
- 1 Department of Biology, University of York, York, UK
- 2 Department of Pharmacology, University of Michigan Medical School, Ann Arbor, MI, USA
Voltage-gated Na+ channels (VGSCs) in mammals contain a pore-forming α subunit and one or more β subunits. There are five mammalian β subunits in total: β1, β1B, β2, β3, and β4, encoded by four genes: SCN1B–SCN4B. With the exception of the SCN1B splice variant, β1B, the β subunits are type I topology transmembrane proteins. In contrast, β1B lacks a transmembrane domain and is a secreted protein. A growing body of work shows that VGSC β subunits are multifunctional. While they do not form the ion channel pore, β subunits alter gating, voltage-dependence, and kinetics of VGSCα subunits and thus regulate cellular excitability in vivo. In addition to their roles in channel modulation, β subunits are members of the immunoglobulin superfamily of cell adhesion molecules and regulate cell adhesion and migration. β subunits are also substrates for sequential proteolytic cleavage by secretases. An example of the multifunctional nature of β subunits is β1, encoded by SCN1B, that plays a critical role in neuronal migration and pathfinding during brain development, and whose function is dependent on Na+ current and γ-secretase activity. Functional deletion of SCN1B results in Dravet Syndrome, a severe and intractable pediatric epileptic encephalopathy. β subunits are emerging as key players in a wide variety of physiopathologies, including epilepsy, cardiac arrhythmia, multiple sclerosis, Huntington’s disease, neuropsychiatric disorders, neuropathic and inflammatory pain, and cancer. β subunits mediate multiple signaling pathways on different timescales, regulating electrical excitability, adhesion, migration, pathfinding, and transcription. Importantly, some β subunit functions may operate independently of α subunits. Thus, β subunits perform critical roles during development and disease. As such, they may prove useful in disease diagnosis and therapy.
Introduction
Mammalian voltage-gated Na+ channels (VGSCs) exist as macromolecular complexes in vivo, comprising, at minimum, one pore-forming α subunit and one or more β subunits in a 1:1 stoichiometry for α:β (Catterall, 1992). Traditionally, VGSC β subunits have been termed “auxiliary.” However, increasing evidence suggests that the β subunits are far from auxiliary, and, in fact, function as critical signaling molecules in their own right, perhaps even independently of α subunits. In this review, we will summarize the latest developments describing the growing, diverse, multifunctional roles of the β subunits, including their contribution to human disease.
Molecular Diversity and Functional Architecture
The topology of the canonical VGSC complex is shown in Figure 1. To date, five β subunits have been identified in mammals: β1, its alternative splice variant β1B (previously called β1A), β2, β3, and β4 (Isom et al., 1992, 1995; Kazen-Gillespie et al., 2000; Morgan et al., 2000; Qin et al., 2003; Yu et al., 2003). Each β subunit is encoded by one of four genes, SCN1B–SCN4B. With the exception of β1B, the β subunits share a similar type I membrane topology, including an extracellular N-terminal region immunoglobulin (Ig) loop, one transmembrane domain, and a small intracellular C-terminal domain (Figure 2). β2 and β4 are disulfide linked to VGSC α subunits, whereas β1 and β3 associate non-covalently (Isom et al., 1992, 1995; Morgan et al., 2000; Yu et al., 2003). The residues(s) responsible for the covalent interaction between β2/β4 and α have not yet been identified. Mutation studies have revealed that the A/A′ strand of the β1 Ig fold contains critical charged residues that interact with, and modulate the activity of, the α subunit whereas the intracellular domain is not involved (Mccormick et al., 1998). Less is known about the β subunit interaction sites on α subunits; however, an epilepsy-causing mutation, D1866Y, in the C-terminal cytoplasmic domain of Nav1.1 disrupts modulation of Na+ current by β1 (Spampanato et al., 2004). β1B shares the same N-terminal Ig domain as β1, but by virtue of retention of intron 3, has a different C-terminal region that lacks a transmembrane domain but contains a stop codon and polyadenylation site (Kazen-Gillespie et al., 2000; Qin et al., 2003). As a result, β1B is unique among the β subunits in that it is a soluble protein (Patino et al., 2011). For reasons that are not understood, the amino acid sequence of the β1B C-terminal domain is species-specific (Patino et al., 2011). Further species-specific alternative splicing events have been discovered within SCN1B, including splice variants of the zebrafish SCN1B ortholog scn1ba, scn1ba_tv1, and scn1ba_tv2 (Fein et al., 2007) with altered protein structure, and β1.2 in rat with an altered 3′ untranslated region (Dib-Hajj and Waxman, 1995). The tissue-specific expression profiles of each of the β subunits are subtly different, but clearly overlapping (Table 1). As with the α subunits, β subunits are highly expressed in excitable cells, including central and peripheral neurons, skeletal and cardiac muscle cells (Isom et al., 1992, 1995; Morgan et al., 2000; Yu et al., 2003; Maier et al., 2004; Lopez-Santiago et al., 2006, 2011; Brackenbury et al., 2010). Importantly, however, increasing evidence points to the expression of β subunits in a broad range of traditionally non-excitable cells, including stem cells, glia, vascular endothelial cells, and carcinoma cells (O’Malley and Isom, manuscript in preparation; Diss et al., 2008; Chioni et al., 2009; Andrikopoulos et al., 2011).
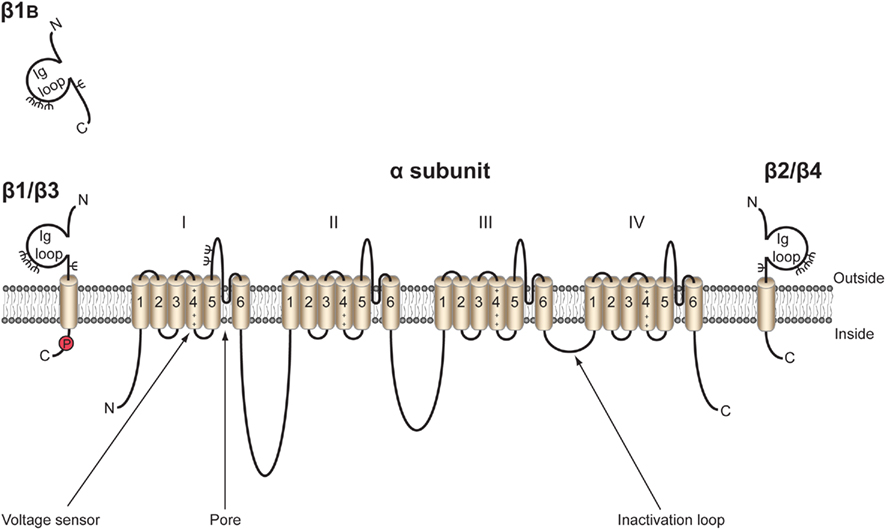
Figure 1. Topology of the voltage-gated Na+ channel α and β subunits. VGSCs contain a pore-forming α subunit consisting of four homologous domains of six transmembrane segments (1–6). Segment 4 contains the voltage sensor (Catterall, 2000). VGSCs also contain one or more β subunits. β1, β2, β3, and β4 contain an extracellular immunoglobulin (Ig) loop, transmembrane domain, and an intracellular C-terminal domain (Isom et al., 1994). β1B also contains an Ig loop, but has a different C-terminus lacking a transmembrane domain, and is thus a soluble, secreted protein (Patino et al., 2011). β1 contains a tyrosine phosphorylation site in its C-terminus (Malhotra et al., 2004) ψ, glycosylation sites. β1 and β3 are non-covalently linked to α, whereas β2 and β4 are covalently linked through disulfide bonds. Figure was produced using Science Slides 2006 software.
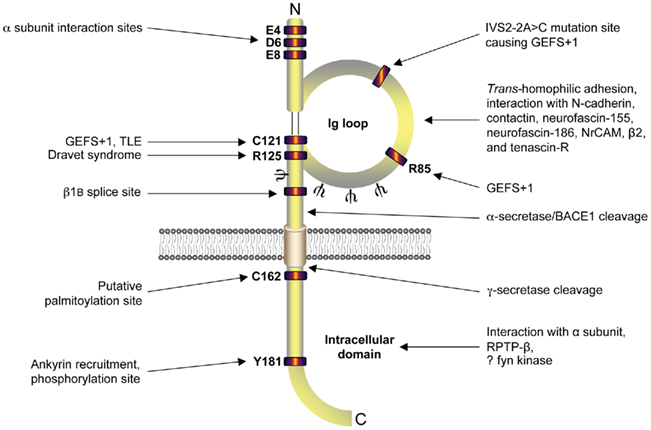
Figure 2. Functional architecture of β1/β1B. β1 contains residues responsible for interaction with α subunit in its intracellular and extracellular domains (Mccormick et al., 1998; Spampanato et al., 2004). Mutation sites responsible for causing genetic epilepsy with febrile seizures plus (GEFS + 1), temporal lobe epilepsy (TLE), and Dravet syndrome are located in the extracellular immunoglobulin loop (Meadows et al., 2002; Wallace et al., 2002; Audenaert et al., 2003; Scheffer et al., 2007; Patino et al., 2009). Alternative splicing site for β1B (Kazen-Gillespie et al., 2000; Qin et al., 2003; Patino et al., 2011), putative palmitoylation site (Mcewen et al., 2004), ankyrin interaction site (Malhotra et al., 2002), tyrosine phosphorylation site (Malhotra et al., 2004), N-glycosylation sites (ψ; Mccormick et al., 1998), α/β/γ-secretase cleavage sites (Wong et al., 2005), receptor protein tyrosine phosphatase β (RPTPβ) interaction (Ratcliffe et al., 2000), and putative fyn kinase interaction (Malhotra et al., 2002, 2004; Brackenbury et al., 2008) are also marked. Figure was produced using Science Slides 2006 software.
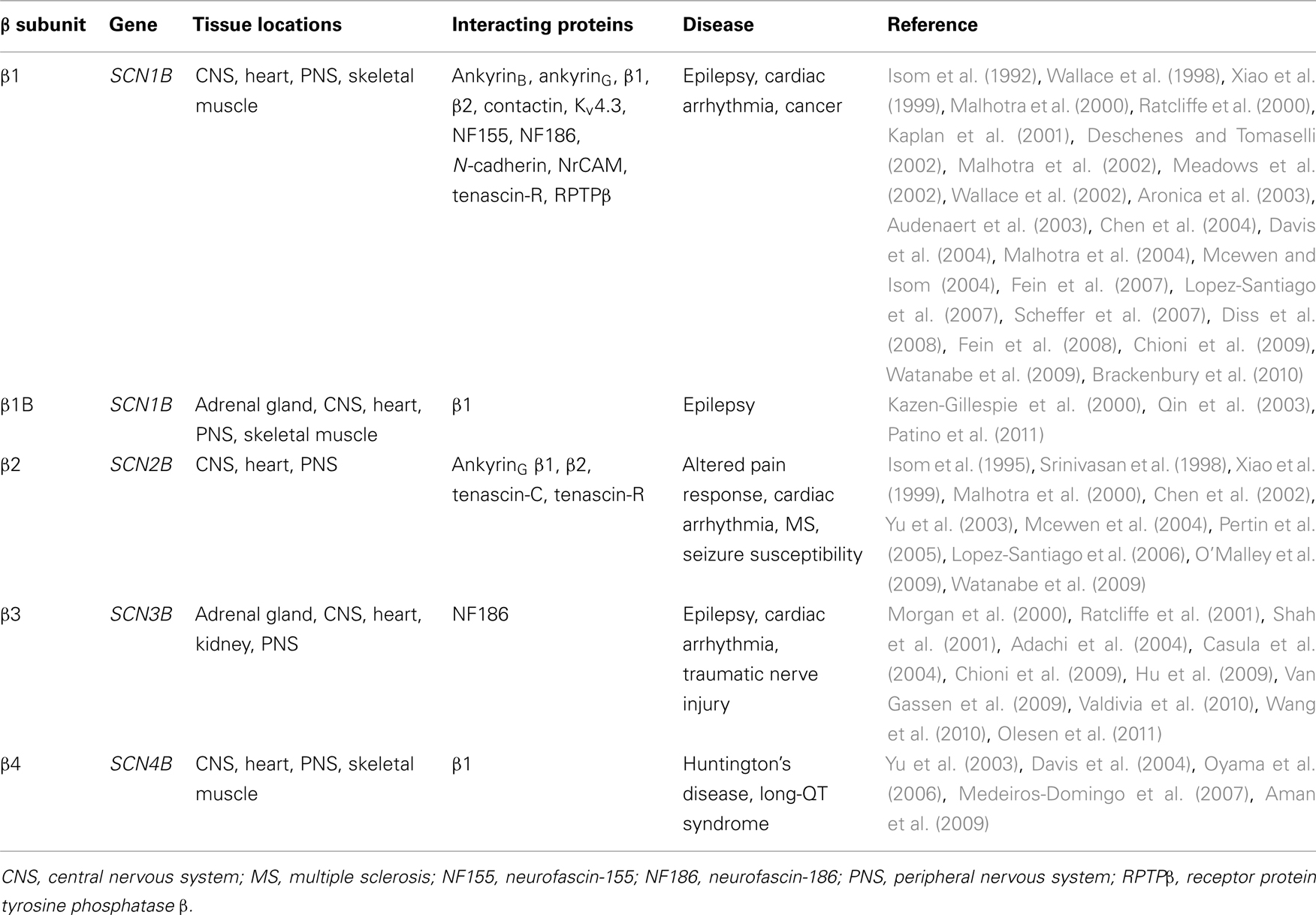
Table 1. The β subunit family: tissue locations, interacting proteins, and disease association.
Regulation of Excitability by Interaction with α Subunits
Beginning with the initial report of β1 cloning in 1992 (Isom et al., 1992), numerous studies have demonstrated that all five β subunits alter gating and kinetics of α subunits expressed in heterologous cells (Catterall, 2000; Kazen-Gillespie et al., 2000; Qin et al., 2003; Yu et al., 2003). For example, in both Xenopus oocytes and mammalian cell lines, β1 and β2 increase the peak Na+ current carried by Nav1.2, accelerate inactivation, and shift the voltage-dependence of activation and inactivation to more negative potentials (Isom et al., 1992, 1994, 1995). However, inconsistencies between different reports documenting the magnitude and types of current modulation suggest that the cell background, including expression of endogenous β subunits and/or other interacting proteins, is a critical factor to consider when interpreting the data (Moran et al., 2000, 2003; Meadows and Isom, 2005). Furthermore, some of the more obvious effects of β1 and β2 on Na+ current gating and kinetics in heterologous cells, especially Xenopus oocytes, do not appear to be reflected in vivo. In fact, results from null mouse models suggest that the effects of β1 and β2 on Na+ currents in vivo are subtle and cell type-specific (Chen et al., 2002, 2004; Aman et al., 2009; Patino et al., 2009; Brackenbury et al., 2010).
Voltage-gated Na+ channel β subunits have major effects on cellular excitability in vivo, suggesting that their subtle effects on Na+ currents are functionally significant. For example, in Scn1b null mice, the fastest components of the compound action potential are slowed in the optic nerve (Chen et al., 2004). The heart rate is also slowed and action potentials in ventricular myocytes are slower to repolarize resulting in QT prolongation (Lopez-Santiago et al., 2007). Scn1b null mice are ataxic and, display frequent spontaneous bilateral myoclonic seizures from postnatal day (P)8–10 (Chen et al., 2004). Action potentials in Scn1b null CA3 neurons fire with a significantly higher peak voltage and significantly greater amplitude compared with wildtype neurons (Patino et al., 2009). In addition, the action potential firing rate is reduced in Scn1b null cerebellar granule neurons (Brackenbury et al., 2010). Reduced action potential firing in inhibitory interneurons (e.g., GABAergic granule neurons) may lead to overall hyperexcitability within the neuronal network, and result in hyperexcitability-related disorders, e.g., seizures (Oakley et al., 2011). In contrast, Scn1b null nociceptive dorsal root ganglion neurons are hyperexcitable (Lopez-Santiago et al., 2011). In the latter example, the hyperexcitability is proposed to be due to modulation of both Na+ and K+ currents by β1 or β1B (Lopez-Santiago et al., 2011). In support of this notion, β1 interacts with, and modulates, the gating of the inward rectifier Kv4.3 in heterologous cells (Deschenes et al., 2002, 2008).
β2 also regulates VGSC α subunits in neurons, and thereby electrical excitability. However, its role is proposed to be somewhat different to β1, and its effects on channel kinetics and voltage-dependence appear even subtler in vivo. Unlike Scn1b null mice, Scn2b null mice appear normal in neurological tests, although they display increased seizure susceptibility, and an elevated action potential threshold in the optic nerve (Chen et al., 2002, 2004). β2 associates with α subunits as the final step in neuronal VGSC biosynthesis, thereby permitting insertion of the complex into the plasma membrane, and increasing Na+ current (Schmidt and Catterall, 1986; Isom et al., 1995). Thus, β2 plays an important role in stabilizing channel expression at the cell surface, and thereby maintaining normal action potential threshold. In agreement with this, there is ∼50% decreased expression of α subunits and Na+ currents at the plasma membrane of Scn2b null hippocampal neurons (Chen et al., 2002). However, Scn2b deletion has no effect on Na+ currents recorded from neurons isolated from the dentate gyrus, suggesting that, similar to β1, its effects are cell type-specific (Uebachs et al., 2010). In Scn2b null small-fast dorsal root ganglion neurons, tetrodotoxin-sensitive Na+ current is reduced by ∼50% and kinetics of activation and inactivation are slowed. Consistent with this, the protein level of Nav1.7 is reduced, whereas tetrodotoxin-resistant Na+ current is unchanged (Lopez-Santiago et al., 2006). β2 may therefore specifically regulate tetrodotoxin-sensitive channels in vivo.
Similar to Scn2b null mice, Scn3b null mice behave normally and have full lifespans (Chen et al., 2002; Hakim et al., 2008). Scn1b may compensate for Scn3b deletion in brain, providing for an apparently normal neurological phenotype (Hakim et al., 2008, 2010a). Scn3b null hearts display ventricular arrhythmogenic properties, including shorter effective refractory periods, induced tachycardia, and shorter action potential durations, corresponding to reduced Na+ current and hyperpolarized inactivation (Hakim et al., 2008). Atrial conduction abnormalities have also been reported in Scn3b null mice (Hakim et al., 2010a). These defects can be, in part, mitigated with the Class I antiarrhythmic agents flecainide and quinidine (Hakim et al., 2010b), supporting the conclusion that β3 modulates α subunit function in the heart.
The β4 intracellular domain may regulate α subunits in cerebellar Purkinje neurons by acting as an open-channel blocker of VGSCs that carry resurgent Na+ current (Grieco et al., 2005). Silencing Scn4b in cerebellar granule neurons reduces resurgent and persistent Na+ currents, hyperpolarizes voltage-dependence of inactivation of transient current, and reduces repetitive action potential firing (Bant and Raman, 2010). Resurgent Na+current is proposed to facilitate repetitive firing in cerebellar neurons (Khaliq et al., 2003; Bant and Raman, 2010). β4 thus appears to play a key role in regulating excitability. Finally, β4 plays an antagonistic role with β1 in regulating hippocampal neuron excitability: β4 slows inactivation and is proposed to promote excitability, whereas β1 promotes inactivation and is proposed to act as a brake on excitability (Aman et al., 2009). In summary, each of the β subunits regulates excitability through interaction with, and modulation of, α subunits in a cell type-specific and channel subtype-specific manner.
Non-Conducting Functions
β subunits are multifunctional (Figure 2). In addition to their “conducting” role in modulating Na+ current kinetics and voltage-dependence, they are members of the Ig superfamily of cell adhesion molecules (CAMs) and participate in a number of “non-conducting” cell adhesion related activities (Isom et al., 1995; Yu et al., 2003). β1 and β2 both participate in trans-homophilic adhesion resulting in cellular aggregation and recruitment of ankyrin to points of cell–cell contact in Drosophila S2 cells (Malhotra et al., 2000). By contrast, β3, in spite of its high homology to β1, does not mediate homophilic adhesion (Mcewen et al., 2009) but participates in heterophilic adhesion (see below). Phosphorylation of tyrosine residue (Y)181 in the intracellular domain of β1 abrogates the recruitment of ankyrinB and ankyrinG in transfected Chinese hamster lung cells (Malhotra et al., 2002). In cardiac myocytes, phosphorylation of Y181 determines localization of β1 to intercalated disks with connexin-43, N-cadherin, and Nav1.5, while non-phosphorylated β1 localizes in the t-tubules with ankyrinB (Malhotra et al., 2004). Thus, the phosphorylation state of Y181 may be important for regulating the subcellular distribution of β1. Interestingly, the intracellular domain of β1 interacts with receptor protein tyrosine phosphatase-β in rat brain neurons (Ratcliffe et al., 2000), potentially providing a mechanism for regulating Y181 phosphorylation. In addition, indirect evidence suggests that β1-mediated trans-homophilic adhesion results in fyn kinase activation in mouse cerebellar granule neurons (Brackenbury et al., 2008), which in turn could further fine tune phosphorylation of Y181.
The β subunits interact heterophilically with several other CAMs and extracellular matrix proteins. β1 interacts with VGSC β2, contactin, neurofascin-155, neurofascin-186, NrCAM, N-cadherin (Kazarinova-Noyes et al., 2001; Ratcliffe et al., 2001; Malhotra et al., 2004; Mcewen and Isom, 2004). Interaction between β1 and contactin, neurofascin-186, or VGSC β2 increases Na+ current in heterologous systems (Kazarinova-Noyes et al., 2001; Mcewen et al., 2004), suggesting that β subunit-dependent adhesion may regulate α subunit function and excitability. Both β1 and β2 interact with the extracellular matrix protein tenascin-R (Xiao et al., 1999). β2 also interacts with tenascin-C (Srinivasan et al., 1998), but does not interact with contactin (Mcewen et al., 2004). Less is known about the heterophilic interactions of the other β subunits. β3, which does not interact with either β1 or contactin, does interact with neurofascin-186 (Ratcliffe et al., 2001; Mcewen et al., 2009). Although similar studies have not been performed for β1B, it has been proposed that its heterophilic binding partners are likely similar to those of β1, given that both molecules share an identical Ig domain (Patino and Isom, 2010).
β1, β2, β3, and β4 subunits are substrates for sequential proteolytic cleavage by enzymes from the secretase family. These β subunits contain cleavage sites for the β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) on the extracellular domain, adjacent to the transmembrane region (Wong et al., 2005; Gersbacher et al., 2010). β2 also contains a cleavage site for the α-secretase enzyme ADAM10 in the extracellular juxtamembrane region (Kim et al., 2005). Cleavage by BACE1 or α-secretase results in shedding of the extracellular Ig domain, leaving transmembrane C-terminal fragments (Kim et al., 2005; Wong et al., 2005). The C-terminal fragments are subsequently processed by γ-secretase at a site in the intracellular juxtamembrane region, thus releasing soluble intracellular domains into the cytoplasm (Kim et al., 2005; Wong et al., 2005). Although these four β subunits are cleaved by BACE1 in vitro, processing in vivo has so far been demonstrated only for β2 and β4 (Wong et al., 2005). Importantly, the signaling events responsible for initiating β subunit processing by the secretases, as well as potential developmental timing of these cleavage events in vivo, have not been investigated.
The functional effects of processing these β subunits appear critical to their in vivo function. For example, both the extracellular domain of β1, and its soluble splice variant, β1B, promote neurite outgrowth (Davis et al., 2004; Patino et al., 2011). Similarly, cleavage of the extracellular domain of β4 by BACE1 also increases neurite outgrowth (Miyazaki et al., 2007). Inhibition of γ-secretase activity reduces β2-dependent cell adhesion and migration, suggesting that the intracellular domain is important for promoting these functions (Kim et al., 2005). The β2 intracellular domain translocates to the nucleus of transfected SH-SY5Y cells and increases expression of SCN1A, suggesting that it may function, directly or indirectly, as a transcriptional regulator of VGSC α subunit expression (Kim et al., 2007). Further, the mRNA and protein levels of Scn1a/Nav1.1 are reduced in the brains of Bace1 null mice (Kim et al., 2011). Altered expression of α subunit mRNA and protein in the peripheral and central nervous systems of Scn1b and Scn2b null mice, as well as the hearts of Scn1b and Scn3b null mice (Chen et al., 2004; Lopez-Santiago et al., 2006, 2007, 2011; Hakim et al., 2008; Brackenbury et al., 2010), suggests that regulation of α subunit expression by β subunits may be widespread. Finally, cleavage of β4 by BACE1 in cerebellar Purkinje cells slows the decay of resurgent Na+ current, thus promoting action potential firing (Huth et al., 2011), suggesting that secretase-mediated β subunit processing may modulate α subunit activity and thus neuronal excitability. However, another study from the same group indicated that BACE1 modulates α subunit gating in transfected human embryonic kidney cells and murine neuroblastoma cells, independent of its proteolytic effect on β2 and β4 (Huth et al., 2009). Thus the effects of BACE1 on α and β subunit function appear complex. Further work is required to understand the regulatory events involved in this putative signaling cascade and to establish whether BACE1 does indeed directly interact with α subunits in addition to β subunits.
Role of β Subunits in Development
β1 promotes neurite outgrowth in cerebellar granule neurons through trans-homophilic cell–cell adhesion (Davis et al., 2004). This mechanism operates through lipid rafts and requires fyn kinase, contactin, and Na+ current (Brackenbury et al., 2008, 2010). β1-mediated neurite outgrowth also requires γ-secretase activity, suggesting that proteolytic processing of the intracellular domain may be important (Figures 3A,B). In contrast, neither β2 nor β4 promote neurite outgrowth in cerebellar granule neurons (Davis et al., 2004). However, β4 enhances neurite extension in neuroblastoma cells, and increases dendritic spine density in hippocampal neurons (Oyama et al., 2006; Miyazaki et al., 2007). In addition, β1 and β2 regulate the migration of fibroblasts away from the extracellular matrix protein tenascin-R (Xiao et al., 1999). Finally, β1 inhibits the migration of metastatic breast cancer cells (Chioni et al., 2009). Thus, β subunit-mediated neurite outgrowth and migration may be subtype and/or cell-specific.
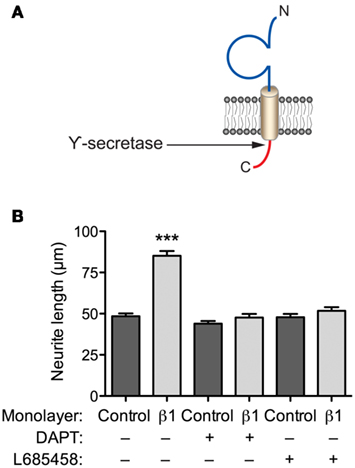
Figure 3. β1-mediated neurite outgrowth requires γ-secretase activity. (A) Location of γ-secretase cleavage site on the intracellular domain of β1 (Wong et al., 2005). (B) Cerebellar granule neurons from postnatal day (P)14 wildtype mice were plated on top of monolayers of control or β1-expressing Chinese hamster lung cells, as described previously (Davis et al., 2004). Cultures were incubated with the either one of the γ-secretase inhibitors, L685458, or DAPT (both 1 μM); or control (DMSO) for 48 h (Kim et al., 2005). Cells were then fixed, processed for GAP43 immunocytochemistry, and neurite lengths measured, as described (Davis et al., 2004). Both L685458 and DAPT inhibited the increase in neurite length caused by β1 expressed in the monolayer. Data are mean + SEM (n = 300). Significance: ***P < 0.001, ANOVA with Tukey’s post hoc test.
The regulation of neurite outgrowth and migration by β subunits has consequences for development and organogenesis. In particular, β1 plays a critical role in neuronal pathfinding in postnatal-developing fiber tracts, coinciding with the onset of its expression from birth (Sutkowski and Catterall, 1990; Sashihara et al., 1995; Brackenbury et al., 2008). Scn1b null mice display a severe phenotype that includes growth retardation, ataxia, spontaneous seizures from P8–10, and death by P21 (Chen et al., 2004). In P14–16 Scn1b null mice, the pathfinding and migration of corticospinal axons is disrupted, leading to significant defasciculation of fibers at the pyramidal decussation (Brackenbury et al., 2008). The cerebellar parallel fibers are also defasciculated in P14 Scn1b null mice. The migration of granule neurons through the cerebellar molecular layer is disrupted, resulting in their accumulation in the external germinal layer, which is consequently thicker in Scn1b null mice than in wildtype littermates (Brackenbury et al., 2008). These cerebellar defects may contribute to the ataxic phenotype (Chen et al., 2004). Consistent with results in mice, abnormal pathfinding has also been reported in the olfactory nerve of zebrafish scn1bb morphants (Fein et al., 2008).
An important next step will be to determine whether defects in Scn1b-mediated cell–cell adhesion and migration in brain occur prior to the onset of convulsive seizures (Chen et al., 2004). It is possible that abnormal neuronal migration and pathfinding in the absence of β1/β1B-mediated cell adhesive interactions may lead to aberrant connections, resulting in neuronal hyperexcitability and epileptogenesis. Further work is required to establish the causational relationship between β1/β1B expression, cell adhesion, migration, and seizures during postnatal development of the nervous system.
Alterations in VGSC Pharmacology by β Subunits
Studies have indicated that β subunits can alter the effect of pharmacological compounds on Na+ currents carried by α subunits. For example, using heterologous systems, co-expression of β1 or β3 with Nav1.3 in Xenopus oocytes attenuates the inhibitory effect of the antiarrhythmic agent and local anesthetic lidocaine on current amplitude and inactivation (Lenkowski et al., 2003). Further, the β1 C121W epilepsy mutation reduces tonic and use-dependent channel block in response to the antiepileptic drug phenytoin (Lucas et al., 2005). This altered sensitivity to phenytoin is proposed to be as a result of the altered gating caused by the mutation (Meadows et al., 2002), rather than a direct effect on the drug receptor. A recent in vivo study showed that the use-dependent reduction of transient Na+ current caused by the anticonvulsant drug carbamazepine in wildtype hippocampal neurons was not observed in Scn1b or Scn2b null hippocampal neurons (Uebachs et al., 2010). However, carbamazepine caused a small hyperpolarizing shift in the voltage-dependence of activation of both transient and persistent Na+ current. The hyperpolarizing shift in persistent Na+ current was significantly increased in Scn1b null neurons at low carbamazepine concentrations, resulting in a complete loss in efficacy of the drug to reduce repetitive action potential firing (Uebachs et al., 2010). Thus, β1/β1B alters the pharmacological response of persistent Na+ current to carbamazepine. Finally, in Scn3b null hearts, the VGSC-blocking antiarrhythmic agents flecainide and quinidine both modify ventricular effective refractory periods, resulting in anti-arrhythmogenic effects, in contrast to their effects in wildtype or Scn5a mutant hearts (Hakim et al., 2010b). Taken together, these findings have important clinical implications, suggesting that function-altering mutations and/or altered expression or localization of β subunits in patients may affect their sensitivity to VGSC-targeting drugs and thus therapeutic efficacy. Further work is required to establish whether or not β subunits alter pharmacological responses to additional VGSC-targeting therapeutics.
Functional Reciprocity between α and β Subunits
Extensive evidence indicates that β subunits modulate channel gating of α subunits (see Regulation of Excitability by Interaction with α Subunits). Similarly, β1-mediated neurite outgrowth is inhibited by the VGSC-blocking toxin tetrodotoxin (Brackenbury et al., 2010). Thus, there is a potential for interplay between β1-mediated modulation of Na+ current carried by α subunits and β1-mediated cell–cell adhesion/migration. β1 is required for normal high-frequency action potential firing in cerebellar granule neurons (Figure 4A). β1-mediated neurite outgrowth is abrogated in Scn8a null cerebellar granule neurons, suggesting that the mechanism requires Na+ current carried by Nav1.6 (Figure 4B). Nav1.6 is vital for high-frequency repetitive firing in cerebellar neurons (Raman and Bean, 1997). Resurgent Na+ current, carried by Nav1.6, and which facilitates repetitive action potential firing (Raman and Bean, 1997; Khaliq et al., 2003; Bant and Raman, 2010), is reduced in Scn1b null cerebellar granule neurons (Brackenbury et al., 2010). The Scn1b null mutation disrupts the expression of Nav1.6 at the axon initial segment (AIS) of cerebellar granule neurons (Figure 4C).
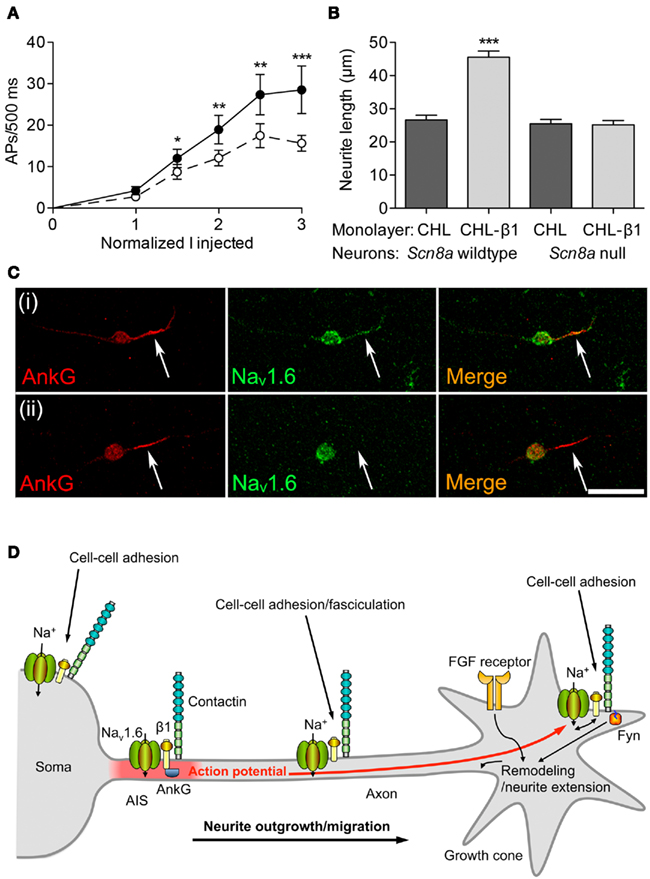
Figure 4. Functional reciprocity between β1 and Nav1.6. (A) Electrical excitability is impaired in Scn1b null cerebellar granule neurons. Action potential firing rate recorded from cerebellar granule neurons in brain slices from 12-day-old mice plotted as a function of injected current, normalized to action potential threshold for wildtype (filled circles) and Scn1b null (open circles). Data are mean ± SEM (n ≥ 15). Significance: *P < 0.05; **P < 0.01; ***P < 0.001; t-test. (B) β1-mediated neurite outgrowth is inhibited by the Scn8a null mutation. Neurite lengths of wildtype and Scn8a null cerebellar granule neurons grown on control Chinese hamster lung or β1-expressing monolayers (n = 300). Data are mean + SEM. Significance: ***P < 0.001, ANOVA with Tukey’s post hoc test. (C) Nav1.6 expression is reduced at the axon initial segment of Scn1b null cerebellar granule neurons. Wildtype and Scn1b null cerebellar granule neurons cultured in vitro for 14 days labeled with anti-ankyrinG (red) and Nav1.6 antibodies (green). Scale bar, 20 μm. Arrows point to axon initial segment expressing ankyrinG. (D) A model for Na+ current involvement in β1-mediated neurite outgrowth. Complexes containing Nav1.6, β1, and contactin are present throughout the neuronal membrane in the soma, neurite and growth cone. Localized Na+ influx is necessary for β1-mediated neurite extension and migration. VGSC complexes along the neurite participate in cell–cell adhesion and fasciculation. β1 is also required for Nav1.6 expression at the axon initial segment, and subsequent high-frequency action potential firing through modulation of resurgent Na+current. Electrical activity may further promote β1-mediated neurite outgrowth at or near the growth cone. Thus, the developmental functions of β1 and Nav1.6 are complementary, such that (1) Na+ influx carried by Nav1.6 is required for β1-mediated neurite outgrowth, and (2) β1 is required for normal expression/activity of Nav1.6 at the axon initial segment. Fyn kinase and ankyrinG are likely also present in all complexes, but are only shown once in each panel for clarity. The FGF-mediated, β1-independent neurite outgrowth pathway is also shown. Figure reproduced with permission (Brackenbury et al., 2010).
Taken together, these data suggest that there is a functional reciprocity between β1 and Nav1.6 in cerebellar neurons, such that, on the one hand, β1 is required for normal localization of Nav1.6 at the AIS, thus permitting resurgent Na+ current, and repetitive action potential firing. On the other hand, Nav1.6 is required for β1-mediated neurite outgrowth. Electrical activity generated at the AIS is proposed to provide a depolarizing signal to open Nav1.6 channels at the growth cone, further promoting β1-mediated neurite outgrowth (Figure 4D; Brackenbury et al., 2008). This reciprocal relationship between β1 and Nav1.6 is critical for postnatal cerebellar development (Chen et al., 2004; Van Wart and Matthews, 2006). Impaired localization of β1 at the AIS may thus lead to altered excitability. In agreement with this, in knock-in mice heterozygous for the β1C121W mutation, which disrupts β1-dependent adhesion and alters channel gating, mutant β1 protein is excluded from the AIS of pyramidal neurons, potentially contributing to febrile seizures (Meadows et al., 2002; Wimmer et al., 2010).
Future work will no doubt establish whether or not further complementary roles exist between VGSC α and β subunits, interacting in a coordinated fashion to regulate processes including excitability and neurite extension. It is already clear however, that β subunits function in macromolecular complexes with α subunits to participate in signaling on multiple timescales to regulate excitability, adhesion, neurite outgrowth, and migration. A critical focus of future work will be to determine whether β subunits that are expressed independently of the ion-conducting pore also play roles in excitability in vivo, perhaps through regulation of axon guidance or fasciculation.
Dysregulation in Disease
Voltage-gated Na+ channel β subunits are implicated in a number of neurological diseases (Table 1) [reviewed extensively in Patino and Isom (2010)]. Of particular note is the growing list of mutations in SCN1B that are associated with genetic epilepsy with febrile seizures plus (GEFS) + 1 (OMIM 604233), a spectrum of disorders that includes mild to severe forms of epilepsy (Wallace et al., 1998, 2002; Audenaert et al., 2003; Burgess, 2005; Yamakawa, 2005; Scheffer et al., 2007; Patino et al., 2009, 2011). No GEFS + 1-causing mutations in the other β subunit genes have yet been identified. However, Scn2b null mice display increased seizure susceptibility (Chen et al., 2002). In addition, SCN3B is reduced in the hippocampus of patients with temporal lobe epilepsy, suggesting that altered β3 expression may contribute to or result from epilepsy (Van Gassen et al., 2009). The mutations in SCN1B may bias neurons toward hyperexcitability and epileptogenesis by one or both of two distinct mechanisms: (1) impaired regulation of α subunit-dependent excitability (Meadows et al., 2002; Chen et al., 2004; Spampanato et al., 2004; Patino et al., 2009; Wimmer et al., 2010); and/or (2) impaired cell–cell adhesive interactions (Meadows et al., 2002; Brackenbury et al., 2008; Fein et al., 2008; Patino et al., 2011). There may also be a causal relationship between VGSC α−β1 interactions, cell–cell adhesion, migration, and epilepsy. Further work is required to establish whether or not disrupted β1-dependent cell adhesion is indeed a prerequisite for seizure activity.
Changes in β2 expression have been implicated in altered pain sensation. Scn2b null mice are more sensitive to noxious thermal stimuli than wildtype mice (Lopez-Santiago et al., 2006). In addition, the spared nerve injury model of neuropathic pain results in increased β2 expression in rat sensory neurons, and mechanical allodynia-like behavior (Pertin et al., 2005). This behavior is absent in Scn2b null mice, suggesting that β2 expression may play an important role in neuropathic pain sensation (Pertin et al., 2005).
The β subunits play roles in neurodegenerative disease. The Scn2b null mutation is neuroprotective in the experimental allergic encephalomyelitis mouse model of multiple sclerosis (O’Malley et al., 2009). In addition, levels of Scn4b are reduced in mouse models of Huntington’s disease prior to onset of motor symptoms, and a similar reduction has also been reported in patients (Oyama et al., 2006).
Indirect evidence suggests that β subunits may be involved in neuropsychiatric disorders. For example, ankyrinG and Nav1.6, which both interact with β subunits, are linked genetically to bipolar disorder (Gargus, 2006; Wang et al., 2008). Migration defects observed in the cerebellum of Scn1b null mice may result in abnormal connections with the prefrontal cortex and posterior parietal cortex, thus providing a possible mechanism for β subunit involvement in mood disorders (Brackenbury et al., 2008; Strick et al., 2009). Similarly, migration defects in other brain areas in patients with SCN1B mutations may contribute to mental disorders. Pathological cellular migration regulated by β subunits extends beyond neurological diseases: β1 regulates cellular adhesion and migration in metastatic breast cancer cell lines (Chioni et al., 2009). β subunit transcripts are also expressed in prostate cancer cells and lung cancer cells (Roger et al., 2007; Diss et al., 2008), suggesting that their involvement in cancer may be widespread.
Finally, mutations in β subunits are associated with cardiac abnormalities (Wilde and Brugada, 2011). Mutations in SCN1B have been reported in patients with idiopathic ventricular fibrillation (Brugada syndrome; Watanabe et al., 2008; Hu et al., 2009; Valdivia et al., 2010). Mutations in SCN1B, SCN2B, and SCN3B are also associated with atrial fibrillation (Watanabe et al., 2009; Wang et al., 2010; Olesen et al., 2011). Mutations in SCN3B and SCN4B are associated with sudden infant death syndrome (Tan et al., 2010). These mutations are proposed to interfere with the ability of β subunits to modulate Nav1.5 currents in vivo (Watanabe et al., 2008, 2009; Hu et al., 2009; Tan et al., 2010). A mutation in SCN4B results in long-QT syndrome (Medeiros-Domingo et al., 2007). Expression of a mutation linked to conduction disease and Brugada syndrome, Scn5a1798insD/+, in a mouse strain (129P2) with markedly reduced Scn4b expression resulted in more severe cardiac conduction slowing than a strain with normal Scn4b levels (FVB/N), suggesting that Scn4b may be a genetic modifier of cardiac conduction (Remme et al., 2009). In summary, abnormal expression and/or function of β subunits appears to play an important role in a number of diseases, ranging from nervous system disorders, to cardiac abnormalities, and cancer.
Conclusion/Outlook
Increasing new evidence supports the hypothesis that the VGSC β subunits are multifunctional. In addition, a growing list of mutations and in vivo studies indicate that the β subunits play important roles in a number of diseases due to abnormal function in both excitable and non-excitable cells. There is no doubt that the classical “conducting” role of β subunits as modulators of Na+current is of paramount importance in regulating ion flux and excitability. However, there is a clear trend in the literature toward an increasingly important role for “non-conducting” functions, including cell adhesion, migration and pathfinding, and putative transcriptional regulation. As a result, the β subunits are integral components of VGSC macromolecular protein complexes, which can direct multiple signaling mechanisms on multiple timescales. Moreover, the cell adhesive, “non-conducting” properties of β subunits observed in vitro suggest that they may play critical functional roles independent of α subunits in vivo. The challenge now will be to clearly delineate the cell adhesive functions of β subunits from their roles in channel modulation during development and in pathophysiology. Clearer understanding of the interaction between the conducting and non-conducting functions of VGSC complexes will hopefully enable the full realization of their therapeutic potential.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Medical Research Council (UK) Career Development Award (to William J. Brackenbury) and National Institutes of Health Grants R01 MH059980 and R01 NS064245 (to Lori L. Isom).
References
Adachi, K., Toyota, M., Sasaki, Y., Yamashita, T., Ishida, S., Ohe-Toyota, M., Maruyama, R., Hinoda, Y., Saito, T., Imai, K., Kudo, R., and Tokino, T. (2004). Identification of SCN3B as a novel p53-inducible proapoptotic gene. Oncogene 23, 7791–7798.
Aman, T. K., Grieco-Calub, T. M., Chen, C., Rusconi, R., Slat, E. A., Isom, L. L., and Raman, I. M. (2009). Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J. Neurosci. 29, 2027–2042.
Andrikopoulos, P., Fraser, S. P., Patterson, L., Ahmad, Z., Burcu, H., Ottaviani, D., Diss, J. K., Box, C., Eccles, S. A., and Djamgoz, M. B. (2011). Angiogenic functions of voltage-gated Na+ channels in human endothelial cells: modulation of vascular endothelial growth factor (VEGF) signaling. J. Biol. Chem. 286, 16846–16860.
Aronica, E., Troost, D., Rozemuller, A. J., Yankaya, B., Jansen, G. H., Isom, L. L., and Gorter, J. A. (2003). Expression and regulation of voltage-gated sodium channel beta1 subunit protein in human gliosis-associated pathologies. Acta Neuropathol. 105, 515–523.
Audenaert, D., Claes, L., Ceulemans, B., Lofgren, A., Van Broeckhoven, C., and De Jonghe, P. (2003). A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology 61, 854–856.
Bant, J. S., and Raman, I. M. (2010). Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc. Natl. Acad. Sci. U.S.A. 107, 12357–12362.
Brackenbury, W. J., Calhoun, J. D., Chen, C., Miyazaki, H., Nukina, N., Oyama, F., Ranscht, B., and Isom, L. L. (2010). Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc. Natl. Acad. Sci. U.S.A. 107, 2283–2288.
Brackenbury, W. J., Davis, T. H., Chen, C., Slat, E. A., Detrow, M. J., Dickendesher, T. L., Ranscht, B., and Isom, L. L. (2008). Voltage-gated Na+ channel beta1 subunit-mediated neurite outgrowth requires Fyn kinase and contributes to postnatal CNS development in vivo. J. Neurosci. 28, 3246–3256.
Burgess, D. L. (2005). Neonatal epilepsy syndromes and GEFS+: mechanistic considerations. Epilepsia 46(Suppl. 10), 51–58.
Casula, M. A., Facer, P., Powell, A. J., Kinghorn, I. J., Plumpton, C., Tate, S. N., Bountra, C., Birch, R., and Anand, P. (2004). Expression of the sodium channel beta3 subunit in injured human sensory neurons. Neuroreport 15, 1629–1632.
Catterall, W. A. (1992). Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 72, S15–S48.
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25.
Chen, C., Bharucha, V., Chen, Y., Westenbroek, R. E., Brown, A., Malhotra, J. D., Jones, D., Avery, C., Gillespie, P. J. III., Kazen-Gillespie, K. A., Kazarinova-Noyes, K., Shrager, P., Saunders, T. L., Macdonald, R. L., Ransom, B. R., Scheuer, T., Catterall, W. A., and Isom, L. L. (2002). Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc. Natl. Acad. Sci. U.S.A. 99, 17072–17077.
Chen, C., Westenbroek, R. E., Xu, X., Edwards, C. A., Sorenson, D. R., Chen, Y., Mcewen, D. P., O’Malley, H. A., Bharucha, V., Meadows, L. S., Knudsen, G. A., Vilaythong, A., Noebels, J. L., Saunders, T. L., Scheuer, T., Shrager, P., Catterall, W. A., and Isom, L. L. (2004). Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J. Neurosci. 24, 4030–4042.
Chioni, A. M., Brackenbury, W. J., Calhoun, J. D., Isom, L. L., and Djamgoz, M. B. (2009). A novel adhesion molecule in human breast cancer cells: voltage-gated Na+ channel beta1 subunit. Int. J. Biochem. Cell Biol. 41, 1216–1227.
Davis, T. H., Chen, C., and Isom, L. L. (2004). Sodium channel β1 subunits promote neurite outgrowth in cerebellar granule neurons. J. Biol. Chem. 279, 51424–51432.
Deschenes, I., Armoundas, A. A., Jones, S. P., and Tomaselli, G. F. (2008). Post-transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J. Mol. Cell. Cardiol. 45, 336–346.
Deschenes, I., Disilvestre, D., Juang, G. J., Wu, R. C., An, W. F., and Tomaselli, G. F. (2002). Regulation of Kv4.3 current by KChIP2 splice variants: a component of native cardiac I(to)? Circulation 106, 423–429.
Deschenes, I., and Tomaselli, G. F. (2002). Modulation of Kv4.3 current by accessory subunits. FEBS Lett. 528, 183–188.
Dib-Hajj, S. D., and Waxman, S. G. (1995). Genes encoding the beta 1 subunit of voltage-dependent Na+ channel in rat, mouse and human contain conserved introns. FEBS Lett. 377, 485–488.
Diss, J. K., Fraser, S. P., Walker, M. M., Patel, A., Latchman, D. S., and Djamgoz, M. B. (2008). Beta-subunits of voltage-gated sodium channels in human prostate cancer: quantitative in vitro and in vivo analyses of mRNA expression. Prostate Cancer Prostatic Dis. 11, 325–333.
Fein, A. J., Meadows, L. S., Chen, C., Slat, E. A., and Isom, L. L. (2007). Cloning and expression of a zebrafish SCN1B ortholog and identification of a species-specific splice variant. BMC Genomics 8, 226.
Fein, A. J., Wright, M. A., Slat, E. A., Ribera, A. B., and Isom, L. L. (2008). scn1bb, a zebrafish ortholog of SCN1B expressed in excitable and nonexcitable cells, affects motor neuron axon morphology and touch sensitivity. J. Neurosci. 28, 12510–12522.
Gargus, J. J. (2006). Ion channel functional candidate genes in multigenic neuropsychiatric disease. Biol. Psychiatry 60, 177–185.
Gersbacher, M. T., Kim, D. Y., Bhattacharyya, R., and Kovacs, D. M. (2010). Identification of BACE1 cleavage sites in human voltage-gated sodium channel beta 2 subunit. Mol. Neurodegener. 5, 61.
Grieco, T. M., Malhotra, J. D., Chen, C., Isom, L. L., and Raman, I. M. (2005). Open-channel block by the cytoplasmic tail of sodium channel β4 as a mechanism for resurgent sodium current. Neuron 45, 233–244.
Hakim, P., Brice, N., Thresher, R., Lawrence, J., Zhang, Y., Jackson, A. P., Grace, A. A., and Huang, C. L. (2010a). Scn3b knockout mice exhibit abnormal sino-atrial and cardiac conduction properties. Acta physiol. 198, 47–59.
Hakim, P., Thresher, R., Grace, A. A., and Huang, C. L. (2010b). Effects of flecainide and quinidine on action potential and ventricular arrhythmogenic properties in Scn3b knockout mice. Clin. Exp. Pharmacol. Physiol. 37, 782–789.
Hakim, P., Gurung, I. S., Pedersen, T. H., Thresher, R., Brice, N., Lawrence, J., Grace, A. A., and Huang, C. L. (2008). Scn3b knockout mice exhibit abnormal ventricular electrophysiological properties. Prog. Biophys. Mol. Biol. 98, 251–266.
Hu, D., Barajas-Martinez, H., Burashnikov, E., Springer, M., Wu, Y., Varro, A., Pfeiffer, R., Koopmann, T. T., Cordeiro, J. M., Guerchicoff, A., Pollevick, G. D., and Antzelevitch, C. (2009). A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ. Cardiovasc. Genet. 2, 270–278.
Huth, T., Rittger, A., Saftig, P., and Alzheimer, C. (2011). Beta-site APP-cleaving enzyme 1 (BACE1) cleaves cerebellar Na+ channel beta4-subunit and promotes Purkinje cell firing by slowing the decay of resurgent Na+ current. Pflugers Arch. 461, 355–371.
Huth, T., Schmidt-Neuenfeldt, K., Rittger, A., Saftig, P., Reiss, K., and Alzheimer, C. (2009). Non-proteolytic effect of beta-site APP-cleaving enzyme 1 (BACE1) on sodium channel function. Neurobiol. Dis. 33, 282–289.
Isom, L. L., De Jongh, K. S., and Catterall, W. A. (1994). Auxiliary subunits of voltage-gated ion channels. Neuron 12, 1183–1194.
Isom, L. L., De Jongh, K. S., Patton, D. E., Reber, B. F. X., Offord, J., Charbonneau, H., Walsh, K., Goldin, A. L., and Catterall, W. A. (1992). Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science 256, 839–842.
Isom, L. L., Ragsdale, D. S., De Jongh, K. S., Westenbroek, R. E., Reber, B. F., Scheuer, T., and Catterall, W. A. (1995). Structure and function of the b2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83, 433–442.
Kaplan, M. R., Cho, M. H., Ullian, E. M., Isom, L. L., Levinson, S. R., and Barres, B. A. (2001). Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron 30, 105–119.
Kazarinova-Noyes, K., Malhotra, J. D., Mcewen, D. P., Mattei, L. N., Berglund, E. O., Ranscht, B., Levinson, S. R., Schachner, M., Shrager, P., Isom, L. L., and Xiao, Z.-C. (2001). Contactin associates with Na+ channels and increases their functional expression. J. Neurosci. 21, 7517–7525.
Kazen-Gillespie, K. A., Ragsdale, D. S., D’andrea, M. R., Mattei, L. N., Rogers, K. E., and Isom, L. L. (2000). Cloning, localization, and functional expression of sodium channel β1A subunits. J. Biol. Chem. 275, 1079–1088.
Khaliq, Z. M., Gouwens, N. W., and Raman, I. M. (2003). The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J. Neurosci. 23, 4899–4912.
Kim, D. Y., Carey, B. W., Wang, H., Ingano, L. A., Binshtok, A. M., Wertz, M. H., Pettingell, W. H., He, P., Lee, V. M., Woolf, C. J., and Kovacs, D. M. (2007). BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 9, 755–764.
Kim, D. Y., Gersbacher, M. T., Inquimbert, P., and Kovacs, D. M. (2011). Reduced sodium channel Nav1.1 levels in BACE1-null mice. J. Biol. Chem. 286, 8106–8116.
Kim, D. Y., Mackenzie Ingano, L. A., Carey, B. W., Pettingell, W. P., and Kovacs, D. M. (2005). Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta 2 subunit regulates cell adhesion and migration. J. Biol. Chem. 280, 23251–23261.
Lenkowski, P. W., Shah, B. S., Dinn, A. E., Lee, K., and Patel, M. K. (2003). Lidocaine block of neonatal Nav1.3 is differentially modulated by co-expression of beta1 and beta3 subunits. Eur. J. Pharmacol. 467, 23–30.
Lopez-Santiago, L. F., Brackenbury, W. J., Chen, C., and Isom, L. L. (2011). Na+ channel Scn1b gene regulates dorsal root ganglion nociceptor excitability in vivo. J. Biol. Chem. 286, 22913–22923.
Lopez-Santiago, L. F., Meadows, L. S., Ernst, S. J., Chen, C., Malhotra, J. D., Mcewen, D. P., Speelman, A., Noebels, J. L., Maier, S. K., Lopatin, A. N., and Isom, L. L. (2007). Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J. Mol. Cell. Cardiol. 43, 636–647.
Lopez-Santiago, L. F., Pertin, M., Morisod, X., Chen, C., Hong, S., Wiley, J., Decosterd, I., and Isom, L. L. (2006). Sodium channel beta2 subunits regulate tetrodotoxin-sensitive sodium channels in small dorsal root ganglion neurons and modulate the response to pain. J. Neurosci. 26, 7984–7994.
Lucas, P. T., Meadows, L. S., Nicholls, J., and Ragsdale, D. S. (2005). An epilepsy mutation in the beta1 subunit of the voltage-gated sodium channel results in reduced channel sensitivity to phenytoin. Epilepsy Res. 64, 77–84.
Maier, S. K., Westenbroek, R. E., Mccormick, K. A., Curtis, R., Scheuer, T., and Catterall, W. A. (2004). Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation 109, 1421–1427.
Malhotra, J. D., Kazen-Gillespie, K., Hortsch, M., and Isom, L. L. (2000). Sodium channel β subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J. Biol. Chem. 275, 11383–11388.
Malhotra, J. D., Koopmann, M. C., Kazen-Gillespie, K. A., Fettman, N., Hortsch, M., and Isom, L. L. (2002). Structural requirements for interaction of sodium channel b1 subunits with ankyrin. J. Biol. Chem. 277, 26681–26688.
Malhotra, J. D., Thyagarajan, V., Chen, C., and Isom, L. L. (2004). Tyrosine-phosphorylated and nonphosphorylated sodium channel beta1 subunits are differentially localized in cardiac myocytes. J. Biol. Chem. 279, 40748–40754.
Mccormick, K. A., Isom, L. L., Ragsdale, D., Smith, D., Scheuer, T., and Catterall, W. A. (1998). Molecular determinants of Na+ channel function in the extracellular domain of the beta1 subunit. J. Biol. Chem. 273, 3954–3962.
Mcewen, D. P., Chen, C., Meadows, L. S., Lopez-Santiago, L., and Isom, L. L. (2009). The voltage-gated Na+ channel beta3 subunit does not mediate trans homophilic cell adhesion or associate with the cell adhesion molecule contactin. Neurosci. Lett. 462, 272–275.
Mcewen, D. P., and Isom, L. L. (2004). Heterophilic interactions of sodium channel beta 1 subunits with axonal and glial cell adhesion molecules. J. Biol. Chem. 279, 52744–52752.
Mcewen, D. P., Meadows, L. S., Chen, C., Thyagarajan, V., and Isom, L. L. (2004). Sodium channel b1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin . J. Biol. Chem. 279, 16044–16049.
Meadows, L. S., and Isom, L. L. (2005). Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc. Res. 67, 448–458.
Meadows, L. S., Malhotra, J., Loukas, A., Thyagarajan, V., Kazen-Gillespie, K. A., Koopman, M. C., Kriegler, S., Isom, L. L., and Ragsdale, D. S. (2002). Functional and biochemical analysis of a sodium channel b1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J. Neurosci. 22, 10699–10709.
Medeiros-Domingo, A., Kaku, T., Tester, D. J., Iturralde-Torres, P., Itty, A., Ye, B., Valdivia, C., Ueda, K., Canizales-Quinteros, S., Tusie-Luna, M. T., Makielski, J. C., and Ackerman, M. J. (2007). SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 116, 134–142.
Miyazaki, H., Oyama, F., Wong, H. K., Kaneko, K., Sakurai, T., Tamaoka, A., and Nukina, N. (2007). BACE1 modulates filopodia-like protrusions induced by sodium channel beta4 subunit. Biochem. Biophys. Res. Commun. 361, 43–48.
Moran, O., Conti, F., and Tammaro, P. (2003). Sodium channel heterologous expression in mammalian cells and the role of the endogenous beta1-subunits. Neurosci. Lett. 336, 175–179.
Moran, O., Nizzari, M., and Conti, F. (2000). Endogenous expression of the β1A sodium channel subunit in HEK-293 cells. FEBS Lett. 473, 132–134.
Morgan, K., Stevens, E. B., Shah, B., Cox, P. J., Dixon, A. K., Lee, K., Pinnock, R. D., Hughes, J., Richardson, P. J., Mizuguchi, K., and Jackson, A. P. (2000). b3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc. Natl. Acad. Sci. U.S.A. 97, 2308–2313.
Oakley, J. C., Kalume, F., and Catterall, W. A. (2011). Insights into pathophysiology and therapy from a mouse model of Dravet syndrome. Epilepsia 52(Suppl. 2), 59–61.
Olesen, M. S., Jespersen, T., Nielsen, J. B., Liang, B., Moller, D. V., Hedley, P., Christiansen, M., Varro, A., Olesen, S. P., Haunso, S., Schmitt, N., and Svendsen, J. H. (2011). Mutations in sodium channel beta-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc. Res. 89, 786–793.
O’Malley, H. A., Shreiner, A. B., Chen, G. H., Huffnagle, G. B., and Isom, L. L. (2009). Loss of Na+ channel beta2 subunits is neuroprotective in a mouse model of multiple sclerosis. Mol. Cell. Neurosci. 40, 143–155.
Oyama, F., Miyazaki, H., Sakamoto, N., Becquet, C., Machida, Y., Kaneko, K., Uchikawa, C., Suzuki, T., Kurosawa, M., Ikeda, T., Tamaoka, A., Sakurai, T., and Nukina, N. (2006). Sodium channel beta4 subunit: down-regulation and possible involvement in neuritic degeneration in Huntington’s disease transgenic mice. J. Neurochem. 98, 518–529.
Patino, G. A., Brackenbury, W. J., Bao, Y., Lopez-Santiago, L. F., O’Malley, H. A., Chen, C., Calhoun, J. D., Lafreniere, R., Cossette, P., Rouleau, G., and Isom, L. L. (2011). Voltage-gated Na channel β1B: a secreted cell adhesion molecule involved in human epilepsy. J. Neurosci. (in press).
Patino, G. A., Claes, L. R. F., Lopez-Santiago, L. F., Slat, E. A., Dondeti, R. S. R., Chen, C., O’Malley, H. A., Gray, C. B. B., Miyazaki, H., Nukina, N., Oyama, F., De Jonghe, P., and Isom, L. L. (2009). A functional null mutation of SCN1B in a patient with Dravet syndrome. J. Neurosci. 29, 10764–10778.
Patino, G. A., and Isom, L. L. (2010). Electrophysiology and beyond: multiple roles of Na+ channel beta subunits in development and disease. Neurosci. Lett. 486, 53–59.
Pertin, M., Ji, R. R., Berta, T., Powell, A. J., Karchewski, L., Tate, S. N., Isom, L. L., Woolf, C. J., Gilliard, N., Spahn, D. R., and Decosterd, I. (2005). Upregulation of the voltage-gated sodium channel beta2 subunit in neuropathic pain models: characterization of expression in injured and non-injured primary sensory neurons. J. Neurosci. 25, 10970–10980.
Qin, N., D’andrea, M. R., Lubin, M. L., Shafaee, N., Codd, E. E., and Correa, A. M. (2003). Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. Eur. J. Biochem. 270, 4762–4770.
Raman, I. M., and Bean, B. P. (1997). Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 17, 4517–4526.
Ratcliffe, C. F., Qu, Y., Mccormick, K. A., Tibbs, V. C., Dixon, J. E., Scheuer, T., and Catterall, W. A. (2000). A sodium channel signaling complex: modulation by associated receptor protein tyrosine phosphatase b. Nat. Neurosci. 3, 437–444.
Ratcliffe, C. F., Westenbroek, R. E., Curtis, R., and Catterall, W. A. (2001). Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J. Cell Biol. 154, 427–434.
Remme, C. A., Scicluna, B. P., Verkerk, A. O., Amin, A. S., Van Brunschot, S., Beekman, L., Deneer, V. H., Chevalier, C., Oyama, F., Miyazaki, H., Nukina, N., Wilders, R., Escande, D., Houlgatte, R., Wilde, A. A., Tan, H. L., Veldkamp, M. W., De Bakker, J. M., and Bezzina, C. R. (2009). Genetically determined differences in sodium current characteristics modulate conduction disease severity in mice with cardiac sodium channelopathy. Circ. Res. 104, 1283–1292.
Roger, S., Rollin, J., Barascu, A., Besson, P., Raynal, P. I., Iochmann, S., Lei, M., Bougnoux, P., Gruel, Y., and Le Guennec, J. Y. (2007). Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int. J. Biochem. Cell Biol. 39, 774–786.
Sashihara, S., Oh, Y., Black, J. A., and Waxman, S. G. (1995). Na+ channel β1 subunit mRNA expression in developing rat central nervous system. Mol. Brain Res. 34, 239–250.
Scheffer, I. E., Harkin, L. A., Grinton, B. E., Dibbens, L. M., Turner, S. J., Zielinski, M. A., Xu, R., Jackson, G., Adams, J., Connellan, M., Petrou, S., Wellard, R. M., Briellmann, R. S., Wallace, R. H., Mulley, J. C., and Berkovic, S. F. (2007). Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 130, 100–109.
Schmidt, J. W., and Catterall, W. A. (1986). Biosynthesis and processing of the alpha subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell 46, 437–445.
Shah, B. S., Stevens, E. B., Pinnock, R. D., Dixon, A. K., and Lee, K. (2001). Developmental expression of the novel voltage-gated sodium channel auxiliary subunit beta3, in rat CNS. J. Physiol. (Lond.) 534, 763–776.
Spampanato, J., Kearney, J. A., De Haan, G., Mcewen, D. P., Escayg, A., Aradi, I., Macdonald, B. T., Levin, S. I., Soltesz, I., Benna, P., Montalenti, E., Isom, L. L., Goldin, A. L., and Meisler, M. H. (2004). A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. J. Neurosci. 24, 10022–10034.
Srinivasan, J., Schachner, M., and Catterall, W. A. (1998). Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R. Proc. Natl. Acad. Sci. U.S.A. 95, 15753–15757.
Strick, P. L., Dum, R. P., and Fiez, J. A. (2009). Cerebellum and nonmotor function. Annu. Rev. Neurosci. 32, 413–434.
Sutkowski, E. M., and Catterall, W. A. (1990). Beta 1 subunits of sodium channels. Studies with subunit-specific antibodies. J. Biol. Chem. 265, 12393–12399.
Tan, B. H., Pundi, K. N., Van Norstrand, D. W., Valdivia, C. R., Tester, D. J., Medeiros-Domingo, A., Makielski, J. C., and Ackerman, M. J. (2010). Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm 7, 771–778.
Uebachs, M., Opitz, T., Royeck, M., Dickhof, G., Horstmann, M. T., Isom, L. L., and Beck, H. (2010). Efficacy loss of the anticonvulsant carbamazepine in mice lacking sodium channel beta subunits via paradoxical effects on persistent sodium currents. J. Neurosci. 30, 8489–8501.
Valdivia, C. R., Medeiros-Domingo, A., Ye, B., Shen, W. K., Algiers, T. J., Ackerman, M. J., and Makielski, J. C. (2010). Loss-of-function mutation of the SCN3B-encoded sodium channel {beta}3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc. Res. 86, 392–400.
Van Gassen, K. L., De Wit, M., Van Kempen, M., Van Der Hel, W. S., Van Rijen, P. C., Jackson, A. P., Lindhout, D., and De Graan, P. N. (2009). Hippocampal Nabeta3 expression in patients with temporal lobe epilepsy. Epilepsia 50, 957–962.
Van Wart, A., and Matthews, G. (2006). Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels. J. Neurosci. 26, 7172–7180.
Wallace, R. H., Scheffer, I. E., Parasivam, G., Barnett, S., Wallace, G. B., Sutherland, G. R., Berkovic, S. F., and Mulley, J. C. (2002). Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology 58, 1426–1429.
Wallace, R. H., Wang, D. W., Singh, R., Scheffer, I. E., George, A. L. Jr., Phillips, H. A., Saar, K., Reis, A., Johnson, E. W., Sutherland, G. R., Berkovic, S. F., and Mulley, J. C. (1998). Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat. Genet. 19, 366–370.
Wang, P., Yang, Q., Wu, X., Yang, Y., Shi, L., Wang, C., Wu, G., Xia, Y., Yang, B., Zhang, R., Xu, C., Cheng, X., Li, S., Zhao, Y., Fu, F., Liao, Y., Fang, F., Chen, Q., Tu, X., and Wang, Q. K. (2010). Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese Gene ID population. Biochem. Biophys. Res. Commun. 398, 98–104.
Wang, Y., Zhang, J., Li, X., Ji, J., Yang, F., Wan, C., Feng, G., Wan, P., He, L., and He, G. (2008). SCN8A as a novel candidate gene associated with bipolar disorder in the Han Chinese population. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 1902–1904.
Watanabe, H., Darbar, D., Kaiser, D. W., Jiramongkolchai, K., Chopra, S., Donahue, B. S., Kannankeril, P. J., and Roden, D. M. (2009). Mutations in sodium channel β1- and β2-subunits associated with atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2, 268–275.
Watanabe, H., Koopmann, T. T., Le Scouarnec, S., Yang, T., Ingram, C. R., Schott, J. J., Demolombe, S., Probst, V., Anselme, F., Escande, D., Wiesfeld, A. C., Pfeufer, A., Kaab, S., Wichmann, H. E., Hasdemir, C., Aizawa, Y., Wilde, A. A., Roden, D. M., and Bezzina, C. R. (2008). Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268.
Wilde, A. A., and Brugada, R. (2011). Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ. Res. 108, 884–897.
Wimmer, V. C., Reid, C. A., Mitchell, S., Richards, K. L., Scaf, B. B., Leaw, B. T., Hill, E. L., Royeck, M., Horstmann, M. T., Cromer, B. A., Davies, P. J., Xu, R., Lerche, H., Berkovic, S. F., Beck, H., and Petrou, S. (2010). Axon initial segment dysfunction in a mouse model of genetic epilepsy with febrile seizures plus. J. Clin. Invest. 120, 2661–2671.
Wong, H. K., Sakurai, T., Oyama, F., Kaneko, K., Wada, K., Miyazaki, H., Kurosawa, M., De Strooper, B., Saftig, P., and Nukina, N. (2005). beta subunits of voltage-gated sodium channels are novel substrates of BACE1 and gamma-secretase. J. Biol. Chem. 280, 23009–23017.
Xiao, Z.-C., Ragsdale, D. S., Malhorta, J. D., Mattei, L. N., Braun, P. E., Schachner, M., and Isom, L. L. (1999). Tenascin-R is a functional modulator of sodium channel β subunits. J. Biol. Chem. 274, 26511–26517.
Yamakawa, K. (2005). Epilepsy and sodium channel gene mutations: gain or loss of function? Neuroreport 16, 1–3.
Yu, F. H., Westenbroek, R. E., Silos-Santiago, I., Mccormick, K. A., Lawson, D., Ge, P., Ferriera, H., Lilly, J., Distefano, P. S., Catterall, W. A., Scheuer, T., and Curtis, R. (2003). Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J. Neurosci. 23, 7577–7585.
Keywords: adhesion, β subunit, development, excitability, voltage-gated Na+ channel
Citation: Brackenbury WJ and Isom LL (2011) Na+ channel β subunits: overachievers of the ion channel family. Front. Pharmacol. 2:53. doi: 10.3389/fphar.2011.00053
Received: 25 July 2011; Paper pending published: 16 August 2011;
Accepted: 12 September 2011; Published online: 28 September 2011.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyCopyright: © 2011 Brackenbury and Isom. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Lori L. Isom, Department of Pharmacology, University of Michigan Medical School, 1150 W. Medical Center Dr., Ann Arbor, MI 48109-5632, USA e-mail: lisom@umich.edu