 Chakrabhavi Dhananjaya Mohan1
Chakrabhavi Dhananjaya Mohan1 Nirvanappa C. Anilkumar2
Nirvanappa C. Anilkumar2 Shobith Rangappa3
Shobith Rangappa3 Muthu K. Shanmugam4Srishti Mishra4Arunachalam Chinnathambi5Sulaiman Ali Alharbi5Atanu Bhattacharjee6Gautam Sethi4Alan Prem Kumar4,7
Muthu K. Shanmugam4Srishti Mishra4Arunachalam Chinnathambi5Sulaiman Ali Alharbi5Atanu Bhattacharjee6Gautam Sethi4Alan Prem Kumar4,7 Basappa2,8*Kanchugarakoppal S. Rangappa9
Basappa2,8*Kanchugarakoppal S. Rangappa9
- 1Department of Studies in Molecular Biology, University of Mysore, Mysore, India
- 2Laboratory of Chemical Biology, Department of Chemistry, Bangalore University, Bangalore, India
- 3Adichunchanagiri Institute for Molecular Medicine, Mandya, India
- 4Department of Pharmacology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
- 5Department of Botany and Microbiology, College of Science, King Saud University, Riyadh, Saudi Arabia
- 6Department of Biotechnology and Bioinformatics, North Eastern Hill University, Shillong, India
- 7Cancer Science Institute of Singapore, National University of Singapore, Singapore, Singapore
- 8Department of Studies in Organic Chemistry, University of Mysore, Mysore, India
- 9Department of Studies in Chemistry, University of Mysore, Mysore, India
Aberrant activation of NF-κB is linked with the progression of human malignancies including hepatocellular carcinoma (HCC), and blockade of NF-κB signaling could be a potential target in the treatment of several cancers. Therefore, designing of novel small molecule inhibitors that target NF-κB activation is of prime importance in the treatment of several cancers. In the present work, we report the synthesis of series of 1,3,4-oxadiazoles, investigated their anticancer potential against HCC cells, and identified 2-(3-chlorobenzo[b]thiophen-2-yl)-5-(3-methoxyphenyl)-1,3,4-oxadiazole (CMO) as the lead compound. Further, we examined the effect of CMO on cell cycle distribution (flow cytometry), apoptosis (annexin V-propidium iodide-FITC staining), and phosphorylation of NF-κB signaling pathway proteins (IκB and p65) in HCC cells. We found that CMO induced antiproliferative effect in dose- and time-dependent manner. Also, CMO significantly increased the percentage of sub-G1 cell population and induced apoptosis. Furthermore, CMO found to decrease the phosphorylation of IκB (Ser 32) in the cytoplasmic extract and p65 (Ser 536) in the nuclear extract of HCC cells. It also abrogated the DNA binding ability and transcriptional activity of NF-κB. CMO induced the cleavage of PARP and caspase-3 in a time-dependent manner. In addition, transfection with p65 small interfering RNA blocks CMO-induced caspase-3/7 activation. Molecular docking analysis revealed that CMO interacts with the hydrophobic region of p65 protein. Thus, we are reporting CMO as an inhibitor of NF-κB signaling pathway.
Introduction
NF-κB is the one of the most widely studied inflammatory mediators associated with several disease conditions including cancer (1, 2). Initially, NF-κB was identified as transcription factor that is essential for the expression of B-cell specific genes and, later, it was demonstrated to present ubiquitously in mammalian cells (3, 4). NF-κB is an inducible transcription factor present in cytoplasm that gets activated in response to inflammation, DNA damage, stress, and viral attack (5). In mammals, RelA, RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2) are NF-κB proteins identified so far (6). During the stimuli, these proteins undergo homo/hetero-dimerization, translocate to nucleus, binds to specific DNA elements, and regulate the expression of more than 500 genes that codes for acute phase proteins (Pentraxin-3, Hepcidin, UPA), stress response proteins (COX, LOX, HSP90), apoptosis regulators (Bcl-2, Bcl-xL, Bax), cell adhesion molecules (CD44, ICAM-1, Fibronectin, P-selectin), cytokines (TNF-α, lymphotoxin-a/b, RANTES), growth factors (G-CSF, SCF, Activin-A), and cell surface receptors (ABCA1, CD23, RAGE) (7, 8). In mammals, NF-κB target genes modulate cell proliferation, apoptosis and survival (9, 10). Baltimore et al. demonstrated the role of NF-κB proteins in cell survival by generating the RelA-deficient mice (11). Their experimental results suggested that RelA disruption leads to embryonic lethality at 15–16 days of gestation, with an extensive degeneration of the liver (11). On the other hand, several cancer-causing mutations have been identified in the genes that signal for the activation of NF-κB pathway. Cancer-causing mutations are most likely to contribute for the persistent activation of NF-κB in turn deregulating of expression of NF-κB target genes, which drive the cells to oppose apoptosis (12). Evidently, aberrant activation of NF-κB is reported in several cancers including solid and liquid tumors (13–15). Moreover, constitutive activation of NF-κB has been observed in hepatocellular carcinoma (HCC) tumor tissues suggesting its critical role in the tumorigenesis (16–18). Therefore, targeting activation NF-κB signaling pathway remains as an attractive therapeutic strategy in the fight against cancer.
Several 1,3,4-oxadiazoles have been reported to possess good anticancer potential against various types of cancer cells (19–21). Some of the reports also suggested that oxadiazoles possibly target NF-κB signaling pathway to induce their anticancer activity (22). Specifically, 3-methyl-1-((5-(5-methyl-1,3,4-oxadiazol-2-yl)-2-(thiophen-2-yl)pyrimidin-4-yl)amino)-1H-pyrrole-2,5-dione was reported to possess the IC50 value of 0.3 µM for AP-1 and NF-κB mediated transcriptional activation in Jurkat T-cells (22). Although anticancer activity of oxadiazoles is well-documented, the comprehensive study on their putative targets and mechanisms of action has not been reported so far. In continuation of our efforts to explore the anticancer potential of heterocycles (23–26), herein, we report the synthesis of series of novel 1,3,4-oxadiazoles and comprehensively demonstrated their mechanism of anticancer activity in vitro against panel of HCC cell lines.
Materials and Methods
All chemicals used were of analytical grade and purchased from Sigma Aldrich, and SRL, Mumbai (India). 1H NMR spectra were recorded on a Agilent (400 MHz) spectrometer in CDCl3 solvent, using TMS as an internal standard, 13C NMR spectra were recorded on a Agilent (100 MHz) spectrometer. Mass spectra were determined on PE Sciex API3000 ESI-MS, elemental analyses were carried out using an Elemental Vario Cube CHNS rapid analyzer. Progress of the reaction was monitored by TLC pre-coated silica gel plates.
HepG2 cell line was initially purchased from ATCC. The cells were cultured in DMEM medium containing 10% fetal bovine serum, 1 mM l-glutamine, 1 mM sodium pyruvate, antibiotic, and antimycotic agent. GAPDH, lamin B, and p65 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against phospho-IκBα (Ser 32), IκBα, phospho-p65 (Ser 536) was purchased from Cell Signaling Technology (Beverly, MA, USA). Nuclear extraction and NF-κB DNA binding kits were purchased from Active motif (USA). Blocking buffer was purchased from Nacalai Tesque (Kyoto, Japan). Chemiluminescence kit was purchased from Advansta (CA, USA). The small interfering RNA (siRNA) for NF-κB and scrambled control was obtained from Santa Cruz Biotechnology. Caspase-Glo 3/7 assay kit and luciferase substrate was purchased from promega (WI, USA).
Chemistry
General Procedure for the Preparation of Acid Hydrazide (3a–c)
The appropriate aromatic acids (0.01 mol) were dissolved in absolute ethanol (10 ml) followed by the addition of hydrazine hydrate (0.02 mol) and 2–3 drops of conc. sulfuric acid. The reaction mixture was refluxed for 7 h. The completion of reaction was monitored by thin layer chromatography, and the resulting solid obtained was filtered, dried, and crystallized (3a-c).
General Procedure for the Synthesis of 1,3,4-Oxadiazole (5a–l)
The aromatic acid hydrazide (0.01 mol) and an appropriate aromatic acid (0.01 mol) were refluxed in phosphorous oxychloride (5 ml) for 8 h, and the reaction mixture was cooled to room temperature. Excess POCl3 was removed through high vacuum, the residue was quenched with ice and made alkaline with potassium carbonate solution. The precipitate was filtered, dried, and crystallized from ethanol. The completion of reaction was monitored by thin layer chromatography. The representative spectra of some of the new compounds are provided as Supplemental Information.
4-(5-(3-Chlorobenzyl)-1,3,4-Oxadiazol-2-yl)-N,N-Dimethylaniline (5a)
Yellow solid; mp 110–112°C; 71% yield; 1H NMR (400 MHz, CDCl3): δ = 7.84–7.81 (d, 2H, Ar-H), 7.34 (s, 1H, Ar-H), 7.22–7.25 (m, 3H, Ar-H), 6.70–6.68 (d, 2H, Ar-H), 4.19 (s, 2H, –CH2), 3.02 (s, 6H, 2CH3); 13C (100 MHz CDCl3): δ = 152.37, 136.19, 134.64, 130.05, 128.96, 128.20, 127.65, 126.96, 111.54, 40.00, and 31.49 ppm; exact mass: 313.0, ESI mass [M + 1] 314.1; Anal. Calcd for C17H16ClN3O: C, 65.07; H, 5.14; N, 13.39; found: C, 65.23; H, 5.11; N, 13.11.
4-(5-Benzyl-1,3,4-Oxadiazol-2-yl)-N,N-Dimethylaniline (5b)
Yellow solid; mp 154–156°C; 79% yield; 1H NMR (400 MHz, CDCl3): δ = 7.84–7.82 (d, 2H, Ar-H), 7.25–7.34 (m, 5H, Ar-H), 6.72–6.69 (d, 2H, Ar-H), 4.23 (s, 2H, -CH2), 3.04 (s, 6H, 2CH3); 13C (100 MHz CDCl3): δ = 134.34, 128.80, 128.77, 128.18, 127.33, 111.68, 40.10, and 31.88 ppm; exact mass: 279.1, ESI mass [M + 1] 280.2; Anal. Calcd for C17H17N3O: C, 73.10; H, 6.13; N, 15.04; found: C, 73.34, H, 5.88, N, 15.24.
4-(5-(4-Bromobenzyl)-1,3,4-Oxadiazol-2-yl)-N,N-Dimethylaniline (5c)
Yellow solid; mp 107–109°C; 82% yield; 1H NMR (400 MHz, CDCl3): δ = 7.82–7.80 (d, 2H, Ar-H), 7.47–7.42 (t, 2H, Ar-H), 7.25–7.21 (t, 2H, Ar-H), 6.70–6.68 (d, 2H Ar-H), 4.18 (s, 2H, -CH2), 3.02 (s, 6H, 2CH3); exact mass: 357.1, ESI mass [M + 2] 359.6; Anal. Calcd for C17H16BrN3O: C, 57.00; H, 4.50; N, 11.73; found: C, 56.98; H, 4.32; N, 11.81.
4-(5-(3-Chlorobenzo[b]thiophen-2-yl)-1,3,4-Oxadiazol-2-yl)-N,N-Dimethylaniline (5d)
Yellow solid; mp 120–123°C; 86% yield; 1H NMR (400 MHz, CDCl3): δ = 8.04–8.02 (d, 1H), 7.96–7.82 (m, 3H), 7.52–7.49 (m, 2H), 6.91–6.88 (d, 2H), 3.08–3.05 (t, 6H); exact mass: 355.1, ESI mass: [M + 1] 356.3, Anal. Calcd for C18H14ClN3OS: C, 60.76; H, 3.97; N, 11.81; found: C, 60.58; H, 3.88, N, 11.52.
3-(5-(4-(Dimethylamino)phenyl)-1,3,4-Oxadiazol-2-yl)-N-(2-Methyl-3-(Trifluoromethyl) phenyl)pyridin-2-amine (5e)
Yellow solid; mp 170–173°C; 78% yield; 1H NMR (400 MHz, CDCl3): δ = 8.31–8.21 (m, 3H, Ar-H), 7.99–7.98 (d, 2H, Ar-H), 7.45–7.25 (m, 2H, Ar-H), 6.88–6.85 (m, 1H, Ar-H), 6.78–6.76 (d, 2H, Ar-H), 3.07–3.05 (d, 6H, 2CH3), 2.53 (s, 3H, -CH3); 13C (100 MHz CDCl3): δ = 164.57, 161.92, 153.26, 152.62, 150.61, 139.56, 136.00, 128.48, 127.16, 125.69, 121.52, 121.47, 113.70, 111.68, 110.18, 103.53, 40.04, 14.28; exact mass: 439.2 [M + 1] 440.3; Anal. Calcd for C23H20F3N5O: C, 62.86; H, 4.59; N, 15.94; found: C, 62.91, H, 4.42; N, 15.63.
2-(4-Bromobenzyl)-5-(6-Chloropyridin-3-yl)-1,3,4-Oxadiazole (5f)
Brown solid; mp 110–112°C; 83% yield; 1H NMR (400 MHz, CDCl3): δ = 8.97–8.96 (d, 1H), 8.26–8.23 (m, 1H), 7.50–7.43 (m, 3H), 7.25–7.21 (t, 2H), 4.24 (s, 2H); exact mass: 348.9, ESI Mass: [M + 2] 351.3; Anal. Calcd for C14H9BrClN3O: C, 47.96; H, 2.59; N, 11.99; found: C, 47.81, H, 2.49, N, 12.13.
2-(4-Bromobenzyl)-5-(Pyridin-3-yl)-1,3,4-Oxadiazole (5g)
Brown solid; mp 133–135°C; 71% yield; 1H NMR (400 MHz, CDCl3): δ = 9.97 (s, 1H), 8.32–8.31 (d, 1H), 8.23–8.21 (t, 1H), 7.99–7.97 (d, 2H), 7.32–7.25 (t, 1H), 6.78–6.76 (d, 2H), 3.08 (s, 2H); exact mass: 315.0, ESI mass: [M + 2] 317.3, Anal. Calcd for C14H10BrN3O: C, 53.19; H, 3.19; N, 13.29; found: C, 53.10; H, 2.99; N, 13.42.
2-Benzyl-5-(4-Bromobenzyl)-1,3,4-Oxadiazole (5h)
Brown solid; 123–124°C; 86% yield; 1H NMR (400 MHz, CDCl3): δ = 7.83–7.81 (d, 2H), 7.47–7.43 (t, 2H), 7.25–7.21 (m, 2H), 6.72–6.70 (d, 2H), 4.02–4.05 (m, 4H); exact mass: 328.0, ESI mass: [M + 2] 330.5; Anal. Calcd for C16H13BrN2O: C, 58.38; H, 3.98; N, 8.51; found: C, 58.19, H, 3.88; N, 8.67.
2-(3-Methoxyphenyl)-5-(4-(Trifluoromethyl)phenyl)-1,3,4-Oxadiazole (5i)
White solid; mp 100–102°C; 77% yield; 1H NMR (400 MHz, CDCl3): δ = 7.98–7.43 (m, 7H), 7.12–7.09 (m, 1H), 3.19 (s, 3H); 13C (100 MHz CDCl3): δ = 160.02, 138.15, 130.29, 127.79, 125.75, 124.56, 123.28, 122.73, 119.59, 119.33, 118.15, 111.91, 55.54; exact mass: 320.1 ESI mass: [M + 1] 321.3; Anal. Calcd for C16H11F3N2O2: C, 60.00; H, 3.46; N, 8.75; found: C, 60.17, H, 3.41, N, 8.69.
2-(3-Chlorobenzo[b]thiophen-2-yl)-5-(3-Methoxyphenyl)-1,3,4-Oxadiazole (CMO, 5j)
White solid; mp 112–114°C; 72% yield; 1H NMR (400 MHz, CDCl3): δ = 7.44–8.00 (m, 7H), 7.12–7.10 (m, 1H), 3.91 (s, 3H); 13C (100 MHz CDCl3): δ = 160.33, 158.67, 155.32, 125.53, 125.43, 122.62, 122.48, 121.47, 121.38, 121.34, 119.91, 114.66, 114.58, 113.66, 113.39, 107.12, and 50.79 ppm; exact mass: 342.0, ESI mass: [M + 1] 343.1; Anal. Calcd for C17H11ClN2O2S: C, 59.56; H, 3.23; N, 8.17; found: C, 59.42; H, 3.32, N, 8.23.
Pharmacology
MTT Assay
The antiproliferative effect of newly synthesized compounds against HCC cells was determined by the MTT dye uptake method as described previously (27, 28). Briefly, HCC cells (4 × 103 cells/well) were incubated in triplicate in a 96-well plate, in the presence of different concentrations of compounds at a volume of 0.2 ml, for different time intervals at 37°C. Thereafter, a 20 µl MTT solution (5 mg/ml in PBS) was added to each well. After a 2 h incubation at 37°C, 0.1 ml lysis buffer (20% SDS, 50% dimethylformamide) was added; incubation was performed for 1 h at 37°C, and the optical density (OD) at 570 nm was measured by Tecan plate reader. 0.01% DMSO was used as the negative control and 0.01% MTT was used as a control agent.
Flow Cytometric Analysis
Flow cytometric analysis was performed to evaluate apoptosis inducing effect of CMO in HepG2 and HCCLM3 cells as described earlier (29, 30). Briefly, HCC cells (5 × 105) were plated in petri dish and, 24 h later, the cells were exposed to compound CMO (50 µM) for 0, 24, 48, and 72 h. Thereafter, cells were washed, fixed with 70% ethanol, and incubated for 30 min at 37°C with 0.1% RNase A in PBS. Cells were washed again, resuspended, and stained with PBS containing 25 µg/ml propidium iodide (PI) for 15 min at room temperature. The cell cycle distribution across the various phases was analyzed using flow cytometer.
Annexin V/PI Apoptosis Assay
Phosphatidylserine exposure and cell death were assessed by FACS analysis using Annexin-V-PI-stained cells as described previously (31). Briefly, 1 × 105 HepG2 cells/well (190 μl/well) were seeded in 96-well plates and incubated with CMO (50 µM) for indicated time points (24, 48, and 72 h), and DMSO treated samples were used as control. Cells were then washed with Annexin V binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2), stained with Annexin V FITC for 30 min at room temperature in the dark, then washed again, and re-suspended in Annexin V binding buffer containing PI. Samples were analyzed immediately.
Immunoblotting Assay
For detection of phophoproteins, control and CMO treated cells, cytoplasmic and nuclear extract were prepared for according to manufacturer’s instructions. Lysates were then spun at 14,000 rpm for 10 min to remove insoluble material and resolved on a 10% SDS gel. After electrophoresis, the proteins were electrotransferred to a nitrocellulose membrane, blocked with blocking buffer, and probed with primary antibodies overnight at 4°C. The blot was washed, exposed to HRP-conjugated secondary antibodies for 1 h, and finally examined by chemiluminescence and imaged using ChemiDoc™ imaging system (BioRad, USA).
NF-κB DNA Binding Assays
To determine NF-κB activation, we performed DNA-binding assay using TransAM NF-κB kit according to the manufacturer’s instructions and as previously described (32). Briefly, 50 µg of nuclear proteins were added into 96-well plate coated with an unlabeled oligonucleotide containing the consensus binding site for NF-κB (5′-GGGACTTTCC-3′) and incubated for 4, 8, and 12 h. The wells were washed and incubated with antibodies against NF-κB p65 subunit. An HRP conjugated secondary antibody was then applied to detect the bound primary antibody and provided the basis for colorimetric quantification. The enzymatic product was measured at 450 nm by microplate reader (Tecan Systems).
NF-κB Luciferase Reporter Assay
The effect of CMO on constitutive a NF-κB-dependent reporter gene transcription in HepG2 and HCCLM3 cells was determined as previously described (32). NF-κB responsive elements linked to a luciferase reporter gene were transfected with wild-type or dominant-negative IκB. The transfected cells were then treated with CMO for 4, 8, and 12 h. Luciferase activity was measured with a Tecan (Durham, NC, USA) plate reader and normalized to β-galactosidase activity. All luciferase experiments were done in triplicate.
p65 siRNA Transfection
HepG2 cells were plated in 6-well plates and allowed to adhere for 24 h. On the day of transfection, lipofectamine was added to control or p65 siRNA in a final volume of 1 ml of culture medium. After 48 h of incubation following transfection, HepG2 cells (1 × 104 cells/well) in 6-well plate were treated with CMO for 24 h. Equal volume of Caspase-Glo® 3/7 reagent was then added to the wells to provide the 1:1 ratio of reagent volume to sample volume. After incubation for 15–30 min at room temperature, the luminescence was measured by Tecan microplate reader and activation of caspase-3/7 was analyzed.
Caspase-Glo® 3/7 Luminescent Assay
Caspase activity was measured using Caspase-Glo® 3/7 assay kit (Promega) according to the manufacturer’s instructions.
In Silico Interaction Analysis
Discovery Studio 2.5 software from Accelrys was used for docking and visualization of the results as described earlier (33, 34). Initially, we retrieved the crystal structure of NF-κB complex (PDB: 1IKN) (35), cleaned, minimized the energy, and identified the spatial region of p65. All the energy calculations were performed using CHARMM force field. The three-dimensional structures of all oxadiazoles were prepared and docked toward the p65 using LIGANDFIT protocol of Discovery Studio. The binding pose of ligands was evaluated using the interaction score function in the Ligand Fit module of Discovery Studio as reported previously (36).
Results and Discussion
Chemistry
Initially, carboxylic acid (1a-c) was converted to their corresponding ester (2a-c) followed by refluxing with hydrazine hydrate in ethanol, which resulted in the formation of acid hydrazides (3a-c). Thereafter, 1,3,4-oxadiazoles (5a-j) were synthesized by refluxing equimolar mixture of acid hydrazide (3a-c), with different aromatic carboxylic acid (4a–h) in phosphorous oxychloride (5 ml) for 7 h (Scheme 1). The structures of all the target compounds (Table 1) were characterized by elemental analysis LCMS, 1H NMR and 13C NMR spectrometry.
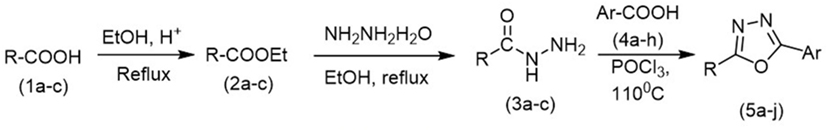
Scheme 1. Schematic representation for the synthesis of 1,3,4-oxadiazoles.
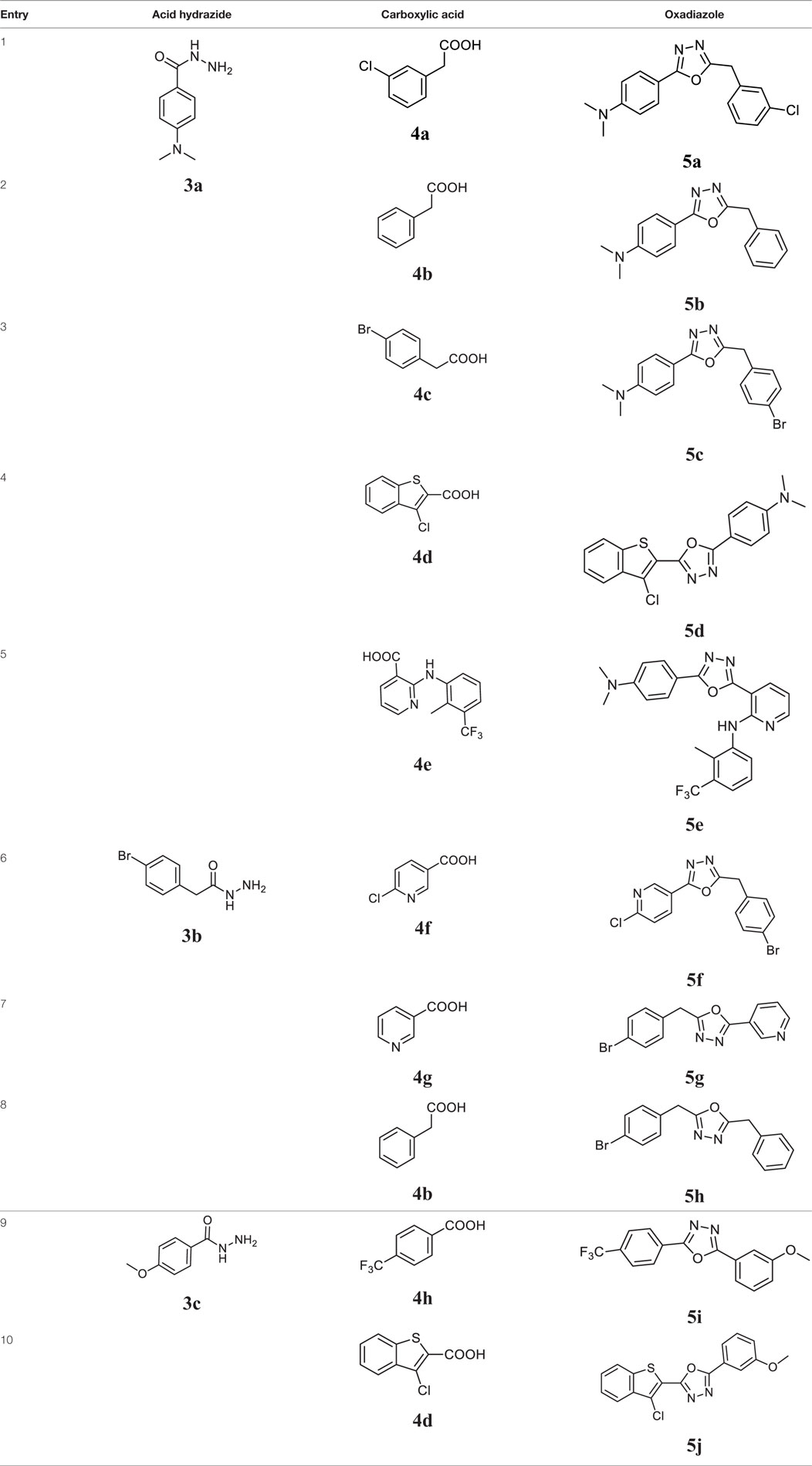
Table 1. Library of newly synthesized 1,3,4-oxadiazoles.
Pharmacology
1,3,4-Oxadiazoles Mitigate the Proliferation of HCC Cells in Time- and Dose-Dependent Manner
Initially, we prepared the library of 10 novel 1,3,4-oxadiazoles and screened the newly synthesized compounds for their antiproliferative potential against HepG2 and HCCLM3 cells by MTT assay (37). Among the screened compounds, CMO was identified as the most potent antiproliferative agent with an IC50 of 27.5 µM against HCCLM3 cells. Our results presented that CMO possess relatively higher antiproliferative efficacy against HCCLM3 than HepG2 cells. We next treated HCCLM3 and HepG2 cells with different concentrations of CMO for different time intervals. We observed a significant reduction in proliferation of cells in a dose- and time-dependent manner (Figures 1A,B).
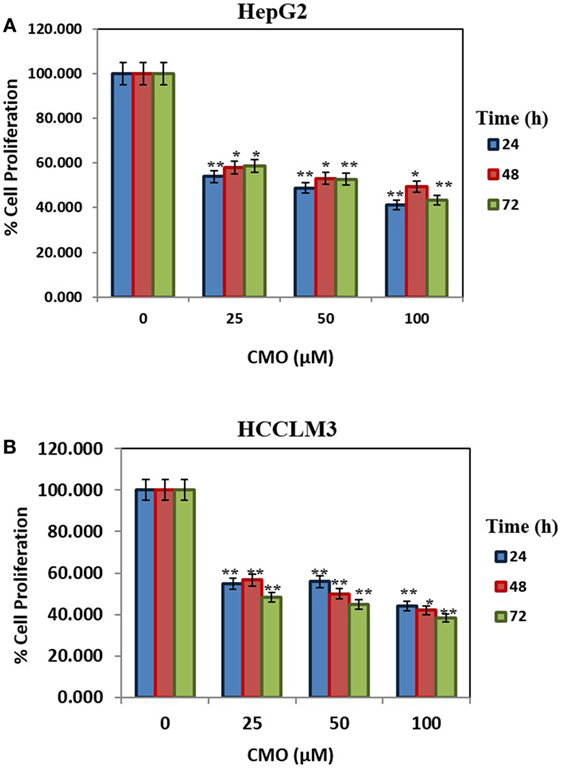
Figure 1. (A,B) CMO induces antiproliferative effect in time- and dose-dependent manner. HepG2 and HCCLM3 cells were plated in triplicate, treated with indicated concentrations of CMO, and then subjected to MTT assay after 24, 48, and 72 h to analyze proliferation of cells. The data are expressed as mean ± SD, compared with the untreated control (*p < 0.05, **p < 0.01).
CMO Causes Increased Accumulation of HCC Cells in subG1 Phase
Caspase-activated DNase-mediated fragmentation of the genomic DNA is remarkable event in the cells committed to undergo apoptosis, which results in the formation of cells with lesser DNA content (38). These cells are termed as hypodiploid cells and can be detected as subG1 cell population in flow cytometric analysis (39). In order to investigate the effect of CMO on distribution of cell cycle, HepG2 and HCCLM3 cells were treated with CMO at 50 µM for different time points up to 72 h and analyzed cell cycle distribution after PI staining. The results demonstrated that CMO significantly increased the subG1 cell population of HCCLM3 to 6, 12.5, and 25.3% at 24, 48, and 72 h, respectively. The cells in subG1 phase of HepG2 were found to be 2.8, 9.66, and 48.6% at 24, 48, and 72 h, respectively (Figures 2A,B).
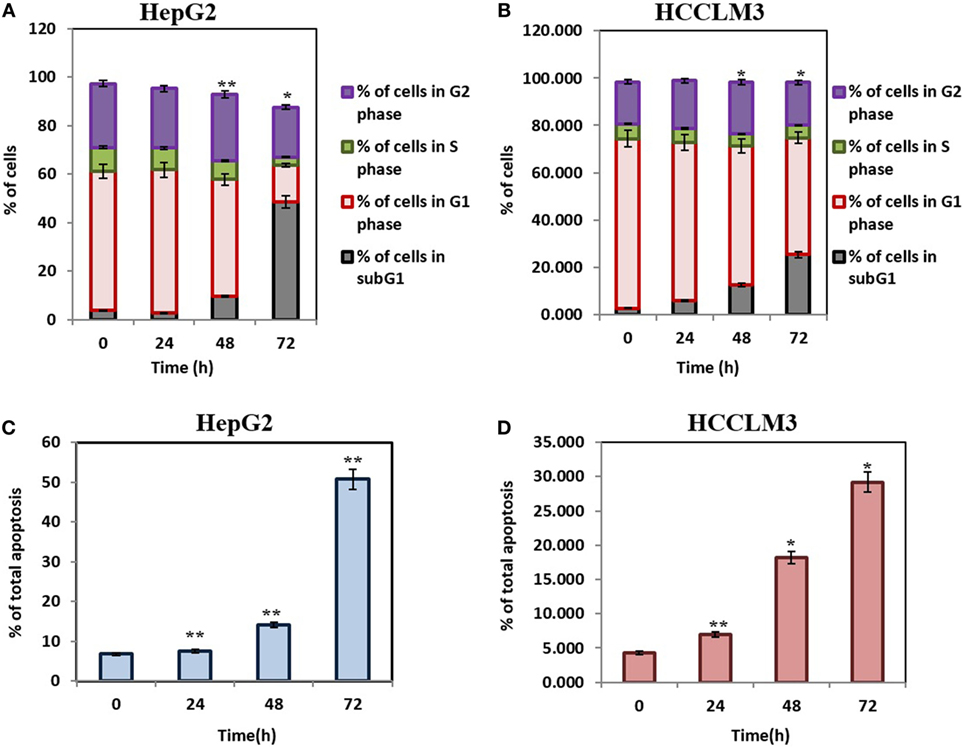
Figure 2. (A,B) CMO induces accumulation of hepatocellular carcinoma (HCC) cells in SubG1 phase. HepG2 and HCCLM3 cells were treated with 50 µM of CMO for 24, 48, and 72 h, after which, the cells were washed, fixed, stained with propidium iodide, and subjected to flow cytometric analysis. (C,D) CMO induces substantial apoptosis in HCC cells. HepG2 and HCCLM3 cells were exposed to 50 µM CMO at indicated times, after which, cells were harvested and stained with Annexin V and propidium iodide. The percentage of early and late apoptosis in the HCC cells treated with 50 µM CMO was examined using flow cytometry. The bar graph shows total percentage of apoptotic cells of HepG2 and HCCLM3 upon treatment with 50 µM of CMO at the indicated time points. The data are expressed as mean ± SD, compared with the untreated control (*p < 0.05, **p < 0.01).
CMO Induces Apoptosis in HCC Cells
The exposure of phosphatidylserine in the outer leaflet of plasma membrane is the most common biochemical change that is observed in the apoptotic cells, which can be detected using annexin V-FITC-PI staining (40). To ensure that CMO induces antiproliferative effect via apoptosis, we next examined the effect of CMO externalization of phosphatidylserine in HCCLM3 cells. Interestingly, the treatment with CMO significantly increased the percentage of early (annexin V-positive and PI-negative cells) and late (annexin V-positive and PI-positive cells) apoptotic cells compared with DMSO-treated cells (Figures 2C,D). These results demonstrate that CMO induce apoptosis in HCC cells.
CMO Inhibits the Phosphorylation of IκB and Depletes the Nuclear Pool of p65 in HCC Cells
Inactive NF-κB in association with inhibitory kappa B (IκB) is present in the cytoplasm (41). Phosphorylation and proteolytic degradation of IκB is essential for posttranslational activation of NF-κB and, upon activation, NF-κB translocate to nucleus to induce the expression of target genes (42). In order to investigate the effect of CMO on NF-κB signaling pathway, HepG2 cells were treated with CMO for different time points up to 12 h, prepared the cytoplasmic extract, and analyzed the levels of phospho-IκB. Interestingly, we found the decrease in phosphorylation of IκB (Ser 32) in a time-dependent manner (Figure 3A). At the same time, IκB and GAPDH protein expression remained unchanged.
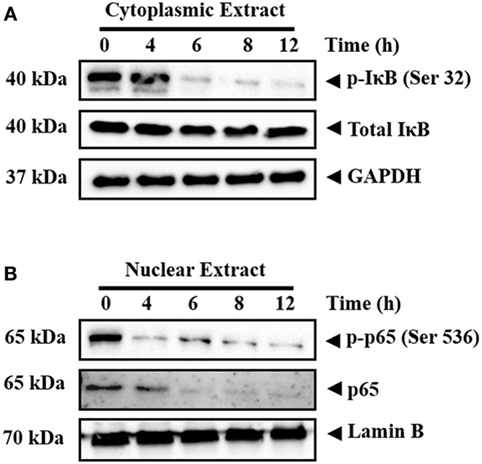
Figure 3. (A,B) CMO inhibits the phosphorylation of IκB and depletes the nuclear pool of p65 in hepatocellular carcinoma cells. HepG2 cells were treated with 25 µM CMO for indicated time point, after which, the cytoplasmic and nuclear extracts were prepared, and protein was resolved on SDS-PAGE gel, electrotransferred onto nitrocellulose membranes, and probed for phospho-IκB, IκB, phospho-p65, p65, GAPDH, and Lamin B antibodies.
The phosphorylation of p65 (Ser 536) defines an IκB-independent NF-κB signaling pathway (43). Therefore, we further examined the levels of phospho-p65 in the nuclear extract of cells treated with CMO at different time points up to 12 h. The results clearly demonstrated the decline in phospho-p65 and p65 in a time-dependent manner (Figure 3B). The expression of lamin B was used as input control, which remained unaltered.
CMO Abrogated NF-κB DNA Binding and Luciferase Activity in HCC Cells
We next investigated the effect of CMO on constitutive NF-κB activity in HCC cells. Cells were preincubated with 25 µM CMO for 4, 8, and 12 h and then nuclear extracts were prepared and tested for NF-κB DNA-binding activity. We noted that treatment with CMO suppressed constitutive NF-κB activity in a time-dependent manner (Figure 4A). To analyze the effect of CMO on constitutive NF-κB-dependent reporter gene expression in HCC cells, transfection was done as described in Section “Materials and Methods.” In the presence of CMO, NF-κB-dependent luciferase expression was significantly reduced in a time-dependent manner with maximum inhibition at 12 h (Figures 4B,C). These results further demonstrate that CMO can abrogate constitutive NF-κB activation in HCC cells.
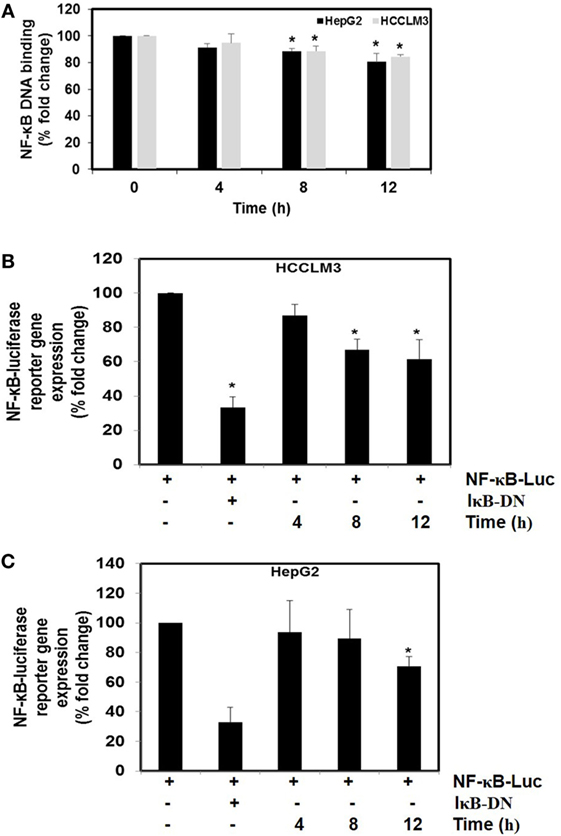
Figure 4. (A) The effect of CMO on constitutive NF-κB DNA-binding activity. HepG2 cells (5 × 105/ml) and HCCLM3 cells (5 × 105/ml) were treated with CMO for 4, 8, and 12 h. Nuclear extracts were prepared, 50 µg of the nuclear extract protein was taken for DNA-binding assay as described in Section “Materials and Methods.” (B,C) CMO inhibits constitutive activation of reporter gene expression. HepG2 (5 × 105/ml) and HCCLM3 (5 × 105/ml) cells were transfected with NF-κB luciferase and β-galactosidase reporter plasmid using lipofectamine, incubated for 24 h, and then treated with CMO for 4, 8, and 12 h. Cells were lysed in reporter lysis buffer and analyzed for luciferase activity and normalized with β-galactosidase activity. Results are expressed as % fold activity over the activity of vector control. *p < 0.05.
Transfection with p65 siRNA Blocks CMO Induced Caspase-3/7 Activation
We determined whether the knockdown of p65 using siRNA could significantly block the increase in CMO induced caspase-3/7 activation in HepG2 cells. In cells transfected with control siRNA, CMO treatment significantly increased caspase-3/7 activation, thereby inducing apoptosis (Figure 5A). The results clearly indicate that the observed increase in caspase-3/7 activation was significantly suppressed in the cells transfected with p65 siRNA when compared to control siRNA treated group.
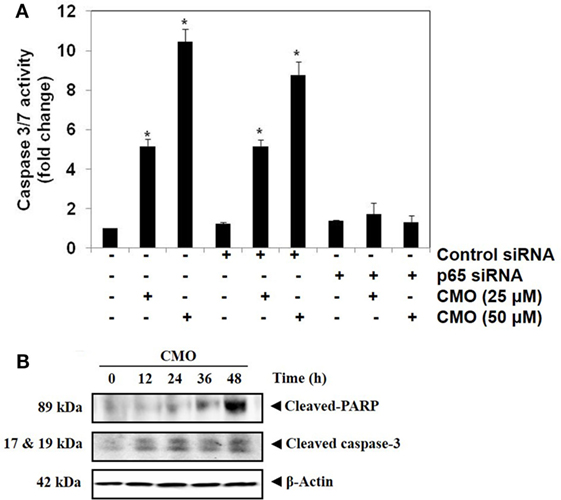
Figure 5. (A) Knockdown of p65 by small interfering RNA (siRNA) reduces the apoptotic effect of CMO. HepG2 cells were transfected with either control or p65 specific siRNA (50 nM). After 48 h, the cells were treated with CMO (25 or 50 µM) for 24 h, and the enzymatic activity of caspase-3/7 was determined by Caspase-Glo® 3/7 assay kit. (B) CMO increases the cleavage of PARP and Caspase 3 in HCCLM3 cells. HCCLM3 cells were treated with 50 µM CMO for 12, 24, 36, and 48 h, after which, the whole-cell extracts were prepared, and 30 µg of protein was resolved on 12% SDS-PAGE gel, electrotransferred onto nitrocellulose membranes, and probed for cleaved PARP and cleaved caspase 3 antibodies. The data are expressed as mean ± SD, compared with the untreated control (*p < 0.05).
CMO Induces the Cleavage of PARP and Caspase-3
During apoptosis, procaspase-3 is cleaved to form active caspase 3 (executioner caspase), which in turn cleaves the full-length PARP (116 kDa) into 85- and 24-kDa fragments. PARP is associated with DNA repair mechanism, and its cleavage drives the cell to apoptosis. We next evaluated whether inhibition of NF-κB by CMO induce PARP and caspase-3 cleavage. We observed the increase in cleaved caspase-3 and PARP demonstrating that CMO induces caspase-3-mediated apoptosis, which is in agreement with the results of previous experiments (Figure 5B).
In Silico Analysis of Oxadiazoles with p65
Compounds of the type 1,3,4-oxadiazoles are known to target NF-κB in several proinflammatory disease models. It was also demonstrated that alkylating agent such as N-ethylmaleimide and oxidizing agent eliminated the DNA binding ability of NF-κB (44). In another study, helenalin (the sesquiterpene lactone) selectively alkylates p65 subunit and inhibits the activation of NF-κB (45). Helenalin is an oxygen containing heterocycle with good NF-κB inhibitory activity. Based on these reports, we predicted that electronegativity of oxygen and nitrogen and electropositivity of other atoms in the oxadiazole ring contributes for inhibiting the activation of NF-κB. To test the hypothesis and in order to understand the interaction of 1,3,4-oxadiazoles toward NF-κB, the crystal structure of NF-κB complex was considered in our study. Accelrys Discovery Studio default tools and settings (version 2.5) were used for the molecular docking procedures. Further, the hydrophobic region near the Cys38 of p65 protein was identified using Accelrys binding site identification tool. Using the LIGANDFIT protocol of the ligand–receptor interaction module of Discovery Studio version 2.5, the oxadiazoles were docked into the hydrophobic region of p65 (Figure 6A), and the docking scores of all the compounds were summarized (Table 2). Docking scores indicated that CMO binds to the p65 with the higher value of (and thus most favorable) score of 61.0 kcal/mol. The interaction pattern revealed that benzothiazole ring of CMO enters the hydrophobic region of p65 and interacts with Asn186, Arg187, Ala188 on one side and Arg30, Gly31, Asp223, Arg274, ser276, Asp277 on the other side. Further, oxadiazole ring of CMO is found to interact with Ala190, Thr191, and Gln220 (Figures 6B,C). In addition, hydrogen bonding is observed with the methoxy phenyl group of CMO and Gln247 and Lys218 of p65. The results of in silico analysis are in agreement with the results of cell based assays.
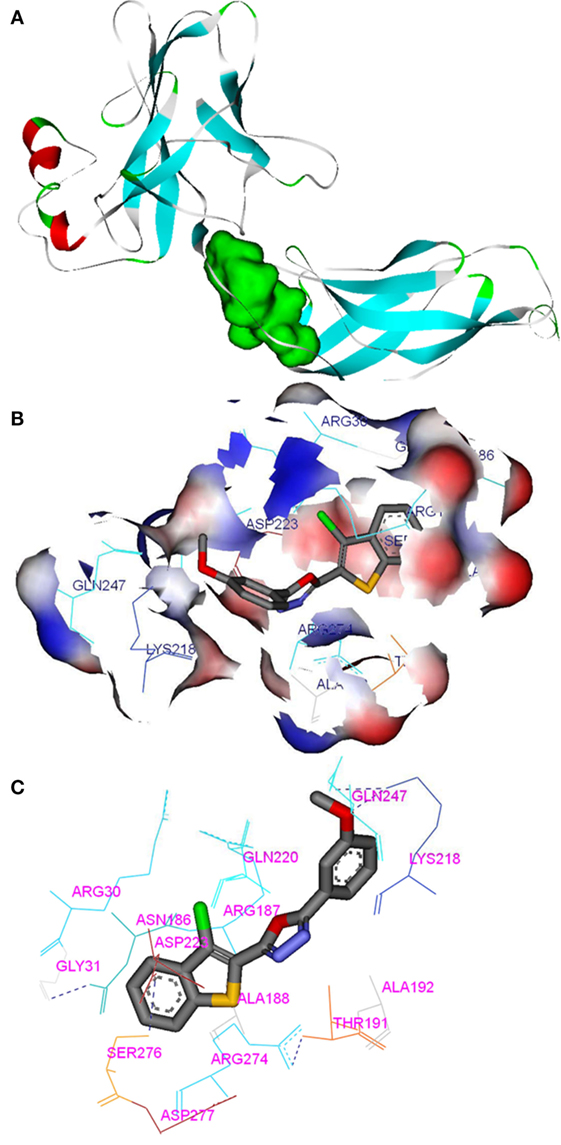
Figure 6. In silico molecular interactions between p65 of NF-κB complex and the oxadiazole derivatives: (A) hydrophobic region of p65 was shown as green mass; (B) surface view of CMO bound p65 with the key amino acids (labeled); (C) interaction map and hydrogen bonding (dotted line) pattern of p65 protein with CMO.
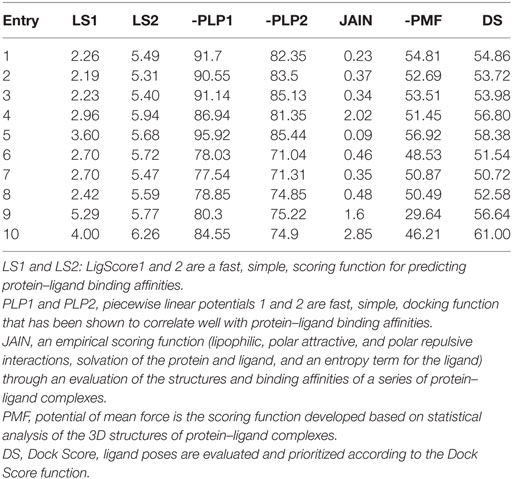
Table 2. Molecular docking results of p65 with oxadiazoles.
Conclusion
In summary, this study aimed at designing a library of chemically novel and biologically active 1,3,4-oxadiazoles. We generated 10 oxadiazole structural analogs and evaluated for their cytotoxic effect against HCC cells, and the lead compound (CMO) displayed good antiproliferative efficacy. Based on the literature, we speculated NF-κB signaling as the putative target of the lead compound, and it is validated via in vitro and in silico approaches. Although, this study identifies NF-κB signaling as the likely target of 1,3,4-oxadiazoles, more comprehensive study on its off-targets, signaling cross talks and in vivo antitumor potential needs to be investigated.
Author Contributions
KR, B, CM, AC, SA, AK, and GS conceived the project. KR, B, CM, GS, and AB designed the experiments. NA, SR, B, MS, and SM carried out the research and analysis of data. KR, B, AK, GS, CM, and NA wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was supported by Council of Scientific and Industrial Research (No.02(0291)17/EMR-II), Department of Biotechnology (No. BT/PR/8064/BID/7/441/2013) to B. AK was supported by grants from National Medical Research Council of Singapore, Medical Science Cluster, Yong Loo Lin School of Medicine, National University of Singapore and by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centers of Excellence initiative to Cancer Science Institute of Singapore, National University of Singapore.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fonc.2018.00042/full#supplementary-material.
References
1. Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab (2011) 13(1):11–22. doi:10.1016/j.cmet.2010.12.008
2. Chai EZ, Siveen KS, Shanmugam MK, Arfuso F, Sethi G. Analysis of the intricate relationship between chronic inflammation and cancer. Biochem J (2015) 468(1):1–15. doi:10.1042/bj20141337
3. Aggarwal BB. Nuclear factor-κB: the enemy within. Cancer Cell (2004) 6(3):203–8. doi:10.1016/j.ccr.2004.09.003
4. Li F, Zhang J, Arfuso F, Chinnathambi A, Zayed ME, Alharbi SA, et al. NF-kappaB in cancer therapy. Arch Toxicol (2015) 89(5):711–31. doi:10.1007/s00204-015-1470-4
5. Nirvanappa AC, Mohan CD, Rangappa S, Ananda H, Sukhorukov AY, Shanmugam MK, et al. Novel synthetic oxazines target NF-kappaB in colon cancer in vitro and inflammatory bowel disease in vivo. PLoS One (2016) 11(9):e0163209. doi:10.1371/journal.pone.0163209
6. Neelgundmath M, Dinesh KR, Mohan CD, Li F, Dai X, Siveen KS, et al. Novel synthetic coumarins that targets NF-kappaB in hepatocellular carcinoma. Bioorg Med Chem Lett (2015) 25(4):893–7. doi:10.1016/j.bmcl.2014.12.065
7. Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta (2010) 1799(10–12):775–87. doi:10.1016/j.bbagrm.2010.05.004
8. Sethi G, Shanmugam MK, Ramachandran L, Kumar AP, Tergaonkar V. Multifaceted link between cancer and inflammation. Biosci Rep (2012) 32(1):1–15. doi:10.1042/bsr20100136
9. Sethi G, Tergaonkar V. Potential pharmacological control of the NF-kappaB pathway. Trends Pharmacol Sci (2009) 30(6):313–21. doi:10.1016/j.tips.2009.03.004
10. Li F, Sethi G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta (2010) 1805(2):167–80. doi:10.1016/j.bbcan.2010.01.002
11. Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature (1995) 376(6536):167–70. doi:10.1038/376167a0
12. Zeligs KP, Neuman MK, Annunziata CM. Molecular pathways: the balance between cancer and the immune system challenges the therapeutic specificity of targeting nuclear factor-kappaB signaling for cancer treatment. Clin Cancer Res (2016) 22(17):4302–8. doi:10.1158/1078-0432.ccr-15-1374
13. Baud V, Karin M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov (2009) 8(1):33. doi:10.1038/nrd2781
14. Kim C, Cho SK, Kim KD, Nam D, Chung WS, Jang HJ, et al. beta-Caryophyllene oxide potentiates TNFalpha-induced apoptosis and inhibits invasion through down-modulation of NF-kappaB-regulated gene products. Apoptosis (2014) 19(4):708–18. doi:10.1007/s10495-013-0957-9
15. Ningegowda R, Shivananju NS, Rajendran P, Basappa , Rangappa KS, Chinnathambi A, et al. A novel 4,6-disubstituted-1,2,4-triazolo-1,3,4-thiadiazole derivative inhibits tumor cell invasion and potentiates the apoptotic effect of TNFalpha by abrogating NF-kappaB activation cascade. Apoptosis (2017) 22(1):145–57. doi:10.1007/s10495-016-1312-8
16. Tai DI, Tsai SL, Chang YH, Huang SN, Chen TC, Chang KS, et al. Constitutive activation of nuclear factor kappaB in hepatocellular carcinoma. Cancer (2000) 89(11):2274–81. doi:10.1002/1097-0142(20001201)89:11<2274::AID-CNCR16>3.0.CO;2-2
17. Qiao L, Zhang H, Yu J, Francisco R, Dent P, Ebert MP, et al. Constitutive activation of NF-kappaB in human hepatocellular carcinoma: evidence of a cytoprotective role. Hum Gene Ther (2006) 17(3):280–90. doi:10.1089/hum.2006.17.280
18. Li W, Tan D, Zenali MJ, Brown RE. Constitutive activation of nuclear factor-kappa B (NF-kB) signaling pathway in fibrolamellar hepatocellular carcinoma. Int J Clin Exp Pathol (2009) 3(3):238–43.
19. Jin L, Chen J, Song B, Chen Z, Yang S, Li Q, et al. Synthesis, structure, and bioactivity of N′-substituted benzylidene-3,4,5-trimethoxybenzohydrazide and 3-acetyl-2-substituted phenyl-5-(3,4,5-trimethoxyphenyl)-2,3-dihydro-1,3,4-oxadiazole derivatives. Bioorg Med Chem Lett (2006) 16(19):5036–40. doi:10.1016/j.bmcl.2006.07.048
20. Kumar D, Sundaree S, Johnson EO, Shah K. An efficient synthesis and biological study of novel indolyl-1,3,4-oxadiazoles as potent anticancer agents. Bioorg Med Chem Lett (2009) 19(15):4492–4. doi:10.1016/j.bmcl.2009.03.172
21. Nandeesh KN, Swarup HA, Sandhya NC, Mohan CD, Kumar CSP, Kumara MN, et al. Synthesis and antiproliferative efficiency of novel bis (imidazol-1-yl) vinyl-1, 2, 4-oxadiazoles. New J Chem (2016) 40(3):2823–8. doi:10.1039/C5NJ02925B
22. Palanki MS, Erdman PE, Manning AM, Ow A, Ransone LJ, Spooner C, et al. Novel inhibitors of AP-1 and NF-kappaB mediated gene expression: structure-activity relationship studies of ethyl 4-[(3-methyl-2,5-dioxo(3-pyrrolinyl))amino]-2-(trifluoromethyl)++ +pyrimidi ne-5-carboxylate. Bioorg Med Chem Lett (2000) 10(15):1645–8. doi:10.1016/S0960-894X(00)00312-7
23. Baburajeev CP, Dhananjaya Mohan C, Ananda H, Rangappa S, Fuchs JE, Jagadish S, et al. Development of novel triazolo-thiadiazoles from heterogeneous “green” catalysis as protein tyrosine phosphatase 1B inhibitors. Sci Rep (2015) 5:14195. doi:10.1038/srep14195
24. Bharathkumar H, Mohan CD, Rangappa S, Kang T, Keerthy HK, Fuchs JE, et al. Screening of quinoline, 1,3-benzoxazine, and 1,3-oxazine-based small molecules against isolated methionyl-tRNA synthetase and A549 and HCT116 cancer cells including an in silico binding mode analysis. Org Biomol Chem (2015) 13(36):9381–7. doi:10.1039/c5ob00791g
25. Rakesh KS, Jagadish S, Swaroop TR, Mohan CD, Ashwini N, Harsha KB, et al. Anti-cancer activity of 2,4-disubstituted thiophene derivatives: dual inhibitors of lipoxygenase and cyclooxygenase. Med Chem (2015) 11(5):462–72. doi:10.2174/1573406411666141210141918
26. Sulaiman NB, Mohan CD, Basappa S, Pandey V, Rangappa S, Bharathkumar H, et al. An azaspirane derivative suppresses growth and induces apoptosis of ER-positive and ER-negative breast cancer cells through the modulation of JAK2/STAT3 signaling pathway. Int J Oncol (2016) 49(3):1221–9. doi:10.3892/ijo.2016.3615
27. Anusha S, Mohan CD, Ananda H, Baburajeev CP, Rangappa S, Mathai J, et al. Adamantyl-tethered-biphenylic compounds induce apoptosis in cancer cells by targeting Bcl homologs. Bioorg Med Chem Lett (2016) 26(3):1056–60. doi:10.1016/j.bmcl.2015.12.026
28. Mohan CD, Srinivasa V, Rangappa S, Mervin L, Mohan S, Paricharak S, et al. Trisubstituted-imidazoles induce apoptosis in human breast cancer cells by targeting the oncogenic PI3K/Akt/mTOR signaling pathway. PLoS One (2016) 11(4):e0153155. doi:10.1371/journal.pone.0153155
29. Keerthy HK, Garg M, Mohan CD, Madan V, Kanojia D, Shobith R, et al. Synthesis and characterization of novel 2-amino-chromene-nitriles that target Bcl-2 in acute myeloid leukemia cell lines. PLoS One (2014) 9(9):e107118. doi:10.1371/journal.pone.0107118
30. Sebastian A, Pandey V, Mohan CD, Chia YT, Rangappa S, Mathai J, et al. Novel adamantanyl-based thiadiazolyl pyrazoles targeting EGFR in triple-negative breast cancer. ACS Omega (2016) 1(6):1412–24. doi:10.1021/acsomega.6b00251
31. Ashwini N, Garg M, Mohan CD, Fuchs JE, Rangappa S, Anusha S, et al. Synthesis of 1,2-benzisoxazole tethered 1,2,3-triazoles that exhibit anticancer activity in acute myeloid leukemia cell lines by inhibiting histone deacetylases, and inducing p21 and tubulin acetylation. Bioorg Med Chem (2015) 23(18):6157–65. doi:10.1016/j.bmc.2015.07.069
32. Shanmugam MK, Rajendran P, Li F, Nema T, Vali S, Abbasi T, et al. Ursolic acid inhibits multiple cell survival pathways leading to suppression of growth of prostate cancer xenograft in nude mice. J Mol Med (Berl) (2011) 89(7):713–27. doi:10.1007/s00109-011-0746-2
33. Chandramohanadas R, Basappa , Russell B, Liew K, Yau YH, Chong A, et al. Small molecule targeting malaria merozoite surface protein-1 (MSP-1) prevents host invasion of divergent plasmodial species. J Infect Dis (2014) 210(10):1616–26. doi:10.1093/infdis/jiu296
34. Srinivas V, Mohan CD, Baburajeev CP, Rangappa S, Jagadish S, Fuchs JE, et al. Synthesis and characterization of novel oxazines and demonstration that they specifically target cyclooxygenase 2. Bioorg Med Chem Lett (2015) 25(15):2931–6. doi:10.1016/j.bmcl.2015.05.047
35. Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell (1998) 95(6):759–70. doi:10.1016/S0092-8674(00)81699-2
36. Sukhorukov AY, Nirvanappa AC, Swamy J, Ioffe SL, Nanjunda Swamy S, Basappa , et al. Synthesis and characterization of novel 1,2-oxazine-based small molecules that targets acetylcholinesterase. Bioorg Med Chem Lett (2014) 24(15):3618–21. doi:10.1016/j.bmcl.2014.05.040
37. Baburajeev CP, Mohan CD, Rangappa S, Mason DJ, Fuchs JE, Bender A, et al. Identification of novel class of triazolo-thiadiazoles as potent inhibitors of human heparanase and their anticancer activity. BMC Cancer (2017) 17(1):235. doi:10.1186/s12885-017-3214-8
38. Roopashree R, Mohan CD, Swaroop TR, Jagadish S, Raghava B, Balaji KS, et al. Novel synthetic bisbenzimidazole that targets angiogenesis in Ehrlich ascites carcinoma bearing mice. Bioorg Med Chem Lett (2015) 25(12):2589–93. doi:10.1016/j.bmcl.2015.04.010
39. Mohan CD, Bharathkumar H, Bulusu KC, Pandey V, Rangappa S, Fuchs JE, et al. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J Biol Chem (2014) 289(49):34296–307. doi:10.1074/jbc.M114.601104
40. Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ (1998) 5(7):551–62. doi:10.1038/sj.cdd.4400404
41. Keerthy HK, Mohan CD, Sivaraman Siveen K, Fuchs JE, Rangappa S, Sundaram MS, et al. Novel synthetic biscoumarins target tumor necrosis factor-alpha in hepatocellular carcinoma in vitro and in vivo. J Biol Chem (2014) 289(46):31879–90. doi:10.1074/jbc.M114.593855
42. Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J (1995) 14(12):2876–83.
43. Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphorylation of RelA/p65 on serine 536 defines an I{kappa}B{alpha}-independent NF-{kappa}B pathway. J Biol Chem (2005) 280(41):34538–47. doi:10.1074/jbc.M504943200
44. Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A (1991) 88(10):4328–32. doi:10.1073/pnas.88.10.4328
Keywords: oxadiazoles, NF-κB, hepatocellular carcinoma, apoptosis, anticancer
Citation: Mohan CD, Anilkumar NC, Rangappa S, Shanmugam MK, Mishra S, Chinnathambi A, Alharbi SA, Bhattacharjee A, Sethi G, Kumar AP, Basappa and Rangappa KS (2018) Novel 1,3,4-Oxadiazole Induces Anticancer Activity by Targeting NF-κB in Hepatocellular Carcinoma Cells. Front. Oncol. 8:42. doi: 10.3389/fonc.2018.00042
Received: 25 September 2017; Accepted: 08 February 2018;
Published: 19 March 2018
Edited by:
Zhi Sheng, Virginia Tech, United StatesReviewed by:
Harikumar KB, Rajiv Gandhi Centre for Biotechnology, IndiaYun Dai, Virginia Commonwealth University, United States
Copyright: © 2018 Mohan, Anilkumar, Rangappa, Shanmugam, Mishra, Chinnathambi, Alharbi, Bhattacharjee, Sethi, Kumar, Basappa and Rangappa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Basappa, salundibasappa@gmail.com