 Kathryn S. Potts1,2
Kathryn S. Potts1,2 Teresa V. Bowman1,2,3*
Teresa V. Bowman1,2,3*
- 1Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, Bronx, NY, United States
- 2Gottesman Institute for Stem Cell Biology and Regenerative Medicine, Albert Einstein College of Medicine, Bronx, NY, United States
- 3Department of Medicine (Oncology), Albert Einstein College of Medicine, Bronx, NY, United States
Human myeloid malignancies represent a substantial disease burden to individuals, with significant morbidity and death. The genetic underpinnings of disease formation and progression remain incompletely understood. Large-scale human population studies have identified a high frequency of potential driver mutations in spliceosomal and epigenetic regulators that contribute to malignancies, such as myelodysplastic syndromes (MDS) and leukemias. The high conservation of cell types and genes between humans and model organisms permits the investigation of the underlying mechanisms of leukemic development and potential therapeutic testing in genetically pliable pre-clinical systems. Due to the many technical advantages, such as large-scale screening, lineage-tracing studies, tumor transplantation, and high-throughput drug screening approaches, zebrafish is emerging as a model system for myeloid malignancies. In this review, we discuss recent advances in MDS and leukemia using the zebrafish model.
Introduction
Myeloid malignancies are clonal disorders of hematopoietic stem and progenitor cells in which there is bone marrow failure, an overgrowth of blasts, differentiation arrest, and lineage skewing. These malignancies include chronic disorders such as myelodysplastic syndrome (MDS), myeloproliferative neoplasm (MPN), and chronic myeloid leukemia (CML) and acute conditions such as acute myeloid leukemia (AML). These disorders are distinguished by the prevailing cell type, pathogenic severity, prognosis, and molecular underpinning. The etiology of most myeloid malignancies is poorly characterized; however, recent large-scale sequencing of patient samples has uncovered key recurrent classes of mutated factors (1–4). Through these studies, researchers identified genetic alterations in factors involved with gene expression regulation including hematopoietic transcription factors, spliceosomal components, and epigenetic regulators. Although genotype–phenotype correlations between a mutated gene and disease state are highly suggestive of causation, model organisms provide a controlled approach to demonstrate the connections between genetic alteration and blood defects as well as to determine the underlying mechanism in more uniform genetic backgrounds. Since the establishment of the first model of transplantable c-myc-driven T-cell acute lymphoblastic leukemia (T-ALL) (5), the zebrafish Danio rerio has emerged as a useful animal model to explore the control of both normal and malignant hematopoiesis.
Zebrafish Hematopoiesis
Most of the core regulators of hematopoiesis are evolutionarily conserved between teleosts, such as zebrafish, and mammals, such that findings in zebrafish can be directly translated into mouse and human systems. According to the recently completed and updated annotation of the zebrafish genome, approximately 70% of protein-coding genes in humans have at least one ortholog in the zebrafish, and 84% of disease-associated genes have a zebrafish equivalent (6). This extent of homology further demonstrates the potential utility of zebrafish to define critical regulators of malignancies and the underlying genetic causes. Having an in-depth understanding of the normal processes and signaling requirements underpinning hematopoietic lineage emergence and development provides a solid framework to understand how genetic perturbations exert their influence in disease states. Studying hematopoiesis during embryonic development can be advantageous to minimize the accumulation of environmental influences acquired through the life of an organism. Utilizing the zebrafish model to study embryonic hematopoiesis has a myriad of advantages including high fecundity, rapid external embryonic development, organismal transparency, numerous hematovascular fluorescent reporter lines, genetic tractability, and similar chronological and spatial lineage emergence kinetics and regulation to mammals.
Like other vertebrates, zebrafish hematopoiesis develops in three discrete waves: primitive, erythro-myeloid progenitor (EMP)-derived, and definitive (7). All three waves of hematopoiesis arise from lateral mesoderm-derived cells that possess different hematopoietic differentiation capacity. The primitive hematopoietic wave arises during the first 24 h post fertilization (hpf) from two locations: the anterior lateral mesoderm generates myeloid lineages, and the intermediate cell mass generates primitive myeloid and erythroid cells. Emergent primitive myeloid cells then migrate around the yolk sac and differentiate into distinct lineages, up-regulating expression of spi/pu.1, colony stimulating factor 1 receptor (csf1r/fms), csf3r, l-plastin, and myeloperoxidase (mpo/mpx), while the primitive gata1-expressing erythroid cells upregulate the levels of erythropoietin receptor (epor) and globin genes then enter circulation (8–14). The transient EMP wave derives from the posterior blood island and differentiates to form definitive erythroid and myeloid cells that lack self-renewal or multilineage differentiation capacity (15, 16). Despite the mostly transient nature of these waves, new findings from the past several years indicate that some myeloid progenitors from the primitive and EMP wave could persist in adulthood and provide the pool for microglia (macrophages in the brain) and other tissue-resident macrophages (17–19). It will be interesting to see if these embryonically derived cells play a role in human disease.
The final wave of hematopoietic specification gives rise to definitive adult-like hematopoietic stem cells (HSCs), which possess both self-renewal capacity and erythroid, myeloid, and lymphoid potential. Starting from approximately 30 hpf, HSCs emerge from kinase insert domain receptor-like (kdrl)-positive endothelium lining the ventral wall of the dorsal aorta, equivalent to the mammalian aorta-gonad-mesonephros region (16, 20, 21). The newly emergent HSCs transiently co-express endothelial markers, such as kdrl, and HSC markers, such as cd41 and the transcription factors cmyb and runt-related transcription factor 1 (runx1) (15, 16). From approximately 48–72 hpf, HSCs then migrate via the circulation to the caudal hematopoietic tissue (CHT) (22), the functional equivalent of the mammalian fetal liver. Between 48–96 hpf, cells from the CHTs will then seed the thymus for T-cell production or the kidney marrow, which functions as the adult hematopoietic niche similar to the mammalian bone marrow (15).
The zebrafish model affords many advantages for investigating mechanisms underlying normal and malignant myelopoiesis [reviewed in Ref. (23)]. The fate determination, differentiation, and maturation of myeloid cells are highly similar from embryonic development to adulthood. As such, studies of myeloid development in zebrafish have been informative not only for understanding embryonic development but also for adult myeloid malignancies [reviewed in Ref. (23)]. Myeloid precursors express conserved transcription factors such as spi1/pu.1, runx1, and ccaat-enhancer binding protein alpha (cebpα) that are critical in myeloid lineage commitment (11, 24, 25). Mature myeloid cell types with similarities or equivalence to well-defined mammalian lineages have been identified in zebrafish development based on their expression of signature genes, histochemical staining properties, and morphology: macrophages that express genes such as l-plastin, lysozyme (lyz), and csfr1/fms; neutrophils that express mpx and matrix metalloproteinase 9 (mmp9); and basophils/eosinophils that have high levels of gata2 expression (9, 24, 26, 27).
Zebrafish also possess many technical advantages. Targeted genetic manipulations allow for rapid alteration of gene function, including anti-sense morpholino knockdown (MO), zinc finger nucleases, transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) technologies for precision genome editing [reviewed in Ref. (28)]. Exogenous expression of proteins of interest is also possible in zebrafish, either through transient introduction of in vitro transcribed mRNA or through stable integration of DNA, most commonly via the Tol2 transposon-based transgenesis system (29, 30). Phenotype-driven genetic or chemical screening approaches are commonly employed due to the large clutch size, rapid generation time, and ease of drug treatment by infusion in the water. External and transparent embryonic development in combination with the multitude of fluorescent reporter lines enables sophisticated in vivo live imaging of lineage emergence and cell dissemination. Transparent mutants, such as the casper line (31), improve imaging capacity in adult zebrafish. Transplantation of zebrafish-derived hematopoietic tumor cells has been utilized to quantify and define subsets of leukemic propagating cells, as well as to image tumor microenvironmental interactions (32–34).
We can therefore take advantage of the technical and genetic advantages of the zebrafish model to study the genetic basis of malignancy and translate the findings to inform a better understanding of human cancer biology for therapeutic application. Current pathways to develop therapeutics for disease treatment are costly, labor and animal intensive, and take 10–30 years from discovery of the molecule or pathway to having a drug in the clinic. It is therefore essential to utilize streamlined processes for in vivo testing of drug targets. The zebrafish model, with high fecundity, conservation of many key genes, and an extensive experimental toolbox, provides a high-throughput model for such in vivo analysis.
Myeloid Malignancies
Myelodysplastic syndrome and AML are among the most common myeloid malignancy of the elderly each affecting 3–5 out of 100,000 people in the USA with approximately 10,000–20,000 newly diagnosed cases per annum (35–39). Both malignancies stem from clonal HSC disorders and are characterized by bone marrow failure and peripheral blood cytopenias. A major distinguishing characteristic of AML is the presence of excessive (>20%) undifferentiated myeloid blast in the bone marrow or peripheral blood, which are generally low in MDS patients. MDS is thought to be a precursor syndrome to AML with up to 30% of MDS cases progressing to secondary AML.
Zebrafish Models of AML
The classic model of AML development states that cells accumulate molecular alterations (large chromosomal rearrangements or genetic point mutations) in two classes: those that promote proliferation (class I) and those that impair differentiation (class II) (40). Prognostic risk is stratified based on the cytogenetic and molecular mutation profile present in the leukemia. For example, cytologically normal FMS-like tyrosine kinase 3 (FLT3)-internal tandem duplication (FLT3-ITD) correlates with an adverse prognosis, while nucleophosmin 1 (NPM1) mutations are linked with favorable outcomes. Investigating the molecular mechanisms driving AML is difficult in human samples, thus disease models are examined in model organisms including mouse and zebrafish. The first cancer model established in zebrafish in the early 2000s was c-myc-driven T-ALL (5), but since then robust myeloid leukemia models have finally been established. Most of these models are based on exogenous expression of prominent human AML fusion oncogenes derived from chromosomal translocations. These oncogenes are generally considered to be potent drivers of AML, with expression of such mutations in zebrafish often resulting in severe, early-arising, embryonic lethal hematologic anomalies, which preclude the study of adult leukemia. Despite this limitation, by employing the pliable genetic and chemical advantages of studies in embryonic zebrafish, much has been discovered regarding underlying mechanisms of these AML-like phenotypes.
Chromosomal Rearrangements in Zebrafish AML Models
The chromosomal translocation t(8;21)(q22;q22) was one of the first molecular alterations identified in AML, with a frequency of 5–15% of all human AML cases. It results in the fusion of two nuclear proteins: acute myeloid leukemia 1 protein (AML1, also called RUNX1/CBFα2/PEBPαB) and eight twenty one [ETO, or myeloid translocation gene on chromosome 8 (MTG8/RUNX1T1)]. AML1 is a master transcriptional regulator of definitive hematopoiesis that binds enhancers and activates hematopoietic gene expression (41). Chromosomal translocations and mutations involving AML1 are associated with several forms of adult leukemia and childhood MDS (42, 43). ETO is broadly expressed in hematopoietic cells, including CD34+ progenitors, and acts as a nuclear localized zinc finger containing protein that normally recruits the nuclear receptor co-repressor/SIN3/histone deacetylase (HDAC) complex to induce transcriptional repression, including of AML1 (44–47). The AML1-ETO molecular subtype of leukemia is characterized by granulocyte precursor accumulation (48, 49). In zebrafish, transient induction of the human AML1-ETO oncogene during development could recapitulate the granulocytic lineage skewing observed in human patients (Figures 1A,B) (50). AML1-ETO expressing embryos displayed a biased expression of spi1/pu.1 in myelo-erythroid progenitors at the expense of gata1, resulting in an expansion of granulocytes (Figure 1C). This largely recapitulated both the differentiation changes observed in human patients and the phenotype in the mouse model (51), demonstrating the utility of the zebrafish system. Mechanistic studies revealed that the observed lineage skewing was mediated via modulation of the early fate choice transcription factor stem cell leukemia (scl). Yeh and colleagues then took advantage of the screening capability of the zebrafish and performed an unbiased chemical suppressor screen to find small molecules that could reverse the myeloid expansion in AML1-ETO-expressing zebrafish. They identified that cyclooxygenase 2 (COX-2) and β-catenin pathways were downstream of AML1-ETO and that HDAC inhibition by trichostatin A could therapeutically target the AML-like effects in the zebrafish model (Figure 1C).
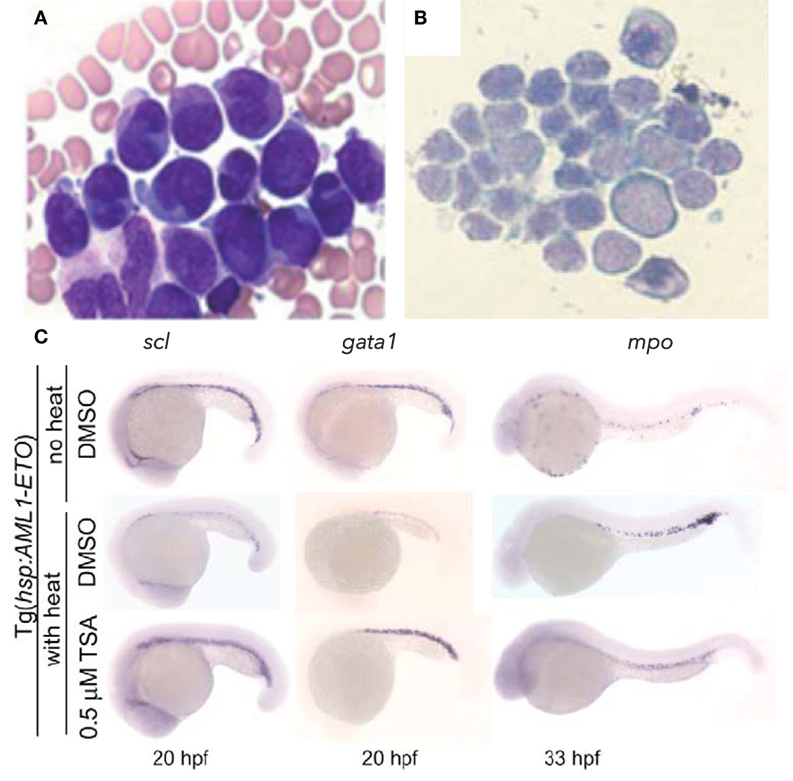
Figure 1. Hematopoietic phenotype conserved in zebrafish model of AML-ETO driven AML. (A,B) Wright–Giemsa stained blood cells from (A) human acute myeloid leukemia (AML) patient bone marrow smear demonstrating accumulation of promyelocytes [modified and published with permission from Ref. (52)] and (B) zebrafish peripheral blood smear showing accumulation of myeloid blasts from AML-ETO overexpressing embryos at 40 hpf. (C) Rescue of hematopoietic phenotype with trichostatin A (TSA) treatment. Inducible Tg[hps:AML1-ETO] line utilized, such that heat-shocked Tg embryos treated with DMSO develop AML-like phenotype, which can be reversed with TSA treatment. scl marks hematopoietic stem and progenitor cells; gata1 marks erythroid lineage; mpo marks myeloid lineage. Panels (B,C) modified and published with permission from Yeh et al. (50).
A zebrafish model of the chromosomal translocation t(9;12)(p24;p13) fusion oncogene ETS leukemia virus 6 (ETV6) and Janus kinase 2 (JAK2) has also been generated. ETV6 (also known as TEL) is an E26 transformation-specific (ETS) family transcription factor involved in early embryonic yolk sac angiogenesis and multilineage adult hematopoiesis including HSC survival (46, 53, 54). JAK2 is a non-receptor tyrosine kinase commonly involved in hematopoietic cytokine signaling cascades and crucial in erythro-myeloid differentiation and HSC maintenance and function (55). The TEL-JAK2 fusion product leads to constitutive activation of JAK2 kinase activity (56). TEL-JAK2 has been identified in lymphoid and myeloid malignancies, with fusion between TEL exon 5 and JAK2 exon 9 occurring in T-ALL, while TEL exon 5 is found to fuse with JAK2 exon 12 in CML (57). To generate a myeloid-restricted mutant zebrafish line, Onnebo and colleagues expressed the tel-jak2a fusion oncogene under the control of the spi1/pu.1 promoter (58). These transgenic animals have disrupted embryonic hematopoiesis, including anemia and expansion of the myeloid compartment. Of note, a subsequent study found that expression of the TEL exon 5-JAK2 exon 9 variant led to lymphoid-restricted defects, while expression of the TEL exon 5-JAK2 exon 12 variant produced myeloid-restricted phenotypes, consistent with prior clinical observations (59). These results indicate that the lineage selection for the specific TEL-JAK2 variant occurs via regulation of the downstream signaling rather than at the level of the chromosomal aberration.
Non-Fusion Oncogenes in Zebrafish AML Models
Gain-of-function mutations in FLT3 occur in ~30% of AML cases and correlate with poor prognosis (60). Mutations include the internal tandem duplication (FLT3-ITD) and point mutations in the tyrosine kinase domain (FLT3-TKD), both of which result in elevated tyrosine kinase activity (61, 62). FLT3 (also known as FLK2 and STK1) is expressed in human HSCs and is essential for adult HSC and immune hemostasis (63). He et al. established the function of zebrafish flt3 in hematopoietic development, demonstrating that MO knockdown of endogenous flt3 in zebrafish significantly impaired progenitor and myeloid differentiation (52). Transient expression of human FLT3-ITD via mRNA injections into embryos resulted in expansion of myeloid progenitors (pu.1+) and mature cells (mpx+ and cebpα+). Elevation of downstream signaling such as phosphorylation of Stat5, Erk1/2, and Akt was also observed indicating human FLT3-ITD can trigger established endogenous signals of Flt3 in the zebrafish. Transient expression of human FLT3-TKD (D835Y) also resulted in myeloid cell expansion, but to a lesser extent than the FLT3-ITD. To demonstrate the ability of the zebrafish to test relevant human drugs, He et al. treated FLT3-ITD and FLT3-TKD expressing zebrafish embryos with AC220, a tyrosine kinase inhibitor shown to have potent selectivity for FLT3 (52, 64). Consistent with inhibiting the kinase domain of FLT3, they found that AC220 partially rescued the myeloid effects of FLT3-ITD; however, this did not abrogate the FLT3-TKD phenotype. Subsequently, Lu and colleagues generated a stable transgenic line with myeloid-restricted (spi1/pu.1 promoter-driven) FLT3-ITD and found that these animals develop adult AML symptoms, further illustrating the conservation of function of this oncogene from zebrafish to humans (65).
Modeling of non-fusion oncogenes is also underway in zebrafish. NPM1 is a ubiquitously expressed nucleolar phosphoprotein that regulates multiple cellular processes and is the most frequently mutated gene in adult AML, occurring in ~30% of cases (4, 66). Mutations in NPM1 result in altered protein localization from the nucleus to the cytoplasm (termed NPMc+). Zebrafish have two NPM1 orthologs, npm1a and npm1b (67). Double MO knockdown of both paralogs results in the production of fewer myeloid cells. Global transient expression of human NPMc+, the human mutant cytoplasmic protein, but not wild-type NPM1, resulted in increased spi1/pu.1+ myeloid precursors, mpx+ granulocytes and csf1r+ macrophages (67). Of note, the myeloid expansion from NPMc+ expression was enhanced in a p53-deficient background, suggesting that too much NPMc+ could trigger apoptosis. In line with this finding, a recent study showed that NPM1 acts as a scaffold for the apoptotic apparatus termed the PIDDosome [p53-induced death domain-containing protein 1—receptor-interacting protein-associated ICH-1/CED-3 homologous protein with a death domain (PIDD-RAIDD)-caspase-2 complex] (68). NPMc+ expression also increased HSC levels within the dorsal aorta, indicating a possible role for mutated NPM1 in leukemic stem cell development. Additionally, NPMc+ was shown to activate canonical Wnt signaling in early zebrafish development, which contributed to hematopoietic cell expansion (69). The elevation of WNT signaling was confirmed in human NPMc+ AML blasts, which was reversed by knockdown of the mutant NPMc+ transcript. Together these finding illustrate how studies in zebrafish embryogenesis can inform mechanism in human AML.
Mutations in isocitrate dehydrogenase 1 and 2 (IDH1/2) are found in ~8% of AML cases (70). IDH1/2 are enzymes that catalyze the oxidative decarboxylation of isocitrate producing α-ketoglutarate. AML-associated mutations in IDH1/2 perturb this function, altering the citric acid cycle, and leading to production of the oncometabolite 2-hydroxyglutarate, which alters DNA methylation via inhibition of ten-eleven translocation 2 (TET2) (70, 71). When idh1 levels were diminished in zebrafish using morpholino or TALEN approaches, Shi et al. observed expansion of pu.1+ precursors, impaired myeloid differentiation and reduced HSC formation (72). In contrast, when idh2 was diminished, the zebrafish displayed similar myeloid cell defects to idh1 mutants, but normal formation of HSCs, indicating a functional redundancy between the two idh factors during early embryonic HSC formation. Expression of human oncogenic IDH1-R132H in wild-type zebrafish induced myeloid compartment expansion that was suppressed by treatment with the potent and selective IDH inhibitor AGI-5198. These studies demonstrate that leukemogenic pathways are conserved between humans and zebrafish and illustrate how zebrafish can be used for therapeutically relevant drug studies.
Adult AML Models in Zebrafish
These embryonic models demonstrate that partial AML phenotypes can be recapitulated in embryonic zebrafish, which can be useful for mechanistic studies and drug discovery, but do not represent a full adult-arising leukemia. The first adult model of AML in zebrafish was based on the inv(8)(p11;q13) chromosomal translocation resulting in the oncogenic fusion of MYST3 (also known as MOZ, YBFR2, SAS2, TIP60 family histone acetyltransferase monocytic leukemia 3) and nuclear co-activator 2 [NCOA2, also called transcriptional mediator/intermediary factor 2 (TIF2)]. MYST3 is in the MYST family of histone acetyltransferases, while NCOA2 is a member of the p160 HAT family [reviewed in Ref. (73)]. To promote AML formation in zebrafish, Zhuravleva and colleagues expressed the human MYST3-NCOA2 (referred to as MOZ-TIF2) oncogene under the zebrafish spi1/pu.1 promoter (74). This resulted in development of AML after 14–26 months with immature myeloid blast accumulation in the kidney marrow, but decreased progenitors and lymphocytes in the spleen. However, such an AML phenotype was a rare event (2/180), indicating inefficient transformation, insufficient expression levels driven from the pu.1 promoter, or perhaps the requirement for a secondary mutation for disease development. Due to the long latency and low penetrance, there have not been further studies with this model.
The t(7;11)(p15;15) chromosomal translocation leading to nuclear pore complex protein 98 (NUP98)-homeobox protein A9 (HOXA9) oncogenic fusion is widely observed in hematological pathologies including MDS, CML, and AML, and correlates with poor prognosis. NUP98 is involved in nuclear trafficking (75), and HOXA9 is a vertebrate transcription factor essential in hematopoiesis with >80% of human AML showing overexpression of HOXA9 (76). Utilizing a novel Cre-LoxP system that allowed for both myeloid-restricted and heat-shock inducible expression [Tg(spi1:loxP-EGFP-loxP:NUP98-HOXA9); Tg(hsp70:cre)], Forrester and colleagues were able to explore the effects of conditional expression of the human NUP98-HOXA9 both in the embryo as in the studies described above, but also in adulthood. The later inducible expression is key to circumvent any deleterious events from early embryonic expression that could preclude analysis of disease in older animals. Similar to the mouse model (77), induction of human NUP98-HOXA9 expression resulted in ~23% of transgenic fish developing preleukemic MPN by 2 years (78). Examination of embryos with NUP98-HOXA9 expression revealed early lineage skewing where pu.1+ myeloid progenitors were enhanced at the expense of gata1+ erythroid progenitors, with perturbed myeloid lineage differentiation. A follow-up study that employed chemical approaches to determine the driving mechanism as well as potential new therapeutics for AML revealed that NUP98-HOXA9 upregulates prostaglandin synthase 2 (ptgs2) to expand HSC numbers (79), a pathway identified in prior studies to be important for normal HSC formation (80, 81). Blocking prostaglandin production with COX inhibitors could reverse the increase in HSCs, suggesting a role for this pathway in leukemic stem cell expansion. Additionally, gene expression analyses showed NUP98-HOXA9 elevated the expression of the epigenetic modifier dna methyltransferase 1 (dnmt1), which lead to hypermethylation. Treatment with an HDAC inhibitor reversed this phenotype. Both of these pathways were also identified as suppressors of myeloid expansion in the AML-ETO model, suggesting that they could play a more general role in AML induction.
Modeling MDS in Zebrafish
Myelodysplastic syndromes are a diverse group of chronic myeloid pathologies defined by perturbed clonal hematopoiesis, impaired differentiation and peripheral blood cytopenias with the potential to transform into AML. Substantial research efforts have been invested in understanding the drivers of MDS toward improving diagnosis and stratification of subtypes, which will improve treatment of patients. Until recently the etiology of the heterogeneous clinical outcomes of MDS was unclear. Extensive genomic analyses in recent years have revealed that some subtypes of MDS correlate strongly with mutations in spliceosomal or epigenetic factors (1–3, 82). Mutations in spliceosomal machinery are common and thought to be critical drivers in MDS pathogenesis. They have been identified in approximately 60% of all MDS cases (2), with mutations in splicing factor 3B, subunit 1 (SF3B1) observed in 80–90% of cases with the refractory anemia with ringed sideroblast (RARS) subtype (1, 3, 83, 84). Epigenetic factors are mutated in approximately 45% of MDS cases (2) with mutations in the methylcytosine dioxygenase TET2 being the most prevalently observed in 30% of MDS (2, 82). The function of splicing and epigenetic factors in MDS is still elusive as their role in normal hematopoietic development is unclear. The best way to clarify their function is to generate and study in vivo animal models to gain an organism-wide context of normal and perturbed gene function throughout lineage emergence, differentiation, and niche interactions.
Spliceosomal Components in MDS
The spliceosome is a large complex within eukaryotic nuclei encompassing five small nuclear ribonucleo-proteins (snRNPs) comprising RNAs and the associated protein molecules [reviewed in Ref. (85)]. The components, structure, and the function of the spliceosome are highly conserved throughout evolution in yeast, teleosts, and mammals. The high conservation of the spliceosome permits experiments from across eukarya to inform human spliceosome function and regulation. The function of the spliceosome is to remove introns from newly transcribed pre-mRNAs, resulting in mature mRNAs that are then translated by ribosomes to generate proteins. Splicing is a dynamic, highly coordinated process, thus its correct action is essential for normal functioning of cells. The major U2-type spliceosome comprises U1, U2, U4, U5, and U6 snRNPs (Figure 2A) and catalyzes the majority of splicing events, while the U12-type minor spliceosome has a specific target subset. Alternative splicing to generate multiple transcript variants for each gene occurs normally throughout development and is regulated in a tissue-specific manner. However, it can also occur as a result of spliceosomal dysfunction. MDS-associated mutations in spliceosomal components can lead to specific alternative splicing events, which correlate with their function in splicing. For example, SF3B1-containing complexes bind the branch point site within introns, and cells with MDS-associated SF3B1 mutations show defects in branch site selection, which can result in alternative proteins or unstable mRNA (Figure 2B) (86–90).
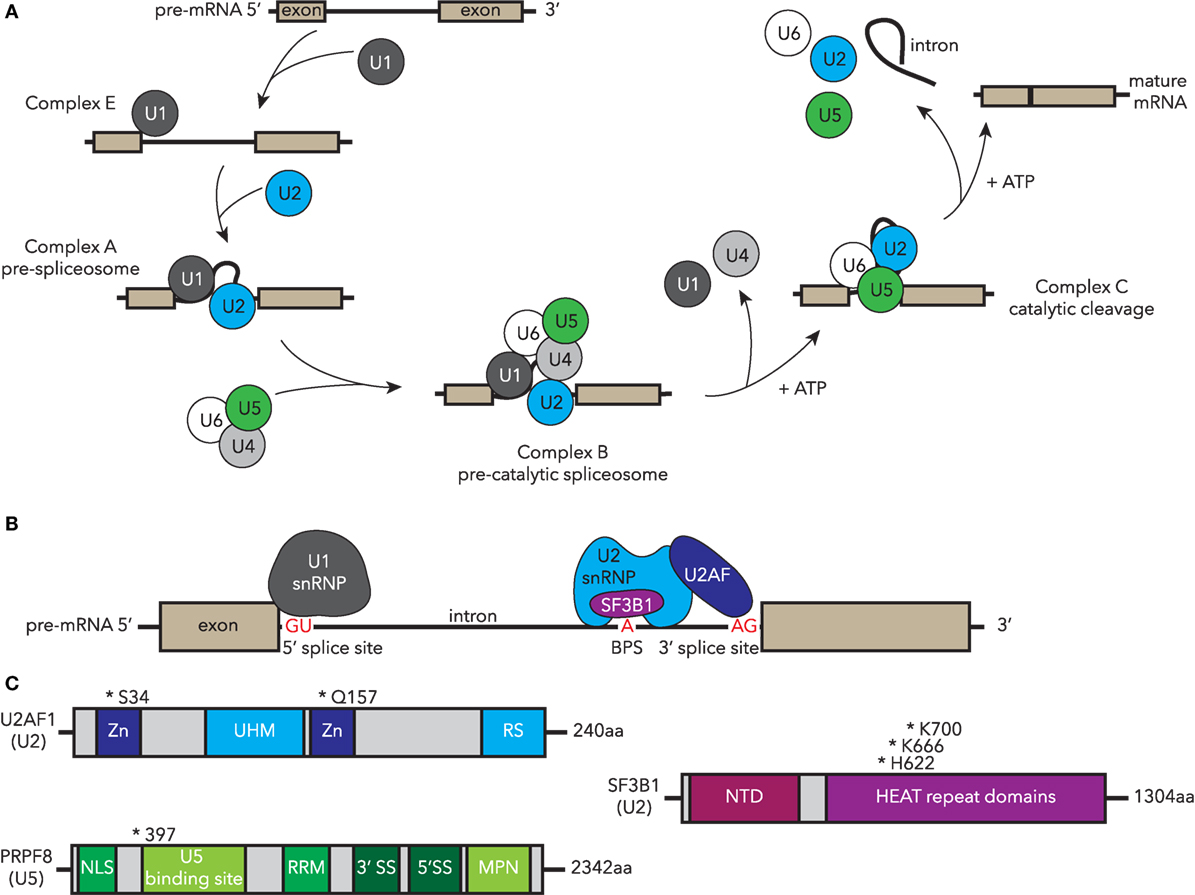
Figure 2. Human MDS-associated mutations in essential components of the spliceosome are conserved in zebrafish disease models. (A) Spliceosomal processing of pre-mRNA to mature transcript, indicating recruitment of snRNP complexes. Complexes containing MDS-mutated factors that have been studied in zebrafish are highlighted in blue (U2) and green (U5). (B) Essential components of complex A, including binding of the U1 snRNP to the 5′ splice site (SS), and U2 snRNP U2AF recognition and binding of AG in the 3′ SS, while SF3B1 recognizes the branch point site (BPS). (C) Structure and common MDS-associated mutations in U2-associated components U2AF1 and SF3B1 and U5 PRPF8. Zn, zinc finger domain; UHM, U2AF homology motif; RS, arginine-serine domain; NTD, N-terminal domain; NLS, nuclear localization sequence; RRM, RNA recognition motif; MDS, myelodysplastic syndrome; snRNP, small nuclear ribonucleo-protein; U2AF1, U2 small auxiliary factor 1; SF3B1, splicing factor 3B, subunit 1; PRPF8, pre-mRNA processing factor 8.
Splicing factor 3B, subunit 1 is a core component of the U2 snRNP and is one of the most highly mutated spliceosomal factors in MDS (2, 82). In addition to MDS, mutations in SF3B1 have been identified in other types of leukemia such as CLL (91, 92), and several solid organ malignancies, including pancreatic cancer (93), breast cancer (94, 95), and uveal melanoma (96, 97). Mutations in SF3B1 are strongly correlated with the ring sideroblast phenotype in MDS and are associated with better prognostic outcomes including a decreased risk of AML evolution (82, 83, 98). In SF3B1, most mutations cluster within the HUNTINGTON-ELONGATION FACTOR 3-PR65/A-TOR (HEAT) repeats in the C-terminus of the protein particularly in residues K700, K666, and H662 (Figure 2C) (83, 99). Recent data suggest that the HEAT repeat domains mediate protein–protein interactions (90). How these point mutations alter SF3B1 function and why this leads to hematologic dysfunction is unclear in part due to the limited understanding of the general signaling mechanism through which SF3B1 usually regulates hematopoiesis. To address this latter question, an sf3b1 loss-of-function zebrafish mutant was studied to understand the normal function of Sf3b1 in hematopoiesis and development (100). The homozygous sf3b1hi3394a loss-of-function mutants displayed an arrest of primitive hematopoiesis in both myeloid and erythroid lineages, which occurred after specification presenting as a block in differentiation and proliferation. In contrast, specification of definitive HSCs was hindered, despite the normal specification and differentiation of the non-hemogenic endothelial cells within the dorsal aorta. The lower production of mature blood cells coupled with poor HSC output was reminiscent of an MDS phenotype. HSC emergence from hemogenic endothelium is a NOTCH-dependent process (101). NOTCH signaling was normal in sf3b1 mutant zebrafish, indicating that the HSC induction defect is downstream or NOTCH-independent. These studies establish the importance of Sf3b1 somewhat selectively in hematopoiesis as other tissues such as the vasculature develop normally. How MDS-associated point mutants behave in this context requires further study. Recently, murine models of the most common MDS-associated point mutation (Sf3b1+/K700E) were generated and can be used to follow-up potential mechanisms identified in unbiased screening systems such as the zebrafish or human cell culture (102, 103).
U2 small auxiliary factor 1 (U2AF1) is mutated in 8–20% of MDS patients with the most common mutations occurring at residues S34 and Q157 (Figures 2B,C) (2, 99, 104, 105). Mutated U2AF1 in MDS causes aberrant splicing and is associated with increased risk of AML evolution. During splicing, SF3B1 interacts with the U2AF complex to help establish the 3′ splice site and splicing fidelity (Figure 2B) (106). Similar to sf3b1 mutants, homozygous loss-of-function u2af1hi199 mutant zebrafish have fewer definitive HSCs, develop anemia, and have elevated tp53 transcript levels, phenotypes which are all observed in MDS (107). Knockdown of tp53 via MO injections suppressed these hematologic defects suggesting it as a downstream mediator of u2af1 phenotypes. This model can therefore be used to further dissect the mechanism underlying U2AF1 and p53 activation in MDS.
Recurrent point mutations in the pre-mRNA processing factor 8 (PRPF8) have been reported in MDS and AML, correlating with increased myeloid progenitors, ring sideroblasts, and overall poor prognosis (108, 109). PRPF8 is a highly conserved component of the U5 snRNP that plays a role in both U2- and U12-spliceosomal processing (Figures 2A,C) (110). A zebrafish loss-of-function prpf8 mutant (called cephalophonus/cphgl1) was identified through a forward genetic screen for factors that regulate embryonic myelopoiesis (111). The cph/prpf8 homozygous mutants have defective myeloid and erythroid development, but unlike sf3b1 and u2af1 mutants, they show normal formation of definitive HSCs.
These spliceosomal mutants have some overlapping, but also distinct phenotypes. These findings are consistent with what is observed clinically; patients harboring mutations in different splicing factors share some disease features, but also have distinguishing characteristics. This suggests that although all factors are part of the spliceosome, their individual functions either within or outside of the spliceosome contribute to specific facets of disease. Using these zebrafish models will permit unbiased mechanistic explorations into these functions.
TET2 in MDS
Epigenetics is the study of changes in gene expression patterns regulated by non-genomic modifications without altering the DNA sequence. Epigenetic marks are transmitted through DNA methylation, histone modifications including acetylation and methylation of histone tails, RNA interference, and nuclear organization, thereby modulating transcriptional activation and silencing [reviewed in Ref. (112)]. Such epigenetic marks are heritable, allowing for transgenerational inheritance of non-genetic traits. Epigenetics has established critical roles in embryonic development, maternal/paternal gene imprinting, X inactivation, and disease. In cancer, there is a high prevalence of DNA hypermethylation and histone modification [reviewed in Ref. (113)]. Specifically in MDS and AML, many of the top class of mutated factors are epigenetic modifiers, which have opened the hematology field to delve into the role of epigenetics in normal and diseased hematopoiesis. The factors controlling epigenetic patterns and inheritance are highly conserved between teleosts and mammals, making zebrafish an excellent model to explore how mutations in this process lead to hematologic dysfunction.
Ten-eleven translocation proteins (TET1/2/3), a family of methylcytosine oxidases, function as epigenetic regulators of the genome methylation state. They catalyze the oxidation of 5-methylcytosine to 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxycytosine (114), which are key intermediates in DNA demethylation. Controlled methylation and de-methylation are crucial for embryonic development and control of gene expression (115). Somatic deletions and loss-of-function mutations in TET2 frequently occur in myeloid malignancies: ~30% of MDS and ~10% of de novo AML cases. Tet2-deficient mouse models have shown the function of TET2 in HSC self-renewal and differentiation, with myeloid defects reminiscent of MDS and AML (116, 117). Zinc finger nuclease technology was utilized to generate a homozygous tet2 loss-of-function zebrafish (118). Consistent with Tet2 null mice, tet2-deficient zebrafish are viable and have intact embryonic hematopoiesis. Similar to murine models and humans, the tet2-mutant zebrafish develop progressive clonal myelodysplasia, anemia, and myeloid progenitor expansion as they age. By 24 months, they develop a more severe MDS phenotype including peripheral blood erythrocyte dysplasia. A study in compound mutants for tet family members uncovered a redundancy of tet2 and tet3 in HSC formation (119). The underlying mechanism for the diminished levels in tet2;tet3 double mutants was via regulation of NOTCH signaling in aortic endothelial cells (119). In mammalian blood cells, TET2 and TET3 are the predominantly expressed TET family members and might act redundantly (120). These data suggest a high degree of similarity in zebrafish and mammalian TET usage in hematopoiesis. Thus, the zebrafish tet2 single and tet2;tet3 double mutants will be useful for screening for new treatment targets of this epigenetic driver of MDS.
5q− Syndrome and Ribosomopathies
5q− syndrome is an MDS subtype with macrocytic anemia arising due to large deletions within chromosome 5 [reviewed in Ref. (121)]. The deleted chromosomal segment includes two common deleted regions (CDRs) encompassing many genes expressed by HSCs including hematopoietic cytokines, protein phosphatase 2, ribosomal protein S14 (RPS14), heat shock protein family A member 9B (HSPA9B), and more distally NPM1, but which factors are involved in disease phenotypes was unknown for quite some time (122).
The zebrafish mutant crimsonless (crs) presents with MDS-like hematological defects from 33 hpf, including anemia with a block in maturation, increased apoptosis and multilineage (erythroid and myeloid) cytopenia (123). The mutation in crs was determined to be a point mutation in the hspa9b (hsp70) gene likely generating a null allele. Hspa9b is a mitochondrial matrix chaperone whose loss leads to blood-restricted oxidative stress and apoptosis. In humans, the HSPA9B gene is located within the 5q31 CDR in human MDS. A recent study in human hematopoietic progenitors showed that similar to the zebrafish mutant depletion of HSPA9B in human hematopoietic progenitors also leads to apoptosis (124). Combined these studies suggest that loss of HSPA9B could contribute to 5q− syndrome MDS.
In 2008, Ebert and colleagues identified RPS14 as a major driver of 5q− anemia (125). A zebrafish homozygous rps14 loss-of-function mutant also develops anemia with a terminal erythroid maturation defect equivalent to that observed in 5q− syndrome (126). The rps14 mutant displayed elevated p53 activity, which was shown to contribute to the later events of the anemia. Mutations in another ribosomal protein RPS19 are linked with the childhood disease Diamond–Blackfan anemia (DBA) (127, 128). Similar to depletion of rps14, zebrafish homozygous rps19 loss-of-function mutants develop a p53-dependent anemia (129). These models will therefore be useful to dissect the signaling pathway intermediates between ribosomal proteins and p53-mediated factors that drive anemia. Indeed, l-leucine, a drug in testing for DBA (130, 131), has already proven effective in treatment of both mutant Rps14- and Rps19-driven anemia in zebrafish (126, 132).
Conclusion and Future Directions
The zebrafish model has great utility for investigating driver mutations underlying disease pathogenesis in MDS and AML. In particular, the high frequency of spliceosomal mutations identified in human MDS and AML and the conservation of myelo-erythroid phenotypes in the mutants studied to date indicates that this is a useful system to investigate the role of the spliceosome and epigenetics in myeloid malignancies. The above discusses some of the zebrafish myeloid disease models currently being studied (Table 1), and with the ability to rapidly generate mutant lines utilizing technologies such as CRISPR/Cas9, many more genes can be investigated in a relatively high-throughput manner. Furthermore, mechanistic studies are faster, cheaper, and higher throughput with in vivo testing of drug pathways feasible. This will facilitate more robust testing of targets in vivo in a whole organism setting. From this, we can identify and test rational, targeted pathways and therapeutics rather than the aggressive, non-specific cytotoxic chemotherapies utilized in current MDS and AML treatment regimes.

Table 1. Zebrafish models of human AML and MDS.
Unlike murine models, which often faithfully recapitulate human leukemias, zebrafish myeloid malignancy models frequently fail to develop the full adult disease state as observed in human MDS and AML, which is a limitation in the system to date. To address this, xenograft models using human leukemic cell lines or primary leukemic cells are now being used in zebrafish to investigate human disease progression (135). As zebrafish younger than a week have an innate immune system, but do not yet have a functioning adaptive immune system, xenografts in larvae are particularly useful to examine cancer progression in vivo without the need for damaging pre-conditioning regimens to permit human cell engraftment (23, 136). Thus, xenografts in zebrafish provide a new dimension for analysis of disease states and causative mechanisms, and will be extremely useful moving forward to screen human cells for drug susceptibility within an in vivo environment.
Currently, ubiquitous loss-of-function mutants are used to investigate the normal function of genes of interest in development. In human myeloid malignancies, mutations in genes often arise somatically and are missense rather than null. Genetic approaches in murine models permit tissue-specific expression of point mutants, which more closely resembles the human condition. With the advent of CRISPR/Cas9 technologies, the next step in zebrafish is to generate specific knock-in models of disease-associated point mutants [reviewed in Ref. (137)] and to induce mutations in a tissue-specific manner (138). These advances will expand our understanding of MDS and AML, including how faithfully the loss-of-function mutants recapitulate the phenotype of point mutants, and for screening of potential treatment molecules. The recent development of clonal lineage tracing capabilities for the blood system in zebrafish (139) opens the door to uncover drivers of the initial clonal events prior to hematologic dysfunction. Additionally, using live animal imaging, the initiation of cancer at the single-cell level was recently demonstrated in zebrafish melanoma (140). Combining these clonal and genetic approaches in myeloid malignancies can help examine the earliest events of disease formation not readily accomplished in other animal models.
Author Contributions
KP and TB designed and wrote the manuscript. TB gave final approval of the manuscript.
Conflict of Interest Statement
This work was supported by the Gabrielle’s Angel Foundation, American Cancer Society RSG-129527-DDC, Kimmel Foundation, the EvansMDS Foundation, and the New York State Department of Health Contract C30292GG. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. All authors declare no conflict of interest.
Funding
This work was funded by Gabrielle’s Angel Foundation, American Cancer Society RSG-129527-DDC, Kimmel Foundation, and the EvansMDS Foundation (to TB) and the Einstein Training Program in Stem Cell Research from the Empire State Stem Cell Fund through the New York State Department of Health Contract C30292GG (to KP).
References
1. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature (2011) 478(7367):64–9. doi:10.1038/nature10496
2. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia (2013) 28(2):241–7. doi:10.1038/leu.2013.336
3. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood (2013) 122(22):3616–27. doi:10.1182/blood-2013-08-518886
4. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med (2016) 374(23):2209–21. doi:10.1056/NEJMoa1516192
5. Langenau DM, Traver D, Ferrando AA, Kutok JL, Aster JC, Kanki JP, et al. Myc-induced T cell leukemia in transgenic zebrafish. Science (2003) 299(5608):887–90. doi:10.1126/science.1080280
6. Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature (2013) 496(7446):498–503. doi:10.1038/nature12111
7. Frame JM, McGrath KE, Palis J. Erythro-myeloid progenitors: “definitive” hematopoiesis in the conceptus prior to the emergence of hematopoietic stem cells. Blood Cells Mol Dis (2013) 51(4):220–5. doi:10.1016/j.bcmd.2013.09.006
8. Detrich HW, Kieran MW, Chan FY, Barone LM, Yee K, Rundstadler JA, et al. Intraembryonic hematopoietic cell migration during vertebrate development. Proc Natl Acad Sci U S A (1995) 92(23):10713–7. doi:10.1073/pnas.92.23.10713
9. Herbomel P, Thisse B, Thisse C. Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development (1999) 126(17):3735–45.
10. Bennett CM, Kanki JP, Rhodes J, Liu TX, Paw BH, Kieran MW, et al. Myelopoiesis in the zebrafish, Danio rerio. Blood (2001) 98(3):643–51. doi:10.1182/blood.v98.3.643
11. Lieschke GJ, Oates AC, Paw BH, Thompson MA, Hall NE, Ward AC, et al. Zebrafish SPI-1 (PU.1) marks a site of myeloid development independent of primitive erythropoiesis: implications for axial patterning. Dev Biol (2002) 246(2):274–95. doi:10.1006/dbio.2002.0657
12. Galloway JL, Wingert RA, Thisse C, Thisse B, Zon LI. Loss of Gata1 but not Gata2 converts erythropoiesis to myelopoiesis in zebrafish embryos. Dev Cell (2005) 8(1):109–16. doi:10.1016/j.devcel.2004.12.001
13. Paffett-Lugassy N, Hsia N, Fraenkel PG, Paw B, Leshinsky I, Barut B, et al. Functional conservation of erythropoietin signaling in zebrafish. Blood (2007) 110(7):2718–26. doi:10.1182/blood-2006-04-016535
14. Ganis JJ, Hsia N, Trompouki E, de Jong J, DiBiase A, Lambert JS, et al. Zebrafish globin switching occurs in two developmental stages and is controlled by the LCR. Dev Biol (2011) 366(2):185–94. doi:10.1016/j.ydbio.2012.03.021
15. Bertrand JY, Kim AD, Teng S, Traver D. CD41+ cmyb+ precursors colonize the zebrafish pronephros by a novel migration route to initiate adult hematopoiesis. Development (2008) 135(10):1853–62. doi:10.1242/dev.015297
16. Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DYR, Traver D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature (2010) 464(7285):108–11. doi:10.1038/nature08738
17. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330(6005):841–5. doi:10.1126/science.1194637
18. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science (2012) 336(6077):86–90. doi:10.1126/science.1219179
19. Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518(7540):547–51. doi:10.1038/nature13989
20. Kissa K, Murayama E, Zapata A, Cortés A, Perret E, Machu C, et al. Live imaging of emerging hematopoietic stem cells and early thymus colonization. Blood (2008) 111(3):1147–56. doi:10.1182/blood-2007-07-099499
21. Boisset J-C, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature (2010) 464(7285):116–20. doi:10.1038/nature08764
22. Murayama E, Kissa K, Zapata A, Mordelet E, Briolat V, Lin H-F, et al. Tracing hematopoietic precursor migration to successive hematopoietic organs during zebrafish development. Immunity (2006) 25(6):963–75. doi:10.1016/j.immuni.2006.10.015
23. Forrester MA, Berman JN, Payne EM. Myelopoiesis and myeloid leukaemogenesis in the zebrafish. Adv Hematol (2012) 2012:358518. doi:10.1155/2012/358518
24. Crowhurst MO, Layton JE, Lieschke GJ. Developmental biology of zebrafish myeloid cells. Int J Dev Biol (2002) 46(4):483–92.
25. Ward AC, McPhee DO, Condron MM, Varma S, Cody SH, Onnebo SMN, et al. The zebrafish spi1 promoter drives myeloid-specific expression in stable transgenic fish. Blood (2003) 102(9):3238–40. doi:10.1182/blood-2003-03-0966
26. Lieschke GJ, Oates AC, Crowhurst MO, Ward AC, Layton JE. Morphologic and functional characterization of granulocytes and macrophages in embryonic and adult zebrafish. Blood (2001) 98(10):3087–96. doi:10.1182/blood.V98.10.3087
27. Balla KM, Lugo-Villarino G, Spitsbergen JM, Stachura DL, Hu Y, Bañuelos K, et al. Eosinophils in the zebrafish: prospective isolation, characterization, and eosinophilia induction by helminth determinants. Blood (2010) 116(19):3944–54. doi:10.1182/blood-2010-03-267419
28. Li M, Zhao L, Page-McCaw PS, Chen W. Zebrafish genome engineering using the CRISPR–Cas9 system. Trends Genet (2016) 32(12):815–27. doi:10.1016/j.tig.2016.10.005
29. Urasaki A, Morvan G, Kawakami K. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics (2006) 174(2):639–49. doi:10.1534/genetics.106.060244
30. Ni J, Wangensteen KJ, Nelsen D, Balciunas D, Skuster KJ, Urban MD, et al. Active recombinant Tol2 transposase for gene transfer and gene discovery applications. Mob DNA (2016) 7(1):6. doi:10.1186/s13100-016-0062-z
31. White R, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell (2008) 2(2):183–9. doi:10.1016/j.stem.2007.11.002
32. Smith ACH, Raimondi AR, Salthouse CD, Ignatius MS, Blackburn JS, Mizgirev IV, et al. High-throughput cell transplantation establishes that tumor-initiating cells are abundant in zebrafish T-cell acute lymphoblastic leukemia. Blood (2010) 115(16):3296–303. doi:10.1182/blood-2009-10-246488
33. Blackburn JS, Langenau DM. Zebrafish as a model to assess cancer heterogeneity, progression and relapse. Dis Model Mech (2014) 7(7):755–62. doi:10.1242/dmm.015842
34. Moore FE, Garcia EG, Lobbardi R, Jain E, Tang Q, Moore JC, et al. Single-cell transcriptional analysis of normal, aberrant, and malignant hematopoiesis in zebrafish. J Exp Med (2016) 213(6):979–92. doi:10.1084/jem.20152013
35. Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood (2010) 115(3):453–74. doi:10.1182/blood-2009-07-235358
36. Troy JD, Atallah E, Geyer JT, Saber W. Myelodysplastic syndromes in the United States: an update for clinicians. Ann Med (2014) 46(5):283–9. doi:10.3109/07853890.2014.898863
37. Cogle CR. Incidence and burden of the myelodysplastic syndromes. Curr Hematol Malig Rep (2015) 10(3):272–81. doi:10.1007/s11899-015-0269-y
38. Kouchkovsky DI, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J (2016) 6(7):e441. doi:10.1038/bcj.2016.50
39. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood (2017) 129(4):424–47. doi:10.1182/blood-2016-08-733196
40. Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia (2014) 28(8):1586–95. doi:10.1038/leu.2014.55
41. Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell (1996) 84(2):321–30. doi:10.1016/S0092-8674(00)80986-1
42. Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, Röck J, Paschka P, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol (2011) 29(10):1364–72. doi:10.1200/JCO.2010.30.7926
43. Migas A, Savva N, Mishkova O, Aleinikova OV. AML1/RUNX1 gene point mutations in childhood myeloid malignancies. Pediatr Blood Cancer (2011) 57(4):583–7. doi:10.1002/pbc.22980
44. Erickson PF, Dessev G, Lasher RS, Philips G, Robinson M, Drabkin HA. ETO and AML1 phosphoproteins are expressed in CD34+ hematopoietic progenitors: implications for t(8;21) leukemogenesis and monitoring residual disease. Blood (1996) 88(5):1813–23.
45. Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR, et al. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol (1998) 18(12):7176–84. doi:10.1128/MCB.18.12.7176
46. Wang L, Swat W, Fujiwara Y, Davidson L, Visvader J, Kuo F, et al. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev (1998) 12(15):2392–402. doi:10.1101/gad.12.15.2392
47. Licht JD. AML1 and the AML1-ETO fusion protein in the pathogenesis of t(8;21) AML. Oncogene (2001) 20(40):5660–79. doi:10.1038/sj.onc.1204593
48. Downing JR. THE AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: biology and clinical significance. Br J Haematol (1999) 106(2):296–308. doi:10.1046/j.1365-2141.1999.01377.x
49. Sanderson RN, Johnson PRE, Moorman AV, Roman E, Willett E, Taylor PR, et al. Population-based demographic study of karyotypes in 1709 patients with adult acute myeloid leukemia. Leukemia (2006) 20(3):444–50. doi:10.1038/sj.leu.2404055
50. Yeh J-RJ, Munson KM, Chao YL, Peterson QP, MacRae CA, Peterson RT. AML1-ETO reprograms hematopoietic cell fate by downregulating scl expression. Development (2008) 135(2):401–10. doi:10.1242/dev.008904
51. Okuda T, Cai Z, Yang S, Lenny N, Lyu CJ, van Deursen JM, et al. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood (1998) 91(9):3134–43.
52. He G, Wang C, Li Q, Tan H, Chen F, Huang Z, et al. Clinical and laboratory features of seven patients with acute myeloid leukemia (AML)-M2/M3 and elevated myeloblasts and abnormal promyelocytes. Cancer Cell Int (2014) 14(1):111. doi:10.1186/s12935-014-0111-y
53. Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, Orkin SH. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the ETS-related factor TEL. EMBO J (1997) 16(14):4374–83. doi:10.1093/emboj/16.14.4374
54. Hock H, Meade E, Medeiros S, Schindler JW, Valk PJ, Fujiwara Y, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev (2004) 18(19):2336–41. doi:10.1101/gad.1239604
55. Akada H, Akada S, Hutchison RE, Sakamoto K, Wagner KU, Mohi G. Critical role of Jak2 in the maintenance and function of adult hematopoietic stem cells. Stem Cells (2014) 32(7):1878–89. doi:10.1002/stem.1711
56. Lacronique V, Boureux A, Valle V, Poirel H, Quang C, Mauchauffé M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science (1997) 278(5341):1309–12. doi:10.1126/science.278.5341.1309
57. Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood (1997) 90(7):2535–40.
58. Onnebo S, Condron MM, McPhee DO, Lieschke GJ, Ward AC. Hematopoietic perturbation in zebrafish expressing a tel-jak2a fusion. Exp Hematol (2005) 33(2):182–8. doi:10.1016/j.exphem.2004.10.019
59. Onnebo SMN, Rasighaemi P, Kumar J, Liongue C, Ward AC. Alternative TEL-JAK2 fusions associated with T-cell acute lymphoblastic leukemia and atypical chronic myelogenous leukemia dissected in zebrafish. Haematologica (2012) 97(12):1895–903. doi:10.3324/haematol.2012.064659
60. Breccia M, Loglisci G, Loglisci M, Ricci R, Diverio D, Latagliata R, et al. FLT3-ITD confers poor prognosis in patients with acute promyelocytic leukemia treated with AIDA protocols: long-term follow-up analysis. Haematologica (2013) 98(12):e161–3. doi:10.3324/haematol.2013.095380
61. Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood (2001) 97(8):2434–9. doi:10.1182/blood.V97.8.2434
62. Liang DC, Shih LY, Hung IJ, Yang CP, Chen SH, Jaing TH, et al. FLT3-TKD mutation in childhood acute myeloid leukemia. Leukemia (2003) 17(5):883–6. doi:10.1038/sj.leu.2402928
63. Kikushige Y, Yoshimoto G, Miyamoto T, Iino T, Mori Y, Iwasaki H, et al. Human FLT3 is expressed at the hematopoietic stem cell and the granulocyte/macrophage progenitor stages to maintain cell survival. J Immunol (2008) 180(11):7358–67. doi:10.4049/jimmunol.180.11.7358
64. Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood (2009) 114(14):2984–92. doi:10.1182/blood-2009-05-222034
65. Lu JW, Hou HA, Hsieh MS, Tien HF, Lin LI. Overexpression of FLT3-ITD driven by spi-1 results in expanded myelopoiesis with leukemic phenotype in zebrafish. Leukemia (2016) 30(10):2098–101. doi:10.1038/leu.2016.132
66. Cancer Genome Atlas Research NetworkLey TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368(22):2059–74. doi:10.1056/NEJMoa1301689
67. Bolli N, Payne EM, Grabher C, Lee J-S, Johnston AB, Falini B, et al. Expression of the cytoplasmic NPM1 mutant (NPMc+) causes the expansion of hematopoietic cells in zebrafish. Blood (2010) 115(16):3329–40. doi:10.1182/blood-2009-02-207225
68. Ando K, Parsons MJ, Shah RB, Charendoff CI, Paris SL, Liu PH, et al. NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J Cell Biol (2017) 216(6):1795–810. doi:10.1083/jcb.201608095
69. Barbieri E, Deflorian G, Pezzimenti F, Valli D, Saia M, Meani N, et al. Nucleophosmin leukemogenic mutant activates Wnt signaling during zebrafish development. Oncotarget (2015) 7(34):55302–12. doi:10.18632/oncotarget.10878
70. Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang H, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med (2010) 207(2):339–44. doi:10.1084/jem.20092506
71. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18(6):553–67. doi:10.1016/j.ccr.2010.11.015
72. Shi X, He B-LL, Ma AC, Guo Y, Chi Y, Man CH, et al. Functions of idh1 and its mutation in the regulation of developmental hematopoiesis in zebrafish. Blood (2015) 125(19):2974–84. doi:10.1182/blood-2014-09-601187
73. Voss AK, Thomas T. MYST family histone acetyltransferases take center stage in stem cells and development. Bioessays (2009) 31(10):1050–61. doi:10.1002/bies.200900051
74. Zhuravleva J, Paggetti J, Martin L, Hammann A, Solary E, Bastie JN, et al. MOZ/TIF2-induced acute myeloid leukaemia in transgenic fish. Br J Haematol (2008) 143(3):378–82. doi:10.1111/j.1365-2141.2008.07362.x
75. Griffis ER, Altan N, Lippincott-Schwartz J, Powers MA. Nup98 is a mobile nucleoporin with transcription-dependent dynamics. Mol Biol Cell (2002) 13(4):1282–97. doi:10.1091/mbc.01-11-0538
76. Lawrence HJ, Rozenfeld S, Cruz C, Matsukuma K, Kwong A, Kömüves L, et al. Frequent co-expression of the HOXA9 and MEIS1 homeobox genes in human myeloid leukemias. Leukemia (1999) 13(12):1993–9. doi:10.1038/sj.leu.2401578
77. Kroon E, Thorsteinsdottir U, Mayotte N, Nakamura T, Sauvageau G. NUP98–HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J (2001) 20(3):350–61. doi:10.1093/emboj/20.3.350
78. Forrester MA, Grabher C, McBride ER, Boyd ER, Vigerstad MH, Edgar A, et al. NUP98-HOXA9-transgenic zebrafish develop a myeloproliferative neoplasm and provide new insight into mechanisms of myeloid leukaemogenesis. Br J Haematol (2011) 155(2):167–81. doi:10.1111/j.1365-2141.2011.08810.x
79. Deveau AP, Forrester AM, Coombs AJ, Wagner GS, Grabher C, Chute IC, et al. Epigenetic therapy restores normal hematopoiesis in a zebrafish model of NUP98–HOXA9-induced myeloid disease. Leukemia (2015) 29(10):2086–97. doi:10.1038/leu.2015.126
80. North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature (2007) 447(7147):1007–11. doi:10.1038/nature05883
81. Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell (2009) 136(6):1136–47. doi:10.1016/j.cell.2009.01.015
82. Cazzola M, Rossi M, Malcovati L, Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood (2013) 121(2):260–9. doi:10.1182/blood-2012-09-399725
83. Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood (2011) 118(24):6239–46. doi:10.1182/blood-2011-09-377275
84. Scott LM, Rebel VI. Acquired mutations that affect pre-mRNA splicing in hematologic malignancies and solid tumors. J Natl Cancer Inst (2013) 105(20):1540–9. doi:10.1093/jnci/djt257
85. Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol (2011) 3(7):a003707. doi:10.1101/cshperspect.a003707
86. Carrocci TJ, Zoerner DM, Paulson JC, Hoskins AA. SF3b1 mutations associated with myelodysplastic syndromes alter the fidelity of branchsite selection in yeast. Nucleic Acids Res (2017) 45(8):4837–52. doi:10.1093/nar/gkw1349
87. Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, et al. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep (2015) 13(5):1033–45. doi:10.1016/j.celrep.2015.09.053
88. DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, et al. Transcriptome sequencing reveals potential mechanism of Cryptic 3’ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol (2015) 11(3):e1004105. doi:10.1371/journal.pcbi.1004105
89. Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun (2016) 7:10615. doi:10.1038/ncomms10615
90. Tang Q, Rodriguez-Santiago S, Wang J, Pu J, Yuste A, Gupta V, et al. SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev (2016) 30(24):2710–23. doi:10.1101/gad.291872.116
91. Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood (2011) 118(26):6904–8. doi:10.1182/blood-2011-08-373159
92. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med (2011) 365(26):2497–506. doi:10.1056/NEJMoa1109016
93. Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature (2016) 531(7592):47–52. doi:10.1038/nature16965
94. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature (2012) 486(7403):353–60. doi:10.1038/nature11143
95. Maguire SL, Leonidou A, Wai P, Marchiò C, Ng CKY, Sapino A, et al. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol (2015) 235(4):571–80. doi:10.1002/path.4483
96. Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov (2013) 3(10):1122–9. doi:10.1158/2159-8290.CD-13-0330
97. Harbour WJ, Roberson EDO, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet (2013) 45(2):133–5. doi:10.1038/ng.2523
98. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med (2011) 365(15):1384–95. doi:10.1056/NEJMoa1103283
99. Hahn CN, Scott HS. Spliceosome mutations in hematopoietic malignancies. Nat Genet (2011) 44(1):9–10. doi:10.1038/ng.1045
100. Garza A, Cameron RC, Nik S, Payne SG, Bowman TV. Spliceosomal component Sf3b1 is essential for hematopoietic differentiation in zebrafish. Exp Hematol (2016) 44(9):826–8370000. doi:10.1016/j.exphem.2016.05.012
101. Kumano K, Chiba S, Kunisato A, Sata M, Saito T, Nakagami-Yamaguchi E, et al. Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity (2003) 18(5):699–711. doi:10.1016/S1074-7613(03)00117-1
102. Mupo A, Seiler M, Sathiaseelan V, Pance A, Yang Y, Agrawal AA, et al. Hemopoietic-specific Sf3b1-K700E knock-in mice display the splicing defect seen in human MDS but develop anemia without ring sideroblasts. Leukemia (2016) 31(3):720–7. doi:10.1038/leu.2016.251
103. Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic expression of Sf3b1K700E causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell (2016) 30(3):404–17. doi:10.1016/j.ccell.2016.08.006
104. Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet (2011) 44(1):53–7. doi:10.1038/ng.1031
105. Je E, Yoo N, Kim Y, Kim M, Lee S. Mutational analysis of splicing machinery genes SF3B1, U2AF1 and SRSF2 in myelodysplasia and other common tumors. Int J Cancer (2013) 133(1):260–5. doi:10.1002/ijc.28011
106. Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res (2014) 25(1):14–26. doi:10.1101/gr.181016.114
107. Danilova N, Kumagai A, Lin J. p53 upregulation is a frequent response to deficiency of cell-essential genes. PLoS One (2010) 5(12):e15938. doi:10.1371/journal.pone.0015938
108. Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood (2012) 119(14):3203–10. doi:10.1182/blood-2011-12-399774
109. Kurtovic-Kozaric A, Przychodzen B, Singh J, Konarska MM, Clemente MJ, Otrock ZK, et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia (2014) 29(1):126–36. doi:10.1038/leu.2014.144
110. Luo HR, Moreau GA, Levin N, Moore MJ. The human Prp8 protein is a component of both U2- and U12-dependent spliceosomes. RNA (1999) 5(7):893–908. doi:10.1017/S1355838299990520
111. Keightley M-C, Crowhurst MO, Layton JE, Beilharz T, Markmiller S, Varma S, et al. In vivo mutation of pre-mRNA processing factor 8 (Prpf8) affects transcript splicing, cell survival and myeloid differentiation. FEBS Lett (2013) 587(14):2150–7. doi:10.1016/j.febslet.2013.05.030
112. Allis DC, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet (2016) 17(8):487–500. doi:10.1038/nrg.2016.59
113. Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet (2016) 17(5):284–99. doi:10.1038/nrg.2016.13
114. He Y-FF, Li B-ZZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (2011) 333(6047):1303–7. doi:10.1126/science.1210944
115. Han J-A, An J, Ko M. Functions of TET proteins in hematopoietic transformation. Mol Cells (2015) 38(11):925–35. doi:10.14348/molcells.2015.0294
116. Li Z, Cai X, Cai C-L, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood (2011) 118(17):4509–18. doi:10.1182/blood-2010-12-325241
117. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell (2011) 20(1):11–24. doi:10.1016/j.ccr.2011.06.001
118. Gjini E, Mansour MR, Sander JD, Moritz N, Nguyen AT, Kesarsing M, et al. A zebrafish model of myelodysplastic syndrome produced through tet2 genomic editing. Mol Cell Biol (2015) 35(5):789–804. doi:10.1128/MCB.00971-14
119. Li C, Lan Y, Schwartz-Orbach L, Korol E, Tahiliani M, Evans T, et al. Overlapping requirements for Tet2 and Tet3 in normal development and hematopoietic stem cell emergence. Cell Rep (2015) 12(7):1133–43. doi:10.1016/j.celrep.2015.07.025
120. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell (2017) 170(6):1079. doi:10.1016/j.cell.2017.07.032
121. Ebert BL. Deletion 5q in myelodysplastic syndrome: a paradigm for the study of hemizygous deletions in cancer. Leukemia (2009) 23(7):1252–6. doi:10.1038/leu.2009.53
122. Boultwood J, Fidler C, Strickson AJ, Watkins F, Gama S, Kearney L, et al. Narrowing and genomic annotation of the commonly deleted region of the 5q− syndrome. Blood (2002) 99(12):4638–41. doi:10.1182/blood.v99.12.4638
123. Craven SE, French D, Ye W, de Sauvage F, Rosenthal A. Loss of Hspa9b in zebrafish recapitulates the ineffective hematopoiesis of the myelodysplastic syndrome. Blood (2005) 105(9):3528–34. doi:10.1182/blood-2004-03-1089
124. Liu T, Krysiak K, Shirai C, Kim S, Shao J, Ndonwi M, et al. Knockdown of HSPA9 induces TP53-dependent apoptosis in human hematopoietic progenitor cells. PLoS One (2017) 12(2):e0170470. doi:10.1371/journal.pone.0170470
125. Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature (2008) 451(7176):335–9. doi:10.1038/nature06494
126. Ear J, Hsueh J, Nguyen M, Zhang Q, Sung V, Chopra R, et al. A zebrafish model of 5q-syndrome using CRISPR/Cas9 targeting RPS14 reveals a p53-independent and p53-dependent mechanism of erythroid failure. J Genet Genomics (2016) 43(5):307–18. doi:10.1016/j.jgg.2016.03.007
127. Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig T, Dianzani I, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet (1999) 21(2):169–75. doi:10.1038/5951
128. Willig TN, Draptchinskaia N, Dianzani I, Ball S, Niemeyer C, Ramenghi U, et al. Mutations in ribosomal protein S19 gene and diamond blackfan anemia: wide variations in phenotypic expression. Blood (1999) 94(12):4294–306.
129. Jia Q, Zhang Q, Zhang Z, Wang Y, Zhang W, Zhou Y, et al. Transcriptome analysis of the zebrafish model of diamond-blackfan anemia from RPS19 deficiency via p53-dependent and -independent pathways. PLoS One (2013) 8(8):e71782. doi:10.1371/journal.pone.0071782
130. Payne EM, Virgilio M, Narla A, Sun H, Levine M, Paw BH, et al. L-leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood (2012) 120(11):2214–24. doi:10.1182/blood-2011-10-382986
131. Steensma DP, Ebert BL. Initial experience with L-leucine therapy in myelodysplastic syndromes with associated chromosome 5q deletion. Blood (2013) 121(21):4428. doi:10.1182/blood-2013-03-493809
132. Narla A, Payne EM, Abayasekara N, Hurst SN, Raiser DM, Look TA, et al. L-leucine improves the anaemia in models of Diamond Blackfan anaemia and the 5q-syndrome in a TP53-independent way. Br J Haematol (2014) 167(4):524–8. doi:10.1111/bjh.13069
133. Yeh J-RJ, Munson KM, Elagib KE, Goldfarb AN, Sweetser DA, Peterson RT. Discovering chemical modifiers of oncogene-regulated hematopoietic differentiation. Nat Chem Biol (2009) 5(4):236–43. doi:10.1038/nchembio.147
134. Ear J, Huang H, Wilson T, Tehrani Z, Lindgren A, Sung V, et al. RAP-011 improves erythropoiesis in zebrafish model of Diamond-Blackfan anemia through antagonizing lefty1. Blood (2015) 126(7):880–90. doi:10.1182/blood-2015-01-622522
135. Pruvot B, Jacquel A, Droin N, Auberger P, Bouscary D, Tamburini J, et al. Leukemic cell xenograft in zebrafish embryo for investigating drug efficacy. Haematologica (2011) 96(4):612–6. doi:10.3324/haematol.2010.031401
136. Novoa B, Figueras A. Zebrafish: model for the study of inflammation and the innate immune response to infectious diseases. Adv Exp Med Biol (2012) 946:253–75. doi:10.1007/978-1-4614-0106-3_15
137. Albadri S, Bene F, Revenu C. Genome editing using CRISPR/Cas9-based knock-in approaches in zebrafish. Methods (2017) 121:77–85. doi:10.1016/j.ymeth.2017.03.005
138. Ablain J, Durand EM, Yang S, Zhou Y, Zon LI. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev Cell (2015) 32(6):756–64. doi:10.1016/j.devcel.2015.01.032
139. Henninger J, Santoso B, Hans S, Durand E, Moore J, Mosimann C, et al. Clonal fate mapping quantifies the number of haematopoietic stem cells that arise during development. Nat Cell Biol (2016) 19(1):17–27. doi:10.1038/ncb3444
Keywords: splicing, myelodysplastic syndrome, acute myeloid leukemia, zebrafish, hematopoiesis, malignancies
Citation: Potts KS and Bowman TV (2017) Modeling Myeloid Malignancies Using Zebrafish. Front. Oncol. 7:297. doi: 10.3389/fonc.2017.00297
Received: 03 October 2017; Accepted: 20 November 2017;
Published: 04 December 2017
Edited by:
Ross L. Levine, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Eirini Trompouki, Max Planck Institute of Immunobiology and Epigenetics (MPG), GermanyPeter Michael Gordon, University of Minnesota, United States
Copyright: © 2017 Potts and Bowman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa V. Bowman, teresa.bowman@einstein.yu.edu