
- Section of Cardiovascular Medicine, Department of Internal Medicine, Yale School of Medicine, Yale University, New Haven, CT, USA
Prostacyclin (PGI2) is a member of the prostaglandin family of bioactive lipids. Its best-characterized role is in the cardiovascular system, where it is released by vascular endothelial cells, serving as a potent vasodilator and inhibitor of platelet aggregation. In recent years, prostacyclin (PGI2) has also been shown to promote differentiation and inhibit proliferation in vascular smooth muscle cells. In addition to these well-described homeostatic roles within the cardiovascular system, prostacyclin (PGI2) also plays an important role as an inflammatory mediator. In this review, we focus on the contribution of prostacyclin (PGI2) as both a pathophysiological mediator and therapeutic agent in three major inflammatory-mediated disease processes, namely rheumatoid arthritis, where it promotes disease progression (“pro-inflammatory”), along with pulmonary vascular disease and atherosclerosis, where it inhibits disease progression (“anti-inflammatory”). The emerging role of prostacyclin (PGI2) in this context provides new opportunities for understanding the complex molecular basis for inflammatory-related diseases, and insights into the development of current and future anti-inflammatory treatments.
Pharmacology of Prostacyclin and Its Receptor
Prostacyclin (PGI2) is a member of the prostaglandin family of bioactive lipids, and is a derivative of the 20-carbon, omega-6 fatty acid, arachidonic acid (AA or 5,8,11,14-eicosatetraenoic acid). Both cyclooxygenase enzymes (COX-1 and COX-2) convert AA into the prostaglandin precursor PGH2, which is subsequently synthesized into prostacyclin (PGI2) via prostacyclin synthase (PGIS; Figure 1). However, the majority of PGI2 produced in vivo, particularly within the systemic and pulmonary vasculature (Moncada et al., 1977; Catella-Lawson et al., 1999; McAdam et al., 1999), and other regions like the synovium (Brodie et al., 1980; Crofford et al., 1994), appears to be derived from COX-2. PGI2 is unstable at physiological pH and, thus, has a very short half-life in vivo (<2 min), rapidly forming the inactive hydration product 6-keto-prostaglandin F1α (6-keto-PGF1α; Lewis and Dollery, 1983; Smyth and FitzGerald, 2002). The actions of PGI2 are mediated through a seven-transmembrane-spanning G-protein coupled receptor (GPCR), referred to as the IP receptor (International Union of Pharmacology nomenclature). The IP receptor is a Class A rhodopsin-like GPCR that couples predominately to the Gs subunit of the heterotrimeric G-protein and mediates intracellular signaling via adenylyl cyclase (AC) activation and cyclic AMP (cAMP) production (Boie et al., 1994). Animal studies have also shown that PGI2 may also signal through alternate Gq- and Gi-related pathways (Lawler et al., 2001), as well as nuclear receptor-mediated pathways, such as the peroxisome proliferator activated receptor gamma (PPARδ) pathway (Lim and Dey, 2002). Stitham et al. (2003) have elucidated the putative binding pocket for the human IP receptor, which has been reported to also accommodate type E prostanoids (i.e., PGE1 and PGE2) in addition to its native ligand PGI2 and its analogs (Boie et al., 1994; Nakagawa et al., 1994). The physiological effects of PGI2 are vast with much remaining to be uncovered. Within the vasculature, PGI2 serves as a potent vasodilator and is the major inhibitory prostanoid in platelet aggregation (Smyth et al., 2009), and has also been shown to inhibit vascular smooth muscle cell (VSMC) proliferation and de-differentiation (Fetalvero et al., 2006, 2007). Within the lungs, PGI2 reduces pulmonary blood pressure as well as bronchial hyper-responsiveness (Idzko et al., 2007). Within the kidneys, PGI2 serves to regulate renal blood flow and glomerular filtration rate, as well as mediates the release of renin (Komhoff et al., 1998). In the nervous system, PGI2 has been shown to elicit nociceptive pain response (Murata et al., 1997).
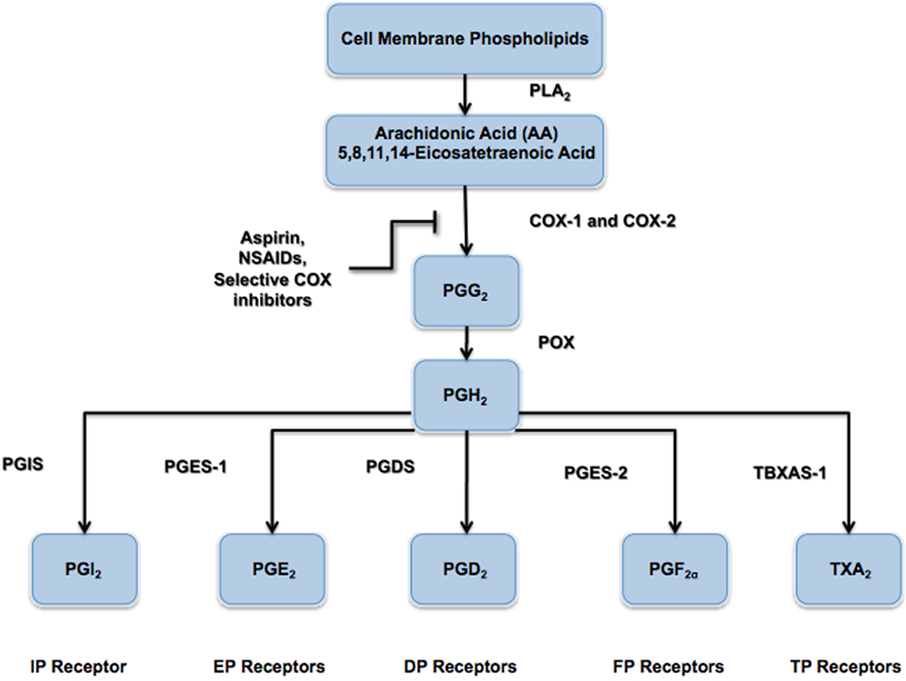
Figure 1. Prostanoid biosynthesis pathway. The enzyme phospholipase A2 (PLA2) hydrolyzes arachidonic acid (AA) from the phospholipids of the extracellular membrane. Arachidonic acid is modified by the cyclooxygenase (COX) enzymes (COX-1 and COX-2) to form the intermediate precursor prostaglandin G2 (PGG2) via the addition of two oxygen (O2) molecules. Prostaglandin H2 (PGH2) is subsequently formed by the actions of peroxidase enzyme, which releases a single oxygen (O2) molecule. As shown, all prostanoids are derived from the parent compound PGH2 and are formed via their respective synthase enzymes, namely prostaglandin I2 synthase (PGIS), prostaglandin E2 synthase (PGES-1), prostaglandin D2 synthase (PGDS), prostaglandin F2α synthase (PGES-2), and thromboxane A2 synthase (TBXAS-1).
Prostacyclin as an Inflammatory Mediator
As described, prostacyclin (PGI2) is best known for its regulatory role within the cardiovascular system, where it promotes VSMC relaxation (vasodilatation) and inhibits platelet aggregation (anti-thrombotic). However, it is also an important inflammatory mediator. The seminal work by Vane (1971) demonstrating the inhibition of prostaglandin biosynthesis as the mechanism of action for aspirin (acetylsalicylic acid) and other aspirin-like drugs first highlighted the importance of the prostaglandin family of molecules, and set the stage for the development of many pharmacologic agents, such as traditional, non-selective non-steroidal anti-inflammatory drugs (tNSAIDs) and the newer selective COX-2 inhibitors. Further work by Davies et al. (1984) pinpointed particular prostaglandins, principally prostaglandin E2 (PGE2) and prostacyclin (PGI2), in the mediation of vascular permeability associated with the hyperemia and edema seen with acute inflammation. Murata et al. (1997) demonstrated the involvement of prostacyclin (PGI2)-mediated inflammatory swelling in vivo, using prostacyclin receptor deficient (IP−/−) mice. In these critical experiments, it was shown that mice lacking the prostacyclin receptor had a reduced inflammatory response, as measured by percent change in vascular permeability using a carrageenan-induced paw-edema model (Murata et al., 1997). Limb edema was decreased by approximately 50% in IP-deficient mice, similar to levels seen in mice pre-treated with the non-steroidal anti-inflammatory agent indomethacin. Moreover, a significant reduction in lung exudate volume, using a carrageenan-induced pleurisy model, was also observed (although data not shown) for IP-deficient mice as well (Murata et al., 1997). In contrast, a study by Takahashi et al. (2002) demonstrated the IP-deficient mice showed higher skin and airway immune responses (relating to increased capillary permeability in these tissues) in antigen-sensitized inflammation, suggesting a protective role for PGI2 in allergic inflammation. These studies (among others) have highlighted prostacyclin as a major endogenous mediator of inflammation – both pro-inflammatory and anti-inflammatory, depending upon the tissue and pathological model being studied (Figure 2).
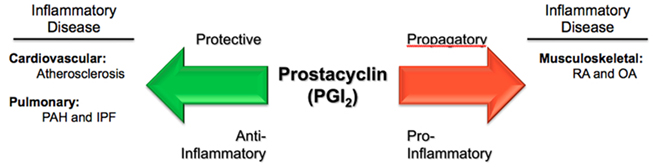
Figure 2. Paradoxical actions of prostacyclin in three inflammatory diseases. Prostacyclin (PGI2) serves as a protective, anti-inflammatory mediator in the processes of atherosclerosis and pulmonary vascular diseases, such as pulmonary arterial hypertension (PAH) and idiopathic pulmonary fibrosis (IPF). Conversely, in rheumatological conditions, such as rheumatoid arthritis (RA) and osteoarthritis (OA), PGI2 acts as a propagatory, pro-inflammatory molecule.
Role of Prostacyclin in Arthritis
While the majority of focus has centered around the role of prostaglandin E2 (PGE2) in rheumatoid arthritis (RA), some studies have shown that the predominate prostaglandin detected within the synovial fluid of patients with RA is in fact prostacyclin (PGI2; Brodie et al., 1980). Moreover, using both collagen-induced arthritis and collagen-antibody-induced arthritis models, Honda et al. (2006) showed that prostacyclin receptor knockout (IP−/−) mice exhibited significantly reduced clinical and histological arthritic scores versus control mice, in both arthritis models, placing further emphasis on receptor-mediated prostacyclin activity in the pathogenesis of RA. Using a K/BxN serum-transfer arthritis model, Chen et al. (2008) administered serum from arthritic K/BxN mice to induce an IgG-mediated autoantibody-induced inflammatory arthritis to recipient mice lacking either prostaglandin E synthase-1 (mPGES-1−/−) or the prostacyclin receptor (IP−/−), in order to determine the relative importance of PGE2 and PGI2, respectively. Findings revealed that mice deficient in prostaglandin E synthase-1 mPGES-1 (and therefore unable to produce PGE2), developed arthritis in normal fashion, whereas mice lacking the receptor for PGI2 demonstrated a significant decrease (31% versus wild-type) in clinical arthritis. Furthermore, using COX-1 and COX-2 knockout animals, it was shown that mice the lacking COX-1 isoform were resistant to the development of arthritic disease, while those lacking the COX-2 isoform remained vulnerable (Wang et al., 2008).
These results convey two important points: (1) a substantial proportion of the prostanoid contribution to joint inflammation (at least in the K/BxN serum-transfer arthritis model) can be accounted for by PGI2 and its interaction with the IP receptor, and (2) selective COX-1 inhibition through genetic knockout prevented the development of disease, suggesting that COX-1-derived PGI2 is the major inflammatory mediator within this arthritis model (Wang et al., 2008). More importantly, such studies lend weight to the involvement of prostacyclin in chronic inflammatory disease processes, as well as being an acute mediator, and also call into question the paradigm regarding COX-1- and COX-2-derived prostaglandin functions in vivo (i.e., regulatory “housekeeping” versus inflammatory induction).
Interestingly, from the perspective of clinical therapies, there does not seem to be a difference in efficacy according to COX selectivity. As Chen et al. (2008) showed, meta-analysis from a systematic review of 145 randomized controlled trials, examining the clinical effectiveness of a variety of COX-2 inhibitors (including etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib, and lumiracoxib), showed similar efficacy compared to non-selective NSAIDs (including naproxen, diclofenac, ibuprofen, loxoprofen, nabumetone, piroxicam, indomethacin, tenoxicam, and nimesulide) in the symptomatic relief of both RA and osteoarthritis (OA), but with superior gastrointestinal tolerability and protection against complicated upper gastrointestinal events (e.g., ulcers, bleeding, perforations) – the majority of evidence coming from OA populations. However, the amount of evidence for this gastro-protective effect varied considerably across individual drugs. Moreover, an increased risk of myocardial infarction (MI) was also observed among those drugs with greater volume of evidence in terms of exposure in patient-years (Chen et al., 2008), presumably as a by-product of the discriminating suppression of COX-2-derived PGI2, which has been shown to be cardioprotective (Murata et al., 1997; Cheng et al., 2002; Egan et al., 2004). Along these same lines, as chronic inflammation has been linked to enhanced development of atherogenesis (Libby et al., 2002), individuals with RA may already be at increased risk for cardiovascular disease. In fact, a recent comparative study involving disease-duration-matched RA and diabetes mellitus (DM) patients found that RA was a substantial and independent cardiovascular risk factor, with similar severity to DM, with significantly worsened preclinical atherosclerotic markers, increased intima-to-media thickness, as well as lower flow-mediated dilatation (measure of endothelial function; Stamatelopoulos et al., 2009). Thus, the combination of chronic RA-mediated inflammation, perpetuating an increased atherosclerotic burden, along with COX inhibition therapy – either selective (COX-2 inhibitor) or non-selective (NSAIDs) – would undoubtedly increase cardiovascular risk drastically, given what we now know about PGI2. Thus, PGI2 is emerging as an important intermediary in inflammatory conditions such as rheumatoid and OA, with the dualistic purpose of pro-inflammatory mediator on the one hand – involved in disease pathophysiology – and cardioprotective factor on the other – both of which are central in the consideration of pharmacologic therapies and adverse effects.
Role of Prostacyclin in Pulmonary Fibrosis and Pulmonary Hypertension
While pulmonary fibrosis and pulmonary hypertension are distinct pathophysiological entities, they do share some commonalities, some of which involve PGI2. Pulmonary arterial hypertension (PAH) is in fact a heterogeneous group of diseases sharing similarities in pathophysiological mechanisms, clinical presentation, and therapeutic approaches (Simonneau et al., 2009). The pathogenesis of PAH is complex and incompletely understood, with both genetic and environmental factors contributing to altered vascular structure and function (Badesch et al., 2007). The main vascular changes in PAH are vasoconstriction, VSMC proliferation, endothelial loss or dysfunction, and thrombosis (Farber and Loscalzo, 2004), which implicates a disruption of vascular hemostasis and its principle mediators, particularly PGI2 and TxA2, among others. This is evidenced by findings in patients with PAH, whereby both the production of prostacyclin synthase (PGIS) within small-to-medium pulmonary arteries, as well as urinary metabolites (6-keto-prostacyclin F2α) of prostacyclin, were shown to be decreased, while levels of thromboxane metabolites (thromboxane B2) were increased (Christman et al., 1992; Tuder et al., 1999). Interestingly, this imbalance of PGI2 and TxA2 within the pulmonary vasculature, leading to increased mean pulmonary artery pressure, mimics that of the cardio-systemic vasculature system. In fact, many of the pathophysiological mechanisms identified in PAH overlap with those involved in atherogenesis, including vascular smooth muscle and endothelial cell dysfunction, enhanced platelet activity and thrombosis, inflammation, and cellular chemotaxis (Essop, 2010). Owing to its potent vasodilatory, anti-thrombotic, and anti-proliferative effects, PGI2 has secured a central role in the treatment of PAH. Continuous intravenous epoprostenol (synthetic PGI2) is the best-studied advanced therapy for PAH and remains a first-line agent, particularly for those with severe disease (WHO functional class IV), as it has been shown to improve overall symptoms, exercise capacity, and hemodynamic status in controlled clinical trials (Barst et al., 2009), as well as confer a survival benefit in both idiopathic and heritable forms of PAH (IPAH and HPAH; Barst et al., 2009). There are limitations to treatment with epoprostenol based upon its pharmacology (plasma half-life = 3–5 min) and long-term use requires a permanent central venous catheter and a portable infusion pump. Analogs of PGI2 have also been used clinically, and are administered by a variety of routes, including intravenously (e.g., treprostinil and iloprost), subcutaneously (e.g., treprostinil), inhalation (e.g., iloprost), and orally (e.g., beraprost). These medications are generally more stable with longer half-lives, but have variable safety and efficacy equivalencies compared to epoprostenol, and clinical trials with these (and other) alternative agents are limited with respect to severe disease classification (Barst et al., 1996). Other vasoactive therapies for PAH include the dual endothelin receptor (ETA and ETB) antagonist (e.g., bosentan) and phosphodiesterase type-5 (PDE-5) inhibitors, which have been proven effective, alone or in combination therapy, in milder forms of PAH (WHO functional class II and III; Rubin et al., 2002; Sitbon et al., 2003; Galie et al., 2005; McLaughlin et al., 2005; Pepke-Zaba et al., 2008). Again, the central role of PGI2, as both an inflammatory and hemodynamic mediator, puts it at the forefront in understanding the pathophysiology and pharmacological treatment of pulmonary vascular diseases, particularly PAH.
Interestingly, PGI2 seems to have a similar safeguarding effect in the chronic inflammatory condition of idiopathic pulmonary fibrosis (IPF), as recent studies have shown that COX-2-derived PGI2 serves a protective role against bleomycin-induced pulmonary fibrosis – a major animal model for IPF that mimics the progressive fibrosis and interstitial inflammation of sub-pleural lung tissue in humans. In a study by Lovgren et al. (2006), IP-deficient mice were more susceptible to bleomycin-induced pulmonary fibrosis, demonstrating increased collagen deposition and cellularity after bleomycin administration compared with the wild-type mice. These observations correlated with increases in quantitative measurements of histological lung scores and hydroxyproline levels within the lung parenchyma. Similar results were found using COX-2-deficient mice, but were not supported using knockouts for either the EP2 or EP4 receptors, which bind COX-2-derived prostaglandin E2 (PGE2; Lovgren et al., 2006). Such findings put direct focus on the loss of COX-2-derived prostacyclin as a protective factor. While COX-2-derived prostacyclin is now well known for its protective effects within the cardiovascular system, such results provide compelling evidence for PGI2-mediated protection against fibrotic pathologies as well.
A more recent animal study by Zhu et al. (2010) also confirms these findings, pharmacologically, and identifies PGI2 as a potential new therapeutic agent for pulmonary fibrotic disease. Using intra-peritoneal injections of iloprost, a stable PGI2 analog, it was demonstrated that a single dose of iloprost (200 μg/kg; prior to bleomycin injection) could preclude pulmonary inflammation and fibrosis in mice (Zhu et al., 2010). Pre-treatment with iloprost seemed to significantly reduce both inflammatory infiltration and collagen deposition with the pulmonary interstitium, as well as improve lung mechanics (reduced static compliance and elevated tissue elastance; Zhu et al., 2010). However, the specific inflammatory cell subtype being suppressed could not be delineated in this current study. Iloprost pre-treatment decreased production of pro-inflammatory and fibrotic cytokines, such as TNF-alpha, IL-6, and TGF-beta-1, and increased release of anti-fibrotic mediators, including IFN-gamma and chemokine CXCL10/IP-10, as measured by mRNA expression levels or ELISA. Moreover, the cumulative mortality in iloprost-treated mice was 10% at day 21 versus 60% in the non-iloprost-treated cohort (Zhu et al., 2010).
In human studies, inhaled iloprost has been proven efficacious in the treatment of various forms of pulmonary hypertension, including pulmonary hypertension secondary to pulmonary fibrosis (Olschewski et al., 1999). In fact, it has been suggested that the majority of vascular resistance in fibrotic lung disease is due to persistent vasoconstriction (Olschewski et al., 1999), which may explain the effectiveness of PGI2 analog therapy. Certain studies also suggest a long-term clinical benefit from continued therapy with inhaled iloprost, which was well tolerated and required no substantial dose increase over a 2-year trial (Olschewski et al., 2010). In the United States, its use has been approved for PAH New York Heart Association (NYHA) functional class III and IV; for individuals with marked limitations or inability to carry on physical activity (Gomberg-Maitland and Olschewski, 2008). Thus, PGI2 seems to be an important effector in both these fibro-proliferative disorders of the lung, playing both a protective role against disease development, as well as a therapeutic role in symptom management.
Role of Prostacyclin in Atherosclerosis
Atherosclerosis is now known as an inflammatory disease, with the same complex cellular interactions involving monocytes, macrophages, lymphocytes, extracellular matrix (ECM) components, and connective tissue cells as seen in other chronic inflammatory and fibro-proliferative diseases (e.g., RA, pulmonary fibrosis, glomerulosclerosis; Ross, 1999). The critical role of PGI2 in atherosclerosis is quickly emerging, with evidence spanning from molecular and cell biology to human clinical trials. Mounting data has demonstrated the protective effect of prostacyclin activity against the development of atherothrombotic cardiovascular disease through the inhibition of various cellular processes, including platelet activation, leukocyte adhesion, as well as VSMC modulation. As such, PGI2 analogs (e.g., iloprost) are able to down-regulate lymphocyte adhesion to endothelial cells, which suggests an ability to block the early events in atherosclerosis (Della Bella et al., 2001). Furthermore, interactions within the realm of lipid metabolism has lent further support toward the atheroprotective properties of PGI2 (Thiemermann, 1991). Specifically, HDL has been shown to induce COX-2 expression and PGI2 production in both endothelial and VSMCs (Pomerantz et al., 1985; Vinals et al., 1997, 1999) while, conversely, PGI2 has been shown to induce cholesterol ester hydrolase activity, which catalyzes the first step in the removal of cholesterol from foam cells, critical components in atherogenesis (Hajjar and Weksler, 1983; Weksler et al., 1983; Hajjar et al., 1989). The effects of PGI2 on VSMCs are becoming an important target for understanding both the pathophysiology of atherothrombosis and the atheroprotective effects of prostacyclin. In mature blood vessels, VSMCs are quiescent and exhibit a differentiated, contractile phenotype. However, in response to vascular injury, these cells have been shown to re-enter the cell cycle, proliferate, migrate toward attractants, down-regulate expression of contractile proteins, and up-regulate synthesis of proteins, particularly ECM (Campbell et al., 1988). PGI2 has been shown to exert both anti-proliferative (Grosser et al., 1995; Zucker et al., 1998) and anti-migratory (Blindt et al., 2002) effects on smooth muscle cells. In advanced atherosclerotic lesions (as well as restenotic lesions), expression levels of smooth-muscle-specific differentiation markers are markedly reduced (Wilcox, 1992; O’Brien et al., 1993), and as Fetalvero et al. (2006) have shown, treatment with the stable PGI2 analog, iloprost, induces VSMC differentiation via a cAMP-PKA-mediated signaling pathway. In similar fashion, Kasza et al. (2009) went on to further show that, in addition to increased contractile protein expression and contractile morphology, iloprost-treated VSMCs up-regulate COX-2 expression, mediated not only by cAMP-PKA, but also novel pathways involving ERK-1/2 activation and Akt-1 inhibition. In turn, the up-regulated COX-2 expression lead to subsequent PGI2 release (i.e., prostacyclin-induced prostacyclin release), which was shown to have a paracrine, positive-feedback effect on neighboring VSMCs (not exposed to iloprost), inducing similar cellular responses (Kasza et al., 2009). Thus, there appears to be a clear link between the major atheroprotective effects of prostacyclin and VSMC modulation. As such, the phenotypic change in VSMCs toward a proliferative and de-differentiated state, which is a hallmark occurrence in the progression of atherosclerosis and restenosis, necessitates a clear understanding of this regulatory process and is an extremely important area of research.
Mouse models have provided valuable insight into the role of prostacyclin in cardiovascular homeostasis and pathogenesis. IP-deficient (IP−/−) mice display increased propensities toward thrombosis (Murata et al., 1997), intimal hyperplasia and restenosis (Cheng et al., 2002), and reperfusion injury (Xiao et al., 2001). Moreover, in both atherogenic apolipoprotein E (apo-E)- and low-density lipoprotein receptor (LDL-R)-deficient backgrounds, mice lacking the IP receptor demonstrated greater atherosclerotic burden with higher platelet reactivity and leukocyte adhesion to endothelial cells (Egan et al., 2004; Kobayashi et al., 2004). Moreover, Egan et al. (2004) went on to further demonstrate that, in female pre-menopausal LDL-R knockout mice, the atheroprotective effect of estrogen is significantly reduced in the absence of the IP receptor (i.e., double LDL-R and IP knockout). Moreover, as long-term estrogen exposure was shown to increase COX-2 expression, as well as the formation of the PGI2 metabolite 6-keto-PGF1α in mouse aortic smooth muscle cells (mASMCs), this study suggests a significant contribution of estrogen-mediated, COX-2-derived PGI2 in the protection against atherogenesis (Egan et al., 2004). Such in vivo findings highlight the important functional presence of prostacyclin activity in the maintenance of cardiovascular homeostasis and, in turn, implicate receptor-ligand (hIP-PGI2) dysfunction in the acceleration of atherogenesis and the subsequent development of related disorders, including stroke, MI, and hypertension. In humans, the importance of PGI2 in atherogenesis has also been shown. Arehart et al. (2008) demonstrated that patients harboring a dysfunctional human prostacyclin receptor variant (R212C) exhibited an enhanced atherothrombotic phenotype, with a higher incidence of triple-vessel coronary artery disease (CAD) and greater number of clinical cardiovascular events (including MI, stroke, PTCA, CABG, PVD, and unstable angina) in high-risk patients versus age- and risk-factor-matched normal-allele patients. Another study by Patrignani et al. (2008) correlated the R212C prostacyclin receptor polymorphism, as well as two other synonymous variants, with intimal hyperplasia and progressive deep venous thrombosis, respectively. Biochemical analyses have revealed potential mechanistic explanations for these R212C disease association studies. Ibrahim et al. (2010) demonstrated that co-expression of the R212C variant, together with wild-type hIP, resulted in dominant-negative inhibition of signaling through receptor homo-dimerization, with enhanced wild-type IP localization to the endoplasmic reticulum. Interestingly, a similar effect was observed with co-expression of the R212C variant and the wild-type thromboxane receptor α (TPα), which revealed R212C hetero-dimerization, with subsequent inhibition of TPα receptor activity (Ibrahim et al., 2010). Since this time, Stitham, Arehart, and colleagues have identified other functionally deficient prostacyclin receptor polymorphisms (i.e., R215C and L104R) associated with increased CAD (Ibrahim et al., 2010). While such mutations are rare, it is notable that single, heterozygous, point mutations within the prostacyclin receptor gene (PTGIR) are associated with clinically significant disease changes.
Similarly, mutations and polymorphisms in the prostacyclin synthase (PGIS) gene have been associated with essential hypertension, MI, and cerebral infarction (Iwai et al., 1999; Nakayama, 2005). The sum of these results correspond not only with the aforementioned IP knockout mice studies, but also parallel findings from the world-wide withdrawal of the selective COX-2 inhibitors (e.g., rofecoxib and valdecoxib) whose discriminating suppression of COX-2-derived prostacyclin (PGI2) resulted in increased risk of cardiovascular events (e.g., MI and thrombotic stroke), particularly in predisposed patients (Fitzgerald, 2004; White et al., 2004; Bresalier et al., 2005). These latter findings relating adverse cardiovascular events to unmatched suppression of COX-2-derived PGI2 have been widely discussed and demonstrate that a disrupted balance between PGI2 and TxA2 – favoring unopposed COX-1-derived TxA2 production – is responsible, at least in fair part, for pro-thrombotic and perhaps pro-atherogenic effects (McAdam et al., 1999; Vane, 2002). In a recent opinion article, Rovati et al. (2010) proposed that concomitant TP receptor antagonism, along with selective COX-2 inhibition (dual COXIB-TP antagonists), may abrogate such adverse cardiovascular events (caused by the imbalance between PGI2 and TxA2) and improve the safety profile of selective COX-2 inhibitors. Other groups have proposed this concept as well, but clinical trials have yet to be pursued. As another approach, Capone et al. (2010) suggest assessment of COX-2 activity in whole blood ex vivo, perhaps in combination with biomarkers – such as biochemical (urinary levels of 6-keto-PGF1α) and genetic (IP receptor and other prostacyclin-related polymorphisms) – as potential surrogate endpoints to assess for prostacyclin synthesis in vivo as a predictor of cardiovascular risk.
Novel Prostacyclin-Related Therapies in Inflammation
As the role of PGI2 is becoming more defined in inflammatory-related diseases, the development of novel agonists and antagonists for the IP receptor is at the forefront of research. As previously stated, the role of PGI2 in arthritic diseases (RA and OA) is one of a pro-inflammatory mediator, and the use of wide-ranging inhibitors of prostaglandin synthesis (NSAIDs and selective COX-2 inhibitors) has remained a mainstay of therapy. However, targeted antagonism of PGI2 activity has proven effective in reducing pain and inflammation in preclinical trials. Using a mono-iodoacetate (MIA)-induced rodent model of chronic OA, Pulichino et al. (2006) have shown that a novel arylamide compound (Keitz et al., 2004) with specific IP receptor antagonism significantly reduced joint discomfort in a dose-dependent manner, and with similar efficacy to diclofenac as well as an MF-tricyclic COX-2 inhibitor. Furthermore, in a collagen-antibody-induced model of RA, the same IP antagonist reduced mean scores for all arthritic parameters by 93% (AUC of the clinical scores) when given in prophylactic mode 1 day prior to collagen antibody injection, in IP+/− mice (Pulichino et al., 2006). Comparably, a 91% reduction in arthritic scores was observed for IP−/− mice and a 98% reduction for COX-2-treated mice. Interestingly, treatment in therapeutic mode (6 days post-injection) had no effect on clinical scores (Pulichino et al., 2006). However, as discussed earlier, targeted PGI2 antagonism has the potential for increased risk of adverse cardiovascular effects, which may be even more pronounced than with broader-spectrum NSAIDs or selective COX-2 inhibitor therapies.
For the treatment of pulmonary vascular disease such as PAH and IPF, the vasoactive PGI2 analog formulations (e.g., epoprostenol, treprostinil, and iloprost) are all in clinical use, but are reserved for advanced therapy for persistent disease (Barst et al., 2009). Moreover, these agents have limitations, including short half-lives, parenteral (non-oral) routes of administration, and heterogeneous therapeutic response. Thus, the quest for novel therapies for these diseases is ongoing. In preclinical studies of both PAH and IPF, a novel, non-prostanoid prostacyclin receptor agonist (ONO-1301, Toray Industries) that also has potent inhibitory activity against thromboxane synthase (TBXAS1) has shown promise. Murakami et al. (2006) have demonstrated attenuation of bleomycin-induced pulmonary fibrosis, as well as improved survival in mice, using ONO-1301. Kataoka et al. (2005) demonstrated similar protective results in a monocrotaline-induced PAH model, whereby rats treated with ONO-1301 showed improved pulmonary hemodynamics and survival, along with a reduction of vascular remodeling (media hypertrophy) and plasma TxA2 metabolites. Moreover, the pharmacokinetic profile (half-life approximately 5.6 h) of ONO-1301 appears to be better than some of the PGI2 analogs in current therapeutic use (Antoniu, 2006) and, thus, if the same clinical efficacy can be proved in humans, ONO-1301 could be a new therapy for PAH and IPF. In lieu of this, several new orally available, non-prostanoid, selective IP receptor agonists have reached clinical trial status for PAH, including APD811 (Arena Pharmaceuticals), which is in Phase I and NS-304, a.k.a., Selexipag (Actelion Pharmaceuticals; Kuwano et al., 2007, 2008), which is currently in Phase III.
With the imbalance between PGI2 and TxA2 playing an important role in atherogenesis and atherothrombosis, the development of dual-acting compounds seems like a promising direction for the development of novel therapeutic agents. In a similar fashion to the aforementioned dual COXIB-TP antagonists, both Miyamoto and Yamada have demonstrated the potential for TRA-418 (Toray Industries), a novel compound with both PGI2 (IP receptor) agonistic and TxA2 (TP receptor) antagonistic activities (Miyamoto et al., 2003; Yamada et al., 2003). In vitro studies showed that TRA-418 inhibited platelet aggregation through impedance of both glycoprotein IIb/IIIa (GPIIb/IIIa) activation and P-selectin expression, which are key markers of platelet activation (Miyamoto et al., 2003). Moreover, the TRA-814 compound was also shown to inhibit platelet–leukocyte complex formation in a dose-dependent manner (Miyamoto et al., 2010). These results suggest that this compound may be useful as both an anti-thrombotic and anti-atherogenic agent. Similarly, Ohno et al. (2005) have developed a dual-acting benzofuran compound (Toray Industries) that possesses similar PGI2 agonism and TxA2 antagonism, which maintains potent anti-platelet activity with minimal effect on blood vessel dilation.
Thus, while the current pool of PGI2-centered compounds is primarily investigational, and clinical safety and efficacy in human disease still largely unproven, their development emphasizes the centrality of PGI2 activity in various inflammatory-mediated pathologies, and provides exciting new directions to steer drug development.
Conclusion
The culmination of data presented in this review reinforces the notion that Bunting et al. (1983) put forth almost 30 years ago – that a dynamic balance between the prostaglandins prostacyclin (PGI2) and thromboxane A2 (TxA2; in addition to many other mediators) is crucial in maintaining cardiovascular homeostasis, and has critical pathophysiological and therapeutic implications. However, we are just now realizing the potential breadth and scope of this seminal proposition with PGI2 playing a central role. Our current knowledge of PGI2, as both a physiological–pathophysiological mediator and therapeutic agent, in a host of inflammatory-related diseases, is growing rapidly. As demonstrated, PGI2 has been shown to play protective roles in atherogenesis – relating to CAD, MI, stroke, and other cardiovascular abnormalities. It has also been shown to be involved in certain fibro-proliferative and pulmonary vascular diseases, such as IPF and PAH, where it serves as both a protective factor and first-line pharmacotherapy. Furthermore, in the setting of RA, PGI2 seems to play a pro-inflammatory role and, as evidence increases, perhaps may one day be considered a therapeutic target in RA and related disorders (Figure 2). Further study of this important prostaglandin, in both the realms of basic science and clinical medicine, is needed and will undoubtedly yield new insights into inflammatory disorders and pharmacological treatments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH), the National Heart, Lung, and Blood Institute (NHLBI), and the American Heart Association (AHA).
References
Antoniu, S. A. (2006). Non-prostanoid prostacyclin agonists for the treatment of pulmonary arterial hypertension. Expert Opin. Investig. Drugs 15, 327–330.
Arehart, E., Stitham, J., Asselbergs, F. W., Douville, K., Mackenzie, T., Fetalvero, K. M., Gleim, S., Kasza, Z., Rao, Y., Martel, L., Segel, S., Robb, J., Kaplan, A., Simons, M., Powell, R. J., Moore, J. H., Rimm, E. B., Martin, K. A., and Hwa, J. (2008). Acceleration of cardiovascular disease by a dysfunctional prostacyclin receptor mutation: potential implications for cyclooxygenase-2 inhibition. Circ. Res. 102, 986–993.
Badesch, D. B., Abman, S. H., Simonneau, G., Rubin, L. J., and Mclaughlin, V. V. (2007). Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest 131, 1917–1928.
Barst, R. J., Gibbs, J. S., Ghofrani, H. A., Hoeper, M. M., Mclaughlin, V. V., Rubin, L. J., Sitbon, O., Tapson, V. F., and Galie, N. (2009). Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J. Am. Coll. Cardiol. 54, S78–S84.
Barst, R. J., Rubin, L. J., Walker, A., Long, W. A., McGoon, M. D., Rich, S., Badesch, D. B., Groves, B.M., Tapson, V.F., Bourge, R. C., Brundage, B. H., Koerner, S. K., Langleben, D., Keller, C. A., Murali, S., Uretsky, B. F., Clayton, L. M., Pharm, D., Jöbsis, M. M., Blackburn, S. D. Jr., Shortino, D., and Crow, J. W. for the Primary Pulmonary Hypertension Study Group. (1996). A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N. Engl. J. Med. 334, 296–302.
Blindt, R., Bosserhoff, A. K., Vom Dahl, J., Hanrath, P., Schror, K., Hohlfeld, T., and Meyer-Kirchrath, J. (2002). Activation of IP and EP(3) receptors alters cAMP-dependent cell migration. Eur. J. Pharmacol. 444, 31–37.
Boie, Y., Rushmore, T. H., Darmon-Goodwin, A., Grygorczyk, R., Slipetz, D. M., Metters, K. M., and Abramovitz, M. (1994). Cloning and expression of a cDNA for the human prostanoid IP receptor. J. Biol. Chem. 269, 12173–12178.
Bresalier, R. S., Sandler, R. S., Quan, H., Bolognese, J. A., Oxenius, B., Horgan, K., Lines, C., Riddell, R., Morton, D., Lanas, A., Konstam, M. A., and Baron, J. A. (2005). Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N. Engl. J. Med. 352, 1092–1102.
Brodie, M. J., Hensby, C. N., Parke, A., and Gordon, D. (1980). Is prostacyclin in the major pro-inflammatory prostanoid in joint fluid? Life Sci. 27, 603–608.
Bunting, S., Moncada, S., and Vane, J. R. (1983). The prostacyclin – thromboxane A2 balance: pathophysiological and therapeutic implications. Br. Med. Bull. 39, 271–276.
Campbell, G. R., Campbell, J. H., Manderson, J. A., Horrigan, S., and Rennick, R. E. (1988). Arterial smooth muscle. A multifunctional mesenchymal cell. Arch. Pathol. Lab. Med. 112, 977–986.
Capone, M. L., Tacconelli, S., Rodriguez, L. G., and Patrignani, P. (2010). NSAIDs and cardiovascular disease: transducing human pharmacology results into clinical read-outs in the general population. Pharmacol. Rep. 62, 530–535.
Catella-Lawson, F., Mcadam, B., Morrison, B. W., Kapoor, S., Kujubu, D., Antes, L., Lasseter, K. C., Quan, H., Gertz, B. J., and Fitzgerald, G. A. (1999). Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J. Pharmacol. Exp. Ther. 289, 735–741.
Chen, Y. F., Jobanputra, P., Barton, P., Bryan, S., Fry-Smith, A., Harris, G., and Taylor, R. S. (2008). Cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib and lumiracoxib) for osteoarthritis and rheumatoid arthritis: a systematic review and economic evaluation. Health Technol. Assess. 12, 1–278, iii.
Cheng, Y., Austin, S. C., Rocca, B., Koller, B. H., Coffman, T. M., Grosser, T., Lawson, J. A., and Fitzgerald, G. A. (2002). Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 296, 539–541.
Christman, B. W., Mcpherson, C. D., Newman, J. H., King, G. A., Bernard, G. R., Groves, B. M., and Loyd, J. E. (1992). An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N. Engl. J. Med. 327, 70–75.
Crofford, L. J., Wilder, R. L., Ristimaki, A. P., Sano, H., Remmers, E. F., Epps, H. R., and Hla, T. (1994). Cyclooxygenase-1 and -2 expression in rheumatoid synovial tissues. Effects of interleukin-1 beta, phorbol ester, and corticosteroids. J. Clin. Invest. 93, 1095–1101.
Davies, P., Bailey, P. J., Goldenberg, M. M., and Ford-Hutchinson, A. W. (1984). The role of arachidonic acid oxygenation products in pain and inflammation. Annu. Rev. Immunol. 2, 335–357.
Della Bella, S., Molteni, M., Mocellin, C., Fumagalli, S., Bonara, P., and Scorza, R. (2001). Novel mode of action of iloprost: in vitro down-regulation of endothelial cell adhesion molecules. Prostaglandins Other Lipid Mediat. 65, 73–83.
Egan, K. M., Lawson, J. A., Fries, S., Koller, B., Rader, D. J., Smyth, E. M., and Fitzgerald, G. A. (2004). Cox-2-derived prostacyclin confers atheroprotection on female mice. Science 306, 1954–1957.
Essop, M. R. (2010). Contemporary insights into the pathogenesis, diagnosis and therapy of pulmonary arterial hypertension. Cardiovasc. J. Afr. 21, 334–337.
Farber, H. W., and Loscalzo, J. (2004). Pulmonary arterial hypertension. N. Engl. J. Med. 351, 1655–1665.
Fetalvero, K. M., Martin, K. A., and Hwa, J. (2007). Cardioprotective prostacyclin signaling in vascular smooth muscle. Prostaglandins Other Lipid Mediat. 82, 109–118.
Fetalvero, K. M., Shyu, M., Nomikos, A. P., Chiu, Y. F., Wagner, R. J., Powell, R. J., Hwa, J., and Martin, K. A. (2006). The prostacyclin receptor induces human vascular smooth muscle cell differentiation via the protein kinase A pathway. Am. J. Physiol. Heart Circ. Physiol. 290, H1337–H1346.
Galie, N., Ghofrani, H. A., Torbicki, A., Barst, R. J., Rubin, L. J., Badesch, D., Fleming, T., Parpia, T., Burgess, G., Branzi, A., Grimminger, F., Kurzyna, M., and Simonneau, G. (2005). Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 353, 2148–2157.
Gomberg-Maitland, M., and Olschewski, H. (2008). Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur. Respir. J. 31, 891–901.
Grosser, T., Bonisch, D., Zucker, T. P., and Schror, K. (1995). Iloprost-induced inhibition of proliferation of coronary artery smooth muscle cells is abolished by homologous desensitization. Agents Actions Suppl. 45, 85–91.
Hajjar, D. P., Marcus, A. J., and Etingin, O. R. (1989). Platelet-neutrophil-smooth muscle cell interactions: lipoxygenase-derived mono- and dihydroxy acids activate cholesteryl ester hydrolysis by the cyclic AMP dependent protein kinase cascade. Biochemistry 28, 8885–8891.
Hajjar, D. P., and Weksler, B. B. (1983). Metabolic activity of cholesteryl esters in aortic smooth muscle cells is altered by prostaglandins I2 and E2. J. Lipid Res. 24, 1176–1185.
Honda, T., Segi-Nishida, E., Miyachi, Y., and Narumiya, S. (2006). Prostacyclin-IP signaling and prostaglandin E2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. J. Exp. Med. 203, 325–335.
Ibrahim, S., Tetruashvily, M., Frey, A. J., Wilson, S. J., Stitham, J., Hwa, J., and Smyth, E. M. (2010). Dominant negative actions of human prostacyclin receptor variant through dimerization: implications for cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 30, 1802–1809.
Idzko, M., Hammad, H., Van Nimwegen, M., Kool, M., Vos, N., Hoogsteden, H. C., and Lambrecht, B. N. (2007). Inhaled iloprost suppresses the cardinal features of asthma via inhibition of airway dendritic cell function. J. Clin. Invest. 117, 464–472.
Iwai, N., Katsuya, T., Ishikawa, K., Mannami, T., Ogata, J., Higaki, J., Ogihara, T., Tanabe, T., and Baba, S. (1999). Human prostacyclin synthase gene and hypertension : the Suita Study. Circulation 100, 2231–2236.
Kasza, Z., Fetalvero, K. M., Ding, M., Wagner, R. J., Acs, K., Guzman, A. K., Douville, K. L., Powell, R. J., Hwa, J., and Martin, K. A. (2009). Novel signaling pathways promote a paracrine wave of prostacyclin-induced vascular smooth muscle differentiation. J. Mol. Cell. Cardiol. 46, 682–694.
Kataoka, M., Nagaya, N., Satoh, T., Itoh, T., Murakami, S., Iwase, T., Miyahara, Y., Kyotani, S., Sakai, Y., Kangawa, K., and Ogawa, S. (2005). A long-acting prostacyclin agonist with thromboxane inhibitory activity for pulmonary hypertension. Am. J. Respir. Crit. Care Med. 172, 1575–1580.
Keitz, P. F., Jahangir, A., Lopez-Tapia, F. J., and O’Yang, C. (2004). Substituted Arylamides as IP Antagonists. US Patent 2004/0220247A1. Palo Alto, CA: Roche.
Kobayashi, T., Tahara, Y., Matsumoto, M., Iguchi, M., Sano, H., Murayama, T., Arai, H., Oida, H., Yurugi-Kobayashi, T., Yamashita, J. K., Katagiri, H., Majima, M., Yokode, M., Kita, T., and Narumiya, S. (2004). Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J. Clin. Invest. 114, 784–794.
Komhoff, M., Lesener, B., Nakao, K., Seyberth, H. W., and Nusing, R. M. (1998). Localization of the prostacyclin receptor in human kidney. Kidney Int. 54, 1899–1908.
Kuwano, K., Hashino, A., Asaki, T., Hamamoto, T., Yamada, T., Okubo, K., and Kuwabara, K. (2007). 2-[4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy]-N-(methylsulfonyl )acetamide (Ns-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J. Pharmacol. Exp. Ther. 322, 1181–1188.
Kuwano, K., Hashino, A., Noda, K., Kosugi, K., and Kuwabara, K. (2008). A long-acting and highly selective prostacyclin receptor agonist prodrug, 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl)acetamide (Ns-304), ameliorates rat pulmonary hypertension with unique relaxant responses of its active form, {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}acetic acid (Mre-269), on rat pulmonary artery. J. Pharmacol. Exp. Ther. 326, 691–699.
Lawler, O. A., Miggin, S. M., and Kinsella, B. T. (2001). Protein kinase A-mediated phosphorylation of serine 357 of the mouse prostacyclin receptor regulates its coupling to G(s)-, to G(i)-, and to G(q)-coupled effector signaling. J. Biol. Chem. 276, 33596–33607.
Lewis, P. J., and Dollery, C. T. (1983). Clinical pharmacology and potential of prostacyclin. Br. Med. Bull. 39, 281–284.
Libby, P., Ridker, P. M., and Maseri, A. (2002). Inflammation and atherosclerosis. Circulation 105, 1135–1143.
Lim, H., and Dey, S. K. (2002). A novel pathway of prostacyclin signaling-hanging out with nuclear receptors. Endocrinology 143, 3207–3210.
Lovgren, A. K., Jania, L. A., Hartney, J. M., Parsons, K. K., Audoly, L. P., Fitzgerald, G. A., Tilley, S. L., and Koller, B. H. (2006). Cox-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 291, L144–L156.
McAdam, B. F., Catella-Lawson, F., Mardini, I. A., Kapoor, S., Lawson, J. A., and Fitzgerald, G. A. (1999). Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of Cox-2. Proc. Natl. Acad. Sci. U.S.A. 96, 272–277.
McLaughlin, V. V., Sitbon, O., Badesch, D. B., Barst, R. J., Black, C., Galie, N., Rainisio, M., Simonneau, G., and Rubin, L. J. (2005). Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur. Respir. J. 25, 244–249.
Miyamoto, M., Ohno, M., Yamada, N., Ohtake, A., and Matsushita, T. (2010). Tra-418, a thromboxane A2 receptor antagonist and prostacyclin receptor agonist, inhibits platelet-leukocyte interaction in human whole blood. Thromb. Haemost. 104, 788–795.
Miyamoto, M., Yamada, N., Ikezawa, S., Ohno, M., Otake, A., Umemura, K., and Matsushita, T. (2003). Effects of Tra-418, a novel TP-receptor antagonist, and IP-receptor agonist, on human platelet activation and aggregation. Br. J. Pharmacol. 140, 889–894.
Moncada, S., Higgs, E. A., and Vane, J. R. (1977). Human arterial and venous tissues generate prostacyclin (prostaglandin x), a potent inhibitor of platelet aggregation. Lancet 1, 18–20.
Murakami, S., Nagaya, N., Itoh, T., Kataoka, M., Iwase, T., Horio, T., Miyahara, Y., Sakai, Y., Kangawa, K., and Kimura, H. (2006). Prostacyclin agonist with thromboxane synthase inhibitory activity (Ono-1301) attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L59–L65.
Murata, T., Ushikubi, F., Matsuoka, T., Hirata, M., Yamasaki, A., Sugimoto, Y., Ichikawa, A., Aze, Y., Tanaka, T., Yoshida, N., Ueno, A., Oh-Ishi, S., and Narumiya, S. (1997). Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature 388, 678–682.
Nakagawa, O., Tanaka, I., Usui, T., Harada, M., Sasaki, Y., Itoh, H., Yoshimasa, T., Namba, T., Narumiya, S., and Nakao, K. (1994). Molecular cloning of human prostacyclin receptor cDNA and its gene expression in the cardiovascular system. Circulation 90, 1643–1647.
Nakayama, T. (2005). Prostacyclin synthase gene: genetic polymorphisms and prevention of some cardiovascular diseases. Curr. Med. Chem. Cardiovasc. Hematol. Agents 3, 157–164.
O’Brien, E. R., Alpers, C. E., Stewart, D. K., Ferguson, M., Tran, N., Gordon, D., Benditt, E. P., Hinohara, T., Simpson, J. B., and Schwartz, S. M. (1993). Proliferation in primary and restenotic coronary atherectomy tissue. Implications for antiproliferative therapy. Circ. Res. 73, 223–231.
Ohno, M., Miyamoto, M., Hoshi, K., Takeda, T., Yamada, N., and Ohtake, A. (2005). Development of dual-acting benzofurans for thromboxane A2 receptor antagonist and prostacyclin receptor agonist: synthesis, structure-activity relationship, and evaluation of benzofuran derivatives. J. Med. Chem. 48, 5279–5294.
Olschewski, H., Ghofrani, H. A., Walmrath, D., Schermuly, R., Temmesfeld-Wollbruck, B., Grimminger, F., and Seeger, W. (1999). Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. Am. J. Respir. Crit. Care Med. 160, 600–607.
Olschewski, H., Hoeper, M. M., Behr, J., Ewert, R., Meyer, A., Borst, M. M., Winkler, J., Pfeifer, M., Wilkens, H., Ghofrani, H. A., Nikkho, S., and Seeger, W. (2010). Long-term therapy with inhaled iloprost in patients with pulmonary hypertension. Respir. Med. 104, 731–740.
Patrignani, P., DI Febbo, C., Tacconelli, S., Douville, K., Guglielmi, M. D., Horvath, R. J., Ding, M., Sierra, K., Stitham, J., Gleim, S., Baccante, G., Moretta, V., DI Francesco, L., Capone, M. L., Porreca, E., and Hwa, J. (2008). Differential association between human prostacyclin receptor polymorphisms and the development of venous thrombosis and intimal hyperplasia: a clinical biomarker study. Pharmacogenet. Genomics 18, 611–620.
Pepke-Zaba, J., Gilbert, C., Collings, L., and Brown, M. C. (2008). Sildenafil improves health-related quality of life in patients with pulmonary arterial hypertension. Chest 133, 183–189.
Pomerantz, K. B., Fleisher, L. N., Tall, A. R., and Cannon, P. J. (1985). Enrichment of endothelial cell arachidonate by lipid transfer from high density lipoproteins: relationship to prostaglandin I2 synthesis. J. Lipid Res. 26, 1269–1276.
Pulichino, A. M., Rowland, S., Wu, T., Clark, P., Xu, D., Mathieu, M. C., Riendeau, D., and Audoly, L. P. (2006). Prostacyclin antagonism reduces pain and inflammation in rodent models of hyperalgesia and chronic arthritis. J. Pharmacol. Exp. Ther. 319, 1043–1050.
Rovati, G. E., Sala, A., Capra, V., Dahlen, S. E., and Folco, G. (2010). Dual COXIB/TP antagonists: a possible new twist in NSAID pharmacology and cardiovascular risk. Trends Pharmacol. Sci. 31, 102–107.
Rubin, L. J., Badesch, D. B., Barst, R. J., Galie, N., Black, C. M., Keogh, A., Pulido, T., Frost, A., Roux, S., Leconte, I., Landzberg, M., and Simonneau, G. (2002). Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 346, 896–903.
Simonneau, G., Robbins, I. M., Beghetti, M., Channick, R. N., Delcroix, M., Denton, C. P., Elliott, C. G., Gaine, S. P., Gladwin, M. T., Jing, Z. C., Krowka, M. J., Langleben, D., Nakanishi, N., and Souza, R. (2009). Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 54, S43–S54.
Sitbon, O., Badesch, D. B., Channick, R. N., Frost, A., Robbins, I. M., Simonneau, G., Tapson, V. F., and Rubin, L. J. (2003). Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary arterial hypertension: a 1-year follow-up study. Chest 124, 247–254.
Smyth, E. M., Grosser, T., Wang, M., Yu, Y., and Fitzgerald, G. A. (2009). Prostanoids in health and disease. J. Lipid Res. 50(Suppl.), S423–S428.
Stamatelopoulos, K. S., Kitas, G. D., Papamichael, C. M., Chryssohoou, E., Kyrkou, K., Georgiopoulos, G., Protogerou, A., Panoulas, V. F., Sandoo, A., Tentolouris, N., Mavrikakis, M., and Sfikakis, P. P. (2009). Atherosclerosis in rheumatoid arthritis versus diabetes: a comparative study. Arterioscler. Thromb. Vasc. Biol. 29, 1702–1708.
Stitham, J., Stojanovic, A., Merenick, B. L., O’Hara, K. A., and Hwa, J. (2003). The unique ligand-binding pocket for the human prostacyclin receptor. Site-directed mutagenesis and molecular modeling. J. Biol. Chem. 278, 4250–4257.
Takahashi, Y., Tokuoka, S., Masuda, T., Hirano, Y., Nagao, M., Tanaka, H., Inagaki, N., Narumiya, S., and Nagai, H. (2002). Augmentation of allergic inflammation in prostanoid IP receptor deficient mice. Br. J. Pharmacol. 137, 315–322.
Thiemermann, C. (1991). Biosynthesis and interaction of endothelium-derived vasoactive mediators. Eicosanoids 4, 187–202.
Tuder, R. M., Cool, C. D., Geraci, M. W., Wang, J., Abman, S. H., Wright, L., Badesch, D., and Voelkel, N. F. (1999). Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 159, 1925–1932.
Vane, J. R. (1971). Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 231, 232–235.
Vinals, M., Martinez-Gonzalez, J., Badimon, J. J., and Badimon, L. (1997). Hdl-induced prostacyclin release in smooth muscle cells is dependent on cyclooxygenase-2 (Cox-2). Arterioscler. Thromb. Vasc. Biol. 17, 3481–3488.
Vinals, M., Martinez-Gonzalez, J., and Badimon, L. (1999). Regulatory effects of HDL on smooth muscle cell prostacyclin release. Arterioscler. Thromb. Vasc. Biol. 19, 2405–2411.
Wang, Y., Chen, X., Lian, L., Tang, T., Stalker, T. J., Sasaki, T., Kanaho, Y., Brass, L. F., Choi, J. K., Hartwig, J. H., and Abrams, C. S. (2008). Loss of PIP5KIbeta demonstrates that PIP5KI isoform-specific PIP2 synthesis is required for IP3 formation. Proc. Natl. Acad. Sci. U.S.A. 105, 14064–14069.
Weksler, B. B., Hajjar, D. P., Eldor, A., Falcone, D. J., Tack-Goldman, K., and Minick, C. R. (1983). Interactions between prostacyclin metabolism and cholesteryl ester metabolism in the vascular wall. Adv. Prostaglandin Thromboxane Leukot. Res. 11, 463–467.
White, W. B., Strand, V., Roberts, R., and Whelton, A. (2004). Effects of the cyclooxygenase-2 specific inhibitor valdecoxib versus nonsteroidal antiinflammatory agents and placebo on cardiovascular thrombotic events in patients with arthritis. Am. J. Ther. 11, 244–250.
Wilcox, J. N. (1992). Analysis of local gene expression in human atherosclerotic plaques. J. Vasc. Surg. 15, 913–916.
Xiao, C. Y., Hara, A., Yuhki Ki, K., Fujino, T., Ma, H., Okada, Y., Takahata, O., Yamada, T., Murata, T., Narumiya, S., and Ushikubi, F. (2001). Roles of prostaglandin i(2) and thromboxane a(2) in cardiac ischemia- reperfusion injury: a study using mice lacking their respective receptors. Circulation 104, 2210–2215.
Yamada, N., Miyamoto, M., Isogaya, M., Suzuki, M., Ikezawa, S., Ohno, M., Otake, A., and Umemura, K. (2003). Tra-418, a novel compound having both thromboxane A(2) receptor antagonistic and prostaglandin I(2) receptor agonistic activities: its antiplatelet effects in human and animal platelets. J. Thromb. Haemost. 1, 1813–1819.
Zhu, Y., Liu, Y., Zhou, W., Xiang, R., Jiang, L., Huang, K., Xiao, Y., Guo, Z., and Gao, J. (2010). A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice. Respir. Res. 11, 34.
Keywords: prostacyclin, IP receptor, inflammation, atherosclerosis, rheumatoid arthritis, pulmonary fibrosis
Citation: Stitham J, Midgett C, Martin KA and Hwa J (2011) Prostacyclin: an inflammatory paradox. Front. Pharmacol. 2:24. doi: 10.3389/fphar.2011.00024
Received: 17 February 2011;
Paper pending published: 10 April 2011;
Accepted: 29 April 2011;
Published online: 13 May 2011.
Edited by:
Angel Lanas, University of Zaragoza, SpainReviewed by:
Emer Smyth, University of Pennsylvania, USASteven W. Kerrigan, Royal College of Surgeons in Ireland, Ireland
Copyright: © 2011 Stitham, Midgett, Martin and Hwa. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: John Hwa, Section of Cardiovascular Medicine, Department of Internal Medicine, Yale School of Medicine, Cardiovascular Research Center, 300 George Street, Room 759H, New Haven, CT 06511, USA. e-mail: john.hwa@yale.edu