Functional Characterization of Novel ATP7B Variants for Diagnosis of Wilson Disease
 Sarah Guttmann1
Sarah Guttmann1  Friedrich Bernick1
Friedrich Bernick1  Magdalena Naorniakowska2 Ulf Michgehl3 Sara Reinartz Groba1 Piotr Socha3 Andree Zibert1 Hartmut H. Schmidt1*
Magdalena Naorniakowska2 Ulf Michgehl3 Sara Reinartz Groba1 Piotr Socha3 Andree Zibert1 Hartmut H. Schmidt1*- 1Medizinische Klinik B für Gastroenterologie und Hepatologie, Universitätsklinikum Münster, Münster, Germany
- 2Department of Gastroenterology, Hepatology, Nutritional Disorders and Pediatrics, The Children's Memorial Health Institute, Warsaw, Poland
- 3Internal Medicine D, Molecular Nephrology, University Hospital of Münster, Münster, Germany
Background: Diagnosis of rare Wilson disease (WD) in pediatric patients is difficult, in particular when hepatic manifestation is absent. Genetic analysis of ATP7B represents the single major determinant of the diagnostic scoring system in WD children having mild symptoms.
Objectives: To assess the impact of molecularly expressed ATP7B gene products in order to assist diagnosis of Wilson disease in pediatric patients having a novel mutation and subtle neuropsychiatric disease.
Methods: The medical history, clinical presentation, biochemical parameters, and the genetic analysis of ATP7B were determined. Due to ambiguous clinical and biochemical findings and identification of a novel compound ATP7B mutation with unknown disease-causing status, a molecular analysis of the ATP7B gene products in a previously well characterized cell model was performed.
Results: The ATP7B variants were transgenically expressed and the respective gene function molecularly characterized. Despite normal mRNA expression, low ATP7B protein expression of the mutants p.L168P and p.S1423N was observed (34.3 ± 8% and 66.0 ± 8%, respectively). Copper exposure did not result in decreased viability of transgenic cells as compared to wild type. Intracellular copper accumulation was reduced (≤47.9 ± 8%) and intracellular protein trafficking was impaired.
Conclusion: Our report suggests that functional characterization of novel ATP7B mutants can assist diagnosis; however mild functional impairments of ATP7B variants may hamper the value of such approaches.
Introduction
Wilson disease (WD; MIM#277900) is a rare, autosomal recessive, monogenetic disorder with a frequency of approximately 1:30,000 [1]. Diagnosis of WD represents a challenge to doctors, including pediatricians [2–4]. The disease manifests at various times throughout life with children representing a major portion of patients. Commonly, disease is diagnosed between 5 and 18 years, however some children below 5 years and elderly (> 60 years) can show first symptoms [5, 6]. WD results from mutations in the ATP7B gene. The gene encodes a large membrane-spanning P-type ATPase [7, 8]. ATP7B is predominately expressed in hepatocytes where it has two main functions: it is responsible for transfer of copper to apoceruloplasmin, which is secreted into the blood for copper supply to other organs, and—in case of excess copper—excretion from the body into the bile [9]. Clinical presentation with pure neurological symptoms in childhood, e.g., tremor or movement disorder, is rare, and most children show hepatic disease due to copper overload of the liver [10]. When left untreated, a severe, life-threatening disease evolves, including liver cirrhosis and/or severe neurodisability. Of note, effective, low-cost therapy has been established with copper chelators, mostly D-penicillamine (DPA) and trientine, or zinc [11]. However, therapy has to be taken lifelong and may result in side effects in a portion of the patients. An early start of the treatment in childhood is suggested to be of clinical benefit.
There is no single biochemical test for diagnosis of WD. A combination of individual assessments is indicative for diagnosis. Respective scores for diagnosis that includes various parameters of liver, including elevated levels of transaminases, and neurological disease have been developed [3, 12, 13]. One hallmark of the disease is the presence of a Kayser-Fleischer ring (KF) which is however absent in a significant portion of patients [14, 15]. Low serum ceruloplasmin and high liver copper are also highly suggestive but could be missed in patients with predominant neurological or psychiatric symptoms [16]. Significant delay of WD diagnosis is therefore not uncommon.
The only single assay to confirm WD is by genetic testing of ATP7B. Detection rates of up to 98% have been reported [17, 18]. More than 650 disease-causing mutations are currently known and novel SNPs are continuously reported. For novel mutations the prediction of penetrance is difficult. Most mutants of ATP7B are missense mutations which are proposed to reduce the function of the protein. To assess ATP7B function, primary specimens derived by biopsy from the patient are limited and also ethically restricted. Consequently, several cellular models reflecting the activity of ATP7B were established to predict the disease-causing impact of a given mutation. Such systems include yeast and several mammalian cell lines [19–22]. In this work we describe a case report of a child with a novel compound ATP7B mutation and relatively mild symptoms, where we have used a previously well characterized cell model to classify the functions of ATP7B for improved diagnosis of WD [23].
Materials and Methods
Clinical Presentation
Medical history, family medical history, clinical presentation, and biochemical parameters were recorded in the Department of Gastroenterology, Hepatology, Nutrition Disorders and Pediatrics of the Children's Memorial Health Institute Warsaw, Poland. Alanine transaminase (ALT), asparganine transaminase (AST), bilirubin, serum ceruloplasmin, rhodanine staining of hepatocytes, Kayser-Fleischer ring (KF), and 24 h urinary copper excretion were determined as reported [24]. After obtaining written informed consent, genomic DNA was extracted from peripheral blood samples and the complete open reading frame and adjacent intron boundaries of ATP7B were sequenced at the Medizinische Klinik B für Gastroenterologie und Hepatologie, Universitätsklinikum Münster, Germany. WD was diagnosed based on the Ferenci scoring system [25].
Cell Culture
HepG2 (human hepatocellular carcinoma) cells were purchased from American Type Culture Collection (ATCC) and ATP7B KO cells were derived as described [26]. RPMI medium (Lonza) containing 10% fetal bovine serum (FBS) was supplemented with 100 U/mL penicillin/streptomycin (PAA). Cells were maintained in 5% CO2 at 37°C in a humidified chamber.
Site-Directed Mutagenesis And Generation of Stable ATP7B Mutant Cell Lines
Wild type ATP7B cDNA was cloned into plasmid pGCsamEN.ATP7B and site-directed mutagenesis was performed using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) [23]. The primer sequences were: p.L168P (5′-3′): GGCAAGGTCCGGAAACCGCAAGGAGTAGTGAG /CTCACTACTCCTTGCGGTTTCCGGACCTTGCC and p.S1423N (5′-3′): CCATGGGACCAGGTCAACTATGTCAGCCAGGT / ACCTGGCTGACATAGTTGACCTGGTCCCATGG. Retroviral vector transduced HepG2 KO cells were selected in media containing 6 μg/ml blasticidin (Invitrogen).
Western Blot
A polyclonal anti-rabbit ATP7B antibody (kind gift of Dr. I. Sandoval, Madrid, Spain) was used for Western blot. HSC70 (Santa Cruz Biotechnology, #sc-1059) staining was used as a protein loading control. Relative expression was normalized to HepG2 KO cells expressing wild type ATP7B [23].
MTT Assay
For cell viability assay triplicates of 104 cells per 96 well were seeded and cultivated overnight in 100 μl RPMI media lacking phenol red (Lonza). Cells were exposed to copper (CuCl2; Sigma Aldrich) for 48 h. Viability was determined by MTT assay [26]. The percentage of viable cells was calculated and compared to untreated (100%).
Copper Accumulation
For copper accumulation assay 105 cells were seeded per 12 well, cultivated overnight and treated with 0.01 mM CuCl2 for 4 h. Cells were washed twice, trypsinized and lyzed in 65% nitric acid (Merck). Bradford Assay (BioRad) was used to determine total protein concentration. Analysis of copper accumulation was performed via inductively coupled plasma mass spectrometry (Thermo Fisher Scientific iCAP Qc).
Real-Time PCR Analysis
RT-qPCR analysis was performed using SYBR Green PCR Core Plus (Eurogentec). Ct values were normalized to the expression of the house-keeping GAPDH gene (ΔΔct method). PCR analysis was conducted on the ABI Prism 7900 HT Sequence Detection System (PE Applied Biosystems). Following primer sequences were used: ATP7B (5′-3′): TCCTCTGTGTCTGTGGTGCTC / ATGCGCCTGTGCCTCATAC and GAPDH (5′-3′): CCCACTCCTCCACCTTTGAC / CCACCACCCTGTTCCTGTAG.
Confocal Staining
For confocal microscopy, cells were maintained in RPMI basal cell culture media or treated by addition of 100 μM copper for 3 h. Primary antibody staining was performed using anti-ATP7B (kind gift of Dr. I. Sandoval, Madrid, Spain) and anti-lamp2 (Santa Cruz Biotechnology, #sc-18822). Three independent experiments were performed. Microscopic images were recorded with an Observer Z1 microscope with Apotome, Axiocam MRm (Zeiss) [27].
Statistical Analysis
Statistical analysis was performed by Kruskal-Wallis 1-way ANOVA and Student's t-test using SPSS 22.0 software. Data are given as mean ± standard error (SE).
Results
Patient History
The clinical characterization of the patient is illustrated in Table 1. At the age of thirteen, the boy was hospitalized following episodes of paranoia, uncommon tics, Tourette syndrome and three attempted suicides. MRI was performed, showing typical images according to age. Serum transaminases were within normal thresholds. Ceruloplasmin was low (0.13 g/L) at one occasion, while a second determination showed normal values (0.22 g/L). Liver biopsy was performed indicating micro- and macrovesicular steatosis with no signs of necrosis, fibrosis or cholestasis. Liver copper was in the normal range. Urine copper concentration was highly elevated (>5 fold) after cuprenil challenge [28]. A previously reported, disease-causing heterozygous p.L168P (c.503T > C) mutation was detected in ATP7B [29]. A second, unknown heterozygous ATP7B variant p.S1423N (c.4268G > A) was observed. The parents were shown to be asymptomatic heterozygotic carriers (Figures 1A–D). Genetic analysis of the patient also revealed a p.TA7/7 mutation in UGT1A1 indicating Gilbert syndrome. DPA and zinc acetate treatment lead to no further reports of neurological and hepatic disease. According to the diagnosing of WD using the Ferenci scoring system an overall score of 4 was determined [25].
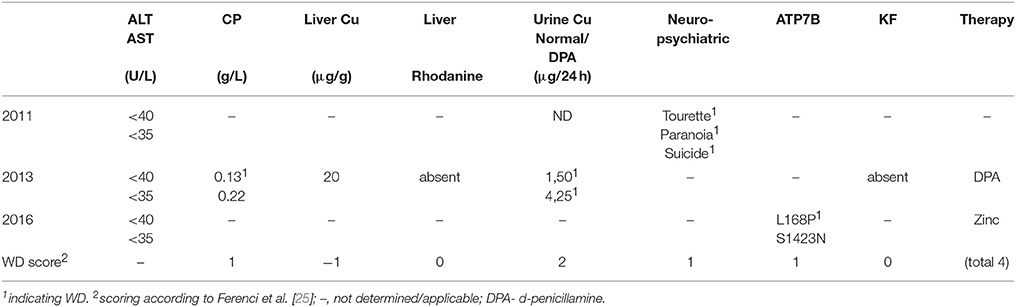
Table 1. Patient characteristics.
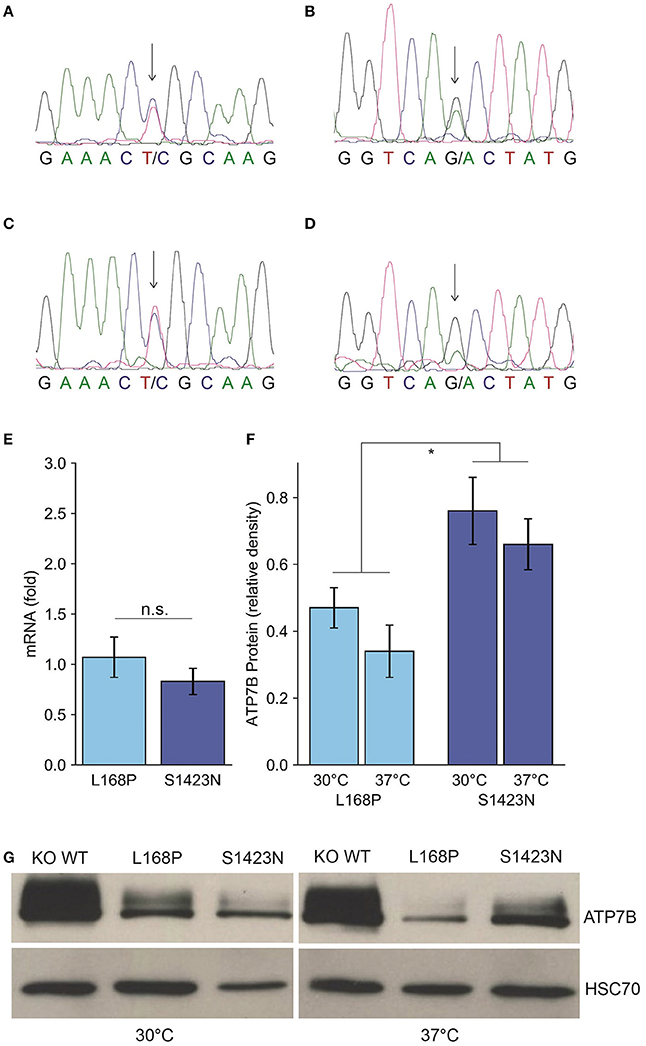
Figure 1. Identification and expression of ATP7B mutants p.L168P and p.S1423N. (A,B) Sequence analysis of the patient shows a compound heterozygote p.L168P (A) and p.S1423N (B) mutation. (C,D) The respective sequences derived from the father (C) and mother (D) are also depicted. (E) ATP7B mRNA expression of both variants was determined by RT-qPCR analysis. Mean/SE are given (n = 3). (F) Densitometry determination of ATP7B protein expression relative to wild type is shown. Mean/SE are given (n = 3). *P < 0.05. (G) One typical Western blot is shown.
Expression Profile of Novel ATP7B Variants
Since functional characterization of ATP7B mutants is not possible using patient materials and biopsy represents a risk especially for children, a previously established cellular model was used to functionally characterize the mutations of the patient [23]. While mRNA expression of the ATP7B variants p.L168P and p.S1423N was in the same range as observed for wild type (Figure 1E), protein expression/stability was significantly affected in both mutants of the mutants p.L168P and p.S1423N (34.3 ± 8% and 66.0 ± 8%, respectively) (Figures 1F,G). Incubation at low temperature (30°C) could marginally increase protein stability for mutants p.L168P and p.S1423N (factor of ≈1.4 and ≈1.2, respectively).
Characterization of ATP7B Mediated Copper Transport
ATP7B activity of both mutants as judged by viability of cells was similar to wild type at most copper concentrations (Figure 2A). IC50 values calculated for p.L168P and p.S1423N were similar (0.87 ± 0.06 mM and 0.89 ± 0.05 mM, respectively) and close to wild type ATP7B. The accumulated cellular copper concentration was significantly reduced (≤47.9 ± 8%) in both ATP7B mutants as compared to wild type (Figure 2B). Values of both mutants were in the same range as observed in ATP7B knockout cells. Confocal microscopy was used to analyze ATP7B trafficking relative to wildtype cells (Supplementary Figure S1). For mutant p.L168P, ATP7B staining was dispersed cytoplasmically at both copper concentrations (Figure 3). A broad co-localization with anti-lamp2, a late endosome-lysosome marker, as observed before for wild type was not detected [23]. For p.S1423N, a co-localization of ATP7B and lamp2 staining was found at low copper concentrations. The ATP7B staining pattern obtained with this mutant moderately responded to elevated copper.
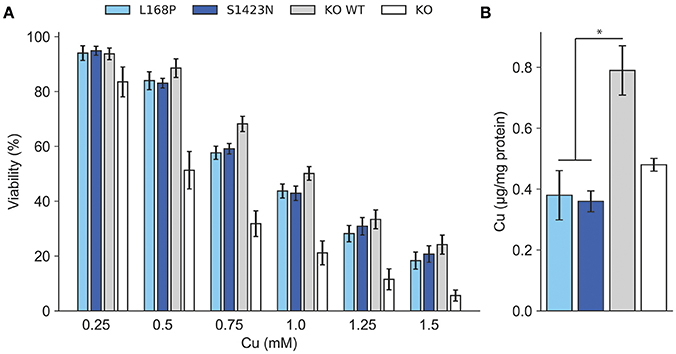
Figure 2. (A) Cell viability was determined after exposure to copper. Viability of cells relative to untreated (100%) is shown. Mean/SE are given (n = 3). *P < 0.05. (B) Analysis of intracellular Cu accumulation. Mean/SE are given (n = 3). *P < 0.05.
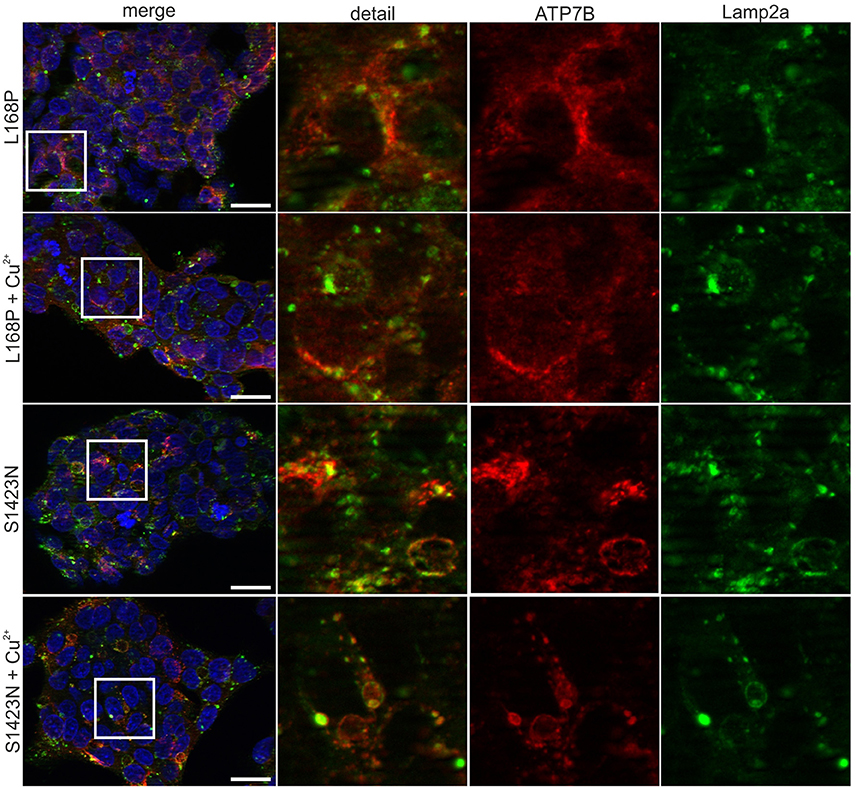
Figure 3. Confocal microscopy before and after addition of copper. For mutant p.L168P, a dispersed cytoplasmically staining of ATP7B was observed at low and high copper. For mutant p.S1423N, a co-localization with late endosome-lysosome marker lamp2 was observed at low copper concentrations. Elevated copper did not show typical trafficking of this mutant. One representative experiment out of three is shown. Scale bar, 20 μm.
Discussion
We here report on the diagnosis of a boy who has a novel compound heterozygote ATP7B mutation, no family history of WD, and subtle neuropsychiatric symptoms without hepatic disease. In this and other cases, WD diagnosis can be delayed due to highly variable symptoms and low awareness of the disease. Symptoms are rarely observed before the age of 5 years [5, 30, 31]. In the first decade, the majority of children present with hepatic symptoms. Liver disease is often accidentally revealed by routine analysis of serum showing elevated levels of hepatic transaminases which represent important first-line indicators of WD. However, the detection rate of elevated transaminases in children having WD is variable [32, 33]. While the Ferenci scoring system for diagnosis of WD includes parameters, e.g., Kayser-Fleischer rings, which are rarely observed in young children, some of the thresholds used for scoring have been adapted to children [28]. The only single parameter confirming WD is by genetic analysis of ATP7B. Genetic testing was proposed for children where liver disease of unknown origin is observed [3, 5]. Irrespective of the presence of liver disease, a family history of WD is highly suggestive for genetic testing. Extrahepatic presentation of WD as observed in the case described here is uncommon at young age. In children presenting with dystonia, tremor, dysarthria and/or impaired school performance, WD should be however carefully taken into account.
The reported case is unusual with respect to a pure psychiatric disease manifestation observed in childhood [34]. All clinical, biochemical, and histological findings considered, the diagnosis of WD for the patient is tentative, since definite hallmarks of the disease, like Kayser-Fleischer ring or elevated liver copper, are missing and overall relatively mild symptoms are observed. One determination of ceruloplasmin was indicative for WD, while a second assay performed in the same year was above thresholds. Of note, around 20% of diagnosed children (and adults) have CP levels above the threshold of 20 mg/dL limiting the value of this diagnostic parameter [35–39]. Elevated urine copper after penicillamine treatment corroborated WD diagnosis. It should be mentioned however that this assay is constrained in children, since collection of urine may be difficult. Tic syndromes are rather uncommon in WD patients but have been occasionally associated with the disease [40]. In addition, the patient displayed a mutation in UGT1A1 indicating Gilbert syndrome and total and indirect bilirubin was elevated. However, jaundice was not reported. Although we cannot fully rule out a multiple disorder of the patient, the general clinical picture seems to be in agreement with WD as the principal underlying disease. Our finding of a borderline positive WD diagnosis score could be significantly modulated by the young age of the patient which precludes a full manifestation of the disease [6, 10].
The genetic analysis of ATP7B in the patient revealed one previously reported disease-causing mutation p.L168P [29], while the second mutation p.S1423N was of unknown status. Variant p.S12423N was not observed in healthy individuals and could not be found in public data banks suggesting that this is not a polymorphism (data not shown). The mutation p.L168P has been reported in a 33 year old female in compound with mutation p.H1069Q, the most frequent mutation found worldwide which is associated with late-onset neurological disease [6]. The woman was diagnosed following the presentation of seizures and episodes of unconsciousness shortly after a cesarean section was performed. Neurological disease was not reported and zinc sulfate treatment resulted in functional improvement suggesting that mutation p.L168P may be associated with a relative mild course of disease. However, apart from this report, a coherent phenotype of p.L168P observed in homozygous WD patients has not been reported. While mutation p.L168P, located in the second copper binding domain of ATPase 7B, is predicted to alter the function of the protein, such analyses gave unambiguous results for p.S1423N (data not shown). The latter mutation is located in the cytosolic portion of ATPase 7B close to the C-terminal end, possibly important for protein trafficking [9].
To molecularly assess the function of the ATP7B gene products, we have employed a hepatic cell model, since hepatocytes represent the best studied cells where various biological functions of the copper transporter have been characterized [20, 26]. Given the almost pure neuropsychiatric symptoms of the patient, it would be interesting to re-address our findings in neuronal cell lines, which are however less established for assessment of WD to date. First, ATP7B-specific protein expression of mutants p.L168P and p.S1423N was found to be greatly reduced. The low protein expression could only be marginally increased by low temperature suggesting that in contrast to other mutations of ATP7B, a decreased translation might be operative [41]. Second, analysis of the cell viability in the presence of toxic copper indicates that mutants p.L168P and p.S1423N are mildly impaired with only marginal functional losses as compared to wild type. Of note, the viability assay employed here can detect various degrees of ATP7B activity, as exemplified by the moderate or deleterious loss-of-function mutations p.H1069Q/p.L795F and p.C271*, respectively [23]. Third, the assessment of intracellular copper accumulation revealed that both mutations have almost completely lost the ability to store copper and showed similar values as observed for knockout cells, whereas overexpression of ATP7B resulted in an increase of copper previously also observed in Chinese Hamster Ovary (CHO) cells [19]. This latter finding suggests that the copper accumulation assay and the viability assay may rely on distinct functions of ATPase 7B. As suggested earlier, copper is translocated to intracellular vesicles via ATP7B leading to increased copper concentrations [19]. Fourth, protein trafficking, a functional hallmark of the ATP7B protein, was significantly disturbed in both mutants. Mutant p.S1423N did not respond to high copper suggesting that the amino acid change at position 1423 might affect the nearby DKWSLL traffic signal that was found to be important for regulation of the transport between the trans-Golgi network (TGN) and the plasma membrane [42]. Trafficking of mutant p.L168P showed a copper dependent response, however lost a specific trans-Golgi localization. Our analysis of protein trafficking is however restricted, since quantification of protein localization relative to marker protein was not performed. Such quantitative analyses awaits further standardization, while different markers and experimental settings have been used [43–45]. Our molecular characterizations therefore indicate that both mutants exhibit distinct, non-overlapping impairments of ATP7B protein function which show different degrees of impact depending on the functional assay used for the determination.
Taken together, molecular characterization of novel ATP7B variants may help to functionally categorize the mutation and to assist in early WD diagnosis. The functional characterization of ATP7B gene products is straightforward and can be achieved within several weeks in order to initiate efficient therapy. However, when the impairment of ATP7B function is relative mild, as in the case reported here, such analysis can be ambiguous. In addition, our molecular analyses has to be subjected to further standardization by a large collection of mutated proteins which is far from being achieved in our and other studies [19–21, 23, 45]. The case reported here thus demonstrates current limitations of genetic analysis even when combined with advanced functional characterizations of ATP7B gene products. Given the evolving methodologies of high-throughput mutagenesis, improved cellular models to predict loss-of-function seem to be on the horizon, especially for monogenetic, severe disorders of childhood where efficient treatment is available.
Ethic Statement
This study was conducted according to the guidelines laid down in the Declaration of Helsinki and all procedures involving human subjects/patients were approved by the Ethics Committee of the Children's Memorial Health Institute (Children's Memorial Health Institute, Warsaw, Poland; approval granted in 2003). All patients and/or parents gave written informed consent.
Author Contributions
Study concept and design: SG, PS, HS, and AZ. Experiment and procedures: SG, FB , MN, UM, SRG, and PS. Results interpretation and drafting of the manuscript: SG, FB, UM, SRG, and AZ. Critical revision of the manuscript for important intellectual content: SG, MN, PS, HS, and AZ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to O. Nadzemova and V. Sauer for technical assistance and U. Karst for copper analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2018.00106/full#supplementary-material
Abbreviations
WD, Wilson disease; DPA, D-Penicillamine; KF, Kayser-Fleischer ring; Cu, copper.
References
1. Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. Lancet (2007) 369:397–408. doi: 10.1016/S0140-6736(07)60196-2
2. Rukunuzzaman M. Wilson's disease in bangladeshi children: analysis of 100 cases. Pediatr Gastroenterol Hepatol Nutr. (2015) 18:121–7. doi: 10.5223/pghn.2015.18.2.121
3. Seo JK. Diagnosis of Wilson disease in young children: molecular genetic testing and a paradigm shift from the laboratory diagnosis. Pediatr Gastroenterol Hepatol Nutr. (2012) 15:197–209. doi: 10.5223/pghn.2012.15.4.197
4. Sturm E, Piersma FE, Tanner MS, Socha P, Roberts EA, Shneider BL. Controversies and variation in diagnosing and treating children with Wilson disease: results of an international survey. J Pediatr Gastroenterol Nutr. (2016) 63:82–7. doi: 10.1097/MPG.0000000000001102
5. Wiernicka A, Dadalski M, Janczyk W, Kaminska D, Naorniakowska M, Husing-Kabar A, et al. Early onset of Wilson disease: diagnostic challenges. J Pediatr Gastroenterol Nutr. (2017) 65:555–60. doi: 10.1097/MPG.0000000000001700
6. Ferenci P, Czlonkowska A, Merle U, Ferenc S, Gromadzka G, Yurdaydin C, et al. Late-onset Wilson's disease. Gastroenterology (2007) 132:1294–8. doi: 10.1053/j.gastro.2007.02.057
7. Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. (1993) 5:327–37. doi: 10.1038/ng1293-327
8. Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. (1993) 5:344–50. doi: 10.1038/ng1293-344
9. Lutsenko S. Copper trafficking to the secretory pathway. Metallomics (2016) 8:840–52. doi: 10.1039/C6MT00176A
10. Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study. Gut (2007) 56:115–20. doi: 10.1136/gut.2005.087262
11. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology (2008) 47:2089–111. doi: 10.1002/hep.22261
12. Noureen N, Rana, MT. Neurological Wilson disease in children: a three years experience from Multan. J Pak Med Assoc. (2011) 61:743–8.
13. Iorio R, D'Ambrosi M, Marcellini M, Barbera C, Maggiore G, Zancan L, et al. Serum transaminases in children with Wilson's disease. J Pediatr Gastroenterol Nutr. (2004) 39:331–6. doi: 10.1097/00005176-200410000-00006
14. European association for study of liver. EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol. (2012) 56:671–85. doi: 10.1016/j.jhep.2011.11.007.
15. Caprai S, Loudianos G, Massei F, Gori L, Lovicu M, Maggiore G. Direct diagnosis of Wilson disease by molecular genetics. J Pediatr. (2006) 148:138–40. doi: 10.1016/j.jpeds.2005.07.036
16. Jesse M, Dempsey R, Eshelman A, Moonka D. Screening for Wilson's disease: which tests are good enough? Liver Transpl. (2014) 20:1525–26. doi: 10.1002/lt.23984
17. Coffey AJ, Durkie M, Hague S, McLay K, Emmerson J, Lo C, et al. A genetic study of Wilson's disease in the United Kingdom. Brain (2013) 136:1476–87. doi: 10.1093/brain/awt035
18. Schmidt HHJ. Role of genotyping in Wilson's disease. J Hepatol. (2009) 50:449–52. doi: 10.1016/j.jhep.2008.11.008
19. Cater MA, La Fontaine S, Shield K, Deal Y, Mercer JFB. ATP7B mediates vesicular sequestration of copper: Insight into biliary copper excretion. Gastroenterology (2006) 130:493–506. doi: 10.1053/j.gastro.2005.10.054
20. Huster D, Hoppert M, Lutsenko S, Zinke J, Lehmann C, Mossner J, et al. Defective cellular localization of mutant ATP7B in Wilson's disease patients and hepatoma cell lines. Gastroenterology (2003) 124:335–45. doi: 10.1053/gast.2003.50066
21. Tsivkovskii R, Eisses JF, Kaplan JH, Lutsenko S. Functional properties of the copper-transporting ATPase ATP7B (the Wilson's disease protein) expressed in insect cells. J Biol Chem. (2002) 277:976–83. doi: 10.1074/jbc.M109368200
22. Iida M, Terada K, Sambongi Y, Wakabayashi T, Miura N, Koyama K, et al. Analysis of functional domains of Wilson disease protein (ATP7B) in Saccharomyces cerevisiae. FEBS Lett. (1998) 428:281–5. doi: 10.1016/S0014-5793(98)00546-8
23. Chandhok G, Horvath J, Aggarwal A, Bhatt M, Zibert A, Schmidt HH. Functional analysis and drug response to zinc and D-penicillamine in stable ATP7B mutant hepatic cell lines. World J Gastroenterol. (2016) 22:4109–19. doi: 10.3748/wjg.v22.i16.4109
24. Wisniewska M, Cremer M, Wiehe L, Becker NP, Rijntjes E, Martitz J, et al. Copper to zinc ratio as disease biomarker in neonates with early-onset congenital infections. Nutrients (2017) 9:343. doi: 10.3390/nu9040343
25. Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease1. Liver Int. (2003) 23:139–42. doi: 10.1034/j.1600-0676.2003.00824.x
26. Chandhok G, Schmitt N, Sauer V, Aggarwal A, Bhatt M, Schmidt HHJ. The effect of zinc and D-penicillamine in a stable human hepatoma ATP7B knockout cell line. PLoS ONE (2014) 9:e98809. doi: 10.1371/journal.pone.0098809
27. Djuric I, Siebrasse JP, Schulze U, Granado D, Schluter MA, Kubitscheck U, et al. The C-terminal domain controls the mobility of Crumbs 3 isoforms. Biochim Biophys Acta (2016) 1863:1208–17. doi: 10.1016/j.bbamcr.2016.03.008
28. Patil M, Sheth KA, Krishnamurthy AC, Devarbhav H. A review and current perspective on Wilson disease. J Clin Exp Hepatol. (2013) 3:321–36. doi: 10.1016/j.jceh.2013.06.002
29. Czlonkowska A, Gromadzka G, Buttner J, Chabik G. Clinical features of hemolysis, elevated liver enzymes, and low platelet count syndrome in undiagnosed Wilson disease: report of two cases. Arch Gynecol Obstet. (2010) 281:129–34. doi: 10.1007/s00404-009-1080-6
30. Wilson DC, Phillips MJ, Cox DW, Roberts EA. Severe hepatic Wilson's disease in preschool-aged children. J Pediatr. (2000) 137:719–22. doi: 10.1067/mpd.2000.108569
31. Lin LJ, Wang DX, Ding NN, Lin Y, Jin Y, Zheng CQ. Comprehensive analysis on clinical features of Wilson's disease: an experience over 28 years with 133 cases. Neurol Res. (2014) 36:157–63. doi: 10.1179/1743132813Y.0000000262
32. O'Connor J, Sokol R. Copper metabolism and copper storage disorders. In: Suchy F, Sokol R, Balistreri W, editors. Liver Diseases in Children 3rd Edn. New York, NY: Cambridge University Press (2007). p. 626–59.
33. Iorio R, D'Ambrosi M, Mazzarella G, Varrella F, Vecchione R, Vegnente A. Early occurrence of hypertransaminasemia in a 13-month-old child with Wilson disease. J Pediatr Gastroenterol Nutr. (2003) 36:637–8. doi: 10.1097/00005176-200305000-00009
34. Svetel M, Potrebic A, Pekmezovic T, Tomic A, Kresojevic N, Jesic R, et al. Neuropsychiatric aspects of treated Wilson's disease. Parkinsonism Relat Disord. (2009) 15:772–5. doi: 10.1016/j.parkreldis.2009.01.010
35. Dhawan A, Taylor RM, Cheeseman P, De Silva P, Katsiyiannakis L, Mieli-Vergani G. Wilson's disease in children: 37-year experience and revised King's score for liver transplantation. Liver Transpl. (2005) 11:441–8. doi: 10.1002/lt.20352
36. Mak CM, Lam CW, Tam S. Diagnostic accuracy of serum ceruloplasmin in Wilson disease: determination of sensitivity and specificity by ROC curve analysis among ATP7B-genotyped subjects. Clin Chem. (2008) 54:1356–62. doi: 10.1373/clinchem.2008.103432
37. Nicastro E, Ranucci G, Vajro P, Vegnente A, Iorio R. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology (2010) 52:1948–56. doi: 10.1002/hep.23910
38. Kim JA, Kim HJ, Cho JM, Oh SH, Lee BH, Kim GH, et al. Diagnostic value of ceruloplasmin in the diagnosis of pediatric Wilson's disease. Pediatr Gastroenterol Hepatol Nutr. (2015) 18:187–92. doi: 10.5223/pghn.2015.18.3.187
39. Wang JS, Lu Y, Wang XH, Zhu QR. Urinary copper/zinc ratio: a promising parameter for replacement of 24-hour urinary copper excretion for diagnosis of Wilson's disease in children. World J Pediatr. (2010) 6:148–53. doi: 10.1007/s12519-010-0023-4
40. Arruda WO, Munhoz RP, de Bem RS, Deguti MM, Barbosa ER, Zavala JA, et al. Pathogenic compound heterozygous ATP7B mutations with hypoceruloplasminaemia without clinical features of Wilson's disease. J Clin Neurosci. (2014) 21:333–6. doi: 10.1016/j.jocn.2013.02.030
41. van den Berghe PV, Stapelbroek JM, Krieger E, Bie P, van de Graaf SF, Groot RE, et al. Reduced expression of ATP7B affected by Wilson disease-causing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology (2009) 50:1783–95. doi: 10.1002/hep.23209
42. Lalioti V, Hernandez-Tiedra S, Sandoval IV. DKWSLLL, a versatile DXXXLL-type signal with distinct roles in the Cu(+)-regulated trafficking of ATP7B. Traffic (2014) 15:839–60. doi: 10.1111/tra.12176
43. Lalioti V, Peiro R, Perez-Berlanga M, Tsuchiya Y, Munoz A, Villalba T, et al. Basolateral sorting and transcytosis define the Cu+-regulated translocation of ATP7B to the bile canaliculus. J Cell Sci. (2016) 129:2190–201. doi: 10.1242/jcs.184663
44. Zhu M, Dong Y, Ni W, Wu ZY. Defective roles of ATP7B missense mutations in cellular copper tolerance and copper excretion. Mol Cell Neurosci. (2015) 67:31–6. doi: 10.1016/j.mcn.2015.05.005
Keywords: delay of diagnosis, copper, neuropsychiatric, WD scoring, rare disease, cell model
Citation: Guttmann S, Bernick F, Naorniakowska M, Michgehl U, Groba SR, Socha P, Zibert A and Schmidt HH (2018) Functional Characterization of Novel ATP7B Variants for Diagnosis of Wilson Disease. Front. Pediatr. 6:106. doi: 10.3389/fped.2018.00106
Received: 16 February 2018; Accepted: 03 April 2018;
Published: 30 April 2018.
Edited by:
Enrico Baruffini, Università degli Studi di Parma, ItalyReviewed by:
Richard Burke, Monash University, AustraliaJulnar A. R. Usta, American University of Beirut, Lebanon
Copyright © 2018 Guttmann, Bernick, Naorniakowska, Michgehl, Groba, Socha, Zibert and Schmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hartmut H. Schmidt, hepar@ukmuenster.de