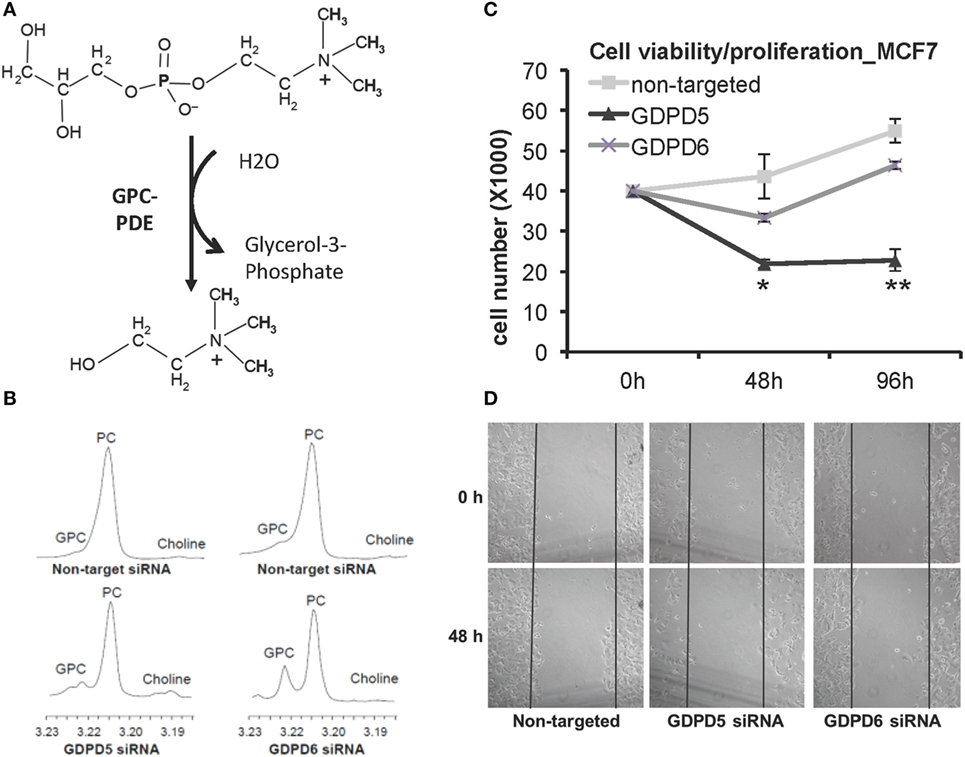
Figure 7. GDPD5 and GDPD6, the two glycerophosphocholine phosphodiesterases (GPC-PDEs) reported to release choline from glycerophosphocholine (GPC), show potential anticancer effects. (A) GPC-PDE enzyme activity is defined as cleaving the choline moiety from GPC and thereby generating glycerol-3-phosphate. (B) Transient knockdown of GDPD5 or GDPD6 by siRNA treatment of MDA-MB-231 cells shows that GDPD6 silencing leads to a significant elevation of the GPC peak in high-resolution 1H MR spectra from water-soluble metabolites, while GDPD5 silencing marginally increases GPC levels in this cell line. (C) GDPD5, but not GDPD6, siRNA shows cytotoxic effect in MCF-7 breast cancer cells indicated by reduced cell numbers following siRNA treatment of MCF-7 cells. (D) Both GDPD5 and GDPD6 silencing decreased MCF-7 cell migration detected by scratch assay. Abbreviations: PC, phosphocholine. *represents P < 0.05, **represents P < 0.01. Adapted from Cao et al. (163).
GDPD5 was first reported in mice as a GPC-PDE that osmotically regulates GPC levels in renal medullary cells, in which GPC is an abundant and important osmoprotective organic osmolyte (161). Phosphorus MRS measurements of MDA-MB-231 breast cancer xenografts with constitutively silenced GDPD5 in nude mice also displayed an increase in GPC (164). GDPD5 expression was found to correlate with breast cancer malignancy (165). GDPD5, as well as PLD1 and ChKα, were highly expressed in estrogen receptor negative (ER−) breast cancers, which also displayed higher PC, tCho, and lower GPC in comparison with ER+ cases (165). High-resolution 1H MRS analysis of breast cancer cell lines in which GDPD5 or GDPD6 were silenced demonstrated that GDPD6 has a leading role in regulating the intracellular GPC level in breast cancer cells (Figure 7B) (163). siRNA silencing of GDPD5 reduced the viability (Figure 7C) and migration (Figure 7D) of ER + MCF-7 breast cancer cells, and decreased the migration and invasion of triple-negative MDA-MB-231 breast cancer cells (163). GDPD5 also has important functions in regulating neuron differentiation through glycosylphosphatidylinositol-anchor cleavage of reversion-inducing cysteine-rich protein with kazal motifs (RECK) (166), which, further downstream, leads to inactivation of the Notch signaling pathway.
GDPD6 was shown to be an Mg2+-dependent GDE that is able to hydrolyze GPC and GPE, but does not show activity with GPG, GPI, and GPS as substrates (167). The role of GDPD6 in cancer was recently identified, demonstrating that GDPD6 expression promotes cancer cell migration and invasion through protein kinase alpha signaling (162). GDPD6 mRNA levels were shown to be increased in metastasizing as compared to non-metastasizing endometrial carcinomas, and GDPD6 expression was negatively associated with relapse-free survival in endometrial and ovarian cancers (162). GDPD6 was overexpressed in endometrial cancers, along with ChKα overexpression, resulting in a 70% increase in PC levels (16). Microarray analysis of GDPD6-silenced MCF-7 and OVCAR-3 cells compared to control samples revealed that the expression of integrin β1 was reduced, which resulted in decreased cell attachment and spreading, while overexpression of GDPD6 had the opposite effect (168). The GDPD6 protein contains two conserved domains, which are a carbohydrate-binding domain and a catalytic domain conferring GDE enzyme activity. GDPD6 was also shown to regulate skeletal muscle development in an enzyme activity-independent domain (167). It is not yet clear whether the choline-releasing enzymatic activity of GDPD6 is important for its function in promoting cancer cell migration and invasion.
Both GDPD5 and GDPD6 are emerging as potential anticancer targets in choline metabolism. Their differing specific roles in conferring cancer aggressiveness and degrading GPC are at a relatively early stage of investigation and will require additional mechanistic and preclinical studies to evaluate their full potential as possible drug targets for anticancer therapy.
Phosphatidylethanolamine N-Methyltransferase (PEMT)
Phosphatidylethanolamine can be directly converted to PtdCho in one step through three methylations of the ethanolamine moiety of PtdEtn, which is catalyzed by the enzyme PEMT. This enzyme was initially isolated from rat liver and was thought to be specific to the liver (169). The first indication that PEMT can suppress cancer was the discovery that the growth of rat hepatoma cells was suppressed by overexpressing the PEMT gene (170). PEMT is highly expressed in normal liver, but its activity was negligible in the two hepatoma cell lines and almost diminished during chemically induced hepatocarcinogenesis (171, 172), and the induced neoplastic phenotype could be partially reversed by PEMT cDNA transfection (172). When growing hepatoma cells in an animal host, PEMT activity and protein expression were barely detected in the fast proliferation stage, while its mRNA and protein reappeared in stationary growth stages (173). Forced expression of PEMT in McArdle-RH7777 hepatoma cells resulted in cell apoptosis (173).
Most early studies of PEMT function were using non-human hepatoma cells or chemically induced hepatocarcinogenesis models in animals. A later study with clinical samples of human hepatocellular carcinomas (HCC) showed that while the PEMT gene was intact, its mRNA level was reduced or even absent compared to peritumoral normal tissue, which was also observed in parallel for PEMT’s enzyme activity (174). PEMT mRNA was found to inversely correlate with HCC histological stage, and the absence of PEMT mRNA in tumor tissue from cancer patients was associated with poor survival (174). While early studies of PEMT’s role in cancer were mostly observed in liver cancers, more recent studies indicated that PEMT polymorphism is also related to breast cancer risk (175). In BRCA1-mutated breast cancer, the PEMT gene undergoes epigenetic repression, and hypermethylation of a specific site in the PEMT promoter is associated with histological grade and estrogen receptor status (176). This finding is consistent with the observation that PtdEtn increased with breast tumor grade (6). Another study in small cell lung cancer indicated that an increase in PEMT expression predicted shorter patient survival time, which is opposite to the observations made in liver and breast cancer (177).
The molecular mechanisms by which PEMT expression suppresses hepatoma growth are not yet fully understood. One study showed that PEMT activity negatively correlated with PtdCho synthesis in the Kennedy pathway (178). When PEMT was overexpressed in McArdle-RH7777 rat hepatoma cells, cellular PtdCho levels did not change even though [methyl-3H]methionine and [3H]ethanolamine were incorporated into PtdCho, suggesting that conversion of PtdEtn to PtdCho occurred (178). Molecular analysis in this study showed that CTP:phosphocholine cytidylyltransferase (CT) activity, which is the rate-limiting step in the Kennedy pathway of PtdCho synthesis, decreased (178). This was most likely the reason for the unchanged PtdCho levels during PEMT overexpression in this study (178). The slowing down of the Kennedy pathway while PEMT activity increased may have contributed to the reduced cell proliferation rate in this study (178). Chemically induced hepatocarcinogenesis and growth of hepatoma cells in host animals also demonstrated opposing activities of PEMT and CT (171, 173). The overexpression of PEMT in CBRH-7919 rat hepatoma cells resulted in a downmodulation of the PI3K/Akt signaling pathway, which was evident by reduced protein levels of c-Met, PDGF receptor, PI3K, Akt, and Bcl-2, in addition to decreased phosphorylation of Akt Thr308, which was accompanied by elevated cell apoptosis (179).
In summary, PEMT, an enzyme that directly converts PtdEtn to PtdCho by sequential methylation, acts as an onco-suppressor. Its function and molecular mechanism of tumor suppression, and its regulation during carcinogenesis are currently starting to emerge and will require more in depth studies.
Ethanolamine Kinase (ETNK)
The first step in the Kenney pathway toward de novo synthesis of PtdEth or PtdCho is the ATP-dependent phosphorylation of free intracellular ethanolamine or free intracellular choline. While ChKα does confer dual choline and ETNK activity with a preference for choline (180), the conversion of free ethanolamine to PE is mainly achieved through ETNK enzyme activity (181, 182). There are two genes encoding ETNK enzymes in the human genome: ETNK1 (or EKI1) and ETNK2 (or EKI2). Forced overexpression of the ETNK1 gene in COS7 cells accelerated the Kennedy pathway of PtdEtn synthesis, but did not result in an accumulation of PtdEtns due to concomitant faster degradation of PtdEtns (183). Interestingly, an ETNK1 recurrent missense mutation was frequently found in systemic mastocytosis with eosinophilia and chronic myelomonocytic leukemia (184, 185). These mutations caused an amino acid change located in the kinase domain, which was predicted to disrupt the enzyme’s catalytic activity (184, 185). Metabolite measurements revealed an averaged 5.2-fold decrease in the PtdEtn/PtdCho ratio of ETNK1-mutated atypical chronic myelomonocytic leukemia samples as compared to ETNK1 wild-type samples (184). In contrast to these findings in leukemia, in different types of human testicular cancers, EKI1 co-amplified with DAD-R and SOX5, all of which reside in close-by chromosomal regions of the short arm of chromosome 12 (12P) (186). The DAD-R gene is thought to be clinically relevant in the malignant transformation of testicular cancers because of elevated DAD-R expression in testicular cancers with 12P amplification, which correlated with invasive growth and a reduced level of apoptosis, as compared to testicular cancers without 12P amplification (186).
While the roles and functions of ETNKs in cancer are still at an early stage of investigation, the product of its enzymatic activity, PE, has been known to be elevated in several cancers for decades (45, 187). Moreover, synthetic PE has recently been suggested to have anticancer effects on melanoma (188), Ehrlich ascites tumor (189), and leukemia (190). PE has also been reported to possess anti-angiogenic and anti-metastatic activity in lung cancer (191) and to induce cell cycle arrest and apoptosis in MCF-7 breast cancer cells (192). While the therapeutic effects of PE still need to be verified by additional studies and eventually clinical trials, ETNK may be an additional target for MRS-monitored anticancer treatment and should be the focus of more research studies in the near future. It is also possible that elevated PE levels in several types of cancer could be the result of elevated ChKα expression and activity levels, which is known to also act on free ethanolamine (180). Additional studies are necessary to clarify the involvement of ChKα in the elevated tumoral PE levels.
The Future: Targeting Multiple Targets and Metabolic Nodes
As our molecular understanding of the different roles of choline and ethanolamine phospholipid metabolism enzymes in cancer is growing, it will become important to study the interactions between these enzymes as well. For example, in aggressive ER− breast cancers, GDPD5, PLD1, and ChKα, were simultaneously highly expressed, leading to elevated PC and tCho levels (165). Moreover, it was recently shown that by silencing ChKα in breast cancer cells, PtdCho–PLD1 was in turn upregulated and vice versa (88). Only silencing of both enzymes simultaneously increased apoptosis in the tested breast cancer cells, supporting the necessity for targeting multiple enzymes in choline phospholipid metabolism as a strategy of choice (88). This is also important as targeting only one choline metabolic enzyme for anticancer treatment may lead to the development of resistance in the treated cancer cells more easily as cancer cells are able to adapt to new growth environments and thereby acquire resistance.
Once the body of knowledge about the regulation of choline and ethanolamine phospholipid enzymes has grown even further, it may be worthwhile pursuing systems biology approaches of network analyses to identify the important nodes in these metabolic pathways that are the most vulnerable for targeting as anticancer treatment strategy. Such types of analyses have already been performed for drug design and development (193) and recently for tissue-specific metabolic networks, including tumors (194).
Conclusion
The aberrant choline and ethanolamine phospholipid metabolism in cancer has recently further been established and solidified as a universal metabolic hallmark of cancer. However, our current knowledge of druggable targets in choline metabolism is still emerging, with only a few enzymes such as ChKα and PLD1 having been explored to a level where a somewhat complete picture of their regulation, oncogenic roles, and interaction networks in cancer are becoming available. For all other enzymes discussed in this review, a lot more basic and translational research is required to evaluate the exact molecular roles of PtdCho-PLC, SMases, choline transporters, GDEs, PEMT, and ETKN in cancer. Metabolic imaging approaches such as MRS and PET are valuable tools to help assess the targeting and regulation outcomes in these studies and to serve later on as monitoring tools in the clinic to assess the response to treatment. Future studies and computational modeling approaches to evaluate the interaction between choline and ethanolamine metabolic enzymes in cancer will be necessary.
Author Contributions
MC drafted the manuscript and the figures. KG conceived of the manuscript and guided MC through writing it and edited the final manuscript and figures. ZB edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors gratefully acknowledge support from NIH R01 CA154725 and P50 CA103175.
References
1. Dobrzyńska I, Szachowicz-Petelska B, Darewicz B, Figaszewski ZA. Characterization of human bladder cell membrane during cancer transformation. J Membr Biol (2015) 248(2):301–7. doi:10.1007/s00232-015-9770-4
2. Dobrzynska I, Szachowicz-Petelska B, Sulkowski S, Figaszewski Z. Changes in electric charge and phospholipids composition in human colorectal cancer cells. Mol Cell Biochem (2005) 276(1–2):113–9. doi:10.1007/s11010-005-3557-3
3. Szachowicz-Petelska B, Dobrzynska I, Skrodzka M, Darewicz B, Figaszewski ZA, Kudelski J. Phospholipid composition and electric charge in healthy and cancerous parts of human kidneys. J Membr Biol (2013) 246(5):421–5. doi:10.1007/s00232-013-9554-7
4. Sakai K, Okuyama H, Yura J, Takeyama H, Shinagawa N, Tsuruga N, et al. Composition and turnover of phospholipids and neutral lipids in human breast cancer and reference tissues. Carcinogenesis (1992) 13(4):579–84. doi:10.1093/carcin/13.4.579
5. Hietanen E, Punnonen K, Punnonen R, Auvinen O. Fatty acid composition of phospholipids and neutral lipids and lipid peroxidation in human breast cancer and lipoma tissue. Carcinogenesis (1986) 7(12):1965–9. doi:10.1093/carcin/7.12.1965
6. Beckonert O, Monnerjahn J, Bonk U, Leibfritz D. Visualizing metabolic changes in breast-cancer tissue using 1H NMR spectroscopy and self-organizing maps. NMR Biomed (2003) 16(1):1–11. doi:10.1002/nbm.797
7. Glunde K, Bhujwalla ZM, Ronen SM. Choline metabolism in malignant transformation. Nat Rev Cancer (2011) 11(12):835–48. doi:10.1038/nrc3162
8. Bolan PJ. Magnetic resonance spectroscopy of the breast: current status. Magn Reson Imaging Clin N Am (2013) 21(3):625–39. doi:10.1016/j.mric.2013.04.008
9. Jacobs MA, Barker PB, Bottomley PA, Bhujwalla Z, Bluemke DA. Proton magnetic resonance spectroscopic imaging of human breast cancer: a preliminary study. J Magn Reson Imaging (2004) 19(1):68–75. doi:10.1002/jmri.10427
10. Meisamy S, Bolan PJ, Baker EH, Bliss RL, Gulbahce E, Everson LI, et al. Neoadjuvant chemotherapy of locally advanced breast cancer: predicting response with in vivo 1H MR spectroscopy – a pilot study at 4 T. Radiology (2004) 233(2):424–31. doi:10.1148/radiol.2332031285
11. Manton DJ, Chaturvedi A, Hubbard A, Lind MJ, Lowry M, Maraveyas A, et al. Neoadjuvant chemotherapy in breast cancer: early response prediction with quantitative MR imaging and spectroscopy. Br J Cancer (2006) 94(3):427–35. doi:10.1038/sj.bjc.6602948
12. Baek HM, Chen JH, Nie K, Yu HJ, Bahri S, Mehta RS, et al. Predicting pathologic response to neoadjuvant chemotherapy in breast cancer by using MR imaging and quantitative 1H MR spectroscopy. Radiology (2009) 251(3):653–62. doi:10.1148/radiol.2512080553
13. Kurhanewicz J, Swanson MG, Nelson SJ, Vigneron DB. Combined magnetic resonance imaging and spectroscopic imaging approach to molecular imaging of prostate cancer. J Magn Reson Imaging (2002) 16(4):451–63. doi:10.1002/jmri.10172
14. Ackerstaff E, Pflug BR, Nelson JB, Bhujwalla ZM. Detection of increased choline compounds with proton nuclear magnetic resonance spectroscopy subsequent to malignant transformation of human prostatic epithelial cells. Cancer Res (2001) 61(9):3599–603.
15. Booth SJ, Pickles MD, Turnbull LW. In vivo magnetic resonance spectroscopy of gynaecological tumours at 3.0 Tesla. BJOG (2009) 116(2):300–3. doi:10.1111/j.1471-0528.2008.02007.x
16. Trousil S, Lee P, Pinato DJ, Ellis JK, Dina R, Aboagye EO, et al. Alterations of choline phospholipid metabolism in endometrial cancer are caused by choline kinase alpha overexpression and a hyperactivated deacylation pathway. Cancer Res (2014) 74(23):6867–77. doi:10.1158/0008-5472.CAN-13-2409
17. Vigneron D, Bollen A, McDermott M, Wald L, Day M, Moyher-Noworolski S, et al. Three-dimensional magnetic resonance spectroscopic imaging of histologically confirmed brain tumors. Magn Reson Imaging (2001) 19(1):89–101. doi:10.1016/S0730-725X(01)00225-9
18. Einstein DB, Wessels B, Bangert B, Fu P, Nelson AD, Cohen M, et al. Phase II trial of radiosurgery to magnetic resonance spectroscopy-defined high-risk tumor volumes in patients with glioblastoma multiforme. Int J Radiat Oncol Biol Phys (2012) 84(3):668–74. doi:10.1016/j.ijrobp.2012.01.020
19. Elkhaled A, Jalbert L, Constantin A, Yoshihara HA, Phillips JJ, Molinaro AM, et al. Characterization of metabolites in infiltrating gliomas using ex vivo 1H high-resolution magic angle spinning spectroscopy. NMR Biomed (2014) 27(5):578–93. doi:10.1002/nbm.3097
20. Aboagye EO, Bhujwalla ZM. Malignant transformation alters membrane choline phospholipid metabolism of human mammary epithelial cells. Cancer Res (1999) 59(1):80–4.
21. Iorio E, Mezzanzanica D, Alberti P, Spadaro F, Ramoni C, D’Ascenzo S, et al. Alterations of choline phospholipid metabolism in ovarian tumor progression. Cancer Res (2005) 65(20):9369–76. doi:10.1158/0008-5472.CAN-05-1146
22. Al-Saffar NM, Troy H, Ramirez de Molina A, Jackson LE, Madhu B, Griffiths JR, et al. Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of the choline kinase inhibitor MN58b in human carcinoma models. Cancer Res (2006) 66(1):427–34. doi:10.1158/0008-5472.CAN-05-1338
23. Beloueche-Babari M, Arunan V, Troy H, te Poele RH, Te Fong A-CW, Jackson LE, et al. Histone deacetylase inhibition increases levels of choline kinase and phosphocholine facilitating noninvasive imaging in human cancers. Cancer Res (2012) 72(4):990–1000. doi:10.1158/0008-5472.can-11-2688
24. Ward CS, Eriksson P, Izquierdo-Garcia JL, Brandes AH, Ronen SM. HDAC inhibition induces increased choline uptake and elevated phosphocholine levels in MCF7 breast cancer cells. PLoS One (2013) 8(4):e62610. doi:10.1371/journal.pone.0062610
25. Beloueche-Babari M, Jackson LE, Al-Saffar NMS, Eccles SA, Raynaud FI, Workman P, et al. Identification of magnetic resonance detectable metabolic changes associated with inhibition of phosphoinositide 3-kinase signaling in human breast cancer cells. Mol Cancer Ther (2006) 5(1):187–96. doi:10.1158/1535-7163.mct-03-0220
26. Chung Y-L, Troy H, Banerji U, Jackson LE, Walton MI, Stubbs M, et al. Magnetic resonance spectroscopic pharmacodynamic markers of the heat shock protein 90 inhibitor 17-allylamino,17-demethoxygeldanamycin (17AAG) in human colon cancer models. J Natl Cancer Inst (2003) 95(21):1624–33. doi:10.1093/jnci/djg084
27. Morse DL, Raghunand N, Sadarangani P, Murthi S, Job C, Day S, et al. Response of choline metabolites to docetaxel therapy is quantified in vivo by localized 31P MRS of human breast cancer xenografts and in vitro by high-resolution 31P NMR spectroscopy of cell extracts. Magn Reson Med (2007) 58(2):270–80. doi:10.1002/mrm.21333
28. Roebuck JR, Cecil KM, Schnall MD, Lenkinski RE. Human breast lesions: characterization with proton MR spectroscopy. Radiology (1998) 209(1):269–75. doi:10.1148/radiology.209.1.9769842
29. Katz-Brull R, Lavin PT, Lenkinski RE. Clinical utility of proton magnetic resonance spectroscopy in characterizing breast lesions. J Natl Cancer Inst (2002) 94(16):1197–203. doi:10.1093/jnci/94.16.1197
30. Tse GM, Cheung HS, Pang LM, Chu WC, Law BK, Kung FY, et al. Characterization of lesions of the breast with proton MR spectroscopy: comparison of carcinomas, benign lesions, and phyllodes tumors. AJR Am J Roentgenol (2003) 181(5):1267–72. doi:10.2214/ajr.181.5.1811267
31. Bartella L, Morris EA, Dershaw DD, Liberman L, Thakur SB, Moskowitz C, et al. Proton MR spectroscopy with choline peak as malignancy marker improves positive predictive value for breast cancer diagnosis: preliminary study. Radiology (2006) 239(3):686–92. doi:10.1148/radiol.2393051046
32. Jagannathan NR, Kumar M, Seenu V, Coshic O, Dwivedi SN, Julka PK, et al. Evaluation of total choline from in vivo volume localized proton MR spectroscopy and its response to neoadjuvant chemotherapy in locally advanced breast cancer. Br J Cancer (2001) 84(8):1016–22. doi:10.1054/bjoc.2000.1711
33. Tozaki M, Sakamoto M, Oyama Y, Maruyama K, Fukuma E. Predicting pathological response to neoadjuvant chemotherapy in breast cancer with quantitative 1H MR spectroscopy using the external standard method. J Magn Reson Imaging (2010) 31(4):895–902. doi:10.1002/jmri.22118
34. Smith TA, Glaholm J, Leach MO, Machin L, Collins DJ, Payne GS, et al. A comparison of in vivo and in vitro 31P NMR spectra from human breast tumours: variations in phospholipid metabolism. Br J Cancer (1991) 63(4):514–6. doi:10.1038/bjc.1991.122
35. Sijens PE, Wijrdeman HK, Moerland MA, Bakker CJ, Vermeulen JW, Luyten PR. Human breast cancer in vivo: 1H and 31P MR spectroscopy at 1.5 T. Radiology (1988) 169(3):615–20. doi:10.1148/radiology.169.3.2847230
36. Glaholm J, Leach MO, Collins DJ, Mansi J, Sharp JC, Madden A, et al. In vivo 31P magnetic resonance spectroscopy for monitoring treatment response in breast cancer. Lancet (1989) 1(8650):1326–7. doi:10.1016/S0140-6736(89)92717-7
37. Leach MO, Verrill M, Glaholm J, Smith TA, Collins DJ, Payne GS, et al. Measurements of human breast cancer using magnetic resonance spectroscopy: a review of clinical measurements and a report of localized 31P measurements of response to treatment. NMR Biomed (1998) 11(7):314–40. doi:10.1002/(SICI)1099-1492(1998110)11:7<314::AID-NBM522>3.0.CO;2-Z
38. Maris JM, Evans AE, McLaughlin AC, D’Angio GJ, Bolinger L, Manos H, et al. 31P nuclear magnetic resonance spectroscopic investigation of human neuroblastoma in situ. N Engl J Med (1985) 312(23):1500–5. doi:10.1056/NEJM198506063122307
39. Narayan P, Jajodia P, Kurhanewicz J, Thomas A, MacDonald J, Hubesch B, et al. Characterization of prostate cancer, benign prostatic hyperplasia and normal prostates using transrectal 31P magnetic resonance spectroscopy: a preliminary report. J Urol (1991) 146(1):66–74.
40. Dixon RM, Angus PW, Rajagopalan B, Radda GK. Abnormal phosphomonoester signals in 31P MR spectra from patients with hepatic lymphoma. A possible marker of liver infiltration and response to chemotherapy. Br J Cancer (1991) 63(6):953–8. doi:10.1038/bjc.1991.208
41. Klomp DW, Wijnen JP, Scheenen TW, Heerschap A. Efficient 1H to 31P polarization transfer on a clinical 3T MR system. Magn Reson Med (2008) 60(6):1298–305. doi:10.1002/mrm.21733
42. van der Kemp WJ, Boer VO, Luijten PR, Wijnen JP, Klomp DW. Increase in SNR for 31P MR spectroscopy by combining polarization transfer with a direct detection sequence. Magn Reson Med (2012) 68(2):353–7. doi:10.1002/mrm.23260
43. Wijnen JP, Scheenen TW, Klomp DW, Heerschap A. 31P magnetic resonance spectroscopic imaging with polarisation transfer of phosphomono- and diesters at 3 T in the human brain: relation with age and spatial differences. NMR Biomed (2010) 23(8):968–76. doi:10.1002/nbm.1523
44. Wijnen JP, Jiang L, Greenwood TR, van der Kemp WJ, Klomp DW, Glunde K. 1H/31P polarization transfer at 9.4 Tesla for improved specificity of detecting phosphomonoesters and phosphodiesters in breast tumor models. PLoS One (2014) 9(7):e102256. doi:10.1371/journal.pone.0102256
45. Podo F. Tumour phospholipid metabolism. NMR Biomed (1999) 12(7):413–39. doi:10.1002/(SICI)1099-1492(199911)12:7<413::AID-NBM587>3.3.CO;2-L
46. Bathen TF, Heldahl MG, Sitter B, Vettukattil R, Bofin A, Lundgren S, et al. In vivo MRS of locally advanced breast cancer: characteristics related to negative or positive choline detection and early monitoring of treatment response. MAGMA (2011) 24(6):347–57. doi:10.1007/s10334-011-0280-9
47. Steen RG. Response of solid tumors to chemotherapy monitored by in vivo 31P nuclear magnetic resonance spectroscopy: a review. Cancer Res (1989) 49(15):4075–85.
48. U.S. Food and Drug Administration. FDA approves 11C-choline for PET in prostate cancer. J Nucl Med (2012) 53(12):11N.
49. De Bari B, Alongi F, Lestrade L, Giammarile F. Choline-PET in prostate cancer management: the point of view of the radiation oncologist. Crit Rev Oncol Hematol (2014) 91(3):234–47. doi:10.1016/j.critrevonc.2014.04.002
50. Aoyama C, Liao H, Ishidate K. Structure and function of choline kinase isoforms in mammalian cells. Prog Lipid Res (2004) 43(3):266–81. doi:10.1016/j.plipres.2004.03.003
51. Gallego-Ortega D, Gomez del Pulgar T, Valdes-Mora F, Cebrian A, Lacal JC. Involvement of human choline kinase alpha and beta in carcinogenesis: a different role in lipid metabolism and biological functions. Adv Enzyme Regul (2011) 51(1):183–94. doi:10.1016/j.advenzreg.2010.09.010
52. Gallego-Ortega D, Ramirez de Molina A, Ramos MA, Valdes-Mora F, Barderas MG, Sarmentero-Estrada J, et al. Differential role of human choline kinase alpha and beta enzymes in lipid metabolism: implications in cancer onset and treatment. PLoS One (2009) 4(11):e7819. doi:10.1371/journal.pone.0007819
53. Ramirez de Molina A, Gallego-Ortega D, Sarmentero J, Banez-Coronel M, Martin-Cantalejo Y, Lacal JC. Choline kinase is a novel oncogene that potentiates RhoA-induced carcinogenesis. Cancer Res (2005) 65(13):5647–53. doi:10.1158/0008-5472.CAN-04-4416
55. Ramirez de Molina A, Gutierrez R, Ramos MA, Silva JM, Silva J, Bonilla F, et al. Increased choline kinase activity in human breast carcinomas: clinical evidence for a potential novel antitumor strategy. Oncogene (2002) 21(27):4317–22. doi:10.1038/sj.onc.1205556
56. Ramirez de Molina A, Rodriguez-Gonzalez A, Gutierrez R, Martinez-Pineiro L, Sanchez J, Bonilla F, et al. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem Biophys Res Commun (2002) 296(3):580–3. doi:10.1016/S0006-291X(02)00920-8
57. Ramirez de Molina A, Sarmentero-Estrada J, Belda-Iniesta C, Taron M, Ramirez de Molina V, Cejas P, et al. Expression of choline kinase alpha to predict outcome in patients with early-stage non-small-cell lung cancer: a retrospective study. Lancet Oncol (2007) 8(10):889–97. doi:10.1016/S1470-2045(07)70279-6
58. Penet MF, Shah T, Bharti S, Krishnamachary B, Artemov D, Mironchik Y, et al. Metabolic imaging of pancreatic ductal adenocarcinoma detects altered choline metabolism. Clin Cancer Res (2015) 21(2):386–95. doi:10.1158/1078-0432.CCR-14-0964
59. Miyake T, Parsons SJ. Functional interactions between choline kinase alpha, epidermal growth factor receptor and c-Src in breast cancer cell proliferation. Oncogene (2012) 31(11):1431–41. doi:10.1038/onc.2011.332
60. Clem BF, Clem AL, Yalcin A, Goswami U, Arumugam S, Telang S, et al. A novel small molecule antagonist of choline kinase-alpha that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene (2011) 30(30):3370–80. doi:10.1038/onc.2011.51
61. Chua BT, Gallego-Ortega D, Ramirez de Molina A, Ullrich A, Lacal JC, Downward J. Regulation of Akt(ser473) phosphorylation by choline kinase in breast carcinoma cells. Mol Cancer (2009) 8:131. doi:10.1186/1476-4598-8-131
62. Sanchez-Lopez E, Zimmerman T, Gomez del Pulgar T, Moyer MP, Lacal Sanjuan JC, Cebrian A. Choline kinase inhibition induces exacerbated endoplasmic reticulum stress and triggers apoptosis via CHOP in cancer cells. Cell Death Dis (2013) 4:e933. doi:10.1038/cddis.2013.453
63. Sher RB, Aoyama C, Huebsch KA, Ji S, Kerner J, Yang Y, et al. A rostrocaudal muscular dystrophy caused by a defect in choline kinase beta, the first enzyme in phosphatidylcholine biosynthesis. J Biol Chem (2006) 281(8):4938–48. doi:10.1074/jbc.M512578200
64. Wu G, Aoyama C, Young SG, Vance DE. Early embryonic lethality caused by disruption of the gene for choline kinase α, the first enzyme in phosphatidylcholine biosynthesis. J Biol Chem (2008) 283(3):1456–62. doi:10.1074/jbc.M708766200
65. Hernandez-Alcoceba R, Fernandez F, Lacal JC. In vivo antitumor activity of choline kinase inhibitors: a novel target for anticancer drug discovery. Cancer Res (1999) 59(13):3112–8.
66. Hernandez-Alcoceba R, Saniger L, Campos J, Nunez MC, Khaless F, Gallo MA, et al. Choline kinase inhibitors as a novel approach for antiproliferative drug design. Oncogene (1997) 15(19):2289–301. doi:10.1038/sj.onc.1201414
67. Rodriguez-Gonzalez A, Ramirez de Molina A, Fernandez F, Ramos MA, del Carmen Nunez M, Campos J, et al. Inhibition of choline kinase as a specific cytotoxic strategy in oncogene-transformed cells. Oncogene (2003) 22(55):8803–12. doi:10.1038/sj.onc.1207062
68. Lacal JC, Campos JM. Preclinical characterization of RSM-932a, a novel anticancer drug targeting the human choline kinase alpha, an enzyme involved in increased lipid metabolism of cancer cells. Mol Cancer Ther (2015) 14(1):31–9. doi:10.1158/1535-7163.MCT-14-0531
69. Glunde K, Raman V, Mori N, Bhujwalla ZM. RNA interference-mediated choline kinase suppression in breast cancer cells induces differentiation and reduces proliferation. Cancer Res (2005) 65(23):11034–43. doi:10.1158/0008-5472.CAN-05-1807
70. Krishnamachary B, Glunde K, Wildes F, Mori N, Takagi T, Raman V, et al. Noninvasive detection of lentiviral-mediated choline kinase targeting in a human breast cancer xenograft. Cancer Res (2009) 69(8):3464–71. doi:10.1158/0008-5472.CAN-08-4120
71. Li C, Penet MF, Wildes F, Takagi T, Chen Z, Winnard PT, et al. Nanoplex delivery of siRNA and prodrug enzyme for multimodality image-guided molecular pathway targeted cancer therapy. ACS Nano (2010) 4(11):6707–16. doi:10.1021/nn102187v
72. Penet MF, Chen Z, Mori N, Krishnamachary B, Bhujwalla ZM. Magnetic resonance spectroscopy of siRNA-based cancer therapy. Methods Mol Biol (2016) 1372:37–47. doi:10.1007/978-1-4939-3148-4_3
73. Mori N, Glunde K, Takagi T, Raman V, Bhujwalla ZM. Choline kinase down-regulation increases the effect of 5-fluorouracil in breast cancer cells. Cancer Res (2007) 67(23):11284–90. doi:10.1158/0008-5472.CAN-07-2728
74. de la Cueva A, Ramirez de Molina A, Alvarez-Ayerza N, Ramos MA, Cebrian A, Del Pulgar TG, et al. Combined 5-FU and ChoKalpha inhibitors as a new alternative therapy of colorectal cancer: evidence in human tumor-derived cell lines and mouse xenografts. PLoS One (2013) 8(6):e64961. doi:10.1371/journal.pone.0064961
75. McDermott M, Wakelam MJ, Morris AJ. Phospholipase D. Biochem Cell Biol (2004) 82(1):225–53. doi:10.1139/o03-079
76. Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science (2001) 294(5548):1942–5. doi:10.1126/science.1066015
77. Hammond SM, Altshuller YM, Sung TC, Rudge SA, Rose K, Engebrecht J, et al. Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J Biol Chem (1995) 270(50):29640–3. doi:10.1074/jbc.270.50.29640
78. Hammond SM, Jenco JM, Nakashima S, Cadwallader K, Gu Q, Cook S, et al. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and rho family monomeric GTP-binding proteins and protein kinase c-alpha. J Biol Chem (1997) 272(6):3860–8. doi:10.1074/jbc.272.6.3860
79. Jang J-H, Lee CS, Hwang D, Ryu SH. Understanding of the roles of phospholipase d and phosphatidic acid through their binding partners. Prog Lipid Res (2012) 51(2):71–81. doi:10.1016/j.plipres.2011.12.003
80. Dall’Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, et al. The phospholipase D1 pathway modulates macroautophagy. Nat Commun (2010) 1:142. doi:10.1038/ncomms1144
81. Bruntz RC, Lindsley CW, Brown HA. Phospholipase D signaling pathways and phosphatidic acid as therapeutic targets in cancer. Pharmacol Rev (2014) 66(4):1033–79. doi:10.1124/pr.114.009217
82. Ahn MJ, Park SY, Kim WK, Cho JH, Chang BJ, Kim DJ, et al. A single nucleotide polymorphism in the phospholipase D1 gene is associated with risk of non-small cell lung cancer. Int J Biomed Sci (2012) 8(2):121–8.
83. Yamada Y, Hamajima N, Kato T, Iwata H, Yamamura Y, Shinoda M, et al. Association of a polymorphism of the phospholipase D2 gene with the prevalence of colorectal cancer. J Mol Med (2003) 81(2):126–31. doi:10.1007/s00109-002-0411-x
84. Min DS, Kwon TK, Park WS, Chang JS, Park SK, Ahn BH, et al. Neoplastic transformation and tumorigenesis associated with overexpression of phospholipase D isozymes in cultured murine fibroblasts. Carcinogenesis (2001) 22(10):1641–7. doi:10.1093/carcin/22.10.1641
85. Henkels KM, Boivin GP, Dudley ES, Berberich SJ, Gomez-Cambronero J. Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene (2013) 32(49):5551–62. doi:10.1038/onc.2013.207
86. Chen Y, Zheng Y, Foster DA. Phospholipase d confers rapamycin resistance in human breast cancer cells. Oncogene (2003) 22(25):3937–42. doi:10.1038/sj.onc.1206565
87. Zheng Y, Rodrik V, Toschi A, Shi M, Hui L, Shen Y, et al. Phospholipase D couples survival and migration signals in stress response of human cancer cells. J Biol Chem (2006) 281(23):15862–8. doi:10.1074/jbc.M600660200
88. Gadiya M, Mori N, Cao MD, Mironchik Y, Kakkad S, Gribbestad IS, et al. Phospholipase D1 and choline kinase-alpha are interactive targets in breast cancer. Cancer Biol Ther (2014) 15(5):593–601. doi:10.4161/cbt.28165
89. Knoepp SM, Chahal MS, Xie Y, Zhang Z, Brauner DJ, Hallman MA, et al. Effects of active and inactive phospholipase D2 on signal transduction, adhesion, migration, invasion, and metastasis in EL4 lymphoma cells. Mol Pharmacol (2008) 74(3):574–84. doi:10.1124/mol.107.040105
90. Kang DW, Choi CY, Cho Y-H, Tian H, Di Paolo G, Choi K-Y, et al. Targeting phospholipase D1 attenuates intestinal tumorigenesis by controlling β-catenin signaling in cancer-initiating cells. J Exp Med (2015) 212(8):1219–37. doi:10.1084/jem.20141254
91. Jang YH, Choi KY, Min DS. Phospholipase D-mediated autophagic regulation is a potential target for cancer therapy. Cell Death Differ (2014) 21(4):533–46. doi:10.1038/cdd.2013.174
92. Zhang Y, Frohman MA. Cellular and physiological roles for phospholipase D1 in cancer. J Biol Chem (2014) 289(33):22567–74. doi:10.1074/jbc.R114.576876
93. Su W, Yeku O, Olepu S, Genna A, Park JS, Ren H, et al. 5-fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase d pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol (2009) 75(3):437–46. doi:10.1124/mol.108.053298
94. Chen Q, Hongu T, Sato T, Zhang Y, Ali W, Cavallo J-A, et al. Key roles for the lipid signaling enzyme phospholipase D1 in the tumor microenvironment during tumor angiogenesis and metastasis. Sci Signal (2012) 5(249):ra79. doi:10.1126/scisignal.2003257
95. Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, et al. Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat Chem Biol (2009) 5(2):108–17. doi:10.1038/nchembio.140
96. Cheol Son J, Woo Kang D, Mo Yang K, Choi K-Y, Gen Son T, Min DS. Phospholipase D inhibitor enhances radiosensitivity of breast cancer cells. Exp Mol Med (2013) 45:e38. doi:10.1038/emm.2013.75
97. Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers MEJ, et al. Impaired α(IIb)β(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci Signal (2010) 3(103):ra1. doi:10.1126/scisignal.2000551
98. Oliveira TG, Chan RB, Tian H, Laredo M, Shui G, Staniszewski A, et al. Phospholipase D2 ablation ameliorates Alzheimer’s disease-linked synaptic dysfunction and cognitive deficits. J Neurosci (2010) 30(49):16419–28. doi:10.1523/JNEUROSCI.3317-10.2010
99. Exton JH. Phosphatidylcholine breakdown and signal transduction. Biochim Biophys Acta (1994) 1212(1):26–42. doi:10.1016/0005-2760(94)90186-4
100. Johansen T, Bjorkoy G, Overvatn A, Diaz-Meco MT, Traavik T, Moscat J. NIH 3T3 cells stably transfected with the gene encoding phosphatidylcholine-hydrolyzing phospholipase C from Bacillus cereus acquire a transformed phenotype. Mol Cell Biol (1994) 14(1):646–54. doi:10.1128/MCB.14.1.646
101. Spadaro F, Ramoni C, Mezzanzanica D, Miotti S, Alberti P, Cecchetti S, et al. Phosphatidylcholine-specific phospholipase C activation in epithelial ovarian cancer cells. Cancer Res (2008) 68(16):6541–9. doi:10.1158/0008-5472.CAN-07-6763
102. Iorio E, Ricci A, Bagnoli M, Pisanu ME, Castellano G, Di Vito M, et al. Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res (2010) 70(5):2126–35. doi:10.1158/0008-5472.CAN-09-3833
103. Abalsamo L, Spadaro F, Bozzuto G, Paris L, Cecchetti S, Lugini L, et al. Inhibition of phosphatidylcholine-specific phospholipase C results in loss of mesenchymal traits in metastatic breast cancer cells. Breast Cancer Res (2012) 14(2):R50. doi:10.1186/bcr3151
104. Cecchetti S, Bortolomai I, Ferri R, Mercurio L, Canevari S, Podo F, et al. Inhibition of phosphatidylcholine-specific phospholipase C interferes with proliferation and survival of tumor initiating cells in squamous cell carcinoma. PLoS One (2015) 10(9):e0136120. doi:10.1371/journal.pone.0136120
105. Paris L, Cecchetti S, Spadaro F, Abalsamo L, Lugini L, Pisanu ME, et al. Inhibition of phosphatidylcholine-specific phospholipase C downregulates HER2 overexpression on plasma membrane of breast cancer cells. Breast Cancer Res (2010) 12(3):R27. doi:10.1186/bcr2575
106. Amtmann E. The antiviral, antitumoural xanthate D609 is a competitive inhibitor of phosphatidylcholine-specific phospholipase C. Drugs Exp Clin Res (1996) 22(6):287–94.
107. Voet DJ, Voet JG, Pratt CW. Lipids, bilayers and membranes. Principles of Biochemistry. 3rd ed. New York, NY: Wiley (2008).
108. Levade T, Jaffrézou J-P. Signalling sphingomyelinases: which, where, how and why? Biochim Biophys Acta (1999) 1438(1):1–17. doi:10.1016/S1388-1981(99)00038-4
109. Henry B, Möller C, Dimanche-Boitrel M-T, Gulbins E, Becker KA. Targeting the ceramide system in cancer. Cancer Lett (2013) 332(2):286–94. doi:10.1016/j.canlet.2011.07.010
110. Santana P, Pena LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, et al. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell (1996) 86(2):189–99. doi:10.1016/S0092-8674(00)80091-4
111. Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science (2003) 300(5622):1155–9. doi:10.1126/science.1082504
112. Garcia-Barros M, Thin TH, Maj J, Cordon-Cardo C, Haimovitz-Friedman A, Fuks Z, et al. Impact of stromal sensitivity on radiation response of tumors implanted in SCID hosts revisited. Cancer Res (2010) 70(20):8179–86. doi:10.1158/0008-5472.CAN-10-1871
113. Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism – a strategy for overcoming drug resistance. J Natl Cancer Inst (2001) 93(5):347–57. doi:10.1093/jnci/93.5.347
114. Lacour S, Hammann A, Grazide S, Lagadic-Gossmann D, Athias A, Sergent O, et al. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res (2004) 64(10):3593–8. doi:10.1158/0008-5472.CAN-03-2787
115. Dumitru CA, Weller M, Gulbins E. Ceramide metabolism determines glioma cell resistance to chemotherapy. J Cell Physiol (2009) 221(3):688–95. doi:10.1002/jcp.21907
116. Dumitru CA, Sandalcioglu IE, Wagner M, Weller M, Gulbins E. Lysosomal ceramide mediates gemcitabine-induced death of glioma cells. J Mol Med (2009) 87(11):1123–32. doi:10.1007/s00109-009-0514-8
117. Morad SAF, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer (2013) 13(1):51–65. doi:10.1038/nrc3398
118. Grassme H, Cremesti A, Kolesnick R, Gulbins E. Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene (2003) 22(35):5457–70. doi:10.1038/sj.onc.1206540
119. Kirkegaard T, Roth AG, Petersen NHT, Mahalka AK, Olsen OD, Moilanen I, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature (2010) 463(7280):549–53. doi:10.1038/nature08710
120. Groth-Pedersen L, Jäättelä M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett (2013) 332(2):265–74. doi:10.1016/j.canlet.2010.05.021
121. Petersen Nikolaj HT, Olsen Ole D, Groth-Pedersen L, Ellegaard A-M, Bilgin M, Redmer S, et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell (2013) 24(3):379–93. doi:10.1016/j.ccr.2013.08.003
122. Dobbelstein M, Moll U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat Rev Drug Discov (2014) 13(3):179–96. doi:10.1038/nrd4201
123. Sawai H, Domae N, Nagan N, Hannun YA. Function of the cloned putative neutral sphingomyelinase as lyso-platelet activating factor-phospholipase C. J Biol Chem (1999) 274(53):38131–9. doi:10.1074/jbc.274.53.38131
124. Reynolds CP, Maurer BJ, Kolesnick RN. Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett (2004) 206(2):169–80. doi:10.1016/j.canlet.2003.08.034
125. Wu BX, Rajagopalan V, Roddy PL, Clarke CJ, Hannun YA. Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J Biol Chem (2010) 285(23):17993–8002. doi:10.1074/jbc.M110.102988
126. Mansat-de Mas V, Bezombes C, Quillet-Mary A, Bettaieb A, D’Orgeix AD, Laurent G, et al. Implication of radical oxygen species in ceramide generation, c-Jun N-terminal kinase activation and apoptosis induced by daunorubicin. Mol Pharmacol (1999) 56(5):867–74.
127. Grazide S, Maestre N, Veldman RJ, Bezombes C, Maddens S, Levade T, et al. Ara-C- and daunorubicin-induced recruitment of Lyn in sphingomyelinase-enriched membrane rafts. FASEB J (2002) 16(12):1685–7. doi:10.1096/fj.01-0794fje
128. Bezombes C, Plo I, Mansat-De Mas V, Quillet-Mary A, Negre-Salvayre A, Laurent G, et al. Oxidative stress-induced activation of Lyn recruits sphingomyelinase and is requisite for its stimulation by Ara-C. FASEB J (2001) 15(9):1583–5.
129. Shamseddine AA, Clarke CJ, Carroll B, Airola MV, Mohammed S, Rella A, et al. P53-dependent upregulation of neutral sphingomyelinase-2: role in doxorubicin-induced growth arrest. Cell Death Dis (2015) 6:e1947. doi:10.1038/cddis.2015.268
130. Duan R-D, Cheng Y, Hansen G, Hertervig E, Liu J-J, Syk I, et al. Purification, localization, and expression of human intestinal alkaline sphingomyelinase. J Lipid Res (2003) 44(6):1241–50. doi:10.1194/jlr.M300037-JLR200
131. Dillehay DL, Webb SK, Schmelz EM, Merrill AH Jr. Dietary sphingomyelin inhibits 1,2-dimethylhydrazine-induced colon cancer in CF1 mice. J Nutr (1994) 124(5):615–20.
132. Hertervig E, Nilsson A, Nyberg L, Duan RD. Alkaline sphingomyelinase activity is decreased in human colorectal carcinoma. Cancer (1997) 79(3):448–53. doi:10.1002/(SICI)1097-0142(19970201)79:3<448::AID-CNCR4>3.0.CO;2-E
133. Hertervig E, Nilsson A, Bjork J, Hultkrantz R, Duan RD. Familial adenomatous polyposis is associated with a marked decrease in alkaline sphingomyelinase activity: a key factor to the unrestrained cell proliferation? Br J Cancer (1999) 81(2):232–6. doi:10.1038/sj.bjc.6690682
134. Chen Y, Zhang P, Xu SC, Yang L, Voss U, Ekblad E, et al. Enhanced colonic tumorigenesis in alkaline sphingomyelinase (NPP7) knockout mice. Mol Cancer Ther (2015) 14(1):259–67. doi:10.1158/1535-7163.MCT-14-0468-T
135. Wu J, Cheng Y, Nilsson A, Duan RD. Identification of one exon deletion of intestinal alkaline sphingomyelinase in colon cancer HT-29 cells and a differentiation-related expression of the wild-type enzyme in Caco-2 cells. Carcinogenesis (2004) 25(8):1327–33. doi:10.1093/carcin/bgh140
136. Cheng Y, Wu J, Hertervig E, Lindgren S, Duan D, Nilsson A, et al. Identification of aberrant forms of alkaline sphingomyelinase (NPP7) associated with human liver tumorigenesis. Br J Cancer (2007) 97(10):1441–8. doi:10.1038/sj.bjc.6604013
137. Groth-Pedersen L, Ostenfeld MS, Høyer-Hansen M, Nylandsted J, Jäättelä M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res (2007) 67(5):2217–25. doi:10.1158/0008-5472.can-06-3520
138. Katz-Brull R, Degani H. Kinetics of choline transport and phosphorylation in human breast cancer cells; NMR application of the zero trans method. Anticancer Res (1996) 16(3B):1375–80.
139. Yuan Z, Tie A, Tarnopolsky M, Bakovic M. Genomic organization, promoter activity, and expression of the human choline transporter-like protein 1. Physiol Genomics (2006) 26(1):76–90. doi:10.1152/physiolgenomics.00107.2005
140. Inazu M. Choline transporter-like proteins CTLs/SLC44 family as a novel molecular target for cancer therapy. Biopharm Drug Dispos (2014) 35(8):431–49. doi:10.1002/bdd.1892
141. Yamada T, Inazu M, Tajima H, Matsumiya T. Functional expression of choline transporter-like protein 1 (CTL1) in human neuroblastoma cells and its link to acetylcholine synthesis. Neurochem Int (2011) 58(3):354–65. doi:10.1016/j.neuint.2010.12.011
142. Eliyahu G, Kreizman T, Degani H. Phosphocholine as a biomarker of breast cancer: molecular and biochemical studies. Int J Cancer (2007) 120(8):1721–30. doi:10.1002/ijc.22293
143. Wang T, Li J, Chen F, Zhao Y, He X, Wan D, et al. Choline transporters in human lung adenocarcinoma: expression and functional implications. Acta Biochim Biophys Sin (2007) 39(9):668–74. doi:10.1111/j.1745-7270.2007.00323.x
144. Kouji H, Inazu M, Yamada T, Tajima H, Aoki T, Matsumiya T. Molecular and functional characterization of choline transporter in human colon carcinoma HT-29 cells. Arch Biochem Biophys (2009) 483(1):90–8. doi:10.1016/j.abb.2008.12.008
145. Inazu M, Yamada T, Kubota N, Yamanaka T. Functional expression of choline transporter-like protein 1 (CTL1) in small cell lung carcinoma cells: a target molecule for lung cancer therapy. Pharmacol Res (2013) 76:119–31. doi:10.1016/j.phrs.2013.07.011
146. Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I. Identification and characterization of the high-affinity choline transporter. Nat Neurosci (2000) 3(2):120–5. doi:10.1038/72059
147. Song P, Sekhon HS, Jia Y, Keller JA, Blusztajn JK, Mark GP, et al. Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res (2003) 63(1):214–21.
148. Song P, Sekhon HS, Lu A, Arredondo J, Sauer D, Gravett C, et al. M3 muscarinic receptor antagonists inhibit small cell lung carcinoma growth and mitogen-activated protein kinase phosphorylation induced by acetylcholine secretion. Cancer Res (2007) 67(8):3936–44. doi:10.1158/0008-5472.CAN-06-2484
149. Cheng K, Samimi R, Xie G, Shant J, Drachenberg C, Wade M, et al. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am J Physiol Gastrointest Liver Physiol (2008) 295(3):G591–7. doi:10.1152/ajpgi.00055.2008
150. Song P, Rekow SS, Singleton CA, Sekhon HS, Dissen GA, Zhou M, et al. Choline transporter-like protein 4 (CTL4) links to non-neuronal acetylcholine synthesis. J Neurochem (2013) 126(4):451–61. doi:10.1111/jnc.12298
151. Song P, Spindel ER. Basic and clinical aspects of non-neuronal acetylcholine: expression of non-neuronal acetylcholine in lung cancer provides a new target for cancer therapy. J Pharmacol Sci (2008) 106(2):180–5. doi:10.1254/jphs.FM0070091
152. Spindel E. Cholinergic targets in lung cancer. Curr Pharm Des (2016) 22(14):2152–9. doi:10.2174/1381612822666160127114237
153. Pingfang S, Jia Y, Singleton C, Shane SR, Eliot RS. Choline transporter-like protein 4 (CTL4) is preferentially linked to acetylcholine (ACh) secretion in small cell lung carcinoma. C66. Molecular Targets for Therapeutic Development in Lung Cancer. Denver, CO: American Thoracic Society International Conference Abstracts; American Thoracic Society (2011). p. A5079–A.
154. Pingfang S, Mark GP, Spindel ER. Knockdown of choline transporter-like protein 1 (CTL1) increases ACh secretion but decreases choline uptake in small cell lung carcinoma. B62. Lung Cancer Biomarkers and Therapeutic Response. New Orleans, LA: American Thoracic Society International Conference Abstracts; American Thoracic Society (2010). p. A3499–A.
155. Albright CD, Liu R, Bethea TC, Da Costa KA, Salganik RI, Zeisel SH. Choline deficiency induces apoptosis in SW40-immortalized CWSV-1 rat hepatocytes in culture. FASEB J (1996) 10(4):510–6.
156. Yen C-LE, Mar M-H, Zeisel SH. Choline deficiency-induced apoptosis in PC12 cells is associated with diminished membrane phosphatidylcholine and sphingomyelin, accumulation of ceramide and diacylglycerol, and activation of a caspase. FASEB J (1999) 13(1):135–42.
157. Ferguson SM, Bazalakova M, Savchenko V, Tapia JC, Wright J, Blakely RD. Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc Natl Acad Sci U S A (2004) 101(23):8762–7. doi:10.1073/pnas.0401667101
158. Gardiner JE. The inhibition of acetylcholine synthesis in brain by a hemicholinium. Biochem J (1961) 81(2):297–303. doi:10.1042/bj0810297
159. Corda D, Mosca MG, Ohshima N, Grauso L, Yanaka N, Mariggio S. The emerging physiological roles of the glycerophosphodiesterase family. FEBS J (2014) 281(4):998–1016. doi:10.1111/febs.12699
160. Zheng B, Berrie CP, Corda D, Farquhar MG. GDE1/MIR16 is a glycerophosphoinositol phosphodiesterase regulated by stimulation of G protein-coupled receptors. Proc Natl Acad Sci U S A (2003) 100(4):1745–50. doi:10.1073/pnas.0337605100
161. Gallazzini M, Ferraris JD, Burg MB. GDPD5 is a glycerophosphocholine phosphodiesterase that osmotically regulates the osmoprotective organic osmolyte GPC. Proc Natl Acad Sci U S A (2008) 105(31):11026–31. doi:10.1073/pnas.0805496105
162. Stewart JD, Marchan R, Lesjak MS, Lambert J, Hergenroeder R, Ellis JK, et al. Choline-releasing glycerophosphodiesterase EDI3 drives tumor cell migration and metastasis. Proc Natl Acad Sci U S A (2012) 109(21):8155–60. doi:10.1073/pnas.1117654109
163. Cao MD, Cheng M, Rizwan A, Jiang L, Krishnamachary B, Bhujwalla ZM, et al. Targeting choline phospholipid metabolism: GDPD5 and GDPD6 silencing decrease breast cancer cell proliferation, migration, and invasion. NMR Biomed (2016) 29(8):1098–107. doi:10.1002/nbm.3573
164. Wijnen JP, Jiang L, Greenwood TR, Cheng M, Dopkens M, Cao MD, et al. Silencing of the glycerophosphocholine phosphodiesterase GDPD5 alters the phospholipid metabolite profile in a breast cancer model in vivo as monitored by 31P MRS. NMR Biomed (2014) 27(6):692–9. doi:10.1002/nbm.3106
165. Cao MD, Döpkens M, Krishnamachary B, Vesuna F, Gadiya MM, Lonning PE, et al. Glycerophosphodiester phosphodiesterase domain containing 5 (GDPD5) expression correlates with malignant choline phospholipid metabolite profiles in human breast cancer. NMR Biomed (2012) 25(9):1033–42. doi:10.1002/nbm.2766
166. Park S, Lee C, Sabharwal P, Zhang M, Meyers CL, Sockanathan S. GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science (2013) 339(6117):324–8. doi:10.1126/science.1231921
167. Okazaki Y, Ohshima N, Yoshizawa I, Kamei Y, Mariggio S, Okamoto K, et al. A novel glycerophosphodiester phosphodiesterase, GDE5, controls skeletal muscle development via a non-enzymatic mechanism. J Biol Chem (2010) 285(36):27652–63. doi:10.1074/jbc.M110.106708
168. Lesjak MS, Marchan R, Stewart JD, Rempel E, Rahnenfuhrer J, Hengstler JG. EDI3 links choline metabolism to integrin expression, cell adhesion and spreading. Cell Adh Migr (2014) 8(5):499–508. doi:10.4161/cam.29284
169. Vance DE, Ridgway ND. The methylation of phosphatidylethanolamine. Prog Lipid Res (1988) 27(1):61–79. doi:10.1016/0163-7827(88)90005-7
170. Cui Z, Houweling M, Vance DE. Suppression of rat hepatoma cell growth by expression of phosphatidylethanolamine N-methyltransferase-2. J Biol Chem (1994) 269(40):24531–3.
171. Tessitore L, Dianzani I, Cui Z, Vance DE. Diminished expression of phosphatidylethanolamine N-methyltransferase 2 during hepatocarcinogenesis. Biochem J (1999) 337(Pt 1):23–7. doi:10.1042/0264-6021:3370023
172. Tessitore L, Sesca E, Vance DE. Inactivation of phosphatidylethanolamine N-methyltransferase-2 in aflatoxin-induced liver cancer and partial reversion of the neoplastic phenotype by PEMT transfection of hepatoma cells. Int J Cancer (2000) 86(3):362–7. doi:10.1002/(SICI)1097-0215(20000501)86:3<362::AID-IJC10>3.3.CO;2-1
173. Tessitore L, Sesca E, Bosco M, Vance DE. Expression of phosphatidylethanolamine N-methyltransferase in Yoshida ascites hepatoma cells and the livers of host rats. Carcinogenesis (1999) 20(4):561–7. doi:10.1093/carcin/20.4.561
174. Tessitore L, Marengo B, Vance DE, Papotti M, Mussa A, Daidone MG, et al. Expression of phosphatidylethanolamine N-methyltransferase in human hepatocellular carcinomas. Oncology (2003) 65(2):152–8. doi:10.1159/000072341
175. Xu X, Gammon MD, Zeisel SH, Lee YL, Wetmur JG, Teitelbaum SL, et al. Choline metabolism and risk of breast cancer in a population-based study. FASEB J (2008) 22(6):2045–52. doi:10.1096/fj.07-101279
176. Li D, Bi FF, Chen NN, Cao JM, Sun WP, Zhou YM, et al. Epigenetic repression of phosphatidylethanolamine N-methyltransferase (PEMT) in BRCA1-mutated breast cancer. Oncotarget (2014) 5(5):1315–25. doi:10.18632/oncotarget.1800
177. Zinrajh D, Horl G, Jurgens G, Marc J, Sok M, Cerne D. Increased phosphatidylethanolamine N-methyltransferase gene expression in non-small-cell lung cancer tissue predicts shorter patient survival. Oncol Lett (2014) 7(6):2175–9. doi:10.3892/ol.2014.2035
178. Cui Z, Houweling M, Vance DE. Expression of phosphatidylethanolamine N-methyltransferase-2 in McArdle-RH7777 hepatoma cells inhibits the CDP-choline pathway for phosphatidylcholine biosynthesis via decreased gene expression of CTP:phosphocholine cytidylyltransferase. Biochem J (1995) 312(Pt 3):939–45. doi:10.1042/bj3120939
179. Zou W, Li ZY, Li YL, Ma KL, Tsui ZC. Overexpression of PEMT2 downregulates the PI3K/AKT signaling pathway in rat hepatoma cells. Biochim Biophys Acta (2002) 1581(1–2):49–56. doi:10.1016/S1388-1981(02)00120-8
180. Aoyama C, Nakashima K, Ishidate K. Molecular cloning of mouse choline kinase and choline/ethanolamine kinase: their sequence comparison to the respective rat homologs. Biochim Biophys Acta (1998) 1393(1):179–85. doi:10.1016/S0005-2760(98)00062-9
181. Aoyama C, Ohtani A, Ishidate K. Expression and characterization of the active molecular forms of choline/ethanolamine kinase-alpha and -beta in mouse tissues, including carbon tetrachloride-induced liver. Biochem J (2002) 363(Pt 3):777–84. doi:10.1042/0264-6021:3630777
182. Kersting MC, Choi HS, Carman GM. Regulation of the yeast EKI1-encoded ethanolamine kinase by inositol and choline. J Biol Chem (2004) 279(34):35353–9. doi:10.1074/jbc.M405704200
183. Lykidis A, Wang J, Karim MA, Jackowski S. Overexpression of a mammalian ethanolamine-specific kinase accelerates the CDP-ethanolamine pathway. J Biol Chem (2001) 276(3):2174–9. doi:10.1074/jbc.M008794200
184. Gambacorti-Passerini CB, Donadoni C, Parmiani A, Pirola A, Redaelli S, Signore G, et al. Recurrent ETNK1 mutations in atypical chronic myeloid leukemia. Blood (2015) 125(3):499–503. doi:10.1182/blood-2014-06-579466
185. Lasho TL, Finke CM, Zblewski D, Patnaik M, Ketterling RP, Chen D, et al. Novel recurrent mutations in ethanolamine kinase 1 (ETNK1) gene in systemic mastocytosis with eosinophilia and chronic myelomonocytic leukemia. Blood Cancer J (2015) 5:e275. doi:10.1038/bcj.2014.94
186. Zafarana G, Gillis AJ, van Gurp RJ, Olsson PG, Elstrodt F, Stoop H, et al. Coamplification of DAD-R, SOX5, and EKI1 in human testicular seminomas, with specific overexpression of DAD-R, correlates with reduced levels of apoptosis and earlier clinical manifestation. Cancer Res (2002) 62(6):1822–31.
187. Negendank W. Studies of human tumors by MRS: a review. NMR Biomed (1992) 5(5):303–24. doi:10.1002/nbm.1940050518
188. Ferreira AK, Meneguelo R, Marques FL, Radin A, Filho OM, Neto SC, et al. Synthetic phosphoethanolamine a precursor of membrane phospholipids reduces tumor growth in mice bearing melanoma B16-F10 and in vitro induce apoptosis and arrest in G2/M phase. Biomed Pharmacother (2012) 66(7):541–8. doi:10.1016/j.biopha.2012.04.008
189. Ferreira AK, Meneguelo R, Pereira A, Mendonca Filho O, Chierice GO, Maria DA. Anticancer effects of synthetic phosphoethanolamine on Ehrlich ascites tumor: an experimental study. Anticancer Res (2012) 32(1):95–104.
190. Ferreira AK, Santana-Lemos BA, Rego EM, Filho OM, Chierice GO, Maria DA. Synthetic phosphoethanolamine has in vitro and in vivo anti-leukemia effects. Br J Cancer (2013) 109(11):2819–28. doi:10.1038/bjc.2013.510
191. Ferreira AK, Freitas VM, Levy D, Ruiz JL, Bydlowski SP, Rici RE, et al. Anti-angiogenic and anti-metastatic activity of synthetic phosphoethanolamine. PLoS One (2013) 8(3):e57937. doi:10.1371/journal.pone.0057937
192. Ferreira AK, Meneguelo R, Pereira A, Filho OM, Chierice GO, Maria DA. Synthetic phosphoethanolamine induces cell cycle arrest and apoptosis in human breast cancer MCF-7 cells through the mitochondrial pathway. Biomed Pharmacother (2013) 67(6):481–7. doi:10.1016/j.biopha.2013.01.012
193. Clancy CE, An G, Cannon WR, Liu Y, May EE, Ortoleva P, et al. Multiscale modeling in the clinic: drug design and development. Ann Biomed Eng (2016) 44(9):2591–610. doi:10.1007/s10439-016-1563-0
Keywords: cancer, target, phospholipid, metabolism, choline, ethanolamine
Citation: Cheng M, Bhujwalla ZM and Glunde K (2016) Targeting Phospholipid Metabolism in Cancer. Front. Oncol. 6:266. doi: 10.3389/fonc.2016.00266
Received: 31 July 2016; Accepted: 14 December 2016;
Published: 27 December 2016
Edited by:
Natalie Julie Serkova, University of Colorado School of Medicine, USAReviewed by:
Aslam Khan, University of Missouri, USAEllen Ackerstaff, Memorial Sloan Kettering Cancer Center, USA
Copyright: © 2016 Cheng, Bhujwalla and Glunde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristine Glunde, kglunde@mri.jhu.edu