
- Pole of Pharmacology and Therapeutics, Institute of Experimental and Clinical Research, University of Louvain Medical School, Brussels, Belgium
Cancer is a metabolic disease and the solution of two metabolic equations: to produce energy with limited resources and to fulfill the biosynthetic needs of proliferating cells. Both equations are solved when glycolysis is uncoupled from oxidative phosphorylation in the tricarboxylic acid cycle, a process known as the glycolytic switch. This review addresses in a comprehensive manner the main molecular events accounting for high-rate glycolysis in cancer. It starts from modulation of the Pasteur Effect allowing short-term adaptation to hypoxia, highlights the key role exerted by the hypoxia-inducible transcription factor HIF-1 in long-term adaptation to hypoxia, and summarizes the current knowledge concerning the necessary involvement of aerobic glycolysis (the Warburg effect) in cancer cell proliferation. Based on the many observations positioning glycolysis as a central player in malignancy, the most advanced anticancer treatments targeting tumor glycolysis are briefly reviewed.
Cancer is a metabolic disease and the solution of two metabolic equations: (i) to produce enough energy to survive when supplies and waste disposal are limited, and (ii) to divert enough metabolic intermediates from energy production to the biosynthetic pathways supporting cell proliferation. This review paper summarizes the major observations positioning glycolysis as a central player in malignancy and justifying the current quest for glycolytic targets in the context of tumor treatment. Our purpose is not to be exhaustive but to briefly summarize the most promising strategies, inviting the reader to gain in-depth knowledge in the referenced papers.
Glycolysis and Cancer Bioenergetics
The first metabolic equation of cancer refers to energy production. It directly calls into play oxygen as the electron acceptor allowing the proper functioning of the respiratory chain at the inner mitochondrial membrane. Most solid tumors are hypoxic with many biological features accounting for the lack of oxygen (Bristow and Hill, 2008). Probably the most comprehensive form of hypoxia is the so-called diffusion-limited hypoxia which arises when cells located at increasing distance from blood vessels eventually fail to receive the minimum amount of oxygen that they would need for an optimal oxidative metabolism in addition to the many non-metabolic redox reactions requiring oxygen (Horsman and Overgaard, 2002). Normal cells in non-malignant tissues are also exposed to various levels of oxygen with respect to their distance from the closest blood vessel and owing to the fact that intermediate layers of cells consume oxygen. Evolution has selected the Pasteur Effect as a system aimed to finely tune cell metabolism in function of the local partial pressure of oxygen (pO2; Wu and Racker, 1959). It relies on the negative feed-back exerted allosterically by energy metabolites [glucose-6-phosphate (G6P), citrate, and ATP] on key glycolytic enzymes, thus accelerating the glycolytic flux when the rate of oxidative phosphorylation (OXPHOS) decreases, and, inversely, improving the coupling between glycolysis and OXPHOS fluxes when oxygen levels increase (Figure 1). Two fundamentals of the Pasteur Effect are that glycolysis is nominally a faster succession of reactions than ATP production through OXPHOS (Curi et al., 1988), and that the ATP yield of OXPHOS is nominally 19-fold higher than that of glycolysis alone. The full oxidation of one molecule of glucose provides up to 38 ATP molecules, whereas glycolysis alone provides only 2 ATP. Although theoretically OXPHOS is the best energy provider, the physiological reality is that both glycolysis and OXPHOS collaborate to produce ATP at a relative level dictated by local oxygen concentration. Tumor hypoxia is an extreme situation under which glycolysis becomes the main source of ATP in tumor cells (Dang and Semenza, 1999). This glycolytic switch, formally corresponding to uncoupling glycolysis from OXPHOS, initially depends on repression of the Pasteur Effect. In this review, unless stated otherwise, the term “glycolysis” refers to “glycolysis uncoupled from OXPHOS of the tricarboxylic acid (TCA) cycle.” Direct molecular consequences are net increases in glucose consumption and lactic acid release (Figure 1). Because switching to glycolysis primarily reflects the metabolic adaptability of tumor cells to extreme environments, it is not surprising that lactate levels positively correlate with the aggressiveness of several types of human cancers (Walenta et al., 1997, 2000, 2004; Brizel et al., 2001; Walenta and Mueller-Klieser, 2004).
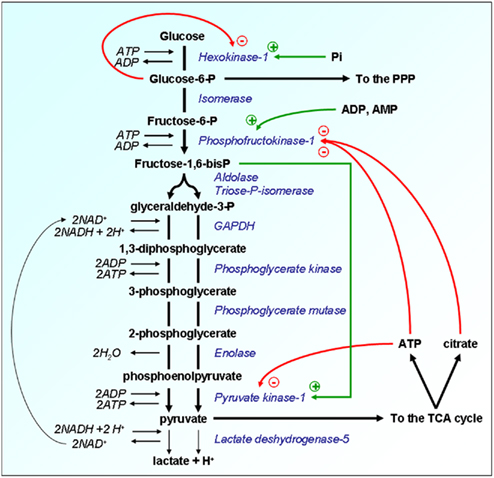
Figure 1. Allosteric regulations of glycolysis confer metabolic plasticity with respect to local pO2. Enzymes are represented in italicized blue font and their substrates in bold black. Because the glycolytic flux is nominally faster than OXPHOS, the Pasteur Effect has been evolutionary selected to couple both metabolic rates. The energy metabolites glucose-6-P, ATP, and citrate restrain the glycolytic flux through allosteric inhibition of key glycolytic enzymes, as represented by the red arrows. Inhibition is at its climax when oxygen is not a limiting substrate for OXPHOS, thus allowing the full oxidation of glucose. When oxygen levels are limited or when the pO2 fluctuates, full glucose oxidation, and consequently the levels of ATP and citrate produced oxidatively are decreased. The Pasteur Effect is reset to less pronounced inhibition, thus allowing accelerated glycolysis to compensate for defective ATP production. An extreme situation characterized by full inhibition of the Pasteur Effect is met under severe hypoxia. The energetic crisis is associated with an increase in the cellular levels of fructose-1,6-bisP, ADP, AMP, and inorganic phosphate (Pi). These molecules exert a series of allosteric stimulations (represented by the green arrows) that accelerate the glycolytic flux. Glycolysis thus becomes the main source of cellular ATP production, a rescue situation allowing short-term cell survival until the pO2 is restored. Other abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; P, phosphate; PPP, pentose phosphate pathway; TCA, tricarboxylic acid (cycle).
A major difference between normal and cancer tissues is that tumors have lost cellular homeostasis. Owing to an agenda of unbridled proliferation, tumor cells perpetually overcome the available oxygen supply when invading regions remote from blood vessels. Growth is further associated with a loosening of the initially dense vascular network. These conditions constitute the initial soil for the establishment of tumor hypoxia in a lasting form that cannot be durably accommodated by the sole suppression of the Pasteur Effect. In nascent tumors, tumor cells at the distal edge of the oxygen gradient have two possible fates: death or metabolic adaptation. Hypoxic death is a typical feature of dormant tumors, i.e., microscopic, asymptomatic lesions characterized by a dynamic equilibrium between the proliferation of oxygenated cells and the death of hypoxic ones (Folkman, 1971). Comparatively (and hopefully for us), durable switch to a glycolytic metabolism is a rare event marking the entry of a tumor into an exponential growth phase. Although this metabolic adaptation initially proceeds through inhibition of the Pasteur Effect, sufficient ATP production for long-term cell survival and to sustain the aggressive phenotype requires further adaptations meant to accelerate the glycolytic flux. A key player is the transcription factor hypoxia-inducible factor-1 (HIF-1). HIF-1 is an αβ-heterodimer: the HIF-1β subunit is constitutively nuclear, whereas HIF-1α is inducible by hypoxia (Figure 2; Pugh and Ratcliffe, 2003). Regulation of HIF-1α expression involves its posttranslational hydroxylation at proline residues 402 and 564 (human sequence) by prolylhydroxylases (PHDs), among which PHD2 is the prominent physiological facilitator of the reaction (Ivan et al., 2001; Jaakkola et al., 2001; Berra et al., 2003). PHD2 is a Fe(II)- and 2-oxoglutarate-dependent dioxygenase which has an absolute requirement for molecular oxygen: because of its low affinity for oxygen (Km = 250 μM, slightly above air pO2; Hirsila et al., 2003), PHD2 is often described as an oxygen sensor. In oxygenated cells, proline hydroxylation addresses HIF-1α to the von Hippel–Lindau (VHL) protein complex for poly-ubiquitylation, followed by destruction by the proteasome (Maxwell et al., 1999; Masson et al., 2001; Yu et al., 2001a). During hypoxia, PHD2 is inactivated, and HIF-1α escapes proteolytic degradation to migrate into the cell nucleus where it binds to HIF-1β. Initiation of transcription further requires the interaction of HIF-1 with the cofactors p300 and the DNA polymerase II complex to bind to hypoxia-responsive elements (HRE) of target genes. Gene products grossly belong to two categories: those such as erythropoietin (EPO), vascular endothelial growth factor (VEGF), and inducible nitric oxide synthase (iNOS) aimed at restoring local pO2; and those involved in the acceleration of the glycolytic flux (Semenza, 2010). Figure 3 highlights the major HIF-1-target gene products directly involved in the perpetuation of a high glycolytic rate in tumor cells. A brief description of their functions follows.
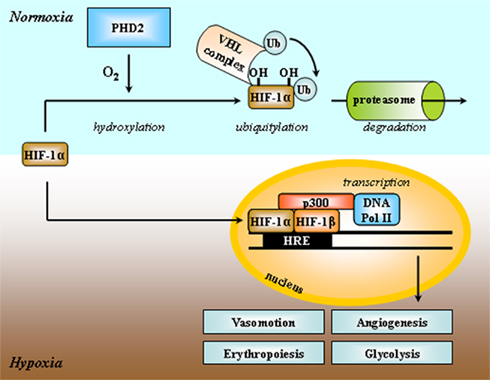
Figure 2. Simplified scheme depicting HIF-1 activation by hypoxia. Both HIF-1α and HIF-1β subunits are constitutively transcribed, but only the HIF-1β protein is stably expressed into the cell nucleus. HIF-1 activity primarily depends on the stability of the HIF-1α protein. Under normoxia, HIF-1α is hydroxylated at two proline residues by the prolylhydroxylase PHD2, and addressed to the von Hippel–Lindau (VHL) complex for ubiquitylation (Ub) and further proteasome-mediated degradation. Oxygen is a limiting substrate for the PHD2 reaction. Under hypoxia, the HIF-1α protein is expressed, migrates to the nucleus, and binds to HIF-1β, the adaptator p300, DNA polymerase II, and to the consensus DNA motif hypoxia-responsive element (HRE) in the promoting region of target genes. HIF-1-target gene products promote glycolysis, angiogenesis, and erythropoiesis, and regulate vasomotion. Adapted from Harris (2002).
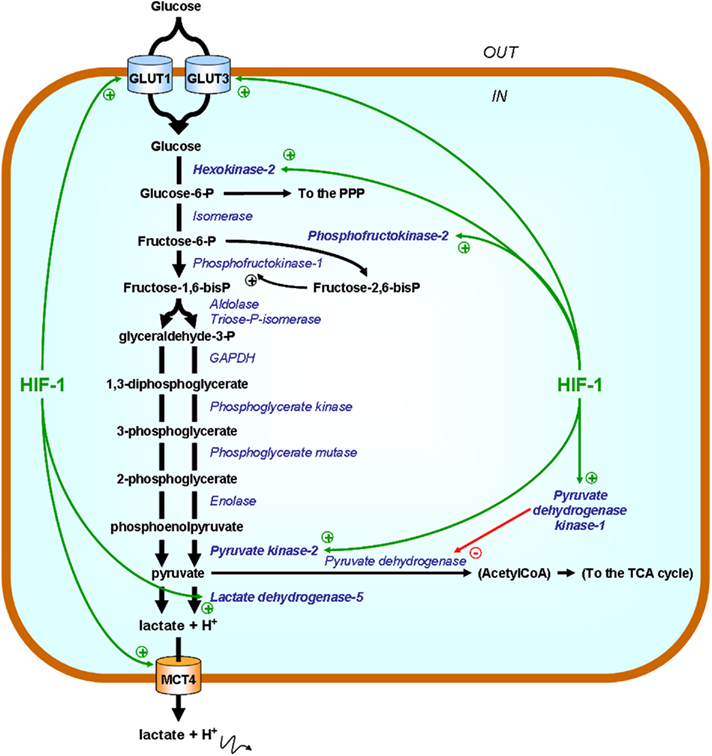
Figure 3. HIF-1 promotes the expression of glycolytic enzymes and transporters. Enzymes are represented in italicized blue font and their substrates in bold black. Green arrows point at HIF-1-target gene products directly involved in the acceleration of the glycolytic flux. In the simplified scheme, only those that have been identified as potential therapeutic targets are highlighted. Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GLUT, glucose transporter; MCT4, monocarboxylate transporter 4; P, phosphate; PPP, pentose phosphate pathway; TCA, tricarboxylic acid (cycle).
Glucose Transporters GLUT1 and GLUT3
GLUT1 and GLUT3 belong to the GLUT/SLC2A family of transporters required for glucose shuttling across cell membranes. GLUT1 (Km glucose = 1–2 mM; Wheeler and Hinkle, 1981) and GLUT3 (Km glucose = 1.4 mM; Gould et al., 1991) have a high affinity for glucose and therefore, once expressed, play the essential role of ensuring efficient glucose uptake even when glucose becomes a limiting resource (Tal et al., 1992; Gatenby and Gillies, 2004). Expression of the two isoforms is HIF-1-inducible (Ebert et al., 1995, 1996), thus coupling the glycolytic switch to increased glucose uptake in hypoxic cancer cells. High expression of GLUT1 and/or GLUT3 is associated with the poor prognosis of several types of human tumors (Younes et al., 1995, 1997; Haber et al., 1998; Baer et al., 2002; Kim et al., 2002; Fenske et al., 2009; Ayala et al., 2010).
Hexokinase 2
Hexokinase 2 is a member of the HK family of enzymes that control the first rate-limiting step of glycolysis, i.e., the phosphorylation of glucose to G6P involving the transfer of phosphate from ATP. Once phosphorylated, negatively charged G6P is literally trapped inside the cell where it fuels both glycolysis and the pentose phosphate pathway (PPP). Compared to other HK isoforms, HK2 is a HIF-1-target gene product (Mathupala et al., 2001) and exists in a phosphorylated form bound to the outer mitochondrial membrane where it interacts with the voltage-dependent anion channel VDAC (Bustamante and Pedersen, 1977; Nakashima et al., 1986, 1988; Gottlob et al., 2001). This strategic localization provides preferential access to ATP (permeating VDAC) and insensitivity to negative feed-back inhibition by G6P (Bustamante and Pedersen, 1977), thus ensuring proficient glucose entrapment by tumor cells expressing mitochondria-bound HK2. The interaction of HK2 with VDAC further interferes with the binding of the pro-apoptotic protein Bax to VDAC, an event that would otherwise form a channel through which cytochrome c can escape from mitochondria an trigger apoptosis (Pastorino et al., 2002). Interestingly, HIF-1 cooperates with the oncogenic transcription factor c-Myc to transactivate HK2 under hypoxia (Kim et al., 2007). Because switching from HK1 to HK2 offers both a metabolic advantage and protection against apoptosis, it is not surprising that HK2 is overexpressed in many cancer types compared with normal tissues, which is of poor prognosis (Lyshchik et al., 2007; Rho et al., 2007; Peng et al., 2008; Palmieri et al., 2009).
Phosphofructokinase 2 (PFK2/PFKFB3)
Fructose-2,6-bisphosphate (F2,6BP) is a key regulator of glycolysis acting as an allosteric activator of PFK1, one of the rate-controlling enzymes of glycolysis. F2,6BP is produced from fructose-6-phosphate (F6P) by a family of homodimeric enzymes known as 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFB). PFKFBs are bifunctional enzymes that catalyze either the ATP-dependent phosphorylation of F6P to F2,6BP (PFK2 activity) or the dephosphorylation of F2,6BP to F6P (FBPase activity). The family comprises four members among which PFKFB1, PFKFB2, and PFKFB4 display equal PFK2 and FBPase activities under basal conditions, whereas PFKFB3 has high PFK2 and almost no FBPase activity (Okar and Lange, 1999; Okar et al., 2001). The transcription of all four PFKFB genes is inducible by hypoxia but the major induction is seen for the PFKFB3 gene which is a target of HIF-1 (Minchenko et al., 2002, 2003). Hypoxic stimulation of the PFK2 activity of PFKFB3 is further enhanced through phosphorylation of a serine residue at position 462 (human sequence), a process involving AMP-activated protein kinase (AMPK; Marsin et al., 2002). PFKFB3 sustains high-rate glycolysis and is highly expressed in several types of human tumors (Atsumi et al., 2002; Minchenko et al., 2005; Kessler et al., 2008).
Pyruvate Kinase 2 (PKM2)
Pyruvate kinase (PK) is a key glycolytic enzyme which catalyzes a rate-limiting step of glycolysis, i.e., the dephosphorylation of phosphoenolpyruvate (PEP) into pyruvate to produce ATP. PK has four isoforms, of which PKM1/M1-PK and PKM2/M2-PK are produced by alternative splicing of transcripts of the PKM gene, a HIF-1-target gene (Luo et al., 2011). Alternative splicing is regulated by the heterogeneous nuclear ribonucleoproteins (hnRNP) I, A1, and A2 (which bind to exon 9 and repress splicing to PKM1; Noguchi et al., 1986; David et al., 2010), in turn controlled by c-Myc (David et al., 2010). Isozyme selection allows for the rapid proliferation observed in tumors. Unlike PKM1, PKM2 is indeed the characteristic isoenzyme of cells with high-rate nucleic acid synthesis, including normal proliferating cells, embryonic cells, adult stem cells, and also importantly tumor cells (Reinacher and Eigenbrodt, 1981; Yamada and Noguchi, 1999). During tissue differentiation in development, embryonic PKM2 is replaced by tissue-specific isoforms. Tumorigenesis, however, is associated with the re-expression of PKM2 together with a down-regulation of the expression of PKM1 and other isozymes (Mazurek et al., 2005). This “glycolytic dedifferentiation” offers a key advantage in terms of metabolic plasticity because, unlike PKM1 (existing only in an active tetrameric form), PKM2 may be expressed either as an active tetramer or as a dimer with low affinity for PEP. PKM2 in its highly active tetrameric conformation (Km PEP = 0.03 mM) provides high yield ATP production from glycolysis, whereas in its nearly inactive dimeric conformation (Km PEP = 0.46 mM) it provides a metabolic bottleneck allowing glycolytic intermediates to be redirected toward biosynthesis, notably fueling through the PPP for DNA synthesis (Mazurek, 2011). The balance between tetrameric and dimeric PKM2 is an oscillating phenomenon subject to allosteric regulation, a topic recently reviewed by Mazurek (2011). Briefly, the tetrameric active form is promoted by accumulation of the upstream glycolytic intermediate F1,6BP and by the biosynthetic byproduct serine; inactivating dimerization is conversely induced when the concentration of downstream biosynthetic products (alanine, other amino-acids, and lipids) increases. The dimeric conformation is further promoted by phosphorylation of tyrosine 105 in response to several oncogenic tyrosine kinases, whereas PEP accumulation can be controlled by a non-ATP-generating phosphate transfer to an histidine residue of phosphoglycerate mutase (Vander Heiden et al., 2010). Most cancer cells express PKM2 (Christofk et al., 2008a), thereby acquiring a finely regulated switch to promote ATP production (switch on) or cell proliferation (switch off; Christofk et al., 2008b). In addition, it has been elegantly demonstrated that, after nuclear translocation, PKM2 cooperates with HIF-1 to transactivate genes further promoting glycolysis and tumor angiogenesis (Luo et al., 2011).
Lactate Dehydrogenase 5
Pyruvate is at a hub between different metabolic pathways: it is the product of glycolysis, the product of malate oxidation in proliferating cells (DeBerardinis et al., 2007), the main fuel of the TCA cycle, the precursor of alanine in a reversible transamination reaction involving glutamate as the nitrogen donor, and the substrate of a redox reaction generating lactate. The latter reaction, coupling the reduction of pyruvate to the oxidation of NADH into NAD+, allows the replenishment of the NAD+ pool required for glycolysis self-sufficiency. NAD+ is indeed mandatory for the OXPHOS of glyceraldehyde-3-phosphate into 1,3-diphosphoglycerate by GAPDH (see Figure 1). Pyruvate reduction into lactate also allows glycolytic cells to maintain the levels of pyruvate low enough to avoid cell death (Thangaraju et al., 2006, 2009). This reversible reaction is catalyzed by the LDH family of tetrameric enzymes. LDHs are formed by the arrangement of up to four copies of two different subunits: subunit LDH-H is encoded by the LDH-B gene and is ubiquitously expressed in healthy tissues, whereas subunit LDH-M is encoded by the HIF-1-target gene LDH-A and is therefore induced by hypoxia (Figure 4). Compared to LDH-H, LDH-M has a higher Km for pyruvate and a higher Vmax for pyruvate reduction (Markert et al., 1975). Consequently, LDH5/LDH-4M preferentially catalyzes the reduction of pyruvate into lactate and plays key roles in the maintenance of a high glycolytic flux and in resistance to apoptosis. Elevated LDH5 expression is of unfavorable prognostic significance to many human tumors (Koukourakis et al., 2003, 2005, 2009). Conversely, LDH1/LDH-4H is most commonly silenced in glycolytic cancer cells, a process involving hypermethylation of the LDH-B gene promoter (Leiblich et al., 2006; Thangaraju et al., 2009).
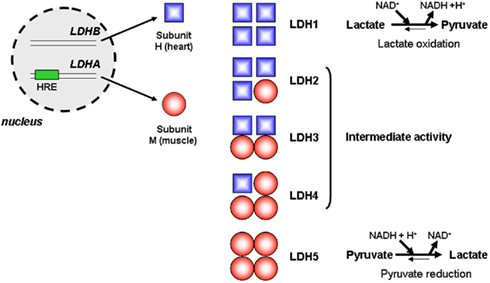
Figure 4. Lactate dehydrogenases. In eukaryote cells, lactate dehydrogenases (LDHs) are tetrameric enzymes catalyzing the reversible reduction of pyruvate into lactate. The LDH-B gene is constitutively transcribed and encodes subunit LDH-H, whereas transcription of the LDH-A gene, which is inducible by hypoxia due to the presence of a consensus HIF-1-binding motif (hypoxia-responsive element, HRE), encodes the LDH-M subunit. Arrangement of the subunits to forms active tetramers may lead to the formation of five distinct enzymes, LDH1 to LDH5. Compared to LDH-H, LDH-M has a higher Km and a higher Vmax for pyruvate reduction. Consequently, LDH5/LDH-4M preferentially catalyzes the reduction of pyruvate into lactate, LDH1/LDH-4H preferentially catalyzes the oxidation of lactate into pyruvate, and LDH2, LDH3, and LDH4 have intermediate enzymatic activities.
Pyruvate Dehydrogenase Kinase 1
Pyruvate dehydrogenase (PDH), the enzyme committing pyruvate to enter into the TCA cycle, is subject to phosphorylative inhibition by PDK1 (Holness and Sugden, 2003). Under hypoxia, PDH inhibition has two main objectives: to orient pyruvate to the LDH5 reaction for NAD+ production, and to prevent the excessive production of reactive oxygen species (ROS) by uncoupled mitochondria (Kim et al., 2006; Papandreou et al., 2006). Indeed, although oxygen is the primary acceptor of the electrons produced by the respiratory chain, electrons are transferred to water to produce ROS when oxygen becomes limiting. As a HIF-1-target gene product, PDK1 couples hypoxia to the attenuation of the respiratory chain activity (Kim et al., 2006). Similar to HK2, it has been shown that HIF-1 cooperates with c-Myc for the transactivation of PDK1 (Kim et al., 2007). High PDK1 expression strongly correlates with poor outcome in head-and-neck cancer (Wigfield et al., 2008).
Monocarboxylate Transporter 4
The LDH5 reaction yields equimolar concentrations of lactate (from pyruvate) and protons (from NADH). To avoid intracellular acidification and death, glycolytic cells must export protons. Several systems are adapted for the transport of protons (see also below) among which MCT1, MCT2, MCT3, and MCT4 are passive lactate–proton symporters (Halestrap and Meredith, 2004). MCT4 (Km lactate = 22 mM) has the lowest affinity for lactate, is encoded by a HIF-1-target gene (Ullah et al., 2006), and is therefore adapted for the export of lactic acid from glycolytic tumor cells (Dimmer et al., 2000). It plays an important contribution to the regulation of intracellular pH (pHi): although it has only a low affinity for lactate, its high turnover rate ensures efficient proton export (Chiche et al., 2011). MCT1 (Km lactate = 3.5–10 mM) has an intermediate affinity for lactate and is ubiquitously expressed in healthy and cancer tissues. In cancer, it facilitates lactate uptake by oxidative tumor cells in a newly described metabolic pathway involving lactate oxidation into pyruvate to fuel the TCA cycle (see below; Sonveaux et al., 2008). MCT2 (Km lactate = 0.5 mM) and MCT3 (Km lactate = 5 mM) have the highest affinity for lactate and are specialized in the import of lactate in very specific tissues such as liver (Cori cycle), kidney, and retina (Garcia et al., 1995; Philp et al., 2001; Perez et al., 2010). It was recently shown that high expression of both MCT1 and MCT4 correlates with the invasiveness of lung cancer cells (Izumi et al., 2011).
In a synthetic form, the full glycolytic reaction is expressed by the equation: Glucose + 2ADP → 2ATP + 2 Lactate + 2H+ + 2H2O. Under hypoxia, the reaction is paced at high-rate to fulfill the energetic needs of tumor cells. Two important questions directly come to mind. How do hypoxic tumor cells often located at distance from blood vessels get access to high level glucose? How do they avoid intracellular acidification?
We have recently proposed metabolic symbiosis as a rationale for efficient glucose delivery to the hypoxic tumor cell compartment (Figure 5; Sonveaux et al., 2008). The symbiosis is based on the exchange of lactate: hypoxic/glycolytic tumor cells produce lactate and normoxic/oxidative tumor cells consume lactate oxidatively. The latter process involves lactate oxidation into pyruvate by LDH1. Core to the symbiosis is the metabolic preference of oxygenated tumor cells for lactate compared to glucose as an oxidative fuel, with as consequence improved distribution of glucose to hypoxic tumor areas. Rationale for the metabolic preference include a competition between LDH1 and the glycolytic enzyme GAPDH for NAD+ (LDH1 being a more efficient pathway; Tanaka et al., 2004) coupled to the fact that lactate inhibits HK and PFK1 activities (Leite et al., 2011). In the symbiotic model, MCT4 serves to export lactate from glycolytic tumor cells (Dimmer et al., 2000) and we have demonstrated MCT1 as the main facilitator of lactate uptake by oxidative tumor cells (Sonveaux et al., 2008).
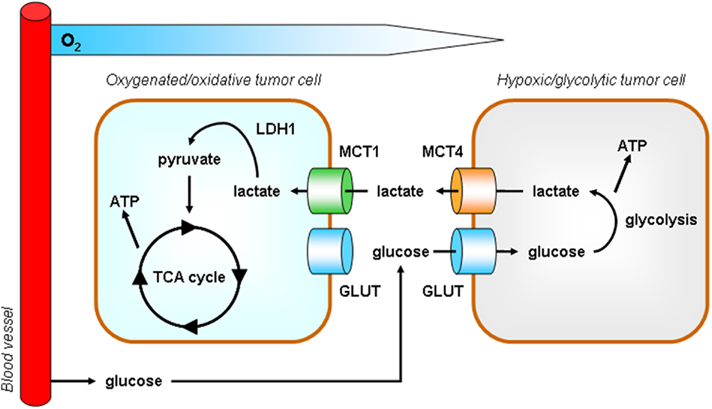
Figure 5. Model according to which tumors behave as metabolic symbionts. Tumor heterogeneity includes metabolism. At a remote location from perfused blood vessels, hypoxic tumors cells rely on glycolysis for survival and proliferation. High ATP production mandatorily depends on high glucose availability and is associated with the release of lactate, a process facilitated by monocarboxylate transporter 4 (MCT4). In contrast, although they also express glucose transporters (GLUT), oxygenated tumor cells have a metabolic preference for lactate versus glucose. MCT1 is a transporter is adapted for lactate uptake (Sonveaux et al., 2008). In the presence of oxygen, lactate is oxidized to pyruvate by lactate dehydrogenase 1 (LDH1) and pyruvate fuels the tricarboxylic acid (TCA) cycle to produce ATP. The metabolic preference of oxidative tumor cells for lactate allows hypoxic tumor cells to get access to high levels of glucose. This metabolic cooperativity is key for tumor cell survival under hypoxia in vivo.
The maintenance of an intracellular pH close to or even slightly above 7.3 is ensured by several pH regulatory systems in addition to MCT4 (see above). Interestingly, most of these transporters and enzymes are also HIF-1-target gene products, thus emphasizing that the regulation of pHi is an integral part of the glycolytic switch (Pouyssegur et al., 2006). Figure 6 proposes a synthetic view of the major pH regulators of cancer cells. Compared to MCT4 which is a fully passive system, the other systems directly [vacuolar ATPase (V-ATPase)] or indirectly require ATP; although they are not sodium-dependent enzymes, CA9 and NHE1 ultimately depend on the export of sodium largely mediated by NaK ATPase to maintain their activities. A brief description of their activities follows.
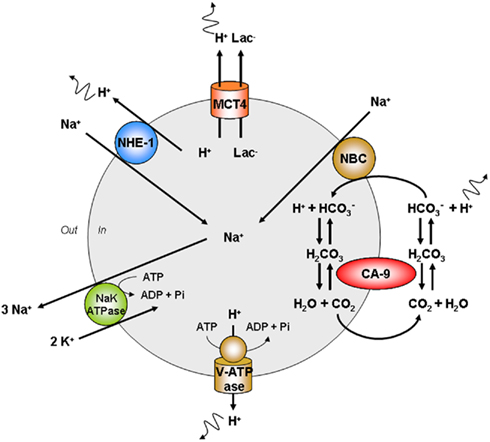
Figure 6. Major cellular pathways involved in the cellular export of protons. The maintenance of high-rate glycolysis requires the export of protons that would otherwise create intracellular acidification leading to cell death. Monocarboxylate transporter 4 (MCT4) is a passive lactate (Lac−)-proton symporter adapted for the export of protons. Carbonic anhydrase-9 (CA9) is a transmembrane enzyme promoting the reversible hydratation of CO2. Acidity is exported under the form of cell-permeable CO2, followed by CA9-facilitated hydratation of CO2 to carbonic acid. Carbonic acid then readily dissociates to proton and bicarbonate extracellularly. Bicarbonate may be recaptured by the sodium bicarbonate cotransporter (NBC) to react with a proton intracellularly. Then, bicarbonate gets dehydrated to yield CO2 for export. V-ATPase is expressed at the plasma membrane of several tumor cell types where it acts as a proton pump fueled by ATP. The sodium–proton exchanger 1 (NHE1) is a passive proton–sodium antiporter. The sodium–potassium (NaK) ATPase promotes the export of sodium that would otherwise accumulate as a consequence of NHE1, CA9, and NBC activities. MCT4 is thus the only truly passive system for proton export. Other abbreviation: Pi, inorganic phosphate.
Carbonic Anhydrase-9
Carbonic anhydrases (CAs) form a large family of zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide into carbonic acid. Although both CA9 and CA12 are transmembrane isoforms with an extracellular-facing catalytic site, CA9 is characterized by the highest H+ transfer rate known among CAs (kcat/Km ∼ 55 μM−1s−1; Wingo et al., 2001). It is a major facilitator of the extracellular trapping of acid in tumors, which is achieved by hydrating cell-generated CO2 into  and H+. Both CA9 and CA12 are HIF-1-target gene products and their expression is therefore significantly induced by hypoxia (Wykoff et al., 2000; Chiche et al., 2009). The two enzymes have been shown to promote tumor cell survival and growth by maintaining pHi within the physiological range, which also confers a survival advantage to tumor cells (compared to non-malignant cells) exposed to an acidic extracellular environment (Chiche et al., 2009). Overexpression of CA9 in several types of malignancies is associated with increased metastatic burden and poor patient survival (Hussain et al., 2007).
and H+. Both CA9 and CA12 are HIF-1-target gene products and their expression is therefore significantly induced by hypoxia (Wykoff et al., 2000; Chiche et al., 2009). The two enzymes have been shown to promote tumor cell survival and growth by maintaining pHi within the physiological range, which also confers a survival advantage to tumor cells (compared to non-malignant cells) exposed to an acidic extracellular environment (Chiche et al., 2009). Overexpression of CA9 in several types of malignancies is associated with increased metastatic burden and poor patient survival (Hussain et al., 2007).
Membrane-Bound Vacuolar ATPase
Vacuolar ATPase is a large heteromultimeric enzyme playing an important role in pH homeostasis. It is composed of two sectors: a catalytic V1 sector and a membrane-bound V0 sector. The V1 sector, comprising eight different subunits, hydrolyzes ATP to ADP to abstract energy for H+ transportation. The V0 sector is composed of five different subunits that form the H+ translocating channel (Forgac, 1989). In the highly invasive human breast cancer cells MDA-MB-231, the extrusion of protons via membrane-bound V-ATPase was shown to cause extracellular acidification and to contribute to the maintenance of a negative pH gradient between the cytosol and the acidic extracellular environment (Sennoune et al., 2004; Hinton et al., 2009). Low extracellular pH (pHe) may induce the increased secretion and activation of proteases such as matrix metalloproteinases (MMP), bone morphogenetic protein-1-type metalloproteinases, tissue serine proteases, and adamalysin-related membrane proteases, resulting in degradation and remodeling of the extracellular matrix. V-ATPase thereby contributes to cancer invasion and metastasis. It is indeed overexpressed in many types of metastatic cancers and positively correlates with invasion and metastasis (Martinez-Zaguilan et al., 1993; Sennoune et al., 2004). Although none of the subunits of V-ATPase was reported as a HIF-1-target gene product so far, subunit c in V0 (ATP6V0C) has been found to directly interact with HIF-1α, suggesting it to be a novel regulator of HIF-1 (Lim et al., 2007).
Sodium–Proton Exchanger (NHE1)
NHE1 is a ubiquitously expressed member of the SLC9A family of Na+/H+ exchangers that mediates the transmembrane exchange of intracellular proton for extracellular sodium. Amiloride-sensitive and growth factor-activated NHE1 is activated by mitogens, integrins and oncogenic transformation (Paris and Pouyssegur, 1984), and has been suggested as a HIF-1-target gene product (Shimoda et al., 2006; Mo et al., 2011). In normal cells, NHE1 is almost quiescent at neutral pH. Proton-dependent activation occurs when pHi becomes acidic, i.e., below a set point of 7.1–6.9 pH units (Reshkin et al., 2000). In mitogen-stimulated normal cells like in cancer cells, an increase in the affinity of the allosteric intracellular proton-binding site thus hyperactivates NHE1 resulting in an increase in pHi and extracellular acidification. Numerous studies with in vitro cancer cell cultures including migration assays have shown that NHE1 is polarized at the leading edge of invadopodia where it promotes a local increase in pHi and a decrease in pHe, both involved in cell extension (Pouyssegur et al., 1984; Cardone et al., 2005; Stuwe et al., 2007; Chiang et al., 2008). Indeed, increased pHi remodels the cytoskeleton, whereas lowered pHe modifies cellular attachment to substrates and disrupts the extracellular matrix (Stuwe et al., 2007; Busco et al., 2010). High NHE1 expression was reported to correlate with poor clinical outcome in cervix and hepatocellular cancers (Chiang et al., 2008; Yang et al., 2010).
Although we emphasized above the prominent role exerted by HIF-1 in the glycolytic switch, several additional pathways collaborate with HIF-1 to promote high-rate glycolysis. An important contributor is c-Myc, a transcription factor normally involved in the regulation of cell metabolism and in the induction of cell proliferation (Grandori et al., 2000). While HIF-1 has evolved to facilitate energy production through glycolysis under hypoxia, c-Myc in contrast promotes mitochondrial biogenesis under normoxic conditions (Li et al., 2005). Through various mechanisms (see Dang et al., 2008 for a complete review), both pathways are mutually exclusive in normal cells. In many tumors, however, c-Myc is found to be overexpressed as a consequence of yet incompletely understood mechanisms involving gene amplification, altered transcriptional control and chromosomal translocation (Oster et al., 2002). Ectopically expressed c-Myc cooperates with HIF-1 to induce the expression of GLUT1, of the glycolytic enzymes HK2, PDK1, and LDH5, and of pro-angiogenic VEGF (Ebert et al., 1995; Osthus et al., 2000; Kim et al., 2007). In addition, c-Myc promotes the selection of PKM2 versus PKM1, as described before (David et al., 2010). c-Myc deregulation in cancer cells thus offers the advantage of cooperativity with HIF-1 to simultaneously promote glycolytic energy production and cataplerosis. Another pathway is initiated when AMPK, the energy-state sensor of the cell, is allosterically activated by the rising levels of AMP associated with the energetic crisis that can be met with hypoxia. Once activated, AMPK directly phosphorylates and activates PFKFB3, supporting accelerated glycolysis as detailed above (Marsin et al., 2002). Interestingly, AMPK activation has also been shown to support the phosphorylative inhibition of mammalian target of rapamycin (mTOR), a kinase stimulating the transcription of HIF-1α and MYC genes (Inoki et al., 2003; Guertin and Sabatini, 2007; Shackelford et al., 2009). While AMPK represses mTOR, mTOR hyperactivation is associated with malignancies characterized by a high glycolytic rate (Inoki et al., 2003; Corradetti et al., 2004; Gwinn et al., 2008). Another feature of AMPK as a cell cycle check-point is its ability to activate p53 (Imamura et al., 2001; Zhang et al., 2010), and recent findings have implicated p53 in the inhibition of glycolysis (Jiang et al., 2011).
Glycolysis and Biosynthesis in Cancer
The second metabolic equation of cancer refers to the biosynthesis of cell constituents. Glucose is a major source of carbohydrates, with as direct consequence that full energy extraction in oxidative pathways would deprive cells from important biosynthetic blocks. Similarly, a glycolytic cell producing lactate from glucose stoichiometrically would fail to proliferate. The second metabolic equation of cancer can therefore be rephrased: which metabolic behavior is compatible with cell proliferation? Or even further: which metabolic behavior would promote cell proliferation? Current knowledge indicates that glycolysis offers the best plasticity to determine the fate of metabolic intermediates, that TCA cycle reactions are an essential providers of biosynthetic precursors in a process termed cataplerosis, and that glucose is not the only source of carbohydrates (DeBerardinis et al., 2008). In fact, any proliferating cell in the body undergoes a metabolic switch primarily consisting of uncoupling the TCA cycle from OXPHOS (Vander Heiden et al., 2010). Organic acids such as citrate, isocitrate, and malate may thereby leak out from mitochondria. The switch is associated with an increased glycolytic rate allowing glycolysis to become a main provider of ATP but also to serve as a hub for several biosynthetic pathways (DeBerardinis et al., 2008). Phenotypically, proliferating cells which have increased glucose uptake and increased lactate production perform aerobic glycolysis, a phenomenon initially described in mouse ascites tumor cells by the 1931 Nobel Prize laureate Otto Warburg and now termed “the Warburg effect” (Warburg et al., 1927). Aerobic glycolysis is a metabolic hallmark of any proliferating cell, being malignant or not (Vander Heiden et al., 2010). What distinguishes cancer cells from normal cells is its persistence, aerobic glycolysis being otherwise naturally reversible in after cell division. Self-autonomous aerobic glycolysis is thus an integral part of the proliferative phenotype of cancers. It is driven by genetic and epigenetic changes that are still incompletely understood. Mutations in enzymes coupling the TCA cycle to OXPHOS have been identified (Pollard et al., 2003; Selak et al., 2005; Dang et al., 2009) but account only marginally for the total population of cell lines performing aerobic glycolysis. Consequently, the Warburg effect may be reverted pharmacologically in many cases (Fantin et al., 2006; Moreno-Sanchez et al., 2007).
Privileged biosynthetic pathways in Warburg-phenotype tumor cells are summarized in Figure 7 and have been extensively reviewed recently (DeBerardinis et al., 2008). Briefly, two HIF-1-target gene products exert key influences: PKM2 and PDK1. PKM2 exists either as an active tetramer that promotes ATP production or as an inactive dimer that redirect carbohydrates to ribulose-5P and NADPH production through the PPP. Ribulose-5P is as an essential precursor for DNA synthesis. NADPH on the other hand is a reductant that may serve either to recycle anti-oxidant glutathione or as an essential reagent for lipid biogenesis (Vander Heiden et al., 2009). As a metabolic sensor, PKM2 is under the positive allosteric regulation of F1,6BP (a glycolytic intermediate signaling the upstream saturation of biosynthetic pathways) and under allosteric repression by downstream biosynthetic products such as alanine, other amino-acids, and lipids (see above). It is further controlled by oncogene-mediated phosphorylation. A second important biosynthetic bottleneck is PDH, under the repressive control of PDK1. PDK1 controls mitochondrial activities: when activated, it prevents pyruvate entry into the TCA cycle; when inactive, pyruvate is converted to acetyl-coA and may serve as a precursor for the production of the cataplerotic products citrate and isocitrate (initiating in lipid biogenesis), glutamate (generating glutamine), and malate (to produce NADPH and pyruvate through the malic enzyme reaction). NADPH, produced either in the oxidative arm of the PPP or through the malic reaction, is a necessary cosubstrate for lipid biogenesis and promotes glutathione turnover for the detoxification of ROS produced from uncoupled mitochondria. Importantly, replenishment of the TCA cycle is ensured by glutamine, taken up from the extracellular environment and recently identified as an important source of organic acids for biosynthesis (DeBerardinis and Cheng, 2010). Glutamine uptake is under the control of c-Myc (Wise et al., 2008; Gao et al., 2009). Although aerobic glycolysis is now widely recognized as an essential component of the proliferative phenotype, key questions still remain unanswered. For instance, several studies report constitutive HIF-1 activity in Warburg-phenotype tumor cells, but, although pathways triggering HIF-1 under normoxia have been known for years (Semenza, 2010), none in particular has been conclusively linked with the Warburg effect yet. Furthermore, the primum movens accounting for OXPHOS inhibition remains elusive, which consequently limits our understanding as to whether or not reversion of the Warburg effect may be therapeutically exploited.
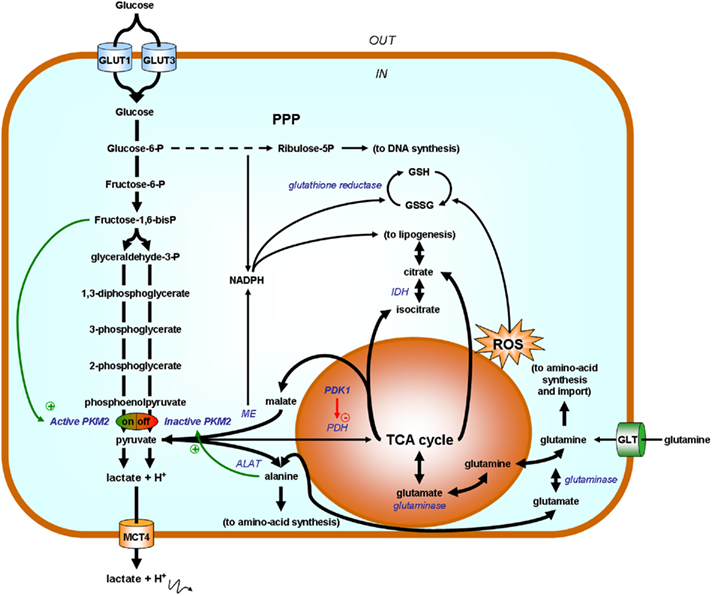
Figure 7. Simplified scheme highlighting glycolysis as a biosynthetic hub. Enzymes are represented in italicized blue font and their substrates in bold black. Tumor cell proliferation relies on aerobic glycolysis for ATP production and also to redirect carbohydrates toward biosynthetic routes. When allosterically activated by fructose-1,6- bisphosphate (F1,6BP), pyruvate kinase M2 (PKM2) promotes pyruvate (and ATP) synthesis. Pyruvate fuels either the production of lactate (for NAD+ production further supporting glycolytic ATP production), alanine [through the reversible alanine aminotransferase (ALAT) reaction during which a nitrogen group is transferred from glutamate onto pyruvate to yield alanine], and/or it can be used to replenish the tricarboxylic acid (TCA) cycle. The fate of pyruvate is oriented by the activity of pyruvate dehydrogenase kinase 1 (PDK1) repressing pyruvate dehydrogenase (PDH) in the mitochondrion. When PKM2 is off (for example when accumulating alanine allosterically promotes dimer formation), glucose-6-P is directed toward the pentose phosphate pathway (PPP) to yield ribulose-5P (for DNA synthesis) and NADPH (2 molecules per molecule of G6P). In addition to glucose, glutamine is an important source of organic acids for proliferating cells. Glutamine uptake is mediated by glutamine transporters (GLT) and glutaminases present in the cytosol or in the mitochondrion generate glutamate. Glutamate fuels the TCA cycle and is also a nitrogen donor for alanine synthesis (ALAT reaction). Uncoupled mitochondria are major providers of biosynthetic precursors and produce reactive oxygen species (ROS). During cataplerosis, citrate, and isocitrate are exported to fuel lipogenesis, malate is exported and converted into pyruvate for NADPH production by the malic enzyme (ME), and glutamate may serve to regenerate glutamine as a precursor for amino-acid synthesis or to be exchanged against extracellular amino-acids. NADPH, produced either from the PPP or from malate, has two main roles: it is a necessary cofactor for lipogenesis (HMG-CoA reductase step) and it is used as a reductant for the regeneration of glutathione (GSH) from its oxidized disulfide form (GSSG). As an anti-oxidant, GSH detoxifies ROS. Other abbreviations: GLUT, glucose transporter; IDH, isocitrate dehydrogenase; MCT, monocarboxylate transporter.
Anticancer Targets in the Glycolytic Metabolism of Tumors
The aforementioned observations position glycolysis as a key contributor to the malignant phenotype and support the quest for new anticancer treatments targeting glycolysis. Indeed, most of the molecular adaptations supporting high-rate glycolysis are either unique to cancer in an otherwise healthy organism or druggable with manageable toxicities. The most advanced clinical applications exploiting tumor glycolysis are briefly summarized in Table 1 and detailed below. Our purpose is not to be exhaustive but to illustrate how the current molecular knowledge may translate into (future) anticancer treatments.
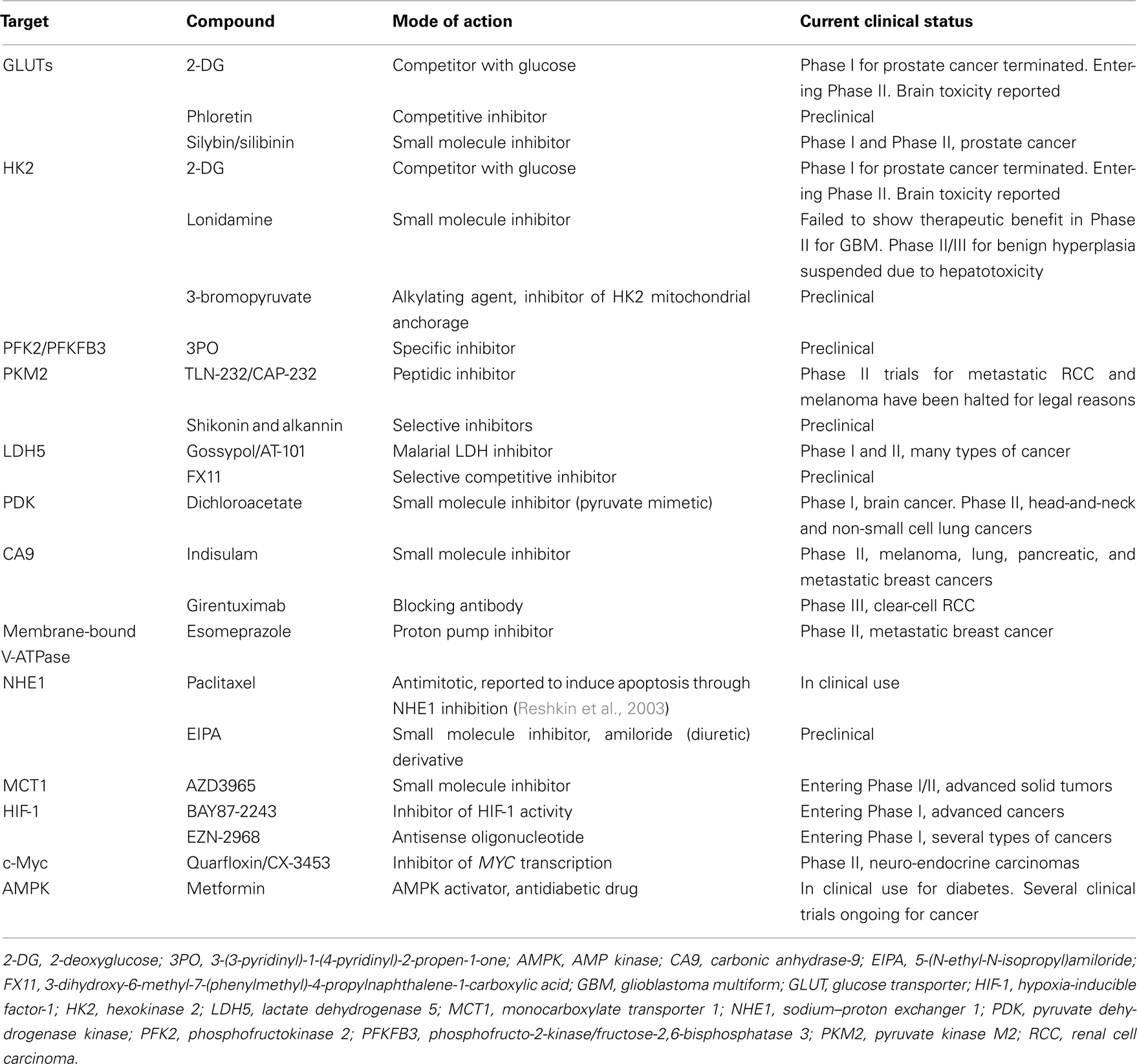
Table 1. Leading therapeutic compounds targeting the glycolytic metabolism of tumors.
Exploiting High Glucose Uptake with 2-Deoxyglucose
High glucose uptake is a direct consequence of the glycolytic switch. While the uptake per se primarily results from overexpression of the high affinity transporters GLUT1 and GLUT3, glucose trapping depends on high-rate glucose phosphorylation by the HK2 reaction (see before). The capability of tumors to take up and sequester glucose even in limiting condition has been broadly imposed for the detection of tumors and their metastases and for tumor staging in the clinics (Gambhir, 2002). The glucose analog [18F]-fluorodeoxyglucose ([18F]-FDG, wherein 18F is a positron emitter) has been developed for positron emission tomography (PET; Som et al., 1980). [18F]-FDG–PET takes advantage of the fact that, although as a glucose analog 2-deoxyglucose (2-DG) is avidly taken up and phosphorylated in tumor cells, lack of the 2′ hydroxyl group does not allow further processing thus leading to 2-DG accumulation. Owing to spatial resolution, the fact that not all tumors are highly glycolytic and because some non-malignant tissues (such as the brain and the bladder) also accumulate the tracer, the overall mean sensitivity and specificity across various applications using [18F]-FDG–PET is of about 85% (Gambhir, 2002).
GLUT Inhibitors
GLUT1 and GLUT3 overexpression in many types of cancer provides a rationale for the anticancer use of GLUT inhibitors. Besides diagnosis and tailored radiotherapy, 2-DG as a GLUT inhibitor has been tested as an anticancer drug. Although its efficacy alone has been questioned (notably because of brain toxicity; Tennant et al., 2010), it has proven efficacy in sensitizing human osteosarcoma and non-small cell lung cancers to adriamycin and paclitaxel (Maschek et al., 2004). A Phase I clinical study for prostate cancer recently terminated defining a dose of 45 mg/kg for Phase II trials (Stein et al., 2010). Phloretin is a natural flavonoid and a competitive GLUT inhibitor which was demonstrated to retard tumor growth in preclinical models (Nelson and Falk, 1993; Kobori et al., 1997). Another natural flavonoid, silybin/silibinin, was recently revealed as a GLUT inhibitor (Zhan et al., 2011). Silybin, already known to inhibit tumor formation and growth in preclinical models (Matsumoto et al., 2008; Garcia-Maceira and Mateo, 2009), is ongoing clinical Phase I (Flaig et al., 2007) and Phase II (ClinicalTrials.gov Identifier: NCT00487721) trials for prostate cancer. Flavonoids have many biological properties and act primarily as anti-oxidants, making it hazardous to conclude that GLUT inhibition is a primary or even a main mechanism accounting for their antitumor effects. To our knowledge, no specific GLUT1 or GLUT3 inhibitor has been identified so far, with as consequence a lack of evidence that GLUT inhibition could be tumor-specific.
HK2 Inhibitors
HK2 has been widely characterized as a facilitator of glycolysis and as a repressor of apoptosis in several types of cancers. Its expression is essential for the growth of glioblastoma multiform (GBM; Wolf et al., 2011). Many efforts have thus been made to identify specific inhibitors. Beside 2-DG, lonidamine has been described since the early eighties as a specific inhibitor of mitochondria-bound HK (Floridi et al., 1981). Lonidamine, which further potentiates the therapeutic efficacy of other anticancer drugs in preclinical models (reviewed in Pelicano et al., 2006), went to Phase II clinical trials for GBM in combination with diazepam, where it failed to show therapeutic benefit in terms of time-to-progression and overall survival (Oudard et al., 2003). The drug was until recently in clinical trials Phase II/III for the treatment of benign prostatic hyperplasia (ClinicalTrials.gov Identifiers: NCT00237536 and NCT00435448), but the trials have been suspended after six patients suffered from severe hepatic adverse effects. The current leading compound is 3-bromopyruvate (3BP), an alkylating agent reacting with cysteine residues in proteins. 3BP was initially identified as an HK inhibitor in an ex vivo model of rabbit liver cancer (Ko et al., 2001), and later confirmed to exert antitumoral activities (including metastatic suppression) in several types of advanced experimental tumors in vivo (see Ganapathy-Kanniappan et al., 2010 for review). The molecular determinants of the cancer selectivity of 3BP are still incompletely understood but could logically depend on the abundance of cysteine-rich glutathione detoxifying 3BP in normal cells and more rapidly exhausted in oxidatively stressed, glycolytic tumor cells (Qin et al., 2010). Because 3BP is chemically highly reactive, several other targets could mediate its antitumor effects (Dell’Antone, 2009). Although preclinical studies led to promising results, 3BP is not yet reported to have entered into clinical trials.
PFKFB3 Inhibitor
As a main provider of the PFK1 allosteric activator F2,6BP, PFKFP3 exerts an important contribution to the glycolytic switch. To our knowledge, 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) is the only specific inhibitor of PFKBP3 identified so far (Clem et al., 2008). 3PO was reported to decrease the concentration of F2,6BP in tumor cell lines, leading to decreased glucose uptake and to growth suppression in several types of experimental tumors in vivo. The same study documented in vitro selectivity for tumor versus non-malignant cells. Independent confirmation is now warranted.
PKM2 Inhibitors
PKM2 is a master switch orienting glycolysis to ATP synthesis or to the production of biosynthetic blocks, making it an attractive target for anticancer treatments. In 2007, Thallion Pharmaceuticals started a Phase II clinical trial with the PKM2 inhibitor TLN-232/CAP-232, a seven amino-acid peptide administered to patients with refractory metastatic renal cell carcinoma. Encouraging results were reported in a poster displayed at the 33rd congress of the European Society for Medical Oncology in September 2008: two out of the 3 patients having completed the study showed stable disease and TLN-232 was generally safe and well tolerated. Recruitment for a second Phase II trial in metastatic melanoma patients started mid 2008 but was halted for legal reasons in June 20101. In 2010, Cantley’s lab screened a huge library of compounds to ultimately identify two water-soluble small molecule inhibitors with selectivity for PKM2 versus PKM1 (Vander Heiden et al., 2010). Both molecules were reported to presumably block the allosteric F1,6BP-binding site of PKM2 absent in PKM1. Recently, shikonin and its enantiomeric isomer alkannin have been shown to inhibit PKM2 at concentrations that resulted in over 50% inhibition of PKM2 without affecting the activities of PKM1 and pyruvate kinase-L (harboring the same F1,6BP-binding site as PKM2; Chen et al., 2011). Both compounds inhibited glucose consumption and lactate release in MCF-7 and A549 tumor cells. These studies collectively demonstrate the possibility to identify specific PKM2 inhibitors that could serve as potent anticancer drugs. Interestingly, a recent publication describes the identification of PKM2 activators intended to be used as antiproliferative agents (Boxer et al., 2010). These drugs would act as oncostatics.
LDH5 Inhibitors
By restoring the NAD+ pool required for the GAPDH reaction, LDH5 plays an essential role in the perpetuation of high-rate glycolysis and is therefore recognized as a therapeutic target for cancer (Xie et al., 2009; Le et al., 2010). High blood and tissue levels are associated with bad prognosis for many types of tumors (discussed in Koukourakis et al., 2011). The significance of LDH5 as a therapeutic target has been documented in recent studies showing that inhibition of its expression using RNA interference impairs tumor initiation, maintenance and progression (Fantin et al., 2006; Le et al., 2010). A selective (with respect to LDH1 and GAPDH) competitive (with respect to NADH binding) inhibitor of LDH5, 3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propylnaphthalene-1-carboxylic acid (FX11), has been identified through screening a bank of compounds derived from the natural product gossypol, a known malarial LDH inhibitor (Yu et al., 2001b). FX11 was recently shown to induce oxidative stress and cell death in vitro, which translated in vivo into inhibition of the progression of human lymphoma and pancreatic cancer xenografts (Le et al., 2010). Although gossypol/AT-101 either alone or in association with chemotherapy is undergoing several Phase I and II clinical trials2, the use of FX11 in clinical studies has not yet been reported. Recently, N-Hydroxy-2-carboxy-substituted indole compounds have been identified as LDH5-specific inhibitors (Granchi et al., 2011). This series of compounds acts as competing inhibitors of LDH5 with respect to both NADH and pyruvate.
PDK Inhibitor
By blocking PDH activity, PDK1 is a major gatekeeper of pyruvate entry into the TCA cycle. Dichloroacetate (DCA) has been known for a long time as a PDK inhibitor (Whitehouse and Randle, 1973) and was already used in the late eighties in clinical trial for lactic acidosis treatment (Stacpoole et al., 1988). This pyruvate mimetic was shown to occupy the pyruvate-binding site of PDK2, which is largely conserved among PDKs, therefore inhibiting non-selectively albeit with different potencies all PDK isoforms (Knoechel et al., 2006). Papandreou et al. (2011) is a recent review describing the preclinical evaluation of DCA. Although the drug showed high heterogeneity in terms of antitumor activity, a prospective clinical trial on five glioblastoma patients treated with surgery, radiation, and temozolomide was completed with promising results (Michelakis et al., 2010). A Phase I clinical trial in brain cancer (ClinicalTrials.gov Identifier: NCT01111097) and two Phase II clinical trials (for head-and-neck, NCT01386632; and for refractory metastatic breast and non-small cell lung cancers, NCT01029925) are ongoing.
Targeting PH Regulation
Low pHe in tumors is the consequence of high metabolic activities and an important determinant of tumor aggressiveness. Indeed, tumor cells contrarily to normal cells at the invasive front of tumors are well equipped for the release of protons and are therefore selectively adapted to survive and proliferate in a moderately acidic environment (Fang et al., 2008; Chiche et al., 2010). Accordingly, Gillies, Gatenby and colleagues have shown that dietary measures that boost bicarbonate levels in the plasma can to some extent induce tumor alkalinization without affecting healthy tissues (Robey et al., 2009). This study also reported a reduced incidence of metastasis in mice in some but not all tumor models. These encouraging results certainly warrant further investigations. On the other hand, many drugs targeting proton transporters have been suggested as anticancer drugs. These drugs should exert maximum toxicity to cancer cells and negligible or minimal toxicity to normal cells. The current leading compound for CA9 inhibition is indisulam, a sulfonamide derivative shown to inhibit CA9 at nanomolar concentrations (Owa et al., 1999; Abbate et al., 2004; Supuran, 2008). Despite no significant efficacy was observed for indisulam as a single agent in a Phase II clinical trial on non-small cell lung cancer (Talbot et al., 2007), additional Phase II trials are ongoing for the treatment of melanoma, lung, pancreatic, and metastatic breast cancers. Blocking antibodies against CA9 or CA12 have been identified (Xu et al., 2010; Battke et al., 2011; Murri-Plesko et al., 2011). Girentuximab, a specific antibody targeting CA9, is now in Phase III clinical trials for treating patients with clear-cell renal cell carcinoma (Deal watch, 2011). Several inhibitors of membrane-bound V-ATPase have been reported and are the topic of a recent review (Perez-Sayans et al., 2009). A blocking antibody has been reported to induce growth retardation in preclinical xenograft models (Wang et al., 2008). Though they have different antitumor efficacies, they all seem to target subunit c in the V0 domain. Esomeprazole (ESOM) is among the leading compounds of the class, currently undergoing Phase II clinical trials in metastatic breast cancer in combination with docetaxel and cisplatin (ClinicalTrials.gov Identifier: NCT01069081). NHE1 can be inhibited by supraclinical concentrations of the diuretic drug amiloride. A derivative of amiloride, 5-(N-ethyl-N-isopropyl)amiloride (EIPA), is 200-times more potent as a specific inhibitor of NHE1 but has not been developed for clinical use (Maidorn et al., 1993). Interestingly, Reshkin et al. (2003) have shown that the pro-apoptotic effects of paclitaxel involved NHE1 inhibition and a decrease in pHi, thus providing additional significance for clinical targeting. For what pertains to MCT4, although MCT4 silencing was shown to reduce tumor cell migration (Gallagher et al., 2007), no specific small molecule inhibitor has been identified so far (Kennedy and Dewhirst, 2010). To our knowledge, no blocking antibody has been identified targeting NHE1 or MCT4. Taken collectively, there is ample evidence that pH regulatory systems constitute promising anticancer targets. Clinical translation will primarily depend on (i) better understanding the relative contribution of each of these systems to the control of tumor pH (target selection), (ii) the identification of isoform-selective inhibitors (to minimize collateral effects), and (iii) the development of non-invasive, sensitive pH imaging modalities. Particular caution (and interest) should be paid to combination treatments with anticancer drugs that are weak acids or bases.
MCT1 Inhibitors
MCT1 inhibitors offer the opportunity to simultaneously target tumor metabolism and angiogenesis within a same molecule. As the main facilitator of lactate uptake, MCT1 is indeed at the core of a metabolic symbiosis in tumors (Figure 5, where MCT1 is expressed in oxidative tumor cells; Sonveaux et al., 2008) and is the most upstream element of a lactate-signaling pathway leading to NF-κB activation and IL-8 production in endothelial cells (Vegran et al., 2011). Expressed at the plasma membrane of oxygenated cells, MCT1 thus offers the advantage of an easy-to-reach target for systemic therapy. Accordingly, the commercially available MCT1 inhibitor α-cyano-4-hydroxycinnamate (CHC) showed potent antitumor affects alone or in combination with radiotherapy in mice, without exerting overt toxicity (Sonveaux et al., 2008; Vegran et al., 2011). Safety is reasonably supported by reports about MCT1-deficient humans who have no symptoms at rest but only develop muscle cramps under intensive exercise; (Fishbein, 1986) and by the use of alternate metabolic substrates by healthy tissues (Halestrap and Meredith, 2004). A first small molecule, AR-C117977, has been developed as a specific MCT1 inhibitor for mild immunosuppression (Bueno et al., 2007; Kennedy and Dewhirst, 2010). A related orally administered compound, AZD3965, is currently entering Phase I/II clinical trials for advanced solid tumors3.
HIF-1 Inhibitors
As a master regulator of the glycolytic and angiogenic switches, HIF-1 has attracted much attention as an anticancer target. Although several drugs have been identified to exert anticancer effects partially through HIF-1 inhibition (extensively reviewed in Onnis et al., 2009), there was until recently no small drug directly and selectively inhibiting HIF-1. BAY87-2243, presented as an inhibitor of HIF-1 activity and of HIF-1α stability and now entering into Phase I clinical trials (ClinicalTrials.gov identifier: NCT01297530) could be the first compound of the class. Patients are also currently recruited for a Phase I trial testing EZN-2968, an antisense oligonucleotide targeting HIF-1α (Greenberger et al., 2008). Conclusively, according to the peer-reviewed information available, validation of HIF-1 as druggable anticancer target is still pending.
C-Myc Inhibitors
c-Myc is a member of the basic helix-loop-helix leucine zipper (bHLH-ZIP) protein family. Its dimerization with another bHLH-ZIP protein, Max, is necessary for various biological activities including cellular transformation, apoptosis, and transcriptional activation (Meyer and Penn, 2008). As recently reviewed in Berg (2011), the c-Myc–Max interaction is an appealing target for drug design having stimulated the development of several small molecule inhibitors. One such small molecule, 10058-F4, inhibited the growth of hepatocellular carcinoma cells in vitro (Lin et al., 2007). Another small molecule compound, Quarfloxin/CX-3453, which inhibits MYC transcription (Brooks and Hurley, 2010), is now in Phase II clinical trials for neuro-endocrine carcinoma (ClinicalTrials.gov Identifier: NCT00780663). Although c-Myc inhibitors are progressing to the clinics, an important limitation is that c-Myc is a pleiotropic transcription factor required for the proliferation of normal cells and for the maintenance of stemness (Wilson et al., 2004). It is not clear to date whether c-Myc inhibitors will display side effects as severe as or even worse than chemo- and radiotherapy.
AMPK Activators
Clinical data and translational research have now revealed that patients with metabolic disorders such as type-2 diabetes or obesity have an increased risk to develop tumors. Hyperglycemia and hyperinsulinemia have indeed been implicated in tumorigenesis by several pathways that either directly or indirectly converge to mTOR activation (Godsland, 2010; Jalving et al., 2010; La Vecchia, 2011). Interestingly, epidemiological studies have also shown reduced incidence of cancer in diabetic patients treated metformin, an AMPK-activating drug currently used for type-2 diabetes treatment (Evans et al., 2005; Libby et al., 2009; Jalving et al., 2010). Ongoing clinical studies are aimed at evaluating whether non-diabetic cancer patients could benefit from metformin as a (neo-)adjuvant or chemopreventive treatment (to limit the risk of cancer recurrence; Muti et al., 2009; Martin-Castillo et al., 2010). Despite the undisputable advantage of using a clinically safe drug, efforts are now indispensable to define the optimal therapeutic dose (being lactic acidosis a side effect of AMPK activators) and the genetic background of cancers suitable to be treated with this drug.
Concluding Remarks
The glycolytic switch occupies a privileged position in the aggressive agenda of most solid tumors. Initially proceeding through suppression of the Pasteur Effect in response to hypoxia, it is indeed an early event marking the entry of dormant tumors into an exponential growth phase. As such, switching to a glycolytic metabolism may precede the evolution of tumors toward the more aggressive angiogenic and metastatic phenotypes. Glycolysis also exerts a pervasive influence throughout tumor growth, making of cancer a metabolic disease and suggesting necessary crosstalks between metabolism, angiogenesis, and metastasis. Persistence of high-rate glycolysis is under the master command of the transcription factor HIF-1 which, in collaboration with other oncogenic signaling pathways including c-Myc, AMPK, and mTOR, promotes the expression of most glycolytic enzymes and transporters. HIF-1 is inducible by hypoxia, thus bridging low pO2 to the glycolytic phenotype for anaerobic energy production. But HIF-1 is also constitutively expressed in Warburg-phenotype tumor cells where it couples high-rate aerobic glycolysis to biosynthesis and cell proliferation. Solid tumors are the result of metabolic selection and a peculiar environment hosting different populations of metabolically overactive cells among which cells with aerobic glycolysis, anaerobic glycolysis and more oxidative phenotypes may cohabit. Our identification of a metabolic symbiosis based on the exchange of lactate between glycolytic and oxidative tumor cells provides a cooperative dimension (Sonveaux et al., 2008), recently extended to non-malignant, supportive stromal cells (Bonuccelli et al., 2010). Metabolism is not static but rather highly adaptive to external influences. The Warburg-phenotype itself is often reversible, as it is for non-malignant cells. The comprehensive (and therefore simplified in its expression) review that we provide here positions tumor metabolism as a key contributor to malignancy and as an attractive target for therapy. What have we learned until know? Researches worldwide have essentially shown that several regulators of glycolysis are amenable for anticancer therapy. The field is still in its infancy, though. Essential questions remain unanswered, as for example regarding the plastic adaptation of tumor metabolism to therapy. Furthermore, although many strategies have been being developed, some of which are currently evaluated in Phase I and Phase II clinical trials, there is still no grounded rationale to select one or several regulator(s) of glycolysis as preferred anticancer target(s) and little clinical information about toxicities. Should the therapy be tailored to a given tumor in a given patient? On which bases? One element to take into account could be the genetic background of the tumor based on experimental evidences that showed different metabolic outcomes deriving from some specific mutation (Cairns et al., 2011). Systemic therapies directly targeting hypoxic tumor cells are generally confronted with difficulties to access to the hypoxic tumor cell compartment remote from blood vessels and to the emergence of resistance due to hypoxia-selected DNA instabilities. It is not clear whether antimetabolic therapies will do better than chemotherapy in this prospect. Alternatively, targeting metabolic cooperativity, which will necessitate a thorough understanding of the functions, regulations, and crosstalks between metabolic transporters, is an avenue recently opened for therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Works at the authors’ lab are supported by the European Research Council (FP7/2007-2013 ERC Independent Researcher Starting Grant 243188 TUMETABO to Pierre Sonveaux), the Belgian Fonds National de la Recherche Scientifique (F.R.S.-FNRS), the Communauté Française de Belgique (ARC 09/14-020), and the Fondation Belge Contre le Cancer (200-2008). Paolo E. Porporato, Rajesh K. Dadhich, Suveera Dhup, and Tamara Copetti contributed equally to this paper. Pierre Sonveaux is a F.R.S.-FNRS Research Associate.
Footnotes
References
Abbate, F., Casini, A., Owa, T., Scozzafava, A., and Supuran, C. T. (2004). Carbonic anhydrase inhibitors: E7070, a sulfonamide anticancer agent, potently inhibits cytosolic isozymes I and II, and transmembrane, tumor-associated isozyme IX. Bioorg. Med. Chem. Lett. 14, 217–223.
Atsumi, T., Chesney, J., Metz, C., Leng, L., Donnelly, S., Makita, Z., Mitchell, R., and Bucala, R. (2002). High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 62, 5881–5887.
Ayala, F. R., Rocha, R. M., Carvalho, K. C., Carvalho, A. L., da Cunha, I. W., Lourenco, S. V., and Soares, F. A. (2010). GLUT1 and GLUT3 as potential prognostic markers for oral squamous cell carcinoma. Molecules 15, 2374–2387.
Baer, S., Casaubon, L., Schwartz, M. R., Marcogliese, A., and Younes, M. (2002). Glut3 expression in biopsy specimens of laryngeal carcinoma is associated with poor survival. Laryngoscope 112, 393–396.
Battke, C., Kremmer, E., Mysliwietz, J., Gondi, G., Dumitru, C., Brandau, S., Lang, S., Vullo, D., Supuran, C., and Zeidler, R. (2011). Generation and characterization of the first inhibitory antibody targeting tumour-associated carbonic anhydrase XII. Cancer Immunol. Immunother. 60, 649–658.
Berg, T. (2011). Small-molecule modulators of c-Myc/Max and Max/Max interactions. Curr. Top. Microbiol. Immunol. 348, 139–149.
Berra, E., Benizri, E., Ginouves, A., Volmat, V., Roux, D., and Pouyssegur, J. (2003). HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 22, 4082–4090.
Bonuccelli, G., Tsirigos, A., Whitaker-Menezes, D., Pavlides, S., Pestell, R. G., Chiavarina, B., Frank, P. G., Flomenberg, N., Howell, A., Martinez-Outschoorn, U. E., Sotgia, F., and Lisanti, M. P. (2010). Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 9, 3506–3514.
Boxer, M. B., Jiang, J. K., Vander Heiden, M. G., Shen, M., Skoumbourdis, A. P., Southall, N., Veith, H., Leister, W., Austin, C. P., Park, H. W., Inglese, J., Cantley, L. C., Auld, D. S., and Thomas, C. J. (2010). Evaluation of substituted N,N′-diarylsulfonamides as activators of the tumor cell specific M2 isoform of pyruvate kinase. J. Med. Chem. 53, 1048–1055.
Bristow, R. G., and Hill, R. P. (2008). Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 8, 180–192.
Brizel, D. M., Schroeder, T., Scher, R. L., Walenta, S., Clough, R. W., Dewhirst, M. W., and Mueller-Klieser, W. (2001). Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 51, 349–353.
Brooks, T. A., and Hurley, L. H. (2010). Targeting MYC Expression through G-quadruplexes. Genes Cancer 1, 641–649.
Bueno, V., Binet, I., Steger, U., Bundick, R., Ferguson, D., Murray, C., Donald, D., and Wood, K. (2007). The specific monocarboxylate transporter (MCT1) inhibitor, AR-C117977, a novel immunosuppressant, prolongs allograft survival in the mouse. Transplantation 84, 1204–1207.
Busco, G., Cardone, R. A., Greco, M. R., Bellizzi, A., Colella, M., Antelmi, E., Mancini, M. T., Dell’Aquila, M. E., Casavola, V., Paradiso, A., and Reshkin, S. J. (2010). NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 24, 3903–3915.
Bustamante, E., and Pedersen, P. L. (1977). High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc. Natl. Acad. Sci. U.S.A. 74, 3735–3739.
Cairns, R. A., Harris, I. S., and Mak, T. W. (2011). Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95.
Cardone, R. A., Casavola, V., and Reshkin, S. J. (2005). The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 5, 786–795.
Chen, J., Xie, J., Jiang, Z., Wang, B., Wang, Y., and Hu, X. (2011). Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. doi: 10.1038/onc.2011.137. [Epub ahead of print].
Chiang, Y., Chou, C. Y., Hsu, K. F., Huang, Y. F., and Shen, M. R. (2008). EGF upregulates Na+/H+ exchanger NHE1 by post-translational regulation that is important for cervical cancer cell invasiveness. J. Cell. Physiol. 214, 810–819.
Chiche, J., Brahimi-Horn, M. C., and Pouyssegur, J. (2010). Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J. Cell. Mol. Med. 14, 771–794.
Chiche, J., Fur, Y. L., Vilmen, C., Frassineti, F., Daniel, L., Halestrap, A. P., Cozzone, P. J., Pouyssegur, J., and Lutz, N. W. (2011). In vivo pH in metabolic-defective Ras-transformed fibroblast tumors: key role of the monocarboxylate transporter, MCT4, for inducing an alkaline intracellular pH. Int. J. Cancer. doi: 10.1002/ijc.26125. [Epub ahead of print].
Chiche, J., Ilc, K., Laferriere, J., Trottier, E., Dayan, F., Mazure, N. M., Brahimi-Horn, M. C., and Pouyssegur, J. (2009). Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 69, 358–368.
Christofk, H. R., Vander Heiden, M. G., Harris, M. H., Ramanathan, A., Gerszten, R. E., Wei, R., Fleming, M. D., Schreiber, S. L., and Cantley, L. C. (2008a). The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233.
Christofk, H. R., Vander Heiden, M. G., Wu, N., Asara, J. M., and Cantley, L. C. (2008b). Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452, 181–186.
Clem, B., Telang, S., Clem, A., Yalcin, A., Meier, J., Simmons, A., Rasku, M. A., Arumugam, S., Dean, W. L., Eaton, J., Lane, A., Trent, J. O., and Chesney, J. (2008). Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 7, 110–120.
Corradetti, M. N., Inoki, K., Bardeesy, N., DePinho, R. A., and Guan, K. L. (2004). Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 18, 1533–1538.
Curi, R., Newsholme, P., and Newsholme, E. A. (1988). Metabolism of pyruvate by isolated rat mesenteric lymphocytes, lymphocyte mitochondria and isolated mouse macrophages. Biochem. J. 250, 383–388.
Dang, C. V., Kim, J. W., Gao, P., and Yustein, J. (2008). The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 8, 51–56.
Dang, C. V., and Semenza, G. L. (1999). Oncogenic alterations of metabolism. Trends Biochem. Sci. 24, 68–72.
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., Fantin, V. R., Jang, H. G., Jin, S., Keenan, M. C., Marks, K. M., Prins, R. M., Ward, P. S., Yen, K. E., Liau, L. M., Rabinowitz, J. D., Cantley, L. C., Thompson, C. B., Vander Heiden, M. G., and Su, S. M. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744.
David, C. J., Chen, M., Assanah, M., Canoll, P., and Manley, J. L. (2010). HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368.
Deal watch: phase III antibody targeting tumour pH acquired in Prometheus deal. (2011). Nat. Rev. Drug Discov. 10, 482.
DeBerardinis, R. J., and Cheng, T. (2010). Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–324.
DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G., and Thompson, C. B. (2008). The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20.
DeBerardinis, R. J., Mancuso, A., Daikhin, E., Nissim, I., Yudkoff, M., Wehrli, S., and Thompson, C. B. (2007). Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 19345–19350.
Dell’Antone, P. (2009). Targets of 3-bromopyruvate, a new, energy depleting, anticancer agent. Med. Chem. 5, 491–496.
Dimmer, K. S., Friedrich, B., Lang, F., Deitmer, J. W., and Broer, S. (2000). The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 350(Pt 1), 219–227.
Ebert, B. L., Firth, J. D., and Ratcliffe, P. J. (1995). Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J. Biol. Chem. 270, 29083–29089.
Ebert, B. L., Gleadle, J. M., O’Rourke, J. F., Bartlett, S. M., Poulton, J., and Ratcliffe, P. J. (1996). Isoenzyme-specific regulation of genes involved in energy metabolism by hypoxia: similarities with the regulation of erythropoietin. Biochem. J. 313(Pt 3), 809–814.
Evans, J. M., Donnelly, L. A., Emslie-Smith, A. M., Alessi, D. R., and Morris, A. D. (2005). Metformin and reduced risk of cancer in diabetic patients. BMJ 330, 1304–1305.
Fang, J. S., Gillies, R. D., and Gatenby, R. A. (2008). Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin. Cancer Biol. 18, 330–337.
Fantin, V. R., St Pierre, J., and Leder, P. (2006). Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9, 425–434.
Fenske, W., Volker, H. U., Adam, P., Hahner, S., Johanssen, S., Wortmann, S., Schmidt, M., Morcos, M., Muller-Hermelink, H. K., Allolio, B., and Fassnacht, M. (2009). Glucose transporter GLUT1 expression is an stage-independent predictor of clinical outcome in adrenocortical carcinoma. Endocr. Relat. Cancer 16, 919–928.
Fishbein, W. N. (1986). Lactate transporter defect: a new disease of muscle. Science 234, 1254–1256.
Flaig, T. W., Gustafson, D. L., Su, L. J., Zirrolli, J. A., Crighton, F., Harrison, G. S., Pierson, A. S., Agarwal, R., and Glode, L. M. (2007). A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest. New Drugs 25, 139–146.
Floridi, A., Paggi, M. G., Marcante, M. L., Silvestrini, B., Caputo, A., and De Martino, C. (1981). Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. J. Natl. Cancer Inst. 66, 497–499.
Forgac, M. (1989). Structure and function of vacuolar class of ATP-driven proton pumps. Physiol. Rev. 69, 765–796.
Gallagher, S. M., Castorino, J. J., Wang, D., and Philp, N. J. (2007). Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer Res. 67, 4182–4189.
Gambhir, S. S. (2002). Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2, 683–693.
Ganapathy-Kanniappan, S., Vali, M., Kunjithapatham, R., Buijs, M., Syed, L. H., Rao, P. P., Ota, S., Kwak, B. K., Loffroy, R., and Geschwind, J. F. (2010). 3-bromopyruvate: a new targeted antiglycolytic agent and a promise for cancer therapy. Curr. Pharm. Biotechnol. 11, 510–517.
Gao, P., Tchernyshyov, I., Chang, T. C., Lee, Y. S., Kita, K., Ochi, T., Zeller, K. I., De Marzo, A. M., Van Eyk, J. E., Mendell, J. T., and Dang, C. V. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765.
Garcia, C. K., Brown, M. S., Pathak, R. K., and Goldstein, J. L. (1995). cDNA cloning of MCT2, a second monocarboxylate transporter expressed in different cells than MCT1. J. Biol. Chem. 270, 1843–1849.
Garcia-Maceira, P., and Mateo, J. (2009). Silibinin inhibits hypoxia-inducible factor-1alpha and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene 28, 313–324.
Gatenby, R. A., and Gillies, R. J. (2004). Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891–899.
Godsland, I. F. (2010). Insulin resistance and hyperinsulinemia in the development and progression of cancer. Clin. Sci. (Lond.) 118, 315–332.
Gottlob, K., Majewski, N., Kennedy, S., Kandel, E., Robey, R. B., and Hay, N. (2001). Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 15, 1406–1418.
Gould, G. W., Thomas, H. M., Jess, T. J., and Bell, G. I. (1991). Expression of human glucose transporters in Xenopus oocytes: kinetic characterization and substrate specificities of the erythrocyte, liver, and brain isoforms. Biochemistry 30, 5139–5145.
Granchi, C., Roy, S., Giacomelli, C., Macchia, M., Tuccinardi, T., Martinelli, A., Lanza, M., Betti, L., Giannaccini, G., Lucacchini, A., Funel, N., Leon, L. G., Giovannetti, E., Peters, G. J., Palchaudhuri, R., Calvaresi, E. C., Hergenrother, P. J., and Minutolo, F. (2011). Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J. Med. Chem. 54, 1599–1612.
Grandori, C., Cowley, S. M., James, L. P., and Eisenman, R. N. (2000). The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 16, 653–699.
Greenberger, L. M., Horak, I. D., Filpula, D., Sapra, P., Westergaard, M., Frydenlund, H. F., Albaek, C., Schroder, H., and Orum, H. (2008). A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol. Cancer Ther. 7, 3598–3608.
Guertin, D. A., and Sabatini, D. M. (2007). Defining the role of mTOR in cancer. Cancer Cell 12, 9–22.
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., Turk, B. E., and Shaw, R. J. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226.
Haber, R. S., Rathan, A., Weiser, K. R., Pritsker, A., Itzkowitz, S. H., Bodian, C., Slater, G., Weiss, A., and Burstein, D. E. (1998). GLUT1 glucose transporter expression in colorectal carcinoma: a marker for poor prognosis. Cancer 83, 34–40.
Halestrap, A. P., and Meredith, D. (2004). The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 447, 619–628.
Harris, A. L. (2002). Hypoxia – a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47.
Hinton, A., Sennoune, S. R., Bond, S., Fang, M., Reuveni, M., Sahagian, G. G., Jay, D., Martinez-Zaguilan, R., and Forgac, M. (2009). Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J. Biol. Chem. 284, 16400–16408.
Hirsila, M., Koivunen, P., Gunzler, V., Kivirikko, K. I., and Myllyharju, J. (2003). Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 278, 30772–30780.
Holness, M. J., and Sugden, M. C. (2003). Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 31, 1143–1151.
Horsman, M. R., and Overgaard, J. (2002). “The oxygen effect and tumour microenvironment,” in Basic Clinical Radiobiology, ed. G. G. Steel (London: Arnold), 156–168.
Hussain, S. A., Ganesan, R., Reynolds, G., Gross, L., Stevens, A., Pastorek, J., Murray, P. G., Perunovic, B., Anwar, M. S., Billingham, L., James, N. D., Spooner, D., Poole, C. J., Rea, D. W., and Palmer, D. H. (2007). Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br. J. Cancer 96, 104–109.
Imamura, K., Ogura, T., Kishimoto, A., Kaminishi, M., and Esumi, H. (2001). Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 287, 562–567.
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590.
Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., Salic, A., Asara, J. M., Lane, W. S., and Kaelin, W. G. Jr. (2001). HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468.
Izumi, H., Takahashi, M., Uramoto, H., Nakayama, Y., Oyama, T., Wang, K. Y., Sasaguri, Y., Nishizawa, S., and Kohno, K. (2011). Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci. 102, 1007–1013.
Jaakkola, P., Mole, D. R., Tian, Y. M., Wilson, M. I., Gielbert, J., Gaskell, S. J., Kriegsheim, A., Hebestreit, H. F., Mukherji, M., Schofield, C. J., Maxwell, P. H., Pugh, C. W., and Ratcliffe, P. J. (2001). Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472.
Jalving, M., Gietema, J. A., Lefrandt, J. D., de Jong, S., Reyners, A. K., Gans, R. O., and de Vries, E. G. (2010). Metformin: taking away the candy for cancer? Eur. J. Cancer 46, 2369–2380.
Jiang, P., Du, W., Wang, X., Mancuso, A., Gao, X., Wu, M., and Yang, X. (2011). p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 13, 310–316.
Kennedy, K. M., and Dewhirst, M. W. (2010). Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. 6, 127–148.
Kessler, R., Bleichert, F., Warnke, J. P., and Eschrich, K. (2008). 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3) is up-regulated in high-grade astrocytomas. J. Neurooncol. 86, 257–264.
Kim, J. W., Gao, P., Liu, Y. C., Semenza, G. L., and Dang, C. V. (2007). Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 27, 7381–7393.
Kim, J. W., Tchernyshyov, I., Semenza, G. L., and Dang, C. V. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185.
Kim, Y. W., Park, Y. K., Yoon, T. Y., and Lee, S. M. (2002). Expression of the GLUT1 glucose transporter in gallbladder carcinomas. Hepatogastroenterology 49, 907–911.
Knoechel, T. R., Tucker, A. D., Robinson, C. M., Phillips, C., Taylor, W., Bungay, P. J., Kasten, S. A., Roche, T. E., and Brown, D. G. (2006). Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry 45, 402–415.
Ko, Y. H., Pedersen, P. L., and Geschwind, J. F. (2001). Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett. 173, 83–91.
Kobori, M., Shinmoto, H., Tsushida, T., and Shinohara, K. (1997). Phloretin-induced apoptosis in B16 melanoma 4A5 cells by inhibition of glucose transmembrane transport. Cancer Lett. 119, 207–212.
Koukourakis, M. I., Giatromanolaki, A., Simopoulos, C., Polychronidis, A., and Sivridis, E. (2005). Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin. Exp. Metastasis 22, 25–30.
Koukourakis, M. I., Giatromanolaki, A., Sivridis, E., Bougioukas, G., Didilis, V., Gatter, K. C., and Harris, A. L. (2003). Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br. J. Cancer 89, 877–885.
Koukourakis, M. I., Giatromanolaki, A., Sivridis, E., Gatter, K. C., Trarbach, T., Folprecht, G., Shi, M. M., Lebwohl, D., Jalava, T., Laurent, D., Meinhardt, G., and Harris, A. L. (2011). Prognostic and predictive role of lactate dehydrogenase 5 (LDH5) expression in colorectal cancer patients treated with PTK787/ZK 222584 (Vatalanib) anti-angiogenic therapy. Clin. Cancer Res. 17, 4892–4900.
Koukourakis, M. I., Giatromanolaki, A., Winter, S., Leek, R., Sivridis, E., and Harris, A. L. (2009). Lactate dehydrogenase 5 expression in squamous cell head and neck cancer relates to prognosis following radical or postoperative radiotherapy. Oncology 77, 285–292.
La Vecchia, C. (2011). Diabetes mellitus, medications for type 2 diabetes mellitus, and cancer risk. Metabolism. PMID: 21550082. [Epub ahead of print].
Le, A., Cooper, C. R., Gouw, A. M., Dinavahi, R., Maitra, A., Deck, L. M., Royer, R. E., Vander Jagt, D. L., Semenza, G. L., and Dang, C. V. (2010). Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. U.S.A. 107, 2037–2042.
Leiblich, A., Cross, S. S., Catto, J. W., Phillips, J. T., Leung, H. Y., Hamdy, F. C., and Rehman, I. (2006). Lactate dehydrogenase-B is silenced by promoter hypermethylation in human prostate cancer. Oncogene 25, 2953–2960.
Leite, T. C., Coelho, R. G., Da Silva, D., Coelho, W. S., Marinho-Carvalho, M. M., and Sola-Penna, M. (2011). Lactate downregulates the glycolytic enzymes hexokinase and phosphofructokinase in diverse tissues from mice. FEBS Lett. 585, 92–98.
Li, F., Wang, Y., Zeller, K. I., Potter, J. J., Wonsey, D. R., O’Donnell, K. A., Kim, J. W., Yustein, J. T., Lee, L. A., and Dang, C. V. (2005). Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol. 25, 6225–6234.
Libby, G., Donnelly, L. A., Donnan, P. T., Alessi, D. R., Morris, A. D., and Evans, J. M. (2009). New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care 32, 1620–1625.
Lim, J. H., Park, J. W., Kim, S. J., Kim, M. S., Park, S. K., Johnson, R. S., and Chun, Y. S. (2007). ATP6V0C competes with von Hippel-Lindau protein in hypoxia-inducible factor 1alpha (HIF-1alpha) binding and mediates HIF-1alpha expression by bafilomycin A1. Mol. Pharmacol. 71, 942–948.
Lin, C. P., Liu, J. D., Chow, J. M., Liu, C. R., and Liu, H. E. (2007). Small-molecule c-Myc inhibitor, 10058-F4, inhibits proliferation, downregulates human telomerase reverse transcriptase and enhances chemosensitivity in human hepatocellular carcinoma cells. Anticancer Drugs 18, 161–170.
Luo, W., Hu, H., Chang, R., Zhong, J., Knabel, M., O’Meally, R., Cole, R. N., Pandey, A., and Semenza, G. L. (2011). Pyruvate kinase M2 Is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145, 732–744.
Lyshchik, A., Higashi, T., Hara, T., Nakamoto, Y., Fujimoto, K., Doi, R., Imamura, M., Saga, T., and Togashi, K. (2007). Expression of glucose transporter-1, hexokinase-II, proliferating cell nuclear antigen and survival of patients with pancreatic cancer. Cancer Invest. 25, 154–162.
Maidorn, R. P., Cragoe, E. J. Jr., and Tannock, I. F. (1993). Therapeutic potential of analogues of amiloride: inhibition of the regulation of intracellular pH as a possible mechanism of tumour selective therapy. Br. J. Cancer 67, 297–303.
Markert, C. L., Shaklee, J. B., and Whitt, G. S. (1975). Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 189, 102–114.
Marsin, A. S., Bouzin, C., Bertrand, L., and Hue, L. (2002). The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem. 277, 30778–30783.
Martin-Castillo, B., Dorca, J., Vazquez-Martin, A., Oliveras-Ferraros, C., Lopez-Bonet, E., Garcia, M., Del Barco, S., and Menendez, J. A. (2010). Incorporating the antidiabetic drug metformin in HER2-positive breast cancer treated with neo-adjuvant chemotherapy and trastuzumab: an ongoing clinical-translational research experience at the Catalan Institute of Oncology. Ann. Oncol. 21, 187–189.
Martinez-Zaguilan, R., Lynch, R. M., Martinez, G. M., and Gillies, R. J. (1993). Vacuolar-type H(+)-ATPases are functionally expressed in plasma membranes of human tumor cells. Am. J. Physiol. 265, C1015–C1029.
Maschek, G., Savaraj, N., Priebe, W., Braunschweiger, P., Hamilton, K., Tidmarsh, G. F., De Young, L. R., and Lampidis, T. J. (2004). 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 64, 31–34.
Masson, N., Willam, C., Maxwell, P. H., Pugh, C. W., and Ratcliffe, P. J. (2001). Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 20, 5197–5206.
Mathupala, S. P., Rempel, A., and Pedersen, P. L. (2001). Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 276, 43407–43412.
Matsumoto, S., Hyodo, F., Subramanian, S., Devasahayam, N., Munasinghe, J., Hyodo, E., Gadisetti, C., Cook, J. A., Mitchell, J. B., and Krishna, M. C. (2008). Low-field paramagnetic resonance imaging of tumor oxygenation and glycolytic activity in mice. J. Clin. Invest. 118, 1965–1973.
Maxwell, P. H., Wiesener, M. S., Chang, G. W., Clifford, S. C., Vaux, E. C., Cockman, M. E., Wykoff, C. C., Pugh, C. W., Maher, E. R., and Ratcliffe, P. J. (1999). The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275.
Mazurek, S. (2011). Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 43, 969–980.
Mazurek, S., Boschek, C. B., Hugo, F., and Eigenbrodt, E. (2005). Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 15, 300–308.
Michelakis, E. D., Sutendra, G., Dromparis, P., Webster, L., Haromy, A., Niven, E., Maguire, C., Gammer, T. L., Mackey, J. R., Fulton, D., Abdulkarim, B., McMurtry, M. S., and Petruk, K. C. (2010). Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2, 31–34.
Minchenko, A., Leshchinsky, I., Opentanova, I., Sang, N., Srinivas, V., Armstead, V., and Caro, J. (2002). Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J. Biol. Chem. 277, 6183–6187.
Minchenko, O., Opentanova, I., and Caro, J. (2003). Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 554, 264–270.
Minchenko, O. H., Ochiai, A., Opentanova, I. L., Ogura, T., Minchenko, D. O., Caro, J., Komisarenko, S. V., and Esumi, H. (2005). Overexpression of 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie 87, 1005–1010.
Mo, X. G., Chen, Q. W., Li, X. S., Zheng, M. M., Ke, D. Z., Deng, W., Li, G. Q., Jiang, J., Wu, Z. Q., Wang, L., Wang, P., Yang, Y., and Cao, G. Y. (2011). Suppression of NHE1 by small interfering RNA inhibits HIF-1alpha-induced angiogenesis in vitro via modulation of calpain activity. Microvasc. Res. 81, 160–168.
Moreno-Sanchez, R., Rodriguez-Enriquez, S., Marin-Hernandez, A., and Saavedra, E. (2007). Energy metabolism in tumor cells. FEBS J. 274, 1393–1418.
Murri-Plesko, M. T., Hulikova, A., Oosterwijk, E., Scott, A. M., Zortea, A., Harris, A. L., Ritter, G., Old, L., Bauer, S., Swietach, P., and Renner, C. (2011). Antibody inhibiting enzymatic activity of tumour-associated carbonic anhydrase isoform IX. Eur. J. Pharmacol. 657, 173–183.
Muti, P., Berrino, F., Krogh, V., Villarini, A., Barba, M., Strano, S., and Blandino, G. (2009). Metformin, diet and breast cancer: an avenue for chemoprevention. Cell Cycle 8, 2661.
Nakashima, R. A., Mangan, P. S., Colombini, M., and Pedersen, P. L. (1986). Hexokinase receptor complex in hepatoma mitochondria: evidence from N,N′-dicyclohexylcarbodiimide-labeling studies for the involvement of the pore-forming protein VDAC. Biochemistry 25, 1015–1021.
Nakashima, R. A., Paggi, M. G., Scott, L. J., and Pedersen, P. L. (1988). Purification and characterization of a bindable form of mitochondrial bound hexokinase from the highly glycolytic AS-30D rat hepatoma cell line. Cancer Res. 48, 913–919.
Nelson, J. A., and Falk, R. E. (1993). The efficacy of phlorizin and phloretin on tumor cell growth. Anticancer Res. 13, 2287–2292.
Noguchi, T., Inoue, H., and Tanaka, T. (1986). The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J. Biol. Chem. 261, 13807–13812.
Okar, D. A., and Lange, A. J. (1999). Fructose-2,6-bisphosphate and control of carbohydrate metabolism in eukaryotes. Biofactors 10, 1–14.
Okar, D. A., Manzano, A., Navarro-Sabate, A., Riera, L., Bartrons, R., and Lange, A. J. (2001). PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem. Sci. 26, 30–35.
Onnis, B., Rapisarda, A., and Melillo, G. (2009). Development of HIF-1 inhibitors for cancer therapy. J. Cell. Mol. Med. 13, 2780–2786.
Oster, S. K., Ho, C. S., Soucie, E. L., and Penn, L. Z. (2002). The myc oncogene: marvelously complex. Adv. Cancer Res. 84, 81–154.
Osthus, R. C., Shim, H., Kim, S., Li, Q., Reddy, R., Mukherjee, M., Xu, Y., Wonsey, D., Lee, L. A., and Dang, C. V. (2000). Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 275, 21797–21800.
Oudard, S., Carpentier, A., Banu, E., Fauchon, F., Celerier, D., Poupon, M. F., Dutrillaux, B., Andrieu, J. M., and Delattre, J. Y. (2003). Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J. Neurooncol. 63, 81–86.
Owa, T., Yoshino, H., Okauchi, T., Yoshimatsu, K., Ozawa, Y., Sugi, N. H., Nagasu, T., Koyanagi, N., and Kitoh, K. (1999). Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J. Med. Chem. 42, 3789–3799.
Palmieri, D., Fitzgerald, D., Shreeve, S. M., Hua, E., Bronder, J. L., Weil, R. J., Davis, S., Stark, A. M., Merino, M. J., Kurek, R., Mehdorn, H. M., Davis, G., Steinberg, S. M., Meltzer, P. S., Aldape, K., and Steeg, P. S. (2009). Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol. Cancer Res. 7, 1438–1445.
Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L., and Denko, N. C. (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197.
Papandreou, I., Goliasova, T., and Denko, N. C. (2011). Anticancer drugs that target metabolism: is dichloroacetate the new paradigm? Int. J. Cancer 128, 1001–1008.
Paris, S., and Pouyssegur, J. (1984). Growth factors activate the Na+/H+ antiporter in quiescent fibroblasts by increasing its affinity for intracellular H+. J. Biol. Chem. 259, 10989–10994.
Pastorino, J. G., Shulga, N., and Hoek, J. B. (2002). Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 277, 7610–7618.
Pelicano, H., Martin, D. S., Xu, R. H., and Huang, P. (2006). Glycolysis inhibition for anticancer treatment. Oncogene 25, 4633–4646.
Peng, S. Y., Lai, P. L., Pan, H. W., Hsiao, L. P., and Hsu, H. C. (2008). Aberrant expression of the glycolytic enzymes aldolase B and type II hexokinase in hepatocellular carcinoma are predictive markers for advanced stage, early recurrence and poor prognosis. Oncol. Rep. 19, 1045–1053.
Perez, d. H., Wood, I. S., and Trayhurn, P. (2010). Hypoxia stimulates lactate release and modulates monocarboxylate transporter (MCT1, MCT2, and MCT4) expression in human adipocytes. Pflugers Arch. 459, 509–518.
Perez-Sayans, M., Somoza-Martin, J. M., Barros-Angueira, F., Rey, J. M., and Garcia-Garcia, A. (2009). V-ATPase inhibitors and implication in cancer treatment. Cancer Treat. Rev. 35, 707–713.
Philp, N. J., Yoon, H., and Lombardi, L. (2001). Mouse MCT3 gene is expressed preferentially in retinal pigment and choroid plexus epithelia. Am. J. Physiol. Cell Physiol. 280, C1319–C1326.
Pollard, P. J., Wortham, N. C., and Tomlinson, I. P. (2003). The TCA cycle and tumorigenesis: the examples of fumarate hydratase and succinate dehydrogenase. Ann. Med. 35, 632–639.
Pouyssegur, J., Dayan, F., and Mazure, N. M. (2006). Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441, 437–443.
Pouyssegur, J., Sardet, C., Franchi, A., L’Allemain, G., and Paris, S. (1984). A specific mutation abolishing Na+/H+ antiport activity in hamster fibroblasts precludes growth at neutral and acidic pH. Proc. Natl. Acad. Sci. U.S.A. 81, 4833–4837.
Pugh, C. W., and Ratcliffe, P. J. (2003). Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9, 677–684.
Qin, J. Z., Xin, H., and Nickoloff, B. J. (2010). 3-Bromopyruvate induces necrotic cell death in sensitive melanoma cell lines. Biochem. Biophys. Res. Commun. 396, 495–500.
Reinacher, M., and Eigenbrodt, E. (1981). Immunohistological demonstration of the same type of pyruvate kinase isoenzyme (M2-Pk) in tumors of chicken and rat. Virchows Arch. B Cell Pathol. 37, 79–88.
Reshkin, S. J., Bellizzi, A., Caldeira, S., Albarani, V., Malanchi, I., Poignee, M., Alunni-Fabbroni, M., Casavola, V., and Tommasino, M. (2000). Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 14, 2185–2197.
Reshkin, S. J., Bellizzi, A., Cardone, R. A., Tommasino, M., Casavola, V., and Paradiso, A. (2003). Paclitaxel induces apoptosis via protein kinase A- and p38 mitogen-activated protein-dependent inhibition of the Na+/H+ exchanger (NHE) NHE isoform 1 in human breast cancer cells. Clin. Cancer Res. 9, 2366–2373.
Rho, M., Kim, J., Jee, C. D., Lee, Y. M., Lee, H. E., Kim, M. A., Lee, H. S., and Kim, W. H. (2007). Expression of type 2 hexokinase and mitochondria-related genes in gastric carcinoma tissues and cell lines. Anticancer Res. 27, 251–258.
Robey, I. F., Baggett, B. K., Kirkpatrick, N. D., Roe, D. J., Dosescu, J., Sloane, B. F., Hashim, A. I., Morse, D. L., Raghunand, N., Gatenby, R. A., and Gillies, R. J. (2009). Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 69, 2260–2268.
Selak, M. A., Armour, S. M., MacKenzie, E. D., Boulahbel, H., Watson, D. G., Mansfield, K. D., Pan, Y., Simon, M. C., Thompson, C. B., and Gottlieb, E. (2005). Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7, 77–85.
Semenza, G. L. (2010). HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56.
Sennoune, S. R., Bakunts, K., Martinez, G. M., Chua-Tuan, J. L., Kebir, Y., Attaya, M. N., and Martinez-Zaguilan, R. (2004). Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am. J. Physiol. Cell Physiol. 286, C1443–C1452.
Shackelford, D. B., Vasquez, D. S., Corbeil, J., Wu, S., Leblanc, M., Wu, C. L., Vera, D. R., and Shaw, R. J. (2009). mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc. Natl. Acad. Sci. U.S.A. 106, 11137–11142.
Shimoda, L. A., Fallon, M., Pisarcik, S., Wang, J., and Semenza, G. L. (2006). HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L941–L949.
Som, P., Atkins, H. L., Bandoypadhyay, D., Fowler, J. S., MacGregor, R. R., Matsui, K., Oster, Z. H., Sacker, D. F., Shiue, C. Y., Turner, H., Wan, C. N., Wolf, A. P., and Zabinski, S. V. (1980). A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): nontoxic tracer for rapid tumor detection. J. Nucl. Med. 21, 670–675.
Sonveaux, P., Vegran, F., Schroeder, T., Wergin, M. C., Verrax, J., Rabbani, Z. N., De Saedeleer, C. J., Kennedy, K. M., Diepart, C., Jordan, B. F., Kelley, M. J., Gallez, B., Wahl, M. L., Feron, O., and Dewhirst, M. W. (2008). Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 118, 3930–3942.
Stacpoole, P. W., Lorenz, A. C., Thomas, R. G., and Harman, E. M. (1988). Dichloroacetate in the treatment of lactic acidosis. Ann. Intern. Med. 108, 58–63.
Stein, M., Lin, H., Jeyamohan, C., Dvorzhinski, D., Gounder, M., Bray, K., Eddy, S., Goodin, S., White, E., and Dipaola, R. S. (2010). Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate 70, 1388–1394.
Stuwe, L., Muller, M., Fabian, A., Waning, J., Mally, S., Noel, J., Schwab, A., and Stock, C. (2007). pH dependence of melanoma cell migration: protons extruded by NHE1 dominate protons of the bulk solution. J. Physiol. 585, 351–360.
Supuran, C. T. (2008). Development of small molecule carbonic anhydrase IX inhibitors. BJU Int. 101(Suppl. 4), 39–40.
Tal, M., Thorens, B., Surana, M., Fleischer, N., Lodish, H. F., Hanahan, D., and Efrat, S. (1992). Glucose transporter isotypes switch in T-antigen-transformed pancreatic beta cells growing in culture and in mice. Mol. Cell Biol. 12, 422–432.
Talbot, D. C., von Pawel, J., Cattell, E., Yule, S. M., Johnston, C., Zandvliet, A. S., Huitema, A. D., Norbury, C. J., Ellis, P., Bosquee, L., and Reck, M. (2007). A randomized phase II pharmacokinetic and pharmacodynamic study of indisulam as second-line therapy in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 13, 1816–1822.
Tanaka, M., Nakamura, F., Mizokawa, S., Matsumura, A., Matsumura, K., Murata, T., Shigematsu, M., Kageyama, K., Ochi, H., and Watanabe, Y. (2004). Role of lactate in the brain energy metabolism: revealed by bioradiography. Neurosci. Res. 48, 13–20.
Tennant, D. A., Duran, R. V., and Gottlieb, E. (2010). Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 10, 267–277.
Thangaraju, M., Carswell, K. N., Prasad, P. D., and Ganapathy, V. (2009). Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem. J. 417, 379–389.
Thangaraju, M., Gopal, E., Martin, P. M., Ananth, S., Smith, S. B., Prasad, P. D., Sterneck, E., and Ganapathy, V. (2006). SLC5A8 triggers tumor cell apoptosis through pyruvate-dependent inhibition of histone deacetylases. Cancer Res. 66, 11560–11564.
Ullah, M. S., Davies, A. J., and Halestrap, A. P. (2006). The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 281, 9030–9037.
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033.
Vander Heiden, M. G., Locasale, J. W., Swanson, K. D., Sharfi, H., Heffron, G. J., Amador-Noguez, D., Christofk, H. R., Wagner, G., Rabinowitz, J. D., Asara, J. M., and Cantley, L. C. (2010). Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329, 1492–1499.
Vegran, F., Boidot, R., Michiels, C., Sonveaux, P., and Feron, O. (2011). Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 71, 2550–2560.
Walenta, S., and Mueller-Klieser, W. F. (2004). Lactate: mirror and motor of tumor malignancy. Semin. Radiat. Oncol. 14, 267–274.
Walenta, S., Salameh, A., Lyng, H., Evensen, J. F., Mitze, M., Rofstad, E. K., and Mueller-Klieser, W. (1997). Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am. J. Pathol. 150, 409–415.
Walenta, S., Schroeder, T., and Mueller-Klieser, W. (2004). Lactate in solid malignant tumors: potential basis of a metabolic classification in clinical oncology. Curr. Med. Chem. 11, 2195–2204.
Walenta, S., Wetterling, M., Lehrke, M., Schwickert, G., Sundfor, K., Rofstad, E. K., and Mueller-Klieser, W. (2000). High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 60, 916–921.
Wang, J., Han, Y., Liang, J., Cheng, X., Yan, L., Wang, Y., Liu, J., Luo, G., Chen, X., Zhao, L., Zhou, X., Wu, K., and Fan, D. (2008). Effect of a novel inhibitory mAb against beta-subunit of F1F0 ATPase on HCC. Cancer Biol. Ther. 7, 1829–1835.
Warburg, O., Wind, F., and Negelein, E. (1927). The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530.
Wheeler, T. J., and Hinkle, P. C. (1981). Kinetic properties of the reconstituted glucose transporter from human erythrocytes. J. Biol. Chem. 256, 8907–8914.
Whitehouse, S., and Randle, P. J. (1973). Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (Short Communication). Biochem. J. 134, 651–653.
Wigfield, S. M., Winter, S. C., Giatromanolaki, A., Taylor, J., Koukourakis, M. L., and Harris, A. L. (2008). PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br. J. Cancer 98, 1975–1984.
Wilson, A., Murphy, M. J., Oskarsson, T., Kaloulis, K., Bettess, M. D., Oser, G. M., Pasche, A. C., Knabenhans, C., Macdonald, H. R., and Trumpp, A. (2004). c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 18, 2747–2763.
Wingo, T., Tu, C., Laipis, P. J., and Silverman, D. N. (2001). The catalytic properties of human carbonic anhydrase IX. Biochem. Biophys. Res. Commun. 288, 666–669.
Wise, D. R., DeBerardinis, R. J., Mancuso, A., Sayed, N., Zhang, X. Y., Pfeiffer, H. K., Nissim, I., Daikhin, E., Yudkoff, M., McMahon, S. B., and Thompson, C. B. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U.S.A. 105, 18782–18787.
Wolf, A., Agnihotri, S., Micallef, J., Mukherjee, J., Sabha, N., Cairns, R., Hawkins, C., and Guha, A. (2011). Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 208, 313–326.
Wu, R., and Racker, E. (1959). Regulatory mechanisms in carbohydrate metabolism. IV. Pasteur effect and Crabtree effect in ascites tumor cells. J. Biol. Chem. 234, 1036–1041.
Wykoff, C. C., Beasley, N. J., Watson, P. H., Turner, K. J., Pastorek, J., Sibtain, A., Wilson, G. D., Turley, H., Talks, K. L., Maxwell, P. H., Pugh, C. W., Ratcliffe, P. J., and Harris, A. L. (2000). Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 60, 7075–7083.
Xie, H., Valera, V. A., Merino, M. J., Amato, A. M., Signoretti, S., Linehan, W. M., Sukhatme, V. P., and Seth, P. (2009). LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol. Cancer Ther. 8, 626–635.
Xu, C., Lo, A., Yammanuru, A., Tallarico, A. S., Brady, K., Murakami, A., Barteneva, N., Zhu, Q., and Marasco, W. A. (2010). Unique biological properties of catalytic domain directed human anti-CAIX antibodies discovered through phage-display technology. PLoS ONE 5, e9625. doi: 10.1371/journal.pone.0009625
Yamada, K., and Noguchi, T. (1999). Regulation of pyruvate kinase M gene expression. Biochem. Biophys. Res. Commun. 256, 257–262.
Yang, X., Wang, D., Dong, W., Song, Z., and Dou, K. (2010). Over-expression of Na+/H+ exchanger 1 and its clinicopathologic significance in hepatocellular carcinoma. Med. Oncol. 27, 1109–1113.
Younes, M., Brown, R. W., Mody, D. R., Fernandez, L., and Laucirica, R. (1995). GLUT1 expression in human breast carcinoma: correlation with known prognostic markers. Anticancer Res. 15, 2895–2898.
Younes, M., Brown, R. W., Stephenson, M., Gondo, M., and Cagle, P. T. (1997). Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer 80, 1046–1051.
Yu, F., White, S. B., Zhao, Q., and Lee, F. S. (2001a). HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. U.S.A. 98, 9630–9635.
Yu, Y., Deck, J. A., Hunsaker, L. A., Deck, L. M., Royer, R. E., Goldberg, E., and Vander Jagt, D. L. (2001b). Selective active site inhibitors of human lactate dehydrogenases A4, B4, and C4. Biochem. Pharmacol. 62, 81–89.
Zhan, T., Digel, M., Kuch, E. M., Stremmel, W., and Fullekrug, J. (2011). Silybin and dehydrosilybin decrease glucose uptake by inhibiting GLUT proteins. J. Cell. Biochem. 112, 849–859.
Keywords: hypoxia, cancer, metabolism, glycolytic switch, Warburg effect, biosynthesis, cataplerosis, HIF-1
Citation: Porporato PE, Dhup S, Dadhich RK, Copetti T and Sonveaux P (2011) Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front. Pharmacol. 2:49. doi: 10.3389/fphar.2011.00049
Received: 12 July 2011; Accepted: 05 August 2011;
Published online: 25 August 2011.
Edited by:
Stephane Germain, INSERM, FranceReviewed by:
Juan Iovanna, INSERM, FranceJacques Pouyssegur, CNRS, France
Olivier Cuvillier, CNRS, France
Copyright: © 2011 Porporato, Dhup, Dadhich, Copetti and Sonveaux. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Pierre Sonveaux, Pole of Pharmacology (FATH 5349), Institute of Experimental and Clinical Research, University of Louvain Medical School, Avenue Emmanuel Mounier 52 Box B1.53.09, Brussels 1200, Belgium. e-mail: pierre.sonveaux@uclouvain.be