 Jackline P. Ayres-Silva1
Jackline P. Ayres-Silva1 Martin H. Bonamino2,3
Martin H. Bonamino2,3 Maria E. Gouveia4
Maria E. Gouveia4 Barbara C. R. Monte-Mor1
Barbara C. R. Monte-Mor1 Diego F. Coutinho1
Diego F. Coutinho1 Adelmo H. Daumas4
Adelmo H. Daumas4 Cristiana Solza5
Cristiana Solza5 Esteban Braggio6
Esteban Braggio6 Ilana Renault Zalcberg1*
Ilana Renault Zalcberg1*
- 1Bone Marrow Transplantation Unit, Specialized Laboratories, Laboratory of Molecular Biology, National Cancer Institute (INCa), Rio de Janeiro, Brazil
- 2Programa de Carcinogênese Molecular, National Cancer Institute (INCa), Rio de Janeiro, Brazil
- 3Vice-presidência de Pesquisa e Coleções Biológicas, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil
- 4Hematology Department, Chemotherapy Unit, Hospital Universitario Antonio Pedro – HUAP, Rio de Janeiro, Brazil
- 5Hematology Unit, Hospital Universitario Pedro Ernesto – HUPE, Rio de Janeiro, Brazil
- 6Department of Hematology and Oncology, Mayo Clinic, Scottsdale, AZ, United States
The genetic events associated with transformation of myeloproliferative neoplasms (MPNs) to secondary acute myeloid leukemia (sAML), particularly in the subgroup of essential thrombocythemia (ET) patients, remain incompletely understood. Deep studies using high-throughput methods might lead to a better understanding of genetic landscape of ET patients who transformed to sAML. We performed array-based comparative genomic hybridization (aCGH) and whole exome sequencing (WES) to analyze paired samples from ET and sAML phases. We investigated five patients with previous history of MPN, which four had initial diagnosis of ET (one case harboring JAK2 p.Val617Phe and the remaining three CALR type II p.Lys385fs*47), and one was diagnosed with MPN/myelodysplastic syndrome with thrombocytosis (SF3B1 p.Lys700Glu). All were homogeneously treated with hydroxyurea, but subsequently transformed to sAML (mean time of 6 years/median of 4 years to transformation). Two of them have chromosomal abnormalities, and both acquire 2p gain and 5q deletion at sAML stage. The molecular mechanisms associated with leukemic progression in MPN patients are not clear. Our WES data showed TP53 alterations recurrently observed as mutations (missense and frameshift) and monoallelic loss. On the other hand, aCGH showed novel chromosome abnormalities (+2p and del5q) potentially associated with disease progression. The results reported here add valuable information to the still fragmented molecular basis of ET to sAML evolution. Further studies are necessary to identify minimal deleted/amplified region and genes relevant to sAML transformation.
Introduction
Essential thrombocythemia (ET) is a myeloproliferative neoplasm (MPN) derived from clonal expansion of hematopoietic progenitor cells carrying a mutation in the cytokine receptor/JAK2-signaling pathway (JAK2, CALR, or MPL). ET patients present platelet overproduction, bone marrow biopsy with enlarged megakaryocytes with hyperlobulated nuclei, with no left shift to granulopoiesis or erythropoiesis, and very rarely reticulin fibers. Other symptoms include transient ischemic attacks, ocular migraine, erythromelalgia, acquired von Willebrand disease, and pseudohyperkalemia due to extreme thrombocytosis, arterial or venous thrombosis (less common), and transformation to bone marrow failure such as myelofibrosis. Around 1 to 5–6% evolve to secondary acute myeloid leukemia (sAML), which has dismal prognosis with most of the patients dying within few months (1–3).
Regarding the MPN/AML transformation, mutations in genes encoding epigenetics modifiers such as ASXL1, IDH1, IDH2, EZH2, and TET2 are associated with nearly 30% of secondary leukemia transformations and de novo acute myeloid leukemia (4, 5). Mutations in IDH1 and IDH2 are observed in around 15% of AML, and specifically detected around 20–30% of sAML samples from MPN patients (5, 6). ASXL1, EZH2, and TET2 are the second largest mutated subgroup of genes in patients with AML and also compose a high-risk subcategory in myeloproliferative and myelodysplasic neoplasms. These mutations are acquired earlier in the disease and have prognostic values (5). Genetic changes and clonal evolution associated with ET to sAML progression remain incompletely understood in nearly 70% of patients. Genome wide analysis is an useful tool to understand the molecular events underlying sAML transformation in those cases.
Here, we describe five MPN patients diagnosed with thrombocytosis characterized by high platelet counts (from 623k × 109/L to 2,395k × 109/L), hyperplasia, and enlarged megakaryocytes with hyperlobulated nuclei at bone marrow biopsy. Physical exam and imaging analysis showed no splenomegalia during follow-up. All patients were homogeneously treated with hydroxyurea and evolved to sAML. Peripheral blood (PB) molecular screening showed three patients with CALR type II, one JAK2 p.Val617Phe, and one with SF3B1 p.Lys700Glu. MPN and sAML samples from all five patients were analyzed by array-based comparative genomic hybridization (aCGH). Three of these patients showed molecular alterations by aCGH and were further investigated by whole exome sequencing (WES).
Materials and Methods
This study was carried out in accordance with the recommendations of the “Institutional Ethics Committee (Brazilian National Institute of Cancer)” with written informed consent in accordance with the Declaration of Helsinki. The clinical data provided in the current case report contain no personal health information, identifier or personal feature, to protect participants’ rights. Written informed consent was obtained from some of the participant close relatives for the publication of this case report. Other participants have from 4 to 8 years since decease and clinical staff has lost follow-up with patient’s relatives, making it impossible to get an informed consent for publication.
Patients
The reported patients are part of a retrospective multicenter study (Hospital Universitário Pedro Ernesto-HUPE/UERJ and Hospital Universitário Antônio Pedro-HUAP/UFF) carried out between January 2007 and August 2015. Among the 158 MPN patients followed up until death, there were 38 with Polycythemia Vera (PV), 91 with ET, and 29 with primary Myelofibrosis (PMF). Six patients transformed to secondary myelofibrosis (secMF) and eight to sAML. From those eight with sAML, three patients were excluded for further analysis because paired samples from MPN and transformation phases were lacking. Five patients were selected for further genomic analysis, and time points analyzed are summarized in Figure 1. Four patients have initial diagnosis of ET and one of MPN/myelodysplastic syndrome (MDS) and clinical and laboratorial features are summarized in Table 1. All the patients were tested for JAK2V617F, CALR, and MPL mutation, and they were classified by the World Health Organization criteria (7).
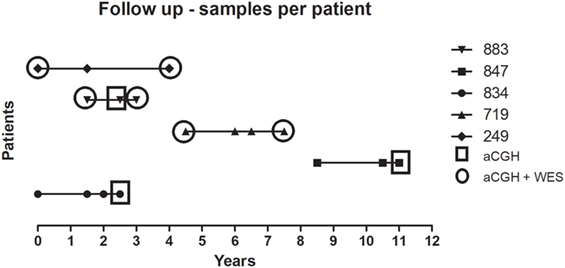
Figure 1. Longitudinal analysis of uniformly treated MPN cases. Five cases were analyzed by aCGH and WES before and after therapy. The horizontal axis represents the timeline (in years) of sample collection. Each case is presented in the right legend. Squares are showing samples analyzed only by aCGH and circles by the two methodologies applied. aCGH, array-based comparative genomic hybridization; MPN, myeloproliferative neoplasm; WES, whole exome sequencing.

Table 1. Clinical and laboratorial features of patients who progressed to sAML.
Samples
DNA Extraction
Peripheral blood samples were collected into EDTA and processed within 24 h of collection in each case. DNA extraction was performed from granulocytes after submitting the blood sample to Ficoll-Hypaque® gradient. The red cells were removed by hypotonic lysis solution. Cell pellets were resuspended in DNAzol® (Invitrogen), and DNA was extracted following the manufacturer’s protocol. Samples cleanup was done using puregene kit (Qiagen) or phenol:chloroform:isoamyl alcohol (25:24:1) protocol.
Mutation Screening
All genomic DNA samples were tested for V617F allele by allelic specific PCR (ASO PCR) as previously reported (8) with minor modifications (9) and also screened for the following mutations: JAK2 exon 12, MPL exon 10, CALR mutations, SF3B1 exons 14 and 15, IDH1/2 exon 4, and TET2 exons 3 and 9. CALR indel analysis was performed by PCR with fluorescent primers followed by fragment analysis and by Sanger sequencing (10). JAK2 exon 12, MPL, TET2, SF3B1, and IDH1 and IDH2 mutations were examined by Sanger sequencing with BigDye Terminator v3.1 kit as described by the manufacturer. We used the following primers to amplify JAK2 exon 12 (F—5′ CTCCTCTTTGGAGCAATTCA 3′/R–5′ GAGAACTTGGGAGTTGCGATA 3′), MPL gene (F—5′ TCCTAGCCTGGATCTCCTTGG 3′/R—5′ CACAGAGCGAACCAAGAATGCC 3′). TET2 mutations were evaluated with primers described by Martínez-Avilés et al. (11), SF3B1 as described by Cui et al. (12), IDH1/2 following Wagner et al. (13) and TP53 by applying IARC database-based method (14).
Array-Based Comparative Genomic Hybridization
Array-based comparative genomic hybridization was performed in five cases using the Human Genome 244A and the Sureprint G3 microarrays (Agilent Technologies). Genomic DNA from PB granulocytes was used. The digestion, labeling, and hybridization steps were done as previously described in Ref. (15, 16). Briefly, 1.2 μg of tumor and reference DNA were independently fragmented with Bovine DNaseI (Ambion, Austin, TX, USA) for 12 min at room temperature. DNA samples from a pool of nine human, female, lymphoblastoid cell lines from the Coriell repository were used as the normal reference in the hybridization experiments. Tumor samples were labeled with Alexa 5 dye, and the normal reference was labeled with Alexa 3 dye. Labeled reactions were cleaned up and hybridized at 65°C for 40 h. Microarrays were scanned in a DNA Microarray Scanner (Agilent), and features were extracted with Feature Extraction software (Agilent) version 10.7.3.1 (Agilent Technologies). Extracted data were imported, and log2 ratios were analyzed using Genomic Workbench software version 5.0.14 (Agilent Technologies). Copy number aberrations were calculated using ADM-1 algorithm with a 6.0 threshold, 5 probe filter, and log2 ratio above/below ±0.25. To identify and eliminate the germline copy number variations (CNVs) from the study, we created a CNV database including the recent higher-resolution copy number (using platform SNP6.0) and sequencing studies available in The Center for Applied Genomics data portal1 as well as our findings in 10 HapMap samples run by Sureprint G3 arrays (16).
Paired-End WES
Genomic DNA from PB granulocytes from each sample was sheared and used for the construction of paired-end sequencing library as described in the protocol provided by Illumina. The exome capture was done using the Sure Select 50Mb Exome Enrichment kit (Agilent) following the manufacturer’s instructions. Next, 100 bp paired-end DNA libraries were prepared, and three samples were run per lane in the HiSeq2000 sequencer (Illumina). An automated workflow for exome-seq data analysis was developed. First, 100 bp paired-end reads were aligned to human genome hg19 using Novoalign (Novocraft Technologies, Malaysia). Quality of sequencing chemistry was evaluated using FastQC.2 Realignment and recalibration were done using Best Practice Variant Detection v3 recommendations implemented in the GATK.3 After alignment, PCR duplication rates and percent reads mapped on target were used to assess the quality of the data. Somatic single nucleotide variants (SNVs) were genotyped using SomaticSniper(17), whereas insertions and deletions were called by GATK Somatic Indel Detector. Each variant in coding regions was functionally annotated by snpEFF4 and PolyPhen-2 (18) to predict biological effects. The variants were annotated using our TREAT workflow (19) whether the gene is associated with disease or phenotypes and any associated pathways. We removed variants found in the 1,000 genomes,5 the Exome Variant Server NHLBI Gene Ontology (GO) Exome Sequencing Project (Seattle, WA, USA),6 and the BGI—Danish Sequencing Project. In addition, we removed variants present in dbSNP data set unless these mutations were also present in the COSMIC database. Variants with read depth less than 10× were excluded from further analysis. Additional germline variants were excluded from analysis after comparing with 25 non-tumoral patient samples. Finally, non-synonymous and non-exonic variants of significant interest were visually inspected using IGV (20).
Clustering Analysis
Clustering analysis was run using clValid package (21) from R software,7 using all somatic mutations (intronic and exonic) that showed more than 50× coverage in the tumor samples. To define the number of clones presented, we combine data from nine clustering models (hierarchical clustering, k-means, DIANA, PAM, CLARA, FANNY, SOM, Expectation-Maximization, SOTA) and elected as the number of clusters presented in each patient the result recurrently found in most of nine models analyzed. Other algorithms were also used: kohonen and mclust for Expectation-Maximization algorithm. Internal validation was analyzed by connectivity, Dunn and Silhouette. We also used a fpc package from R software and run a prediction strength and nselectboot function for selection of the number of clusters via bootstrap and computes the prediction strength of a clustering of a data set into different numbers of components.
Gene Set Enrichment Analysis
Enrichment analysis on gene sets of functional gene classes or ontology terms was performed using the analysis tool provided by Database for Annotation, Visualization and Integrated Discovery8 (22, 23) maintained by National Institute of Allergy and Infectious Diseases (NIH) using Protein Information Resource, Kyoto Encyclopedia of Genes and Genomes, or GO Consortium databases to execute enrichment examination. We analyzed genes contained in each chromosome abnormality region (gain or loss) or genes set from each patient containing somatic mutations [by point or insertions–deletions (INDELS)] detected by WES.
Statistical Analysis
Hematological data were analyzed using two-sided Wilcoxon signed-rank test with p = 0.05 using Prism 6 (Graphpad, La Jolla, CA, USA).
Results
Clinical Presentation
Clinical and hematological data are summarized in Table 1. All the patients were female, and median follow–up time for those five patients included were 1,527 days (range, 978–4,171 days) from diagnosis until transformation and 56 days (range, 31–299 days) during sAML. Weight loss, hemorrhage, and thrombosis were the main clinical presentations observed during follow-up. All patients were screened for mutations previously described to be more frequently found upon transformation such IDH1/IDH2 (exon 4) and TET2 (exon 3) but were absent at MPN and sAML samples. Other clinical relevant features from each patient are described as follows:
Case 1 (UPN834): A patient diagnosed with ET, CALR type II positive (p.Lys385fs*47) with bone marrow biopsy at diagnosis showing proliferation of all lineages with increased number of mature megakaryocytes. The patient was treated with hydroxyurea, and no adverse events or thrombotic events were reported during follow-up. Three years after diagnosis, a new bone marrow biopsy was performed, which showed 44% blasts and presence of megakaryoblasts. At that time, the patient was still JAK2 p.Val617Phe negative, CALR type II positive and aCGH analysis in the sAML sample showed no chromosome abnormalities.
Case 2 (UPN847): A patient was diagnosed with ET, showed CALR type II mutation (p.Lys385fs*47), with bone marrow biopsy at diagnosis showing megakaryocyte proliferation with hyperplasia and absence of fibrosis. The patient was treated with hydroxyurea. A new bone marrow biopsy was performed 12 years after diagnosis and revealed atypic cells and presence of micromegakaryocytes. No chromosome abnormalities were observed in the granulocyte fraction at sAML blood sample by aCGH.
Case 3 (UPN719): A patient was diagnosed with ET and was JAK2 p.Val617Phe positive. The patient was treated with hydroxyurea, but during follow-up presented a thrombosis in left lower limb, epistaxis, and weight loss. A new bone marrow biopsy was performed 7.5 years after diagnostic and showed hyperplasia of megacaryocytes, elevated myeloid/erythroid ratio, and a presence of myeloblasts. Paired aCGH analysis of initial diagnosis and sAML samples evidenced eight chromosome abnormalities at chromosomes 2, 3, 5, 6, and 8 at progression (Table 2). WES analysis of these same samples showed 171 single-nucleotide variants/INDELS (SNV/INDELS) with variant allele frequencies (VAFs) ratio higher than 1.5 between the two samples analyzed (Table S1 in Supplementary Material).
Case 4 (UPN883): A patient was diagnosed with ET, CALR type II (p.Lys385fs*47) positive with bone marrow biopsy showing bizarre megakaryocytes, hypocellularity in the myeloid and erythroid lineages, and grade I fibrosis. The patient was treated with hydroxyurea and showed no adverse events during follow-up. Since diagnosis, the patient remained with high platelet counts, which required unusual hydroxyurea doses of 2–3 g/day for more than 2/3 of treatment (1,200 days). After 2 years, cytopenia forced the reduction of hydroxyurea to 0.5 g/day, until the leukemic transformation, about 140 days after the onset of cytopenia. Three years after diagnosis new bone marrow biopsy was performed and showed myeloblasts. aCGH analysis of ET and sAML samples showed 10 chromosome abnormalities found at both chronic and acute phases and six other abnormalities found only at sAML progression (Table 2). WES analysis of two paired samples revealed 211 SNV/INDELS with chronic/sAML VAF ratio at least more than 1.5 (Table S1 in Supplementary Material).
Case 5 (UPN249): A patient with molecular profile showing SF3B1 (p.Lys700Glu) positive. Bone marrow biopsy could not evidence ring sideroblats and was diagnosed with MPN/MDS. The patient was treated with hydroxyurea and had no adverse events during follow-up. A new bone marrow biopsy 4.7 years after diagnosis showed myeloblasts. Paired ET/sAML analysis by aCGH detected one chromosome abnormality found in both samples (12q24.31) and five other abnormalities found upon leukemic transformation (Table 2). WES screening identified 206 differentials SNV/INDELS with VAF ratio chronic/sAML at least more than 1.5 (Table S1 in Supplementary Material).
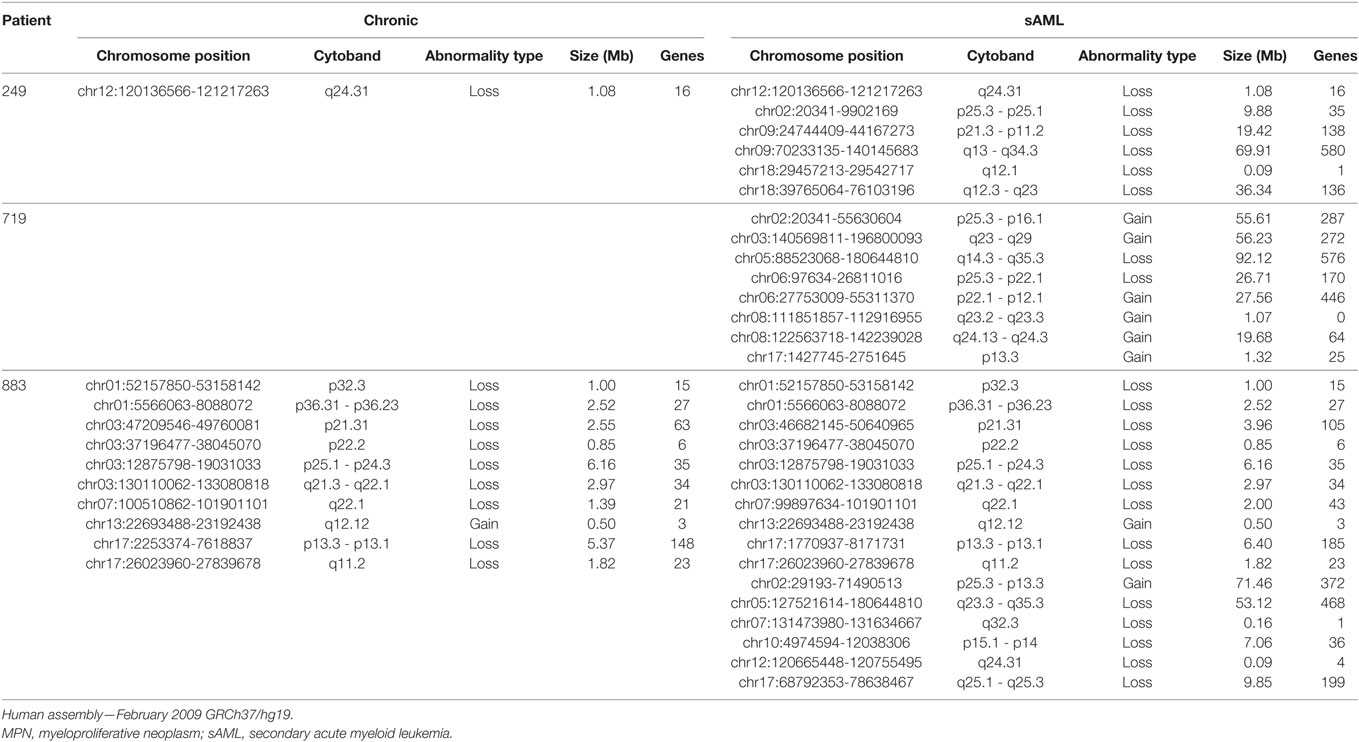
Table 2. Comparative list of chromosome abnormalities found since MPN phase, showing differences in chromosome gain and losses in the two samples analyzed per patient.
Genomic Analysis
ET transformation to sAML is a rare event. In our 91 ET patients cohort, we observed five cases of sAML transformation reported herein.
Comprehensive genomic characterization of paired samples by aCGH identified two common genetic abnormalities—gain of 2p and deletion of 5q—not identified in initial ET but present in the sAML phase, suggesting that they were acquired during disease progression (Table 2; Figures 2A,B). The common deleted region in chromosome five covers cytobands q23.3–q35.3 and the 2p minimal gain region comprised cytobands p13.3–p25.3.
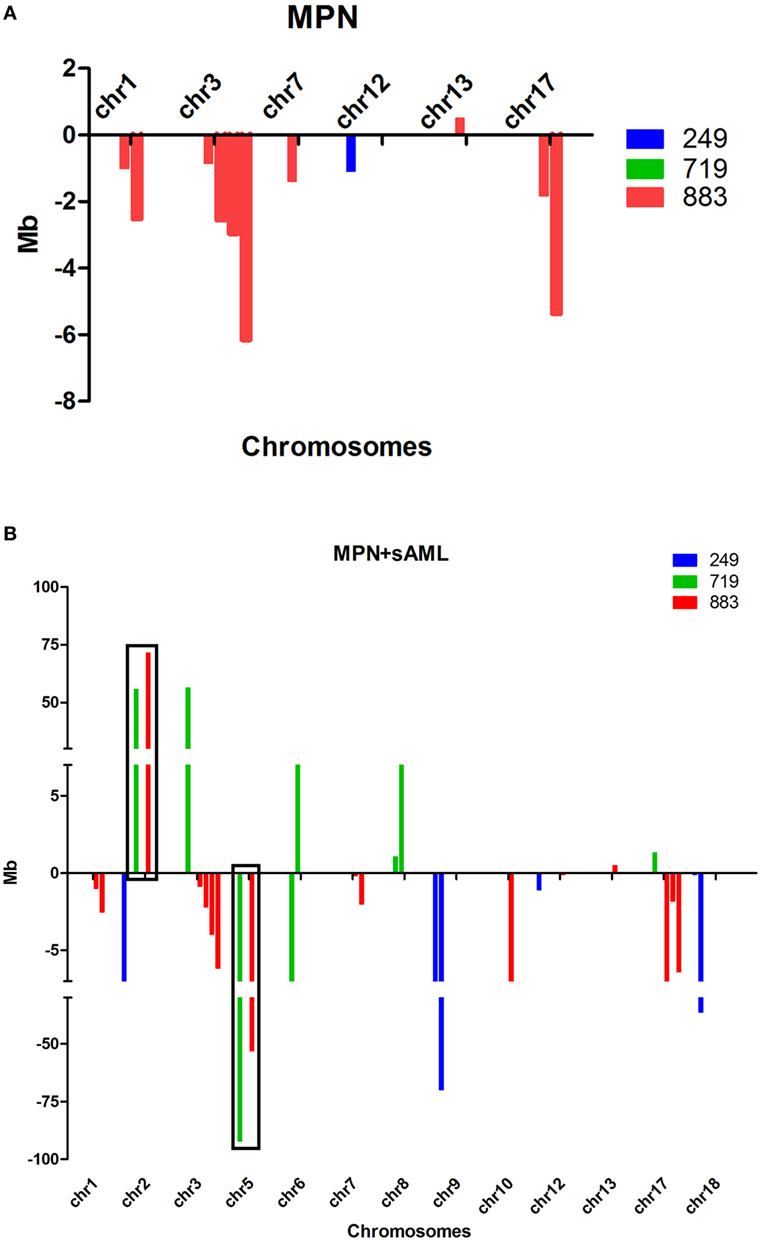
Figure 2. (A,B) Size and type of the abnormalities found at chronic phase (A) and sAML (B) samples from three patients analyzed by aCGH. Abnormalities found at chronic phase remained until sAML. Gains are represented above and losses below X-axis; note that there are more abnormalities found at sAML, and they are larger compared to chronic phase. aCGH, array-based comparative genomic hybridization; chr, chromosome; MPN, myeloproliferative neoplasms; sAML, secondary acute myeloid leukemia.
The three patients with chromosomal abnormalities were subsequently analyzed by WES. Mutations in paralogous genes already associated with MPN phenotype such as CALR3, ASXL2, and TET3 were found augmented at sAML sample from ET patient positive for CALR (p.Lys385fs*47) (Figure 3C; Table S1 in Supplementary Material). A novel JAK2 mutation p.Arg1063Cys was found at MPN/MDS patient who carries a SF3B1 p.Lys700Glu since the first sample analyzed.
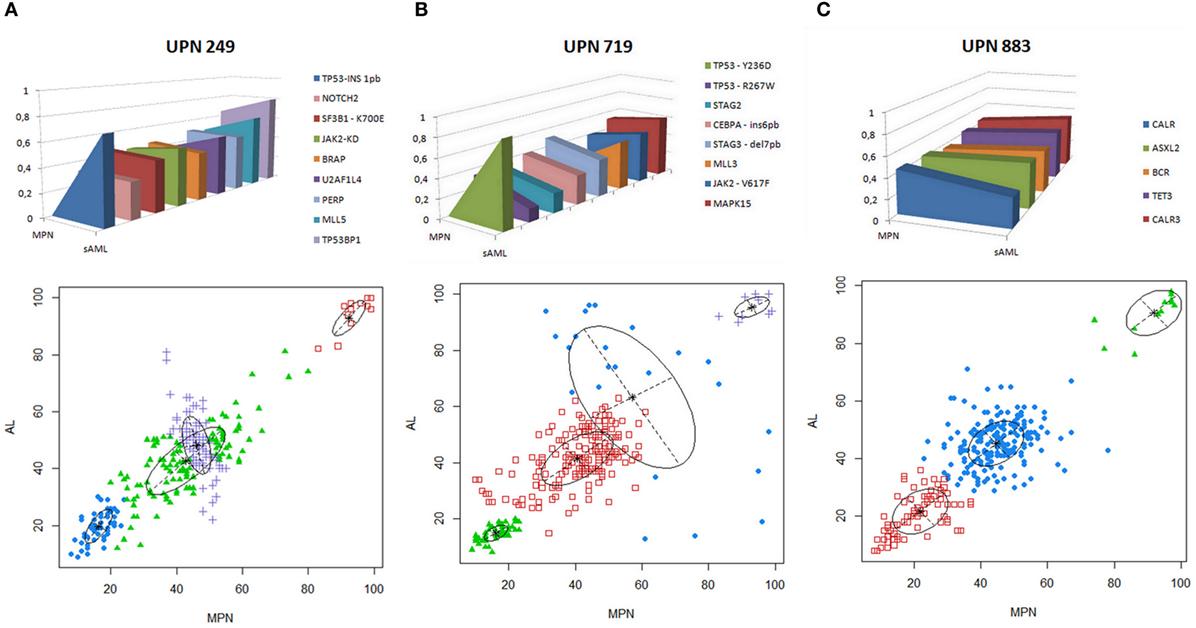
Figure 3. (A–C) Genomic landscape of thrombocythosis (ET and MPN/MDS) that progressed to sAML. VAF of candidate driver mutations or paralogous genes already associated with MPN phenotype in each patient analyzed are depicted in upper panels. Model of clonal evolution of MPN to sAML showing average VAF of each subclone in the stages analyzed. Clonal evolution complexity showing VAF of mutations found in each clone per patient in MPN (abcissa) and sAML (ordinate) stages is depicted in the lower panels. VAF plots were generated by mclust after running fpc and nselectboot functions. ET, essential thrombocythemia; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasm; sAML, secondary acute myeloid leukemia.; UPN, unique patient number; VAFs, variant allele frequencies.
TP53 alterations were recurrently observed with mutations in two cases (UPN719 and UPN249) and monoallelic loss (UPN883). UPN719 was characterized by the presence of JAK2 p.Val617Phe and TP53 p.Arg267Trp at initial ET stage and subsequent acquisition of TP53 p.Tyr236Asp at sAML phase. UPN249 had a SF3B1 p.Lys700Glu at chronic phase and subsequently acquired a TP53 p.Leu106ArgfsTer25 in the progression to sAML. UPN883 had a monoallelic loss of TP53 and a CALR p.Lys385fs*47 (Figures 3A–C).
The analysis of clonal architecture showed a founding dominant clone and the subsequent rise of a minor genetic subclone growing overtime in all the three cases analyzed (Figures 3A–C). No recurrent gene mutations were found among patients and samples analyzed, besides the TP53 and JAK2 alterations aforementioned (Table S1 in Supplementary Material).
Discussion
Genomic studies evaluating serial samples of MPNs are few (24, 25), especially in the subgroup of ET (26, 27). Our work is one of the few reports associating aCGH and WES data in sequential paired samples from ET diagnosis to sAML progression.
Among the identified chromosomal alterations, we could identify +2p and −5q only at sAML samples. Chr2 gain is a novel genetic change described in the subgroup of MPN patients. Whether +2p and −5q abnormalities are derived from a pre-existing MPN small subclone or acquired in the sAML transformation is still a matter of debate, particularly in ET, where very few reports describing 2p gain (28) and 5q deletions associated with MPN exists (Table 3). Other possibility is that chromosome alterations are present but at low frequency, thus undetectable by the methods used in our approach. In contrast, −5q MDS represents a well-known subgroup with cellular and molecular mechanisms well understood, in contrast to MPN, which requires further studies.
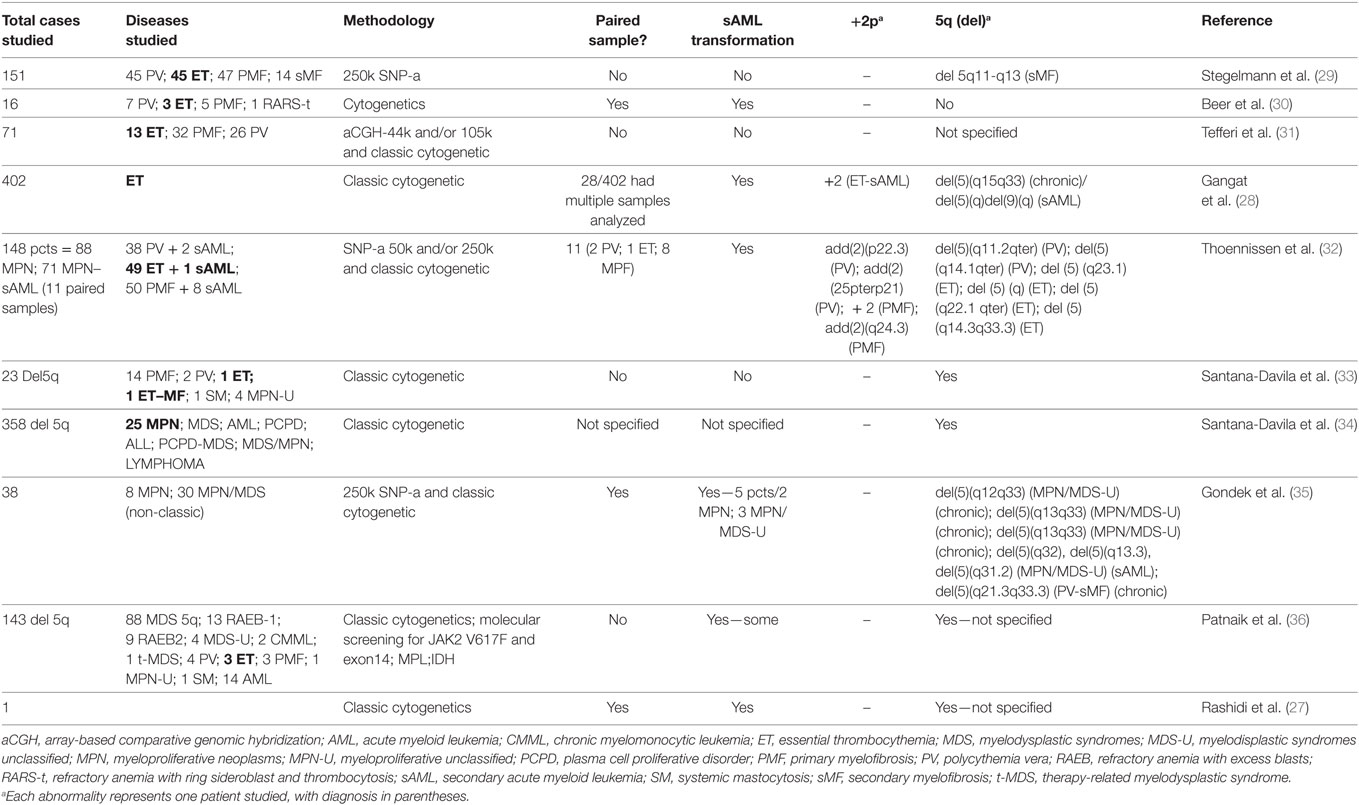
Table 3. Comparative studies showing previous reports in +2p and del5q in myeloproliferative and myelodysplasia neoplasms.
At MPN, we observed a cytokine cluster in the minimal 5q deleted genomic region compassing nine genes (IL4, CSF2, TSLP, IL3, IL5, IL9, IL13, IL12B, and SPRY4) that are part of the JAK-STAT signaling pathway associated with proliferation and differentiation of the hematopoietic compartment. The 2p minimal gain region includes known regulators of epigenetic processes such as DNMT3A, ASXL2 e DPY30. There are few reports showing chromosome 2 gain in MPN cases, and only one study showing this alteration in ET (28). A comparative analysis of published data describing alterations in this locus is presented in Table 3.
Genomic abnormalities targeting cytokines and cytokine receptors clusters are described in several types of tumors, such as loss of chromosome 4 and the subsequent decreased IL15 expression in colorectal cancer (37). The potential role of cytokine cluster deletions in MPNs depends on the description of such molecular phenomena in larger cohorts and on mechanistic studies on how these deletions can impact the leukemic niche and/or the sAML transformation process.
We identified a novel JAK2 mutation at kinase domain that is predicted to result in STAT5 activation, as suggested by a previous study that showed a mutation in the same residue, JAK2 p.Arg1063His, associated with STAT5 phosphorylation and increase of CFU-E formation, in addition to in silico modeling, which predicted a facilitation of the active conformation of JH1 (38).
Our work reinforces the relevance of TP53 mutations in sAML progression and supports the previous descriptions that JAK2-negative patients can also acquire TP53 mutations (39, 40). TP53 mutations in AML occurs around 10–20% and is attributed to poor prognosis. In recent years, diagnosis of TP53 dysfunction is growing as therapeutics drugs to target TP53 mutations is increasing and getting promising results (41, 42). Association of TP53 loss with 5q haploinsufficiency in mice promoting myeloid leukemia was observed, and studies associating other abnormalities such as +2p need further attention (43).
Clonal analysis showed the molecular heterogeneity among ET patients, as no recurrent gene mutation was found. Even comparing our data with other WES findings, we could not find any recurrent gene mutation, corroborating Engle et al. (25) findings describing that most mutations found in a patient with secondary myelofibrosis who progressed to sAML are passenger mutations, with a clonal architecture similar to the ones described in our patients.
Conclusion
Secondary Acute Myeloid Leukemia progression is a rare event, especially in ET, with incidence rate of less than 5%. Because ET is the more indolent MPN, with a long natural disease history, studies with paired sample analysis are scarce. Genomic analysis of paired samples, as reported here, adds valuable information to understand the molecular basis of sAML transformation. Our data showed chromosome abnormalities potentially associated with disease progression not previously described, and those abnormalities (+2p and del5q) have common regions that should be further screened.
Ethics Statement
This study was carried out in accordance with the recommendations of the Institutional Ethics Committee (Brazilian National Institute of Cancer), with written informed consent in accordance with the Declaration of Helsinki. The clinical data provided in the current case report contains no personal health information, identifier, or personal feature to protect participants’ rights. Written informed consent was obtained from some of the participant close relatives for the publication of this case report. Other participants have from 4 to 8 years since decease and clinical staff has lost follow-up with patient’s relatives, making it impossible to get an informed consent for publication.
Author Contributions
JA-S, MB, IZ, and EB designed the study. JA-S, BM-M, MB, IZ, and EB analyzed molecular data and wrote the manuscript. JA-S and DC performed and developed molecular assays. MG, AD, and CS took care of the patients, performed clinical review of the cases, and contributed to the interpretation of data. All authors made a substantial contribution, reviewed, and approved the final version of the manuscript. All the authors are in agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer PB and the handling editor declared their shared affiliation.
Acknowledgments
The authors would like to thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) and Mayo Clinic for supporting this study. We would like to thank Scott A. Van Wier and Greg J. Ahmann for technical support.
Funding
The authors would like to thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ). JPAS was supported by a scholarship of the Brazilian Ministry of Health and CNPq (Sandwich PhD-Science without borders).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fonc.2018.00032/full#supplementary-material.
Footnotes
- ^http://dgv.tcag.ca/dgv/app/home.
- ^http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- ^https://software.broadinstitute.org/gatk/best-practices/.
- ^http://snpeff.sourceforge.net/.
- ^www.1000genomes.org/.
- ^http://evs.gs.washington.edu/EVS/.
- ^http://www.R-project.org/.
- ^http://david.abcc.ncifcrf.gov/.
References
1. Abdulkarim K, Girodon F, Johansson P, Maynadié M, Kutti J, Carli P-M, et al. AML transformation in 56 patients with Ph− MPD in two well defined populations. Eur J Haematol (2009) 82:106–11. doi:10.1111/j.1600-0609.2008.01163.x
2. Spivak JL. Myeloproliferative Neoplasms. N Engl J Med (2017) 376:2168–81. doi:10.1056/NEJMra1406186
3. Kennedy JA, Atenafu EG, Messner HA, Craddock KJ, Brandwein JM, Lipton JH, et al. Treatment outcomes following leukemic transformation in Philadelphia-negative myeloproliferative neoplasms. Blood (2013) 121:2725–33. doi:10.1182/blood-2012-10-464248
4. Medinger M, Passweg JR. Acute myeloid leukaemia genomics. Br J Haematol (2017) 179(4):530–42. doi:10.1111/bjh.14823
5. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med (2016) 374:2209–21. doi:10.1056/NEJMoa1516192
6. Green A, Beer P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med (2010) 362:369–70. doi:10.1056/NEJMc0910063
7. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood (2009) 114:937–51. doi:10.1182/blood-2009-03-209262
8. Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood (2005) 106:2162–8. doi:10.1182/blood-2005-03-1320
9. Xavier SG, Gadelha T, Pimenta G, Eugenio AM, Ribeiro DD, Gomes FM, et al. JAK2V617F mutation in patients with splanchnic vein thrombosis. Dig Dis Sci (2010) 55:1770–7. doi:10.1007/s10620-009-0933-y
10. Monte-Mor Bda C, Ayres-Silva Jde P, Correia WD, Coelho AC, Solza C, Daumas AH, et al. Clinical features of JAK2V617F- or CALR-mutated essential thrombocythemia and primary myelofibrosis. Blood Cells Mol Dis (2016) 60:74–7. doi:10.1016/j.bcmd.2016.03.003
11. Martínez-Avilés L, Besses C, Álvarez-Larrán A, Torres E, Serrano S, Bellosillo B. TET2, ASXL1, IDH1, IDH2, and c-CBL genes in JAK2- and MPL-negative myeloproliferative neoplasms. Ann Hematol (2012) 91:533–41. doi:10.1007/s00277-011-1330-0
12. Cui R, Gale RP, Xu Z, Qin T, Fang L, Zhang H, et al. Clinical importance of SF3B1 mutations in Chinese with myelodysplastic syndromes with ring sideroblasts. Leuk Res (2012) 36:1428–33. doi:10.1016/j.leukres.2012.08.015
13. Wagner K, Damm F, Göhring G, Görlich K, Heuser M, Schäfer I, et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol (2010) 28:2356–64. doi:10.1200/JCO.2009.27.6899
14. IARC TP53 Database. (2014). Available from: http://p53.iarc.fr/ProtocolsAndTools.aspx
15. Braggio E, Keats JJ, Leleu X, Van Wier S, Jimenez-Zepeda VH, Valdez R, et al. Identification of copy number abnormalities and inactivating mutations in two negative regulators of nuclear factor-kappaB signaling pathways in Waldenstrom’s macroglobulinemia. Cancer Res (2009) 69:3579–88. doi:10.1158/0008-5472.CAN-08-3701
16. Kay NE, Eckel-Passow JE, Braggio E, VanWier S, Shanafelt TD, Van Dyke DL, et al. Progressive but previously untreated CLL patients with greater array CGH complexity exhibit a less durable response to chemoimmunotherapy. Cancer Genet Cytogenet (2010) 203:161–8. doi:10.1016/j.cancergencyto.2010.09.003
17. Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics (2012) 28:311–7. doi:10.1093/bioinformatics/btr665
18. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods (2010) 7:248–9. doi:10.1038/nmeth0410-248
19. Asmann YW, Middha S, Hossain A, Baheti S, Li Y, Chai H-S, et al. TREAT: a bioinformatics tool for variant annotations and visualizations in targeted and exome sequencing data. Bioinformatics (2011) 28:277–8. doi:10.1093/bioinformatics/btr612
20. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol (2011) 29:24–6. doi:10.1038/nbt.1754
21. Brock G, Pihur V, Datta S, Datta S. clValid, an R package for cluster validation. J Stat Softw (2011). Available from: ftp://ftp2.it.postgresql.org/pub/mirrors/CRAN/web/packages/clValid/vignettes/clValid.pdf Accessed March 12, 2014
22. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc (2009) 4:44–57. doi:10.1038/nprot.2008.211
23. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res (2009) 37:1–13. doi:10.1093/nar/gkn923
24. Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood (2014) 123(14):2220–8. doi:10.1182/blood-2013-11-537167
25. Engle EK, Fisher DA, Miller CA, McLellan MD, Fulton RS, Moore DM, et al. Clonal evolution revealed by whole genome sequencing in a case of primary myelofibrosis transformed to secondary acute myeloid. Leukemia (2014) 29(4):869–76. doi:10.1038/leu.2014.289
26. Zaidi U, Shahid S, Fatima N, Ahmed S, Sufaida G, Nadeem M, et al. Genomic profile of a patient with triple negative essential thrombocythemia, unresponsive to therapy: a case report and literature review. J Adv Res (2017) 8:375–8. doi:10.1016/j.jare.2017.04.001
27. Rashidi A, Nelson AC, Linden MA, Weisdorf DJ, Dolan MM, Ustun C. Genomics of clonal evolution in a case of essential thrombocythemia. Leuk Lymphoma (2017) 59(2):497–500. doi:10.1080/10428194.2017.1337113
28. Gangat N, Tefferi A, Thanarajasingam G, Patnaik M, Schwager S, Ketterling R, et al. Cytogenetic abnormalities in essential thrombocythemia: prevalence and prognostic significance. Eur J Haematol (2009) 83:17–21. doi:10.1111/j.1600-0609.2009.01246.x
29. Stegelmann F, Bullinger L, Griesshammer M, Holzmann K, Habdank M, Kuhn S, et al. High-resolution single-nucleotide polymorphism array-profiling in myeloproliferative neoplasms identifies novel genomic aberrations. Haematologica (2009) 95:666–9. doi:10.3324/haematol.2009.013623
30. Beer PA, Delhommeau F, LeCouedic JP, Dawson MA, Chen E, Bareford D, et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood (2009) 115:2891–900. doi:10.1182/blood-2009-08-236596
31. Tefferi A, Sirhan S, Sun Y, Lasho T, Finke CM, Weisberger J, et al. Oligonucleotide array CGH studies in myeloproliferative neoplasms: comparison with JAK2V617F mutational status and conventional chromosome analysis. Leuk Res (2009) 33:662–4. doi:10.1016/j.leukres.2008.09.009
32. Thoennissen NH, Krug UO, Lee DHT, Kawamata N, Iwanski GB, Lasho T, et al. Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of Philadelphia chromosome-negative myeloproliferative neoplasms. Blood (2010) 115:2882–90. doi:10.1182/blood-2009-07-235119
33. Santana-Davila R, Tefferi A, Holtan SG, Ketterling RP, Dewald GW, Knudson RA, et al. Primary myelofibrosis is the most frequent myeloproliferative neoplasm associated with del(5q): clinicopathologic comparison of del(5q)-positive and -negative cases. Leuk Res (2008) 32:1927–30. doi:10.1016/j.leukres.2008.04.022
34. Santana-Davila R, Holtan SG, Dewald GW, Ketterling RP, Knudson RA, Hanson CA, et al. Chromosome 5q deletion: specific diagnoses and cytogenetic details among 358 consecutive cases from a single institution. Leuk Res (2008) 32:407–11. doi:10.1016/j.leukres.2007.07.007
35. Gondek LP, Dunbar AJ, Szpurka H, McDevitt MA, Maciejewski JP. SNP array karyotyping allows for the detection of uniparental disomy and cryptic chromosomal abnormalities in MDS/MPD-U and MPD. PLoS One (2007) 2:e1225. doi:10.1371/journal.pone.0001225
36. Patnaik MM, Lasho TL, Finke CM, Knudson RA, Ketterling RP, Chen D, et al. Isolated del(5q) in myeloid malignancies: clinicopathologic and molecular features in 143 consecutive patients. Am J Hematol (2011) 86:393–98. doi:10.1002/ajh.21984
37. Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci Transl Med (2014) 6:ra37–228. doi:10.1126/scitranslmed.3007240
38. Kapralova K, Horvathova M, Pecquet C, Fialova Kucerova J, Pospisilova D, Leroy E, et al. Cooperation of germline JAK2 mutations E846D and R1063H in hereditary erythrocytosis with megakaryocytic atypia. Blood (2016) 128(10):1418–23. doi:10.1182/blood-2016-02-698951
39. Harutyunyan A, Klampfl T, Cazzola M, Kralovics R. p53 lesions in leukemic transformation. N Engl J Med (2011) 364:488–90. doi:10.1056/NEJMc1012718
40. Rampal R, Ahn J, Abdel-Wahab O, Nahas M, Wang K, Lipson D, et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci U S A (2014) 111:E5401–10. doi:10.1073/pnas.1407792111
41. Prokocimer M, Molchadsky A, Rotter V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood (2017) 130:699–712. doi:10.1182/blood-2017-02-763086
42. Ohgami RS, Ma L, Merker JD, Gotlib JR, Schrijver I, Zehnder JL, et al. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod Pathol (2015) 28:706–14. doi:10.1038/modpathol.2014.160
Keywords: essential thrombocythemia, secondary acute myeloid leukemia, array-based comparative genomic hybridization, whole exome sequencing, myeloproliferative neoplasms, +2p
Citation: Ayres-Silva JP, Bonamino MH, Gouveia ME, Monte-Mor BCR, Coutinho DF, Daumas AH, Solza C, Braggio E and Zalcberg IR (2018) Genetic Alterations in Essential Thrombocythemia Progression to Acute Myeloid Leukemia: A Case Series and Review of the Literature. Front. Oncol. 8:32. doi: 10.3389/fonc.2018.00032
Received: 27 September 2017; Accepted: 31 January 2018;
Published: 19 February 2018
Edited by:
Naval Daver, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Raajit Rampal, Memorial Sloan Kettering Cancer Center, United StatesPrithviraj Bose, University of Texas MD Anderson Cancer Center, United States
Copyright: © 2018 Ayres-Silva, Bonamino, Gouveia, Monte-Mor, Coutinho, Daumas, Solza, Braggio and Zalcberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilana Renault Zalcberg, zalcberg@inca.gov.br