
- School of Pharmacy and Division of BioMedical Sciences, Faculty of Medicine, Memorial University of Newfoundland, St. John’s, NL, Canada
Cell death and dysfunction after traumatic brain injury (TBI) is caused by a primary phase, related to direct mechanical disruption of the brain, and a secondary phase which consists of delayed events initiated at the time of the physical insult. Arguably, the calcium ion contributes greatly to the delayed cell damage and death after TBI. A large, sustained influx of calcium into cells can initiate cell death signaling cascades, through activation of several degradative enzymes, such as proteases and endonucleases. However, a sustained level of intracellular free calcium is not necessarily lethal, but the specific route of calcium entry may couple calcium directly to cell death pathways. Other sources of calcium, such as intracellular calcium stores, can also contribute to cell damage. In addition, calcium-mediated signal transduction pathways in neurons may be perturbed following injury. These latter types of alterations may contribute to abnormal physiology in neurons that do not necessarily die after a traumatic episode. This review provides an overview of experimental evidence that has led to our current understanding of the role of calcium signaling in death and dysfunction following TBI.
Introduction
The calcium ion (Ca2+), and maintenance of cellular Ca2+ homeostasis, is essential for the proper function of the nervous system. For example, Ca2+ is critical for growth and development (Ghosh and Greenberg, 1995; Spitzer and Ribera, 1998; Takei et al., 1998), is a necessary component for neurotransmission (Berridge, 1998; Iwasaki et al., 2000), and contributes to distinct patterns of differential gene expression in neurons (Bading et al., 1993; Lerea and McNamara, 1993; Finkbeiner and Greenberg, 1998). Calcium also appears to be required for most forms of activity-dependent synaptic plasticity, generally believed to be the cellular correlate for learning and memory (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999; Lamont and Weber, 2012; Kawamoto et al., 2012).
As the importance of Ca2+ in a wide range of physiological functions in the nervous system is now well established, it is understandable that changes in the levels of intracellular Ca2+ or alterations in Ca2+ signaling could contribute to a variety of pathologies. For example, changes in Ca2+ metabolism and homeostasis are well noted during the aging process (Kirischuk and Verkhratsky, 1996; Oliveira and Bading, 2011). Altered Ca2+ homeostasis is postulated to contribute to neurodegenerative diseases such as Huntington’s, Parkinson’s, and Alzheimer’s disease, as well as amyotrophic lateral sclerosis (Mattson et al., 2000; Heath and Shaw, 2002; Wojda et al., 2008; Nakamura and Lipton, 2010; Surmeier et al., 2010). Disrupted signaling through Ca2+ channels also contributes to epilepsy and migraine (Cain and Snutch, 2011). Perhaps two of the most studied pathologies that are believed to involve disturbed Ca2+ homeostasis are cerebral ischemia and traumatic brain injury (TBI; Young, 1992; Tymianski and Tator, 1996; Kristián and Siesjö, 1998; Weber, 2004). The purpose of this review is to provide an overview of our current understanding of disrupted Ca2+ homeostasis following TBI, primarily at a cellular level with a focus on neurons. Although the role of Ca2+ in TBI was arguably of considerably more attention to scientists a few decades ago, some important studies have been conducted in recent years that have advanced our understanding in this research field. These studies have provided more data not only on the role of Ca2+ in neuronal death, but on the role that Ca2+ could play in altered physiology in cells that do not die, such as the effects of disturbed signal transduction mechanisms or synaptic plasticity. These findings are highlighted in latter sections of the review.
Pathophysiology of Traumatic Brain Injury
Traumatic brain injury is an insult to the brain caused by an external physical force, resulting in functional disability. Falls and motor vehicle accidents are the primary causes of TBI, while sports, assaults, and gunshot wounds also contribute significantly to these types of injuries (Centers for Disease Control and Prevention, 2010). TBI is one of the leading causes of death and disability worldwide, including the developing world (Reilly, 2007). TBI is characterized as mild, moderate or severe. Mild TBI, i.e., concussion, accounts for 70–90% of all TBI cases and 15–20% of individuals with a mild TBI have long-term dysfunction (Ryu et al., 2009). The principle mechanisms of damage from a TBI are due either to contact or acceleration/deceleration types of injury (Gennarelli, 1994). Contact injury results from an object striking the head and is generally associated with focal brain damage, which includes surface contusions, intracranial hematoma, and raised intracranial pressure (McIntosh et al., 1996; Patt and Brodhun, 1999). Acceleration/deceleration brain injury results from unrestricted head movement in the instant after injury and leads to shear, tensile, and compressive strain. These types of injuries are generally associated with diffuse brain damage which includes widespread damage to axons, ischemic brain injury, and diffuse brain swelling (Gennarelli, 1994).
The pathophysiology of TBI consists of two main phases, a primary (mechanical) phase of damage and secondary (delayed) damage. Primary damage occurs at the moment of insult and includes contusion and laceration, diffuse axonal injury, and intracranial hemorrhage (McIntosh et al., 1996; Patt and Brodhun, 1999). Secondary damage includes processes that are initiated at the time of insult, but do not appear clinically for hours or even days after injury. Such processes include brain damage due to ischemia, swelling (edema), altered neurochemical mechanisms, and activation of degradative enzymes. The overall events that constitute secondary or delayed cell damage after TBI are complex, and still only partially understood.
A wide variety of mechanisms are proposed to contribute to secondary damage following trauma (for reviews, see Leker and Shohami, 2002; Weber, 2004; Slemmer et al., 2008; McAllister, 2011). Ischemia, caused by a reduction in cerebral blood flow (CBF), is an important mechanism underlying secondary brain damage, especially in severely injured individuals (Maas et al., 2000; van den Brink et al., 2000). A reduction of CBF damages mitochondria, which is followed by a shift from aerobic to anaerobic metabolism in neurons. In addition, ionic homeostasis becomes compromised and increases in intracellular Na+, K+, and Ca2+ can lead to cell swelling and an even greater reduction in CBF. Damage to mitochondria leads to the release of reactive oxygen species (ROS), such as superoxide and hydroxyl radicals. These oxygen free radicals can damage cellular membranes by causing lipid peroxidation, can cause DNA strand breaks and damage purine and pyrimidine bases, and can also feedback and further damage mitochondrial membranes leading to altered function (Novack et al., 1996; Raghupathi and McIntosh, 1998; Slemmer et al., 2008). Inflammation also likely plays an important role in secondary brain damage after TBI. Neurons and glia synthesize and release cytokines such as tumor necrosis factor and interleukins, which may contribute to the immunologic response after TBI (Morganti-Kossmann et al., 1992; Stelmasiak et al., 2000). Enhancement of inflammation due to cytokine release and accumulation of immune cells could result in a further decrease in CBF due to swelling of the brain.
The extracellular level of glutamate, the major excitatory neurotransmitter in the mammalian nervous system, is known to be dramatically elevated after TBI in vivo (Faden et al., 1989; Katayama et al., 1990; Palmer et al., 1994). Glutamate activates a variety of receptors on neurons including Ca2+-permeable ion channels. Excessive release of glutamate and over-stimulation of excitatory amino acid receptors, a process known as “excitotoxicity” as originally coined by Olney (1969), is believed to play a major role in secondary injury and death of cells of the CNS after traumatic injury (Hayes et al., 1992; Young, 1992; Weber, 2004; Lau and Tymianski, 2010). Furthermore, activation of voltage-gated calcium channels (VGCCs) or leakage through damaged cell membranes, can lead to a large elevation of Ca2+ within cells after injury (Young, 1992). This influx of Ca2+ into cells causes a corresponding elevation of intracellular free calcium ([Ca2+]i), which can activate a variety of enzymes, setting in motion a cascade of events which eventually leads to cell injury and death (Trump and Berezesky, 1995; Wojda et al., 2008). This alteration in cellular Ca2+ homeostasis is likely one of the key mechanisms contributing to secondary neuronal damage and altered physiology following TBI.
Mechanisms of Maintaining Calcium Homeostasis in Neurons
Mechanisms for Increasing [Ca2+]i Levels
Arguably, Ca2+ is the most ubiquitous signal transduction molecule in the body as it acts as an important intracellular second messenger that regulates vital processes in almost all mammalian cells (Petersen et al., 2005). Calcium is so important to normal neuronal function in particular, that these cells have developed several homeostatic mechanisms to control the subcellular location of Ca2+ and the level of [Ca2+]i. These mechanisms include Ca2+ influx through voltage-operated and receptor-operated channels (ROCs), Ca2+ buffering by the plasma membrane and cytosolic proteins, Ca2+ storage in intracellular organelles and Ca2+ efflux (Miller, 1991; Kostyuk and Verkhratsky, 1994; Wojda et al., 2008; see Figure 1). These homeostatic mechanisms maintain [Ca2+]i at low levels, usually about 100 nM in neurons compared to an extracellular concentration of approximately 1 mM. Maintaining [Ca2+]i at such low levels in the cell allows relatively small or localized increases in [Ca2+]i to be used as a trigger to activate signal transduction pathways which lead to physiological processes such as activation of specific enzymes or modulation of ion channels (Racay and Lehotsky, 1996; Tymianski and Tator, 1996; Barish, 1998; Weber, 2004).
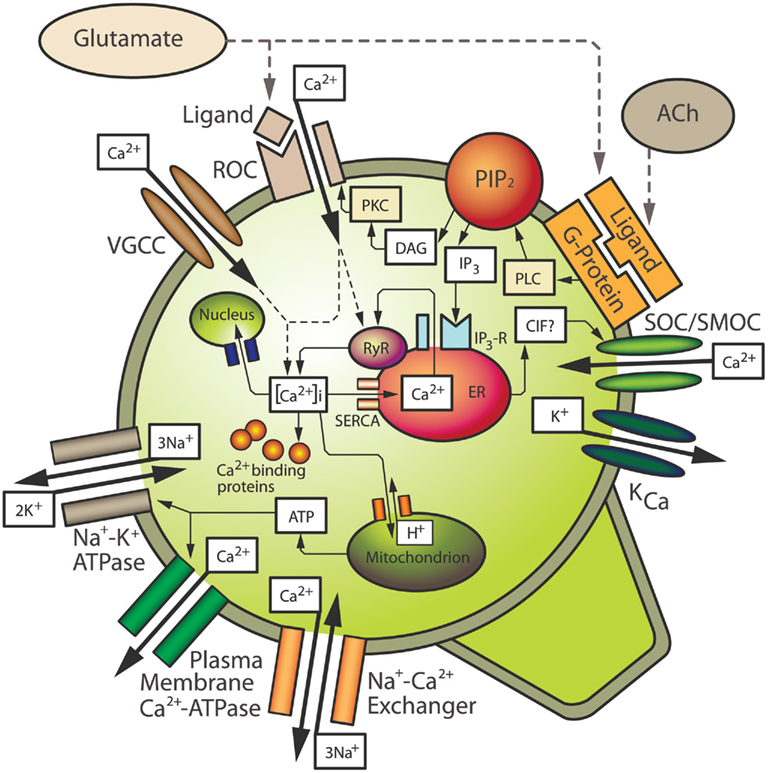
Figure 1. Mechanisms contributing to intracellular calcium homeostasis and signaling in neurons. See text for details. ACh, acetylcholine; CIF, calcium influx factor; DAG, diacylglycerol; ER, endoplasmic reticulum; IP3, inositol 1,4,5-trisphosphate; IP3-R, IP3 receptor; KCa, calcium-sensitive potassium channel; PIP2, phosphatidylinositol bisphosphate; PKC, protein kinase C; PLC, phospholipase C; ROC, receptor-operated channel; RyR, ryanodine receptor; SERCA, sarcoplasmic–endoplasmic reticulum Ca2+-ATPase; SOC/SMOC, store-operated/second messenger-operated channel; VGCC, voltage-gated calcium channel.
In order to activate various signal transduction mechanisms leading to a physiological response, an elevation in [Ca2+]i is often required. Neurons have various ways of increasing [Ca2+]i. One such way is by ROCs, which are Ca2+ ion channels activated by ligands (Figure 1). An important example of a ROC is the N-methyl-D-aspartate (NMDA) receptor. Activation of NMDA receptors by the endogenous ligand, glutamate, leads to the opening of its associated ion channel and an influx of Ca2+ from the extracellular space into the cytosol. Another major route of Ca2+ influx into neurons is through several types of VGCCs (Mintz et al., 1992; Snutch and Reiner, 1992; Takahashi and Momiyama, 1993; Westenbroek et al., 1998; Iwasaki et al., 2000; Schmolesky et al., 2002; Cain and Snutch, 2011). The activation of VGCCs is dependent on the membrane voltage of the cell and they are generally in a closed state when the neuronal membrane is polarized. When the membrane becomes depolarized, e.g., due to Na+ influx through ion channels or due to Ca2+ influx through ROCs, these VGCCs allow the influx of Ca2+ into the cell. An individual neuron may have several different types of VGCCs and ROCs, and Ca2+ influx through specific channels is quite important. For example, certain classes of VGCCs trigger neurotransmitter release at synaptic terminals (Robitaille et al., 1990; Augustine et al., 1991; Robitaille et al., 1993; Wu et al., 1998), and specific ionotropic glutamate receptors can lead to Ca2+-dependent gene induction (Bading et al., 1993; Lerea and McNamara, 1993; Barish, 1998).
Another important mechanism by which neurons increase [Ca2+]i is through the release of Ca2+ from intracellular stores located on the endoplasmic reticulum (ER). As with Ca2+ influx, an elevation of [Ca2+]i through the release of stored Ca2+ is also believed to play an important role in the normal physiological function of neurons (Kostyuk and Verkhratsky, 1994). The ER consists of inositol 1,4,5-trisphosphate (IP3)-sensitive Ca2+ stores, and ryanodine-sensitive Ca2+ stores, so named because of their sensitivity to the plant alkaloid ryanodine, which is generally an agonist at low doses and an antagonist at higher doses (Ehrlich et al., 1994). Ryanodine receptors (RyRs) are associated with Ca2+-induced Ca2+ release (CICR) channels. Both IP3-sensitive stores and ryanodine-sensitive stores have been identified in neuronal somata and dendrites in various areas of the brain including the hippocampus, cerebellum, and the neocortex (Challiss et al., 1990; Sharp et al., 1993; Simpson et al., 1996; Kurumatani et al., 1997; Nakashima et al., 1997; Oberdorf et al., 1997).
Release of Ca2+ from IP3-sensitive stores occurs upon the activation of G protein-coupled receptors that activate phospholipase C (PLC), e.g., Group I metabotropic glutamate receptors (mGluRs), or the M1 or M3 type of acetylcholine (ACh) receptors. Upon activation, PLC cleaves phosphatidylinositol bisphosphate (PIP2) from cell membranes, resulting in the release of diacylglycerol (DAG) and IP3 (Figure 1). IP3 binds to receptors (IP3Rs) on IP3-sensitive intracellular Ca2+ stores located on the ER, resulting in release of Ca2+ (Berridge, 1993; Ozawa et al., 1998). In addition, depletion of Ca2+ stores stimulates influx of extracellular Ca2+ through store-operated or second messenger-operated channels (SOCs/SMOCs) in the plasma membrane, a process known as “capacitative calcium influx” (CCI) as originally postulated by Putney (1986). Capacitative Ca2+ influx is analogous with a capacitor in an electrical circuit in which Ca2+ stores prevent entry when they are filled but promote entry when they become depleted. Capacitative Ca2+ entry has been extensively studied in non-excitable cells (Putney and Bird, 1993; Berridge, 1995), and more recently, capacitative Ca2+ influx has been described in neurons (e.g., Bouron, 2000; Emptage et al., 2001; Weber et al., 2001; Baba et al., 2003; Bouron et al., 2005). Influx likely occurs through members of the transient receptor potential channel (TRPC) family, which is a group of membrane bound ion channels (see Salido et al., 2011 for review). Ca2+ influx through TRPCs thereby represents a way in which neurons can further increase [Ca2+]i. CICR can be triggered by Ca2+ entering through VGCCs or ROCs, however the putative second messenger, cyclic ADP ribose, may also contribute to CICR in neurons (Galione, 1994; Lee, 1994), most likely by increasing the sensitivity of RyR to Ca2+ (Hua et al., 1994). In addition, both IP3Rs and RyRs are sensitive to Ca2+ that has entered through membrane channels or has been released from adjacent receptors on the stores (Kano et al., 1995; Verkhratsky and Shmigol, 1996). A buildup of Ca2+ within the ER also seems to increase the sensitivity of IP3Rs and RyRs (Berridge, 1998). It should also be noted that release of Ca2+ from intracellular stores, or mGluR activation alone, can alter the sensitivity of membrane ROCs or VGCCs, further contributing to Ca2+ elevation in the cytosol (Bruno et al., 1995; Choi and Lovinger, 1996; Rahman and Neuman, 1996; Pisani et al., 2001).
Mechanisms for Decreasing [Ca2+]i Levels
The maintenance of Ca2+ homeostasis in neurons requires mechanisms that can compensate for the influx of extracellular Ca2+ and release of Ca2+ from intracellular stores, once a target protein or biological process has been activated. These mechanisms of lowering [Ca2+]i include a balance of Ca2+ extrusion systems in the plasma membrane, buffering by Ca2+ binding proteins, and uptake and storage by various intracellular organelles (Figure 1). Extrusion of Ca2+ from the cell to the extracellular space occurs by membrane pumps such as the plasma membrane Ca2+-ATPase (PMCA), and a Na+/Ca2+ exchanger which is activated by high levels of [Ca2+]i and is dependent on the Na+ gradient, which in turn is dependent on the membrane Na+/K+ ATPase (Garcia and Strehler, 1999; Wojda et al., 2008; Brittain et al., 2012). In addition to these cell extrusion mechanisms for lowering [Ca2+]i, elevated Ca2+ is buffered by Ca2+ binding proteins in the cytosol (Baimbridge et al., 1992; Racay and Lehotsky, 1996), which is believed to be the major way of decreasing elevated [Ca2+]i in neuronal cells (Neher and Augustine, 1992; Zhou and Neher, 1993). Some cytoplasmic proteins, such as calbindin and parvalbumin, act primarily as Ca2+ acceptors; these proteins however, are thought to deliver Ca2+ ions to sites of extrusion, such as the PMCA, or to sites of storage such as the ER. The plasma membrane also contains several Ca2+ binding proteins, which can buffer Ca2+ but also play a structural role in the cell by contributing to the maintenance of cytoskeletal elements (Baimbridge et al., 1992; Racay and Lehotsky, 1996). Some of these buffering proteins may also play a physiological role in addition to being able to reduce [Ca2+]i. For example, calmodulin, once bound to Ca2+, can activate several protein kinases, which can trigger a wide variety of physiological responses in neurons (Braun and Schulman, 1995).
Although Ca2+-binding proteins can buffer much of the Ca2+ in neurons, these proteins can become saturated. Therefore, neurons also sequester Ca2+ into intracellular organelles such as the ER, mitochondria, and the nucleus in order to maintain homeostatic levels of [Ca2+]i (Miller, 1991; Kostyuk and Verkhratsky, 1994). However, excess Ca2+ in the mitochondria can lead to altered energy metabolism (Budd and Nicholls, 1996; Schinder et al., 1996; White and Reynolds, 1996) and elevated Ca2+ in the nucleus can activate endonucleases, which can cause DNA damage (Trump and Berezesky, 1995; McConkey and Orrenius, 1996). Therefore, these storage methods may only be utilized as secondary mechanisms in cases of excess [Ca2+]i levels not achieved under normal physiological circumstances. In addition to contributing to an elevation of [Ca2+]i for signal transduction events, Ca2+ stores on the ER also take up Ca2+ from the cytosol in order to return [Ca2+]i to normal resting levels. This is accomplished by a sarcoplasmic–endoplasmic reticulum Ca2+-ATPase (SERCA) on the ER membrane (Figure 1). Once stores of the ER are filled, Ca2+ is again available for second messenger signaling events mediated by IP3 and CICR. Based on the essential role that calcium ions play in normal neuronal function, it is easy to envision the profound effects that an imbalance in Ca2+ homeostasis would have in these cells, which ultimately could be manifested at a behavioral level. Also, given the complexity of the mechanisms by which neurons maintain [Ca2+]i within a very small range, an alteration to just one component of the homeostatic system could result in altered [Ca2+]i levels and abnormal physiology.
Role of Elevated Calcium in the Pathology of TBI
Mechanisms of Elevated Intracellular [Ca2+]i Levels after TBI
One of the major causes of secondary injury following TBI is likely related to an excessive influx of Ca2+ into cells of the brain, which can cause cell death, especially after moderate or severe TBI (Young, 1992; Farooqui and Horrocks, 1994). This original “calcium hypothesis” for TBI is based in part on findings that Ca2+ in the cerebrospinal fluid (CSF) decreases from 1 to 0.01 mM following traumatic injury (Young, 1992; Nilsson et al., 1993), and that total tissue 45Ca2+ uptake is increased after injury in vivo (Hovda et al., 1992; Fineman et al., 1993). The hypothesis is also based on previous evidence from other studies in the CNS not directly related to TBI. For example, Schlaepfer and Bunge (1973) showed that amputated axons in tissue culture would degenerate only in the presence of extracellular Ca2+. Additional studies demonstrated that the toxicity of excitatory amino acids appeared to be Ca2+-dependent (Berdichevsky et al., 1983; Choi, 1985; Garthwaite et al., 1986). Therefore, the prevailing hypothesis three decades ago was that Ca2+ overload following head injury triggered a cascade of events leading to cell damage and eventual cell death.
There are several mechanisms by which neuronal Ca2+ becomes elevated following TBI (see Figure 1). Because TBI is caused by a physical insult it is widely proposed that a major route of Ca2+ entry is through plasma membrane pores formed by tear-shearing forces during the primary injury (Maxwell et al., 1993; Gennarelli and Graham, 1998; Büki et al., 1999). This concept is supported by the increased permeability of neuronal membranes to large molecules following experimental TBI in vivo (Pettus et al., 1994; Pettus and Povlishock, 1996) and after mechanical stretch in vitro (McKinney et al., 1996; Pike et al., 2000; Slemmer et al., 2002). In addition to producing pores in cell membranes, mechanical strain can cause disturbances to existing membrane proteins, such as voltage-gated Na+ channels, which are known to be sensitive to mechanical stress (Shcherbatko et al., 1999; Tabarean et al., 1999). An increased influx of Na+ into the cell would cause membrane depolarization, which could then activate VGCCs leading to Ca2+ influx (Wolf et al., 2001). Also, a high level of Na+ in the cell could cause the Na+/Ca2+ exchanger to operate in reverse, further contributing to a buildup of Ca2+ inside the cell (Stys et al., 1992; Kiedrowski et al., 1994; Stys and Lopachin, 1998; Brittain et al., 2012). Membrane depolarization causes the release of neurotransmitters such as glutamate and ACh from presynaptic terminals. The extracellular concentration of glutamate has long been known to be elevated after TBI (Faden et al., 1989; Katayama et al., 1990; Palmer et al., 1994; Zauner et al., 1996) and levels of ACh are also increased in CSF (Hayes et al., 1992).
Upon release, glutamate can activate a variety of functionally distinct receptors on neurons in the CNS (for reviews, see Ozawa et al., 1998; Coutinho and Knöpfel, 2002; Slemmer et al., 2005). These include ionotropic receptors such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA), kainate, and NMDA receptors, as well as mGluRs. NMDA receptors are well-distributed in the brain, and over-activation of this receptor subtype is thought to be a major route of Ca2+ influx into cells. Under normal resting membrane potential, NMDA receptors are subject to a blockade of its ion pore by Mg2+, even in the presence of glutamate and glycine, which is a coagonist of the receptor. When the cell membrane becomes depolarized, the Mg2+ block is decreased or lost leading to Ca2+ influx through the ion pore of the receptor. NMDA receptors are also sensitive to mechanical deformation, as membrane stretch causes potentiated responses mediated by these receptors (Paoletti and Ascher, 1994; Casado and Ascher, 1998). Stretch injury also causes a reduction in the voltage-dependent Mg2+ blockade of NMDA receptors, which leads to elevated [Ca2+]i levels (Zhang et al., 1996). AMPA receptors probably play less of a role in Ca2+ influx into neurons following injury than do NMDA receptors, because these channels are primarily permeable to Na+ (Ozawa et al., 1998). However, some types of neurons do express Ca2+-permeable AMPA receptors (Iino et al., 1990; Lu et al., 1996; Carriedo et al., 1998). AMPA receptors are also susceptible to stretch injury, which causes an enhancement in current through these channels (Goforth et al., 1999). Increased Na+ flux through AMPA receptors could indirectly lead to further Ca2+ influx by membrane depolarization, and a corresponding increase in the activation of NMDA receptors and VGCCs. There are also several types of mGluRs, divided into three groups, which are activated directly by binding of glutamate (see Coutinho and Knöpfel, 2002 for a review); however only Group I mGluRs produce a direct increase in [Ca2+]i. Once activated, these G protein-coupled receptors cause an increase in IP3 which binds to IP3Rs on the ER, resulting in a release of Ca2+ from the stores (Figure 1). This depletion of Ca2+ stores also stimulates influx of extracellular Ca2+ through channels (SOCs/SMOCs) in the plasma membrane in neurons (Bouron, 2000; Weber et al., 2001; Baba et al., 2003), which are likely members of the TRPC family. In addition, some studies have shown that activation of mGluRs coupled to intracellular Ca2+ stores can enhance [Ca2+]i elevation mediated by NMDA receptors (Bruno et al., 1995; Rahman and Neuman, 1996; Pisani et al., 2001), and mGluRs in cerebellar Purkinje cells can directly couple to TRPC1, to cause Ca2+ influx through these channels (Kim et al., 2003). Besides glutamate, ACh can also activate G protein receptors coupled to IP3 (muscarinic receptor types 1 and 3) and contribute to intracellular Ca2+ elevation. ACh also activates nicotinic receptors on some neurons, which are permeable to both Ca2+ and Na+ (Le Novère et al., 2002). As with other types of Na+ channels, influx of Na+ leads to depolarization and subsequent activation of other types of Ca2+ channels. Elevated Ca2+ levels can also cause over-activation of Ca2+-sensitive potassium channels (KCa), which could alter the membrane potential of cells and alter signaling cascades (Wulff et al., 2007).
Elevated intracellular Ca2+ after TBI may also be a result of reduced ATP levels. This may be due to a reduction in glucose levels from decreased CBF, or direct mechanical damage to mitochondria (Slemmer et al., 2008). For example, mechanically induced injury can lead to altered mitochondrial membrane potential and decreased ATP levels in neurons in the absence of hypoxia (Tavalin et al., 1995, 1997; Ahmed et al., 2000, 2002). A reduction in ATP is a serious consequence for neurons, i.e., as the supply of ATP to the PMCA and the SERCA of intracellular stores is reduced, the cells are unable to pump Ca2+ either into the extracellular space or into stores and thus Ca2+ accumulates in the cytosol (Parsons et al., 1997; Limbrick et al., 2001). Other transmembrane pumps are compromised, such as the Na+/K+-ATPase, furthering contributing to a breakdown of the ionic gradient of the membrane (Tavalin et al., 1997). The cell becomes further depolarized, again with subsequent activation of glutamatergic ion channels and VGCCs, leading to elevated [Ca2+]i. This can initiate a cyclical process whereby many depolarized neurons release additional glutamate leading to further over-activation of all classes of glutamate receptors on adjacent cells.
Mechanisms of Calcium-Mediated Cell Damage and Death
Elevated neuronal [Ca2+]i leads to damage and death through several mechanisms following TBI; the activation of Ca2+-dependent enzymes and Ca2+ overload of the mitochondria appearing to be major causes of neuronal damage (for reviews, see Trump and Berezesky, 1996; Kermer et al., 1999; Weber, 2004; Slemmer et al., 2008). Depending on the Ca2+ load and route of Ca2+ entry, these mechanisms can cause cell death by rapid necrotic means or by a more delayed cell death (apoptosis).
Several cysteine proteases, such as calpains, are activated by high levels of [Ca2+]i. In neurons, specific calpain isoforms are activated in the low micromolar [Ca2+]i range, a level that is achieved by hypoxic or excitotoxic insults, and is believed to be reached after TBI (Wang and Yuen, 1994; Tymianski and Tator, 1996). Several cytoskeletal elements serve as protein substrates for calpain, including spectrin, tubulin, tau, microtubule-associated protein, and neurofilaments (Wang and Yuen, 1994; Kermer et al., 1999). Calpain is also reported to be involved in the degradation of membrane proteins such as glutamate receptors and transporters (Takahashi, 1990). Cell-adhesion molecules and other enzymes such as protein kinases and phosphatases can also serve as substrates for calpain (Takahashi, 1990). Therefore, injury-induced over-activation of calpain can lead to abnormal and possibly irreversible changes to the membrane and cytoskeleton of cells in the CNS (Wang and Yuen, 1994; Bartus, 1997). These changes can compromise the structural integrity of the cell, leading to increased membrane permeability to ions and even macromolecules. More recent data has suggested that calpain activation may not occur due the rapid, initial increase in [Ca2+]i after injury or exposure to high levels of glutamate (Araújo et al., 2010). For example, some studies investigating excitotoxicity have shown that calpain activation occurs only after there is a delayed Ca2+ deregulation in neurons (Gerencser et al., 2009; Brustovetsky et al., 2010). Therefore, calpain activation may occur only after a neuron is physiologically compromised, rather than directly contributing to initial cell damage.
Activation of the caspase gene family is also associated with elevated intracellular Ca2+. The caspases are a family of cysteine proteases that are distinct from the calpains (Toescu, 1998). These proteins undergo proteolytic processing which leads to the induction of apoptotic cascades. Caspases have been intensely studied following TBI, with enhanced activation being noted following trauma (for reviews, see Büki and Povlishock, 2006; Saatman et al., 2010). Ca2+-dependent endonucleases can also be activated following TBI, which generate DNA damage in the form of the orderly chromatin cleavage patterns that are typical of apoptosis or delayed cell death (Trump and Berezesky, 1995; McConkey and Orrenius, 1996; Toescu, 1998).
An increase in [Ca2+]i after injury can activate phospholipases, most notably PLC and cytosolic phospholipase A2 (cPLA2). Both PLC (Wei et al., 1982; Dhillon et al., 1999) and cPLA2 (Shohami et al., 1989) are reported to be elevated after TBI. cPLA2 activation leads to the release and accumulation of free arachidonic acid (AA), which itself may alter membrane permeability and increase ROS (Slemmer et al., 2008). The production of ROS such as the oxygen free radicals, superoxide and hydroxyl radicals is part of the normal metabolism of the brain. Under normal physiological conditions, oxygen free radicals are kept under strict control by cellular defense mechanisms. Following a traumatic episode, however, these defense mechanisms may be compromised leading to an abundance of free radicals which can cause cellular damage by peroxidation of proteins, DNA, and lipid membranes (Slemmer et al., 2008). As previously described, PLC cleaves PIP2 from cell membranes, resulting in the release of DAG and IP3, which can cause release of Ca2+ from intracellular stores, and contribute further to an increase in [Ca2+]i. Injury-induced formation of DAG has been associated with post-traumatic edema (Dhillon et al., 1994, 1995). In addition, DAG activates protein kinase C (PKC), which can modulate other signal transduction pathways. PKC has been shown to be elevated after experimental TBI (Sun and Faden, 1994) and its activation can directly modulate NMDA and VGCCs leading to potentiated activity and an enhanced influx of Ca2+ into neurons (Choi and Lovinger, 1996; Zhang et al., 1996).
Another toxic enzymatic pathway in neurons regulated by Ca2+ is the production of nitric oxide (NO). Upon the activation of NMDA receptors and subsequent Ca2+ influx, Ca2+ binds to the enzyme calmodulin, which then regulates NO synthase (NOS), thereby producing NO from L-arginine (Dawson and Dawson, 1996). Under physiological conditions, the level of NO is low and it contributes to several important biological processes including cerebral vasodilation, neurotransmission, and synaptic plasticity (Dawson and Dawson, 1996). However, excessive NO can combine with superoxide to produce peroxynitrite (ONOO−), which is highly toxic to proteins, membrane lipids, and DNA (Dawson, 1999; Liu et al., 2002; Slemmer et al., 2008). NOS itself produces superoxide, but levels are generally kept in check by endogenous free radical scavengers like superoxide dismutase (SOD). However following trauma, when there may be over-activation of NOS and additional mechanisms producing superoxide (i.e., metabolism of AA and damage to mitochondria), endogenous scavengers like SOD may become saturated, allowing high levels of peroxynitrite to form, ultimately leading to neuronal damage.
Calcium Load Versus Source Specificity
The available current evidence strongly supports the fact that intracellular Ca2+ levels are elevated following traumatic neuronal injury. However, the levels of Ca2+ in cells may differ based on the severity of injury and the temporal Ca2+ dynamics may also depend on the level of TBI. The exact mechanisms by which Ca2+ causes neuronal dysfunction are obviously complex, and exactly how Ca2+ causes toxicity to neurons is still not completely understood. One of the original major hypotheses in this field postulated that there is simply a correlation of neuronal death with the amount of Ca2+ that enters the cytosol (Hartley et al., 1993; Eimerl and Schramm, 1994; Lu et al., 1996). This “calcium load” hypothesis is in line with the original calcium hypothesis for TBI, suggesting that after a traumatic insult, neurons are simply overwhelmed with a high Ca2+ load that activates all of the processes discussed previously. However, additional data led to the formation of the refined “source specificity” hypothesis, which suggests that Ca2+-induced neurotoxicity is caused by specific pathways of influx (Sattler and Tymianski, 2000), or by release of Ca2+ from distinct intracellular sources such as the ER (Paschen, 2000). Much of the data in support of this latter hypothesis has been generated by studies of excitotoxicity or ischemia in neurons, however, because TBI can contain both excitotoxic and ischemic components, this idea can be translated to traumatic neuronal injury.
Determining Ca2+ accumulation in cells or measuring [Ca2+]i in vivo is technically challenging, therefore most studies aimed at achieving these goals have been conducted in vitro. Several reports using cultured neuronal cells have exhibited a strong relationship between the intracellular Ca2+ load and neuronal death following excitotoxic or ischemic challenges (e.g., Manev et al., 1989; Marcoux et al., 1990; Hartley et al., 1993; Eimerl and Schramm, 1994; Lu et al., 1996). However, several other studies have resulted in findings that seem to oppose this correlation between Ca2+ concentration and cell death. For example, Turner et al. (2002) showed that reducing intracellular Ca2+ levels induces injury in developing neurons in vitro and in vivo. In a model of neurite transection, injury caused cell death but was independent of the level of extracellular Ca2+ in rat sympathetic ganglion cells (Chu et al., 2001). Important studies conducted by Nilsson et al. (1993, 1996) in vivo demonstrated a decrease in extracellular Ca2+ after injury and a resulting increase in tissue 45Ca2+ uptake, however, neuronal damage did not correlate with the extent of intracellular Ca2+ accumulation (Nilsson et al., 1996).
Due to these conflicting findings, several investigators have suggested that cell death is due to the distinct type of Ca2+ pool that is elevated. Despite the evidence that intracellular Ca2+ is elevated in cells of the CNS after trauma, the relative distribution of Ca2+ throughout the cell after traumatic injury is still unknown. Much of the Ca2+ influx after injury may be buffered by the plasma membrane, cytoskeletal elements, and by cytoplasmic proteins such as calmodulin, calbindin, and parvalbumin (Baimbridge et al., 1992), which could provide rapid protection from such dramatic elevations in intracellular Ca2+ levels. Another major pool that absorbs a high Ca2+ load under times of stress is the mitochondria (Kristián and Siesjö, 1998), and as discussed earlier, this uptake of Ca2+ by mitochondria and resulting energy failure may ultimately lead to neuronal degeneration.
An essential form of Ca2+ within the cell is [Ca2+]i, the type of Ca2+ necessary to activate the degradative enzymes proposed to lead to neuronal injury and death. Several studies using in vitro models of mechanical stretch or shear strain have measured levels of [Ca2+]i after injury in neuronal cell lines (Cargill and Thibault, 1996; LaPlaca et al., 1997; LaPlaca and Thibault, 1998; Geddes and Cargill, 2001; Tárnok and Ulrich, 2001), primary cultures of cortical neurons (Weber et al., 1999, 2001, 2002; Spaethling et al., 2008; Shahlaie et al., 2010; Staal et al., 2010; Goforth et al., 2011), and hippocampal neurons (Geddes et al., 2003; Lusardi et al., 2004). These studies generally show that [Ca2+]i is acutely elevated following a mechanical insult, and although somewhat dependent on the strain level, estimates of [Ca2+]i levels after injury are generally in the nanomolar range. However, most of these studies used the high affinity Ca2+ indicator fura-2 for [Ca2+]i measurements and fura-2 may underestimate [Ca2+]i levels when reaching the micromolar range, which is considered to be a toxic level (Hyrc et al., 1997; Stout and Reynolds, 1999). Therefore, it is possible that [Ca2+]i does in fact reach micromolar levels following traumatic injury. Despite the absolute levels achieved, [Ca2+]i returns to basal levels within a few hours after injury in cortical neurons and remains normal through 24 h except after very high magnitudes of stretch (Weber et al., 1999). A topic of considerable debate is whether a sustained level of [Ca2+]i is needed to cause neurotoxicity, or if a rapid, transient increase is enough to activate degradative enzymes and other damaging mechanisms, even if [Ca2+]i levels return to normal. A sustained elevation of [Ca2+]i would likely be detrimental to neurons, however it is also possible that an initial transient elevation of [Ca2+]i is sufficient to cause neuronal death. Some cells also exhibit a delayed secondary increase in [Ca2+]i levels after glutamate exposure, which represents a loss of the ability to regulate Ca2+ levels, leading to cell death (Glaum et al., 1990; DeCoster et al., 1992; Randall and Thayer, 1992; Tymianski et al., 1993a; Brustovetsky et al., 2010; Brittain et al., 2012). In a few studies, removal of extracellular Ca2+ eliminated the delayed Ca2+ elevation but neurons still died (Tymianski et al., 1993a,b). Therefore, the secondary Ca2+ rise does not appear to cause neuronal death, suggesting that neurodegenerative processes may have been initiated with the first Ca2+ rise. These findings implicate that an initial [Ca2+]i rise may trigger pathways of neuronal death following TBI. However, the effects of Ca2+ on cell death may also be injury dependent, e.g., Lusardi et al. (2004), found that mechanically injured hippocampal cells exhibited increased death in Ca2+-free media at low levels of stretch. In a few recent studies (Deshpande et al., 2008; Sun et al., 2008), [Ca2+]i was measured in hippocampal neurons that were isolated from rats that had received a moderate TBI in vivo. When measured at 24 h or 7 days after injury, [Ca2+]i levels were elevated even in the absence of cell death, which suggests that at least some cells may be able to tolerate long-lasting elevations in [Ca2+]i. Therefore, based on all of the current evidence it is fairly evident that there is no strong correlation with absolute [Ca2+]i levels in neurons and resulting cell death.
Another idea related to Ca2+-mediated neurodegeneration is that this process is regulated through specific Ca2+ influx signaling pathways (Sattler and Tymianski, 2000). This hypothesis is based on the fact that a multitude of data supports the role of specific Ca2+ influx pathways in regulating normal physiological processes. Therefore, therapeutic strategies aimed at completely blocking Ca2+ load may cause alterations to essential physiological processes, even in an injured brain. For example, although antagonists of VGCCs and glutamate receptors have shown some efficacy in experimental TBI (Tymianski and Tator, 1996; Obrenovitch and Urenjak, 1997), in the clinic, these drugs have not been successful (Reinert and Bullock, 1999; Ikonomidou and Turski, 2002; Schouten, 2007). Sattler et al. (1998) demonstrated that equivalent Ca2+ loads through VGCCs and NMDA receptors do not produce equal toxicity, as influx through NMDA receptors caused neurotoxicity while influx through VGCCs was not toxic. In addition, glutamate-mediated neurotoxicity appears to be due to increases in [Ca2+]i in microdomains located near NMDA receptors (Tymianski et al., 1993a, 1994). Further studies were conducted to specifically target Ca2+ toxicity mediated by NMDA receptors. These receptors are coupled to intracellular signaling molecules through a post-synaptic density protein (PSD-95; Sattler et al., 1999). Specifically, PSD-95 couples NMDA receptors to neuronal NOS (nNOS), which produces NO subsequently leading to neurotoxicity. By uncoupling this pathway, toxicity to cortical neurons triggered through NMDA receptors was significantly attenuated, while normal function of NMDA receptors remained intact (Sattler et al., 1999). When extended to a model of cerebral ischemia, protection from hypoxic damage was elicited when NMDA receptor-PSD-95 interactions were disrupted as late as 1 h after the insult (Aarts et al., 2002). These findings may translate to TBI as well, especially if there is a hypoxic component involved. In another recent study, Shahlaie et al. (2010) showed that blockade of N-type Ca2+ channels specifically was neuroprotective in vitro. Therefore, although VGCC blockade and glutamate receptor antagonism in general has not been useful therapeutically, it is possible that more specific targeting of Ca2+ channels could prove to be a viable means of clinical neuroprotection after TBI.
Another source specificity hypothesis is that Ca2+ released specifically from intracellular stores on the ER is toxic to neurons, and a fair amount of data supports this notion. Release of Ca2+ from internal stores causes apoptosis in developing cerebellar granule neurons (Levick et al., 1995), and depletion of Ca2+ stores is toxic to neuronal cell lines (Nguyen et al., 2002). The injurious effects of Ca2+ in traumatic injury of spinal cord white matter are mediated specifically through RyR and IP3-mediated Ca2+ release (Thorell et al., 2002). Recently, Staal et al. (2010) found that a release of Ca2+ from intracellular stores was associated with degeneration of mechanically injured axons. Pharmacological studies have indicated that dantrolene, which blocks RyR-mediated Ca2+ release, is neuroprotective against various insults. For example, dantrolene is protective against neuronal loss in the hippocampus after in vivo ischemia (Wei and Perry, 1996; Nakayama et al., 2002). Blocking the release of Ca2+ from stores can prevent glutamate and NMDA-induced neurotoxicity even though [Ca2+]i is elevated (Frandsen and Schousboe, 1991; Lei et al., 1992). Traumatically injured hippocampal neurons in vitro are also protected by dantrolene, independent of Ca2+ level (Yoon et al., 1996). In addition to these experimental findings, low molecular weight heparin, an antagonist of IP3-mediated Ca2+ release, has been demonstrated to be neuroprotective in stroke patients (Jonas et al., 1997). Lastly, IP3-induced Ca2+ mobilization causes a corresponding increase in mitochondrial Ca2+ concentration (Rizzuto et al., 1993); therefore, an important interplay seems to exist between these Ca2+ storage mechanisms. Also, NMDA receptor activation can also lead to a significant amount of Ca2+ release from intracellular stores (Mody and MacDonald, 1995). These findings suggest that it is unlikely that a single source of Ca2+ is responsible for neurotoxicity, and that several signaling pathways may contribute to neuronal dysfunction following TBI.
Alterations to Ca2+-Mediated Signal Transduction Pathways
There is strong evidence in support of the concept that abnormal neurotransmission and Ca2+-mediated signal transduction pathways could be responsible for neuronal dysfunction following trauma. After moderate TBI in vivo, extracellular glutamate is cleared within 5 min, but antagonists of glutamate receptors and presynaptic glutamate release inhibitors are efficacious even if administered 30 min after the insult (Obrenovitch and Urenjak, 1997). This finding suggests that immediate release of glutamate and over-activation of glutamatergic receptors alone may not be responsible for cellular damage after TBI, and that alterations in glutamatergic neurotransmission persist after injury. A possible abnormality of neurotransmission at glutamate receptors is a deficient Mg2+ blockade of Ca2+-permeable NMDA receptors. Using an in vitro model of stretch-induced injury (Ellis et al., 1995), a reduction in the voltage-dependent Mg2+ blockade of the NMDA current was discovered in cortical neurons (Zhang et al., 1996). An enhanced elevation of [Ca2+]i in response to NMDA or glutamate is also observed after injury, even if basal [Ca2+]i levels are above normal (Weber et al., 1999). Although the response to these agonists appeared to recover to control levels after injury, 24 h post-injury some of these cells again display enhanced glutamate-mediated responses, suggesting a biphasic enhanced response to glutamate.
Signaling through G protein-coupled receptors that elevate Ca2+ by IP3 production also appears to be perturbed following TBI in vivo (Delahunty et al., 1995) and mechanical injury in vitro (Rzigalinski et al., 1998; Weber et al., 1999, 2001). However, unlike ionotropic glutamate receptors, immediately after stretch injury Ca2+ elevation mediated by mGluRs is actually decreased upon agonist stimulation. When measured at later time points (3 or 24 h post-injury), the mGluR-mediated [Ca2+]i response is potentiated. mGluRs appear to contribute to enhanced NMDA current as modulation of these receptors can attenuate enhanced NMDA currents after injury (Lea et al., 2002). In addition, downstream activation of PKC also contributes to the enhanced NMDA response (Zhang et al., 1996). Signaling through muscarinic ACh receptors is also disrupted following mechanical stretch (Weber et al., 2002), and many neurons that have low [Ca2+]i levels after injury are still unresponsive to M1 receptor agonists, suggesting an uncoupling of signal transduction mechanisms, possibly at the receptor or G protein level. Another interesting finding is that differential levels of [Ca2+]i and responses to muscarinic receptor agonists were observed even within the same culture well of neurons after injury. Similar to these findings, Goforth et al. (1999) showed a decreased desensitization of the AMPA receptor subtype of glutamate receptors in a subpopulation of cortical neurons following stretch injury. A subsequent study showed that this reduced desensitization was mediated through NMDA receptors and activation of Ca2+ calmodulin-dependent protein kinase II (CaMKII; Goforth et al., 2004), which is generally elevated after TBI (Folkerts et al., 2007; Zhang et al., 2012). Interestingly, using another in vitro injury model, cortical neurons display an injury-induced increase in Ca2+-permeable AMPA receptors, which is mediated by CaMKIIα phosphorylation (Spaethling et al., 2008). This form of AMPA receptor upregulation could lead to a greater Ca2+ influx into cells upon activation by glutamate, and would thus be a form of altered Ca2+ signaling.
Disrupted mGluR-mediated Ca2+ signaling also involves alterations in intracellular Ca2+ stores. The status of intracellular Ca2+ stores in neurons can be determined by using SERCA inhibitors, such as thapsigargin, which causes a slow leak of Ca2+ from the stores, resulting in an elevation of [Ca2+]i (Thastrup et al., 1989). Fifteen minutes after stretch-induced injury, thapsigargin-stimulated elevation of [Ca2+]i is abolished in cortical neurons, suggesting that intracellular Ca2+ stores are depleted (Weber et al., 1999). This finding may be due to increased levels of IP3, as has been demonstrated in astrocytes following stretch injury (Floyd et al., 2001). Also, inhibition of PLC maintains Ca2+ replete stores after injury (Weber et al., 2001), as does antagonism of Group I mGluRs (Chen et al., 2004). Taken together, these results suggest that elevated levels of IP3 may maintain Ca2+ stores in an empty state in the early period after injury. Also contributing to store depletion is the possibility of enhanced CICR. After stretch injury, the number of neurons that respond to caffeine, which stimulates CICR, is dramatically reduced (Weber et al., 2002).
When measured at later time points following stretch-induced injury (3 and 24 h), mGluR and thapsigargin-stimulated elevation of [Ca2+]i is potentiated, a response which appears to be due to an enhancement of CCI (Weber et al., 2001) As earlier described, depletion of intracellular Ca2+ stores leads to further influx of extracellular Ca2+ into cells through SOCs/SMOCs on the plasma membrane. The exact mechanism for the activation of CCI is still a topic of considerable debate. The “conformational coupling” hypothesis as first proposed by Irvine (1990) suggests a direct physical connection between the IP3 receptor and plasma membrane channels which controls CCI. When the IP3 receptor is activated, it is proposed that an alteration in the physical coupling causes influx of Ca2+ into the cell via plasma membrane channels. An alternate hypothesis suggests that when IP3-sensitive stores are activated and depleted of Ca2+, a diffusible messenger termed “calcium influx factor” (CIF) is released from the stores and binds to SOCs/SMOCs on the plasma membrane and activates CCI (Randriamampita and Tsien, 1993; Rzigalinski et al., 1999). There are several findings in support of a physical coupling and also the possible involvement of a secretory mechanism in CCI (Kiselyov et al., 1998, 1999; Patterson et al., 1999; Yao et al., 1999), however there is also evidence of a soluble CIF that may be produced from the release of AA from phospholipid stores by cPLA2 and subsequent cytochrome P450 metabolism of AA (Rzigalinski et al., 1999). Using the SOC/SMOC antagonist SKF 96365, the potentiation in thapsigargin-stimulated elevation of [Ca2+]i at 3 h post-injury in neurons is blocked, a result that mirrored findings in Ca2+-free conditions (Weber et al., 2001). This delayed enhancement in store-mediated signaling could have detrimental effects on neuronal physiology and may be due to alterations in CIF production, the sensitivity of SOCs/SMOCs to CIF, a damaged physical connection of the stores with the plasma membrane, or to a combination of these factors. As previously mentioned, SOC/SMOCs are likely members of the TRPC family, and they contribute to cell death following excitotoxic insults (Aarts and Tymianski, 2005). These channels therefore represent potential future therapeutic targets.
Ca2+-mediated intercellular signaling may also be perturbed after traumatic neuronal injury. Neurons in pure culture often display spontaneous oscillations in [Ca2+]i that are generally considered to be indicative of synaptic connectivity and intercellular communication (Wang and Gruenstein, 1997). Immediately after stretch injury, oscillations in [Ca2+]i were inhibited in pure neuronal cultures (unpublished data), therefore injury may inhibit intercellular communication in neurons. This effect was later observed in neurons grown in the presence of glia, where the amplitude of neuronal oscillations was decreased following injury (Goforth et al., 2011). Interestingly, Geddes-Klein et al. (2006) showed that pharmacological induction of neuronal oscillations with bicuculline (a GABAA receptor antagonist) resulted in a lower [Ca2+]i transient induced by stretch, which conferred protection. It is known that IP3-sensitive stores play an important role in intercellular communication between neurons, but also between neurons and glia (Berridge, 1993; Sanderson, 1996; Winder and Conn, 1996). Intercellular waves appear to be mediated by the diffusion of IP3, thereby providing a mechanism of multicellular cooperativity (Sanderson, 1996). In relationship to this, inositol polyphosphates are released from injured astrocytes into the extracellular medium, suggesting that release of IP3 from glial cells could affect neighboring neurons (Floyd et al., 2001). Gap junctions may also play a role in the injured brain, e.g., gap junctions can increase the vulnerability of hippocampal neurons to in vitro trauma (Frantseva et al., 2002). In a finding opposite to this, blocking gap junctions can increase glutamate-induced toxicity in glial-neuron co-cultures from mouse cortices (Ozog et al., 2002). Therefore, the exact role of gap junctions in contributing to secondary injury appears to be complex.
Physiological Consequences of Altered Signal Transduction Mechanisms
It is not conclusively known whether neurons displaying altered Ca2+ signaling in the early post-traumatic period eventually die after injury, while those that display or have recovered normal signaling survive. It is likely that many cells that survive TBI have a prolonged alteration in their physiology, and this may contribute to functional impairment after a traumatic insult (Cohen et al., 2007). Recent evidence supports this notion, e.g., Witgen et al. (2005) demonstrated alterations in inhibitory synaptic transmission in the hippocampus after TBI in vivo. As previously discussed, Sun et al. (2008) and Deshpande et al. (2008) measured [Ca2+]i levels in hippocampal neurons at 1, 7, or 30 days after fluid percussion injury in rats. [Ca2+]i levels were elevated at 1 and 7 days post-injury, and these animals also displayed cognitive deficits on these days as measured by Morris water maze performance. At 30 days post-injury [Ca2+]i levels were no longer significantly elevated and cognitive function had also returned to normal. This is strong evidence of a correlation between Ca2+ homeostasis and cognitive function. Extended disruption of intracellular signal transduction could alter the normal response to neurotransmitters such as glutamate and ACh, which may have detrimental consequences for cells due to their importance in vital functions such as the control of neuronal membrane excitability, intercellular communication, and further neurotransmitter release. Injury of cortical neurons in vitro has shown that synaptic transmission is in fact depressed at 4 h and 2 days post-injury (Goforth et al., 2011), which could have long-term effects on plasticity. [Ca2+]i signaling also plays a pivotal role in the maintenance of cytoskeletal integrity in neurons, so a sustained alteration in neuronal physiology may affect spine morphology, which could have profound effects on synaptic communication (Slemmer et al., 2005). Consistent with this idea is the finding that cerebellar cells exhibit synaptic alterations after TBI in vivo (Ai et al., 2007).
Specific disruption of Ca2+ signaling through intracellular stores may also contribute to abnormal neuronal function, as depletion of IP3-sensitive Ca2+ stores and inhibition of IP3-mediated signaling can inhibit neurite extension and axon guidance (Takei et al., 1998). Thus, interference with IP3-mediated signaling may inhibit neuronal recovery and plasticity after injury. Group I mGluRs coupled to IP3-sensitive stores are reported to modulate VGCCs in cortical neurons, thereby contributing to the control of neuronal membrane excitability (Choi and Lovinger, 1996). IP3-sensitive Ca2+ stores also play a role in modulating pre- and post-synaptic neurotransmission. Group I mGluR are reported to both amplify (Bruno et al., 1995; Rahman and Neuman, 1996; Pisani et al., 2001) and attenuate (Yu et al., 1997) elevation of [Ca2+]i elicited by NMDA receptors. IP3-linked receptors also contribute to the modulation of neurotransmitter release (Ozawa et al., 1998). The physiological functions of CICR in neurons are not as well understood, however, evidence suggests that quantal release of Ca2+ from ryanodine-sensitive stores may contribute to Ca2+ oscillations or waves in neurons (Bootman and Berridge, 1995; Verkhratsky and Shmigol, 1996). These mechanisms may serve as spatial or temporal signals contributing to physiological functions within cells, or they may contribute to intercellular communication. CICR may also facilitate the release of neurotransmitters in hippocampal neurons (Savic and Sciancalepore, 1998; Sharma and Vijayaraghavan, 2003). Therefore, altered Ca2+ store-mediated signaling could have adverse effects on neuronal function.
In addition to altered signal transduction events mediated by release of Ca2+ from internal stores, a low level of Ca2+ in the ER can have further debilitating effects at the subcellular level. Maintenance of Ca2+ homeostasis is important for normal processing of proteins, and Ca2+ store depletion can result in decreased protein synthesis (Doutheil et al., 1997). A low level of Ca2+ in the ER lumen can also cause misfolded proteins and disaggregation of ribosomes (Doutheil et al., 1997; Lehotsky et al., 2003). These observations are also documented during ischemia, which causes disrupted ER Ca2+ homeostasis (Paschen and Doutheil, 1999; Paschen and Frandsen, 2001; DeGracia et al., 2002). Ischemia-induced alterations in Ca2+ store homeostasis also leads to the induction of a cellular stress response, in which several stress-related genes become activated (Paschen and Doutheil, 1999; DeGracia et al., 2002). It is thus possible that documented changes in intracellular stores after traumatic neuronal injury initiate a similar response, leading to dysfunctional protein synthesis after TBI.
Ca2+ is believed to play a major role in activity-dependent changes in synaptic strength in neurons, thought to represent the underlying cellular mechanisms of learning and memory (Lamont and Weber, 2012; also see Kawamoto et al., 2012). An increase in [Ca2+]i in neuronal dendrites through NMDA channels is the critical trigger in the most intensively studied form of synaptic plasticity, hippocampal long-term potentiation (LTP; Bliss and Collingridge, 1993; Malenka and Nicoll, 1999). A rise in cytoplasmic Ca2+ is also required for other types of synaptic plasticity, for example long-term depression (LTD) in the hippocampus (Bear and Abraham, 1996; Reyes and Stanton, 1996), and LTD in the cerebellum, at both parallel fiber (PF-LTD) and climbing fiber (CF-LTD) synapses with Purkinje cells (Hansel and Linden, 2000; Hansel et al., 2001). In addition, CF-LTD is associated with a long-term reduction in calcium transients (Weber et al., 2003), and low levels of Ca2+ influx can result in PF-LTP (Coesmans et al., 2004). Given the important role of Ca2+ in synaptic plasticity, it is logical to assume that altered Ca2+ homeostasis following traumatic neuronal injury could contribute to changes in the ability to induce LTP or LTD, which could underlie memory impairments following trauma (Cohen et al., 2007).
Because of the importance of the hippocampus in memory formation, the majority of research aimed at evaluating synaptic plasticity after TBI has been conducted in this brain region (Albensi, 2001). Several studies have reported an impairment in the induction of hippocampal LTP following experimental TBI in vivo (Miyazaki et al., 1992; Reeves et al., 1995; D’Ambrosio et al., 1998; Sick et al., 1998; Albensi et al., 2000; Sanders et al., 2000; Schwarzbach et al., 2006). However, the effects of TBI on hippocampal LTD are less clear, with some studies showing either no effect (D’Ambrosio et al., 1998) or an enhancement in LTD following injury (Albensi et al., 2000). The underlying cellular mechanisms by which different forms of synaptic plasticity are altered following TBI are not understood. It is possible that perturbed Ca2+ signaling contributes to these alterations. For example, the release of Ca2+ from internal stores contributes to synaptic plasticity in the hippocampus and cerebellum (Svoboda and Mainen, 1999; Mattson et al., 2000; Rose and Konnerth, 2001), thus altered Ca2+ store-mediated signaling could impair the ability to induce LTP or LTD. Blockade of CCI has been shown to attenuate hippocampal LTP (Baba et al., 2003). In hippocampal neurons, CCI may directly activate CaMKII (Scholz and Palfrey, 1998), which plays a vital role in the induction of LTP. Therefore, altered CCI may also contribute to changes in synaptic plasticity after injury. Alterations in synaptic plasticity following TBI could have long-lasting effects in the brain, which may be manifested at the behavioral level. Accordingly, a further understanding of the underlying mechanisms that contribute to injury-induced changes in hippocampal plasticity is warranted, as well as additional studies on the effects of TBI on synaptic plasticity in other brain areas, such as the cerebellum and cortex.
Conclusion
The relationship of the calcium ion to neurotoxicity has been intensely studied in the past decades. Originally, it appeared that a high Ca2+ load within neurons was solely responsible for cell damage and death following insults such as ischemia, excitotoxicity, and TBI. Therefore, therapeutic strategies were aimed at inhibiting Ca2+ influx into cells. However, the failure of VGCC and glutamate receptor antagonists in the clinical setting demonstrates that treatment strategies need to be redesigned. Research in recent years demonstrates more clearly that altered neuronal Ca2+ homeostasis, rather than absolute intracellular Ca2+ levels per se, may lead to perturbed cellular physiology and neuronal death. Although an elevated level of intracellular Ca2+ can still initiate destructive pathways, this probably occurs at very defined regions within neurons, and the severity of TBI is also likely important in determining the extent of Ca2+-dependent cell death and altered physiology. How do we translate this recently gained knowledge into viable new therapeutic strategies for TBI patients? Drugs aimed at specific intracellular Ca2+ signaling pathways or subcellular components show some promise for the future treatment of TBI, as well as other disease states of the brain such as neurodegenerative disorders. These agents may be particularly useful for mild TBI, i.e., concussion, where altered physiology in surviving cells may be a more prominent portion of pathology than cell death. The goal of future therapeutic agents should not only be to inhibit cell death, but to establish normal neuronal physiology in cells that do not die following a traumatic episode.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I am currently supported by research grants from the Natural Sciences and Engineering Research Council (NSERC) and the Canada Foundation for Innovation. I would also like to thank Suzanne van Niekerk for preparing the figure in this manuscript.
References
Aarts, M., Liu, Y., Liu, L., Besshoh, S., Arundine, M., Gurd, J. W., Wang, Y.-T., Salter, M. W., and Tymianski, M. (2002). Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298, 846–850.
Ahmed, S. M., Rzigalinski, B. A., Willoughby, K. A., Sitterding, H. A., and Ellis, E. F. (2000). Stretch-induced injury alters mitochondrial membrane potential and cellular ATP in cultured astrocytes and neurons. J. Neurochem. 74, 1951–1960.
Ahmed, S. M., Weber, J. T., Liang, S., Willoughby, K. A., Sitterding, H. A., Rzigalinski, B. A., and Ellis, E. F. (2002). NMDA receptor activation contributes to a portion of the decreased mitochondrial membrane potential and elevated intracellular free calcium in strain-injured neurons. J. Neurotrauma 19, 1619–1629.
Ai, J., Liu, E., Park, E., and Baker, A. J. (2007). Structural and functional alterations of cerebellum following fluid percussion injury in rats. Exp. Brain Res. 177, 95–112.
Albensi, B. C. (2001). Models of brain injury and alterations in synaptic plasticity. J. Neurosci. Res. 65, 279–283.
Albensi, B. C., Sullivan, P. G., Thompson, M. B., Scheff, S. W., and Mattson, M. P. (2000). Cyclosporin ameliorates traumatic brain-injury-induced alterations of hippocampal synaptic plasticity. Exp. Neurol. 162, 385–389.
Araújo, I. M., Carreira, B. P., Carvalho, C. M., and Carvalho, A. P. (2010). Calpains and delayed calcium deregulation in excitotoxicity. Neurochem. Res. 35, 1966–1969.
Augustine, G. J., Adler, E. M., and Charlton, M. P. (1991). The calcium signal for transmitter secretion from presynaptic nerve terminals. Ann. N. Y. Acad. Sci. 635, 365–381.
Baba, A., Yasui, T., Fujisawa, S., Yamada, R. X., Yamada, M. K., Nishiyama, N., Matsuki, N., and Ikegaya, Y. (2003). Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J. Neurosci. 23, 7737–7741.
Bading, H., Ginty, D. D., and Greenberg, M. E. (1993). Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260, 181–186.
Baimbridge, K. G., Celio, M. R., and Rogers, J. H. (1992). Calcium-binding proteins in the nervous system. Trends Neurosci. 15, 303–308.
Barish, ME. (1998). Intracellular calcium regulation of channel and receptor expression in the plasmalemma: potential sites of sensitivity along the pathways linking transcription, translation, and insertion. J. Neurobiol. 37, 146–157.
Bartus, R. T. (1997). The calpain hypothesis of neurodegeneration: evidence for a common cytotoxic pathway. Neuroscientist 3, 314–327.
Bear, M. F., and Abraham, W. C. (1996). Long-term depression in hippocampus. Annu. Rev. Neurosci. 19, 437–462.
Berdichevsky, E., Riveros, N., Sanchez-Armass, S., and Orrego, F. (1983). Kainate, N-methylaspartate and other excitatory amino acids increase calcium influx into rat brain cortex cells in vitro. Neurosci. Lett. 36, 75–80.
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39.
Bootman, M. D., and Berridge, M. J. (1995). The elemental principles of calcium signaling. Cell 83, 675–678.
Bouron, A. (2000). Activation of a capacitative Ca(2+) entry pathway by store depletion in cultured hippocampal neurones. FEBS Lett. 470, 269–272.
Bouron, A., Altafaj, X., Boisseau, S., and De Waard, M. (2005). A store-operated Ca2+ influx activated in response to the depletion of thapsigargin-sensitive Ca2+ stores is developmentally regulated in embryonic cortical neurons from mice. Brain Res. Dev. Brain Res. 159, 64–71.
Braun, A. P., and Schulman, H. (1995). The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu. Rev. Physiol. 57, 417–445.
Brittain, M. K., Brustovetsky, T., Sheets, P. L., Brittain, J. M., Khanna, R., Cummins, T. R., and Brustovetsky, N.. (2012). Delayed calcium dysregulation in neurons requires both the NMDA receptor and the reverse Na+/Ca(2+) exchanger. Neurobiol. Dis. 46, 109–117.
Bruno, V., Copani, A., Knöpfel, T., Kuhn, R., Casabona, G., Dell’Albani, P., Condorelli, D. F., and Nicoletti, F. (1995). Activation of metabotropic glutamate receptors coupled to inositol phospholipid hydrolysis amplifies NMDA-induced neuronal degeneration in cultured cortical cells. Neuropharmacology 34, 1089–1098.
Brustovetsky, T., Bolshakov, A., and Brustovetsky, N. (2010). Calpain activation and Na+/Ca2+ exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. J. Neurosci. Res. 88, 1317–1328.
Budd, S. L., and Nicholls, D. G. (1996). Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurochem. 67, 2282–2291.
Büki, A., and Povlishock, J. T. (2006). All roads lead to disconnection?–Traumatic axonal injury revisited. Acta Neurochir. (Wien). 148, 181–193; discussion 193–194.
Büki, A., Siman, R., Trojanowski, J. Q., and Povlishock, J. T. (1999). The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J. Neuropathol. Exp. Neurol. 58, 365–375.
Cain, S. M., and Snutch, T. P. (2011). Voltage-gated calcium channels and disease. Biofactors 37, 197–205.
Cargill, R. S. II, and Thibault, L. E. (1996). Acute alterations in [Ca2+]i in NG108-15 cells subjected to high strain rate deformation and chemical hypoxia: an in vitro model for neural trauma. J. Neurotrauma 13, 395–407.
Carriedo, S. G., Yin, H. Z., Sensi, S. L., and Weiss, J. H. (1998). Rapid Ca2+ entry through Ca2+-permeable AMPA/kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J. Neurosci. 18, 7727–7738.
Casado, M., and Ascher, P. (1998). Opposite modulation of NMDA receptors by lysophospholipids and arachidonic acid: common features with mechanosensitivity. J. Physiol. 513, 317–330.
Centers for Disease Control and Prevention. (2010). Injury Prevention and Control: Traumatic Brain Injury. Available at: http://www.cdc.gov/ncipc/tbi/TBI.htm
Challiss, R. A., Chilvers, E. R., Willcocks, A. L., and Nahorski, S. R. (1990). Heterogeneity of [3H]inositol 1,4,5-trisphosphate binding sites in adrenal-cortical membranes. Characterization and validation of a radioreceptor assay. Biochem. J. 265, 421–427.
Chen, T., Willoughby, K. A., and Ellis, E. F. (2004). Group I metabotropic receptor antagonism blocks depletion of calcium stores and reduces potentiated capacitative calcium entry in strain-injured neurons and astrocytes. J. Neurotrauma 21, 271–281.
Choi, D. W. (1985). Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci. Lett. 58, 293–297.
Choi, S., and Lovinger, D. M. (1996). Metabotropic glutamate receptor modulation of voltage-gated Ca2+ channels involves multiple receptor subtypes in cortical neurons. J. Neurosci. 16, 36–45.
Chu, G. K., Tawadros, P. S., Kulbatski, I., and Tator, C. H. (2001). Death of rat sympathetic ganglion cells in vitro caused by neurite transection: effect of extracellular calcium. J. Neurotrauma 18, 699–710.
Coesmans, M., Weber, J. T., De Zeeuw, C. I., and Hansel, C. (2004). Bidirectional parallel fibre plasticity in the cerebellum under climbing fibre control. Neuron 428, 856–860.
Cohen, A. S., Pfister, B. J., Schwarzbach, E., Grady, M. S., Goforth, P. B., and Satin, L. S. (2007). “Injury-induced alterations in CNS electrophysiology,” in Progress in Brain Research, Vol. 161, Neurotrauma: New Insights into Pathology and Treatment, eds J. T. Weber, and A. I. R. Maas (Amsterdam: Elsevier), 142–169.
Coutinho, V., and Knöpfel, T. (2002). Metabotropic glutamate receptors: electrical and chemical signaling properties. Neuroscientist 8, 551–561.
D’Ambrosio, R., Maris, D. O., Grady, M. S., Winn, H. R., and Janigro, D. (1998). Selective loss of hippocampal long-term potentiation, but not depression, following fluid percussion injury. Brain Res. 786, 64–79.
Dawson, V. L. (1999). Potent neuroprotectants linked to bifunctional inhibition. Proc. Natl. Acad. Sci. U.S.A. 96, 10557–10558.
Dawson, V. L., and Dawson, T. M. (1996). Nitric oxide actions in neurochemistry. Neurochem. Int. 29, 97–110.
DeCoster, M. A., Koenig, M. L., Hunter, J. C., and Tortella, F. C. (1992). Calcium dynamics in neurons treated with toxic and non-toxic concentrations of glutamate. Neuroreport 3, 773–776.
DeGracia, D. J., Kumar, R., Owen, C. R., Krause, G. S., and White, B. C. (2002). Molecular pathways of protein synthesis inhibition during brain reperfusion: implications for neuronal survival or death. J. Cereb. Blood Flow Metab. 22, 127–141.
Delahunty, T. M., Jiang, J. Y., Black, R. T., and Lyeth, B. G. (1995). Differential modulation of carbachol and trans-ACPD-stimulated phosphoinositide turnover following traumatic brain injury. Neurochem. Res. 20, 405–411.
Deshpande, L. S., Sun, D. A., Sombati, S., Baranova, A., Wilson, M. S., Attkisson, E., Hamm, R. J., and DeLorenzo, R. J. (2008). Alterations in neuronal calcium levels are associated with cognitive deficits after traumatic brain injury. Neurosci. Lett. 441, 115–119.
Dhillon, H. S., Carbary, T., Dose, J., Dempsey, R. J., and Prasad, M. R. (1995). Activation of phosphatidylinositol bisphosphate signal transduction pathway after experimental brain injury: a lipid study. Brain Res. 698, 100–106.
Dhillon, H. S., Carman, H. M., and Prasad, R. M. (1999). Regional activities of phospholipase C after experimental brain injury in the rat. Neurochem. Res. 24, 751–755.
Dhillon, H. S., Donaldson, D., Dempsey, R. J., and Prasad, M. R. (1994). Regional levels of free fatty acids and Evans blue extravasation after experimental brain injury. J. Neurotrauma 11, 405–415.
Doutheil, J., Gissel, C., Oschlies, U., Hossmann, K. A., and Paschen, W. (1997). Relation of neuronal endoplasmic reticulum calcium homeostasis to ribosomal aggregation and protein synthesis: implications for stress-induced suppression of protein synthesis. Brain Res. 775, 43–51.
Ehrlich, B. E., Kaftan, E., Bezprozvannaya, S., and Bezprozvanny, I. (1994). The pharmacology of intracellular Ca2+-release channels. Trends Pharmacol. Sci. 15, 145–149.
Eimerl, S., and Schramm, M. (1994). The quantity of calcium that appears to induce neuronal death. J. Neurochem. 62, 1223–1226.
Ellis, E. F., McKinney, J. S., Willoughby, K. A., Liang, S., and Povlishock, J. T. (1995). A new model for rapid stretch-induced injury of cells in culture. J. Neurotrauma 12, 325–339.
Emptage, N. J., Reid, C. A., and Fine, A. (2001). Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29, 197–208.
Faden, A. I., Demediuk, P., Panter, S. S., and Vink, R. (1989). The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244, 798–800.
Farooqui, A. A., and Horrocks, L. A. (1994). Involvement of glutamate receptors, lipases, and phospholipases in long-term potentiation and neurodegeneration. J. Neurosci. Res. 38, 6–11.
Fineman, I., Hovda, D. A., Smith, M., Yoshino, A., and Becker, D. P. (1993). Concussive brain injury is associated with a prolonged accumulation of calcium: a 45Ca autoradiographic study. Brain Res. 624, 94–102.
Finkbeiner, S., and Greenberg, M. E. (1998). Ca2+ channel-regulated neuronal gene expression. J. Neurobiol. 37, 171–189.
Floyd, C. L., Rzigalinski, B. A., Weber, J. T., Sitterding, H. A., Willoughby, K. A., and Ellis, E. F. (2001). Traumatic injury of cultured astrocytes alters inositol (1,4,5)-trisphosphate-mediated signaling. Glia 33, 12–23.
Folkerts, M. M., Parks, E. A., Dedman, J. R., Kaetzel, M. A., Lyeth, B. G., and Berman, R. F. (2007). Phosphorylation of calcium calmodulin-dependent protein kinase II following lateral fluid percussion brain injury in rats. J. Neurotrauma 24, 638–650.
Frandsen, A., and Schousboe, A. (1991). Dantrolene prevents glutamate cytotoxicity and Ca2+ release from intracellular stores in cultured cerebral cortical neurons. J. Neurochem. 56, 1075–1078.
Frantseva, M. V., Kokarovtseva, L., Naus, C. G., Carlen, P. L., MacFabe, D., and Perez Velazquez, J. L. (2002). Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J. Neurosci. 22, 644–653.
Galione, A. (1994). Cyclic ADP-ribose, the ADP-ribosyl cyclase pathway and calcium signalling. Mol. Cell. Endocrinol. 98, 125–131.
Garcia, M. L., and Strehler, E. E. (1999). Plasma membrane calcium ATPases as critical regulators of calcium homeostasis during neuronal cell function. Front. Biosci. 4, D869–D682.
Garthwaite, G., Hajos, F., and Garthwaite, J. (1986). Ionic requirements for neurotoxic effects of excitatory amino acid analogues in rat cerebellar slices. Neuroscience 18, 437–447.
Geddes, D. M., and Cargill, R. S. II (2001). An in vitro model of neural trauma: device characterization and calcium response to mechanical stretch. J. Biomech. Eng. 123, 247–255.
Geddes, D. M., LaPlaca, M. C., and Cargill, R. S. II (2003). Susceptibility of hippocampal neurons to mechanically induced injury. Exp. Neurol. 184, 420–427.
Geddes-Klein, D. M., Serbest, G., Mesfin, M. N., Cohen, A. S., and Meaney, D. F. (2006). Pharmacologically induced calcium oscillations protect neurons from increases in cytosolic calcium after trauma. J. Neurochem. 97, 462–474.
Gennarelli, T. A., and Graham, D. I. (1998). Neuropathology of the head injuries. Semin. Clin. Neuropsychiatry 3, 160–175.
Gerencser, A. A., Mark, K. A., Hubbard, A. E., Divakaruni, A. S., Mehrabian, Z., Nicholls, D. G., and Polster, B. M. (2009). Real-time visualization of cytoplasmic calpain activation and calcium deregulation in acute glutamate excitotoxicity. J. Neurochem. 110, 990–1004.
Ghosh, A., and Greenberg, M. E. (1995). Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268, 239–247.
Glaum, S. R., Scholz, W. K., and Miller, R. J. (1990). Acute- and long-term glutamate-mediated regulation of [Ca++]i in rat hippocampal pyramidal neurons in vitro. J. Pharmacol. Exp. Ther. 253, 1293–1302.
Goforth, P. B., Ellis, E. F., and Satin, L. S. (1999). Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J. Neurosci. 19, 7367–7374.
Goforth, P. B., Ellis, E. F., and Satin, L. S. (2004). Mechanical injury modulates AMPA receptor kinetics via an NMDA receptor-dependent pathway. J. Neurotrauma 21, 719–732.
Goforth, P. B., Ren, J., Schwartz, B. S., and Satin, L. S. (2011). Excitatory synaptic transmission and network activity are depressed following mechanical injury in cortical neurons. J. Neurophysiol. 105, 2350–2363.
Hansel, C., and Linden, D. J. (2000). Long-term depression of the cerebellar climbing fiber – Purkinje neuron synapse. Neuron 26, 473–482.
Hansel, C., Linden, D. J., and D’Angelo, E. (2001). Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat. Neurosci. 4, 467–475.
Hartley, D. M., Kurth, M. C., Bjerkness, L., Weiss, J. H., and Choi, D. W. (1993). Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J. Neurosci. 13, 1993–2000.
Hayes, R. L., Jenkins, L. W., and Lyeth, B. G. (1992). Neurotransmitter-mediated mechanisms of traumatic brain injury: acetylcholine and excitatory amino acids. J. Neurotrauma 9, S173–S187.
Heath, P. R., and Shaw, P. J. (2002). Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 26, 438–458.
Hovda, D. A., Becker, D. P., and Katayama, Y. (1992). Secondary injury and acidosis. J. Neurotrauma 9, S47–S60.
Hua, S. Y., Tokimasa, T., Takasawa, S., Furuya, Y., Nohmi, M., Okamoto, H., and Kuba, K. (1994). Cyclic ADP-ribose modulates Ca2+ release channels for activation by physiological Ca2+ entry in bullfrog sympathetic neurons. Neuron 12, 1073–1079.
Hyrc, K., Handran, S. D., Rothman, S. M., and Goldberg, M. P. (1997). Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. J. Neurosci. 17, 6669–6677.
Iino, M., Ozawa, S., and Tsuzuki, K. (1990). Permeation of calcium through excitatory amino acid receptor channels in cultured rat hippocampal neurones. J. Physiol. 424, 151–165.
Ikonomidou, C., and Turski, L. (2002). Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 1, 383–386.
Irvine, R. F. (1990). ‘Quantal’ Ca2+ release and the control of Ca2+ entry by inositol phosphates – a possible mechanism. FEBS Lett. 263, 5–9.
Iwasaki, S., Momiyama, A., Uchitel, O. D., and Takahashi, T. (2000). Developmental changes in calcium channel types mediating central synaptic transmission. J. Neurosci. 20, 59–65.
Jonas, S., Sugimori, M., and Llinas, R. (1997). Is low molecular weight heparin a neuroprotectant? Ann. N. Y. Acad. Sci. 825, 389–393.
Kano, M., Garaschuk, O., Verkhratsky, A., and Konnerth, A. (1995). Ryanodine receptor-mediated intracellular calcium release in rat cerebellar Purkinje neurones. J. Physiol. 487, 1–16.
Katayama, Y., Becker, D. P., Tamura, T., and Hovda, D. A. (1990). Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 73, 889–900.
Kawamoto, E. M., Vivar, C., and Camandola, S. (2012). Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 3:61. doi:10.3389/fphar.2012.00061
Kermer, P., Klocker, N., and Bahr, M. (1999). Neuronal death after brain injury. Models, mechanisms, and therapeutic strategies in vivo. Cell Tissue Res. 298, 383–395.
Kiedrowski, L., Brooker, G., Costa, E., and Wroblewski, J. T. (1994). Glutamate impairs neuronal calcium extrusion while reducing sodium gradient. Neuron 12, 295–300.
Kim, S. J., Kim, Y. S., Yuan, J. P., Petralia, R. S., Worley, P. F., and Linden, D. J. (2003). Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 426, 285–291.
Kirischuk, S., and Verkhratsky, A. (1996). Calcium homeostasis in aged neurones. Life Sci. 59, 451–459.
Kiselyov, K., Mignery, G., Zhu, M. X., and Muallem, S. (1999). The N-terminal domain of the IP3 receptor gates store-operated hTrp3 channels. Mol. Cell 4. 423–429.
Kiselyov, K., Xu, X., Mozhayeva, G., Kuo, T., Pessah, I., Mignery, G., Zhu, X., Birnbaumer, L., and Muallem, S. (1998). Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature 396, 478–482.
Kostyuk, P., and Verkhratsky, A. (1994). Calcium stores in neurons and glia. Neuroscience 63, 381–404.
Kurumatani, T., Cowburn, R. F., Bogdanovic, N., Winblad, B., and Fastbom, J. (1997). Autoradiographic characterization of [3H]inositol (1,4,5) trisphosphate and [3H]inositol (1,3,4,5) tetrakisphosphate binding sites in human brain. J. Neural Transm. 104, 175–189.
Lamont, M. G., and Weber, J. T. (2012). The role of calcium in synaptic plasticity and motor learning in the cerebellar cortex. Neurosci. Biobehav. Rev. 36, 1153–1162.
LaPlaca, M. C., Lee, V. M., and Thibault, L. E. (1997). An in vitro model of traumatic neuronal injury: loading rate-dependent changes in acute cytosolic calcium and lactate dehydrogenase release. J. Neurotrauma 14, 355–368.
LaPlaca, M. C., and Thibault, L. E. (1998). Dynamic mechanical deformation of neurons triggers an acute calcium response and cell injury involving the N-methyl-D-aspartate glutamate receptor. J. Neurosci. Res. 52, 220–229.
Lau, A., and Tymianski, M. (2010). Glutamate receptors, neurotoxicity and neurodegeneration. Pflügers Arch. 460, 525–542.
Le Novère, N., Corringer, P. J., and Changeux, J. P. (2002). The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J. Neurobiol. 53, 447–456.
Lea, P. M., Custer, S. J., Vicini, S., and Faden, A. I. (2002). Neuronal and glial mGluR5 modulation prevents stretch-induced enhancement of NMDA receptor current. Pharmacol. Biochem. Behav. 73, 287–298.
Lee, H. C. (1994). Cyclic ADP-ribose: a calcium mobilizing metabolite of NAD+. Mol. Cell. Biochem. 138, 229–235.
Lehotsky, J., Kaplan, P., Babusikova, E., Strapkova, A., and Murin, R. (2003). Molecular pathways of endoplasmic reticulum dysfunctions: possible cause of cell death in the nervous system. Physiol. Res. 52, 269–274.
Lei, S. Z., Zhang, D., Abele, A. E., and Lipton, S. A. (1992). Blockade of NMDA receptor-mediated mobilization of intracellular Ca2+ prevents neurotoxicity. Brain Res. 598, 196–202.
Leker, R. R., and Shohami, E. (2002). Cerebral ischemia and trauma-different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res. Brain Res. Rev. 39, 55–73.
Lerea, L. S., and McNamara, J. O. (1993). Ionotropic glutamate receptor subtypes activate c-fos transcription by distinct calcium-requiring intracellular signaling pathways. Neuron 10, 31–41.
Levick, V., Coffey, H., and D’Mello, S. R. (1995). Opposing effects of thapsigargin on the survival of developing cerebellar granule neurons in culture. Brain Res. 676, 325–335.
Limbrick, D. D. Jr., Pal, S., and DeLorenzo, R. J. (2001). Hippocampal neurons exhibit both persistent Ca2+ influx and impairment of Ca2+ sequestration/extrusion mechanisms following excitotoxic glutamate exposure. Brain Res. 894, 56–67.
Liu, P. K., Robertson, C. S., and Valadka, A. (2002). The association between neuronal nitric oxide synthase and neuronal sensitivity in the brain after brain injury. Ann. N. Y. Acad. Sci. 962, 226–241.
Lu, Y. M., Yin, H. Z., Chiang, J., and Weiss, J. H. (1996). Ca(2+)-permeable AMPA/kainate and NMDA channels: high rate of Ca2+ influx underlies potent induction of injury. J. Neurosci. 16, 5457–5465.
Lusardi, T. A., Wolf, J. A., Putt, M. E., Smith, D. H., and Meaney, D. F. (2004). Effect of acute calcium influx after mechanical stretch injury in vitro on the viability of hippocampal neurons. J. Neurotrauma 21, 61–72.
Maas, A. I. R., Dearden, M., Servadei, F., Stocchetti, N., and Unterberg, A. (2000). Current recommendations for neurotrauma. Curr. Opin. Crit. Care 6, 281–292.
Malenka, R. C., and Nicoll, R. A. (1999). Long-term potentiation – a decade of progress? Science 285, 1870–1874.
Manev, H., Favaron, M., Guidotti, A., and Costa, E. (1989). Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol. Pharmacol. 36, 106–112.
Marcoux, F. W., Probert, A. W. Jr., and Weber, M. L. (1990). Hypoxic neuronal injury in tissue culture is associated with delayed calcium accumulation. Stroke 21, III71–III74.
Mattson, M. P., LaFerla, F. M., Chan, S. L., Leissring, M. A., Shepel, P. N., and Geiger, J. D. (2000). Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 23, 222–229.
Maxwell, W. L., Watt, C., Graham, D. I., and Gennarelli, T. A. (1993). Ultrastructural evidence of axonal shearing as a result of lateral acceleration of the head in non-human primates. Acta Neuropathol. (Berl.) 86, 136–144.
McAllister, T. W. (2011). Neurobiological consequences of traumatic brain injury. Dialogues Clin. Neurosci. 13, 287–230.
McConkey, D. J., and Orrenius, S. (1996). The role of calcium in the regulation of apoptosis. J. Leukoc. Biol. 59, 775–783.
McIntosh, T. K., Smith, D. H., Meaney, D. F., Kotapka, M. J., Gennarelli, T. A., and Graham, D. I. (1996). Neuropathological sequelae of traumatic brain injury: relationship to neurochemical and biomechanical mechanisms. Lab. Invest. 74, 315–342.
McKinney, J. S., Willoughby, K. A., Liang, S., and Ellis, E. F. (1996). Stretch-induced injury of cultured neuronal, glial, and endothelial cells. Effect of polyethylene glycol-conjugated superoxide dismutase. Stroke 27, 934–940.
Mintz, I. M., Adams, M. E., and Bean, B. P. (1992). P-type calcium channels in rat central and peripheral neurons. Neuron 9, 85–95.
Miyazaki, S., Katayama, Y., Lyeth, B. G., Jenkins, L. W., DeWitt, D. S., Goldberg, S. J., Newlon, P. G., and Hayes, R. L. (1992). Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Res. 585, 335–339.
Mody, I., and MacDonald, J. F. (1995). NMDA receptor-dependent excitotoxicity: the role of intracellular Ca2+ release. Trends Pharmacol. Sci. 16, 356–359.
Morganti-Kossmann, M. C., Kossmann, T., and Wahl, S. M. (1992). Cytokines and neuropathology. Trends Pharmacol. Sci. 13, 286–291.
Nakamura, T., and Lipton, S. A. (2010). Preventing Ca2+-mediated nitrosative stress in neurodegenerative diseases: possible pharmacological strategies. Cell Calcium 47, 190–197.
Nakashima, Y., Nishimura, S., Maeda, A., Barsoumian, E. L., Hakamata, Y., Nakai, J., Allen, P. D., Imoto, K., and Kita, T. (1997). Molecular cloning and characterization of a human brain ryanodine receptor. FEBS Lett. 417, 157–162.
Nakayama, R., Yano, T., Ushijima, K., Abe, E., and Terasaki, H. (2002). Effects of dantrolene on extracellular glutamate concentration and neuronal death in the rat hippocampal CA1 region subjected to transient ischemia. Anesthesiology 96, 705–710.
Neher, E., and Augustine, G. J. (1992). Calcium gradients and buffers in bovine chromaffin cells. J. Physiol. 450, 273–301.
Nguyen, H. N., Wang, C., and Perry, D. C. (2002). Depletion of intracellular calcium stores is toxic to SH-SY5Y neuronal cells. Brain Res. 924, 159–166.
Nilsson, P., Hillered, L., Olsson, Y., Sheardown, M. J., and Hansen, A. J. (1993). Regional changes in interstitial K+ and Ca2+ levels following cortical compression contusion trauma in rats. J. Cereb. Blood Flow Metab. 13, 183–192.
Nilsson, P., Laurensen, H., Hillered, L., and Hansen, A. J. (1996). Calcium movements in traumatic brain injury: the role of glutamate receptor-operated ion channels. J. Cereb. Blood Flow Metab. 16, 262–270.
Novack, T. A., Dillon, M. C., and Jackson, W. T. (1996). Neurochemical mechanisms in brain injury and treatment: a review. J. Clin. Exp. Neuropsychol. 18, 685–706.
Oberdorf, J., Vallano, M. L., and Wojcikiewicz, R. J. (1997). Expression and regulation of types I and II inositol 1,4,5-trisphosphate receptors in rat cerebellar granule cell preparations. J. Neurochem. 69, 1897–1903.
Obrenovitch, T. P., and Urenjak, J. (1997). Is high extracellular glutamate the key to excitotoxicity in traumatic brain injury? J. Neurotrauma 14, 677–698.
Oliveira, A. M., and Bading, H. (2011). Calcium signaling in cognition and aging-dependent cognitive decline. Biofactors 37, 168–174.
Olney, J. W. (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164, 719–721.
Ozawa, S., Kamiya, H., and Tsuzuki, K. (1998). Glutamate receptors in the mammalian central nervous system. Prog. Neurobiol. 54, 581–618.
Ozog, M. A., Siushansian, R., and Naus, C. C. (2002). Blocked gap junctional coupling increases glutamate-induced neurotoxicity in neuron-astrocyte co-cultures. J. Neuropathol. Exp. Neurol. 61, 132–141.
Palmer, A. M., Marion, D. W., Botscheller, M. L., Bowen, D. M., and DeKosky, S. T. (1994). Increased transmitter amino acid concentration in human ventricular CSF after brain trauma. Neuroreport 6, 153–156.
Paoletti, P., and Ascher, P. (1994). Mechanosensitivity of NMDA receptors in cultured mouse central neurons. Neuron 13, 645–655.
Parsons, J. T., Churn, S. B., and DeLorenzo, R. J. (1997). Ischemia-induced inhibition of calcium uptake into rat brain microsomes mediated by Mg2+/Ca2+ ATPase. J. Neurochem. 68, 1124–1134.
Paschen, W. (2000). Role of calcium in neuronal cell injury: which subcellular compartment is involved? Brain Res. Bull. 53, 409–413.
Paschen, W., and Doutheil, J. (1999). Disturbances of the functioning of endoplasmic reticulum: a key mechanism underlying neuronal cell injury? J. Cereb. Blood Flow Metab. 19, 1–18.
Paschen, W., and Frandsen, A. (2001). Endoplasmic reticulum dysfunction – a common denominator for cell injury in acute and degenerative diseases of the brain? J. Neurochem. 79, 719–725.
Patt, S., and Brodhun, M. (1999). Neuropathological sequelae of traumatic injury in the brain. An overview. Exp. Toxicol. Pathol. 51, 119–123.
Patterson, R. L., van Rossum, D. B., and Gill, D. L. (1999). Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell 98, 487–499.
Petersen, O. H., Michalak, M., and Verkhratsky, A.. (2005). Calcium signalling: past, present and future. Cell Calcium 38, 161–169.
Pettus, E. H., Christman, C. W., Giebel, M. L., and Povlishock, J. T. (1994). Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J. Neurotrauma 11, 507–522.
Pettus, E. H., and Povlishock, J. T. (1996). Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 722, 1–11.
Pike, B. R., Zhao, X., Newcomb, J. K., Glenn, C. C., Anderson, D. K., and Hayes, R. L. (2000). Stretch injury causes calpain and caspase-3 activation and necrotic and apoptotic cell death in septo-hippocampal cell cultures. J. Neurotrauma 17, 283–298.
Pisani, A., Gubellini, P., Bonsi, P., Conquet, F., Picconi, B., Centonze, D., Bernardi, G., and Calabresi, P. (2001). Metabotropic glutamate receptor 5 mediates the potentiation of N-methyl-D-aspartate responses in medium spiny striatal neurons. Neuroscience 106, 579–587.
Putney, J. W. Jr., and Bird, G. S. (1993). The inositol phosphate-calcium signaling system in nonexcitable cells. Endocrinol. Rev. 14, 610–631.
Racay, P., and Lehotsky, J. (1996). Intracellular and molecular aspects of Ca (2+)-mediated signal transduction in neuronal cells. Gen. Physiol. Biophys. 15, 273–289.
Raghupathi, R., and McIntosh, T. K. (1998). Pharmacotherapy for traumatic brain injury: a review. Proc. West. Pharmacol. Soc. 41, 241–246.
Rahman, S., and Neuman, R. S. (1996). Characterization of metabotropic glutamate receptor-mediated facilitation of N-methyl-D-aspartate depolarization of neocortical neurones. Br. J. Pharmacol. 117, 675–683.
Randall, R. D., and Thayer, S. A. (1992). Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J. Neurosci. 12, 1882–1895.
Randriamampita, C., and Tsien, R. Y. (1993). Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature 364, 809–814.
Reeves, T. M., Lyeth, B. G., and Povlishock, J. T. (1995). Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp. Brain Res. 106, 248–256.
Reilly, P. (2007). “The impact of neurotrauma on society: an international perspective,” in Progress in Brain Research, Vol. 161, Neurotrauma: New Insights into Pathology and Treatment, eds J. T. Weber, and A. I. R. Maas (Amsterdam: Elsevier), 3–9.
Reyes, M., and Stanton, P. K. (1996). Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J. Neurosci. 16, 5951–5960.
Rizzuto, R., Brini, M., Murgia, M., and Pozzan, T. (1993). Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262, 744–747.
Robitaille, R., Adler, E. M., and Charlton, M. P. (1990). Strategic location of calcium channels at transmitter release sites of frog neuromuscular synapses. Neuron 5, 773–779.
Robitaille, R., Garcia, M. L., Kaczorowski, G. J., and Charlton, MP. (1993). Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron 11, 645–655.
Rose, C. R., and Konnerth, A. (2001). Stores not just for storage. intracellular calcium release and synaptic plasticity. Neuron 31, 519–522.
Ryu, W. H., Feinstein, A., Colantonio, A., Streiner, D. L., and Dawson, D. R. (2009). Early identification and incidence of mild TBI in Ontario. Can. J. Neurol. Sci. 36, 429–435.
Rzigalinski, B. A., Weber, J. T., Willoughby, K. A., and Ellis, E. F. (1998). Intracellular free calcium dynamics in stretch-injured astrocytes. J. Neurochem. 70, 2377–2385.
Rzigalinski, B. A., Willoughby, K. A., Hoffman, S. W., Falck, J. R., and Ellis, E. F. (1999). Calcium influx factor, further evidence it is 5, 6-epoxyeicosatrienoic acid. J. Biol. Chem. 274, 175–182.
Saatman, K. E., Creed, J., and Raghupathi, R. (2010). Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics 7, 31–42.
Salido, G. M., Jardín, I., and Rosado, J. A.. (2011). The TRPC ion channels: association with Orai1 and STIM1 proteins and participation in capacitative and non-capacitative calcium entry. Adv. Exp. Med. Biol. 704, 413–433.
Sanders, M. J., Sick, T. J., Perez-Pinzon, M. A., Dietrich, W. D., and Green, E. J. (2000). Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Res. 861, 69–76.
Sattler, R., Charlton, M. P., Hafner, M., and Tymianski, M. (1998). Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J. Neurochem. 71, 2349–2364.
Sattler, R., and Tymianski, M. (2000). Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 78, 3–13.
Sattler, R., Xiong, Z., Lu, W. Y., Hafner, M., MacDonald, J. F., and Tymianski, M. (1999). Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284, 1845–1848.
Savic, N., and Sciancalepore, M. (1998). Intracellular calcium stores modulate miniature GABA-mediated synaptic currents in neonatal rat hippocampal neurons. Eur. J. Neurosci. 10, 3379–3386.
Schinder, A. F., Olson, E. C., Spitzer, N. C., and Montal, M. (1996). Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 16, 6125–6133.
Schlaepfer, W. W., and Bunge, R. P. (1973). Effects of calcium ion concentration on the degeneration of amputated axons in tissue culture. J. Cell Biol. 59, 456–470.
Schmolesky, M. T., Weber, J. T., De Zeeuw, C. I., and Hansel, C. (2002). The making of a complex spike: ionic composition and plasticity. Ann. N. Y. Acad. Sci. 978, 359–390.
Scholz, W. K., and Palfrey, H. C. (1998). Activation of Ca2+/calmodulin-dependent protein kinase II by extracellular calcium in cultured hippocampal pyramidal neurons. J. Neurochem. 71, 580–591.
Schouten, J. W. (2007). Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr. Opin. Crit. Care 13, 134–134.
Schwarzbach, E., Bonislawski, D. P., Xiong, G., and Cohen, A. S. (2006). Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus 16, 541–550.
Shahlaie, K., Lyeth, B. G., Gurkoff, G. G., Muizelaar, J. P., and Berman, R. F. (2010). Neuroprotective effects of selective N-type VGCC blockade on stretch-injury-induced calcium dynamics in cortical neurons. J. Neurotrauma 27, 175–187.
Sharma, G., and Vijayaraghavan, S. (2003). Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 38, 929–939.
Sharp, A. H., McPherson, P. S., Dawson, T. M., Aoki, C., Campbell, K. P., and Snyder, S. H. (1993). Differential immunohistochemical localization of inositol 1,4,5-trisphosphate- and ryanodine-sensitive Ca2+ release channels in rat brain. J. Neurosci. 13, 3051–3063.
Shcherbatko, A., Ono, F., Mandel, G., and Brehm, P. (1999). Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophys. J. 77, 1945–1959.
Shohami, E., Shapira, Y., Yadid, G., Reisfeld, N., and Yedgar, S. (1989). Brain phospholipase A2 is activated after experimental closed head injury in the rat. J. Neurochem. 53, 1541–1546.
Sick, T. J., Perez-Pinzon, M. A., and Feng, Z. Z. (1998). Impaired expression of long-term potentiation in hippocampal slices 4 and 48 h following mild fluid-percussion brain injury in vivo. Brain Res. 785, 287–292.
Simpson, P. B., Nahorski, S. R., and Challiss, R. A. (1996). Agonist-evoked Ca2+ mobilization from stores expressing inositol 1,4,5-trisphosphate receptors and ryanodine receptors in cerebellar granule neurones. J. Neurochem. 67, 364–373.
Slemmer, J. E., De Zeeuw, C. I., and Weber, J. T. (2005). “Don’t get too excited: mechanisms of glutamate-mediated Purkinje cell death” in Progress in Brain Research, Vol. 148, Creating Coordination in the Cerebellum, eds C. I. DeZeeuw, and F. Cicirata (Amsterdam: Elsevier), 367–390.
Slemmer, J. E., Matser, E. J. T., De Zeeuw, C. I., and Weber, J. T. (2002). Repeated mild injury causes cumulative damage to hippocampal cells. Brain 125, 2699–2709.
Slemmer, J. E., Shacka, J. J., Sweeney, M. I., and Weber, J. T. (2008). Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr. Med. Chem. 15, 404–414.
Snutch, T. P., and Reiner, P. B. (1992). Ca2+ channels: diversity of form and function. Curr. Opin. Neurobiol. 2, 247–253.
Spaethling, J. M., Klein, D. M., Singh, P., and Meaney, D. F. (2008). Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J. Neurotrauma 25, 1207–1216.
Spitzer, N. C., and Ribera, A. B. (1998). Development of electrical excitability in embryonic neurons: mechanisms and roles. J. Neurobiol. 37, 190–197.
Staal, J. A., Dickson, T. C., Gasperini, R., Liu, Y., Foa, L., and Vickers, J. C. (2010). Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J. Neurochem. 112, 1147–1155.
Stelmasiak, Z., Dudkowska-Konopa, A., and Rejdak, K. (2000). Head trauma and neuroprotection. Med. Sci. Monit. 6, 426–432.
Stout, A. K., and Reynolds, I. J. (1999). High-affinity calcium indicators underestimate increases in intracellular calcium concentrations associated with excitotoxic glutamate stimulations. Neuroscience 89, 91–100.
Stys, P. K., and Lopachin, R. M. (1998). Mechanisms of calcium and sodium fluxes in anoxic myelinated central nervous system axons. Neuroscience 82, 21–32.
Stys, P. K., Waxman, S. G., and Ransom, B. R. (1992). Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+–Ca2+ exchanger. J. Neurosci. 12, 430–439.
Sun, D. A., Deshpande, L. S., Sombati, S., Baranova, A., Wilson, M. S., Hamm, R. J., and DeLorenzo, R. J. (2008). Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur. J. Neurosci. 27, 1659–1672.
Sun, F.-Y., and Faden, A. I. (1994). N-methyl-D-aspartate receptors mediate post-traumatic increases of protein kinase C in rat brain. Brain Res. 661, 63–69.
Surmeier, D. J., Guzman, J. N., and Sanchez-Padilla, J. (2010). Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell Calcium 47, 175–182.
Svoboda, K., and Mainen, Z. F. (1999). Synaptic [Ca2+]: intracellular stores spill their guts. Neuron 22, 427–430.
Tabarean, I. V., Juranka, P., and Morris, C. E. (1999). Membrane stretch affects gating modes of a skeletal muscle sodium channel. Biophys. J. 77, 758–774.
Takahashi, K. (1990). “Calpain substrate specificity,” in Intracellular Calcium-dependent Proteolysis, eds R. L. Mellgren, and T. Murachi (Boca Raton, FL: CRC Press), 55–74.
Takahashi, T., and Momiyama, A. (1993). Different types of calcium channels mediate central synaptic transmission. Nature 366, 156–158.
Takei, K., Shin, R. M., Inoue, T., Kato, K., and Mikoshiba, K. (1998). Regulation of nerve growth mediated by inositol 1,4,5-trisphosphate receptors in growth cones. Science 282, 1705–1708.
Tárnok, A., and Ulrich, H. (2001). Characterization of pressure-induced calcium response in neuronal cell lines. Cytometry 43, 175–181.
Tavalin, S. J., Ellis, E. F., and Satin, L. S. (1995). Mechanical perturbation of cultured cortical neurons reveals a stretch-induced delayed depolarization. J. Neurophys. 74, 2767–2773.
Tavalin, S. J., Ellis, E. F., and Satin, L. S. (1997). Inhibition of the electrogenic Na pump underlies delayed depolarization of cortical neurons after mechanical injury or glutamate. J. Neurophys. 77, 632–638.
Thastrup, O., Dawson, A. P., Scharff, O., Foder, B., Cullen, P. J., Drobak, B. K., Bjerrum, P. J., Christensen, S. B., and Hanley, M. R. (1989). Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions 27, 17–23.
Thorell, W. E., Leibrock, L. G., and Agrawal, S. K. (2002). Role of RyRs and IP3 receptors after traumatic injury to spinal cord white matter. J. Neurotrauma 19, 335–342.
Toescu, E. C. (1998). Apoptosis and cell death in neuronal cells: where does Ca2+ fit in? Cell Calcium 24, 387–403.
Trump, B. F., and Berezesky, I. K. (1995). Calcium-mediated cell injury and cell death. FASEB J. 9, 219–228.
Trump, B. F., and Berezesky, I. K. (1996). The mechanisms of calcium-mediated cell injury and cell death. New Horizons 4, 139–150.
Turner, C. P., Pulciani, D., and Rivkees, S. A. (2002). Reduction in intracellular calcium levels induces injury in developing neurons. Exp. Neurol. 178, 21–32.
Tymianski, M., Charlton, M. P., Carlen, P. L., and Tator, C. H. (1993a). Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J. Neurosci. 13, 2085–2104.
Tymianski, M., Charlton, M. P., Carlen, P. L., and Tator, C. H. (1993b). Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 607, 319–323.
Tymianski, M., Charlton, M. P., Carlen, P. L., and Tator, C. H. (1994). Properties of neuroprotective cell-permeant Ca2+ chelators: effects on [Ca2+]i and glutamate neurotoxicity in vitro. J. Neurophys. 72, 1973–1992.
Tymianski, M., and Tator, C. H. (1996). Normal and abnormal calcium homeostasis in neurons: a basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery 38, 1176–1195.
van den Brink, W. A., van Santbrink, H., Steyerberg, E. W., Avezaat, C. J., Suazo, J. A., Hogesteeger, C., Jansen, W. J., Kloos, L. M., Vermeulen, J., and Maas, A. I. (2000). Brain oxygen tension in severe head injury. Neurosurgery 46, 868–876.
Verkhratsky, A., and Shmigol, A. (1996). Calcium-induced calcium release in neurones. Cell Calcium 19, 1–14.
Wang, K. K. W., and Yuen, P. W. (1994). Calpain inhibition: an overview of its therapeutic potential. Trends Pharmacol. Sci. 15, 412–419.
Wang, X., and Gruenstein, E. I. (1997). Mechanism of synchronized Ca2+ oscillations in cortical neurons. Brain Res. 767, 239–249.
Weber, J. T. (2004). Calcium homeostasis following traumatic neuronal injury. Curr. Neurovasc. Res. 1, 151–171.
Weber, J. T., De Zeeuw, C. I., Linden, D. J., and Hansel, C. (2003). Long-term depression of climbing fiber-evoked calcium transients in Purkinje cell dendrites. Proc. Natl. Acad. Sci. U.S.A. 100, 2878–2883.
Weber, J. T., Rzigalinski, B. A., and Ellis, E. F. (2001). Traumatic injury of cortical neurons causes changes in intracellular calcium stores and capacitative calcium influx. J. Biol. Chem. 276, 1800–1807.
Weber, J. T., Rzigalinski, B. A., and Ellis, E. F. (2002). Calcium responses to caffeine and muscarinic receptor agonists are altered in traumatically injured neurons. J. Neurotrauma 19, 1433–1443.
Weber, J. T., Rzigalinski, B. A., Willoughby, K. A., Moore, S. F., and Ellis, E. F. (1999). Alterations in calcium-mediated signal transduction after traumatic injury of cortical neurons. Cell Calcium 26, 289–299.
Wei, E. P., Lamb, R. G., and Kontos, H. A. (1982). Increased phospholipase activity after experimental brain injury. J. Neurosurgery 56, 695–698.
Wei, H., and Perry, D. C. (1996). Dantrolene is cytoprotective in two models of neuronal cell death. J. Neurochem. 67, 2390–2398.
Westenbroek, R. E., Hoskins, L., and Catterall, W. A. (1998). Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J. Neurosci. 18, 6319–6330.
White, R. J., and Reynolds, I. J. (1996). Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J. Neurosci. 16, 5688–5697.
Winder, D. G., and Conn, P. J. (1996). Roles of metabotropic glutamate receptors in glial function and glial-neuronal communication. J. Neurosci. Res. 46, 131–137.
Witgen, B. M., Lifshitz, J., Smith, M. L., Schwarzbach, E., Liang, S. L., Grady, M. S., and Cohen, A. S. (2005). Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience 133, 1–15.
Wojda, U., Salinska, E., and Kuznicki, J. (2008). Calcium ions in neuronal degeneration. IUBMB Life 60, 575–590.
Wolf, J. A., Stys, P. K., Lusardi, T., Meaney, D., and Smith, D. H. (2001). Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci. 21, 1923–1930.
Wu, L. G., Borst, J. G., and Sakmann, B. (1998). R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc. Natl. Acad. Sci. U.S.A. 95, 4720–4725.
Wulff, H., Kolski-Andreaco, A., Sankaranarayanan, A., Sabatier, J. M., and Shakkottai, V. (2007). Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr. Med. Chem. 14, 1437–1457.
Yao, Y., Ferrer-Montiel, A. V., Montal, M., and Tsien, R. Y. (1999). Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell 98, 475–485.
Yoon, K. W., Mitchell, H. L., Broder, L. D., Brooker, R. W., and Delisle, R. K. (1996). Trauma-induced neurotoxicity in rat hippocampal neurons. Stroke 27, 122–126.
Yu, S. P., Sensi, S. L., Canzoniero, L. M., Buisson, A., and Choi, D. W. (1997). Membrane-delimited modulation of NMDA currents by metabotropic glutamate receptor subtypes 1/5 in cultured mouse cortical neurons. J. Physiol. 499, 721–732.
Zauner, A., Bullock, R., Kuta, A. J., Woodward, J., and Young, H. F. (1996). Glutamate release and cerebral blood flow after severe human head injury. Acta Neurochir. Suppl. 67, 40–44.
Zhang, L., Rzigalinski, B. A., Ellis, E. F., and Satin, L. S. (1996). Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science 274, 1921–1923.
Zhang, M., Shan, H., Gu, Z., Wang, D., Wang, T., Wang, Z., and Tao, L. (2012). Increased expression of calcium/calmodulin-dependent protein kinase type II subunit delta after rat traumatic brain injury. J. Mol. Neurosci. 46, 631–643.
Keywords: calcium stores, excitotoxicity, in vitro, trauma, signal transduction
Citation: Weber JT (2012) Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 3:60. doi: 10.3389/fphar.2012.00060
Received: 11 March 2012; Accepted: 24 March 2012;
Published online: 12 April 2012.
Edited by:
Amalia M. Dolga, Philipps-Universität Marburg, GermanyReviewed by:
Muzamil Ahmad, Indian Institute of Integrative Medicine, IndiaUlrich Eisel, University of Groningen, Netherlands
Copyright: © 2012 Weber. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: John T. Weber, School of Pharmacy, Health Sciences Centre, Memorial University of Newfoundland, 300 Prince Philip Drive, St. John’s, NL, Canada A1B 3V6. e-mail: jweber@mun.ca