
- Faculty of Health Sciences, University of Eastern Finland, Kuopio, Finland
In silico toxicology in its broadest sense means “anything that we can do with a computer in toxicology.” Many different types of in silico methods have been developed to characterize and predict toxic outcomes in humans and environment. The term non-testing methods denote grouping approaches, structure–activity relationship, and expert systems. These methods are already used for regulatory purposes and it is anticipated that their role will be much more prominent in the near future. This Perspective will delineate the basic principles of non-testing methods and evaluate their role in current and future risk assessment of chemical compounds.
Introduction
We live in a world of chemicals. More than 60 million chemical compounds were known to exist as of 26 May 2011 (CAS1). Fortunately we are exposed to only a fraction of these during our lifetime. These include inadvertent exposures, e.g., pesticide residues and products of chemical industries, and deliberate exposures, e.g., cosmetics and drugs. Toxicology as a science and regulatory tool has the goal of ensuring the safety of humans, animals, and the environment. Assessment or risks of all categories of chemicals foreign to the body (xenobiotics) is still mainly based on animal experimentation. However, developments in knowledge of general pathophysiology, cellular pathways, genetics, and computer-supported modeling, have resulted in a better understanding of the molecular mechanisms of xenobiotic action and toxicity. Key elements in the integrated risk assessment approach are the evaluation of chemical functionalities representing structural alerts for toxic actions, the construction of kinetic models on the basis of non-animal data, and more specific tests for evaluating cell toxicity (Boobis et al., 2002; Nigsch et al., 2009).
In this Perspective I will introduce the concept of in silico toxicology and have a more detailed look into the so-called non-testing methods and their application in predicting biokinetic and target organ toxicity properties of compounds. I will also attempt to predict how these methods will be implemented in the near and more distant future in chemical safety evaluation. Technical jargon is avoided as this Perspective is aimed to the reader not familiar with the field.
What is in silico Toxicology?
In silico, a phrase coined as an analogy to the familiar phrases in vivo and in vitro, is an expression used to denote “performed on computer or via computer simulation.” In silico or computational toxicology is an area of very active development and great potential. In silico toxicology is difficult to define exactly, as today practically all toxicological research and risk assessment have major in silico components. The United States Environmental Protection Agency (US EPA) defines in silico toxicology as the “integration of modern computing and information technology with molecular biology to improve agency prioritization of data requirements and risk assessment of chemicals” (US EPA, 2003).
Hartung and Hoffmann (2009) defined in silico methodologies more liberally as “anything we can do with a computer in toxicology, and there are few tests that would not fall into this category, as most make use of computer-based planning and/or analysis.” They identified nine different types of in silico approaches, of which I will concentrate on what are called non-testing methods in the European Union (EU) vocabulary and the application of these methods in toxicity testing and regulation of chemicals.
In silico toxicology differs from traditional toxicology in many ways, but perhaps the most important is that of scale. Scale in the numbers of chemicals that are studied, breadth of endpoints and pathways covered, levels of biological organization examined, and range of exposure conditions considered simultaneously (Kavlock et al., 2008).
Drivers of in silico Toxicology
There are several scientific, economical, and societal drivers involving governments, academia, and industry, which promote development and use of in silico methods in toxicology.
A major portion of in silico technology was developed by the pharmaceutical industry for use in drug discovery. Environmental chemicals differ from drug candidates in many crucial ways. Drugs are developed toward specific targets in the human (and animal) body, are designed to possess physico-chemical properties that augment absorption, distribution, metabolism, and excretion, and have use patterns that are known and quantifiable. In contrast, environmental chemicals generally are not designed with biological activity in mind, cover extremely diverse chemical space, have poorly understood biokinetic profiles, and are generally evaluated at exposures levels well in excess of likely real world situations.
Several different kinds of in silico methods have been developed and applied in the academia and pharmaceutical industry to model pharmacodynamic, pharmacokinetic, and toxicological hypothesis development and testing. These in silico methods include databases, different kinds of quantitative SAR (QSAR) methods, pharmacophores, homology models and other molecular modeling approaches, machine learning, data mining, network analysis tools, and data analysis tools using computers. Different types of physiologically based pharmacokinetic (PBPK) models require also extensive computing. One should also make a distinction between fundamentally statistical methods and modeling approaches that attempt to describe the underlying chemical or biological system and use this approach to obtain predictions of system-level behavior. A vast and rapidly growing literature and a range of in silico tools are now available. Excellent recent reviews on the use of in silico methods in drug development include (Ekins et al., 2007a,2007b; Muster et al., 2008; Kortagere et al., 2009; Valerio, 2009; Cook, 2010; Merlot, 2010).
One of the goals of the European public–private initiative Innovative Medicines Initiative (IMI) is to develop in silico methods for predicting conventional and recently recognized types of toxicity (IMI, 2010). The IMI project eTOX (Integrating bioinformatics and chemoinformatics approaches for the development of expert systems allowing the in silico prediction of toxicities) aims to develop a drug safety database from the pharmaceutical industry toxicology reports and public toxicology data, innovative methodological strategies, and novel software tools to better predict the toxicological profiles of small molecules in early stages of the drug development process.
Currently, there is no specific US Food and Drug Administration (FDA) guidance dedicated to the use of these tools in the development of drugs. Computational toxicology data are submitted on a voluntary basis and are not required (Valerio, 2009). The situation is very similar with the European Medicines Agency (EMA).
In 2006, the EU completely revised its regulatory framework for chemicals with the passage of the regulation (EC, 2006. EuropeanComm:2006) concerning registration, evaluation, authorization, and restriction of chemicals (REACH). Alternative testing methods are urgently needed to fulfill the goal of reducing animal testing in REACH. The REACH regulation mentions non-testing methods for “predictive toxicology” in risk assessment of commercial chemicals in the EU. The regulation (EC, 2008. EuropeanComm:2008) on test methods to be applied for the purposes of REACH embraces a very limited set of in silico methods, but the Guidance on information requirements and chemical safety assessment issued by the European Chemicals Agency (ECHA) gives very detailed background information and recommendations for use of non-testing methods and grouping of chemicals (ECHA, 2008).
The seventh amendment to the EU Cosmetics Directive (EC, 1976. EuropeanComm:1976) foresees a regulatory framework with the aim of phasing out animal testing. The marketing ban has been in effect since March 2009 for all cosmetic ingredients investigated by animal studies intended to predict human health effects, with the exception of repeated-dose toxicity (including skin sensitization and carcinogenicity), reproductive, and developmental toxicity and toxicokinetics, for which the deadline is extended to 2013.
In the United States, the US National Research Council (NRC, 2007) report proposed an approach to assessing environmental chemicals based on identifying human toxicity pathways, modeling and interpreting these pathways using cellular and computational systems, and predicting levels of human exposure that are not likely to be associated with adverse effects. A key issue in the report is the use of systems biology and the “omics” approaches in identifying toxicity pathways as the basis for new methodologies in toxicity testing and dose response modeling, together with the application of computational modeling of toxicokinetics and toxicodynamics (Blaauboer, 2008; Andersen and Krewski, 2009; Valerio, 2009).
The NRC report envisions a future in which virtually all routine toxicity testing would be conducted in human cells or cell lines in vitro by evaluating cellular responses in a battery of toxicity pathway assays using high-throughput tests. Obviously, challenges exist in ensuring that the suite of assays provides sufficient coverage of toxicity pathways so as to capture the broad range of possible pathway perturbations. The identification and characterization of these pathways is not the sole province of the toxicology community; most research into these pathways comes from, and will continue to arise from, contemporary cell biology. These cell-signaling pathways are being enumerated through mechanistic studies from front-line biologists interested in responses of cells and organisms to various stressors (Kavlock et al., 2008; Andersen and Krewski, 2009).
Non-Testing Methods
According to the ECHA Guidance on information requirements and chemical safety assessment (ECHA, 2008), non-testing data can be generated by three main approaches: (1) grouping approaches, which include read-across and chemical category formation; (2) structure–activity relationship (SAR) and quantitative SAR (QSAR, a term used in the following text to imply both); and (3) expert systems. The development and application of all kinds of non-testing methods is based on the similarity principle, i.e., the hypothesis that similar compounds should have similar biological activities.
In more general terms, non-testing methods can be divided into two main classes, i.e., comprehensive (global) and specific (local) ones. Comprehensive methods, also called expert systems, mimic human reasoning and formalize existing knowledge. Predictive expert systems portray properties of a compound based on knowledge rules. Expert systems have an advantage over QSAR methods in that prediction is related to specific mechanisms. Specific systems generally apply to a narrow range of targets, e.g., specific receptors or enzymes.
Specific methods can be divided to ligand-based and target-based techniques. Ligand-based modeling such as QSAR involves active ligands without considering the 3-dimensional (3D) structure of the protein and the possible sites of interaction. QSAR is a mathematical model that correlates a quantitative measure of chemical structure to either a physical property or a biological effect (e.g., toxic outcome). The term quantitative in QSAR refers to the nature of the parameters (also called descriptors) used to make the prediction. A molecular descriptor provides a means of representing molecular structures in a numerical form. The number may be a theoretical attribute (e.g. relating to size or shape) or a measurable property. Linear regression analysis is often used but a variety of other multivariate statistical techniques are also used.
Structure-based methods calculate atomic interactions between ligands and their target macromolecules. They require 3D structures of both ligands and macromolecules and need more computational power. Algorithms from target- and ligand-based approaches can be integrated to gain additional structural insights and to further validate the individual models. Today ligand- and target-based methods are often combined (Höltje et al., 2003; van de Waterbeemd and Gifford, 2003; Johnson and Rodgers, 2006; Lewis and Ito, 2008).
Registration, evaluation, authorization, and restriction of chemicals opens the possibility of evaluating chemicals not only on a one-by-one basis, but by grouping chemicals in categories. In the ECHA Guidance (ECHA, 2008), the terms category approach and analog approach are used to describe techniques for grouping chemicals, whilst the term read-across is reserved for a technique of filling data gaps in either approach. These approaches involve categorizing potential chemical analogs based upon their degree of structural, reactivity, metabolic, and physico-chemical similarity to the chemical with missing toxicological data. It extends beyond structural similarity by including differentiation based upon chemical reactivity and addressing the potential that an analog and target molecule could show toxicologically significant metabolic convergence or divergence. In addition, it identifies differences in physico-chemical properties, which could affect bioavailability and consequently biological responses observed in vitro or in vivo. The approach provides a stepwise decision tree for categorizing the suitability of analogs, which qualitatively characterizes the strength of the evidence supporting the hypothesis of similarity and level of uncertainty associated with their use for read-across. The chemical category and QSAR concepts are strongly connected. The concept of forming chemical categories and then using measured data on a few category members to estimate the missing values for the untested members is a common sense application of QSAR. The reason this concept is so compatible with QSAR is that this broad description of the categories concept and the historical description of QSAR are one and the same (ECHA, 2008; Wu et al., 2010).
Toxicokinetics, Xenobiotic Metabolism, and Structural Alerts
The term ADME refers to absorption, distribution, metabolism, and excretion, the four processes related to the pharmacokinetic profile of substances interacting with living organisms. In toxicology, ADME is often called toxicokinetics or biokinetics. Collectively, these processes determine the fate of the substance inside the body. The term ADMET is used to express the overall profiling of ADME properties and toxic effects of a substance. The ADME properties of compounds are important in discriminating between the toxicological profiles of parent compounds and their metabolites or degradation products. Metabolism plays a crucial role in pharmacological and toxicological effects caused by xenobiotics. There are two facets to metabolism. First, metabolism leads to termination of the action of a compound and renders it excretable from the body. Second, the same system sometimes produces metabolites that are toxic. In these cases parent compounds are often transformed into reactive electrophiles, which bind covalently with proteins and DNA. Formation of reactive metabolites is the main initial mechanism of chemical carcinogenesis and also explains a large proportion of the so-called idiosyncratic drug adverse reactions (Liebler and Guengerich, 2005; Walker et al., 2009).
The cytochrome P450 (CYP) enzymes constitute a large superfamily of heme proteins capable of metabolizing a vast number of exogenous and endogenous compounds. About 10 CYP forms are responsible for the metabolism of xenobiotics in humans. The CYPs metabolize, e.g., polycyclic aromatic hydrocarbons (ambient air), aromatic amines (occupational exposure), heterocyclic amines (food), pesticides and herbicides, and virtually all drugs. Ligands to CYP enzymes are either substrates, i.e., are metabolized by the enzyme, or act as inhibitors and thus block substrate turnover (Pelkonen et al., 2008). Recent progress in molecular modeling of CYP enzymes demonstrate that it is possible to generate realistic models for the xenobiotic metabolizing human CYPs that compare favorably with crystal structures, and thus may be used to derive substrate binding energies that agree closely with experimental kinetic values (Stjernschantz et al., 2008; Pelkonen et al., 2009). A workshop sponsored by European Centre for the Validation of Alternative Methods (ECVAM; Coecke et al., 2006) identified lack of knowledge in metabolism aspects as a key bottleneck in the development of in vitro toxicity tests.
Many functional groups in chemical structures are known to be associated with formation of reactive metabolites, very often catalyzed by the CYP enzymes. However, not all compounds with such functional groups are toxic, since formation of reactive intermediates is limited by the ability of CYPs to activate them. In this process, the orientation of the compound in the enzyme active site is critical, as only compounds with specific steric and electrostatic properties will orientate critical atoms toward the heme iron (Hollenberg et al., 2008). The crystal structures are now available for several mammalian CYP enzymes. These structures allow visualizing the docking of ligands in CYP proteins. However, these snapshots do not provide a complete picture. Based on comparisons between substrate-free and substrate-bound CYP structures, it can be concluded that substrate binding causes conformational changes in the enzyme, the extent of which is dependent on the nature of the substrate and the CYP involved (Isin and Guengerich, 2008).
The need to assess the ability of a chemical to act as a mutagen or a genotoxic carcinogen (collectively termed genotoxicity) is one of the primary requirements in regulatory toxicology. A key step in the development of chemical categories for genotoxicity is defining the organic chemistry associated with the formation of a covalent bond between DNA and an exogenous chemical. This organic chemistry is typically defined as structural alerts. In a recent paper, Enoch and Cronin (2010) identified 57 structural alerts associated with mutagenicity and genotoxic carcinogenicity. Importantly, the identification of 22 new structural alerts identified from idiosyncratic drug toxicity data has significantly improved the coverage of the chemical universe to which these alerts can be applied. In contrast to the historical alert compilations, this work has taken an additional step, and compiled the detailed mechanistic chemistry reaction associated with each of the alerts into a single source.
Several commercial and public in silico methods are available for assessing ADMET properties. These methods predict compound physico-chemical properties, gastrointestinal permeability, blood–brain barrier permeability, binding to plasma proteins, affinity for transporter proteins, metabolic clearance, potential to inhibit or induce drug metabolizing enzymes (especially CYPs), and generation of reactive metabolites. A report on alternative methods for cosmetics testing (Adler et al., 2011) concluded that a whole array of in vitro/in silico methods at various levels of development is available for most of the steps and mechanisms which govern the toxicokinetics of cosmetic substances. One exception is renal excretion for which until now no in vitro/in silico methods are available; thus there is an urgent need for further developments in this area. The same limitations apply to testing all other chemical compounds to which we are exposed.
Biokinetic Modeling and Integration of Data
In drug development and environmental toxicology, PBPK modeling has become a cornerstone approach to organize and integrate input from in vivo, in vitro, and in silico studies. PBPK models can be used to predict concentration–time profiles if their parameter values can be determined from in vitro data, or in vivo data in humans, from QSAR models, or from the scientific literature. PBPK models are evolved compartmental models which try to use realistic biological descriptions of the determinants of the disposition of a chemical compound in the body (Loizou et al., 2008).
Analogously, the advancements made in physiologically based biokinetic (PBBK) modeling have substantially increased the possibilities to give a better interpretation of in vitro and in silico toxicity data for their relevance in terms of a toxic dose in the in vivo situation. These models are usually used to estimate the concentration in a particular tissue, given a certain dose (or external exposure pattern). And vice versa, an effective toxic concentration determined in a relevant in vitro system (i.e., relevant for the toxic effect in the target tissue) can be used as input in the PBBK model to estimate the external dose that would result in the effective concentration in the target tissue (Blaauboer, 2008; Pelkonen, 2010).
The concept of integrated testing as defined by Blaauboer et al. (1999) applies also today: “An integrated testing strategy is any approach to the evaluation of toxicity which is based on the use of two or more of the following: physicochemical, in vitro, human (e.g., epidemiological, clinical case reports), animal data (as available or if unavoidable), computational methods, structural alerts and kinetic models.”
In practice, in silico predictions are not used in isolation. They already constitute a part of the weight of evidence approach, or an integrated testing strategy. An evidence based approach involves an assessment of the relative values/weights of different pieces of the available information that have been retrieved and gathered. These weights/values can be assigned either in an objective way by using a formalized procedure or by using expert judgment. The weight given to the available evidence will be influenced by factors such as the quality of the data, consistency of results, nature and severity of effects, relevance of the information for the given regulatory endpoint (ECHA, 2010).
Validation of Methods
Models and simulations without experimental data are empty exercises, so the experimental data must be correct and reproducible (Pelkonen, 2010). There is widespread agreement that in silico models should be scientifically valid or validated if they are to be used in the regulatory assessment of chemicals. In the EU, the concept of scientifically valid model is incorporated into the REACH text. Since the concept of validation is incorporated into legal texts and regulatory guidelines, it is important to clearly define what it means, and to describe what the validation process might entail. For the purposes of REACH, an assessment of QSAR model validity should be performed by reference to the internationally agreed principles for the validation of QSARs. The validation exercise itself may be carried out by any person or organization, but it will be the industry registrant of the chemical who needs to argue the case for using the QSAR data in the context of the registration process. This is consistent with a key principle of REACH that the responsibility for demonstrating the safe use of chemicals lies with industry (ECHA, 2008). It should be emphasized that the ECVAM is not mandated to carry out validation of in silico methods.
The principles for QSAR validation by the Organisation for Economic Co-operation and Development (OECD, 2004. OECDPrinci:2004) state that in order: “to facilitate the consideration of a (Q)SAR model for regulatory purposes, it should be associated with the following information:
1. a defined endpoint;
2. an unambiguous algorithm;
3. a defined domain of applicability;
4. appropriate measures of goodness-of-fit, robustness and predictivity;
5. a mechanistic interpretation, if possible.”
A guidance document has been produced to provide practical guidance on the interpretation of these OECD principles (OECD, 2007).
An important issue in model validation is the definition of its applicability domain. The applicability domain of a QSAR is the physico-chemical, structural or biological space, knowledge or information on which the training set of the model has been developed, and for which it is applicable to make predictions for new compounds. Therefore QSAR models are associated with limitations as to what they can reliably be used (Jaworska et al., 2005). A valid QSAR will be associated with at least one defined applicability domain in which the model makes estimations with a defined level of accuracy (reliability). When applied to chemicals within its applicability domain, the model is considered to give reliable results (ECHA, 2008). The ultimate case of falling out of the applicability domain is stated in the old saying in computing sciences: “garbage in, garbage out.” In other words, input of inappropriate (or poor quality) data will give false results.
Despite the significant advancements of several independent validation studies, the use of in silico tools must be taken cautiously in the context of their current capability due to the quality of available bioassay data, lack of widespread understanding of model construction (the black box dilemma), limited chemical space of training data sets (i.e., limited applicability domain), and high potential for multiple mechanisms of compound toxicity that cannot at present be modeled. These uncertainties in the technology do not render the approaches useless but point out they should be cautiously taken into consideration. There may be a workable balance in terms of customized computational platforms amenable to the hazard identification and risk characterization processes for obtaining desired performance of a model (e.g., sensitivity vs. specificity; Valerio, 2009).
Implementation
Although technical progress has been rapid, regulatory acceptance of QSAR approaches has been slow. The situation is changing as there is now a clear regulatory need for the use of QSAR. The improved in silico technologies in regulatory toxicology presents an opportunity to bridge the communication gap between toxicologists and QSAR experts. The extensive work which is going on to further develop mechanistic understanding of toxicological effects seen in vivo and in vitro is a reason for optimism.
There are currently no internationally accepted in silico alternative in the sense of a full replacement for all testing of a specific hazard. Regulatory use so far has been limited mainly to prioritization of test demands or filling data gaps in some cases, especially for low risk chemicals (Hartung and Hoffmann, 2009). It should be stressed that QSAR estimates are already used routinely for predicting some key environmental fate parameters of organic substances, partly because the experimental determination of these parameters can be difficult and/or expensive, and partly because the information is not normally required in the regulatory submissions (ECHA, 2008).
The REACH regulation leaves a lot of room to practical implementation of in silico tools (EC, 2006. EuropeanComm:2006; Schaafsma et al., 2009). Classification of substances, especially for existing high-production volume substances, based only on computational toxicology is at this stage highly unlikely, especially since regulatory implementation is a consensus process. Broad waiving of testing for REACH because of negative in silico results is also rather unlikely, since not even validated cell systems are generally accepted for this. The most likely use in the mid-term will be the intelligent combination of in vitro, in silico, and in vivo information (Hartung and Hoffmann, 2009).
The process of QSAR acceptance under REACH will involve initial acceptance by industry and subsequent evaluation by the authorities, on a case-by-case basis. It is not foreseen that there will be a formal adoption process, in the same way that test methods are currently adopted in the EU and OECD. In other words, it is not anticipated that there will be an official, legally binding list of QSAR methods (ECHA, 2008).
The REACH principles induced the emergence of a comparable program from the US EPA named ToxCast (US EPA2). ToxCast promotes “the application of Computational Toxicology to assess the risk chemicals poses to human health and environment.” The current emphasis in the ToxCast program, which uses a variety of high-throughput tests and in silico methods, is on prioritization of compounds for targeted testing in animals.
Thus, regulatory officials in the EU and US agree on the need to modernize toxicological testing, but they have taken somewhat different approaches. Several trans-Atlantic cooperative efforts are already in place to help bridge the gap. Research collaboration was between the US Environmental Protection Agency (EPA) and the European Commission Joint Research Centre’s Institute for Health and Consumer Protection (IHCP). The common theme of this collaboration, based on sharing high-throughput toxicological profile data and data from integrated testing strategies based on computational and in vitro methods, is the shift toward a “toxicological pathway based hazard assessment” (IHCP, 2010. Institutef:2010).
A wide variety of publicly available and commercial computational tools has been developed that are suitable for the development and application of QSARs. Such tools include methods for a range of QSAR-related tasks, including data management and data mining, descriptor generation, molecular similarity analysis, analog searching, and hazard assessment. Due to the limited availability of freely accessible in silico software, there is a need to develop a range of transparent and open-source tools, which should eventually be available to all stakeholders in the regulatory process (especially industry and authorities). A prime example of such an open-access tool is the (Q)SAR Application Toolbox developed by OECD.
Table 1 lists examples of projects, organizations, and open-access software promoting use of in silico methods in toxicology. More comprehensive lists are available in review articles (Hou and Wang, 2008; Pelkonen et al., 2009; Valerio, 2009; Wang, 2009; Mostrag-Szlichtyng and Worth, 2010) and the ECHA guidance (ECHA, 2008). An extensive list of in silico tools can be found at the Swiss Institute of Bioinformatics Click2Drug homepage3.
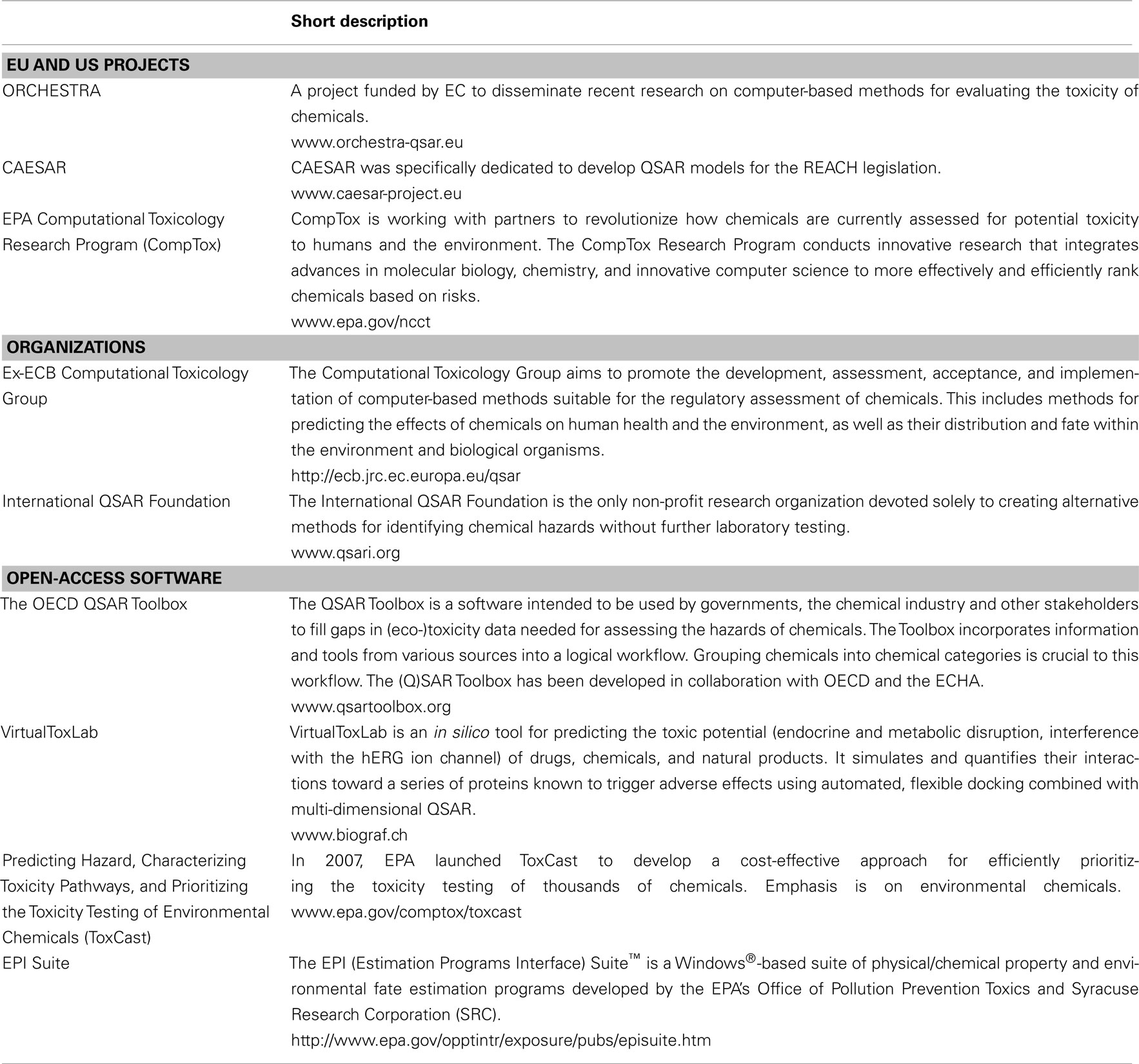
Table 1. Examples of in silico toxicology projects and tools.
Pros and Cons of in silico Approaches
As with any testing approaches, non-testing methods have their pros and cons. According to Valerio (2009), the advantages of these methods compared with in vitro and especially in vivo approaches include the following:
• higher throughput
• less expensive
• less time consuming
• constant optimization possible
• have higher reproducibility if the same model is used
• have low compound synthesis requirements
• have potential to reduce the use of animals
• Limitations include (Weaver and Gleeson, 2008; Valerio, 2009):
• quality and transparency of training set experimental data
• transparency of the program (what is being modeled?)
• descriptors sometimes confusing
• applicability domain sometimes not clear
• ADME features, especially metabolism, not taken into account
• carcinogenicity prediction does not work on non-genotoxic compounds
To date, positive experiences with QSAR approaches (>70% correct predictions, notably mostly not validated with external datasets) have been reported mainly for mutagenicity, sensitization, and aquatic toxicity, i.e., areas with relatively well understood mechanisms, not for complex/multiple endpoints. Hepatotoxicity, neurotoxicity, and developmental toxicity cannot be accurately predicted with in silico methods. Here, the perspective lies in breaking down complex endpoints into different steps or pathways with the common problem of how to validate these and put them together to make one prediction (Hartung and Hoffmann, 2009; Merlot, 2010).
Conclusion
“In silico tools have a bright future in toxicology. They add the objectivity and the tools to appraise our toolbox. They help to combine various approaches in more intelligent ways than a battery of tests” (Hartung and Hoffmann, 2009).
It is evident that the requirements for reducing animal testing in REACH and similar laws elsewhere in the world, together with rapid technological progress and economic incentives, are the main drivers of promoting use of in silico methods. Currently they play a role in providing mechanistic information and thus explaining underlying systems, but also in identifying testing needs and setting testing priorities. The whole process of implementing in silico methods into regulatory use is rapidly evolving. In the near future, we will see more and more results of in silico methods being included in risk assessment documents as supporting data.
The current risk assessment methodologies for chemicals regarding human toxicity endpoints are often derived from those for preclinical studies of pharmaceuticals. Obviously, the methods for hazard assessment are largely the same for industrial chemicals, pesticides, and drug candidates. It is likely that in silico methods will be used increasingly for the direct replacement of test data, as relevant and reliable models become available, and as experience in their use becomes more widespread. In the more distant future, we may well witness a gradual replacement of some classical in vivo tests by in vitro and in silico methods. Some in silico tools will be useful, but not widely available, due to their proprietary nature. Other tools are currently under development, or will need to be developed in the near future.
Ideally, in silico results can be used as stand-alone evidence for regulatory purposes if they are considered relevant, reliable and adequate for the purpose, and if they are documented properly. In practice, there may be uncertainty in one or more of these aspects, but this does not preclude the use of the in silico estimate in the context of a weight of evidence approach, in which additional information compensates for uncertainties resulting from a lack of information on the in silico method.
Final note: the term non-testing method, although probably politically correct, is misleading in the sense that a lot of in vivo and in vitro testing has gone into generating these methods, and will need to do so in the future also.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Adler, S., Basketter, D., Creton, S., Pelkonen, O., van Benthem, J., Zuang, V., Andersen, K. E., Angers-Loustau, A., Aptula, A., Bal-Price, A., Benfenati, E., Bernauer, U., Bessems, J., Bois, F. Y., Boobis, A., Brandon, E., Bremer, S., Broschard, T., Casati, S., Coecke, S., Corvi, R., Cronin, M., Daston, G., Dekant, W., Felter, S., Grignard, E., Gundert-Remy, U., Heinonen, T., Kimber, I., Kleinjans, J., Komulainen, H., Kreiling, R., Kreysa, J., Leite, S. B., Loizou, G., Maxwell, G., Mazzatorta, P., Munn, S., Pfuhler, S., Phrakonkham, P., Piersma, A., Poth, A., Prieto, P., Repetto, G., Rogiers, V., Schoeters, G., Schwarz, M., Serafimova, R., Tähti, H., Testai, E., van Delft, J., van Loveren, H., Vinken, M., Worth, A., and Zaldivar, J. M. (2011). Alternative (non-animal) methods for cosmetics testing: current status and future prospects-2010. Arch. Toxicol. 85, 367–485.
Andersen, M. E., and Krewski, D. (2009). Toxicity Testing in the 21st century: bringing the vision to life. Toxicol. Sci. 107, 324–330.
Blaauboer, B. J. (2008). The contribution of in vitro toxicity data in hazard and risk assessment: current limitations and future perspectives. Toxicol. Lett. 180, 81–84.
Blaauboer, B. J., Barratt, M. D., and Houston, J. B. (1999). The integrated use of alternative methods in toxicological risk evaluation. ECVAM integrated testing strategies task force report 1. Altern. Lab. Anim. 27, 229–237.
Boobis, A., Gundert-Remy, U., Kremers, P., Macheras, P., and Pelkonen, O. (2002). In silico prediction of ADME and pharmacokinetics. Report of an expert meeting organised by COST B15. Eur. J. Pharm. Sci. 17, 183–193.
Coecke, S., Ahr, H., Blaauboer, B. J., Bremer, S., Casati, S., Castell, J., Combes, R., Corvi, R., Crespi, C. L., Cunningham, M. L., Elaut, G., Eletti, B., Freidig, A., Gennari, A., Ghersi-Egea, J. F., Guillouzo, A., Hartung, T., Hoet, P., Ingelman-Sundberg, M., Munn, S., Janssens, W., Ladstetter, B., Leahy, D., Long, A., Meneguz, A., Monshouwer, M., Morath, S., Nagelkerke, F., Pelkonen, O., Ponti, J., Prieto, P., Richert, L., Sabbioni, E., Schaack, B., Steiling, W., Testai, E., Vericat, J. A., and Worth, A. (2006). Metabolism: a bottleneck in in vitro toxicological test development. The report and recommendations of ECVAM workshop 54. Altern. Lab. Anim. 34, 49–84.
EC. (1976). European Commission, Directive of 27 July 1976 on the Approximation of the Laws of the Member States Relating to Cosmetic Products (76/768/EEC). Available at: http://ec.europa.eu/consumers/ sectors/cosmetics/documents/ directive/
EC. (2006). European Commission, Regulation (EC) No 1907/2006. Available at: http://eur-lex.europa.eu/LexUriServ /LexUriServ.do?uri=CELEX:32006R 1907:EN:NOT
EC. (2008). European Commission, Regulation (EC) No 440/2008. Available at: http://eur-lex.europa.eu/LexUriServ/LexUri Serv.do?uri=CELEX:32008R0440: EN:HTML
ECHA. (2008). Guidance on Information Requirements and Chemical Safety Assessment Chapter R.6: QSARs and Grouping of Chemicals. Available at: http://guidance.echa.europa.eu/docs/ guidance_document/information_ requirements_en.htm
ECHA. (2010). Practical Guide 2: How to Report Weight of Evidence. Available at: http://echa.europa.eu/doc/ publications/practical_guides/pg_ report_weight_of_evidence.pdf
Ekins, S., Mestres, J., and Testa, B. (2007a). In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br. J. Pharmacol. 152, 9–20.
Ekins, S., Mestres, J., and Testa, B. (2007b). In silico pharmacology for drug discovery: applications to targets and beyond. Br. J. Pharmacol. 152, 21–37.
Enoch, S. J., and Cronin, M. T. (2010). A review of the electrophilic reaction chemistry involved in covalent DNA binding. Crit. Rev. Toxicol. 40, 728–748.
Hartung, T., and Hoffmann, S. (2009). Food for thought on in silico methods in toxicology. ALTEX 36, 155–166.
Hollenberg, P. F., Kent, U. M., and Bumpus, N. N. (2008). Mechanism-based inactivation of human cytochromes P450s: experimental characterization, reactive intermediates, and clinical implications. Chem. Res. Toxicol. 21,189–205.
Höltje, H. D., Sippl, W., Rognan, D., and Folkers, G. (2003). Molecular Modelling – Basic Principles and Applications. Heidelberg: Wiley- VCH.
Hou, T., and Wang, J. (2008). Structure-ADME relationship: still a long way to go? Expert Opin. Drug Metab. Toxicol. 4, 759–770.
IHCP. (2010). Institute for Health and Consumer Protection, European Commission Joint Research Centre. Available at: http://ihcp.jrc.ec.europa.eu/ agreement-ihcp-ncct-epa
IMI. (2010). Innovative Medicines Initiative. Available at: www.imi.europa.eu
Isin, E. M., and Guengerich, F. P. (2008). Substrate binding to cytochromes P450. Anal. Bioanal. Chem. 392,1019–1030.
Jaworska, J., Nikolova-Jeliazkova, N., and Aldenberg, T. (2005). Review of methods for QSAR applicability domain estimation by the training set. Altern. Lab. Anim. 33, 445–459.
Johnson, D. E., and Rodgers, A. D. (2006). Computational toxicology: heading toward more relevance in drug discovery and development. Curr. Opin. Drug Discov. Devel. 9, 29–37.
Kavlock, R. J., Ankley, G., Blancato, J., Breen, M., Conolly, R., Dix, D., Houck, K., Hubal, E., Judson, R., Rabinowitz, J., Richard, A., Setzer, R. W., Shah, I., Villeneuve, D., and Weber, E. (2008). Computational toxicology-a state of the science mini review. Toxicol. Sci. 103, 14–27.
Kortagere, S., Krasowski, M. D., and Ekins, S. (2009). The importance of discerning shape in molecular pharmacology. Trends Pharmacol. Sci. 30, 138–147.
Lewis, D. F., and Ito, Y. (2008). Human cytochromes P450 in the metabolism of drugs: new molecular models of enzyme-substrate interactions. Expert Opin. Drug Metab. Toxicol. 4, 1181–1186.
Liebler, D. C., and Guengerich, F. P. (2005). Elucidating mechanisms of drug-induced toxicity. Nat. Rev. Drug Discov. 4, 410–420.
Loizou, G., Spendiff, M., Barton, H. A., Bessems, J., Bois, F. Y., d’Yvoire, M. B., Buist, H., Clewell, H. J. III, Meek, B., Gundert-Remy, U., Goerlitz, G., and Schmitt, W. (2008). Development of good modelling practice for physiologically based pharmacokinetic models for use in risk assessment: the first steps. Regul. Toxicol. Pharmacol. 50, 400–411.
Merlot, C. (2010). Computational toxicology – a tool for early safety evaluation. Drug Discov. Today 15, 16–22.
Mostrag-Szlichtyng, A., and Worth, A. (2010). Review of QSAR Models and Software Tools for Predicting Biokinetic Properties. Luxembourg: European Commission, Joint Research Centre, Institute for Health and Consumer Protection.
Muster, W., Breidenbach, A., Fischer, H., Kirchner, S., Müller, L., and Pähler, A. (2008). Computational toxicology in drug development. Drug Discov. Today 13, 303–310.
Nigsch, F., Macaluso, N. J., Mitchell, J. B., and Zmuidinavicius, D. (2009). Computational toxicology: an overview of the sources of data and of modelling methods. Expert Opin. Drug Metab. Toxicol. 5, 1–14.
NRC, National Research Council. (2007). Toxicity Testing in the 21st Century: A Vision and A Strategy. Washington, DC: National Academy Press.
OECD. (2004). OECD Principles for the Validation, for Regulatory Purposes, of (Quantitative) Structure-Activity Relationship Models. Available at: http://www.oecd.org/document/23/ 0,2340,en_2649_34379_33957015_ 1_1_1_1,00.html
OECD. (2007). “Guidance document on the validation of (quantitative) structure-activity relationships [(Q)SAR] models,” in OECD Series on Testing and Assessment No. 69. ENV/JM/MONO (2007) 2 (Paris: Organisation for Economic Cooperation and Development), 154.
Pelkonen, O. (2010). Predictive toxicity: grand challenges. Front. Pharmacol. 1:3. doi:10.3389/fphar.2010.00003
Pelkonen, O., Tolonen, A., Korjamo, T., Turpeinen, M., and Raunio, H. (2009). From known knowns to known unknowns: predicting in vivo drug metabolites. Bioanalysis 1, 393–414.
Pelkonen, O., Turpeinen, M., Hakkola, J., Honkakoski, P., Hukkanen, J., and Raunio, H. (2008). Inhibition and induction of human cytochrome P450 enzymes – current status. Arch. Toxicol. 82, 667–715.
Schaafsma, G., Kroese, E. D., Tielemans, E. L., Van de Sandt, J. J., and Van Leeuwen, C. J. (2009). REACH, non-testing approaches and the urgent need for a change in mind set. Regul. Toxicol. Pharmacol. 53, 70–80.
Stjernschantz, E., Vermeulen, N. P. E., and Oostenbrink, C. (2008). Computational prediction of drug binding and rationalisation of selectivity towards cytochromes P450. Expert Opin. Drug Metab. Toxicol. 4, 513–527.
US EPA. (2003). A Framework for a Computational Toxicology Research Program. Washington, D.C: Office of Research and Development, U.S. Environmental Protection Agency, [EPA600/R-03/65].
Valerio, L. G. (2009). In silico toxicology for the pharmaceutical sciences. Toxicol. Appl. Pharmacol. 241, 356–370.
van de Waterbeemd, H., and Gifford, E. (2003). ADMET in silico modelling: towards prediction paradise? Nat. Rev. Drug Discov. 2, 192–204.
Walker, D., Brady, J., Dalvie, D., Davis, J., Dowty, M., Duncan, N., Nedderman, A., Obach, R. C., and Wright, P. (2009). A holistic strategy for characterizing the safety of metabolites through drug discovery and development. Chem. Res. Toxicol. 22, 1653–1662.
Wang, J. (2009). Comprehensive assessment of ADMET risks in drug discovery. Curr. Pharm. Des. 15, 2195–2219.
Weaver, S., and Gleeson, M. P. (2008). The importance of the domain of applicability in QSAR modeling. J. Mol. Graph. Model. 26, 1315–1326.
Keywords: in silico, modeling, REACH, toxicity
Citation: Raunio H (2011) In silico toxicology – non-testing methods. Front. Pharmacol. 2:33. doi: 10.3389/fphar.2011.00033
Received: 04 January 2011;
Paper pending published: 25 May 2011;
Accepted: 15 June 2011;
Published online: 30 June 2011.
Edited by:
Kirsi Vähäkangas, University of Kuopio, FinlandReviewed by:
Paavo Honkakoski, University of Eastern Finland, FinlandRory Conolly, United States Environmental Protection Agency, USA
Copyright: © 2011 Raunio. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Hannu Raunio, Division of Pharmacology, Faculty of Health Sciences, University of Eastern Finland, Box 1627, Kuopio, Finland. e-mail: hannu.raunio@uef.fi