
- The Massachusetts General Hospital, Cancer Center-Harvard Medical School, Boston, MA, USA
More than a decade ago, sirtuins were discovered as a highly conserved family of NAD+-dependent enzymes that extend lifespan in lower organisms. In mammals, sirtuins are key regulators of stress responses and metabolism, influencing a range of diseases, including diabetes, neurodegeneration, and cancer. In recent years, new functions of sirtuins have been characterized, uncovering the underlying mechanisms of their multifaceted role in metabolism. Here, we specifically review recent progress on the role of sirtuins in DNA repair and energy metabolism, further discussing the implication of sirtuins in the biology of cancer.
Introduction
Cancer can be defined as an uncontrolled proliferation of cells in the body. Cells become malignant by the acquisition of a succession of capabilities that allows them to evade strict regulatory circuits governing cell proliferation and homeostasis (Hanahan and Weinberg, 2011). On their way to tumorigenesis, genomic instability fuels the acquisition of these tumoral capabilities by generating a genomic environment highly propitious to mutations. Among the hallmarks of cancer cells, metabolic reprogramming stands high, allowing these cells to face up to new energetic requirements. While under normal conditions cells rely on mitochondrial oxidative phosphorylation to produce energy from glucose, cancer cells exhibit enhanced glycolysis. Simply put, pyruvate generated from glucose is directed toward production of lactate in the cytosol instead of entering the TCA cycle in the mitochondria. Because oxygen is limiting during oxidation of pyruvate to CO2 in the mitochondria, anaerobic glycolysis is usually activated in normal cells under low oxygen conditions. However, cancer cells switch their metabolism to glycolysis even under normoxic conditions, in a process that only generates 2 mol of ATP per mol of glucose whereas oxidative phosphorylation generates up to 36 mol of ATP per mol of glucose. This process is known as the Warburg effect after Otto Warburg, who observed in the 1920s that cancer cells metabolize tenfold more glucose to lactate than normal cells, even in the presence of sufficient oxygen to support oxidative phosphorylation (Warburg et al., 1924). Although aerobic glycolysis is highly inefficient for ATP production, it is thought to be an anaplerotic reaction that supplies critical intermediate metabolites for biomass production (amino acids, lipids, and nucleotides), imperative in highly proliferating cells (Vander Heiden et al., 2009). In addition, such metabolic shift may also confer a survival advantage to tumor cells, given their characteristic hypoxic environment.
Cells in an organism are subjected to stress arising from nutrient fluctuations as well as from genotoxic damage that menace genome integrity. A proper cellular response to stress is essential to avoid transformation. Studies over the last decade strongly indicate that sirtuins may play a fundamental role in this process. Sirtuins are a highly conserved family of proteins, homologs of the Saccharomyces cerevisiae protein Sir2 (Finkel et al., 2009). They are nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases and/or mono-[ADP-ribosyl] transferases. Mammals contain seven sirtuins (SIRT1–7) with different subcellular localization; SIRT1, SIRT6, and SIRT7 are nuclear, SIRT2 is mainly cytoplasmic and SIRT3, SIRT4, and SIRT5 are located in the mitochondria (Finkel et al., 2009). Nowadays, sirtuins are recognized as crucial regulators of energy metabolism, working as stress adaptors (oxidative, genotoxic and metabolic stress) likely through sensing changes in levels of intracellular NAD+, an obligated cofactor for their enzymatic activity. As the best-studied sirtuin, SIRT1 has been implicated in a large variety of metabolic processes, in particular in the regulation of glucose and lipid metabolism during fasting and caloric restriction. Several lines of evidence also point to a fundamental role of SIRT3 and SIRT6 in the regulation of glucose utilization (Zhong and Mostoslavsky, 2011). These three sirtuins have also been shown to protect from genomic instability upon genotoxic and oxidative stress, protecting the genome from mutations that can drive tumorigenesis. In this review we will summarize current advances in the biology of these sirtuins.
Sirtuins in Glucose Metabolism and DNA Damage
SIRT1
During fasting, an increase in pyruvate and NAD+ levels induces expression and activity of SIRT1 in hepatocytes promoting the deacetylation of PGC-1α, a transcriptional co-activator of a large number of genes (Figure 1). Deacetylation of PGC-1α results in the induction of gluconeogenic genes and repression of glycolytic genes (Rodgers et al., 2005). In parallel, SIRT1 activates the transcription factor FOXO1, which induces the same switch in transcription, reinforcing gluconeogenesis (Frescas et al., 2005) (Figure 1). In muscle, like in liver, SIRT1-mediated activation of PGC-1α during fasting activates peroxisome proliferators-activated receptor α (PPARα)-mediated transcription of fatty-acid catabolic genes (Gerhart-Hines et al., 2007; Purushotham et al., 2009). Concretely, the activation of PGC-1α in muscle induces the transcription of genes involved in mitochondrial oxidative phosphorylation and electron transport, coupling fatty-acid oxidation with energy production. In this manner, activation of SIRT1 in muscle coordinates a shift toward preservation of glucose in the cell, while lipids are used to obtain energy. In white adipose tissue, SIRT1 represses PPARγ during fasting, in turn inducing lipolysis and reducing fat storage (Picard et al., 2004). Lastly, SIRT1 also protects from the negative effects of high fat diet (HFD), stimulating secretion of insulin by pancreatic β-cells through PPARγ-mediated transcriptional repression of uncoupling protein 2 (UCP2), improving in this way glucose tolerance (Moynihan et al., 2005; Bordone et al., 2006) (Figure 1).
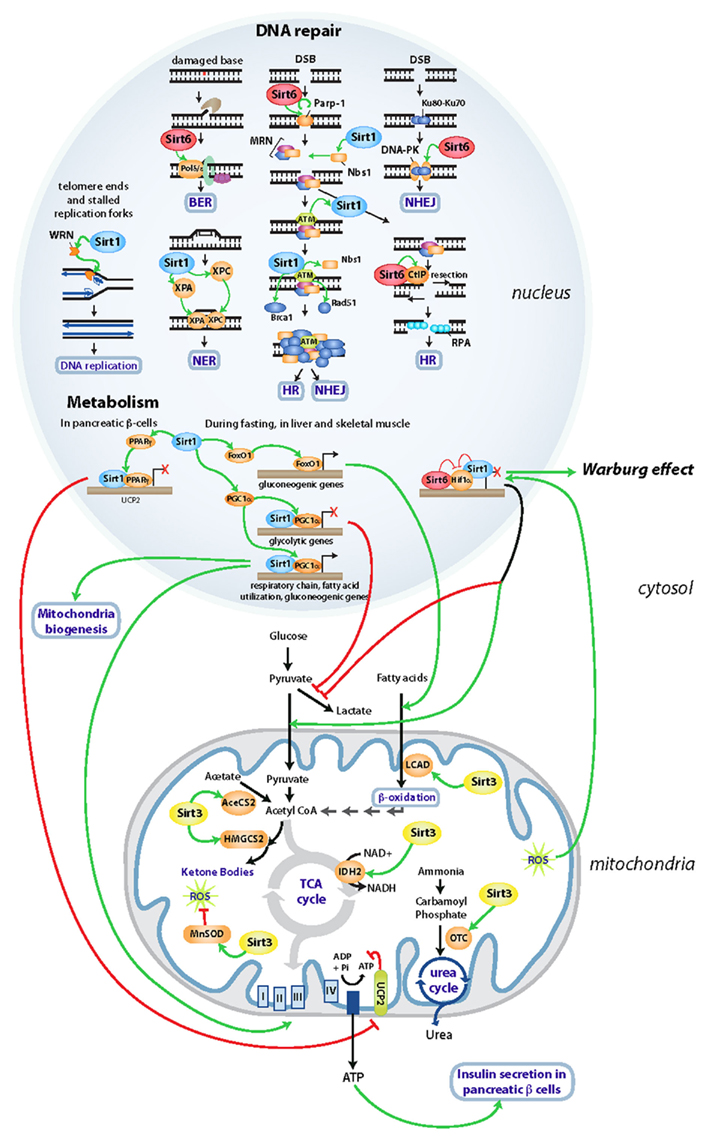
Figure 1. Sirtuins’ regulation of DNA repair and metabolism. Diagram indicating the main DNA repair and metabolic pathways regulated by SIRT1, SIRT3, and SIRT6 in the nucleus and in the mitochondria. Activating and inhibitory functions are depicted in green and red, respectively.
Although SIRT1 null mice die perinatally due to developmental defects, absence of SIRT1 provokes a large amount of chromosome abnormalities in embryos, impairing progression of cells through mitosis (Wang et al., 2008). A defective signaling of DNA double-strand breaks (DSBs) seems to be the cause behind this genomic instability. SIRT1 associates with and deacetylates the repair factor NBS1, a modification required for its subsequent phosphorylation by the Ataxia Telangiectasia Mutated (ATM) kinase in the first steps of the DNA damage response (Yuan et al., 2007) (Figure 1). In addition, SIRT1 is also recruited to DNA DSBs in an ATM-dependent manner. The recruitment of SIRT1 at the breaks is important for the accumulation of proteins involved in signaling and repair, and SIRT1-deficient MEFs (mouse embryonic fibroblasts) exhibit a marked reduction of Rad51-, NBS1-, and BRCA1-foci following γ-irradiation (Oberdoerffer et al., 2008; Wang et al., 2008) (Figure 1). SIRT1 appears to be involved in multiple DNA-repair pathways. When a DSB occurs during S or G2 phases of the cell cycle, it will be preferentially repaired by homologous recombination (HR) using the sister chromatid as a template for DNA repair. SIRT1 modulates homologous recombination (HR) by deacetylating WRN –the gene mutated in the progeroid Werner Syndrome- and promoting its relocalization to DNA lesions (Li et al., 2008), where it repairs DNA strand breaks that arise from replication arrest. In this context, transgenic mice expressing increased levels of SIRT1 exhibit longer telomeres and higher frequency of sister chromatid HR events throughout the entire genome (Palacios et al., 2010). In addition to the regulation of DSB repair, SIRT1 is involved in the repair of DNA single strand breaks as well. SIRT1 null MEFs are hyper-sensitive to UV damage, which is mainly repaired by the nucleotide excision repair (NER) pathway. Recent studies demonstrated that SIRT1 deacetylates two components of NER: XPA and XPC (Xeroderma Pigmentosum A and C) upon UV damage. This deacetylation is essential for the recognition of DNA lesions by XPA and XPC, affecting the recruitment of downstream NER factors (Fan and Luo, 2010; Ming et al., 2010). Overall, it appears that SIRT1 has evolved to coordinate both proper genomic integrity and adequate metabolic adaptation, in this way allowing cells to adapt against stress (Figure 1). Failure on such mechanisms could clearly lead to both metabolic diseases and tumorigenesis, as described in further detail below.
SIRT6
The role of sirtuins in metabolism and maintenance of genomic integrity is not restricted to SIRT1. SIRT6 deficient mice appear relatively normal at birth, but beginning at 2 weeks of age they develop an acute degenerative phenotype, reminiscent of a progeroid syndrome (Mostoslavsky et al., 2006). The most striking phenotype observed in SIRT6 knockout mice is a severe hypoglycemia that causes death before 1 month of age (Mostoslavsky et al., 2006). Deletion of SIRT6 in mice triggers an increase in glucose uptake that is preferentially used for glycolysis instead of mitochondrial respiration, a metabolic switch similar to the Warburg effect in cancer cells. Indeed, SIRT6 maintains deacetylation of H3K9 in promoters of glycolytic genes, co-repressing Hif1α on these promoters (Figure 1). Under conditions of nutrient scarcity, SIRT6 is likely inactivated, causing activation of Hif1α and transcription of glycolytic genes (Zhong et al., 2010). These results support a model whereby SIRT6 works as a sensor of nutrient availability and as a critical modulator of glucose homeostasis.
In addition to its metabolic functions, SIRT6 knockout cells exhibit hypersensitivity to DNA-damaging agents and genomic instability. According to the selective sensitivity among the spectrum of DNA damage agents tested, SIRT6 was proposed to be an important regulator of base excision repair (BER), a mechanism that repairs the damage of single bases in the DNA (Mostoslavsky et al., 2006). Interestingly, in human cells, SIRT6 appears to regulate repair of DNA DSBs as well. Together with ATM and ATR, DNA-dependent protein kinase (DNA-PK) is a sensor of DSBs, promoting DNA repair by NHEJ. SIRT6 stabilizes DNA-PK at DSBs promoting repair (McCord et al., 2009). Notably, HR is prompted by the resection of a DSB through the action of different proteins like CtIP and PARP1, in turn generating single-stranded DNA that will invade the homologous strand for repair. SIRT6 has been shown to bind to DSBs and deacetylate CtIP. Although the mechanism remains unclear, deacetylation of CtIP would enhance the resection of the DSB, promoting HR (Kaidi et al., 2010). In human cancer cell lines, SIRT6 deacetylation of H3K9 at telomeres promotes the stable association of WRN protein at these regions, critical for the processing of telomeres during S phase. Thus, SIRT6 downregulation leads to telomere dysfunction and premature senescence, as in Werner Syndrome cells (Michishita et al., 2008). Finally, recent studies have demonstrated that upon oxidative stress, SIRT6 associates with PARP1 at the DSBs and stimulates its activity through ADP-ribosylation. PARP1 activity will promote NHEJ as well as HR repair (Mao et al., 2011). This evidence suggests an additional mechanism for SIRT6-mediated genome protection during stress. While studies in vitro have implicated SIRT6 in several DNA repair pathways (Figure 1), a protective role for SIRT6 in vivo remains as yet poorly understood, and future genetic studies in mice will be needed to obtain a clearer picture.
SIRT3
SIRT3 works as the major protein deacetylase within the mitochondrial matrix (Lombard et al., 2007) (Figure 1). In low nutrient conditions, SIRT3 stimulates alternative pathways for energy production in the mitochondria. On one hand, SIRT3 functions as a positive modulator of fatty-acid oxidation through deacetylation of long-chain acyl coenzyme A dehydrogenase (LCAD; Hirschey et al., 2010). In parallel, SIRT3 helps to detoxify the ammonia generated during amino acid catabolism promoting the urea cycle through the deacetylation of ornithine transcarbamoylase (OTC), the enzyme that catalyzes the second step in the urea cycle (Hallows et al., 2011). Finally, it promotes acetate recycling by activating acetyl-CoA synthetase 2 (AceCS2), the enzyme that converts acetate into acetyl-CoA, that can now enter the TCA cycle (Hallows et al., 2006; Schwer et al., 2006). Under fasting, low-carbohydrate diet as well as in diabetic patients, ketone bodies produced in the liver are used by the brain and skeletal muscle as main energy sources. SIRT3 promotes ketone body production from fatty-acid oxidation by deacetylating 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS2) (Shimazu et al., 2010) (Figure 1).
Although SIRT3 is mainly mitochondrial, and therefore no role in protection of the genome could be presumed for this sirtuin, SIRT3-deficient MEFs exhibit increased cellular superoxide levels and chromosomal instability when stressed (Kim et al., 2010). In fact, metabolic functions of SIRT3 are closely coupled with a protective role against oxidative damage. SIRT3 decreases mitochondrial reactive oxygen species (ROS) generated upon cellular stress by regulating the expression and activity of MnSOD, the main scavenger of superoxide in the mitochondria (Kim et al., 2010; Qiu et al., 2010; Tao et al., 2010; Chen et al., 2011) (Figure 1). This seems to play a key role in the cellular response against calorie restriction, where SIRT3 reduces oxidative stress by enhancing glutathione antioxidant defense mechanisms through deacetylation of Idh2 (Someya et al., 2010). Moreover, SIRT3 regulates the electron transport chain Complex III, that can direct ROS out of the mitochondria and into both the matrix and the cytoplasm (Kim et al., 2010). In this manner, SIRT3 management of ROS from mitochondria at different levels might protect the cell from oxidative damage, in turn inhibiting genomic instability.
The aforementioned roles of sirtuins in DNA repair, together with their ability to regulate both glucose homeostasis and ROS levels suggest the possibility that these sirtuins may act as putative tumor suppressors, as discussed in detail below.
Sirtuins in Cancer
The first evidences linking SIRT1 and tumorigenesis came from two cell-based studies, which uncovered the ability of SIRT1 to deacetylate and inhibit the tumor suppressor p53 (Luo et al., 2001; Vaziri et al., 2001). In this manner, SIRT1 promotes cell survival under stress conditions by specifically repressing p53 dependent apoptosis. However, as we already mentioned, an increase in SIRT1 associates with enhanced DNA repair, so this phenomena could also be a protecting mechanism to give priority to repair over apoptosis. SIRT1 regulation of p53 was confirmed in vivo in SIRT1 knockout mice that exhibit hyperacetylation of p53 and increased apoptosis of thymocytes after DNA damage (Cheng et al., 2003). In this context, it would be interesting to explore if the enhancement of p53 activity could as well be secondary to increased DNA damage in the absence of SIRT1. In addition, SIRT1 knockout mice did not show enhanced p21 induction or sensitivity upon DNA damage, suggesting that only the apoptotic function of p53 is regulated by SIRT1 (Cheng et al., 2003). Although SIRT1-mediated deacetylation of p53 has been confirmed by other groups, the biological relevance is a matter of debate, since alterations in p53 biological activities have not been observed in the absence of SIRT1 in vivo (Kamel et al., 2006). Thus, the role for SIRT1 on p53-mediated tumor suppression still remains to be elucidated. In humans, SIRT1 expression is higher in several types of cancer, including acute myeloid leukemia, primary colon cancer, prostate cancers, and non-melanoma skin cancers (reviewed in Deng, 2009), suggesting again a potential role of SIRT1 as an oncogene. This result is also true for several cell lines. Depletion of SIRT1 induces growth arrest and reduces drug resistance of cancer cells in vitro (Ota et al., 2006; Liang et al., 2008) and increases tumor cell death with no toxic effects in normal cells in culture (Ford et al., 2005) proposing SIRT1 as a potential target in cancer therapy. Also, overexpression or activation of SIRT1 promotes cellular proliferation, impairs cellular senescence and increases growth rate via ERK/S6K1 signaling pathway (Huang et al., 2008). SIRT1 overexpression also represses epigenetically the activity or expression of tumor suppressor and DNA repair genes including FOXO family members (FOXO1, FOXO3a, and FOXO4) (Motta et al., 2004), p73 (Dai et al., 2007), Rb (Wong and Weber, 2007), MLH1 (Pruitt et al., 2006), and Ku70 (Cohen et al., 2004). Reciprocally, the activity or expression of SIRT1 is inhibited by at least two tumor suppressor genes; the transcriptional repressors HIC1 (hypermethylated in cancer-1) and DBC1 (deleted in breast cancer-1; Chen et al., 2005; Kim et al., 2008; Zhao et al., 2008).
The current data available for SIRT1 expression in cancer contrasts with previous hypothesis pointing toward a role for SIRT1 as a tumor suppressor. Although a variety of cancer cell lines show SIRT1 overexpression, SIRT1 expression is reduced in different human cancers, like glioblastoma, bladder carcinoma, prostate carcinoma, and ovarian cancer (Wang et al., 2008). Moreover, an increase in SIRT1 levels has been shown to inhibit growth of BRCA mutant tumors in vivo. Thus far, the data from cell-based studies do not clear up whether SIRT1 acts as an oncogene or a tumor suppressor. Only the generation of mice with genetically modified SIRT1 is finally shedding some light on this matter.
The studies of cancer in mouse models of SIRT1 support the idea that SIRT1 has, indeed, a tumor suppressor activity. In cancer prone mouse models, like p53+/− and APC+/min mice, SIRT1 expression protects from tumor development. Increased expression of SIRT1 has been shown to protect from ionizing radiation-induced cancer in p53+/− mice (Oberdoerffer et al., 2008) whereas SIRT1+/− p53+/− mice show an earlier onset of tumor development in comparison to SIRT1+/+ p53+/− littermates (Oberdoerffer et al., 2008). It has been proposed that SIRT1 relocalization to DNA breaks represses transcription of these regions and promotes repair (Oberdoerffer et al., 2008), protecting from genome instability. A different mechanism underlies SIRT1 protection from intestinal tumors in the APC+/min mouse, where SIRT1 expression promotes the cytoplasmic relocalization of the oncogenic form of β-catenin inhibiting its ability to activate transcription and drive cell proliferation (Firestein et al., 2008). Herranz and colleagues demonstrated that SIRT1 overexpression provides strong protection against metabolic syndrome-associated liver cancer by reducing DNA damage and by preventing inflammation and fatty liver. In addition, they demonstrated that a mild increase in SIRT1 levels protects mice from spontaneous and aging-associated cancers (Herranz et al., 2010). Additionally, a recent work demonstrated that overexpression of SIRT1 reduces growth and angiogenesis of xenograft tumors through the interaction and repression of Hif1α (Lim et al., 2010).
Despite the important role of SIRT6 in maintaining genome stability and energy metabolism, it remains as yet unexplored whether SIRT6 plays a role in tumorigenesis. There are no studies about SIRT6 expression in human cancer either, besides the observation that SIRT6 chromosomal locus is a region prone to chromosomal breaks in human acute myeloid leukemia (Mahlknecht et al., 2006). It has been recently shown that overexpression of SIRT6 induces apoptosis in cancer cell lines but not in non-transformed cells through its ADP-ribosyltransferase activity, suggesting that SIRT6 could be a target for overexpression in cancer therapy (Van Meter et al., 2011).
Increased levels of SIRT3 associate with node-positive breast cancer versus non-malignant breast tissue (Ashraf et al., 2006) as well as with oral squamous cell carcinoma, suggesting that SIRT3 could function as a tumor promoter. However, studies in genetically modified mouse models of SIRT3 question this hypothesis. SIRT3-deficient MEFs are transformed by the expression of a single oncogene (Ras) whereas wildtype MEFs also require inactivation of a tumor-suppressor gene. In addition, loss of SIRT3 in vivo results in a mild tumor proneness; higher incidence of spontaneous mammary tumors in 24 months old mice (Kim et al., 2010). How SIRT3 protects from tumorigenesis is a matter of debate. A current hypothesis poses that SIRT3 tumor suppressive role comes from its ability to regulate cellular ROS levels, an idea that fits in the free radical theory that proposes the accumulation of oxidative stress as a major cause of ageing and cancer. Such a model is clearly supported by the above mentioned role for SIRT3 in regulating MnSOD (Kim et al., 2010; Qiu et al., 2010; Tao et al., 2010; Chen et al., 2011). However, the recent observation that SIRT3-deficient cells become highly glycolytic, suggests another possibility (Bell et al., 2011; Finley et al., 2011). SIRT3 loss generates an increase in cellular ROS that would stabilize Hif1α. Stabilization of Hif1α, as it occurs in SIRT6-null cells, will lead to a metabolic reprogramming where cells will prioritize glycolysis over oxidative phosphorylation, a shift that confers metabolic advantages in highly proliferative cells. Therefore, it has been suggested that SIRT3 works as a tumor suppressor by opposing reprogramming of cancer cell metabolism (Bell et al., 2011; Finley et al., 2011), in other words, repressing the Warburg effect. According to a tumor suppressive role for this sirtuin, expression of SIRT3 was found significantly reduced in human breast cancer samples (Finley et al., 2011). It remains to be determined whether oxidative stress or glycolytic metabolism plays a dominant role in this phenotype.
Sirtuins: Tumor Suppressors or Oncogenes
The above-discussed results suggest that SIRT1 and SIRT3 are tumor suppressor genes. In the multistep development of cancer, loss of SIRT1 or SIRT3 might be a late phenomenon in carcinogenesis allowing the growing tumor to evade apoptosis or acquire different metabolic requirements. Thus, SIRT1 and SIRT3 might be lost in late stages of tumor progression, conferring proliferative advantage to cells with already multiple genetic alterations. In addition, the analysis of updated databases (such as Oncomine and Tumorscape), as well as upcoming results from our laboratory, appears to suggest that SIRT6 is downregulated in tumors, therefore functioning as a tumor suppressor as well (Table 1). Much work will be needed to pinpoint the precise molecular mechanisms governing sirtuins’ functions in cancer. But one thing is sure: these proteins clearly link DNA repair and metabolism, two hallmarks of cancer. As such, it is tempting to envision that modulators of sirtuin activity could provide future beneficial alternatives against this devastating disease.

Table 1. Sirtuins in cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ashraf, N., Zino, S., Macintyre, A., Kingsmore, D., Payne, A. P., George, W. D., and Shiels, P. G. (2006). Altered sirtuin expression is associated with node-positive breast cancer. Br. J. Cancer 95, 1056–1061.
Bell, E. L., Emerling, B. M., Ricoult, S. J. H., and Guarente, L. (2011). SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30, 2986–2996.
Bordone, L., Motta, M. C., Picard, F., Robinson, A., Jhala, U. S., Apfeld, J., McDonagh, T., Lemieux, M., McBurney, M., Szilvasi, A., Easlon, E. J., Lin, S-J., and Guarente, L. (2006). Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 4, e31. doi:10.1371/journal.pbio.0040031
Chen, W. Y., Wang, D. H., Yen, R. C., Luo, J., Gu, W., and Baylin, S. B. (2005). Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123, 437–448.
Chen, Y., Zhang, J., Lin, Y., Lei, G., Guan, K-L., Zhao, S., and Xiong, Y. (2011). Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 12, 534–541.
Cheng, H.-L., Mostoslavsky, R., Saito, S., Manis, J. P., Gu, Y., Patel, P., Bronson, R., Appella, E., Alt, F. W., and Chua, K. F. (2003). Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 100, 10794–10799.
Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., Howitz, K. T., Gorospe, M., de Cabo, R., and Sinclair, D. A. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392.
Dai, J. M., Wang, Z. Y., Sun, D. C., Lin, R. X., and Wang, S. Q. (2007). SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. J. Cell. Physiol. 166, 161–166.
Deng, C.-X. (2009). SIRT1, is it a tumor promoter or tumor suppressor? Int. J. Biol. Sci. 5, 147–152.
Fan, W., and Luo, J. (2010). SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol. Cell 39, 247–258.
Finkel, T., Deng, C. X., and Mostoslavsky, R. (2009). Recent progress in the biology and physiology of sirtuins. Nature 260, 587–591.
Finley, L. W. S., Carracedo, A., Lee, J., Souza, A., Egia, A., Zhang, J., Teruya-Feldstein, J., Moreira, P. I., Cardoso, S. M., Clish, C. B., Pandolfi, P. P., and Haigis, M. C. (2011). SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 19, 416–428.
Firestein, R., Blander, G., Michan, S., Oberdoerffer, P., Ogino, S., Campbell, J., Bhimavarapu, A., Luikenhuis, S., de Cabo, R., Fuchs, C., Hahn, W. C., Guarente, L. P., and Sinclair, D. A. (2008). The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 3, e2020. doi:10.1371/journal.pone.0002020
Ford, J., Jiang, M., and Milner, J. (2005). Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 65, 10457–10463.
Frescas, D., Valenti, L., and Accili, D. (2005). Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 280, 20589–20595.
Gerhart-Hines, Z., Rodgers, J. T., Bare, O., Lerin, C., Kim, S-H., Mostoslavsky, R., Alt, F. W., Wu, Z., and Puigserver, P. (2007). Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 26, 1913–1923.
Hallows, W. C., Lee, S., and Denu, J. M. (2006). Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. U.S.A. 103, 10230–10235.
Hallows, W. C., Yu, W., Smith, B. C., Devires, M. K., Ellinger, J. J., Someya, S., Shortreed, M. R., Prolla, T., Markley, J. L., Smith, L. M., Zhao, S., Guan, K.-L., and Denu, J. M. (2011). Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol. Cell 41, 139–149.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Herranz, D., Muñoz-Martin, M., Cañamero, M., Mulero, F., Martinez-Pastor, B., Fernandez-Capetillo, O., and Serrano, M. (2010). Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 1, 3.
Hirschey, M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D. B., Grueter, C. A., Harris, C., Biddinger, S., Ilkayeva, O. R., Stevens, R. D., Li, Y., Saha, A. K., Ruderman, N. B., Bain, J. R., Newgard, C. B., Farese, R. V., Alt, F. W., Kahn, C. R., and Verdin, E. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125.
Huang, J., Gan, Q., Han, L., Li, J., Zhang, H., Sun, Y., Zhang, Z., and Tong, T. (2008). SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE 3, e1710. doi:10.1371/journal.pone.0001710
Kaidi, A., Weinert, B. T., Choudhary, C., and Jackson, S. P. (2010). Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 329, 1348–1353.
Kamel, C., Abrol, M., Jardine, K., He, X., and McBurney, M. W. (2006). SirT1 fails to affect p53-mediated biological functions. Aging Cell 5, 81–88.
Kim, H.-S., Patel, K., Muldoon-Jacobs, K., Bisht, K. S., Aykin-Burns, N., Pennington, J. D., van der Meer, R., Nguyen, P., Savage, J., Owens, K. M., Vassilopoulos, A., Ozden, O., Park, S.-H., Singh, K. K., Abdulkadir, S. A., Spitz, D. R., Deng, C.-X., and Gius, D. (2010). SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17, 41–52.
Kim, J.-E., Chen, J., and Lou, Z. (2008). DBC1 is a negative regulator of SIRT1. Nature 451, 583–586.
Li, K., Casta, A., Wang, R., Lozada, E., Fan, W., Kane, S., Ge, Q., Gu, W., Orren, D., and Luo, J. (2008). Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J. Biol. Chem. 283, 7590–7598.
Liang, X.-J., Finkel, T., Shen, D.-W., Yin, J.-J., Aszalos, A., and Gottesman, M. M. (2008). SIRT1 contributes in part to cisplatin resistance in cancer cells by altering mitochondrial metabolism. Mol. Cancer Res. 6, 1499–1506.
Lim, J.-H., Lee, Y.-M., Chun, Y.-S., Chen, J., Kim, J.-E., and Park, J.-W. (2010). Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 38, 864–878.
Lombard, D. B., Alt, F. W., Cheng, H.-L., Bunkenborg, J., Streeper, R. S., Mostoslavsky, R., Kim, J., Yancopoulos, G., Valenzuela, D., Murphy, A., Yang, Y., Chen, Y., Hirschey, M. D., Bronson, R. T., Haigis, M., Guarente, L. P., Farese, R. V., Weissman, S., Verdin, E., and Schwer, B. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814.
Luo, J., Nikolaev, A. Y., Imai, S.-I., Chen, D., Su, F., Shiloh, A., Guarente, L., and Gu, W. (2001). Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107, 137–148.
Mahlknecht, U., Ho, A. D., and Voelter-Mahlknecht, S. (2006). Chromosomal organization and fluorescence in situ hybridization of the human Sirtuin 6 gene. Int. J. Oncol. 28, 447–456.
Mao, Z., Hine, C., Tian, X., Van Meter, M., Au, M., Vaidya, A., Seluanov, A., and Gorbunova, V. (2011). SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446.
McCord, R. A., Michishita, E., Hong, T., Berber, E., Boxer, L. D., Kusumoto, R., Guan, S., Shi, X., Gozani, O., Burlingame, A. L., Bohr, V. A., and Chua, K. F. (2009). SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging 1, 109–121.
Michishita, E., McCord, R. A., Berber, E., Kioi, M., Padilla-Nash, H., Damian, M., Cheung, P., Kusumoto, R., Kawahara, T. L., Barret, J. C., Chang, H. Y., Bohr, V. A., Ried, T., Gozani, O., and Chua, K. F. (2008). SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452, 492–496.
Ming, M., Shea, C. R., Guo, X., Li, X., Soltani, K., Han, W., and He, Y.-Y. (2010). Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc. Natl. Acad. Sci. U.S.A. 107, 22623–22628.
Mostoslavsky, R., Chua, K. F., Lombard, D. B., Pang, W. W., Fischer, M. R., Gellon, L., Liu, P., Mostoslavsky, G., Franco, S., Murphy, M. M., Mills, K. D., Patel, P., Hsu, J. T., Hong, A. L., Ford, E., Cheng, H.-L., Kennedy, C., Nunez, N., Bronson, R., Frendewey, D., Auerbach, W., Valenzuela, D., Karow, M., Hottiger, M. O., Hursting, S., Barrett, J. C., Guarente, L., Mulligan, R., Demple, B., Yancopoulos, G. D., and Alt, F. W. (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329.
Motta, M. C., Divecha, N., Lemieux, M., Kamel, C., Chen, D., Gu, W., Bultsma, Y., McBurney, M., and Guarente, L. (2004). Mammalian SIRT1 represses forkhead transcription factors. Cell 116, 551–563.
Moynihan, K. A., Grimm, A. A., Plueger, M. M., Bernal-Mizrachi, E., Ford, E., Cras-Méneur, C., Permutt, M. A., and Imai, S-I. (2005). Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2, 105–117.
Oberdoerffer, P., Michan, S., McVay, M., Mostoslavsky, R., Vann, J., Park, S.-K., Hartlerode, A., Stegmuller, J., Hafner, A., Loerch, P., Wright, S. M., Mills, K. D., Bonni, A., Yankner, B. A., Scully, R., Prolla, T. A., Alt, F. W., and Sinclair, D. A. (2008). SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918.
Ota, H., Tokunaga, E., Chang, K., Hikasa, M., Iijima, K., Eto, M., Kozaki, K., Akishita, M., Ouchi, Y., and Kaneki, M. (2006). Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 25, 176–185.
Palacios, J. A., Herranz, D., De Bonis, M. L., Velasco, S., Serrano, M., and Blasco, M. A. (2010). SIRT1 contributes to telomere maintenance and augments global homologous recombination. J. Cell Biol. 191, 1299–1313.
Picard, F., Kurtev, M., Chung, N., Topark-Ngarm, A., Senawong, T., Machado De Oliveira, R., Leid, M., McBurney, M. W., and Guarente, L. (2004). Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429, 771–776.
Pruitt, K., Zinn, R. L., Ohm, J. E., McGarvey, K. M., Kang, S.-H. L., Watkins, D. N., Herman, J. G., and Baylin, S. B. (2006). Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2, e40. doi:10.1371/journal.pgen.0020040
Purushotham, A., Schug, T. T., Xu, Q., Surapureddi, S., Guo, X., and Li, X. (2009). Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9, 327–338.
Qiu, X., Brown, K., Hirschey, M. D., Verdin, E., and Chen, D. (2010). Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12, 662–667.
Rodgers, J. T., Lerin, C., Haas, W., Gygi, S. P., Spiegelman, B. M., and Puigserver, P. (2005). Nutrient control of glucose homeostasis through a complex of PGC-1a and SIRT1. Nature 434, 3–8.
Schwer, B., Bunkenborg, J., Verdin, R. O., Andersen, J. S., and Verdin, E. (2006). Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. U.S.A. 103, 10224–10229.
Shimazu, T., Hirschey, M. D., Hua, L., Dittenhafer-Reed, K. E., Schwer, B., Lombard, D. B., Li, Y., Bunkenborg, J., Alt, F. W., Denu, J. M., Jacobson, M. P., and Verdin, E. (2010). SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661.
Someya, S., Yu, W., Hallows, W. C., Xu, J., Vann, J. M., Leeuwenburgh, C., Tanokura, M., Denu, J. M., and Prolla, T. A. (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812.
Tao, R., Coleman, M. C., Pennington, J. D., Ozden, O., Park, S.-H., Jiang, H., Kim, H.-S., Flynn, C. R., Hill, S., Hayes McDonald, W., Olivier, A. K., Spitz, D. R., and Gius, D. (2010). Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 40, 893–904.
Van Meter, M., Mao, Z., Gorbunova, V., and Seluanov, A. (2011). Sirt6 overexpression induces massive apoptosis in cancer cells but not in normal cells. Cell Cycle 10, 1–6.
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033.
Vaziri, H., Dessain, S. K., Ng Eaton, E., Imai, S.-I., Frye, R. A., Pandita, T. K., Guarente, L., and Weinberg, R. A. (2001). hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159.
Wang, R.-H., Sengupta, K., Li, C., Kim, H.-S., Cao, L., Xiao, C., Kim, S., Xu, X., Zheng, Y., Chilton, B., Jia, R., Zheng, Z.-M., Appella, E., Wang, X. W., Ried, T., and Deng, C.-X. (2008). Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14, 312–323.
Warburg, O., Posener, K., and Negelein, E. (1924). Uber den Stoffwechsel der Carcinomzelle. Biochem. Z. 152, 309–344.
Wong, S., and Weber, J. D. (2007). Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochem. J. 407, 451–460.
Yuan, Z., Zhang, X., Sengupta, N., Lane, W. S., and Seto, E. (2007). SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 27, 149–162.
Zhao, W., Kruse, J.-P., Tang, Y., Jung, S. Y., Qin, J., and Gu, W. (2008). Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451, 587–590.
Zhong, L., D’Urso, A., Toiber, D., Sebastian, C., Henry, R. E., Vadysirisack, D. D., Guimaraes, A., Marinelli, B., Wikstrom, J. D., Nir, T., Clish, C. B., Vaitheesvaran, B., Iliopoulos, O., Kurland, I., Dor, Y., Weissleder, R., Shirihai, O. S., Ellisen, L. W., Espinosa, J. M., and Mostoslavsky, R. (2010). The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140, 280–293.
Keywords: SIRT1, SIRT6, SIRT3, Warburg effect, cancer metabolism, DNA repair, genomic instability, sirtuin biology
Citation: Martinez-Pastor B and Mostoslavsky R (2012) Sirtuins, metabolism, and cancer. Front. Pharmacol. 3:22. doi: 10.3389/fphar.2012.00022
Received: 29 November 2011; Paper pending published: 23 December 2011;
Accepted: 07 February 2012; Published online: 21 February 2012.
Edited by:
Aleksey G. Kazantsev, Harvard Medical School and Massachusetts General Hospital, USAReviewed by:
Markus Kalkum, Beckman Research Institute of the City of Hope, USAWei Ni, Michigan State University, USA
Copyright: © 2012 Martinez-Pastor and Mostoslavsky. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Raul Mostoslavsky, The Massachusetts General Hospital, Cancer Center-Harvard Medical School, 185 Cambridge Street, Boston, MA, USA. e-mail: rmostoslavsky@mgh.harvard.edu