MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches
 Amanda C. Winters
Amanda C. Winters Kathrin M. Bernt
Kathrin M. Bernt- Division of Pediatric Hematology/Oncology/BMT, University of Colorado School of Medicine and Children’s Hospital Colorado, Aurora, CO, USA
The mixed-lineage leukemia 1 (MLL1) gene (now renamed Lysine [K]-specific MethylTransferase 2A or KMT2A) on chromosome 11q23 is disrupted in a unique group of acute leukemias. More than 80 different partner genes in these fusions have been described, although the majority of leukemias result from MLL1 fusions with one of about six common partner genes. Approximately 10% of all leukemias harbor MLL1 translocations. Of these, two patient populations comprise the majority of cases: patients younger than 1 year of age at diagnosis (primarily acute lymphoblastic leukemias) and young- to-middle-aged adults (primarily acute myeloid leukemias). A much rarer subgroup of patients with MLL1 rearrangements develop leukemia that is attributable to prior treatment with certain chemotherapeutic agents—so-called therapy-related leukemias. In general, outcomes for all of these patients remain poor when compared to patients with non-MLL1 rearranged leukemias. In this review, we will discuss the normal biological roles of MLL1 and its fusion partners, how these roles are hypothesized to be dysregulated in the context of MLL1 rearrangements, and the clinical manifestations of this group of leukemias. We will go on to discuss the progress in clinical management and promising new avenues of research, which may lead to more effective targeted therapies for affected patients.
Structure and Function of Wild-Type MLL1
Mixed-Lineage Leukemia 1 (MLL1) Protein Structure and Binding Partners
The normal MLL1 gene at the 11q23 locus encodes an approximately 500-kDa nuclear protein with multiple functional domains and binding partners (Figure 1A), whose structure was first described by both Tkachuk et al. and Gu et al. (1, 2) and which is expressed in a wide variety of normal human tissues (3). The N-terminal portion of the protein contains a domain for binding Menin, a protein that serves as a link between MLL1 and the chromatin-binding protein lens epithelium-derived growth factor (LEDGF). LEDGF is a binder of dimethylated H3K36 (placed by ASH1L). The association of MLL1 with Menin/LEDGF is particularly critical for the function of MLL fusions, but also affects wild-type MLL1 (4–10). The N-terminus also contains AT-hook motifs (DNA-binding domains), speckled nuclear localization domains 1 and 2 (SNL-1 and SNL-2), and two repression domains (RD1 and RD2), the first of which (RD1) also contains a CxxC domain (1, 11–13). The CxxC domain has homology to DNA methyltransferase 1 (DNMT1), which methylates cytosine residues of DNA (11, 14). Although DNMT1 preferentially targets hemimethylated CpG motifs, the MLL1 CxxC domain binds non-methylated CpG DNA (15). All of these domains are typically conserved in chimeric MLL1 fusion proteins (12). The middle portion of MLL1 contains four plant homeodomain (PHD) fingers (which mediate protein–protein interactions) and a bromodomain (which mediates binding to histones with acetylated lysine residues). The C-terminal portion contains a transcriptional activation domain and a SET domain (1, 12, 16). The third PHD finger allows association between MLL1 and the cyclophilin CYP33, which is important for negative regulation of certain MLL1 target genes (17). The SET (Su(var)3-9, enhancer of zeste, trithorax) domain is homologous to that of Drosophila trithorax and catalyzes mono-, di-, and trimethylation of lysine 4 on histone 3 (H3K4) in vitro (1, 18). These latter four domains (PHD finger, bromodomain, activation domain, and SET domain) are all lost in most MLL1 fusion proteins (12) (Figure 1B).
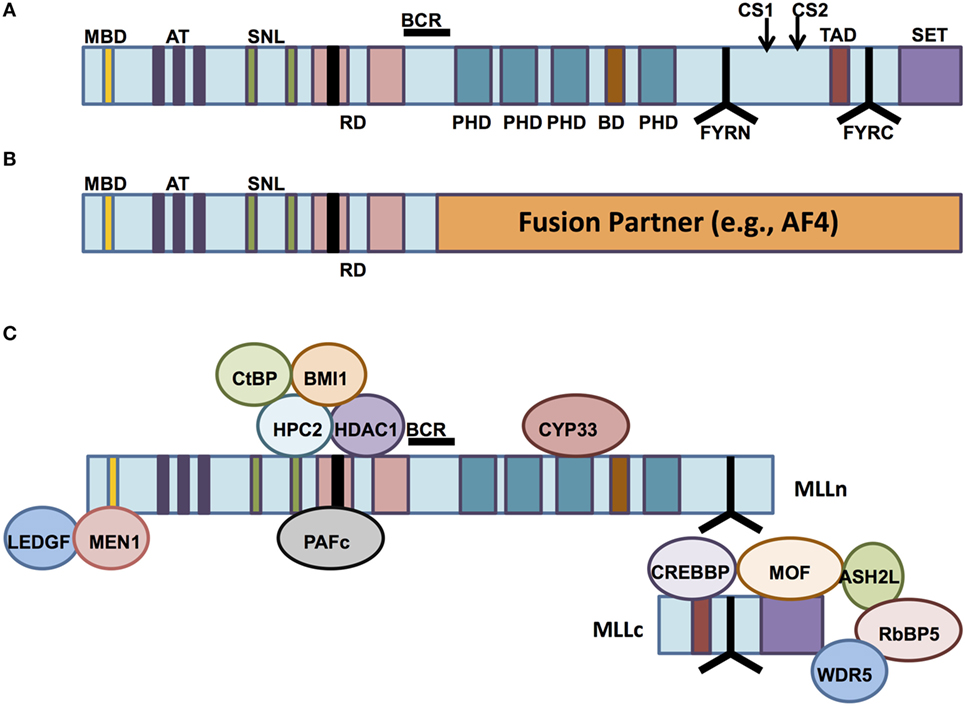
Figure 1. The structure of mixed-lineage leukemia (MLL) and normal vs aberrant MLL complexes. (A) The structure of the wild-type MLL protein, emphasizing the functional domains. MBD, Menin-binding domain; AT, AT hooks; SNL, speckled nuclear localization domains; RD, repression domains (black box in first RD represents the CXXC domain); BCR, breakpoint cluster region; PHD, PHD fingers; BD, bromodomain. CS1 and CS2 are the taspase-1 cleavage sites, and FYRN and FYRC are the domains whereby MLL-N and MLL-C interact after cleavage. TAD, transactivation domain; SET, H3K4 histone methyltransferase domain. (B) MLL fusion proteins are caused by chromosomal rearrangements leading to in-frame fusions between N-terminal MLL (to the BCR) and any of 80 different fusion partners. PHD domains, transactivation domains, and the SET domain are lost. (C) MLL-interacting proteins. Proteins involved in repressive functions of MLL are grouped above the MLL protein (regulated by CYP33), whereas proteins involved in activation of MLL-dependent transcription are grouped below the MLL protein schematic.
After its translation, wild-type MLL1 is proteolytically cleaved by the enzyme taspase-1 (19, 20). The resulting 320-kDa N-terminal fragment (MLL-N) contains all domains except the transcriptional activation domain and the SET domain, both of which are retained by the 180-kDa C-terminal fragment (MLL-C). MLL-N and MLL-C normally associate with one another as components of a multiprotein complex that regulates chromatin modification and gene expression (19, 21) (Figure 1C). Other essential proteins that make up the core of the MLL1 complex include RbBP5, Ash2L, and WDR5 (21). These three proteins form a complex that is able to bind a variety of H3K4 methytransferases with SET domains, including MLL1. Recent biochemical and structural analyses of the interactions between the complex members reveal that the RbBP5-Ash2L heterodimer interaction with MLL1 stabilizes it in the catalytic conformation, whereas WDR5 acts as a bridge between the RbBP5-Ash2L complex and MLL1 itself (22). The WDR5 bridge is not needed for the interaction between RbBP5-Ash2L and other MLL family members, but it is essential for the H3K4 methyltransferase activity of MLL1. MLL1 recruits these other components, along with other chromatin remodeling proteins such as the histone acetyltransferases CBP/p300 and hMOF, to specific target genes (21, 23, 24). In fact, recruitment of these other histone-modifying proteins, particularly hMOF, has recently been shown to be crucial for MLL1 target gene expression, whereas the H3K4 methyltransferase activity of MLL1 is dispensable in this regard (24).
The N-terminal portion of MLL1 present in translocation-encoded fusions loses its ability to interact with MLL-C (19). The functional consequence of this feature is not clear. In most leukemias, residual core complex including MLL-C would be expected to be present and retain its histone methyltransferase activity, either from expression of the reciprocal fusion (although this probably happens only in a minority of patients) or from the second, non-rearranged MLL1 allele. There is debate whether the second allele is required—on one hand, experimental data from knockout mice suggest that it might be (25, 26), on the other hand, deletion of the second MLL1 allele has been reported in patients (27) and also occurs in the ML2 cell line. Whether leukemias with deletions of the MLL1 wild-type allele retain residual wild-type function through expression and cleavage of a reciprocal fusion is unclear, as is the role of the reciprocal fusion in general. Wilkinson et al. reported that the MLL-AF4 fusion activates expression of RUNX1 and that the RUNX1 protein then interacts with the AF4-MLL reciprocal fusion and the MLL-C complex proteins (28). The authors hypothesized that interaction of AF4-MLL enhances its coactivation of RUNX1 target genes, although they were not able to successfully target AF4-MLL via siRNA for functional confirmation of this theory. Furthermore, a reciprocal translocation predicted to result in the expression of a reciprocal fusion transcript was found in only 24 of 182 MLL-rearranged (MLL-r) patients (29). The fact that in most patients the reciprocal fusion is likely not expressed strongly argues against a critical role.
Physiologic Functions of MLL1
MLL1 is both structurally and functionally homologous to the Drosophila melanogaster protein trithorax (1), which is involved in epigenetic regulation of defined developmental genes [reviewed in Ref. (30)]. Homozygous deletion of Mll1 in murine embryos results in lethality at E10.5–E12.5, with null embryos showing abnormal facial development and innervation of embryonic structures, as well as abnormal fetal hematopoiesis (31–33). Mll1 ± (heterozygous) embryos display both body segmentation abnormalities and decreased numbers of cells of several hematopoietic lineages. Many of these defects closely resemble those seen upon knockout of developmental patterning genes, such as the homeobox (Hox) genes, many of which (Hoxa9, Hoxa7, and Hoxc8) have been identified as Mll1 target genes. Although Hox genes are expressed in Mll1−/− embryos before the E9.0 stage, their expression is not maintained at later time points in the absence of Mll1 (34). These findings indicate that Mll1 is required for the maintenance, and not the initiation, of Hox gene expression. In steady-state adult murine hematopoiesis, hematopoiesis-specific knockout of Mll1 resulted in moderate to severe impairment of stem cell function (35, 36).
Identification of MLL1 target genes involved in embryogenesis and hematopoiesis has been the goal of multiple studies. MLL1 has been reported to occupy as much as 5,000 genes in leukemia cell lines and cultured lymphoblasts (37) and a smaller number of genes in fibroblasts (38). MLL1 binding correlated with the presence of H3K4me3 and occupancy of RNA polymerase II, suggesting that despite the presence of multiple negative regulatory domains in the MLL1 protein, the net outcome of MLL1 binding is typically transcriptional activation. Despite correlation of MLL1 binding with H3K4 trimethylation, MLL1 is not the methyltransferase responsible for the deposition of the majority of H3K4 trimethylation in any tissue examined to date, as knockout does not result in decreased global levels of H3K4me3 (24).
Normal Functions of the Common MLL1 Fusion Partners
Leukemia-associated translocations involving 11q23 have been shown to generate in-frame fusions of the MLL1 gene to more than 80 different partner genes (29). N-terminally truncated MLL1 alone is not sufficient to transform cells (39, 40). This finding argues for a crucial contribution on the part of the fusion partner proteins to leukemogenesis. Although the proteins encoded by the 80 + MLL1 partner genes seemingly have diverse structures and functions, two common features have emerged that likely have importance for the oncogenic potential of the chimeric protein. First, many of the partners, including the ones most frequently encountered in MLL1 fusions, are nuclear proteins involved in the regulation of transcriptional elongation [though interaction with the positive transcription elongation factor b (pTEFb) complex and phosphorylation of Pol II] and direct or indirect recruitment of the H3K79 histone methyltransferase DOT1L (41–49). Second, many partners, including those that are cytoplasmic, have been shown to form complexes in the nucleus as fusion proteins (50). A revealing study demonstrated that a fusion construct of Mll exons 1–8 and lacZ, the gene encoding the non-oncogenic enzyme β-galactosidase, was able to cause leukemias in mice, albeit with longer latency and lower incidence than the more traditional Mll-Af9 fusion (51). Importantly, the formation of a tetramer is essential for the functionality of the enzyme β-galactosidase, and all of the leukemia cells demonstrated β-galactosidase activity, suggesting that the fusion proteins also oligomerized. Martin et al. confirmed that dimerization of MLL1 is transforming through the fusion of MLL1 to FKBP12, an inducible dimerizer (52). A final interesting consideration is that N-terminal MLL1 is normally destabilized by the loss of interaction with MLL-C (19). The MLL1 fusion construct loses the domain necessary for MLL-C binding and therefore would be expected to be degraded—since it is not, the fusion partner may also play a role in enhancing the stability of the fusion protein.
The AF4 Protein Family
The AF4 protein (ALL1-fused gene from chromosome 4) is fused in-frame to MLL1 as a result of a t(4,11)(q21,q23) translocation (2). This fusion is responsible for approximately 50% of cases of infant acute lymphoblastic leukemia (ALL) with MLL1 rearrangement and more than 75% of adult MLL-r ALLs (29). AF4 is a member of the ALF (AF4, LAF-4, FMR-2) family of nuclear proteins (41, 42). Two additional family members (LAF-4 and AF5q31) have been identified in MLL1 fusions from patient samples (53, 54). These proteins share several regions of homology, including a region rich in serine residues that has been shown to have transactivation properties in reporter assays and which is conserved in fusions with MLL1 (55, 56). The functions of the known family members remain incompletely characterized. However, AF4 knockout mice display significant delays in lymphopoiesis (generation of B and T cells) (57). AF4 has been shown to interact with pTEFb and DOT1L (44–46). pTEFb is a complex of cyclin T1/T2 and cyclin-dependent kinase 9 (CDK9), which phosphorylates the C-terminal domain of RNA polymerase II and thus promotes transcriptional elongation (58). AF4 binding to pTEFb enhances PolII-CTD phosphorylation and promotes gene transcription. AF4 family members may also interact with another transcriptional complex, selectivity factor 1 (SL1), which is composed of TATA-binding protein and four associated factors, and this association may play a role in direct recruitment of RNA polymerase II to target genes (59).
AF9 and ENL
ALL1-fused gene from chromosome 9 (AF9) and eleven-nineteen leukemia (ENL) are the second and third most common fusion partners of MLL1, and these fusions arise from the t(9,11)(p22,q23) and t(11,19)(q23,p13.3) translocations, respectively (29). MLL-AF9 is most commonly associated with myeloid leukemias, while MLL-ENL is prevalent in both lymphoid and myeloid leukemias (60). AF9 and ENL have highly similar structures. Both proteins have a conserved C-terminal coiled coil region with transactivation properties that is necessary and sufficient for leukemic transformation in the context of MLL1 fusions (40). Furthermore, AF9 and ENL have also been shown to interact with AF4 via their C-termini and thus be part of AF4 containing complexes that also bind pTEFb and DOT1L (44–49). The C-terminal domains mediating this interaction are conserved in MLL1 fusions (43) and mutation of the DOT1L-binding domain of ENL in MLL-ENL cells abrogated colony formation and reduced Hox gene expression typically associated with transformation (45).
Similar to MLL1, AF9 and ENL have roles in the epigenetic/transcriptional control of developmental pathways (31, 61). Wild-type AF9 in mice and humans seems to have a regulatory function specifically in megakaryocyte/erythrocyte lineages (61, 62). AF9 and ENL have been shown to interact with the protein Polycomb 3, also known as CBX8, a component of polycomb repressive complex 1 (PRC1), which is implicated in maintenance of stable repression of genes, and with certain isoforms of the BCL-6 corepressor (45, 63–65). However, rather than mediating transcriptional repression, the role of CBX8 in the context of MLL fusions appears to mediate the recruitment of the histone acetyl transferase Tip60, thereby promoting fusion target gene expression (66). Finally, the N-terminal YEATS domain of ENL and AF9 have reader function recognizing histone proteins 1 and 3 (H1 and H3) acetylation and, as recently demonstrated, crotonylation (49, 67–69). The wild-type AF9 YEATS domain has been reported to be critically involved in the recruitment of DOT1L to chromatin and H3K79 methylation-mediated transcriptional control [(49) and see “DOT1L Inhibitors” section]; however, in the MLL1 fusion, the YEATS domain is typically excluded (40). It is possible that the N-terminal MLL1 fragment supplies this function; however, this has been difficult to experimentally confirm. The precise function of these various binding partners to the function of AF9 or ENL in their wild-type or MLL1-fused states will still require more investigation.
AF10 and AF17
AF10 was the first MLL1 fusion partner to be shown to interact with DOT1L (70). AF10 and the structurally related AF17 (also a rare fusion partner) are consistently co-purified with DOT1L and part of the canonical DOT-complex (47). AF10 is required for di- and tri- (although not mono-) methylation of H3K79 by DOT1L (71). The PHD finger of AF10 specifically binds to unmodified H3K27 (72). Although both AF10/AF17 and AF9/ENL co-purify with DOT1L, it is unclear whether all these proteins reside in one or in two separate complexes and what the relationship of these complexes is to elongation complexes containing AF4, AF5, and pTEFb or SL1 (46–48, 59).
Transcriptional Dysregulation in the Context of MLL1 Fusions
Controversies Around and Potential Roles of an Oncogenic Multiprotein Complex
The cooperation of most major MLL1 fusion partners in a single elongation regulatory complex, termed “super elongation complex” (SEC), “AF4/ENL family protein complex,” or “ENL-associated protein complex”, offered an elegant explanation for the large number of different partners: translocation of any of the members of a large complex containing AF10, AF17, AF9, ENL, ELL, AF4, AF5, pTEFb, and DOT1L would cause aberrant transcriptional elongation and similar phenotypes. However, such a super complex containing an MLL fusion has remained elusive, and careful mapping of binding sites has shown that binding of several of these members is mutually exclusive, suggesting several smaller, rather than one large complex [(46–48, 73, 74); Figure 2].
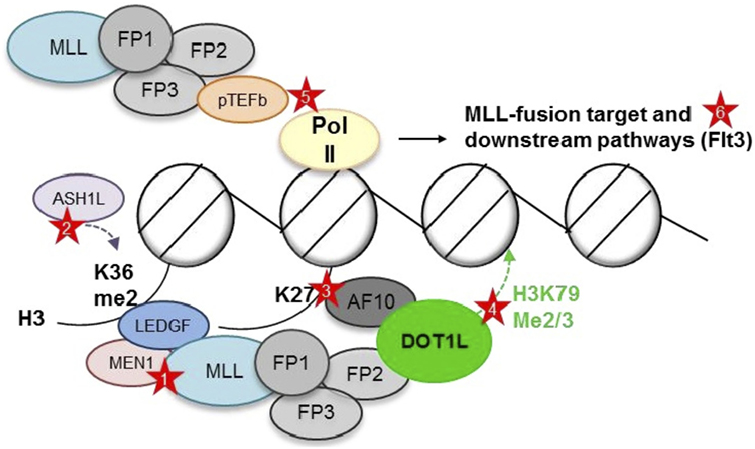
Figure 2. Putative complexes between mixed-lineage leukemia (MLL) fusions and nuclear proteins involved in histone modifications and transcriptional elongation. MEN1, Menin; FP, fusion partner. AF10, AF17, AF9, ENL, ELL, AF4, and AF5 have all been reported as MLL1 fusion partner, as well as interaction partner with each other, DOT1L, and pTEFb. However, DOT1L and pTEFb most likely do not reside within one large complex (73, 74). Red stars depict opportunities for targeted inhibition—Protein–protein: (1) Menin-MLL1 interaction. Chromatin: (2) LEDGF—H3K36me2 interaction (blocking reader domain or ASH1L). (3) AF10—unmodified H3K27 interaction (blocking reader domain or demethylases?). (4) DOT1L—placement of H3K79me2/3 (blocking methyltransferase domain). Pol II phosphorylation: (5) Inhibition of pTEFb. Downstream targets: (6) FLT3.
Furthermore, this theory did not explain the different clinical phenotypes observed in dependence of the fusion partner (discussed below). It also did not provide an explanation for the transforming activity of MLL fusions with partners such as cytosolic coiled coil domain proteins, CBP, septins, and the MLL partial tandem duplication (PTDs). Despite a vast amount of mechanistic knowledge, MLL rearrangements thus still defy a simple and unifying theory of how they cause leukemia.
The Gene Expression Program Controlled by MLL1 Fusions
Genome-wide comparisons of gene expression in MLL-r vs MLL wild-type leukemias have consistently demonstrated that this set of leukemias—irrespective of fusion partner or myeloid vs lymphoid differentiation—is distinct from all other leukemia subtypes with respect to its gene expression signature (75–77). The most frequently overexpressed genes in MLL-r leukemias are the later HOX cluster genes (particularly HOXA7-HOXA10) and the HOX cofactor MEIS1 (78, 79). HOX genes encode transcription factors whose activities control developmental processes such as segmentation and hematopoiesis (31, 34, 78, 80). In these functions, they appear to have somewhat redundant roles (80, 81). In the hematopoietic system, both HOX genes and MEIS1 are expressed at highest levels in the stem cells and early lineage progenitor cells, and expression levels are downregulated with differentiation (80, 81). Persistent expression of both MEIS1and HOX genes has been observed in a wide variety of leukemias (78, 82). Investigations into the dependence of MLL-r leukemias on upregulation of these genes have shown that MEIS1 is necessary for leukemia growth and proliferation and that levels of expression of MEIS1 correlate inversely with disease latency (83). The dependence of MLL-r leukemias for individual Hox genes appeared somewhat less consistent, likely due to functional redundancy among HOX members (81, 84, 85). However, it seems safe to say that dysregulated expression of the HOX developmental regulators and their cofactor MEIS1 contributes critically to the stem cell-like characteristics of MLL-r leukemias and confers or maintains on these cells self-renewal properties, growth, and survival advantages that promote their oncogenic potential.
These stem cell-like properties—which may also in part depend on the developmental stage at which the leukemia arose (stem cell vs early progenitor)—have been proposed to contribute to the high level of resistance to programmed cell death frequently observed in the clinic (86–91). In addition, frequent dysregulation of prosurvival pathways such as BCL-2, which counteracts the intrinsic mitochondria-mediated apoptotic pathway, may contribute to the therapeutic difficulties many of these leukemias pose in the clinical setting (89, 92).
Clinical Features of MLL-r Leukemias
Demographics and Common Features
As the name suggests, MLL rearrangements are found in mixed-lineage leukemias [now named mixed phenotype acute leukemia (MPAL) (93)]. For the most part, however, leukemias arising from rearrangements of the MLL gene manifest as either acute lymphoid or acute myeloid leukemias (ALL or AML, respectively), and only a minority of MPAL actually carry MLL rearrangements. MLL-r leukemias make up approximately 10% of acute leukemias in all age groups (94). There is a bimodal distribution of affected patients, with MLL rearrangements most commonly found in ALL in infants less than 12 months of age and in a much broader age range of older children or adults, with AML slightly more common than ALL in this age range (94). Finally, there is a rare entity known as “therapy-related leukemia,” which typically occurs after exposure to topoisomerase II inhibitors (e.g., etoposide, doxorubicin) (95, 96).
In the case of infant leukemias, the incidence of MLL rearrangements is 70–80% (29, 97). Therapy-related leukemias secondary to the aforementioned chemotherapeutic agents also harbor MLL translocations in at least 70% of cases (98, 99). Of all patients treated with topoisomerase II inhibitors, between 2 and 12% go on to develop secondary leukemias (100). The majority of these are AML, although a smaller number of cases of ALL have also been reported (96). The latency period for this group of leukemias, in contrast to leukemias secondary to other types of carcinogens, is extremely short—as early as 6 months postexposure, and generally within 24–48 months of exposure, to topo-II inhibitors (95, 96, 98, 100). The mechanisms behind the development of MLL-r leukemias will be explored in the “Environmental and Genetic Risks” section.
MLL-r as a subgroup of acute leukemias is associated with certain phenotypic features that set it apart from other classes of leukemias. MLL-r acute leukemias, particularly in infants, are more likely to present with hyperleukocytosis and CNS involvement (101–103). In cases of MLL-r B-ALL, the blasts are typically of the pro-B phenotype and lack expression of CD10/common acute lymphoblastic leukemia antigen and frequently show coexpression of myeloid markers (104). This is also true in many cases of MLL-r leukemias in adults (105). In vitro, MLL-r blasts often have resistance to commonly used chemotherapeutic drugs such as prednisone and l-asparaginase, but typically have acute sensitivity to cytarabine (106). It has been reported that the transporter protein that imports Ara-C across the cell membrane, the human equilibrative nucleoside transporter 1, was expressed at 2.5-fold higher levels in a cohort of leukemia cells with MLL rearrangements than in MLL wild-type leukemias (107). It is possible that enhanced transport of Ara-C across cell membranes leads to preferential accumulation of the drug in MLL-r cells, which contributes to their specific sensitivity.
Common MLL Fusion Partners and Lineage Plasticity
The majority of MLL-r leukemias involve fusions of MLL with one of six common partner genes: AF4 [t(4,11)], AF9 [t(9,11)], ENL [t(11,19)(q23,p13.3)], AF10 [t(10,11)], ELL [t(11,19)(q23,p13.1)], or AF6 [t(6,11)] (29). The relative frequency of these fusions with respect to leukemia subtype and age are shown in Figure 3 [data adapted from the study by Meyer et al. (29)]. Translocations may or may not be observable on karyotype analysis, but are more reliably identified by fluorescence in situ hybridization (101, 104).
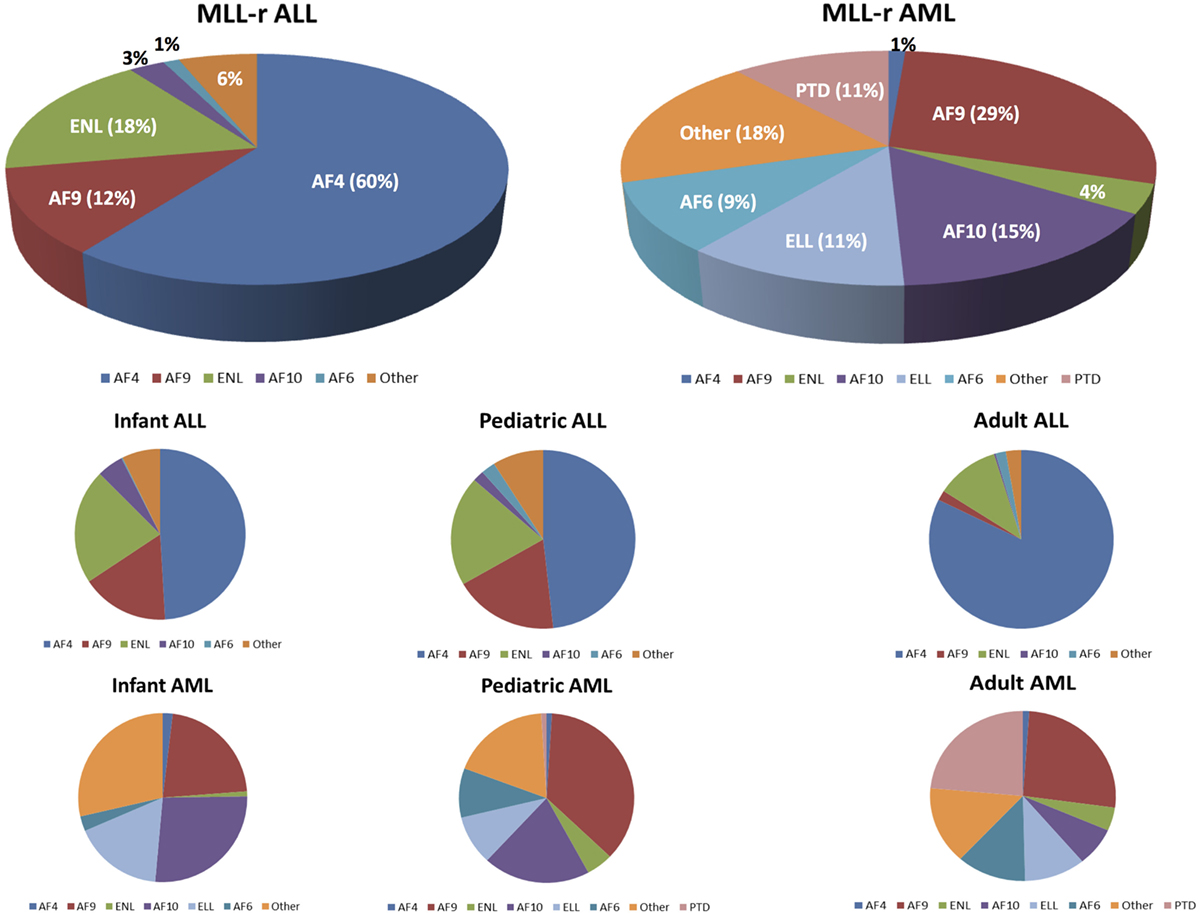
Figure 3. Mixed-lineage leukemia (MLL)-rearranged leukemias involve fusions of 11q23 with 1 of more than 80 different partner genes. Six or seven fusion partners are responsible for the majority of cases. The pie chart illustrates the relative frequencies of the different fusion partners in acute lymphoblastic leukemia (ALL) and acute myeloid leukemias (AML), respectively. Numbers are adapted from the published data by Meyer et al. (29). The bottom half of the figure shows the breakdown of the relative frequencies of MLL fusion partners based on the leukemia type (ALL vs AML) and age group.
Clinical evidence suggests that the fusion partner of MLL1 is a major determinant of the ultimate leukemia phenotype. In patients, MLL-AF4 is predominantly associated with lymphoid malignancies, whereas MLL-AF9 more often results in myeloid malignancies (29). At the same time, particularly, the lymphoid MLL-r leukemias retain a substantial amount of lineage infidelity and lineage plasticity. This is evident in the frequent co-expression of myeloid markers and is the phenomenon familiar to every clinician of MLL-r B-ALL patients who relapse with apparent AML that is cytogenetically related or even identical to the initial lymphoid disease. This phenomenon is likely to increase, as therapies directed against B-lymphoid cell surface markers enter expanded clinical use (antibodies, antibody–drug conjugates, bispecific antibodies such as blinatumomab, and CAR-T). Relapse with leukemia that has adopted a myeloid fate was recently reported for two of seven patients treated with a CD19 directed CAR-T (108) and in an infant with t(4,11) ALL treated with blinatumomab (109). This plasticity is also reflected the recurrent finding of MLL rearrangements in leukemias of ambiguous lineage (MPAL) (93, 110). Experimentally, Wei et al. demonstrated that the microenvironment can play a role in lineage determination. On transduction of human HSCs with a retroviral MLL-AF9 construct, transformed cells propagated in culture with cytokines that promote myeloid differentiation invariably expressed myeloid surface markers (111). Despite the association of MLL-AF9 with myeloid features, transformed cells exposed to cytokines that promote lymphoid differentiation expressed both B cell and myeloid markers. Importantly, leukemia cells of different phenotype from lymphoid or myeloid culture were found to be clonally related, suggesting that they arose from a single leukemia stem cell. Therefore, although the fusion partner affects the leukemia phenotype, environmental cues and selective pressure can also contribute.
In addition to translocations, in-frame PTD of exons 5-12 or a portion thereof can be seen in acute leukemias (112, 113). This type of MLL mutation was originally described in adult de novo AML patients with normal karyotype and has since been demonstrated in both childhood and adult ALL and AML as well as in therapy-related leukemia (112), with an overall incidence of 5–10% (113). MLL-PTD has also been found in a number of leukemias with extra copies of chromosome 11 (114). The presence of this abnormality is associated with early relapse of disease following initial remission (113, 114).
Environmental and Genetic Risks
When translocated, disruption of the MLL gene typically occurs within the breakpoint cluster region (BCR), which spans an 8.3-kb region from exon 8 to exon 14, inclusive (115, 116). A number of sites within this portion of MLL are vulnerable to damage. Among them are the scaffold attachment regions (SARs), which are areas of contact between DNA and non-histone proteins of the chromatin scaffold. Two such SARs have been identified within the MLL coding region—one 5’ to the BCR and a stronger one within the 3’ part of the BCR (117). Cleavage sites of topoisomerase II are also found scattered throughout the MLL BCR, with a higher density in the SAR that overlaps the 3’ region of the BCR. Topoisomerase II is an enzyme that is essential for the relaxation of supercoiled DNA during chromatin remodeling processes (100, 116). Drugs that inhibit the enzyme, such as epipodophyllotoxins and certain alkylating agents, typically do so by forming a stable ternary complex with the enzyme and DNA. The resulting double-strand breaks are most likely to be repaired by non-homologous end joining.
Topoisomerase II inhibitors such as etoposide are known to be associated with development of MLLs in therapy-related cases, and the breakpoints within the MLL gene are frequently adjacent to known cleavage sites of the enzyme (116, 118). Potential MLL cleavage by unknown apoptosis-activated proteases that seem to act independently of topoisomerase II has also been reported in response to stimuli such as ionizing radiation (119). Interestingly, the majority of MLL breakpoints identified in infant leukemias lie toward the 3’ end of the BCR, similar to those seen in leukemias secondary to treatment with topoisomerase II inhibitors (115–117). This finding suggests a possible common mechanism for these two groups of leukemias. Along those lines, investigations into prenatal exposures of infants with MLL-r leukemia have suggested that bioflavonoids found in foods and herbal remedies such as dipyrone (“Mexican aspirin”); senna in herbal teas; quercetin, a bioflavonoid found in onions, red wine, and other foods; and genistein found in soybean products could act as inhibitors of topoisomerase II (100, 120, 121) and promote rearrangement of the MLL locus in a variety of cells, including CD34+ hematopoietic progenitor cells (120, 122). Therefore, it seems possible that, at least in some instances, in utero exposure to environmentally occurring topoisomerase II poisons could contribute to the development of MLL-rearrangements. However, most of these agents are very common, and infant leukemia is a very rare disease. This discrepancy has not well resolved and suggests additional stochastic or genetic mechanisms.
It is also important to note that, in very young patients, there is substantial evidence that the gene rearrangement usually, if not always, occurs in utero. Polymerase chain reaction (PCR) testing of neonatal blood spots has demonstrated the presence of translocations involving the MLL gene even in babies whose disease was diagnosed months later (123). Twin studies offer further support for the prenatal origin of these leukemias. The concordance rate for infant leukemia in identical twins is predicted to be close to 100%, and siblings typically have identical MLL breakpoints (123, 124). These observations suggest a transplacental transfer of leukemia cells from one twin to the other. Despite a rate of twin–twin transfusions of 8% in dichorionic twins, the rate of transplacental seeding of leukemia is much lower in this situation. Both immune-mediated and genetic mechanisms may be responsible for this discrepancy. In fact, there is emerging evidence that genetic risk factors contribute to MLL-r leukemogenesis. In a remarkable GWAS study, a rare polymorphism in MLL3 was present in 100% of infants with MLL-r leukemia (125). The mechanistic implications of this variant are currently being explored.
Treatments and Outcomes for MLL-r Leukemias
Principles and Outcomes of Multiagent Chemotherapy
Historically, the 5-year event-free survival (EFS) for infant ALL has ranged from 20 to 40% for those with MLL rearrangements, vs 60% or higher for those with wild-type MLL (102, 104). The most recent studies for whom published long-term survival data exist have indicated only modest improvement in these numbers [4-year EFS of 40–50% and overall survival (OS) of 50–55%] (102, 126, 127). Most patients (~80–90%) will go into remission initially, but relapse rates of 50–60% are reported, the most common site of relapse being the bone marrow (128). Intensification of chemotherapy may reduce the risks of relapse, but comes at the cost of significant therapy-related morbidity and mortality, mostly of infectious etiology (104). In contrast, the cases of MLL-r AML in infants do not generally have worse outcomes than their non-MLL-r AML counterparts (129). Pediatric patients greater than 1 year of age with MLL-r ALL are better than infants, although not as well as their non-MLL-r counterparts. Most recent data estimate a 5-year EFS of ~60% (129) compared to ~92% in pediatric ALL overall (130). In a European study of 85 adult ALL patients with t(4,11) rearrangements (105), 5-year EFS and OS were 34 and 35%, respectively, which is slightly diminished compared to ~40–45% long-term survival in adult ALL overall (131). Again, most MLL-r patients achieve an initial remission (>90%), but many patients ultimately relapse.
The relationship between MLL rearrangements and outcome in AML is less straightforward than in ALL. The most common MLL fusion in AML, MLL-AF9, has been reported to be associated with an intermediate to good prognosis (132, 133). In contrast when analyzing a large cohort of pediatric de novo AML with a variety of different MLL rearrangements, 5-year EFS and OS were poorer [44% EFS and 56% OS (133)] when compared to pediatric AML in general [55% EFS and 70% OS (134)], with substantial differences depending on the fusion partner.
Clinical features that have been shown to be predictive of outcome in infant MLL-r ALL include age at diagnosis, total white blood cell count at diagnosis, presence or absence of CD10 on blast cells, and initial response to steroid therapy (97, 104, 126, 128, 129). Age cutoff predictive of poorest outcomes varies based on the study (<90 days vs <6 months). WBC count >300K, lack of CD10 expression, and poor response to prednisone (defined as >1,000 blast cells per microliter in the peripheral blood) all confer particularly dismal outcomes as well (97, 104, 126, 128, 129). In adult MLL-r ALL, older age (>25 years) was the only independent factor associated with decreased survival [<35 vs 71% (105)].
Historically, it was thought that t(4,11) fusions in ALL were associated with poorer survival compared to other translocations (128). However, despite the association of t(4,11) and t(11,19) fusions with younger age groups, more recent trials have failed to find any significant association between relapse or survival in MLL-r ALL and any particular fusion partner (97, 104, 126, 129). This may be related to the fact that despite a myriad of fusion partners being reported, only a few dominate the clinical experience. Furthermore, MLL-r leukemias are typically not treated on a unified protocol, but managed largely based on phenotype (AML vs ALL) and age (infant leukemia). Most current clinical risk stratifications do not take the fusion partner into account. However, several studies investigating the relationship between fusion partner and outcome have suggested that there is a correlation. A meta-analysis of the association between fusion partner and outcome in 756 children with MLL-r AML from 11 study groups operating in 15 countries suggested massively divergent OS: while 24 children with the t(1,11)(q21,q23) translocation (MLL-AF1q) had an OS of 100% (event free survival, EFS 92%), EFS and OS were 11 and 22%, respectively, for patients with the t(6,11)(q27,q23) translocation (MLL-AF6) (133). This study did not confirm the possible “good risk” feature of the common MLL-AF9 translocation that was previously reported (132). The dismal outcome for MLL-AF6 mutant disease had previously been reported in adults (135). Also, children older than 1 year with MLL-AF4 (and interestingly, MLL-AF9) mutant B-ALL were reported to have a worse outcome than children with other MLL translocation partners (129). In an even more fascinating twist, MLL-AF9 might be predictive of a good OS when it occurs in FAB M5-AML as opposed to other FAB subgroup AML or ALL PIMD (133). Whether this reflects statistical outliers in an increasingly smaller and more finely sliced “pie,” genetic/pharmacogenomics differences, or underlying biology (possibly reflecting cell of origin) is unclear.
One interesting correlation is that both infant ALL and therapy-related leukemias, which have overall the worst outcomes of MLL-r leukemias, are associated with breakpoints in intron 11 rather than intron 9 or 10 (115). Emerenciano et al. recently demonstrated that the presence of MLL breakpoint in intron 11 was also an independent predictor of poor survival in a cohort of 30 MLL-r pediatric leukemia patients (136). Fragmentation of the MLL gene at intron 11 is predicted to generate an MLL C-terminal truncated protein whose PHD fingers are misfolded, eliminating the ability to associate with its repressive complex (137). The authors of this study theorize that, in these cases, unbalanced activation functions of the resultant MLL fusion protein lead to more aggressive leukemia phenotypes. Whether this can be mechanistically proven in future experiments remains to be seen.
The Role of Hematopoietic Stem Cell Transplant (HSCT) in the Treatment of MLL-r Leukemia
The role of HSCT in the treatment of MLL-r leukemias continues to be a matter of intense debate, with several studies and meta-analyses suggesting that HSCT does not improve survival in MLL-r leukemias at any age group or lineage, with the exception of therapy-associated AML (126, 129, 133, 138–141). The combined analysis of the North American CCG 1953 and POG 9407 infant ALL trials concluded that HSCT failed to show any benefit (142). Initial chemotherapy was identical on the two protocols. In the later phases, chemotherapy was very similar, with the main difference being methotrexate dosing. Patients on the CCG also received maintenance therapy, while patients on the POG trial did not. On the CCG trial, HSCT in CR1 was the preferred mode of treatment if a suitable donor could be identified, whereas on the POG trial, this was left to the judgment of the investigator. The recommended conditioning consisted of Ara-C/Cy/TBI, although only about half of the patients received this conditioning. Transplant-related mortality, particularly in children receiving TBI, was high. This study included 132 infants with MLL rearrangement, although after adjustment for time to transplant, only 100 children were evaluable. Fifty-three underwent HSCT, 47 did not. Five-year EFS for children who were alive at the time of transplant was similar between the HSCT and chemotherapy groups (48.85 vs 48.7%), prompting the authors to conclude that HSCT did not improve survival in MLL-r infant ALL. In addition, there were no differences in subgroups based on WBC, age, or CD10 expression. However, the comparatively smaller number of patients with high-risk features and variability in transplant regimen made the subgroup analysis for this patient population difficult.
No benefit for HSCT for infants with MLL-r leukemia was also shown by two retrospective analyses (129, 143) and in a report of children treated in Europe (144). In contrast, the analysis of a larger cohort of 297 infants with MLL rearrangement treated on Interfant99 identified a group of patients less than 6 months of age with either a WBC of >300,000, prednisone poor response, or high end consolidation MRD that had an extremely poor survival with chemotherapy only (97, 145, 146). On Interfant99, high-risk patients did benefit from HSCT: the survival for children <6 months with either a WBC > 300,00 or PPR who were alive at the time of HSCT was only 22.2% when treated with chemotherapy and 59% on the HSCT arm (97). The number of patients who received HSCT was small, but given the dismal outcome of this subgroup, a more aggressive approach seems justified. The outcome of a similar group of infants on other trials such as CCG 1953 and POG 9407 is not known, since the number of patients was smaller, prednisone response and MRD were not assessed or reported, and WBC criteria for subgroup analysis were different from the Interfant99 study (97, 104).
In summary, although numbers are small, HSCT is likely beneficial for a defined subgroup of high-risk infants, particularly if conditioning regimen and donor choice allow for a low transplant-related mortality. There are no data supporting HSCT in CR1 in older children with MLL-r ALL or de novo AML. As discussed earlier, the study by Balgobind et al. suggests that defined fusion partners may be associated with a particularly poor prognosis (133); however, currently, this is not used for treatment stratification in either ALL or AML. In contrast to de novo leukemia, outcomes for therapy-related AML are substantially worse, and HSCT in first CR is standard of care (141). In addition to tAML, treatment-related MLL-r ALL is occasionally seen. Although outcomes for tAML are inferior to de novo AML, no such data exist for MLL-r tALL vs de novo ALL, and chemotherapy only may be the treatment of choice, particularly for patients who show a good response to therapy (as measured by MRD).
Role of FLT-3 in MLL-r Leukemias
One of the most significantly upregulated genes in the transcriptional profile of MLL-r leukemias is Fms-like receptor tyrosine kinase-3 (FLT-3) (75). This gene encodes a class III receptor tyrosine kinase, which is closely related to KIT, FMS, and platelet-derived growth factor receptor (147). Under physiologic conditions, binding of the FLT-3 ligand leads to dimerization and phosphorylation of the receptor, which activates downstream signaling pathways such as PI3K/Akt, Ras/MAPK, and Stat5 (147). Activating mutations in FLT-3 have been described in a variety of hematologic malignancies, but have primarily been characterized in pediatric and adult AML (147, 148). The two types of activating mutations commonly seen in these contexts are internal tandem duplications in the juxtamembrane domain of FLT-3 [so-called FLT-3 internal tandem duplications (ITD) mutations] and point mutations within the tyrosine kinase domain (TKD) that confer constitutive activity to the enzyme (147). The presence of a FLT-3 ITD mutation confers an extremely poor prognosis (148).
FLT-3 gene upregulation in MLL-r leukemias correlates to overexpression of the FLT-3 protein in these ALL samples compared to non-MLL-r leukemias, which has been demonstrated by several groups (149–152). On average, MLL-r infant ALL (which is the most thoroughly studied subtype of MLL-r leukemias) expresses 37-fold higher FLT-3 protein compared to normal bone marrow and 2- and 16-fold higher expression of FLT-3 compared to non-MLL-r ALL in children less than 1 year of age and older children, respectively (150, 152). By contrast, the data have been less consistent with respect to activating mutations of FLT-3, with most of the recent studies suggesting that they are rare (149–151, 153). FLT-3 ITD have not been demonstrated in recent cohorts of MLL-r ALL patients, and FLT-3 TKD mutations have an approximate incidence of only 3–18% in MLL-r ALL (149–151). Most recently, Andersson et al. investigated a cohort of 85 infant and pediatric patients with ALL, of which 67 had MLL rearrangements. Of these, only four patients had FLT-3 mutations, two of which were TKD mutations and two of which were present only in a minor clone (154). This finding is also consistent with the general finding by this group that MLL-r leukemias, in particular those arising in infants, have one of the lowest frequencies of somatic non-silent mutations of any other type of cancer (mean 1.3 per major clone). Nevertheless, in vitro studies have demonstrated that high levels of FLT-3 expression, even in the absence of these activating mutations, are associated with phosphorylation and activation of the protein (149, 150).
Mouse models of MLL-r leukemias have suggested cooperation between the MLL fusion oncoprotein and FLT-3 in the progression to the leukemia phenotype (155). In addition, a retrospective study showed correlation of high levels of FLT-3 expression with poor outcomes in 32 MLLr infants treated on Interfant99 (36 vs 71% 1-year EFS in high vs low FLT-3 expressing leukemias) (156). A later study by Chillon et al. confirmed these findings—of 17 patients with MLL-AF4 B-ALL, none of those with high FLT-3 expression were alive at 1 year, compared to 71% of patients with low FLT-3 expression (152). FLT-3 expression levels were not predictive of outcomes in patients with non-MLL-r ALL. These findings suggested that targeting of FLT-3 in MLL-r patients might be a beneficial therapeutic approach.
In vitro cytotoxicity experiments with MLL-r ALL patient samples demonstrated in vitro sensitivity to the FLT-3 kinase inhibitors, with response correlating with the amount of FLT-3 overexpression [PKC412 (150) and CEP-701/lestaurtinib (151)). Furthermore, synergy studies between CEP-701 and standard chemotherapeutic agents (e.g., etoposide, daunomycin) suggested that timing is critical—administration of CEP-701 after cytotoxic agents yielded synergistic cytotoxicity, whereas administration of CEP-701 before cytotoxic chemotherapy was antagonistic (157). These studies laid the groundwork for the design of clinical trials to test the efficacy of FLT-3 inhibitors.
Several clinical trials involving FLT-3 inhibitors have been conducted in both adult and pediatric leukemias. Of primary relevance to MLL-r leukemias is the Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) study, whose results have just been published (158). This study was a phase 1 trial evaluating the safety of quizartinib (AC220) in combination with high-intensity chemotherapy for relapsed childhood leukemia. Quizartinib is a second-generation kinase inhibitor designed to be potently active against FLT-3 and is more selective than first-generation inhibitors such as lestaurtinib (159, 160). Twenty-two patients were enrolled, of which 18 had relapsed AML (9 with FLT-3 mutations) and 4 had relapsed MLL-r ALL (3 infants and 1 teenager). Patients received combination chemotherapy with cytarabine and etoposide (days 1–5) followed by quizartinib (days 7–28) for 1–2 cycles. In all cases, target-specific activity of quizartinib was demonstrated with near-maximal (>95%) suppression of FLT-3 phosphorylation in plasma inhibitory assays (PIAs). Dose-limiting toxicities attributable to the targeted agent involved primarily GI toxicities such as elevated lipase or transaminases or nausea/vomiting/diarrhea. Of the 17 evaluable patients for response, better response correlated with the presence of FLT-3 ITD mutations in the AML patients. Three of the four MLL-r ALL patients could be evaluated for disease response—one had stable disease and two had progressive disease. The study was not powered to make conclusions about statistically significant impacts on OS.
Another trial specific to infant leukemia, the Children’s Oncology Group (COG) trial AALL0631, has been recently closed, and data analysis is ongoing. This trial was a randomized, phase III trial of the FLT-3 inhibitor lestaurtinib in combination with intensive cytotoxic chemotherapy for newly diagnosed infants with MLL-r ALL. Although the final results of the trial have not yet been published, the results from the TACL trial raise concern that FLT3 inhibition may not be the breakthrough that is so desperately needed for these patients. However, final results on the outcomes and the depth of FLT-3 inhibition achieved in AALL0631 regimen is not (yet) known, thus failure to achieve sufficient target inhibition remains a possible explanation for the lack of efficacy. It is also critical to keep in mind that in both AALL0631 and the TACL study, the assays used to determine the degree of FLT3 inhibition measures the inhibitory effect of patient serum on BAF3 cells that are Flt3 dependent (PIA). The threshold at which this very sensitive indicator cell line responds may be different from responses in patients’ leukemia cells. It may be necessary to determine on-target activity in actual patient cells and correlate that with response to get a better sense for whether FLT3 inhibition is of therapeutic value in MLL-r leukemia.
Proteasome Inhibitors
Proteasome inhibitors are increasingly being integrated into therapeutic regimens for a variety of malignancies. The rationale behind their use has traditionally been that cancer cells, due to increased cell turnover, are more dependent on the proteasome machinery for protein recycling than are normal cells. These drugs have yielded mixed results when used alone or in combination with cytotoxic chemotherapy for a variety of malignancies and are associated with significant toxicities, particularly neurotoxicity [reviewed in Ref. (161)].
Accumulating data now suggest that proteasome inhibitors may be promising agents to supplement the treatment of MLL-r leukemias. Liu et al. noted that the expression levels of MLL fusion proteins was not excessive in leukemic cells and hypothesized that tight regulation of fusion protein expression might be achieved through the proteasome machinery. Indeed, they demonstrated that proteasome inhibitor treatment increased the protein levels of both wild-type MLL and, to a greater extent, MLL fusion proteins (162). Stabilization of MLL fusions activated transcription of CDKN1B, which encodes p27, via PAX5. Of note, PAX5 is selectively expressed in pro-B cells (163). Consistent with this model, proteasome inhibitor treatment was associated with a dose-dependent decrease in cell viability in lymphoid, but not myeloid, MLL-r leukemia cell lines. A cohort of five adult patients with MLL-r leukemias were then treated compassionately with single agent bortezomib. Three patients (two with pro-B phenotype and one with biphenotypic leukemia) had transient hematologic responses to the drug, one for over a year. Two patients with myeloid phenotype had no response to proteasome inhibitor therapy.
Bortezomib was also identified through high-throughput drug screens as an active agent against models of infant MLL-r leukemias (164, 165). Mechanistically, Koss et al. found that bortezomib treatment led to decreased histone 2B ubiquitination (H2Bub). H2Bub is required for the methylation of H3K79 mono- to di- and trimethylation (166), and knockdown of the H2B ubiquitin ligase RNF20 led to decrease in H3K79me2 mark at MLL target gene sites and in vitro and in vivo compromise of leukemia cell viability (167). Both the study by Liu et al. (162) and Wang et al. (167) also explored the effect of bortezomib in NF-kB signaling. Intact NF-kB has been reported to be required for MLL-fusion-mediated leukemogenesis in a murine model (168, 169), and bortezomib has been implicated in negatively regulating NF-kB via the accumulation of IkBa in the absence of proteasomal degradation (170). However, both Liu et al. (162) and Koss et al. (164) found no evidence of NF-kB modulation, suggesting that this pathway is not mechanistically involved in therapeutic effect. Taken together, proteasome inhibitors may be useful as adjunctive therapy for MLL-r leukemias, and bortezomib in combination with standard chemotherapy and HDAC inhibition is currently being evaluated in clinical trials for MLL-r ALL (NCT02419755, see next section).
HDAC Inhibitors
Similar to proteasome inhibitors, HDAC inhibitors (HDACi) have been reported by multiple groups to be active in MLL-r leukemias, with effects being attributed to diverging mechanisms (165, 171–174). HDACs are a large family of proteins named for the ability of the founding member, HDAC1, to deacetylate histones. However, many members of the family are cytosolic proteins named HDAC due to structural homology, but without possessing histone deacetylase function. Several HDACs have been reported to be overexpressed in pediatric ALL; however, there is no agreement as to which members are specifically deregulated (175–177).
The earliest functional relevance of HDACs was suggested by a study investigating the activity of valproic acid in MLL-r leukemia. Valproic acid induced growth inhibition and cell-cycle arrest in MLL-r leukemia cell lines and primary samples. The authors proposed upregulation of p21 as a mechanism (171). Stumpel et al. showed that romidepsin (FK288) and vorinostat had activity in 2 t(4,11) B-ALL cell lines and 15 infant B-ALL patient samples (172). Good, although not as profound, sensitivity to HDAC inhibition was also found for non-MLL rearranged B-ALL. They also demonstrated decrease in expression of MLL-AF4 at both the transcript and protein levels, raising the possibility that effect of these drugs is primarily due to downregulation of the MLL fusion itself.
In contrast, Stubbs et al. found that several HDACis were broadly active against a variety of different cytogenetic subtypes of B-ALL, including (but not exclusively) MLL-r leukemias (174). Genetic knockdown as well as class-specific inhibitors suggested that HDAC1 and 2 are the critical HDACs in B-ALL, with knockdown or inhibition resulting in direct cytotoxicity and DNA damage. DNA damage as a result of HDAC inhibition has been suggested to underlie the frequently observed synergy with chemotherapy. Stubbs et al. proposed that the particular sensitivity of B-ALL to HDAC inhibition could also relate to the role of HDAC1 and HDAC2 in early B-cell development (178). On the other hand, two studies suggested that inhibition of HDAC3 is critically responsible for the activity of HDACis in B-ALL (179, 180).
Bhatla et al. published sensitivity of the t(4,11) B-ALL cell line RS4,11 to the HDACi vorinostat, and vorinostat was shown to be synergistic with standard chemotherapeutic agents such as prednisolone and cytarabine (173). The authors of this study also specifically investigated relapsed B-ALL (irrespective of karyotype) and proposed the reversal of the “relapse gene signature” as a mechanism. Finally, an interesting mechanism was proposed by Ahmad et al., who proposed that HDACis reactivate wild-type MLL to counteract the transcriptional functions of MLL-AF4 or other fusions (181). However, the role of wild-type MLL in MLL-r leukemia is controversial as discussed above, and the experiments by Ahmad et al. were performed in HeLa cells, with unclear implications for the context of leukemia.
From a clinical standpoint, a 2011 case study reported a sustained complete cytogenetic response to single-agent panobinostat in an elderly man with therapy-related MLL-r leukemia (182). St. Jude Children’s Research Hospital has an ongoing phase II clinical trial combining a proteasome inhibitor (bortezomib) and an HDACi (vorinostat) in combination with cytotoxic chemotherapy for pediatric patients with relapsed or refractory MLL-r leukemias (NCT02419755). Chemotherapy backbone varies depending on the leukemia phenotype (ALL vs AML), and all drugs are intended as a bridge to transplant. A report of six “pilot” patients with relapsed/refractory MLL-r leukemia was presented at the 2014 American Society of Hematology Annual Meeting. The overall response rate of this cohort to chemotherapy in combination with bortezomib and vorinostat was 83%: four patients had complete response, one patient had partial response, and one patient had stable disease (164). Whether this regimen can achieve durable responses without excess toxicity in these patients remains an ongoing question, but initial results are certainly promising.
Hypomethylating Agents
Two separate groups have investigated the methylation status of MLL-r leukemias compared to MLL-wild-type leukemias and normal controls (183, 184). Global promoter hypermethylation was seen in the MLL-r leukemias relative to both non-MLL-r leukemias and normal samples, leading to downregulation or silencing of a subset of tumor suppressor genes. Retrospective analysis demonstrated a statistically significant correlation between degree of methylation and risk of relapse (183). Furthermore, hypomethylating agents zebularine and decitabine showed preferential cytotoxicity to MLL-r cells compared to other leukemic cells. Both decitabine and another hypomethylating agent, 5-azacitidine, are FDA approved for treatment of myelodysplastic syndromes and AML in adult patients, particularly those with co-morbidities limiting other therapeutic options [reviewed in Ref. (185)]. 5-azacitidine, but not decitabine, usage was associated with increased OS in these patients. These data form the basis for a clinical trial run by the National Cancer Institute (NCT02828358), and for a new COG therapy trial (AALL15P1), both for infant MLL-r ALL, which test the tolerability of 5-azacitidine in combination with standard cytotoxic chemotherapy.
Similarly to decitabine and 5-azacitidine, other nucleoside analogs may also target the methylation status of MLL-r leukemias. Clofarabine, an powerful cytotoxic adenosine analog, is thought to also block DNA methylation through depletion of S-adenosyl methionine, which donates methyl groups to DNA methyltransferase enzymes (186), as has been demonstrated for the related molecule cladribine (187). In a recent study by Stumpel et al., low doses of clofarabine were cytotoxic to MLL-r leukemias in vitro, and clofarabine was synergistically active with cytarabine against these cells (188). A variety of clinical trials, none specific to MLL-r leukemias, currently incorporate clofarabine into study therapy.
Immunotherapy
One of the most exciting new therapeutic approaches in B-ALL has been the development of immunotherapies, particularly the use of bispecific antibodies (blinatumomab) and engineered T-cells (CAR-T) in B-ALL. Most clinical trials using CAR-T cells in B-ALL to date have allowed children >1 year and adults with MLL rearrangements, but have excluded infants, mostly due to difficulties around efficient collection and expansion of autologous T-cells. Infants were included in the early clinical trials with blinatumomab (189), and despite theoretical concerns about the immaturity of T-cell responses very early in life, some encouraging responses were seen [Lia Gore, personal communication (109)]. However, MLL-r B-ALL may have “built-in” mechanisms to evade immune recognition and/or destruction through their lineage plasticity. As mentioned earlier, relapse with leukemia that has adopted a myeloid fate has been observed in two out of seven patients treated with a CD19-directed CAR-T (108) and in an infant with t(4;11) ALL treated with blinatumomab (109). It is not yet clear whether this will remain a rare occurrence or emerge into a common mechanism of relapse and resistance.
MLL Specific Pathways and Targeted Inhibitors in Early Clinical Trials
Role of RAS Pathway Mutations in MLL-r Leukemias
Mutations in RAS pathway members have been frequently described in MLL-r leukemias and are perhaps more prevalent than mutations in FLT-3. Prelle et al. evaluated the incidence of secondary mutations in a cohort of 144 pediatric and adult patients with MLL-r leukemias, of which 100 individuals had t(4;11) mutations and the remaining 44 patients had a variety of other fusions. NRAS or KRAS mutations were present in 16 patients (11.1%) (153). In an independent cohort of 109 infant ALL patients screened for NRAS, KRAS, or BRAF, 15 patients (13.8%) had mutations in either NRAS or KRAS (190). This group also reported a significant decrease in OS was specifically seen in patients with RAS mutations in the t(4;11) cohort, but not in the overall study group. Additional studies have cited frequency of RAS pathway mutations in MLL-r leukemia patients ranging from 22 to 45% (191–194).
Recently genome-wide analysis of infant MLL-r and MLL-wild-type ALLs, in addition to pediatric MLL-r ALL in older age groups, was performed as a part of the Pediatric Cancer Genome Project (154). This study confirmed that although MLL-r leukemias in general carry a paucity of additional mutations, the most commonly seen mutations involve the RAS pathway. However, the variant allele frequencies (VAF) for these mutations in the majority of the infant cases were <30%, indicating that the individual mutations were present in minor clones within the leukemia population. This was also noted for the NRAS and KRAS mutations described in the study by Driessen et al. (190). Furthermore, of five RAS pathway-mutated patients with matched diagnosis and relapse samples, the mutations were lost in two cases and the VAF decreased in one case, suggesting a gradual depletion of the RAS-mutated subclone (154). Again, this mirrors previous studies by Prelle et al. and Emerenciano et al., both of whom documented loss of RAS mutations in two of three and in five of 18 relapse samples evaluated, respectively (153, 192). Furthermore, Emerenciano et al. documented the presence of RAS mutations in DNA samples from newborn blood spots for two patients in their cohort, one with higher allele frequency than at diagnosis of leukemia. This finding suggests the possibility that RAS pathway mutations provide a proliferative advantage during onset of leukemogenesis, but are not necessary for leukemia maintenance in the context of MLL rearrangements.
With the advent of multiple inhibitors of the RAS pathway that are either FDA approved or in clinical trials, the question whether RAS pathway activation plays a role in MLL-r leukemia receives new urgency. It is also possible, similar to FLT3, that activation of the pathway can occur in the absence of mutations. Kampen et al., using peptide arrays of normal bone marrow and leukemia cells, demonstrated increased phosphorylation of MAPK pathway proteins in MLL-r AML samples compared to either normal bone marrow or non-MLL-r AML’s (195). MEK inhibitors have shown selective activity against MLL-r leukemia cell lines and primary samples in vitro in several studies, although in almost every case those cells with RAS mutations were more sensitive to these drugs than were cells without RAS mutations (193–195). The possible exception to this rule lies in leukemia cells harboring t(6;11), leading to an MLL-AF6 fusion. Manara et al. have shown that the normally cytoplasmic protein AF6 is instead localized to the nucleus in the presence of MLL-AF6, which is associated with increased RAS pathway activity. The AF6 protein has RAS-association domains, and genetic silencing of MLL-AF6 leads to decreased RAS activity and decreased phosphorylation of ERK (196). Furthermore, chemical inhibition of RAS signaling by either PD98059 (MEK inhibitor) or tipifarnib (farnesyltransferase inhibitor) was selectively toxic to t(6;11) leukemia cells. Therefore, although RAS inhibition may not be of benefit in the majority of MLL-r leukemias, where mutations are subclonal and not likely to impact the survival of the leukemia, it may be of benefit in the context of leukemias with MLL-AF6 fusions, which are notorious for their particularly poor outcomes.
Dot1L Inhibitors
The histone 3 lysine 79 methyltransferase Dot1L has been shown to be necessary for MLL fusion-mediated transformation in a variety of experimental models (45, 70, 197–200), and increased levels of H3K79 dimethylation have been demonstrated at MLL fusion target gene loci [(70, 198, 201); see also Figure 2]. DOT1L contributes to the maintenance of the MLL leukemic gene expression program at least in part by antagonizing Sirtuin-1-mediated repressive epigenetic modifications to H3K9 (202).
A small molecule inhibitor of Dot1L (EPZ-5676 or pinometostat) has been developed by Epizyme, Inc. and is being studied in early clinical trials in both adult and pediatric patients with MLL-r leukemias (NCT01684150 and NCT02141828). Preclinical studies demonstrated target specificity and downregulation of MLL target genes upon treatment of MLL-r cell lines with EPZ-5676 (203). EPZ-5676 also demonstrated synergy with other chemotherapeutic agents known to target MLL-r leukemias, regardless of the order of administration (204). Continuous intravenous infusion of the compound caused tumor regression and prolonged survival in mice and rat xenograft models of MLL-r leukemias (203). Unfortunately, the lack of oral bioavailability and short half-life of the drug currently mandate the continuous IV infusion, resulting in efforts to develop alternative Dot1L inhibitors, which maintain specificity and efficacy but are easier to administer (205). Data from the phase I/II clinical trials are forthcoming, but it will be crucial to correlate mechanistic effect (i.e., reduction of H3K79 methylation) with outcomes in these patients. Preliminary data published in abstract form suggest sustained single-agent efficacy in two patients, but also a substantial number of patients with only transient or no response, and who did not achieve profound depletion of H3K79 methylation at MLL-fusion target loci at the dose used (206, 207).
Bromodomain Inhibitors
The bromodomain and extra terminal (BET) family of proteins, which includes BRD2, BRD3, and BRD4, are a family of chromatin adaptor proteins that recognize and bind to acetyl-lysine residues. A global proteomic screen identified interaction of these proteins with components of the SEC, many of whom are MLL fusion partners (208, 209). Simultaneously, an shRNA screen identified the BET protein BRD4 as a therapeutic target in MLL-AF9,NrasG12D murine AML model (210). Inhibitors of BRD4 had efficacy in MLL-AF9,NrasG12D model and on a variety of leukemia cell lines, with MLL-r leukemias preferentially affected (209, 210). Efficacy of BRD4 inhibition was confirmed in primary MLL-r patient samples in vitro (165, 209).
In addition to the bromodomain of BET family proteins, the bromodomain of CBP/p300 bromodomain has emerged as a potential therapeutic target for leukemia, including MLL-r leukemia. A small molecule inhibitor of the CBP/p300 bromodomain led to decreased colony formation and promoted differentiation in MLL-CBP and MLL-AF9 leukemia models as well as primary MLL-r patient cells (211). This latter inhibitor was found to have synergistic inhibition of MLL-r cells when combined with the bromodomain inhibitor JQ1 or doxorubicin.
One recent study suggested a sequential recruitment of DOT1L and BRD4 to a subset of genes located adjacent to super enhancers (212). Dimethylation of H3K79 by DOT1L allowed binding of histone acetyltransferases including EP500 and CREBBP to these regions, which leads to acetylation of H4K5 and subsequent binding of BRD4 and the SEC. Accordingly, inhibition of DOT1L led to dramatically decreased binding of BRD4 to chromatin, and the combination of a bromodomain inhibitor and a DOT1L inhibitor was synergistically active against MLL-r leukemias both in vitro and in vivo. In contrast, regulation of distinct programs by DOT1L and BRD4 were reported by Garcia-Cuellar et al. (213). DOT1L-dependent loci were characterized by promoter-centered binding of MLL-ENL, while BRD4-dependent loci exhibited fusion binding beyond the termination site. Despite the discrepancies in proposed molecular mechanisms, the combination of DOT1L and BRD4 inhibition may be promising to explore further.
Lysine-Specific Demethylase-1 (LSD1) Inhibitors
Lysine-specific demethylase-1, also known as KDM1A, has been shown to be important for maintenance of MLL target gene expression (214) and was identified in an RNAi screen as a gene whose repression inhibited growth of MLL-AF9,NrasG12D murine cells (215). It has enzymatic specificity for lysines 4 and 9 on histone 3. Pharmacologic inhibition of LSD1 with tranylcypromine (TCP) was shown to result in a decreased expression of MLL target genes; it also impaired colony-forming potential and leukemic engraftment in immunodeficient mice (214). However, there were significant toxicities to the mice from TCP and related inhibitors, particularly related to thrombocytopenia and anemia. Using newer generation small-molecule inhibitors of LSD1, these results were confirmed in vitro and in vivo without any significant toxicities to mice (216). Furthermore, in vitro synergy was demonstrated when LSD1 inhibitors were combined with the DOT1L inhibitor SYC-522. However, Shi et al. were only able to demonstrate a detrimental effect of LSD1 inhibition in vitro, whereas no disadvantage of LSD1 inhibition could be shown in competitive engraftment experiments in mice (215). Pharmacologic inhibitors of LSD1 are in early clinical trials in adult AML/MDS (GSK GSK2879552 single agent, NCT02177812, Tranylcypromine + ATRA, NCT02717884, NCT02261779, and NCT02273102). It remains to be seen whether LSD1 inhibitors will have efficacy in patients with MLL-r leukemias.
Polycomb Protein Inhibitors
Polycomb repressive complexes 1 and 2 are two protein complexes involved in chromatin modulation and transcriptional repression. Both have been implicated in MLL-r leukemias. PRC1 contains core components BMI1, RING2A, and RING2B and mediates H2Ak119 monoubiquitination. Several groups have investigated the functional requirement of these PRC1 components in MLLr leukemia. Initial reports investigating the role of BMI1 using BMI1 knockdown and/or MLL-AF9 leukemias generated on a Bmi1−/− background suggested that PRC1 canonical function is not required for MLLr leukemogenesis, although some transcriptional and minor functional effects on leukemia initiating cell frequency were observed (66, 217, 218). In contrast, combined knockout of Ring1a/b in a murine model of MLL-AF9-induced leukemia was not tolerated (219). The stark discrepancy in phenotypic consequences between knockout of different PRC1 components have not been well resolved and may relate to differently composed subcomplexes and/or functions outside of canonical PCR1.
In addition, the non-canonical PRC1 member CBX8 has been implicated leukemogenesis particularly mediated by MLL-AF9 and MLL-ENL. As mentioned earlier, AF9 and ENL bind CBX8 (63, 64), and leukemia initiation and maintenance in murine models MLL-AF9 and MLL-ENL AML were dependent on CBX8 (66). CBX function in MLLr leukemia appears to be independent of its role in PRC1, however, and instead involve the recruitment of the histone acetyl transferase Tip60 to fusion target loci (66). This is further supported by the finding that binding of CBX8 to ENL reverses the repressor activity of CBX8 (220). In mouse models, deletion of CBX8 had no detrimental effect on normal hematopoiesis, suggesting CBX8 and/or Tip60 could be interesting target for future development of inhibitors.
PRC2 consists of the canonical components EZH2, EED, and SUZ12. EZH2 is a histone methyltransferase targeting H3K27. Multiple groups have documented decreased proliferation, differentiation, and loss of stem cell potential in MLL fusion leukemias when any component of the complex was genetically knocked down or deleted (215, 221–223). This effect was most prominent with depletion of the universal components of the complex, EED or SUZ12, whereas EZH1 and EZH2 have somewhat redundant functions (215, 221). Impairment of the leukemia phenotype was largely attributable to de-repression of INK4A and ARF, although deregulation of other PRC2 target genes such as GATA2 and EGR1 were also implicated (223). Two inhibitors (DZNep and UNC1999) have shown efficacy in vitro against MLL-r leukemias and have prolonged survival in xenograft models of disease (224, 225). Another small molecule inhibitor, which disrupts the protein–protein interaction between EZH2 and EED, has had similar utility in MLL-AF9 models without any effect on non-transformed cells (222). However, inactivating mutations in PRC2 components has also been observed in AML and MDS patients and at least in MDS correlates with poor prognosis. As an added wrinkle to the story, the observation that AF10, a necessary cofactor for H3K79 di- and trimethylation by DOT1L, binds unmodified H3K27 (72) suggests that inhibition of EZH2 or PRC2 components could also facilitate H3K79 methylation on MLL-fusion target genes, although there is currently no experimental data to document that this is the case. Pharmacologic inhibitors of EZH2 are in clinical trials for diseases where PRC2/EZH2 hyperfunction is clearly linked to malignant transformation (such as lymphomas with activating EZH2 mutations or INI1-negative solid tumors). EZH2 inhibition is not currently investigated in clinical trials for AML or MLL-r leukemias.
Agents that Counteract Antiapoptotic Mechanisms
As previously mentioned, MLL-r leukemias have been reported to be resistant to programmed cell death (86–89). Leukemias with t(4,11) translocations (MLL-AF4) tend to have elevated levels of prosurvival BCL-2 protein, which counteracts the intrinsic mitochondria-mediated apoptotic pathway (89). In vitro cytotoxicity studies of the pan-BCL-2 family inhibitor obatoclax demonstrated efficacy of this agent against a panel of MLL-r infant leukemias as well as MLL-r cell lines (165, 226); obatoclax also synergized with multiple standard chemotherapeutic agents (226). Recent work has suggested that t(4,11) leukemias tend to have highest expression of BCL-2 of multiple classes of acute leukemias and that the MLL-AF4 protein upregulates BCL-2 expression via DOT1L-mediated H3K79 methylation (92). The selective BCL-2 inhibitor, ABT199 (venetoclax), which has shown promise in clinical trials against chronic lymphocytic leukemia and other hematopoietic malignancies (227–229), was effective both in vitro and in xenograft models against MLL-r leukemias in combination with cytotoxic chemotherapies and with a DOT1L inhibitor. Further work in xenograft models confirms not only the enhanced sensitivity of MLL-r leukemias to BCL-2 inhibition compared to other subgroups of ALL but also the enhanced efficacy of combined inhibition of BCL-2 and BCL-XL (230). These data suggest yet another class of targeted agents that may prove useful adjuncts to therapy in MLL-r leukemias.
Cell Cycle Checkpoint Inhibitors
Recent studies have identified cyclin-dependent kinase 6 (CDK6) as a target gene of MLL fusion proteins (231). CDK6 binds to D cyclins and promotes cell cycle progression through phosphorylation and inhibition of target genes such as RB1. MLL-r leukemias seem to be dependent on CDK6, but not on CDK4, for growth and proliferation (231, 232). This dependence on CDK6 was not seen in non-MLL-r leukemias. Furthermore, treatment of either MLL-r cell lines or primary patient AML cells with the CDK6 inhibitor palbociclib (PD0332991) led to both growth inhibition, decreased colony formation, and a differentiated phenotype (232). Third-generation transplant recipient mice given palbociclib-treated MLL-AF9 cells had decreased disease burden and prolonged survival compared to mice given MLL-AF9 control cells. These preclinical data suggest that CDK6 is a potential target for MLL-r leukemias; accordingly, there is an active phase Ib/IIa clinical trial out of the University of Ulm (NCT02310243) of palbociclib as monotherapy for adults with MLL-r leukemias.
Menin Inhibitors
As mentioned earlier, the protein Menin interacts with the N-terminal portion of the MLL1 protein and has been shown to be essential for MLL fusion protein leukemogenesis [(6–9); see also Figure 2]. Menin also interacts with wild-type MLL1, and studies in mice have shown that genetic deletion of Menin affects long-term hematopoietic stem cell potential and B-lineage lymphoid progenitors (233). The therapeutic window of Menin inhibition for MLL-r leukemias is therefore uncertain. Nonetheless, several groups have developed small molecule inhibitors that disrupt the interaction between MLL1 and Menin and have shown in vitro and in vivo impairment of leukemia growth and proliferation, irrespective of the MLL fusion partner (234–237). As these studies were all short-term experiments with murine models of MLL-r leukemias, longer-term preclinical models will be essential studies to perform before development of clinical trials with these agents.
Dinaciclib
Due to the association of common MLL fusion partners in the SEC with pTEFb, the role of specific pTEFb inhibitors has also been examined as potentially useful in targeting MLL-r leukemias (Figure 2). The efficacy of the CDK9 inhibitor (part of the pTEFb complex) Flavopiridol on MLL-r leukemia cells has long been recognized (46). Dinaciclib, which inhibits the CDK9 component of pTEFb, showed efficacy in preclinical models both in vitro and in vivo, inducing apoptotic cell death in MLL-r leukemia models and inhibition of MLL target genes (165, 238). No toxicity data were reported in these studies, so it remains to be seen whether this inhibitor will demonstrate appropriate specificity for MLL target genes without causing inordinate toxicity due to global repression of RNA polymerase II.
Final Thoughts
MLL translocations lead to aberrant expression of stem cell genetic programs in hematopoietic cells, which leads to a particularly aggressive subtype of leukemias in children and adults. Outcomes with conventional chemotherapy remain suboptimal to dismal, and hematopoietic stem cell transplantation has not proven to be beneficial except in the most high-risk infant patients. Despite extensive resources and manpower devoted to a better understanding of MLL fusion biology, we still possess an inadequate understanding of the pathophysiology of this disease. Accompanying the ever increasing number of fusion partners identified is the ever widening circle of epigenetic regulators thought to be involved in genetic dysregulation upon expression of MLL translocations. However, in the era of targeted therapies, we may finally be at the cusp of discovering combinations of therapeutic agents that can improve the outcomes for these patients.
Author Contributions
Both authors conducted extensive literature review and co-wrote the final manuscript.
Conflict of Interest Statement
KB: I hold two patents with respect to the DOT1L inhibitor discussed in this manuscript (pertaining to it’s use in leukemias with high MN1 expression, and pertaining to the role of MDR1 mediated drug resistance). Neither is directly relevant to the discussion in this review article. My husband has just accepted a job as a medical director at Janssen. His work will not involve any of the agents discussed in this review article. AW declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH/NCI R01CA201230 (salary support to KB), 5T32CA082086-16 (salary support to AW), and funds from the Children’s Hospital of Colorado (salary support to KB and AW).
References
1. Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell (1992) 71:691–700. doi: 10.1016/0092-8674(92)90602-9
2. Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, et al. The t(4,11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell (1992) 71:701–8. doi:10.1016/0092-8674(92)90603-A
3. Butler LH, Slany R, Cui X, Cleary ML, Mason DY. The HRX proto-oncogene product is widely expressed in human tissues and localizes to nuclear structures. Blood (1997) 89(9):3361–70.
4. Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, et al. Menin associates with a trithorax family histone methyltransferase complex and with the Hoxc8 locus. Mol Cell (2004) 13:587–97. doi:10.1016/S1097-2765(04)00081-4
5. Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A (2005) 102(3):749–54. doi:10.1073/pnas.0408836102
6. Yokoyama A, Somervaille TCP, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The Menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell (2005) 123:207–18. doi:10.1016/j.cell.2005.09.025
7. Chen YX, Yan J, Keeshan K, Tubbs AT, Wang H, Silva A, et al. The tumor suppressor Menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci U S A (2006) 103:1018–23. doi:10.1073/pnas.0510347103
8. Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with Menin is required for transformation. Cancer Res (2007) 67(15):7275–83. doi:10.1158/0008-5472.CAN-06-2369
9. Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell (2008) 14:36–46. doi:10.1016/j.ccr.2008.05.003
10. Zhu L, Li Q, Wong SH, Huang M, Klein BJ, Shen J, et al. ASH1L links histone H2 lysine 36 dimethylation to MLL leukemia. Cancer Discov (2016) 7:770–83. doi:10.1158/2159-8290.CD-16-0058
11. Ma Q, Alder H, Nelson KK, Chatterjee D, Gu Y, Nakamura T, et al. Analysis of the murine All-1 gene reveals conserved domains with human ALL-1 and identifies a motif shared with DNA methyltransferases. Proc Natl Acad Sci U S A (1993) 90:6350–4. doi:10.1073/pnas.90.13.6350
12. Zeleznik-Le NJ, Harden AM, Rowley JD. 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc Natl Acad Sci U S A (1994) 91:10610–4. doi:10.1073/pnas.91.22.10610
13. Yano T, Nakamura T, Blechman J, Sorio C, Dang CV, Geiger B, et al. Nuclear punctate distribution of ALL-1 is conferred by distinct elements at the N-terminus of the protein. Proc Natl Acad Sci U S A (1997) 94:7286–91. doi:10.1073/pnas.94.14.7286
14. Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet (2000) 24:88–91. doi:10.1038/71750
15. Birke M, Schreiner S, Garcia-Cuellar MP, Mahr K, Titgemeyer F, Slany RK. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res (2002) 30:958–65. doi:10.1093/nar/30.4.958
16. Tschiersch B, Hofmann A, Krauss V, Dorn R, Korge G, Reuter G. The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J (1994) 13:3822–31.
17. Chen J, Santillan DA, Koonce M, Wei W, Luo R, Thirman JM, et al. Loss of MLL PHD finger 3 is necessary for MLL-ENL-induced hematopoietic stem cell immortalization. Cancer Res (2008) 68(15):6199–207. doi:10.1158/0008-5472.CAN-07-6514
18. Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis D, et al. MLL targets SET methyltransferase activity to Hox gene promoters. Mol Cell (2002) 10:1107–17. doi:10.1016/S1097-2765(02)00741-4
19. Hsieh JJD, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol (2003) 23(1):186–94. doi:10.1128/MCB.23.1.186-194.2003
20. Hsieh JJD, Cheng EHY, Korsmeyer SJ. Taspase-1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell (2003) 115:293–303. doi:10.1016/S0092-8674(03)00816-X
21. Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol (2006) 13(8):713–9. doi:10.1038/nsmb1128
22. Li Y, Han J, Zhang Y, Cao F, Liu Z, Li S, et al. Structural basis for activity regulation of MLL family methyltransferases. Nature (2016) 530:447–52. doi:10.1038/nature16952
23. Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol (2001) 21:2249–58. doi:10.1128/MCB.21.7.2249-2258.2001
24. Mishra BP, Zaffuto KM, Artinger EL, Org T, Mikkola HKA, Cheng C, et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep (2014) 7:1239–47. doi:10.1016/j.celrep.2014.04.015
25. Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell (2010) 17:148–59. doi:10.1016/j.ccr.2009.12.034
26. Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb SJ, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukeomgenesis. Mol Cell (2010) 38:853–63. doi:10.1016/j.molcel.2010.05.011
27. Lim JH, Jang S, Park CJ, Chi HS, Lee JO, Seo EJ. FISH analysis of MLL gene rearrangements: detection of the concurrent loss or gain of the 3’ signal and its prognostic significance. Int J Lab Hematol (2014) 36:571–9. doi:10.1111/ijlh.12192
28. Wilkinson AC, Ballabio E, Geng H, North P, Tapia M, Kerry J, et al. RUNX1 is a key target in t(4,11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell Rep (2013) 3:116–27. doi:10.1016/j.celrep.2012.12.016
29. Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, et al. The MLL recombinome of acute leukemias in 2013. Leukemia (2013) 27:2165–76. doi:10.1038/leu.2013.135
30. Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol (2011) 12:799–814. doi:10.1038/nrm3230
31. Yu BD, Hess JL, Horning SE, Brown GAJ, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature (1995) 378(6556):505–8. doi:10.1038/378505a0
32. Yagi H, Deguchi K, Aono A, Tani Y, Kishimoto T, Komori T. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood (1998) 92:108–17.
33. Yokoyama A, Ficara F, Murphy MJ, Meisel C, Naresh A, Kitabayashi I, et al. Proteolytically cleaved MLL subunits are susceptible to distinct degradation pathways. J Cell Sci (2011) 124:2208–19. doi:10.1242/jcs.080523
34. Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci U S A (1998) 95:10632–6. doi:10.1073/pnas.95.18.10632
35. McMahon KA, Hiew SYL, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, et al. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell (2007) 1:338–45. doi:10.1016/j.stem.2007.07.002
36. Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell (2007) 1:324–37. doi:10.1016/j.stem.2007.05.019
37. Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, et al. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A (2005) 102(24):8603–8. doi:10.1073/pnas.0503072102
38. Milne TA, Dou Y, Martin ME, Brock HW, Roeder RG, Hess JL. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci U S A (2005) 102(41):14765–70. doi:10.1073/pnas.0503630102
39. Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, et al. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell (1996) 85:853–61. doi:10.1016/S0092-8674(00)81269-6
40. Slany RK, Lavau C, Cleary ML. The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol Cell Biol (1998) 18(1):122–9. doi:10.1128/MCB.18.1.122
41. Ma C, Staudt LM. LAF-4 encodes a lymphoid nuclear protein with transactivation potential that is homologous to AF-4, the gene fused to MLL in t(4,11) leukemias. Blood (1996) 87:734–45.
42. Li Q, Frestedt JL, Kersey JH. AF4 encodes a ubiquitous protein that in both native and MLL-AF4 fusion types localizes to subnuclear compartments. Blood (1998) 92:3841–7.
43. Erfurth F, Hemenway CS, de Erkenez AC, Domer PH. MLL fusion partners AF4 and AF9 interact at subnuclear foci. Leukemia (2004) 18:92–102. doi:10.1038/sj.leu.2403200
44. Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet (2007) 16(1):92–106. doi:10.1093/hmg/ddl444
45. Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood (2007) 110(13):4445–54. doi:10.1182/blood-2007-05-090514
46. Mueller D, Garcia-Cuellar MP, Bach C, Buhl S, Maethner E, Slany RK. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol (2009) 7(11):1–14. doi:10.1371/journal.pbio.1000249
47. Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, et al. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev (2010) 24:574–89. doi:10.1101/gad.1898410
48. Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell (2010) 17(2):198–212. doi:10.1016/j.ccr.2009.12.040
49. Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell (2014) 159:558–71. doi:10.1016/j.cell.2014.09.049
50. So CW, Lin M, Ayton PM, Chen EH, Cleary ML. Dimerization contributes to oncogenic activation of MLL chimeras in acute leukemias. Cancer Cell (2003) 4:99–110. doi:10.1016/S1535-6108(03)00188-0
51. Dobson CL, Warren AJ, Pannell R, Forster A, Rabbitts TH. Tumorigenesis in mice with a fusion of the leukaemia oncogene Mll and the bacterial lacZ gene. EMBO J (2000) 19(5):843–51. doi:10.1093/emboj/19.5.843
52. Martin ME, Milne TA, Bloyer S, Galoian K, Shen W, Gibbs D, et al. Dimerization of MLL fusion proteins immortalizes hematopoietic cells. Cancer Cell (2003) 4:197–207. doi:10.1016/S1535-6108(03)00214-9
53. Taki T, Kano H, Taniwaki M, Sako M, Yanagisawa M, Hayashi Y. AF5q31, a newly identified AF4-related gene, is fused to MLL in infant acute lymphoblastic leukemia with ins(5,11)(q31,q13q23). Proc Natl Acad Sci U S A (1999) 96:14535–40. doi:10.1073/pnas.96.25.14535
54. von Bergh AR, Beverloo HB, Rombout P, van Wering ER, van Weel MH, Beverstock GC, et al. LAF4, an AF4-related gene, is fused to MLL in infant acute lymphoblastic leukemia. Genes Chromosomes Cancer (2002) 35:92–6. doi:10.1002/gcc.10091
55. Prasad R, Yano T, Sorio C, Nakamura T, Rallapalli R, Gu Y, et al. Domains with transcriptional regulatory activity within the ALL1 and AF4 proteins involved in acute leukemia. Proc Natl Acad Sci U S A (1995) 92:12160–4. doi:10.1073/pnas.92.26.12160
56. Bitoun E, Davies KE. The robotic mouse: unraveling the function of AF4 in the cerebellum. Cerebellum (2005) 4:250–60. doi:10.1080/14734220500325897
57. Isnard P, Core N, Naquet P, Djabali M. Altered lymphoid development in mice deficient for the mAF4 proto-oncogene. Blood (2000) 96(2):705–10.
58. Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell (2006) 23:297–305. doi:10.1016/j.molcel.2006.06.014
59. Okuda H, Kanai A, Ito S, Matsui H, Yokoyama A. AF4 uses the SL1 components of RNAP1 machinery to initiate MLL fusion- and AEP-dependent transcription. Nat Commun (2015) 6:8869. doi:10.1038/ncomms9869
60. Drynan LF, Pannell R, Forster A, Chan NMM, Cano F, Daser A, et al. Mll fusions generated by Cre-loxP-mediated de novo translocations can induce lineage reassignment in tumorigenesis. EMBO J (2005) 24:3136–46. doi:10.1038/sj.emboj.7600760
61. Collins EC, Appert A, Ariza-McNaughton L, Pannell R, Yamada Y, Rabbitts TH. Mouse Af9 is a controller of embryo patterning, like Mll, whose human homologue fuses with AF9 after chromosomal translocation in leukemia. Mol Cell Biol (2002) 22(20):7313–24. doi:10.1128/MCB.22.20.7313-7324.2002
62. Pina C, May G, Soneji S, Hong D, Enver T. MLLT3 regulates early human erythroid and megakaryocytic cell fate. Cell Stem Cell (2008) 2:264–73. doi:10.1016/j.stem.2008.01.013
63. Garcia-Cuellar MP, Zilles O, Schreiner SA, Birke M, Winkler TH, Slany RK. The ENL moiety of the childhood leukemia-associated MLL-ENL oncoprotein recruits human Polycomb 3. Oncogene (2001) 20:411–9. doi:10.1038/sj.onc.1204108
64. Hemenway CS, de Erkenez AC, Gould GCD. The polycomb protein MPc3 interacts with AF9, and MLL fusion partner in t(9,11)(p22,q23) acute leukemias. Oncogene (2001) 20:3798–805. doi:10.1038/sj.onc.1204478
65. Srinivasan RS, de Erkenez AC, Hemenway CS. The mixed lineage leukemia fusion partner AF9 binds specific isoforms of the BCL-6 corepressor. Oncogene (2003) 22:3395–406. doi:10.1038/sj.onc.1206361
66. Tan J, Jones M, Koseki H, Nakayama M, Muntean AG, Maillard I, et al. CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer Cell (2011) 20:563–75. doi:10.1016/j.ccr.2011.09.008
67. Zeisig DT, Bittner CB, Zeisig BB, Garcia-Cuellar MP, Hess JL, Slany RK. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene (2005) 24:5525–32. doi:10.1038/sj.onc.1208699
68. Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, et al. Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol Cell (2016) 62:181–93. doi:10.1016/j.molcel.2016.03.028
69. Andrews FH, Shanle EK, Strahl BD, Kutateladze TG. The essential role of acetyllysine binding by the YEATS domain in transcriptional regulation. Transcription (2016) 7:14–20. doi:10.1080/21541264.2015.1125987
70. Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, et al. hDOT1L links histone methylation to leukemogenesis. Cell (2005) 121:167–78. doi:10.1016/j.cell.2005.02.020
71. Deshpande AJ, Deshpande A, Sinha AU, Chen L, Chang J, Cihan A, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell (2014) 26:896–908. doi:10.1016/j.ccell.2014.10.009
72. Chen S, Yang Z, Wilkinson AW, Desphande AJ, Sidoli S, Krajewski K, et al. The PZP domain of AF10 senses unmodified H3K27 to regulate DOT1L mediated methylation of H3K79. Mol Cell (2015) 60(2):319–27. doi:10.1016/j.molcel.2015.08.019
73. Biswas D, Milne TA, Basrur V, Kim J, Elenitoba-Johnson KSJ, Allis CD, et al. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc Natl Acad Sci U S A (2011) 108(38):15751–6. doi:10.1073/pnas.1111498108
74. He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, et al. Human polymerase-associated factor complex (PAFc) connects the super elongation complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A (2011) 108(36):E636–45. doi:10.1073/pnas.1107107108
75. Armstrong SA, Staunton JE, Silverman LB, Pieters R, Den Boer ML, Minden MD, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet (2002) 30:41–7. doi:10.1038/ng765
76. Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell (2002) 1:133–43. doi:10.1016/S1535-6108(02)00032-6
77. Rozovskaia T, Ravid-Amir O, Tillib S, Getz G, Feinstein E, Agrawal H, et al. Expression profiles of acute lymphoblastic and myeloblastic leukemias with ALL-1 rearrangements. Proc Natl Acad Sci U S A (2003) 100(13):7853–8. doi:10.1073/pnas.1132115100
78. Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J (1998) 17:3714–25. doi:10.1093/emboj/17.13.3714
79. Li Z, Luo RT, Mi S, Sun M, Chen P, Bao J, et al. Consistent deregulation of gene expression between human and murine MLL rearrangement leukemias. Cancer Res (2009) 69:1109–16. doi:10.1158/0008-5472.CAN-08-3381
80. Lawrence HJ, Helgason CD, Sauvageau G, Fong S, Izon DJ, Humphries RK, et al. Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood (1997) 89:1922–30.
81. So CW, Karsunky H, Wong P, Weissman IL, Cleary ML. Leukemic transformation of hematopoietic progenitors by MLL-GAS7 in the absence of Hoxa7 or Hoxa9. Blood (2004) 103:3192–9. doi:10.1182/blood-2003-10-3722
82. Kawagoe H, Humphries RK, Blair A, Sutherland HJ, Hogge DE. Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia (1999) 13:687–98. doi:10.1038/sj.leu.2401410
83. Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev (2007) 21:2762–74. doi:10.1101/gad.1602107
84. Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M, et al. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood (2009) 113:2375–85. doi:10.1182/blood-2007-09-113597
85. Kumar AR, Li Q, Hudson WA, Chen W, Sam T, Yao Q, et al. A role for MEIS1 in MLL-fusion gene leukemia. Blood (2009) 113:1756–8. doi:10.1182/blood-2008-06-163287
86. Kersey JH, Wang D, Oberto M. Resistance of t(4,11) (MLL-AF4 fusion gene) leukemias to stress-induced cell death: possible mechanism for extensive extramedullary accumulation of cells and poor prognosis. Leukemia (1998) 12:1561–4. doi:10.1038/sj.leu.2401148
87. Inukai T, Zhang X, Goto M, Hirose K, Uno K, Akahane K, et al. Resistance of infant leukemia with MLL rearrangement to tumor necrosis factor-related apoptosis-inducing ligand: a possible mechanism for poor sensitivity to antitumor immunity. Leukemia (2006) 20:2119–29. doi:10.1038/sj.leu.2404429
88. Gaussmann A, Wenger T, Eberle I, Bursen A, Bracharz S, Herr I, et al. Combined effects of the two reciprocal t(4,11) fusion proteins MLL.AF4 and AF4.MLL confer resistance to apoptosis, cell cycling capacity and growth transformation. Oncogene (2007) 26:3352–63. doi:10.1038/sj.onc.1210125
89. Robinson BW, Behling KC, Gupta M, Zhang AY, Moore JS, Bantly AD, et al. Abundant anti-apoptotic BCL-2 is a molecular target in leukaemias with t(4,11) translocation. Br J Haematol (2008) 141:827–39. doi:10.1111/j.1365-2141.2008.07100.x
90. Bindels EM, Havermans M, Lugthart S, Erpelinck C, Wocjtowicz E, Krivtsov AV, et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9-rearranged AMLs. Blood (2012) 119:5838–49. doi:10.1182/blood-2011-11-393827
91. Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia (2013) 27:852–60. doi:10.1038/leu.2012.363
92. Benito JM, Godfrey L, Kojima K, Hogdal L, Wunderlich M, Geng H, et al. MLL-rearranged acute lymphoblastic leukemias activate BCL-2 through H3K79 methylation and are sensitive to the BCL-2-specific antagonist ABT-199. Cell Rep (2015) 13:2715–27. doi:10.1016/j.celrep.2015.12.003
93. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127:2391–405. doi:10.1182/blood-2016-03-643544
94. Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol (2012) 7:283–301. doi:10.1146/annurev-pathol-011811-132434
95. Super HJG, McCabe NR, Thirman MJ, Larson RA, LeBeau MM, Pederson-Bjergaard J, et al. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood (1993) 82(12):3705–11.
96. Andersen MK, Christiansen DH, Jensen BA, Ernst P, Hauge G, Pedersen-Bjergaard J. Therapy-related acute lymphoblastic leukaemia with MLL rearrangements following DNA topoisomerase II inhibitors, an increasing problem: report on two new cases and review of the literature since 1992. Br J Haematol (2001) 114:539–43. doi:10.1046/j.1365-2141.2001.03000.x
97. Mann G, Attarbaschi A, Schrappe M, De Lorenzo P, Peters C, Hann I, et al. Improved outcome with hematopoietic stem cell transplantation in a poor progenostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 study. Blood (2010) 116(15):2644–50. doi:10.1182/blood-2010-03-273532
98. Blanco JG, Dervieux T, Edick MJ, Mehta PK, Rubnitz JE, Shurtleff S, et al. Molecular emergence of acute myeloid leukemia during treatment for acute lymphoblastic leukemia. Proc Natl Acad Sci U S A (2001) 98(18):10338–43. doi:10.1073/pnas.181199898
99. Chowdhury T, Brady HJM. Insights from clinical studies into the role of the MLL gene in infant and childhood leukemia. Blood Cells Mol Dis (2008) 40(2):192–9. doi:10.1016/j.bcmd.2007.07.005
100. Felix CA. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim Biophys Acta (1998) 1400:233–55. doi:10.1016/S0167-4781(98)00139-0
101. Silverman LB. Acute lymphoblastic leukemia in infancy. Pediatr Blood Cancer (2007) 49:1070–3. doi:10.1002/pbc.21352
102. Tomizawa D, Koh K, Sato T, Kinukawa N, Morimoto A, Isoyama K, et al. Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia (2007) 21:2258–63. doi:10.1038/sj.leu.2404903
103. Tauchi H, Tomizawa D, Eguchi M, Eguchi-Ishimae M, Koh K, Hirayama M, et al. Clinical features and outcome of MLL gene rearranged acute lymphoblastic leukemia in infants with additional chromosomal abnormalities other than 11q23 translocation. Leuk Res (2008) 32(10):1523–9. doi:10.1016/j.leukres.2008.03.018
104. Hilden JM, Dinndorf PA, Meerbaum SO, Sather H, Villaluna D, Heerema NA, et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children’s Oncology Group. Blood (2006) 108:441–51. doi:10.1182/blood-2005-07-3011
105. Marks DI, Moorman AV, Chilton L, Paietta E, Enshaie A, DeWald G, et al. The clinical characteristics, therapy and outcome of 85 adults with acute lymphoblastic leukemia and t(4,11)(q21,q23)/MLL-AFF1 prospectively treated in the UKALLXII/ECOG2993 trial. Haematologica (2013) 98(6):945–52. doi:10.3324/haematol.2012.081877
106. Ramakers-van Woerden NL, Beverloo HB, Veerman AJ, Camitta BM, Loonen AH, van Wering ER, et al. In vitro drug-resistance profile in infant acute lymphoblastic leukemia in relation to age, MLL rearrangements and immunophenotype. Leukemia (2004) 18(3):521–9. doi:10.1038/sj.leu.2403253
107. Stam RW, den Boer ML, Meijerink JPP, Ebus MEG, Peters GJ, Noordhuis P, et al. Differential mRNA expression of Ara-C-metabolizing enzymes explains Ara-C sensitivity in MLL gene-rearranged infant acute lymphoblastic leukemia. Blood (2003) 101(4):1270–6. doi:10.1182/blood-2002-05-1600
108. Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood (2016) 127:2406–10. doi:10.1182/blood-2015-08-665547
109. Rayes A, McMasters RL, O’Brien MM. Lineage switch in MLL-rearranged infant leukemia following CD19-directed therapy. Pediatr Blood Cancer (2016) 63:1113–5. doi:10.1002/pbc.25953
110. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood (2009) 114:937–51. doi:10.1182/blood-2009-03-209262
111. Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell (2008) 13:483–95. doi:10.1016/j.ccr.2008.04.020
112. Whitman SP, Liu S, Vukosavljevic T, Rush LJ, Yu L, Liu C, et al. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood (2005) 106(1):345–52. doi:10.1182/blood-2005-01-0204
113. Basecke J, Whelan JT, Griesinger F, Bertrand FE. The MLL partial tandem duplication in acute myeloid leukaemia. Br J Haematol (2006) 135:438–49. doi:10.1111/j.1365-2141.2006.06301.x
114. Dohner K, Tobis K, Ulrich R, Frohling S, Benner A, Schlenk RF, et al. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol (2002) 20(15):3254–61. doi:10.1200/JCO.2002.09.088
115. Reichel M, Gillert E, Angermuller S, Hensel JP, Heidel F, Lode M, et al. Biased distribution of chromosomal breakpoints involving the MLL gene in infants versus children and adults with t(4,11) ALL. Oncogene (2001) 20:2900–7. doi:10.1038/sj.onc.1204401
116. Zhang Y, Rowley JD. Chromatin structural elements and chromosomal translocations in leukemia. DNA Repair (Amst) (2006) 5(9–10):1282–97. doi:10.1016/j.dnarep.2006.05.020
117. Broeker PLS, Super HG, Thirman MJ, Pomykala H, Yonebayashi Y, Tanabe S, et al. Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood (1996) 87(5):1912–22.
118. Scharf S, Zech J, Bursen A, Schraets D, Oliver PL, Kliem S, et al. Transcription linked to recombination: a gene-internal promoter coincides with the recombination hot spot II of the human MLL gene. Oncogene (2007) 26:1361–71. doi:10.1038/sj.onc.1209948
119. Betti CJ, Villalobos MJ, Jiang Q, Cline E, Diaz MO, Loredo G, et al. Cleavage of the MLL gene by activators of apoptosis is independent of topoisomerase II activity. Leukemia (2005) 19:2289–95. doi:10.1038/sj.leu.2403966
120. Strick R, Strissel PL, Borgers S, Smith SL, Rowley JD. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc Natl Acad Sci U S A (2000) 97(9):4790–5. doi:10.1073/pnas.070061297
121. Alexander FE, Patheal SL, Biondi A, Brandalise S, Cabrera ME, Chan LC, et al. Transplacental chemical exposure and risk of infant leukemia with MLL gene fusion. Cancer Res (2001) 61:2542–6.
122. Khosrovani SB, Janssen J, Maas LM, Godschalk RWL, Nijhuis JG, van Schooten FJ. Dietary flavonoids induce MLL translocations in primary human CD34+ cells. Carcinogenesis (2007) 28(8):1703–9. doi:10.1093/carcin/bgm102
123. Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood (2003) 102(7):2321–33. doi:10.1182/blood-2002-12-3817
124. Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, et al. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature (1993) 363:358–60. doi:10.1038/363358a0
125. Valentine MC, Linabery AM, Chasnoff S, Hughes AE, Mallaney C, Sanchez N, et al. Excess congenital non-synonymous variation in leukemia-associated genes in MLL-infant leukemia: a Children’s Oncology Group report. Leukemia (2014) 28:1235–41. doi:10.1038/leu.2013.367
126. Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet (2007) 370:240–50. doi:10.1016/S0140-6736(07)61126-X
127. Dreyer ZE, Hilden JM, Jones TL, Devidas M, Winick NJ, Willman CL, et al. Intensified chemotherapy without Sct in Infant All: results from COG P9407 (cohort 3). Pediatr Blood Cancer (2015) 62:419–26. doi:10.1002/pbc.25322
128. Reaman GH, Sposto R, Sensel MG, Lange BJ, Feusner JH, Heerema NA, et al. Treatment outcome and prognostic factors for infants with acute lymphoblastic leukemia treated on two consecutive trials of the Children’s Cancer Group. J Clin Oncol (1999) 17:445–55.
129. Pui CH, Gaynon PS, Boyett JM, Chessells JM, Baruchel A, Kamps W, et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet (2002) 359:1909–15. doi:10.1016/S0140-6736(02)08782-2
130. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. New Engl J Med (2015) 373:1541–52. doi:10.1056/NEJMra1400972
131. Jabbour E, O’Brien S, Konopleva M, Kantarjian H. New insights into the pathophysiology and therapy of adult acute lymphoblastic leukemia. Cancer (2015) 121:2517–28. doi:10.1002/cncr.29383
132. Rubnitz JE, Raimondi SC, Tong X, Srivastava DK, Razzouk BI, Shurtleff SA, et al. Favorable impact of the t(9,11) in childhood acute myeloid leukemia. J Clin Oncol (2002) 20:2302–9. doi:10.1200/JCO.2002.08.400
133. Balgobind BV, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood (2009) 114:2489–96. doi:10.1182/blood-2009-04-215152
134. Zwaan CM, Kolb EA, Reinhardt D, Abrahamsson J, Adachi S, Aplenc R, et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J Clin Oncol (2015) 33:2949–62. doi:10.1200/JCO.2015.62.8289
135. Blum W, Mrozek K, Ruppert AS, Carroll AJ, Rao KW, Pettenati MJ, et al. Adult de novo acute myeloid leukemia with t(6,11)(q27,q23): results from Cancer and Leukemia Group B Study 8461 and review of the literature. Cancer (2004) 101:1420–7. doi:10.1002/cncr.20489
136. Emerenciano M, Meyer C, Mansur MB, Marschalek R, Pombo-de-Oliveira MS. Brazilian Collaborative Study Group of Infant Acute Leukaemia. The distribution of MLL breakpoints correlates with outcome in infant acute leukaemia. Br J Haematol (2013) 161:224–36. doi:10.1111/bjh.12250
137. Marschalek R. Systematic classification of mixed-lineage leukemia fusion partners predicts additional cancer pathways. Ann Lab Med (2016) 36:85–100. doi:10.3343/alm.2016.36.2.85
138. Hale GA, Heslop HE, Bowman LC, Rochester RA, Pui CH, Brenner MK, et al. Bone marrow transplantation for therapy-induced acute myeloid leukemia in children with previous lymphoid malignancies. Bone Marrow Transplant (1999) 24:735–9. doi:10.1038/sj.bmt.1701962
139. Tomizawa D, Koh K, Hirayama M, Miyamura T, Hatanaka M, Saikawa Y, et al. Outcome of recurrent or refractory acute lymphoblastic leukemia in infants with MLL gene rearrangements: a report from the Japan Infant Leukemia Study Group. Pediatr Blood Cancer (2009) 52:808–13. doi:10.1002/pbc.21975
140. Litzow MR, Tarima S, Perez WS, Bolwell BJ, Cairo MS, Camitta BM, et al. Allogeneic transplantation for therapy-related myelodysplastic syndrome and acute myeloid leukemia. Blood (2010) 115:1850–7. doi:10.1182/blood-2009-10-249128
141. Finke J, Schmoor C, Bertz H, Marks R, Wasch R, Zeiser R, et al. Long-term follow-up of therapy-related myelodysplasia and AML patients treated with allogeneic hematopoietic cell transplantation. Bone Marrow Transplant (2016) 51:771–7. doi:10.1038/bmt.2015.338
142. Dreyer ZE, Dinndorf PA, Camitta B, Sather H, La MK, Devidas M, et al. Analysis of the role of hematopoietic stem-cell transplantation in infants with acute lymphoblastic leukemia in first remission and MLL gene rearrangements: a report from the Children’s Oncology Group. J Clin Oncol (2011) 29:214–22. doi:10.1200/JCO.2009.26.8938
143. Pui CH, Chessells JM, Camitta B, Baruchel A, Biondi A, Boyett JM, et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia (2003) 17:700–6. doi:10.1038/sj.leu.2402883
144. Chessells JM, Harrison CJ, Watson SL, Vora AJ, Richards SM. Treatment of infants with lymphoblastic leukaemia: results of the UK Infant Protocols 1987–1999. Br J Haematol (2002) 117:306–14. doi:10.1046/j.1365-2141.2002.03442.x
145. van der Velden VHJ, Corral L, Valsecchi MG, Jansen MWJC, De Lorenzo P, Cazzaniga G, et al. Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia (2009) 23:1073–9. doi:10.1038/leu.2009.17
146. van der Linden MH, Valsecchi MG, De Lorenzo P, Moricke A, Janka G, Leblanc TM, et al. Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-99 protocol. Blood (2009) 114:3764–8. doi:10.1182/blood-2009-02-204214
147. Annesley CE, Brown P. The biology and targeting of FLT3 in pediatric leukemia. Front Oncol (2014) 4:263. doi:10.3389/fonc.2014.00263
148. Meshinchi S, Woods WG, Stirewalt DL, Sweetser DA, Buckley JD, Tjoa TK, et al. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood (2001) 97:89–94. doi:10.1182/blood.V97.1.89
149. Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, et al. Inhibition of FLT3 in MLL: validation of a therapeutic target identified by gene expression based classification. Cancer Cell (2003) 3:173–83. doi:10.1016/S1535-6108(03)00003-5
150. Stam RW, Den Boer ML, Schneider P, Nollau P, Horstmann M, Beverloo HB, et al. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood (2005) 106:2484–90. doi:10.1182/blood-2004-09-3667
151. Brown P, Levis M, Shurtleff S, Campana D, Downing J, Small D. FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood (2005) 105:812–20. doi:10.1182/blood-2004-06-2498
152. Chillon MC, Gomez-Casares MT, Lopez-Jorge CE, Rodriguez-Medina C, Molines A, Sarasquete ME, et al. Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-germline acute lymphoblastic leukemia. Leukemia (2012) 26:2360–6. doi:10.1038/leu.2012.161
153. Prelle C, Bursen A, Dingermann T, Marschalek R. Secondary mutations in t(4,11) leukemia patients. Leukemia (2013) 27:1425–7. doi:10.1038/leu.2012.365
154. Andersson AK, Ma J, Wang J, Chen X, Larson Gedman A, Dang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet (2015) 47(4):330–7. doi:10.1038/ng.3230
155. Stubbs MC, Kim YM, Krivtsov AV, Wright RD, Feng Z, Agarwal J, et al. MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: development of a model for rapid therapeutic assessment. Leukemia (2008) 22:66–77. doi:10.1038/sj.leu.2404951
156. Stam RW, Schneider P, de Lorenzo P, Valsecchi MG, Den Boer ML, Pieters R. Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood (2007) 110(7):2774–5. doi:10.1182/blood-2007-05-091934
157. Brown P, Levis M, McIntyre E, Griesemer M, Small D. Combinations of the FLT3 inhibitor CEP-701 and chemotherapy synergistically kill infant and childhood MLL-rearranged ALL cells in a sequence-dependent manner. Leukemia (2006) 20:1368–76. doi:10.1038/sj.leu.2404277
158. Cooper TM, Cassar J, Eckroth E, Malvar J, Sposto R, Gaynon P, et al. A phase I study of quizartinib combined with chemotherapy in relapsed childhood leukemia: a Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) study. Clin Cancer Res (2016) 22:4014–22. doi:10.1158/1078-0432.CCR-15-1998
159. Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood (2009) 114:2984–92. doi:10.1182/blood-2009-05-222034
160. Kampa-Schittenhelm KM, Heinrich MC, Akmut F, Dohner H, Dohner K, Schittenhelm MM. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA, and –KIT isoforms. Mol Cancer (2013) 12:19. doi:10.1186/1476-4598-12-19
161. Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol (2012) 19:99–115. doi:10.1016/j.chembiol.2012.01.003
162. Liu H, Westergard TD, Cashen A, Piwnica-Worms DR, Kunkle L, Vij R, et al. Proteasome inhibitors evoke latent tumor suppression programs in pro-B MLL leukemias through MLL-AF4. Cancer Cell (2014) 25:530–42. doi:10.1016/j.ccr.2014.03.008
163. Xue K, Song J, Yang Y, Li Z, Wu C, Jin J, et al. PAX5 promotes pre-B cell proliferation by regulating the expression of pre-B cell receptor and its downstream signaling. Mol Immunol (2016) 73:1–9. doi:10.1016/j.molimm.2016.03.007
164. Koss C, Nance S, Connelly M, Ma J, Shelat A, Cotton A, et al. Targeted inhibition of the MLL transcriptional complex by proteasome inhibitors elicits a high response rate in relapsed/refractory MLL rearranged leukemia. Poster Abstract, American Society of Hematology Annual Meeting. San Francisco (2014).
165. Cruickshank MN, Ford J, Cheung LC, Heng J, Singh S, Wells J, et al. Systematic chemical and molecular profiling of MLL-rearranged infant acute leukemia reveals efficacy of romidepsin. Leukemia (2017) 31(1):40–50. doi:10.1038/leu.2016.165
166. McGinty RK, Kim J, Chatterjee C, Roeder RG, Muir TW. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature (2008) 453:812–9. doi:10.1038/nature06906
167. Wang E, Kawaoka S, Yu M, Shi J, Ni T, Yang W, et al. Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc Natl Acad Sci U S A (2013) 110(10):3901–6. doi:10.1073/pnas.1301045110
168. Kuo HP, Wang Z, Lee DF, Iwasaki M, Duque-Afonso J, Wong SHK, et al. Epigenetic roles of MLL oncoproteins are dependent on NF-kB. Cancer Cell (2013) 24:423–37. doi:10.1016/j.ccr.2013.08.019
169. Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S, et al. Positive feedback between NF-kappaB and TNF-alpha promotes leukemia-initiating cell capacity. J Clin Invest (2014) 124:528–42. doi:10.1172/JCI68101
170. Sun XF, Zhang H. NFKB and NFKBI polymorphisms in relation to susceptibility of tumor and other diseases. Histol Histopathol (2007) 22:1387–98.
171. Tonelli R, Sartini R, Fronza R, Freccero F, Franzoni M, Dongiovanni D, et al. G1 cell-cycle arrest and apoptosis by histone deacetylase inhibition in MLL-AF9 acute myeloid leukemia cells is p21 dependent and MLL-AF9 independent. Leukemia (2006) 20:1307–10. doi:10.1038/sj.leu.2404221
172. Stumpel DJPM, Schneider P, Seslija L, Osaki H, Williams O, Pieters R, et al. Connectivity mapping identifies HDAC inhibitors for the treatment of t(4,11)-positive infant acute lympoblastic leukemia. Leukemia (2012) 26:682–92. doi:10.1038/leu.2011.278
173. Bhatla T, Wang J, Morrison DJ, Raetz EA, Burke MJ, Brown P, et al. Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood (2012) 119(22):5201–10. doi:10.1182/blood-2012-01-401687
174. Stubbs MC, Kim W, Bariteau M, Davis T, Vempati S, Minehart J, et al. Selective inhibition of HDAC1 and HDAC2 as a potential therapeutic option for B-ALL. Clin Cancer Res (2015) 21(10):2348–58. doi:10.1158/1078-0432.CCR-14-1290
175. Moreno DA, Scrideli CA, Cortez MAA, de Paula Queiroz R, Valera ET, da Silva Silveira V, et al. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol (2010) 150:665–73. doi:10.1111/j.1365-2141.2010.08301.x
176. Tao YF, Pang L, Du XJ, Sun LC, Hu SY, Lu J, et al. Differential mRNA expression levels of human histone-modifying enzymes in normal karyotype B cell pediatric acute lymphoblastic leukemia. Int J Mol Sci (2013) 14(2):3376–94. doi:10.3390/ijms14023376
177. Gruhn B, Naumann T, Gruner D, Walther M, Wittig S, Becker S, et al. The expression of histone deacetylase 4 is associated with prednisone poor-response in childhood acute lymphoblastic leukemia. Leuk Res (2013) 37(10):1200–7. doi:10.1016/j.leukres.2013.07.016
178. Yamaguchi T, Cubizolles F, Zhang Y, Reichert N, Kohler H, Seiser C, et al. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev (2010) 24:455–69. doi:10.1101/gad.552310
179. Jones CL, Bhatla T, Blum R, Wang J, Paugh SW, Wen X, et al. Loss of TBL1XR1 disrupts glucocorticoid receptor recruitment to chromatin and results in a glucocorticoid resistance in a B-lymphoblastic leukemia model. J Biol Chem (2014) 289(30):20502–15. doi:10.1074/jbc.M114.569889
180. Matthews GM, Mehdipour P, Cluse LA, Falkenberg KJ, Wang E, Roth M, et al. Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood (2015) 126(21):2392–403. doi:10.1182/blood-2015-03-632984
181. Ahmad K, Katryniok C, Scholz B, Merkens J, Loscher D, Marschalek R, et al. Inhibition of class I HDACs abrogates the dominant effect of MLL-AF4 by activation of wild-type MLL. Oncogenesis (2014) 3:e127. doi:10.1038/oncsis.2014.39
182. Burbury KL, Bishton MJ, Johnstone RW, Dickinson MJ, Szer J, Prince HM. MLL-aberrant leukemia: complete cytogenetic remission following treatment with a histone deacetylase inhibitor (HDACi). Ann Hematol (2011) 90:847–9. doi:10.1007/s00277-010-1099-6
183. Stumpel DJPM, Schneider P, van Roon EHJ, Boer JM, de Lorenzo P, Valsecchi MG, et al. Specific promoter methylation identifies different subgroups of MLL-rearranged infant acute lymphoblastic leukemia, influences clinical outcome, and provides therapeutic options. Blood (2009) 114:5490–8. doi:10.1182/blood-2009-06-227660
184. Schafer E, Irizarry R, Negi S, McIntyre E, Small D, Figueroa ME, et al. Promoter hypermethylation in MLL-r infant acute lymphoblastic leukemia: biology and therapeutic targeting. Blood (2010) 115(23):4798–809. doi:10.1182/blood-2009-09-243634
185. Yun S, Vincelette ND, Abraham I, Robertson KD, Fernandez-Zapico ME, Patnaik MM. Targeting epigenetic pathways in acute myeloid leukemia and myelodysplastic syndrome: a systematic review of hypomethylating agents trials. Clin Epigenetics (2016) 8:68–76. doi:10.1186/s13148-016-0233-2
186. Lubecka-Pietruszewska K, Kaufman-Szymczyk A, Stefanska B, Cebula-Obrzut B, Smolewski P, Fabianowska-Majewska K. Clofarabine, a novel adenosine analogue, reactivates DNA methylation-silenced tumor suppressor genes and inhibits cell growth in breast cancer cells. Eur J Pharmacol (2014) 723:276–87. doi:10.1016/j.ejphar.2013.11.021
187. Wyczechowska D, Fabianowska-Majewska K. The effects of cladribine and fludarabine on DNA methylation in K562 cells. Biochem Pharmacol (2003) 65(2):219–25. doi:10.1016/S0006-2952(02)01486-7
188. Stumpel DJPM, Schneider P, Pieters R, Stam RW. The potential of clofarabine in MLL-rearranged infant acute lymphoblastic leukaemia. Eur J Cancer (2015) 51:2008–21. doi:10.1016/j.ejca.2015.06.117
189. Stackelberg AV, Locatelli F, Zugmaier G, Handgretinger R, Trippet TM, Rizzari C, et al. Phase I/phaseII study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol (2016) 34(36):4381–9.
190. Driessen EMC, van Roon EHJ, Spijkers-Hagelstein JAP, Schneider P, de Lorenzo P, Valsecchi MG, et al. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica (2013) 98:937–44. doi:10.3324/haematol.2012.067983
191. Chandra P, Luthra R, Zuo Z, Yao H, Ravandi F, Reddy N, et al. Acute myeloid leukemia with t(9;11)(p21-22;q23): common properties of dysregulated Ras pathway signaling and genomic progression characterize de novo and therapy-related cases. Am J Clin Pathol (2010) 133:686–93. doi:10.1309/AJCPGII1TT4NYOGI
192. Emerenciano M, Barbosa TC, de Almeida Lopes B, Meyer C, Marschalek R, Pombo-de-Oliveira MS. Subclonality and prenatal origin of RAS mutations in KMT2A (MLL)-rearranged infant acute lymphoblastic leukaemia. Br J Haematol (2015) 170:268–87. doi:10.1111/bjh.13279
193. Lavallee VP, Baccelli I, Krosl J, Wilhelm B, Barabe F, Gendron P, et al. The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat Genet (2015) 47(9):1030–7. doi:10.1038/ng.3371
194. Kerstjens M, Driessen EMC, Willekes M, Pinhancos SS, Schneider P, Pieters R, et al. MEK inhibition is a promising therapeutic strategy for MLL-rearranged infant acute lymphoblastic leukemia patients carrying RAS mutations. Oncotarget (2016). doi:10.18632/oncotarget.11730
195. Kampen KR, Elst A, Mahmud H, Scherpen FJG, Diks SH, Peppelenbosch MP, et al. Insights in dynamic kinome reprogramming as a consequence of MEK inhibition in MLL-rearranged AML. Leukemia (2014) 28:589–99. doi:10.1038/leu.2013.342
196. Manara E, Baron E, Tregnago C, Aveic S, Bisio V, Bresolin S, et al. MLL-AF6 fusion oncogene sequesters AF6 into the nucleus to trigger RAS activation in myeloid leukemia. Blood (2014) 124(2):263–72. doi:10.1182/blood-2013-09-525741
197. Chang MJ, Wu H, Achille NJ, Reisenauer MR, Chou CW, Zeleznik-Le NJ, et al. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res (2010) 70(24):10234–42. doi:10.1158/0008-5472.CAN-10-3294
198. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell (2011) 20:66–78. doi:10.1016/j.ccr.2011.06.010
199. Jo SY, Granowicz EM, Maillard I, Thomas D, Hess JL. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood (2011) 117(18):4759–68. doi:10.1182/blood-2010-12-327668
200. Nguyen AT, Taranova O, He J, Zhang Y. Dot1l, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood (2011) 117(25):6912–22. doi:10.1182/blood-2011-02-334359
201. Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell (2008) 14:355–68. doi:10.1016/j.ccr.2008.10.001
202. Chen CW, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat Med (2015) 21(4):335–43. doi:10.1038/nm.3832
203. Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood (2013) 122(6):1017–25. doi:10.1182/blood-2013-04-497644
204. Klaus CR, Iwanowicz D, Johnston D, Campbell CA, Smith JJ, Moyer MP, et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J Pharmacol Exp Ther (2014) 350:646–56. doi:10.1124/jpet.114.214577
205. Luo M, Wang H, Zou Y, Zhang S, Xiao J, Jiang G, et al. Identification of phenoxyacetamide derivatives as novel DOT1L inhibitors via docking screening and molecular dynamics simulation. J Mol Graph Model (2016) 68:128–39. doi:10.1016/j.jmgm.2016.06.011
206. Shukla N, O’Brien MM, Silverman LB, Pauly M, Wetmore C, Loh ML, et al. Preliminary report of the phase 1 study of the DOT1L inhibitor, Pinometostat, EPZ-5676, in children with relapsed or refractory MLL-r acute leukemia: safety, exposure and target inhibition. Blood (2015) 126:3792.
207. Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Jongen-Lavrencic M, et al. A phase 1 study of the DOT1L inhibitor, Pinometostat (EPZ-5676), in adults with relapsed or refractory leukemia: safety, clinical activity, exposure and target inhibition. Blood (2015) 126:2547.
208. Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, et al. AFF4, a component of the ELL-P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell (2010) 37:429–37. doi:10.1016/j.molcel.2010.01.026
209. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukemia. Nature (2011) 478:529–33. doi:10.1038/nature10509
210. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature (2011) 478:524–8. doi:10.1038/nature10334
211. Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J, et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res (2015) 75(23):5106–19. doi:10.1158/0008-5472.CAN-15-0236
212. Gilan O, Lam EYN, Becher I, Lugo D, Cannizzaro E, Joberty G, et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat Struct Mol Biol (2016) 23(7):673–81. doi:10.1038/nsmb.3249
213. Garcia-Cuellar MP, Buttner C, Bartenhagen C, Dugas M, Slany RK. Leukemogenic MLL-ENL fusions induce alternative chromatin states to drive a functionally dichotomous group of target genes. Cell Rep (2016) 15:310–22. doi:10.1016/j.celrep.2016.03.018
214. Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell (2012) 21:473–87. doi:10.1016/j.ccr.2012.03.014
215. Shi J, Wang E, Zuber J, Rappaport A, Taylor M, Johns C, et al. The polycomb complex PRC2 supports aberrant self-renewal in a mouse model of MLL-AF9,NrasG12D acute myeloid leukemia. Oncogene (2013) 32:930–8. doi:10.1038/onc.2012.110
216. Feng Z, Yao Y, Zhou C, Chen F, Wu F, Wei L, et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol (2016) 9:24–36. doi:10.1186/s13045-016-0252-7
217. Smith LL, Yeung J, Zeisig BB, Popov N, Huijbers I, Barnes J, et al. Functional crosstalk between Bmi1 and MLL/Hoxa9 axis in establishment of normal hematopoietic and leukemic stem cells. Cell Stem Cell (2011) 8:649–62. doi:10.1016/j.stem.2011.05.004
218. Yuan J, Takeuchi M, Negishi M, Oguro H, Ichikawa H, Iwama A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia (2011) 25:1335–43. doi:10.1038/leu.2011.85
219. Rossi A, Ferrari KJ, Piunti A, Jammula S, Chiacchiera F, Mazzarella L, et al. Maintenance of leukemic cell identity by the activity of the Polycomb complex PRC1 in mice. Sci Adv (2016) 2(10):e1600972. doi:10.1126/sciadv.1600972
220. Maethner E, Garcia-Cuellar MP, Breitinger C, Takacova S, Divoky V, Hess JL, et al. MLL-ENL inhibits polycomb repressive complex 1 to achieve efficient transformation of hematopoietic cells. Cell Rep (2013) 3:1553–66. doi:10.1016/j.celrep.2013.03.038
221. Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A (2012) 109(13):5028–33. doi:10.1073/pnas.1202258109
222. Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol (2013) 9:643–50. doi:10.1038/nchembio.1331
223. Danis E, Yamauchi T, Echanique K, Haladyna J, Kalkur R, Riedel S, et al. Inactivation of Eed impedes MLL-AF9-mediated leukemogenesis through Cdkn2a-dependent and Cdkn2a-independent mechanisms in a murine model. Exp Hematol (2015) 43:930–5. doi:10.1016/j.exphem.2015.06.005
224. Ueda K, Yoshimi A, Kagoya Y, Nishikawa S, Marquez VE, Nakagawa M, et al. Inhibition of histone methyltransferase EZH2 depletes leukemia stem cell of mixed lineage leukemia fusion leukemia through upregulation of p16. Cancer Sci (2014) 105:512–9. doi:10.1111/cas.12386
225. Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood (2015) 125(2):346–57. doi:10.1182/blood-2014-06-581082
226. Urtishak KA, Edwards AYZ, Wang LS, Hudome A, Robinson BW, Barrett JS, et al. Potent obatoclax cytotoxicity and activation of triple death mode killing across infant acute lymphoblastic leukemia. Blood (2013) 121:2689–703. doi:10.1182/blood-2012-04-425033
227. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med (2013) 19:202–8. doi:10.1038/nm.3048
228. Souers AJ. Phase I study of ABT-199 (GDC-0199) in patients with relapsed/refractory non-Hodgkin lymphoma: responses observed in diffuse large B-cell (DLBCL) and follicular lymphoma (FL) at higher cohort doses. Clin Adv Hematol Oncol (2014) 12(8 Suppl 16):18–9.
229. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med (2016) 374:311–22. doi:10.1056/NEJMoa1513257
230. Khaw SL, Suryani S, Evans K, Richmond J, Robbins A, Kurmasheva RT, et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood (2016) 128(10):1382–95. doi:10.1182/blood-2016-03-707414
231. van der Linden MH, Willekes M, van Roon E, Seslija L, Schneider P, Pieters R, et al. MLL fusion-driven activation of CDK6 potentiates proliferation in MLL-rearranged infant ALL. Cell Cycle (2014) 13(5):834–44. doi:10.4161/cc.27757
232. Placke T, Faber K, Nonami A, Putwain SL, Salih HR, Heidel FH, et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood (2014) 124(1):13–23. doi:10.1182/blood-2014-02-558114
233. Maillard I, Chen YX, Friedman A, Yang Y, Tubbs AT, Shestova O, et al. Menin regulates the function of hematopoietic stem cells and lymphoid progenitors. Blood (2009) 113:1661–9. doi:10.1182/blood-2009-01-135012
234. Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol (2012) 8(3):277–84. doi:10.1038/nchembio.773
235. Borkin D, Pollock J, Kempinska K, Purohit T, Li X, Wen B, et al. Property focused structure-based optimization of small molecule inhibitors of the protein-protein interaction between Menin and mixed-lineage leukemia (MLL). J Med Chem (2016) 59(3):892–913. doi:10.1021/acs.jmedchem.5b01305
236. He S, Malik B, Borkin D, Miao H, Shukla S, Kempinska K, et al. Menin-MLL inhibitors block oncogenic transformation by MLL-fusion partners in a fusion partner-independent manner. Leukemia (2016) 30(2):508–13. doi:10.1038/leu.2015.144
237. Xu Y, Yue L, Wang Y, Xing J, Chen Z, Shi Z, et al. Discovery of novel inhibitors targeting the Menin-mixed lineage leukemia interface using pharmacophore- and docking-based virtual screening. J Chem Inf Model (2016) 56(9):1847–55. doi:10.1021/acs.jcim.6b00185
Keywords: mixed lineage leukemia, infant leukemia, epigenetics, targeted inhibitor, chemotherapy, HSCT
Citation: Winters AC and Bernt KM (2017) MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 5:4. doi: 10.3389/fped.2017.00004
Received: 09 November 2016; Accepted: 09 January 2017;
Published: 09 February 2017
Edited by:
Alex Kentsis, Memorial Sloan Kettering Cancer Center, USAReviewed by:
Yana Pikman, Dana-Farber Cancer Institute, USALeo Wang, City of Hope National Medical Center, USA
Copyright: © 2017 Winters and Bernt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathrin M. Bernt, berntk@email.chop.edu