 Virginia Mirra1,2
Virginia Mirra1,2- 1Department of Translational Medical Sciences, Federico II University, Naples, Italy
- 2Department of Pediatrics, Federico II University, Naples, Italy
- 3Department of General Pediatrics, University Children’s Hospital Muenster, Muenster, Germany
Primary ciliary dyskinesia (PCD) is an orphan disease (MIM 244400), autosomal recessive inherited, characterized by motile ciliary dysfunction. The estimated prevalence of PCD is 1:10,000 to 1:20,000 live-born children, but true prevalence could be even higher. PCD is characterized by chronic upper and lower respiratory tract disease, infertility/ectopic pregnancy, and situs anomalies, that occur in ≈50% of PCD patients (Kartagener syndrome), and these may be associated with congenital heart abnormalities. Most patients report a daily year-round wet cough or nose congestion starting in the first year of life. Daily wet cough, associated with recurrent infections exacerbations, results in the development of chronic suppurative lung disease, with localized-to-diffuse bronchiectasis. No diagnostic test is perfect for confirming PCD. Diagnosis can be challenging and relies on a combination of clinical data, nasal nitric oxide levels plus cilia ultrastructure and function analysis. Adjunctive tests include genetic analysis and repeated tests in ciliary culture specimens. There are currently 33 known genes associated with PCD and correlations between genotype and ultrastructural defects have been increasingly demonstrated. Comprehensive genetic testing may hopefully screen young infants before symptoms occur, thus improving survival. Recent surprising advances in PCD genetic designed a novel approach called “gene editing” to restore gene function and normalize ciliary motility, opening up new avenues for treating PCD. Currently, there are no data from randomized clinical trials to support any specific treatment, thus, management strategies are usually extrapolated from cystic fibrosis. The goal of treatment is to prevent exacerbations, slowing the progression of lung disease. The therapeutic mainstay includes airway clearance maneuvers mainly with nebulized hypertonic saline and chest physiotherapy, and prompt and aggressive administration of antibiotics. Standardized care at specialized centers using a multidisciplinary approach that imposes surveillance of lung function and of airway biofilm composition likely improves patients’ outcome. Pediatricians, neonatologists, pulmonologists, and ENT surgeons should maintain high awareness of PCD and refer patients to the specialized center before sustained irreversible lung damage develops. The recent creation of a network of PCD clinical centers, focusing on improving diagnosis and treatment, will hopefully help to improve care and knowledge of PCD patients.
Introduction
Primary ciliary dyskinesia (PCD) is a clinically and genetically heterogeneous group of disorders of ciliary motility (MIM 244400) (1). In most cases of PCD, inheritance is autosomal recessive, but X-linked PCD caused by mutations in RPGR gene, which is responsible for 20% of all cases with retinitis pigmentosa, or also in PIH1D3 gene have been reported (2–4).
History of PCD starts with Kartagener who first described a syndrome that included the triad of chronic sinusitis, bronchiectasis, and situs viscerum inversus (SI) (5). Approximately 40 years later, Afzelius reported on four subjects with recurrent bronchitis and pneumonia associated with recurrent upper airways infections who also had SI in 50% of the cases, then known as Kartagener’s syndrome (6). In that case series, sperm tails and respiratory cilia lacked dynein arms and showed impaired motility. This report clarified that a congenital defect in cilia and sperm tails can result in the association of chronic respiratory tract infections and male sterility, and the term “immotile-cilia syndrome” was eventually coined (7). The term “primary” was used to distinguish this condition from secondary ciliary abnormalities caused by inflammation and infection.
The goal of this review is to provide an update on the genetics, the diagnosis, and current and future treatment of PCD in order to increase the clinicians’ awareness of the disorder and hopefully improve final outcome.
Cilia Biology: Structure and Function
Cilia are hair-like organelles that project from cells. Traditionally, cilia are distinguished into thre classes: primary cilia, which are not motile and are expressed on most cells during development, when they play important roles in sensing and transducing environmental signals (8); nodal cilia, which are found in the embryonic node; and motile cilia, which are long thin protrusions that extend up to 20 mm from the cell surface and propel fluids along surfaces of respiratory epithelium, brain ependyma, and falloppian tubes. Syndromes associated with defects in cilia of either classes are termed ciliopathies (9).
Each ciliated cell has approximately 200 motile cilia projecting from its surface that beat in a coordinated fashion. Motile cilia are dysfunctional in PCD. They are mainly immotile, but stiff, uncoordinated, and/or ineffective ciliary beats have also been reported (10).
Motile cilia are found in the apical surface of the upper and lower respiratory tract, on the ependymal cells that line the ventricles of the central nervous system, in the oviducts of the female reproductive system, and in the flagellum of male spermatozoa (11).
The motile cilium structure is made of nine peripheral doublet microtubules and two central single microtubules (central pair complex) and includes inner and outer dynein arms (ODAs), radial spokes, and nexin links (9 + 2 axonemes). Nexin links connect the nine peripheral doublets which are connected to the central pair by radial spokes. Outer and inner dynein arms (IDAs) are motor proteins that are attached to the outer microtubules providing energy for ciliary movement.
Cilia play a fundamental role in mucociliary clearance. Ciliary ultrastructure and orientation are critical for enhancing clearance of the lower respiratory tract as they help move fluids, mucus, and inhaled foreign materials vectorially from distal to more proximal airways. In normal airways, cilia beat with a rapid frequency that ranges from approximately 8–20 Hz and mobilizes the mucus that sits atop the cilia (12).
During embryogenesis, the motile 9 + 0 monocilia generate a whirling, rotational movement that directs leftward flow of extracellular fluid (nodal flow). The nodal cilia play a vital role in establishing left–right body orientation, and abnormalities can lead to laterality defects that include SI and a spectrum of situs ambiguous condition, that may be also associated with congenital heart abnormalities (13). The association of cilia dysfunction and SI, formerly described as Kartagener syndrome (5), may occur even in less than 50% of all PCD as some defects, in particular those associated with mutations in HYDIN, RSPH9, RSPH4A, and RSPH1 genes, do not cause SI (14).
Sperm flagella and motile cilia have a similar, although not identical, axonemal structure, which might explain why sperm flagella dyskinesia is often, but not necessarily, associated with PCD and vice versa (15).
Epidemiology
In 2010, Kuehni et al. conducted the largest international survey of pediatric PCD patients ever undertaken, which included 1,192 patients from 26 European countries (16). They concluded that the prevalence of diagnosis ranged from 1:10,000 to 1:20,000 live-born children. Actually, PCD prevalence shows large variations, with estimates ranging from 1:2,200 to 1:40,000 due to different methods of analysis (17, 18). The highest prevalence was reported in Cyprus, Switzerland, and Denmark. The wide variation of doctor-diagnoses in different countries is likely due, at least in part, to geographic differences in mutational data, founder effects for certain gene mutations, high proportions of consanguineous marriages, or to differences in the diagnostic work-up of PCD among the participating countries.
Diagnosis of PCD may be delayed or missed completely, due to lack of awareness and/or difficulties in confirming it (19). In Europe, median age at diagnosis is 5.3 years, with cases with SI being confirmed as PCD at significantly lower age than those without (3.5 years versus 5.8 years) (16). Many patients may also experience a extraordinarily high number of physicians visits (50–100) before PCD is confirmed, thus indicating that also in specialized centers the awareness of the disorder may be poor (20).
Registries of patients with rare disorders are increasingly recognized as crucial tools to achieve a collection of phenotypic data, to understand the pathophysiology of the underlying condition, and to facilitate multicentre collection of data for research studies. In order to systematically collect data on PCD incidence, clinical presentation, and treatment, a registry was launched in January 2014, that provides epidemiological data and clinical information of 201 patients with PCD from several European and North-American centers (21).
Disease Manifestations
At all ages, the clinical phenotype of PCD is very wide (Figure 1). Respiratory manifestations are part of the classic description of the disease and are considered “sine qua non” features for the diagnosis. Main PCD manifestations include recurrent to chronic upper and lower respiratory tract infections that eventually complicate with bronchiectasis at older ages. Most symptoms occur on a chronic, daily basis and start soon after birth (1). Unfortunately, most of the symptoms or signs of PCD upper and lower airway disease are very common also in healthy children, and this is why the diagnosis is often made beyond infancy or childhood, with delayed start of follow-up and/or adequate treatment (19, 22).
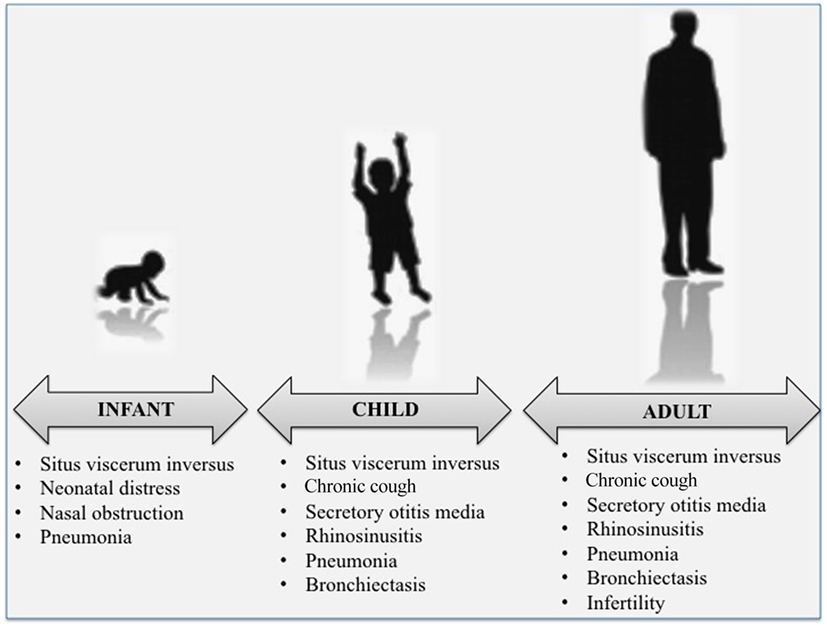
Figure 1. The classical clinical phenotypes of primary ciliary dyskinesia at various ages.
Unexplained neonatal respiratory distress is a possible manifestation of PCD. Transient tachypnea of the newborn, starting soon after birth, is a well-known cause of respiratory distress in term and near-term newborns, and resolution by the fifth day of life is generally reported (23). Conversely, more than 75% of full-term neonates with PCD require continuous supplemental oxygen for days to weeks (24). The most stricking finding of a recent study was that pneumonia and multiple lobar collapse that require prolonged hospital stay may be very severe in newborns who will be later confirmed as PCD (25).
Persistent nasal obstruction is very common at all ages, as children and adults refer a daily year-round nose congestion evident yet in the neonatal period or in the first years of life (26, 27).
Chronic rhinorrhea complicated by anosmia, associated with recurrent secretory type otitis (glue ear), occur in 76–100% of PCD children (19, 28) and may lead to sleep-disordered breathing (29, 30). Chronic rhinosinusitis is frequently associated with hypoplastic frontal and sphenoid sinuses (31). Recurrent otitis media is a troublesome complaint in many PCD patients, with as much as 38% of the cases requiring more than 30 antibiotic courses in their life (20).
Lower airways are commonly involved in PCD.
In preschool and school-age children daily wet cough due to repeated episodes of bronchitis and/or recurrent pneumonia is a universal finding (32), that may result in the development of chronic obstructive suppurative lung disease, with localized-to-diffuse bronchiectasis (1). The underlying cause of bronchiectasis was PCD in 1–17% of several pediatric case series (33–35). Although the development of bronchiectasis increases with aging (28), it has been reported even in toddlers with PCD (36). High-resolution computed tomography (HRCT) is a highly sensitive imaging modality for investigating PCD lung disease, and in particular to detect bronchiectasis (35, 37, 38) (Figure 2). However, HRCT involves larger radiation doses than the conventional X-ray procedure, and therefore its use in the follow-up of pediatric chronic lung disorders is controversial (39). Chest magnetic resonance imaging may be a valid alternative with a good-to-excellent agreement with HRCT findings (40, 41). In addition to wet cough and bronchiectasis, chronic asthma, generally unresponsive to maintenance treatment, is frequently reported at school-age and during adolescence (42). A mild to moderate obstructive pattern is a common finding at spirometry. Possible pathological changes mainly include bronchial obstruction, and altered lung mechanics secondary to repeated endobronchial infection (43).
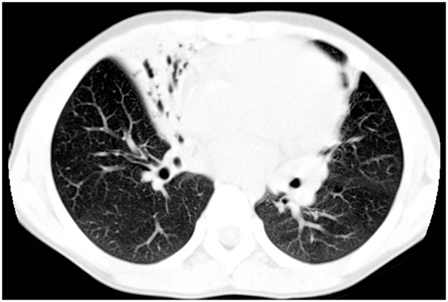
Figure 2. High-resolution computed tomography findings from a 7-year-old girl with primary ciliary dyskinesia. The scan demonstrates an area of consolidation both in the lingula and in the middle lobe, the latter also including bronchiectasis.
Sputum cultures tipically yield oropharyngeal flora including Haemophilus influenzae, Streptococcus pneumoniae, and Staphylococcus aureus in the early childhood, while Pseudomonas aeruginosa (first smooth and then mucoid) and other Gram-negative pathogens such as Klebsiella species are prevalent in older PCD (44). Actually, mucoid P. aeruginosa may be found in 5% of PCD patients younger than 19 years (45). Although rare in childhood, non-tuberculous mycobacteria are reported in more than 10% of PCD adults (28).
As a consequence of abnormal sperm structure some, but not all, male patients with PCD have fertility problems (46). Sperm flagellum is a type of cilia. Therefore, abnormal ciliary structure may lead to the reduction or loss of the ability of the flagellum to swing, causing ultimately male infertility (7, 47). The most frequent ultrastructural defects of the sperm flagella are missing dynein arms, microtubular translocations, and lack of radial spokes (48). Having immotile sperm is common among affected males, and spontaneous pregnancy is rarely achieved unless through artificial insemination, including in vitro fertilization and intracytoplasmic sperm injection (49). For this reason, genetic counseling to couples pursuing assisted reproductive technology is mandatory, and genetic assessment of sperm is highly recommended prior to any clinical action. Females with PCD may experience an increased rate of ectopic pregnancy and decreased fertilization ability, because of impaired ciliary function in the oviduct (50, 51).
Situs inversus totalis is present in 50% of individuals with PCD (6). Heterotaxy, defined as an abnormality where the internal thoracoabdominal organs demonstrate abnormal arrangement across the left–right axis of the body is described in approximately 6% of the cases (52). Patients with heterotaxy may also have complex cardiac defects such as double outlet right ventricle, atrioventricular canal defects, atrial and ventricular septal defects, L-transposition of the great arteries, and tetralogy of Fallot (52, 53). The respiratory phenotypes of the PCD patients with heterotaxy are not different than those without heterotaxy (28). Other conditions as complex congenital heart disease, polycystic kidney and liver disease, hydrocephalus, biliary atresia, severe esophageal disease (esophageal atresia, severe reflux), and retinal degeneration, including retinitis pigmentosa, could also be associated in patients with PCD (54).
Data on growth of PCD patients are controversial. Chronic respiratory disease and long-term inflammation decrease IGF-I levels and compromise children’s growth, as demonstrated in cystic fibrosis (CF) (55, 56). At present, few studies that investigated growth in PCD using national and international reference values show conflicting results, with some suggesting impaired growth (57–59), and others reporting no differences (45, 60).
Despite it is well known that micronutrients and vitamins play a role in respiratory infections, data on their contribution in the inception or maintenance of PCD-associated airway infections are very scarce. Children and adults with stable PCD have deficient-to-insufficient serum vitamin D levels (61). Since, vitamin D has immunomodulatory properties and its deficiency may contribute to an increased risk of respiratory infections in PCD, studies aimed to evaluate the efficacy of vitamin D supplementation on the rate or severity of PCD infections exacerbations should be proposed at a multicenter level.
Compared to CF, the natural history of PCD lung disease is much less clear. Information on PCD disease progression is still incomplete, even though the mortality data are hard to interpret, as they are not age standardized. A recent retrospective study of 151 PCD adults with a median age of 35 years longitudinally followed for 7 years found an incidence of all-cause mortality of nearly 5%, and a respiratory mortality of 3.3% (62). Authors showed that older age at diagnosis was associated with impaired baseline FEV1 and increased P. aeruginosa colonization. Lung function decline, estimated at FEV1 decline of 0.49% pred per year, was positively associated with ciliary ultrastructure abnormalities, mainly microtubular defects (62).
The severity of lung disease in adults with PCD is highly variable, but is generally milder than in CF (14). However, a progressive course of PCD pulmonary disease is possible in mid-adulthood, with some patients developing an end-stage lung disease who may eventually require lung transplantation (28). Early studies have suggested relatively stable lung disease, in the absence of significant lung function decline (63, 64). Conversely, a recent study showed that only 57% of PCD patients followed at a single center over 5–30 years have a stable FEV1 and that lung function may progressively decline in approximately one-third of these (65). This finding has been recently confirmed by Werner et al. who documented progressive decline of FEV1 in 71 PCD cases from the international registry (21). Surprisingly, early referral to a PCD center may not be associated with better spirometry (60).
Traditionally, pulmonary function testing is the best non-invasive way of tracking the progression of the disease in chronic lung disorders, also including PCD. Spirometry results correlate with lung structure changes at HRCT, but the latter may progress despite little or no change in lung function (66). In recent years, there has been increasing focus on the lung clearance index (LCI), a measure of ventilation inhomogeneity that appears more sensitive than FEV1 in detecting early airway disease (67). Nevertheless, data in PCD on the relationship among LCI, spirometry, and lung structure changes at HRCT are conflicting (68, 69), and further investigation should be provided to clarify the role of LCI in the medium- to long-term progression of the disease.
Diagnosis
A complete diagnostic work-up of PCD is mandatory if a positive family history of PCD is reported, and the latter can account up to 10% of all PCD diagnoses (19). Siblings of probands should have PCD excluded, particularly if they exhibit mild respiratory features that may not indicate PCD (1).
A 7-point questionnaire-based prediction tool (PICADAR) has been recently developed to predict the likelihood that a patient referred for evaluation of persistent wet cough has PCD (70). Authors proposed a final score that includes seven predictive variables, such as full-term gestational age, admittance to a neonatal unit, neonatal chest symptoms, persistent perennial rhinitis, chronic ear and hearing symptoms, situs abnormalities, and presence of a cardiac defect. Patients with a PICADAR score ≥10 have more than 90% probability of testing positive for PCD, while a score ≥5 indicates more than 11% chances of being diagnosed as PCD.
There is no single gold standard diagnostic test for PCD. Current diagnosis requires a combination of technically demanding investigations, including nasal nitric oxide (nNO), high-speed video microscopy analysis (HVMA), and transmission electron microscopy (TEM) (71).
Among the earliest diagnostic tests for PCD evaluation, the saccharine test and the investigation of mucociliary clearance by a radioactive tracer have been long used either inside or outside of specialized centers to demonstrate that mucociliary transport is impaired as a result of abnormal ciliary motion (1). However, the saccharin test may miss cases with dyskinetically beating cilia and the radioaerosol mucociliary clearance techniques are associated with radiation exposure albeit quite low. Therefore, the evidence appears too limited to recommend them (71).
Measurement of nNO is a helpful tool for screening PCD. Its levels are extremely low in PCD compared to healthy and disease controls (72). Possible explanation include a reduced biosynthesis of NO by paranasal sinuses or a possible increased consumption by superoxide anions or, alternatively, a sequestration in the upper respiratory tract within blocked paranasal sinuses or, finally, its biosynthesis or storage capacity is limited due to agenesis of the sinuses (73). nNO measurement should be used as part of the diagnostic work-up of schoolchildren over 6 years and of adults suspected of having PCD, preferably using a chemiluminescence analyzer and the velum closure technique, that achieves palate closure by exhaling through the mouth into a disposable resistor (74). This test is sensitive, rapid, non-invasive, and results are immediately available. Unfortunately, standardized methods to measure nNO are not appropriate for younger children, precisely the age group that would need urgent targeting for diagnostic measurement. In preschool children, nNO should be preferably measured using tidal breathing. Available data suggest that measurements of nNO correlate well with the values obtained at the plateau, but values are lower (71). The limitation is that breath-hold with velum maneuver can be difficult to obtain particularly by young children, and there is mounting evidence that simpler measurements, such as breath-hold without velum closure or sampling during tidal breathing, can discriminate between PCD and non-PCD also in younger children (75, 76). Diagnostic cutoff values for tidal techniques from preschool children are not currently available (77).
In older children, nNO analysis includes multiple methods of measurement and different cutoff values, making it difficult to provide definite thresholds in that age range. It has been reported that nNO cutoff value less than 77 nl/min strongly suggest PCD, with 98% sensitivity and 99% specificity (78). However, cases with demonstrated PCD may exhibit normal or even raised nNO levels (79). This indicates that patients with high clinical suspicion of PCD should be evaluated by additional diagnostic procedures other than nNO measurement (71).
Measurement of nNO can be obtained by stationary or handheld devices. Stationary devices are very commonly used, but are expensive and need frequent technical assistance (78). A handheld device simple to use and cheap has been developed, and a study found no difference between nNO obtained from stationary or handheld analyzer during silent and humming exhalation (80). A portable device equipped also for nNO analysis through the aspiration method is currently available (81, 82), but more experience is needed to validate its use.
Historically, a PCD diagnosis was based on analysis at TEM of ciliary cross sections from a nasal respiratory epithelium sample (83). This is usually obtained from the inferior turbinate of the nose by brush or curette biopsy or from the lower respiratory tract during bronchoscopy. Nasal brushing represents an elegant, simple, well-tolerated, and only minimally invasive way to collect the ciliated epithelium (84). The sample is chemically fixed with glutaraldehyde, processed, and cilia are analyzed using a transmission electron microscope (83, 85). Examination of the ciliary ultrastructure by electron microscopy remains a definitive diagnostic test for PCD (71). Nevertheless, TEM analysis can confirm but does not always exclude the diagnosis (86). It usually allows to identify PCD variants exhibiting a complete or partial absence of ODAs, combined ODA and IDA defects, and microtubular disorganization defects. Figure 3 shows cilia ultrastructure from a PCD patient, compared to the normal ultrastructure from a healthy subject. It has been reported that TEM fails to identify at least 30% of all PCD variants (57, 87), such as the nexin link (88, 89) or the central pair components defects (90), or those associated with DNAH11 mutations (80, 91, 92). Moreover, only a subset of radial spokes defects are identified by TEM (93), while false-positive diagnosis has been reported in cases with isolated IDA (94). Importantly, a number of ciliary abnormalities including absence of the central microtubular pair, disorientation of the cilia, and disarrangement of microtubules may be secondary to infection or inflammation as well (1). As secondary defects are absent after ciliogenesis in culture, this procedure has been recommended in order to distinguish between primay and secondary cilia defects (71). Finally, further diagnostic investigations should be performed in all cases with normal ultrastructure if the clinical history strongly suggests PCD (91).
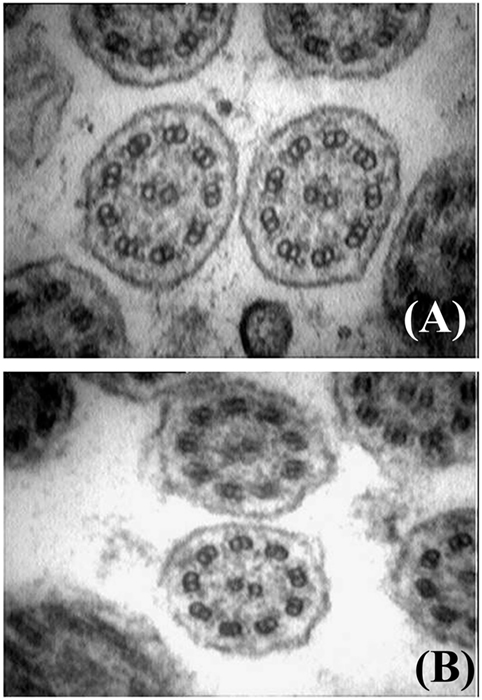
Figure 3. Electron microscopy findings showing normal cilia ultrastructure from an healthy subject (A), and outer and inner dynein arms defect from a patient with primary ciliary dyskinesia (B) (courtesy of Dr. Mariarosaria Cervasio, Department of Advanced Biomedical Sciences, Anatomo-Pathology Unit, Federico II University, Naples, Italy).
Direct visualization of ciliary beat pattern (CBP) and frequency (CBF) by HVMA should be used as part of the diagnostic work-up of patients suspected of having PCD and in order to improve diagnostic accuracy of HVMA, CBF/CBP assessment should be repeated after air–liquid interface culture (71). CBF should not be used without assessment of CBP in diagnosing PCD. HVMA protocols differ among centers in many respects including sampling techniques, microscopes and cameras, temperature during analysis, software, and evaluation criteria. Videos are recorded using a digital high-speed video camera attached to an inverted phase-contrast microscope. Digital image sampling was performed at 120–150 frames per second (fps) and a 640 × 480 pixel resolution. A CBF of less than 11 beats per second (<11 Hz) has been suggested as a cutoff value, with only those with lower beat frequency proceeding to EM (95). On the other hand, HVMA is not sufficiently standardized to rule in or rule out PCD in isolation.
High-resolution immunofluorescence (IF) analysis is an emerging tool to investigate the subcellular localization of ciliary proteins in respiratory epithelia (96) (Figure 4). It reliably identifies all ultrastructural abnormalities which are detectable by TEM (96–98), and additionally abnormalities of nexin links components (89) and radial spoke head proteins (10, 99–101). This technology has been adopted by an increasing number of laboratories and it is likely that further development will allow to recognize an increasing number of PCD variants.
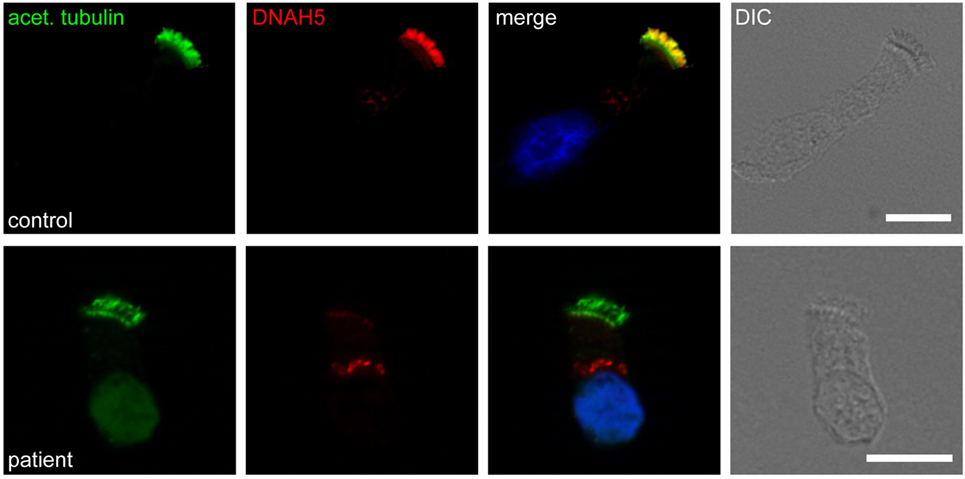
Figure 4. Immunofluorescence staining of human respiratory epithelial cells with DNAH5-specific antibodies (red) and antibodies against acetylated α-tubulin (green). Nuclei were stained with Hoechst 33342 (blue). Overlays and bright-field images are shown on the right. Whereas in healthy human respiratory epithelial cells (control; upper panel), both antibodies colocalize along the entire length of the ciliary axonemes, in an individual with an outer dynein arm defect (patient; lower panel), DNAH5 is absent.
Genetics
Most PCD variants follow an autosomal recessive inheritance trait. The number of genes associated with PCD is still growing rapidly (Table 1).1 Some mutations leading to PCD are loss-of-function variants (102). Missense mutations can be found in a minority of cases. In these instances, it is often difficult to distinguish disease-causing mutations from rare polymorphisms. Most mutations are private. Clustering of mutations in specific genetic regions, as it is known from other genetic disorders, is less common. There is a good correlation between specific genetic mutations and their TEM, IF, and video microscopic phenotype (10). To date, only preliminary data have been published correlating genetic findings with distinct clinical phenotypes. Mutations in genes affecting central pair or radial spoke components (RSPH1, RSPH3, RSPH4A, RSPH9) as well as genes involved in the generation of multiple motile cilia (MCIDAS, CCNO) do not result in randomization of left/right body asymmetry. Therefore, affected individuals of those disease variants do not display situs abnormalities. This feature can be explained by the physiologic absence of the central pairs in the motile monocilia of the embryonic node. Patients with mutations in RSPH1 are reported to exhibit a milder clinical course (92); males with PCD due to CCDC114 mutations do not suffer from sperm immotility and therefore are not infertile (103). Subjects with reduced generation of multiple motile cilia are likely to have a more severe respiratory disease phenotype with lung failure at younger age (104, 105). Finally, a study has showed that lung disease is heterogeneous across all ultrastructural and genotype groups in 118 PCD patients from North America, but worse in those with biallelic mutations in CCDC39 or CDCC40 (45).
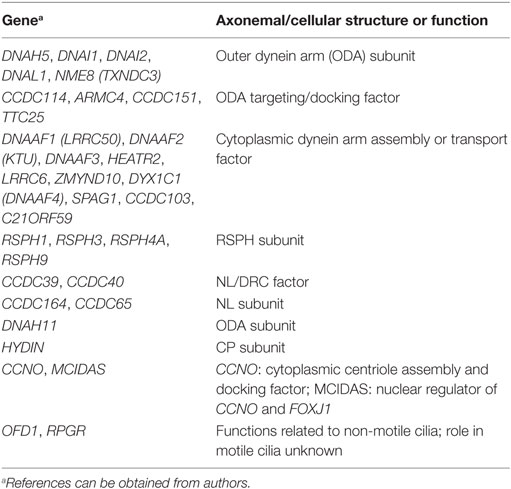
Table 1. Genes associated with primary ciliary dyskinesia and corresponding ultrastructure.
With the support of modern high-throughput genetic technologies, it is possible to identify disease-causing biallelic mutations in ~70% of affected individuals. However, given the complexity of diagnosing PCD using multiple different and repetitive tests, next-generation sequencing is a cost-efficient and effective diagnostic approach in many instances. It is very likely that further advances in PCD molecular genetics will continue to facilitate early diagnosis and treatment.
Current and Future Treatment Strategies
Currently, therapeutic strategies of PCD are not based on validated disease-specific recommendations. Usually, patients are treated according expert opinion or to available evidence for CF, despite differences in the pathophysiology of the two disorders are evident.
The mainstay of treatment for PCD involves airway clearance, infection control and prevention, and the elimination of exposure to inflammatory triggers, also including passive smoke.
Different techniques guarantee airway clearance, including manual chest physiotherapy, postural drainage, autogenic drainage, active cycle breathing, and exercise (106).
Chest physiotherapists conduct deep breathing exercises, such as postural drainage combined with percussion and vibration and forced expirations, but the exclusive need for technical assistance may be time-consuming or uncomfortable (107). In addition to forced cough and breathing techniques, a variety of manual devices also exists that aid patients in improving mucus clearance. These include positive expiratory pressure (PEP) valves, and mouthpiece or chest wall oscillating devices (108). PEP devices, which give a constant back pressure to the airways during expiration, provide a pressure behind the mucus that push it out of the lungs and is widely used also in CF patients (109). There is no clear evidence that PEP is a more or less effective intervention than other forms of physiotherapy (110). High-frequency chest wall oscillation involves an inflatable vest that is attached to a machine, generating extrathoracic oscillations at variable frequencies and intensities, which are transmitted to the airways, promoting coughs or huffs (107). In conclusion, irrespective of the chosen modality and despite the lack of evidence-based comparisons of the various techniques, routine daily physiotherapy is strongly recommended in PCD (71).
Physical exercise should be prescribed to all subjects with obstructive pulmonary disease for improving respiratory muscle strength and maintaining lung health. It has been reported that a high proportion of PCD cases (79%) have limitations in performing vigorous activities, and approximately 50% spend less than 3 h per week doing physical activity, thus suggesting that PCDs are quite inactive (61). Performing exercise prior to airway clearance may more significantly enhance mucociliary clearance and is more effective as bronchodilator stimulus than β2-agonists drugs (111). Actually, compared to healthy individuals, patients with PCD have also significantly lower peak oxygen uptake measured by cicloergometry (112, 113).
Nebulized inhalation is a common procedure to help moisten and dilute viscous airway secretions, and thereby facilitates mucoclearance techniques (114). Inhaled hypertonic saline is used in the treatment of bronchiectasis for enhancing mucociliary clearance (115). In a randomized controlled trial of 7% hypertonic saline versus isotonic saline, adults with non-CF bronchiectasis reported increased ease of expectoration, reduction in antibiotics use and emergency health care visits over a 3-month period (116). Conversely, no significant change of spirometry, as well of sputum colonization or quality of life, was reported (117). A recent randomized controlled study of a small sample of adult PCD patients treated with inhaled hypertonic saline for 12 weeks neither improved quality of life nor significantly affected spirometry or airway inflammation (118). Further, larger studies also including children are needed to confirm these results.
During infections, DNA and actin released by neutrophils accumulation increase sputum viscosity in the airways. Recombinant human DNase I (rhDNase) cleaves extracellular DNA, decreasing the DNA concentration, and thereby decreasing sputum viscosity (119–121). Inhaled rhDNase improves FEV1 percent predicted in CF patients, and CF physicians often recommend it in their clinical practice (122). Neutrophilic airway inflammation has been reported in PCD (123). Until now, few PCD studies have showed significant clinical benefits of a trial with inhaled DNase (124–126). At present, rhDNase is not recommended in PCD, and larger studies are needed to confirm its efficacy in PCD. Uridine-59-triphosphate (UTP) may enhance clearance during cough stimulating chloride− secretion and mucin releasing by goblet cells. Several years ago, a small study demonstrated that aerosolized UTP improves whole lung clearance measured by gamma scintigraphy during forced cough in 12 adolescents and adults with PCD, without any adverse effects (127). Unfortunately, no further studies were published on this issue. Mannitol also affects mucociliary clearance and is often prescribed to CF patients because it creates an osmotic drive for water to move into the airway and hydrate secretions (128). Data that sustain inhaled mannitol in PCD are lacking. Mannitol 400 mg inhaled twice daily for 12 months in adults with clinically significant non-CF bronchiectasis did not reduce exacerbation rates, but quality of life significantly improved (129). These findings indicate that a randomized clinical trial of inhaled mannitol might be proposed to PCD patients as well.
All patients with PCD should have routine clinical visits for spirometry monitoring and respiratory culture surveillance through sputum or oropharyngeal cultures (130). At any age, a minimum of two to four visits per year are recommended (131), and in case of respiratory exacerbations, antibiotics selected should be prescribed accordingly to culture history and microbial sensitivity.
Studies of CF and non-CF bronchiectasis also including some patients with PCD have demonstrated that systemic antibiotics are effective at treating “exacerbations” of lung disease (132, 133). Either respiratory tract symptoms including changes in cough, sputum production, respiratory rate, and work of breathing, or a decline in FEV1% predicted may be considered as reliable markers of a respiratory exacerbation in PCD. While mild exacerbations may be treated with oral antibiotics and increased aggressive airway clearance, severe or refractory exacerbations may require intravenous antibiotics and inpatient hospitalization. A duration of 14–21 days of antibiotic therapy is recommended in PCD, according to what is reported in CF and non-CF bronchiectasis (134–136). The selection of antibiotics should be made on the basis of the most recent sputum culture results and would take into account the airways colonization history of the individual patient. Macrolides are a class of antibiotics that deserves particular attention by pulmonologists. Macrolides play antibacterial activity at concentrations lower than those required to kill the infecting or colonizing bacteria (137). In addition to this, macrolides anti-inflammatory and immunomodulatory properties are also well recognized (138). Three randomized, double-blind, placebo-controlled studies of non-CF bronchiectasis, also including few cases with PCD demonstrated that azithromycin or erythromycin taken for 6–12 months led to significant decrease in exacerbation rate and reduced the decline in lung function (139–141). A PCD multicenter, double-blind, randomized, placebo-controlled trial is currently evaluating the efficacy of oral azithromycin administered three times a week for 6 months on the frequency of respiratory infectious exacerbations (142). Results will hopefully clarify whether macrolides may play a role also in PCD.
Cycled or regular inhaled or oral antibiotics may be a treatment option in patients with moderate to severe lung disease that fail eradication strategies and continue to be symptomatic. Despite there are no published studies, inhaled antibiotics are also an option for PCD respiratory exacerbations, but these are usually reserved for patients with P. aeruginosa infection. The use of inhaled tobramycin (300 mg nebulized twice daily) for a 28-day period should be considered upon the first evidence of P. aeruginosa growth (143).
Pulmonary surgical resection (i.e., segmentectomy or lobectomy) may be considered with caution in the presence of diffuse lung disease and can be considered only when a disproportionately burdened region of the lung has failed medical management of bronchiectasis, and there is a significant decline in patient’s health due for instance to severe hemoptysis.
If end-stage lung disease develops, lung transplantation may be an option in PCD. Particular attention must be payed in lung transplant evaluation of patients with PCD as situs abnormalities may pose a barrier in donor lung selection and require advanced surgical planning (144, 145).
The management of PCD ear and nose disease does not differ from that of the lung disease. Close follow-up also of ear–nose–throat district may help undoubtedly to avoid local or systemic complications. Recurrent or persistent otitis media with effusion may lead to chronic otitis media and hearing loss, and frequent use of antibiotics or even middle ear surgery may be ultimately decided (146). Whether or not tympanostomy tube placement may improve hearing loss is controversial, as it significantly increased the risk of chronic otorrhea and infection (147). Patients with sinus disease refractory to medical management may benefit from endoscopic sinus surgery (148).
It is also critical to avoid exposure to inflammatory triggers such as tobacco smoke, and therefore, patients and their family members should receive smoking cessation counseling.
Infection prevention is strongly recommended in PCD, as in all chronic respiratory diseases. Children and adults with PCD have increased risk for pneumococcal disease (148), and therefore, CV13 vaccinations are recommended followed by PPSV23 vaccination. Influenza vaccines are also recommended on an annual basis (149), and additional vaccinations are recommended as per the routine schedules of patients’ geographic regions of treatment.
As far as the future, an improved understanding of the underlying genetics and phenotyping of PCD will also hopefully lead to novel therapeutic strategies. A great expectation has originated from the recent study by Pifferi et al. who first applied the “gene editing” to PCD; thus, they restored DNAH11 gene function ex vivo by replacing the inactivating mutation with wild-type sequence in the diseased cell (150). A new exciting era is cheerfully rising from genetic studies that will result in improving the outcome of affected patients.
The optimal integration of multiple skills in an enlarged team, which hopefully includes pediatricians, pulmonologists, chest physiotherapists, geneticists, biologists, cardiologists, radiologists, andrologists, and ENT surgeons, is essential to provide the most appropriate care to children and adults with PCD. All these specialists do have the unique opportunity to play an integrated role in the multidisciplinary approach to the disease.
Psychological Issues
Physicians who take care of children with PCD should take into valuable account the psychosocial impact of the disease. PCD leads to chronic respiratory symptoms and progressive loss of lung function, and this has a great impact on patients’ health and on style and quality of life of their families (151). As a chronic disorder, PCD is a stressful condition particular during adolescence and young adulthood because of the psychological effects of the chronic burden on the intrafamiliar relationships. Fortunately, patients who are diagnosed early, and hence receive more treatment for their condition, have better clinical outcome (152).
Ethics Statement
The authors declare that written informed consent was obtained by the patients or healthy controls or their legal guardians for images to be published.
Author Contributions
VM has made substantial contributions to conception and design, has been involved in drafting the manuscript, and has given final approval of the version to be published. CW has been involved in drafting the manuscript and has given final approval of the version to be published. FS has made substantial contributions to conception and design, has been involved in drafting the manuscript and revising it critically for important intellectual content, and has given final approval of the version to be published.
Conflict of Interest Statement
The research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors of this manuscript are participants in BEAT-PCD (COST Action 1407).
Footnote
- ^Available from: https://www.omim.org/entry/244400 (2017).
References
1. Shapiro AJ, Zariwala MA, Ferkol T, Davis SD, Sagel SD, Dell SD, et al. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr Pulmonol (2016) 51:115–32. doi: 10.1002/ppul.23304
2. Boaretto F, Snijders D, Salvoro C, Spalletta A, Mostacciuolo ML, Collura M, et al. Diagnosis of primary ciliary dyskinesia by a targeted next-generation sequencing panel: molecular and clinical findings in italian patients. J Mol Diagn (2016) 18:912–22. doi:10.1016/j.jmoldx.2016.07.002
3. Moore A, Escudier E, Roger G, Tamalet A, Pelosse B, Marlin S, et al. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J Med Genet (2006) 43:326–33. doi:10.1136/jmg.2005.034868
4. Paff T, Loges NT, Aprea I, Wu K, Bakey Z, Haarman EG, et al. Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am J Hum Genet (2017) 100:160–8. doi:10.1016/j.ajhg.2016.11.019
5. Kartagener M. Zur pathogenese der bronkiectasien. Bronkiectasien bei situs viscerum inversus. Beitr Klin Tuberk Spezif Tuberkuloseforsch (1933) 83:489–501. doi:10.1007/BF02141468
6. Afzelius BA. A human syndrome caused by immotile cilia. Science (1976) 193:317–9. doi:10.1126/science.1084576
7. Eliasson R, Mossberg B, Camner P, Afzelius BA. The immotile-cilia syndrome. A congenital ciliary abnormality as an etiologic factor in chronic airway infections and male sterility. N Engl J Med (1977) 297:1–6. doi:10.1056/NEJM197707072970101
8. Zimmerman K, Yoder BK. SnapShot: sensing and signaling by cilia. Cell (2015) 161:692. doi:10.1016/j.cell.2015.04.015
9. Mitchison HM, Valente EM. Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol (2017) 241:294–309. doi:10.1002/path.4843
10. Raidt J, Wallmeier J, Hjeij R, Onnebrink JG, Pennekamp P, Loges NT, et al. Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. Eur Respir J (2014) 44:1579–88. doi:10.1183/09031936.00052014
11. Horani A, Ferkol TW, Dutcher SK, Brody SL. Genetics and biology of primary ciliary dyskinesia. Paediatr Respir Rev (2016) 18:18–24. doi:10.1016/j.prrv.2015.09.001
12. Ferkol TW, Puffenberger EG, Lie H, Helms C, Strauss KA, Bowcock A, et al. Primary ciliary dyskinesia-causing mutations in Amish and Mennonite communities. J Pediatr (2013) 163:383–7. doi:10.1016/j.jpeds.2013.01.061
13. Kennedy MP, Omran H, Leigh MW, Dell S, Morgan L, Molina PL, et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation (2007) 115:2814–21. doi:10.1161/CIRCULATIONAHA.106.649038
14. Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med (2013) 188:913–22. doi:10.1164/rccm.201301-0059CI
15. Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol (2007) 8:880–93. doi:10.1038/nrm2278
16. Kuehni CE, Frischer T, Strippoli MP, Maurer E, Bush A, Nielsen KG, et al. Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. Eur Respir J (2010) 36:1248–58. doi:10.1183/09031936.00001010
17. Lucas JS, Walker WT, Kuehni CE, Lazor R. Primary ciliary dyskinesia. In: Courdier J-F, editor. Orphan Lung Diseases (Vol. 54), Lausanne: ERS Monograph (2011). p. 201–17.
18. O’Callaghan C, Chetcuti P, Moya E. High prevalence of primary ciliary dyskinesia in a British Asian population. Arch Dis Child (2010) 95:51–2. doi:10.1136/adc.2009.158493
19. Coren ME, Meeks M, Morrison I, Buchdahl RM, Bush A. Primary ciliary dyskinesia: age at diagnosis and symptom history. Acta Paediatr (2002) 91:667–9. doi:10.1111/j.1651-2227.2002.tb03299.x
20. Sommer JU, Schäfer K, Omran H, Olbrich H, Wallmeier J, Blum A, et al. ENT manifestations in patients with primary ciliary dyskinesia: prevalence and significance of otorhinolaryngologic co-morbidities. Eur Arch Otorhinolaryngol (2011) 268:383–8. doi:10.1007/s00405-010-1341-9
21. Werner C, Lablans M, Ataian M, Raidt J, Wallmeier J, Große-Onnebrink J, et al. An international registry for primary ciliary dyskinesia. Eur Respir J (2016) 47:849–59. doi:10.1183/13993003.00776-2015
22. Werner C, Onnebrink JG, Omran H. Diagnosis and management of primary ciliary dyskinesia. Cilia (2015) 4:2. doi:10.1186/s13630-014-0011-8
23. Fedakar A, Aydogdu C. Clinical features of neonates treated in the intensive care unit for respiratory distress. Turk J Pediatr (2011) 53:173–9.
24. Ferkol T, Leigh M. Primary ciliary dyskinesia and newborn respiratory distress. Semin Perinatol (2006) 30:335–40. doi:10.1053/j.semperi.2005.11.001
25. Mullowney T, Manson D, Kim R, Stephens D, Shah V, Dell S. Primary ciliary dyskinesia and neonatal respiratory distress. Pediatrics (2014) 134:1160–6. doi:10.1542/peds.2014-0808
26. Fretzayas A, Moustaki M. Clinical spectrum of primary ciliary dyskinesia in childhood. World J Clin Pediatr. (2016) 5:57–62. doi:10.5409/wjcp.v5.i1.57
27. Bush A, Hogg C. Primary ciliary dyskinesia: recent advances in epidemiology, diagnosis, management and relationship with the expanding spectrum of ciliopathy. Expert Rev Respir Med (2012) 6:663–82. doi:10.1586/ers.12.60
28. Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, et al. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med (2004) 169:459–67. doi:10.1164/rccm.200303-365OC
29. Oktem S, Karadag B, Erdem E, Gokdemir Y, Karakoc F, Dagli E, et al. Sleep disordered breathing in patients with primary ciliary dyskinesia. Pediatr Pulmonol (2013) 48:897–903. doi:10.1002/ppul.22710
30. Santamaria F, Esposito M, Montella S, Cantone E, Mollica C, De Stefano S, et al. Sleep disordered breathing and airway disease in primary ciliary dyskinesia. Respirology (2014) 19:570–5. doi:10.1111/resp.12273
31. Pifferi M, Bush A, Caramella D, Di Cicco M, Zangani M, Chinellato I, et al. Agenesis of paranasal sinuses and nasal nitric oxide in primary ciliary dyskinesia. Eur Respir J (2011) 37:566–71. doi:10.1183/09031936.00068810
32. Montella S, Corcione A, Santamaria F. Recurrent pneumonia in children: a reasoned diagnostic approach and a single centre experience. Int J Mol Sci (2017) 18(2):E296. doi:10.3390/ijms18020296
33. Kumar A, Lodha R, Kumar P, Kabra SK. Non-cystic fibrosis bronchiectasis in children: clinical profile, etiology and outcome. Indian Pediatr (2015) 52:35–7. doi:10.1007/s13312-015-0563-8
34. Kim HY, Kwon JW, Seo J, Song YH, Kim BJ, Yu J, et al. Bronchiectasis in children: 10-year experience at a single institution. Allergy Asthma Immunol Res (2011) 3:39–45. doi:10.4168/aair.2011.3.1.39
35. Eastham KM, Fall AJ, Mitchell L, Spencer DA. The need to redefine non-cystic fibrosis bronchiectasis in childhood. Thorax (2004) 59:324–7. doi:10.1136/thx.2003.011577
36. Brown DE, Pittman JE, Leigh MW, Fordham L, Davis SD. Early lung disease in young children with primary ciliary dyskinesia. Pediatr Pulmonol (2008) 43:514–6. doi:10.1002/ppul.20792
37. Kennedy MP, Noone PG, Leigh MW, Zariwala MA, Minnix SL, Knowles MR, et al. High-resolution CT of patients with primary ciliary dyskinesia. AJR Am J Roentgenol (2007) 188:1232–8. doi:10.2214/AJR.06.0965
38. Santamaria F, Montella S, Tiddens HA, Guidi G, Casotti V, Maglione M, et al. Structural and functional lung disease in primary ciliary dyskinesia. Chest (2008) 134:351–7. doi:10.1378/chest.07-2812
39. Brenner DJ, Hall EJ. Computed tomography – an increasing source of radiation exposure. N Engl J Med (2007) 357:2277–84. doi:10.1056/NEJMra072149
40. Montella S, Santamaria F, Salvatore M, Pignata C, Maglione M, Iacotucci P, et al. Assessment of chest high-field magnetic resonance imaging in children and young adults with noncystic fibrosis chronic lung disease: comparison to high-resolution computed tomography and correlation with pulmonary function. Invest Radiol (2009) 44:532–8. doi:10.1097/RLI.0b013e3181b4c1ba
41. Montella S, Maglione M, Bruzzese D, Mollica C, Pignata C, Aloj G, et al. Magnetic resonance imaging is an accurate and reliable method to evaluate non-cystic fibrosis paediatric lung disease. Respirology (2012) 17:87–91. doi:10.1111/j.1440-1843.2011.02067.x
42. Sagel SD, Davis SD, Campisi P, Dell SD. Update of respiratory tract disease in children with primary ciliary dyskinesia. Proc Am Thorac Soc (2011) 8:438–43. doi:10.1513/pats.201103-024SD
43. Patella V, Bocchino M, Steinhilber G. Asthma is associated with increased susceptibility to infection. Minerva Med (2015) 106:1–7.
44. Alanin MC, Nielsen KG, von Buchwald C, Skov M, Aanaes K, Høiby N, et al. A longitudinal study of lung bacterial pathogens in patients with primary ciliary dyskinesia. Clin Microbiol Infect (2015) 21:1093.e1–7. doi:10.1016/j.cmi.2015.08.020
45. Davis SD, Ferkol TW, Rosenfeld M, Lee HS, Dell SD, Sagel SD, et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am J Respir Crit Care Med (2015) 191:316–24. doi:10.1164/rccm.201409-1672OC
46. Munro NC, Currie DC, Lindsay KS, Ryder TA, Rutman A, Dewar A, et al. Fertility in men with primary ciliary dyskinesia presenting with respiratory infection. Thorax (1994) 49:684–7. doi:10.1136/thx.49.7.684
47. Mossberg B, Afzelius BA, Eliasson R, Camner P. On the pathogenesis of obstructive lung disease. Scand J Respir Dis (1978) 59:55–65.
48. Afzelius BA. Genetic and ultrastructural aspects of the immotile-cilia syndrome. Am J Hum Genet (1981) 33:852–64.
49. Yan-Wei S, Ding L, Li P. Management of primary ciliary dyskinesia/Kartagener’s syndrome in infertile male patients and current progress in defining the underlying genetic mechanism. Asian J Androl (2014) 16:101–6. doi:10.4103/1008-682X.122192
51. Halbert SA, Patton DL, Zarutskie PW, Soules MR. Function and structure of cilia in the fallopian tube of an infertile woman with Kartagener’s syndrome. Hum Reprod (1997) 12:55–8. doi:10.1093/humrep/12.1.55
52. Kennedy MP, Ostrowski LE. Primary ciliary dyskinesia and upper airway diseases. Curr Allergy Asthma Rep (2006) 6:513–7. doi:10.1007/s11882-006-0030-7
53. Nakhleh N, Francis R, Giese RA, Tian X, Li Y, Zariwala MA, et al. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation (2012) 125:2232–42. doi:10.1161/CIRCULATIONAHA.111.079780
54. Goutaki M, Meier AB, Halbeisen FS, Lucas JS, Dell SD, Maurer E, et al. Clinical manifestations in primary ciliary dyskinesia: systematic review and meta-analysis. Eur Respir J (2016) 48:1081–95. doi:10.1183/13993003.00736-2016
55. De Benedetti F, Alonzi T, Moretta A, Lazzaro D, Costa P, Poli V, et al. Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I. A model for stunted growth in children with chronic inflammation. J Clin Invest (1997) 99:643–50. doi:10.1172/JCI119207
56. Lebl J, Zahradníková M, Bartosová J, Zemková D, Pechová M, Vávrová V. Insulin-like growth factor-I and insulin-like growth factor-binding protein-3 in cystic fibrosis: a positive effect of antibiotic therapy and hyperalimentation. Acta Paediatr (2001) 90:868–72. doi:10.1111/j.1651-2227.2001.tb02447.x
57. Boon M, Smits A, Cuppens H, Jaspers M, Proesmans M, Dupont LJ, et al. Primary ciliary dyskinesia: critical evaluation of clinical symptoms and diagnosis in patients with normal and abnormal ultrastructure. Orphanet J Rare Dis (2014) 9:11. doi:10.1186/1750-1172-9-11
58. Cohen-Cymberknoh M, Simanovsky N, Hiller N, Gileles Hillel A, Shoseyov D, Kerem E. Differences in disease expression between primary ciliary dyskinesia and cystic fibrosis with and without pancreatic insufficiency. Chest (2014) 145:738–44. doi:10.1378/chest.13-1162
59. Svobodova T, Djakow J, Zemkova D, Cipra A, Pohunek P, Lebl J. Impaired growth during childhood in patients with primary ciliary dyskinesia. Int J Endocrinol (2013) 2013:731423. doi:10.1155/2013/731423
60. Maglione M, Bush A, Nielsen KG, Hogg C, Montella S, Marthin JK, et al. Multicenter analysis of body mass index, lung function, and sputum microbiology in primary ciliary dyskinesia. Pediatr Pulmonol (2014) 49:1243–50. doi:10.1002/ppul.22984
61. Mirra V, Caffarelli C, Maglione M, Valentino R, Perruolo G, Mazzarella C, et al. Hypovitaminosis D: a novel finding in primary ciliary dyskinesia. Ital J Pediatr (2015) 41:14. doi:10.1186/s13052-015-0119-5
62. Shah A, Shoemark A, MacNeill SJ, Bhaludin B, Rogers A, Bilton D, et al. A longitudinal study characterising a large adult primary ciliary dyskinesia population. Eur Respir J (2016) 48:441–50. doi:10.1183/13993003.00209-2016
63. Hellinckx J, Demedts M, De Boeck K. Primary ciliary dyskinesia: evolution of pulmonary function. Eur J Pediatr (1998) 157:422–6. doi:10.1007/s004310050843
64. Ellerman A, Bisgaard H. Longitudinal study of lung function in a cohort of primary ciliary dyskinesia. Eur Respir J (1997) 10:2376–9. doi:10.1183/09031936.97.10102376
65. Marthin JK, Petersen N, Skovgaard LT, Nielsen KG. Lung function in patients with primary ciliary dyskinesia: a cross-sectional and 3-decade longitudinal study. Am J Respir Crit Care Med (2010) 181:1262–8. doi:10.1164/rccm.200811-1731OC
66. Maglione M, Bush A, Montella S, Mollica C, Manna A, Esposito A, et al. Progression of lung disease in primary ciliary dyskinesia: is spirometry less accurate than CT? Pediatr Pulmonol (2012) 47:498–504. doi:10.1002/ppul.21569
67. Fuchs SI, Ellemunter H, Eder J, Mellies U, Grosse-Onnebrink J, Tümmler B, et al. Feasibility and variability of measuring the lung clearance index in a multi-center setting. Pediatr Pulmonol (2012) 47:649–57. doi:10.1002/ppul.21610
68. Boon M, Vermeulen FL, Gysemans W, Proesmans M, Jorissen M, De Boeck K. Lung structure-function correlation in patients with primary ciliary dyskinesia. Thorax (2015) 70:339–45. doi:10.1136/thoraxjnl-2014-206578
69. Irving SJ, Ives A, Davies G, Donovan J, Edey AJ, Gill SS, et al. Lung clearance index and high-resolution computed tomography scores in primary ciliary dyskinesia. Am J Respir Crit Care Med (2013) 188:545–9. doi:10.1164/rccm.201304-0800OC
70. Behan L, Dunn G, Rubbo B, Masefield S, Copeland F, Manion M, et al. Diagnosing primary ciliary dyskinesia; an international patient perspective. Eur Respir J (2016) 48:1096–107. doi:10.1183/13993003.02018-2015
71. Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, et al. European respiratory society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J (2017) 4:49. doi:10.1183/13993003.01090-2016
72. Marthin JK, Nielsen KG. Choice of nasal nitric oxide technique as firstline test for primary ciliary dyskinesia. Eur Respir J (2011) 37:559–65. doi:10.1183/09031936.00032610
73. Walker WT, Jackson CL, Lackie PM, Hogg C, Lucas JS. Nitric oxide in primary ciliary dyskinesia. Eur Respir J (2012) 40:1024–32. doi:10.1183/09031936.00176111
74. American Thoracic Society; European Respiratory Society. ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide. Am J Respir Crit Care Med (2005) 171:912–30. doi:10.1164/rccm.200406-710ST
75. Lucas JS, Walker WT. Nasal nitric oxide is an important test in the diagnostic pathway for primary ciliary dyskinesia. Ann Am Thorac Soc (2013) 10:645–7. doi:10.1513/AnnalsATS.201309-328ED
76. Mateos-Corral D, Coombs R, Grasemann H, Ratjen F, Dell SD. Diagnostic value of nasal nitric oxide measured with non-velum losure techniques for children with primary ciliary dyskinesia. J Pediatr (2011) 159:420–4. doi:10.1016/j.jpeds.2011.03.007
77. Manna A, Montella S, Maniscalco M, Maglione M, Santamaria F. Clinical application of nasal nitric oxide measurement in pediatric airway diseases. Pediatr Pulmonol (2014) 50:85–99. doi:10.1002/ppul.23094
78. Leigh MW, Hazucha MJ, Chawla KK, Baker BR, Shapiro AJ, Brown DE, et al. Genetic disorders of mucociliary clearance consortium. Standardizing nasal nitric oxide measurement as a test for primary ciliary dyskinesia. Ann Am Thorac Soc (2013) 10:574–81. doi:10.1513/AnnalsATS.201305-110OC
79. Fokkens WJ, Lund VJ, Mullol J, Bachert C, Alobid I, Baroody F, et al. European position paper on rhinosinusitis and nasal polyps 2012. Rhinol Suppl (2012) 23:3.
80. Montella S, Alving K, Maniscalco M, Sofia M, De Stefano S, Raia V, et al. Measurement of nasal nitric oxide by hand-held and stationary devices. Eur J Clin Invest (2011) 41:1063–70. doi:10.1111/j.1365-2362.2011.02501.x
81. Marthin JK, Nielsen KG. Hand-held tidal breathing nasal nitric oxide measurement-a promising targeted case-finding tool for the diagnosis of primary ciliary dyskinesia. PLoS One (2013) 8:e57262. doi:10.1371/journal.pone.0057262
82. Abete P, Adlbrecht C, Assimakopoulos SF, Côté N, Dullaart RP, Evsyukova HV, et al. Research update for articles published in EJCI in 2011. Eur J Clin Invest (2013) 43:1097–110. doi:10.1111/eci.12131
83. Sturgess JM, Turner JA. Ultrastructural pathology of cilia in the immotile cilia syndrome. Perspect Pediatr Pathol (1984) 8:133–61.
84. Papon JF, Coste A, Roudot-Thoraval F, Boucherat M, Roger G, Tamalet A, et al. A 20-year experience of electron microscopy in the diagnosis of primary ciliary dyskinesia. Eur Respir J (2010) 35:1057–63. doi:10.1183/09031936.00046209
85. Rutland J, Dewar A, Cox T, Cole P. Nasal brushing for the study of ciliary ultrastructure. J Clin Pathol (1982) 35:357–9. doi:10.1136/jcp.35.3.357
86. Shoemark A, Dixon M, Corrin B, Dewar A. Twenty-year review of quantitative transmission electron microscopy for the diagnosis of primary ciliary dyskinesia. J Clin Pathol (2012) 65:267–71. doi:10.1136/jclinpath-2011-200415
87. Knowles MR, Leigh MW, Carson JL, Davis SD, Dell SD, Ferkol TW, et al. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax (2012) 67:433–41. doi:10.1136/thoraxjnl-2011-200301
88. Horani A, Brody SL, Ferkol TW, Shoseyov D, Wasserman MG, Ta-shma A, et al. CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PLoS One (2013) 8:e72299. doi:10.1371/journal.pone.0072299
89. Wirschell M, Olbrich H, Werner C, Tritschler D, Bower R, Sale WS, et al. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat Genet (2013) 45:262–8. doi:10.1038/ng.2533
90. Olbrich H, Schmidts M, Werner C, Onoufriadis A, Loges NT, Raidt J, et al. Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am J Hum Genet (2012) 91:672–84. doi:10.1016/j.ajhg.2012.08.016
91. Schwabe GC, Hoffmann K, Loges NT, Birker D, Rossier C, de Santi MM, et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mutat (2008) 29:289–98. doi:10.1002/humu.20656
92. Bartoloni L, Blouin JL, Pan Y, Gehrig C, Maiti AK, Scamuffa N, et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci U S A (2002) 99:10282–6. doi:10.1073/pnas.152337699
93. Knowles MR, Ostrowski LE, Leigh MW, Sears PR, Davis SD, Wolf WE, et al. Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am J Respir Crit Care Med (2014) 189:707–17. doi:10.1164/rccm.201311-2047OC
94. O’Callaghan C, Rutman A, Williams GM, Hirst RA. Inner dynein arm defects causing primary ciliary dyskinesia: repeat testing required. Eur Respir J (2011) 38:603–7. doi:10.1183/09031936.00108410
95. Bush A, Cole P, Hariri M, Mackay I, Phillips G, O’Callaghan C, et al. Primary ciliary dyskinesia: diagnosis and standards of care. Eur Respir J (1998) 12:982–8. doi:10.1183/09031936.98.12040982
96. Fliegauf M, Olbrich H, Horvath J, Wildhaber JH, Zariwala M, Kennedy M, et al. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am J Respir Crit Care Med (2005) 171:1343–9. doi:10.1164/rccm.200411-1583OC
97. Omran H, Kobayashi D, Olbrich H, Tsukahara T, Loges NT, Hagiwara H, et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature (2008) 456:611–6. doi:10.1038/nature07471
98. Merveille A-C, Davis EE, Becker-Heck A, Legendre M, Amirav I, Bataille G, et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat Genet (2011) 43:72–8. doi:10.1038/ng.726
99. Jeanson L, Copin B, Papon JF, Dastot-Le Moal F, Duquesnoy P, Montantin G, et al. RSPH3 mutations cause primary ciliary dyskinesia with central-complex defects and a near absence of radial spokes. Am J Hum Genet (2015) 97:153–62. doi:10.1016/j.ajhg.2015.05.004
100. Kott E, Legendre M, Copin B, Papon J-F, Dastot-Le Moal F, Montantin G, et al. Loss-of-function mutations in RSPH1 cause primary ciliary dyskinesia with central-complex and radial-spoke defects. Am J Hum Genet (2013) 93:561–70. doi:10.1016/j.ajhg.2013.07.013
101. Onoufriadis A, Shoemark A, Schmidts M, Patel M, Jimenez G, Liu H, et al. Targeted NGS gene panel identifies mutations in RSPH1 causing primary ciliary dyskinesia and a common mechanism for ciliary central pair agenesis due to radial spoke defects. Hum Mol Genet (2014) 23:3362–74. doi:10.1093/hmg/ddu046
102. Frommer A, Hjeij R, Loges NT, Edelbusch C, Jahnke C, Raidt J. Immunofluorescence analysis and diagnosis of primary ciliary dyskinesia with radial spoke defects. Am J Respir Cell Mol Biol (2015) 53:563–73. doi:10.1165/rcmb.2014-0483OC
103. Onoufriadis A, Paff T, Antony D, Shoemark A, Micha D, Kuyt B, et al. Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am J Hum Genet (2013) 92:88–98. doi:10.1016/j.ajhg.2012.11.002
104. Wallmeier J, Al-Mutairi D, Chen C-T, Loges NT, Pennekamp P, Menchen T, et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet (2014) 46:646–51. doi:10.1038/ng.2961
105. Boon M, Wallmeier J, Ma L, Loges NT, Jaspers M, Olbrich H, et al. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Commun (2014) 5:4418. doi:10.1038/ncomms5418
106. Bradley J, Moran F, Greenstone M. Physical training for bronchiectasis. Cochrane Database Syst Rev (2002) 3:CD002166. doi:10.1002/14651858.CD002166
107. Rogers D, Doull IJM. Physiological principles of airway clearance techniques used in the physiotherapy management of cystic fibrosis. Curr Paediatr (2005) 15:233–8. doi:10.1016/j.cupe.2005.02.007
108. Gokdemir Y, Karadag-Saygi E, Erdem E, Bayindir O, Ersu R, Karadag B, et al. Comparison of conventional pulmonary rehabilitation and high-frequency chest wall oscillation in primary ciliary dyskinesia. Pediatr Pulmonol (2014) 49:611–6. doi:10.1002/ppul.22861
109. Marks JH. Airway clearance devices in cystic fibrosis. Paediatr Respir Rev (2007) 8:17–23. doi:10.1016/j.prrv.2007.02.003
110. Elkins MR, Jones A, van der Schans C. Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis. Cochrane Database Syst Rev (2006) 2:CD003147.
111. Phillips GE, Thomas S, Heather S, Bush A. Airway response of children with primary ciliary dyskinesia to exercise and beta2-agonist challenge. Eur Respir J (1998) 11:1389–91. doi:10.1183/09031936.98.11061389
112. Valerio G, Giallauria F, Montella S, Vaino N, Vigorito C, Mirra V, et al. Cardiopulmonary assessment in primary ciliary dyskinesia. Eur J Clin Invest (2012) 42:617–22. doi:10.1111/j.1365-2362.2011.02626.x
113. Madsen A, Green K, Buchvald F, Hanel B, Nielsen KG. Aerobic fitness in children and young adults with primary ciliary dyskinesia. PLoS One (2013) 8:e71409. doi:10.1371/journal.pone.0071409
114. Boe J, Dennis JH, O’Driscoll BR, Bauer TT, Carone M, Dautzenberg B, et al. European respiratory society guidelines on the use of nebulizers. Eur Respir J (2001) 18:228–42.
115. Hart A, Sugumar K, Milan SJ, Fowler SJ, Crossingham I. Inhaled hyperosmolar agents for bronchiectasis. Cochrane Database Syst Rev (2014) 5:CD002996. doi:10.1002/14651858.CD002996.pub3
116. Kellett F, Robert NM. Nebulised 7% hypertonic saline improves lung function and quality of life in bronchiectasis. Respir Med (2011) 105:1831–5. doi:10.1016/j.rmed.2011.07.019
117. Nicolson CH, Stirling RG, Borg BM, Button BM, Wilson JW, Holland AE. The long term effect of inhaled hypertonic saline 6% in non-cystic fibrosis bronchiectasis. Respir Med (2012) 106:661–7. doi:10.1016/j.rmed.2011.12.021
118. Paff T, Daniels JM, Weersink EJ, Lutter R, Vonk Noordegraaf A, Haarman EG. A randomised controlled trial on the effect of inhaled hypertonic saline on quality of life in primary ciliary dyskinesia. Eur Respir J (2017) 49:1601770. doi:10.1183/13993003.01770-2016
119. Chernick WS, Barbero GJ. Composition of tracheobronchial secretions in cystic fibrosis of the pancreas and bronchiectasis. Pediatrics (1959) 24:739–45.
120. Potter JL, Spector S, Matthews LW, Lemm J. Studies on pulmonary secretions. 3. The nucleic acids in whole pulmonary secretions from patients with cystic fibrosis, bronchiectasis, and laryngectomy. Am Rev Respir Dis (1969) 99:909–16.
121. Picot R, Das I, Reid L. Pus, deoxyribonucleic acid, and sputum viscosity. Thorax (1978) 33:235–42. doi:10.1136/thx.33.2.235
122. Konstan MW, Ratjen F. Effect of dornase alfa on inflammation and lung function: potential role in the early treatment of cystic fibrosis. J Cyst Fibros (2012) 11:78–83. doi:10.1016/j.jcf.2011.10.003
123. Mackerness KJ, Jose PJ, Bush A. Differences in airway inflammation in cystic fibrosis and primary ciliary dyskinesia. Pediatr Asthma Allergy Immunol (2009) 22:163–8. doi:10.1089/pai.2009.0022
124. El-Abiad NM, Clifton S, Nasr SZ. Long-term use of nebulized human recombinant DNase1 in two siblings with primary ciliary dyskinesia. Respir Med (2007) 101:2224–6. doi:10.1016/j.rmed.2007.05.014
125. Desai M, Weller PH, Spencer DA. Clinical benefit from nebulized human recombinant DNase in Kartagener’s syndrome. Pediatr Pulmonol (1995) 20:307–8. doi:10.1002/ppul.1950200509
126. ten Berge M, Brinkhorst G, Kroon AA, de Jongste JC. DNase treatment in primary ciliary dyskinesia-assessment by nocturnal pulse oximetry. Pediatr Pulmonol (1999) 27:59–61. doi:10.1002/(SICI)1099-0496(199901)27:1<59::AID-PPUL11>3.0.CO;2-D
127. Noone PG, Bennett WD, Regnis JA, Zeman KL, Carson JL, King M. Effect of aerosolized uridine-5’-triphosphate on airway clearance with cough in patients with primary ciliary dyskinesia. Am J Respir Crit Care Med (1999) 160:144–9. doi:10.1164/ajrccm.160.1.9806146
128. Daviskas E, Rubin BK. Effect of inhaled dry powder mannitol on mucus and its clearance. Expert Rev Respir Med (2013) 7:65–75. doi:10.1586/ers.12.72
129. Bilton D, Tino G, Barker AF, Chambers DC, De Soyza A, Dupont LJ, et al. Inhaled mannitol for non-cystic fibrosis bronchiectasis: a randomised, controlled trial. Thorax (2014) 69:1073–9. doi:10.1136/thoraxjnl-2014-205587
130. Lucas JS, Burgess A, Mitchison HM, Moya E, Williamson M, Hogg C, et al. Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child (2014) 99:850–6. doi:10.1136/archdischild-2013-304831
131. Pasteur MC, Bilton D, Hill AT, British Thoracic Society Bronchiectasis non CFGG. British Thoracic Society guideline for non-CF bronchiectasis. Thorax (2010) 65:i1–58. doi:10.1136/thx.2010.136119
132. Flume PA, O’Sullivan BP, Robinson KA, Goss CH, Mogayzel PJ Jr, Willey-Courand DB, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med (2007) 176:957–69. doi:10.1164/rccm.200705-664OC
133. King PT, Holmes PW. Use of antibiotics in bronchiectasis. Rev Recent Clin Trials (2012) 7:24–30. doi:10.2174/157488712799363280
134. Bhatt JM. Treatment of pulmonary exacerbations in cystic fibrosis. Eur Respir Rev (2013) 22:205–16. doi:10.1183/09059180.00006512
135. Stenbit AE, Flume PA. Pulmonary exacerbations in cystic fibrosis. Curr Opin Pulm Med (2011) 17:442–7. doi:10.1097/MCP.0b013e32834b8c04
136. Hill AT, Pasteur M, Cornford C, Welham S, Bilton D. Primary care summary of the British Thoracic Society guideline on the management of non-cystic fibrosis bronchiectasis. Prim Care Respir J (2011) 20:135–40. doi:10.4104/pcrj.2011.00007
137. Cole PJ. Inflammation: a two-edged sword – the model of bronchiectasis. Eur J Respir Dis Suppl (1986) 147:6–15.
138. Spagnolo P, Fabbri LM, Bush A. Long-term macrolide treatment for chronic respiratory disease. Eur Respir J (2013) 42:239–51. doi:10.1183/09031936.00136712
139. Altenburg J, de Graaff CS, Stienstra Y, Sloos JH, van Haren EH, Koppers RJ, et al. Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: the BAT randomized controlled trial. JAMA (2013) 309:1251e9. doi:10.1001/jama.2013.1937
140. Serisier DJ, Martin ML, McGuckin MA, Lourie R, Chen AC, Brain B, et al. Effect of long-term, low-dose erythromycin on pulmonary exacerbations among patients with non-cystic fibrosis bronchiectasis: the BLESS randomized controlled trial. JAMA (2013) 309:1260e7. doi:10.1001/jama.2013.2290
141. Wong C, Jayaram L, Karalus N, Eaton T, Tong C, Hockey H, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet (2012) 380:660e7. doi:10.1016/S0140-6736(12)60953-2
142. Kobbernagel HE, Buchvald FF, Haarman EG, Casaulta C, Collins SA, Hogg C. Study protocol, rationale and recruitment in a European multi-centre randomized controlled trial to determine the efficacy and safety of azithromycin maintenance therapy for 6 months in primary ciliary dyskinesia. BMC Pulm Med (2016) 16:104. doi:10.1186/s12890-016-0261-x
143. Stillwell PC, Wartchow EP, Sagel SD. Primary ciliary dyskinesia in children: a review for pediatricians, allergists, and pediatric pulmonologists. Pediatr Allergy Immunol Pulmonol (2011) 4:191–6. doi:10.1089/ped.2011.0099
144. Deuse T, Reitz BA. Heart-lung transplantation in situs inversus totalis. Ann Thorac Surg (2009) 88:1002–3. doi:10.1016/j.athoracsur.2009.01.060
145. Macchiarini P, Chapelier A, Vouhe P, Cerrina J, Ladurie FL, Parquin F, et al. Double lung transplantation in situs inversus with Kartagener’s syndrome. Paris-Sud University Lung Transplant Group. J Thorac Cardiovasc Surg (1994) 108:86–91.
146. Prulière-Escabasse V, Coste A, Chauvin P, Fauroux B, Tamalet A, Garabedian EN, et al. Otologic features in children with primary ciliary dyskinesia. Arch Otolaryngol Head Neck Surg (2010) 136:1121–6. doi:10.1001/archoto.2010.183
147. Campbell R. Managing upper respiratory tract complications of primary ciliary dyskinesia in children. Curr Opin Allergy Clin Immunol (2012) 12:32–8. doi:10.1097/ACI.0b013e32834eccc6
148. Chang CC, Singleton RJ, Morris PS, Chang AB. Pneumococcal vaccines for children and adults with bronchiectasis. Cochrane Database Syst Rev (2009) 2:CD006316. doi:10.1002/14651858.CD006316.pub3
149. Chang CC, Morris PS, Chang AB. Influenza vaccine for children and adults with bronchiectasis. Cochrane Database Syst Rev (2007) 2:CD006218. doi:10.1002/14651858.CD006218.pub2
150. Lai M, Pifferi M, Bush A, Piras M, Michelucci A, Di Cicco M, et al. Gene editing of DNAH11 restores normal cilia motility in primary ciliary dyskinesia. J Med Genet (2016) 53:242–9. doi:10.1136/jmedgenet-2015-103539
151. Carotenuto M, Esposito M, Di Pasquale F, De Stefano S, Santamaria F. Psychological, cognitive and maternal stress assessment in children with primary ciliary dyskinesia. World J Pediatr (2013) 9:312–7. doi:10.1007/s12519-013-0441-1
Keywords: primary ciliary dyskinesia, Kartagener’s syndrome, bronchiectasis, ciliopathy, mucociliary clearance
Citation: Mirra V, Werner C and Santamaria F (2017) Primary Ciliary Dyskinesia: An Update on Clinical Aspects, Genetics, Diagnosis, and Future Treatment Strategies. Front. Pediatr. 5:135. doi: 10.3389/fped.2017.00135
Received: 15 December 2016; Accepted: 22 May 2017;
Published: 09 June 2017
Edited by:
Michael David Shields, Queen’s University Belfast, IrelandReviewed by:
Antonio Martinez-Gimeno, Complejo Hospitalario de Toledo, SpainMieke Boon, KU Leuven, Belgium
Copyright: © 2017 Mirra, Werner and Santamaria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Santamaria, santamar@unina.it