 Giannoula L. Klement1,3 David Goukassian2 Lynn Hlatky1 Joseph Carrozza2 James P. Morgan2 Xinhua Yan2*
Giannoula L. Klement1,3 David Goukassian2 Lynn Hlatky1 Joseph Carrozza2 James P. Morgan2 Xinhua Yan2*- 1 Center of Cancer Systems Biology, St. Elizabeth’s Medical Center, Tufts University School of Medicine, Boston, MA, USA
- 2 Cardiovascular Medicine, St. Elizabeth’s Medical Center, Tufts University School of Medicine, Boston, MA, USA
- 3 Floating Hospital for Children at Tufts Medical Center, Boston, MA, USA
The HER2-PI3K pathway is the one of the most mutated pathways in cancer. Several drugs targeting the major kinases of this pathway have been approved by the Food and Drug Administration and many are being tested in clinical trials for the treatment of various cancers. However, the HER2-PI3K pathway is also pivotal for maintaining the physiological function of the heart, especially in the presence of cardiac stress. Clinical studies have shown that in patients treated with doxorubicin concurrently with Trastuzumab, a monoclonal antibody that blocks the HER2 receptor, the New York Heart Association class III/IV heart failure was significantly increased compared to those who were treated with doxorubicin alone (16 vs. 3%). Studies in transgenic mice have also shown that other key kinases of this pathway, such as PI3Kα, PDK1, Akt, and mTOR, are important for protecting the heart from ischemia-reperfusion and aortic stenosis induced cardiac dysfunction. Studies, however, have also shown that inhibition of PI3Kγ improve cardiac function of a failing heart. In addition, results from transgenic mouse models are not always consistent with the outcome of the pharmacological inhibition of this pathway. Here, we will review these findings and discuss how we can address the cardiac side-effects caused by inhibition of this important pathway in both cancer and cardiac biology.
Introduction
HER receptor tyrosine kinases (RTKs) and the major components of the phosphoinositide 3-kinase (PI3K) pathway are frequently mutated or aberrantly expressed in a wide variety of cancers (Luo et al., 2003; Yuan and Cantley, 2008). Therefore, they are major targets for cancer therapy (Garcia-Echeverria and Sellers, 2008; Yap et al., 2008). Trastuzumab (Herceptin), a monoclonal antibody that blocks the HER2 receptor, was one of the first drugs of this class approved by the US Food and Drug Administration (FDA) for cancer therapy. Subsequent clinical trials had shown that Trastuzumab significantly improved survival in breast cancer patients (Slamon et al., 2001). Since then, multiple drugs targeting the HER2-PI3K pathway have been approved by FDA for cancer treatment. These drugs include lapatinib (Tykerb), a small molecule that inhibits EGFR (HER1) and HER2, Erlotinib (Tarceva), a small molecule that inhibits EGFR, Cetuximab (Erbitux), a chimeric IgG1 monoclonal antibody that blocks EGFR, Temsirolimus (Torisel), and Everolimus (Afinitor), small molecules that inhibit mTOR (a nodal kinase of the PI3K pathway). In addition, more than 20 new drugs targeting this pathway are currently being tested in clinical trials1.
Compared to traditional cancer chemotherapy, targeted cancer therapy was designed to target molecules that were aberrantly activated in cancer. This strategy was therefore thought to be more specific and restricted to the tumor tissues. It was expected that these agents would cause much less damage to normal tissues (Hait and Hambley, 2009). While this may still be true, but because Tyrosine Kinase Inhibitors (TKIs) are presently introduced to clinics as adjuvant therapies and are tested in combination with standard chemotherapeutic regimens, unexpected toxicities are being observed. This should not be surprising considering that most targeted therapies, and particularly TKIs, are chemotherapy sensitizers (Ueno et al., 2000; Dickerson et al., 2010), and both cancer cells as well as highly metabolic organs such as heart, lungs, and kidney should have been expected to have increased sensitivity to these drugs. Cardiovascular toxicities, have been frequently reported in patients treated with targeted cancer therapies, and have resulted in widespread concern regarding the cardiac safety of using these drugs. An early concern was first raised in clinical studies of Trastuzumab. Patients treated with combination of Trastuzumab and doxorubicin, had an increase rate in the New York Heart Association class III/IV heart failure compared to those who were treated with doxorubicin alone (16 vs. 3%; Slamon et al., 2001). Similarly, the FDA recently revoked the approval of Bevacizumab (Avastin), a drug that blocks VEGF (vascular endothelial growth factor), for breast cancer treatment. This was due at least in part to severe cardiovascular events2. The anthracyclines are perhaps the most notorious offenders in the setting of clinical use of growth factor pathway inhibitors such as TKIs (Hershman and Shao, 2009). The results of early clinical trials suggest that the introduction of targeted therapies to clinical settings must include a careful consideration for the mechanism of action of these agents, and future development of cardio, lung, and kidney protective strategies. Results from clinical studies of Trastuzumab and animal studies using transgenic mouse models or pharmacological approaches have demonstrated that major kinases of the HER2-PI3K pathway are important for regulating and maintaining cardiac physiological function, especially in the presence of cardiac stress (Heineke and Molkentin, 2006). Preclinical studies also suggest that this pathway is delicately regulated (Klein and Dybdal, 2003), and depending on the type of nodal kinase inhibited within this pathway cardiac function may be either augmented or depressed. The cardiac outcome of inhibiting the nodal kinases within this pathway also depends on the specific disease setting. In this review, we will revisit the specific findings in transgenic mice with cardiomyocyte-specific expression of mutants of key kinases of the HER2-PI3K pathway.
The HER2-PI3K Signaling Pathway
The HER receptors belong to the epidermal growth factor receptor tyrosine kinase family, which include four receptors HER1 (also known as EGFR-epidermal growth factor receptor), HER2 (ErbB2/Neu), HER3 (ErbB3), and HER4 (ErbB4; Yarden and Sliwkowski, 2001; Citri and Yarden, 2006). The HER receptors are composed of an extracellular ligand-binding domain, a transmembrane domain and a cytoplasmic region with kinase activity. Upon ligand-binding, HER receptors form hetero- or homo-dimers, followed by auto-phosphorylation of the tyrosine kinase residues on the receptors. These residues then serve as docking sites for recruiting cytosolic signaling molecules to the cell membrane. Each receptor has a unique pattern of binding partners. HER2 and HER3 are unique receptors. There are no-known ligands for the HER2 receptor, while HER3 lacks intrinsic kinase activity. However, HER2 and HER3 can form potent heterodimers to propagate signals and induce cancer cell proliferation (Guy et al., 1994; Klapper et al., 1999; Citri et al., 2003).
In growth factor signaling, multiple signaling pathways can be activated simultaneously. Two most identified pathways are the PI3K pathway and the MAPK pathway. Other pathways include STATs, JNK, and PLCs (Yarden and Sliwkowski, 2001; Citri and Yarden, 2006). Systems biology studies revealed that this signaling network is highly organized and precisely regulated through a network of positive and negative feed-back loops and cross-talk among pathways. The activation and integration of all signaling pathways lead to the regulation of key functions of the cell, which include growth, proliferation, differentiation, survival, and metabolism (Yarden and Sliwkowski, 2001; Citri and Yarden, 2006).
Phosphoinositide 3-kinases (PI3Ks) are conserved lipid kinases that phosphorylate the 3′-hydroxyl group of phosphoinositides (Cantley, 2002). The best studied are class I PI3Ks, and these also represent the major targets for cancer therapy (Zhao and Vogt, 2008). They are further divided into class IA and class IB. Class IA are heterodimers comprised of a regulatory subunit (p85α, p55α, p50α, p85β, p55γ) and a catalytic subunit (p110α, p110β, p110δ). In response to the RTKs activation, class IA PI3Ks are recruited, and bind, to the tyrosine phosphate motifs on the activated RTKs via the regulatory subunits. Class IB PI3Ks are composed of a regulatory subunit p101 and a catalytic subunit p110γ, and activated by G-protein-coupled receptors (GPCRs; Shaw and Cantley, 2006; Liu et al., 2009). The catalytic subunit of the PI3Ks produces phosphatidylinositol-3,4,5-triphosphate (PIP3), a key signaling messenger that recruits and activates a spectrum of signaling molecules. The PIP3 signal is negatively regulated by PTEN (phosphatase and tensin homolog), which converts PIP3 back to PIP2 (Shaw and Cantley, 2006).
In addition to class I PI3Ks, a number of other key components of the PI3K pathway that are mutated in cancer, have been identified as targets for intervention in cancer therapy. These include Akt, PDK1 (3-phosphoinositide-dependent kinase-1), and mTOR (mammalian target of rapamycin; Garcia-Echeverria and Sellers, 2008).
Akt, also known as protein kinase B, is a serine-threonine protein kinase. There are three isoforms – Akt1, Akt2, and Akt3 (Brazil et al., 2004; Dummler and Hemmings, 2007). PIP3 recruits Akt to the cell membrane, where it is phosphorylated by PDK1 on Thr308 and kinases such as mTORC2 on Ser473. Phosphorylation of both residues is necessary for full activation of Akt. Activated Akt then regulates a wide variety of transcription factors and signaling molecules, including FOXO1 (forkhead box O1), GSK3β (glycogen synthase kinase 3β), NF-κB (nuclear factor-κB), and mTOR (Luo et al., 2003; Shaw and Cantley, 2006).
mTOR is central for cell growth, nutrients, and energy metabolism (Zoncu et al., 2011). There are two mTOR complexes: mTORC1 and mTORC2. In addition to mTOR, the mTORC1 contains RAPTOR (regulatory associated protein of mTOR), PRAS40 (proline-rich Akt substrate 40 kDa), mLST8 (mammalian lethal with SEC13 protein 8), and DEPTOR (DEP domain-containing mTOR-interacting protein). The mTORC2 is composed of mTOR, RICTOR (rapamycin-insensitive companion of mTOR), mSIN1 (mammalian stress-activated MAP kinase interacting protein 1), mLST8, DEPTOR, and PROTOR (protein observed with RICTOR; Zoncu et al., 2011). Akt activates mTORC1 by releasing the inhibitory effects of PRAS40 and TSC2 (tuberous sclerosis 2 protein, also known as tuberin; Shaw and Cantley, 2006). mTORC1 promotes protein synthesis and cell growth by activating ribosomal protein S6 kinase-1 (S6K1) and inhibiting eukaryotic translation initiation factor 4E-binding protein (4E-BP). mTORC2, on the other hand, is an upstream signaling molecule of Akt. It activates Akt by phosphorylating Ser473 (Sarbassov et al., 2005; Zoncu et al., 2011).
The HER2-PI3K Pathway in Cancer
The HER2-PI3K pathway is the most frequently mutated or aberrantly amplified oncogenic pathway in cancer (Yuan and Cantley, 2008; Lin et al., 2010). HER2 is overexpressed in 25–30% of invasive breast and ovarian cancers. PIK3CA, the gene encoding the p110α subunit of PI3K, is mutated in 27% of breast, 24% of endometrial, and 15% of colorectal cancers. Amplification of p110α was found in 53% of squamous cell lung cancer and 69% of cervical cancer. p110β of PI3K is amplified in 5% of breast and ovarian cancers. PDK1 is amplified in 20% of breast cancers. Akt1, Akt2, and Akt3 mutations were found in breast, colon, ovarian, lung, gastric, pancreas, and skin cancers in the range of 2–20%. mTORC1, a downstream target of PI3K and MAPK pathways, are frequently overactivated in a wide range of cancers.
HER-PI3K Pathway Inhibitors
Inhibitors targeting HER receptors and nodal kinases of the PI3K pathway have been developed. Some of them are approved by the FDA and many are in clinical trials (Table 1). The FDA approved EGFR and HER2 inhibitors include Gefitinib (AstraZeneca), Cetuximab (ImClone), a chimeric IgG1 monoclonal antibody that binds the extracellular domain of the EGFR, Erlotinib (Genentech, OSI, and Roche), a small molecule tyrosine kinase inhibitor, Vandetanib (AstraZeneca), small molecule tyrosine kinase inhibitor of VEGFR and EGFR, Trastuzumab (Genentech), a monoclonal antibody blocks HER2, and Lapatinib (GSK), a small TKI of EGFR and HER2.

Table 1. Drugs targeting the HER-PI3K pathway for cancer treatment.
At least 10 inhibitors target PI3Ks are in clinical Phase I–II trials for treatment of various cancers, including breast, lung, ovarian cancers, and hematological malignancies. These inhibitors can be divided into the following categories: isoform-specific, class IA PI3K, pan-PI3K, and PI3K-mTOR dual inhibitor. PI3Kα-specific inhibitors include BYL719 (Novartis) and INK1117 (Intellikine). PI3Kδ inhibitors include CAL-101 (Calistoga). PI3Kα, δ, and γ specific inhibitors include GDC-0032 (Genentech) and PX-866 (Oncothyreon). Pan-PI3K inhibitors include GSK1059615 (GlaxoSmithKline). Class I PI3K inhibitors include XL147 (Exelixis), BKM120 (Novartis), and GDC0941 (Genentech). PI3K/mTOR dual inhibitors include BEZ235 (Novartis), BGT226 (Novartis), GDC-0980 (Genentech), and PF-4691502 (Pfizer).
Inhibitors of Akt include MK2206 (Merck), GDC-0068 (Genentech), AZD5363 (Astrazeneca), Perifosine (Keryx), VQD-002 (VioQuest), and XL418 (Exelixis). Rapamycin, Temsirolimus, and Everolimus are inhibitors of mTOR and approved by the FDA for treatment of certain types of tumors. Clinical trials are ongoing to test these drugs in other tumors. In addition, new inhibitors of mTOR are in development. They are AZD8055 (Astrazeneca), INK128 (Intellikine), AP23573 (Merck/Ariad), and OSI-027 (OSI).
Inhibition of Major Kinases of the HER2-PI3K Pathway in the Heart: Lessons from Animal Studies
The role of the HER2-PI3K pathway in cardiac physiology and pathophysiology has been extensively studied during the past 10 years. Transgenic mice with cardiomyocyte-specific overexpression of the mutants of nodal kinases of this pathway were generated. The biological functions and complex signal transduction in the cell, including the cardiomyocytes, of this network have been comprehensively reviewed (Dorn and Force, 2005; Yuan and Cantley, 2008; Oudit and Penninger, 2009; Aoyagi and Matsui, 2011; Chaanine and Hajjar, 2011; Ghigo et al., 2011; Hers et al., 2011). Here, we will focus on the cardiac physiology in mice with perturbation of key nodal kinases of this network, with emphasis on those where the activities of these kinases are inhibited. These findings may provide clues of whether inhibitors of these kinases may cause significant impact on the heart.
Mice with Cardiac Expression of HER2 Mutants
Mice carrying an HER2 null allele died around E10.5 (HER2KO). The mutant embryos exhibited malformation of LV trabeculae (Lee et al., 1995). Cardiomyocyte-specific overexpression of the HER2 gene in the HER2KO mice restored normal ventricular trabeculation and prolonged survival of HER2KO mice (Morris et al., 1999).
To study the effects of HER2 inhibition in the adult heart, two mouse models with cardiac ventricular myocyte-specific conditional deletion of the HER2 gene were generated (HER2-CKO). Mice that harbor the loxP-flanked HER2 allele were cross-bred with mice that carry Cre coding sequence under the control of myosin light chain 2v (MLC2v) locus, which drives ventricular-restricted gene deletions (Crone et al., 2002; Ozcelik et al., 2002). HER2-CKO mice survived to the adulthood, but progressively developed dilated cardiomyopathy, with increased LV hypertrophy, chamber dilation, and dysfunction. The HER2 protein expression was decreased in HER2-CKO hearts with no changes of HER4 expression. Electron microscopy studies showed an increase in the numbers of mitochondria and vacuoles in the HER2-CKO myocardium. Although TUNEL staining was not increased in HER2-CKO hearts, expression of Bcl-xL partially rescued dilated cardiomyopathy in HER2-CKO mice. Transverse aortic constriction (TAC) induced a similar LV hypertrophic response with increased mortality rate in one HER2-CKO model (Crone et al., 2002), but a de-compensated response and worsened cardiac dysfunction in another model (Ozcelik et al., 2002). This discrepancy may be caused by the baseline cardiac function of mice before surgery. Cardiomyocytes isolated from HER2-CKO mice were more susceptible to doxorubicin (Crone et al., 2002) (Table 2).
Mice with Cardiac Expression of PDK1 Mutants
Two types of transgenic mouse models with conditional deletion of the PDK1 gene have been generated. The first model was generated by cross-breeding mice harboring a “floxed” PDK1 allele (PDK1fl/fl) with mice expressing Cre recombinase under the control of the muscle creatine kinase (MCK) promoter (PDK1-MckCre; Mora et al., 2003). The MCK promoter induces expression of Cre specifically in skeletal muscle and heart just prior to birth (Bruning et al., 1998). The second model was generated by cross-breeding PDK1fl/fl with mice expressing tamoxifen-inducible Cre recombinase under the control of the α-MHC promoter (PDK1-MerCre; Ito et al., 2009; Di et al., 2010).
Both PDK1-MckCre and PDK1-MerCre mice developed dilated cardiomyopathy within weeks after the onset of PDK1 gene deletion. Insulin-induced activations of Akt, mTOR, and S6K were decreased in the PDK1 knockout hearts (Mora et al., 2003; Ito et al., 2009; Di et al., 2010). LV mass and cardiomyocyte volume were decreased in PDK1-MckCre, but not in PDK1-MerCre mice. On the other hand, apoptosis was increased in PDK1-MerCre, but not in PDK1-MckCre hearts. Studies in PDK1-MerCre mice further showed that β-adrenergic responsiveness was impaired in the heart. This was associated with increased internalization of β1-adrenergic receptor (β1-AR) and increased β-adrenergic receptor kinase-1 (β-ARK1)-PI3Kp110γ complex formation. Disruption of this complex reduced cardiac dysfunction in PDK1-MerCre mice (Ito et al., 2009). These results suggest that inhibition of PDK1 signaling can cause heart failure. This may be related to increased apoptosis and β-AR desensitization (Table 2).
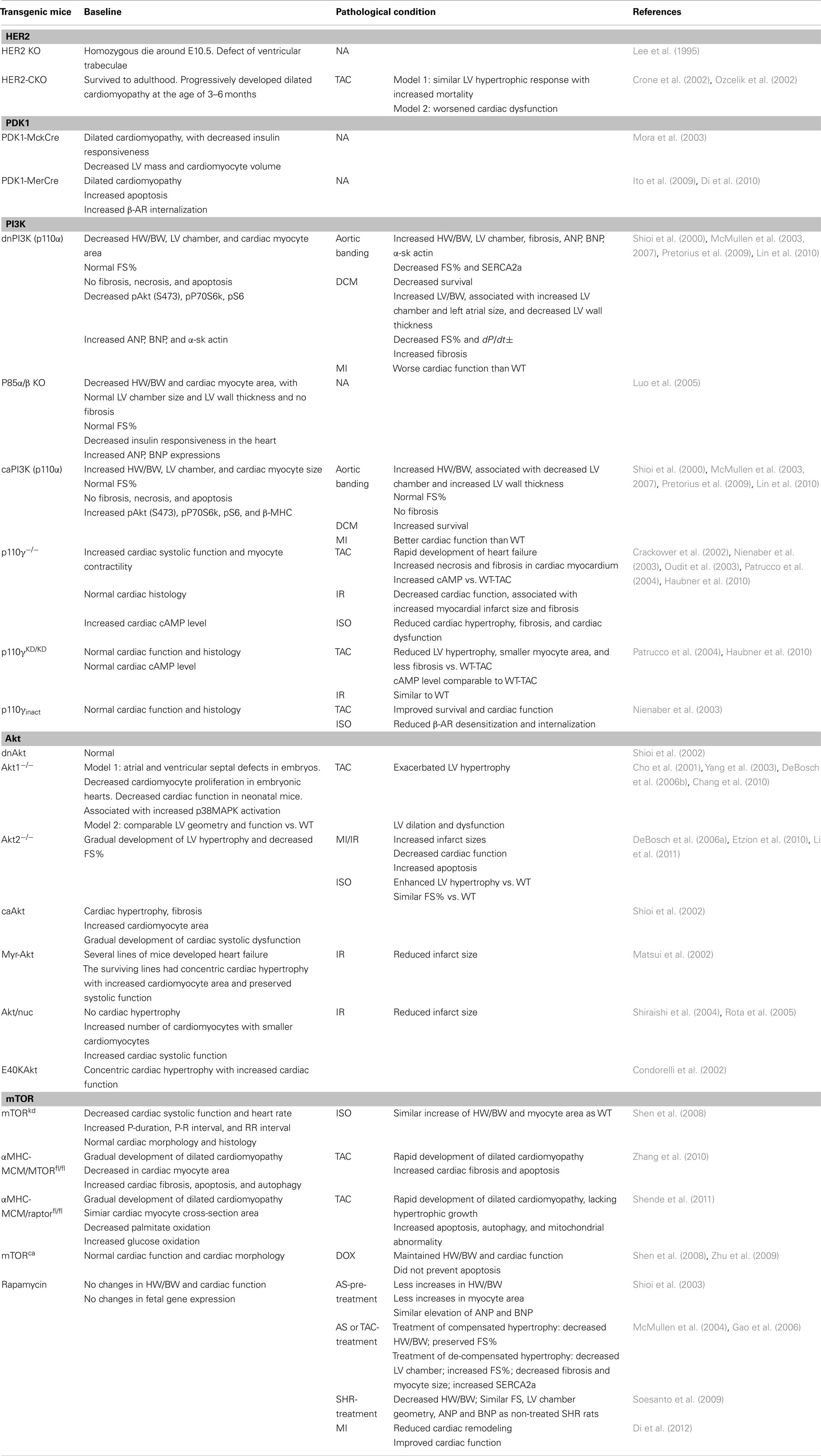
Table 2. Cardiac phenotypes in mice with inhibition/activation of nodal kinases of the HER2-PI3K pathway.
Mice with Cardiac Expression of PI3K Mutants
At least three class I PI3Ks can be found in the heart, which are p110α, p110β (class IA) and p110γ (class IB). The class IA PI3Ks are activated by RTKs in cardiomyocytes, whereas the class IB PI3Ks are activated by GPCRs. To understand the physiological role of class I PI3Ks in the heart, transgenic mice with cardiomyocyte-specific expression of PI3K isoform-specific mutants were generated (Table 2).
Mice with Cardiac Expression of Class IA PI3K Mutants
Shioi et al. (2002) reported their studies in mice with cardiomyocyte-specific overexpression of a dominant negative PI3Kp110α (dnPI3K) and mice with constitutively active PI3Kp110α (caPI3K; Shioi et al., 2000). To generate dnPI3K mice, a truncated p110α mutant that has p85 binding domains but is devoid of the kinase domain was cloned downstream of the α-MHC promoter, which drives transgene expression exclusively in cardiomyocytes. To generate caPI3K mice, iSH2p110, a chimeric molecule that contains the iSH2 domain of p85, was fused to the N-terminus of the bovine p110α by a flexible glycine linker. This molecule was then constructed downstream of the α-myosin heavy chain (α-MHC) promoter. In dnPI3K mouse hearts, PI3K activity was decreased by 77%. In caPI3K mice, PI3K activity was increased 6.5-fold compared to wild type mice (WT).
One specific finding was that dnPI3K mice had smaller hearts while caPI3K mice had bigger hearts (Shioi et al., 2000). The heart weight to body weight ratio (HW/BW), an index for cardiac hypertrophy, was decreased in dnPI3K mice, which was associated with decreased LV chamber and cardiomyocyte area. Conversely, HW/BW was increased in caPI3K mice, associated with increased LV chamber and cardiomyocyte area. No abnormalities of cardiac morphology and function were observed. mTORC1 (pP70S6K and pS6) and mTORC2 (pAktS473) activities were decreased in dnPI3K mouse hearts but increased in caPI3K mouse hearts.
When mice were subjected to aortic banding (McMullen et al., 2003, 2007), myocardial infarction (MI; Lin et al., 2010), or cross-bred with DCM mice (dilated cardiomyopathy; McMullen et al., 2007; Pretorius et al., 2009), the dnPI3K mice rapidly developed dilated cardiomyopathy. The LV chamber of dnPI3K mice became dilated with thinning of the LV wall, increased fibrosis and decreased LV systolic function. On the other hand, aortic banding induced concentric hypertrophy in caPI3K mice, with preserved LV function. MI in caPI3K induced less cardiac remodeling and LV dysfunction compared to WT mice and dnPI3K mice. Cross-bred caPI3K mice with DCM mice prolonged survival of DCM mice (Table 2).
The p85 regulatory subunit of the class 1A PI3Ks is essential for stabilizing p110 catalytic subunit and recruiting it to activated RTKs at the cell membrane (Fruman et al., 1998). There are five p85 isoforms, which are encoded by three genes. The p85α isoforms (p85α, p55α, and p50α) are encoded by gene pik3r1, the p85β isoform is encoded by gene pik3r2 and p55γ isoform is encoded by the pik3r3 gene (Luo et al., 2005). The p85α and p85β are ubiquitously expressed, while the p55γ isoform is expressed mainly in the brain and the testis (Fruman et al., 1998).
Luo et al. (2005) generated mice with striated muscle-specific deletion of both p85α and p85β. The HW/BW ratio was significantly lower in p85α/β KO mice compared to WT, associated with a smaller cardiomyocyte area. The size of the cardiac chamber and the LV wall thickness were comparable to WT mice. No increase of fibrosis was found in p85α/β KO hearts. The systolic function was preserved in p85α/β KO mice. However, ANP and BNP were significantly increased, and the response to insulin signaling was attenuated, in p85α/β KO hearts.
These results suggest that class IA PI3Ks determine the size of the heart. It is also crucial for maintaining normal cardiac function in the presence of cardiac stress (Table 2).
Mice with Cardiac Expression of Class IB PI3K Mutants
The class IB PI3Kp110γ is activated by GPCRs by binding to Gβγ directly or via its regulatory subunit p101 (Stoyanov et al., 1995; Stephens et al., 1997). The recruitment of PI3Kp110γ to GPCRs is also mediated by β-ARK1 which forms a complex with PI3Kp110γ. Upon ligand-binding to GPCRs, the heterotrimeric G-proteins dissociate into Gα and Gβγ subunits. The termination of GPCR signals is initiated by phosphorylation of agonist occupied GPCRs by GPCR kinase, such as β-ARK1, followed by binding to β-arrestins and AP-2 which mediate receptor internalization. Studies suggested that PI3Kp110γ negatively regulates cardiac contractile function by least two mechanisms: (1) it serves as a scaffold protein to stabilize phosphodiesterase 3B (PDE3B), which degrades cAMP. This effect of PI3Kp110γ is kinase-independent; (2) it forms a complex with β-ARK1, which contributes to internalization of β-ARs. This effect of PI3Kp110γ is kinase-dependent. The PI3Kp110γ proteins generate PIP3, which can recruit β-arrestins and AP-2 to the cell membrane (Gaidarov and Keen, 1999; Naga Prasad et al., 2002).
Transgenic mouse models with PI3Kp110γ knockout (p110γ−/−), expression of PI3Kp110γ kinase-dead (p110γKD/KD), or cardiomyocyte-specific overexpression of an inactive mutant of PI3Kp110γ (p110γinact) were generated (Hirsch et al., 2000; Li et al., 2000; Nienaber et al., 2003; Patrucco et al., 2004). The major difference of p110γ−/− mice with p110γKD/KD or p110γinact mice is that p110γ proteins are depleted in p110γ−/− hearts, but not in p110γKD/KD or p110γinact hearts; whereas in p110γKD/KD or p110γinact hearts the PI3Kp110γ kinase activity was impaired.
The p110γ−/− mice had normal LV chamber geometry and histology under the baseline condition. However, the cardiac systolic function and cardiomyocyte contractility were increased which were associated with increased cAMP in cardiomyocytes. TAC induced a rapid development of heart failure in p110γ−/− mice showing LV chamber dilation and wall thinning, decreases in FS% and dP/dt±, as well as increases in necrosis, fibrosis, and infiltration of inflammatory cells in the myocardium. Similarly, ischemia/reperfusion (IR) induced a significant increase in infarct size, collagen deposition, and scar formation in p110γ−/− mice, which was associated with severe reduction of cardiac systolic function (Crackower et al., 2002; Nienaber et al., 2003; Patrucco et al., 2004; Haubner et al., 2010).
Conversely, the cardiac function was normal in p110γKD/KD or p110γinact mice at the baseline. TAC induced concentric hypertrophy in p110γKD/KD −TAC mice, which is comparable to WT-TAC mice. The cardiac myocyte diameter was smaller in p110γKD/KD −TAC mice with less fibrosis in the cardiac myocardium (Patrucco et al., 2004). IR induced a similar infarct size, fibrosis, and cardiac dysfunction in p110γKD/KD mice compared to WT mice. Similar to the findings in p110γKD/KD mice, TAC induced a comparable degree of LV hypertrophy in p110γinact vs. WT mice 1 week after TAC. In addition, 12 weeks after TAC, survival was improved in p110γinact –TAC mice, with less cardiac dysfunction and LV dilation (Crackower et al., 2002; Nienaber et al., 2003; Patrucco et al., 2004; Haubner et al., 2010).
Further studies revealed that cAMP was excessively elevated in p110γ−/− −TAC, while maintained in p110γKD/KD –TAC mouse hearts. Injections of propranolol, a non-selective β-blocker, lowered cAMP levels, and reverse the adverse cardiac remodeling in p110γ−/− −TAC hearts. In addition, the activity of phosphodiesterase (PDE) was significantly lower in p110γ−/− −TAC hearts compared to p110γKD/KD −TAC mouse hearts. Co-immunoprecipitation showed that p110γ and PDE3B are physically associated with each other. These results suggest that p110γ serves as a scaffold protein that stabilizes PDE3B, leading to the degradation of cAMP (Patrucco et al., 2004).
In addition, βARK1-associated PI3K activity was decreased in p110γinact hearts, but not in p110γ−/− hearts. Chronic isoproterenol infusion caused desensitization and downregulation of β-ARs in p110γ−/− hearts, but not in p110γinact hearts (Nienaber et al., 2003).
Taken together, p110γ proteins are necessary for maintaining cAMP homeostasis in cardiomyocytes. On the other hand, blockade of p110γ kinase activity may be beneficial for preventing β-AR desensitization in a failing heart (Table 2).
Mice with Cardiac Expression of Akt Mutants
Akt is a key regulator of multiple aspects of cardiomyocyte functions, including survival, hypertrophy, calcium homeostasis, and metabolism (Lawlor and Alessi, 2001; Ceci et al., 2004). All three isoforms of Akt are expressed in the heart, Akt1 and Akt2 are most abundant (Matsui and Rosenzweig, 2005; DeBosch et al., 2006b). Akt can be activated by RTKs (such as insulin and IGF) and by GPCRs (such as β-ARs) in cardiomyocytes (Tian, 2005). Studies have shown that Akt1 is important for exercise-induced cardiac hypertrophy, while Akt2 is pivotal for normal glucose metabolism of the heart (DeBosch et al., 2006b; Muslin, 2011).
Several transgenic mouse models were generated with either systematic disruption of the Akt1 gene (Akt1−/−) or cardiomyocyte-specific overexpression of a kinase-dead Akt1 (kdAkt; Cho et al., 2001; Shioi et al., 2002; Yang et al., 2003). In the first Akt1−/− model, mice developed atrial and ventricular septal defects in embryos, associated with a high early mortality rate. Neonatal Akt1−/− mice developed dilated cardiomyopathy. The phosphorylated and total Akt proteins were decreased in Akt1−/− hearts; While Akt2 and Akt3 proteins were comparable to WT. The activity of p38MAPK was increased in Akt1−/− hearts. Cross-breeding of Akt1−/− with null p38α mice partially rescued the cardiac defects (Chang et al., 2010).
In the second Akt1−/− model, no embryonic or early mortality were found. Mice had a normal lifespan and were fertile. The cardiac geometry and function were similar to WT. Adult cardiomyocytes isolated from Akt1−/− mice had an impaired response to IGF1 stimulation, as assessed by phosphorylation of key signaling molecules of IGF1 signaling and IGF1-induced protein synthesis. Accordingly, swimming induced cardiac hypertrophy was blunted in Akt1−/− mice; instead, LV dilation and decreased cardiac function was observed. On the other hand, cardiomyocytes from Akt1−/− mice retained normal response to GPCR ligand endothelin-1. In response to pressure overload, Akt1−/− mice developed increased LV hypertrophy compared to WT, which was associated with LV dilation and dysfunction (DeBosch et al., 2006b) (Table 2).
In kdAkt mice, a kinase-dead Akt (K179M), in which the ATP binding site of Akt1 was mutated, was conditionally overexpressed in cardiomyocytes. The activity of Akt at baseline and that induced by IGF1 stimulation was decreased in kdAkt hearts. The phosphorylation of GSK3β, p70S6K, and S6 were lower in kdAkt hearts. However, cardiac geometry, function, and cardiomyocyte size were preserved with no fibrosis and apoptosis in the cardiac myocardium (Shioi et al., 2002).
Akt2 knockout mice (Akt2−/−), had normal cardiac function and geometry at the age of 2 months. However, these mice gradually developed LV hypertrophy and heart failure and this was associated with hyperglycemia (Etzion et al., 2010). Cardiomyocytes isolated from Akt2−/− mice showed impaired insulin-induced glucose uptake, but enhanced palmitate uptake and oxidation. The responses to IGF1 and endothelin-1 were comparable to WT (DeBosch et al., 2006a). TAC induced a similar LV hypertrophy in both Akt2−/− and WT mice (DeBosch et al., 2006a); whereas isoproterenol infusion induced more LV hypertrophy in Akt2−/− mice (Etzion et al., 2010). Myocardial infarction-induced a larger infarction area, increased apoptosis in the myocardium, and decreased FS% in Akt2−/− mice (Li et al., 2011).
Several transgenic mouse models with α-MHC promoter driven cardiomyocyte-specific overexpression of constitutively active mutants of Akt1 (caAkt, myr-Akt, and E40KAkt) and nuclear-targeted Akt (Akt/nuc) were generated. The caAkt mice had LV hypertrophy with increased cardiomyocyte size and myocardial fibrosis; and gradually developed LV systolic dysfunction (Shioi et al., 2002). The myr-Akt mouse lines had different phenotypes. Several lines of myr-Akt mice developed heart failure associated with early mortality. Survived lines had concentric cardiac hypertrophy and preserved cardiac function. Ischemia-reperfusion induced a smaller infarct size in myr-Akt hearts vs. WT, suggesting a protective role of Akt (Matsui et al., 2002). The E40KAkt mice also developed LV hypertrophy with increased cardiomyocyte size and cardiac contractility (Condorelli et al., 2002). On the other hand, the Akt/nuc mice did not develop LV hypertrophy. Rather, cardiomyocyte volume was smaller and the number of cardiomyocytes was increased. The cardiac function and myocyte contractility were enhanced in Akt/nuc mice at the baseline. Ischemia-reperfusion induced a smaller infarct size in Akt/nuc hearts (Shiraishi et al., 2004; Rota et al., 2005) (Table 2).
Mice with Inhibition/Activation of mTOR in the Heart
The cardiac effects of mTOR inhibition were tested in transgenic mice with cardiomyocyte-specific overexpression of a dominant negative or constitutively active mTOR, a common component of mTORC1 and mTORC2 (Shen et al., 2008), in mice with cardiomyocyte-specific conditional deletion of mTOR, in mice with cardiomyocyte-specific deletion of raptor, a specific subunit of mTORC1, and in mice that treated with rapamycin (Shioi et al., 2003; Boluyt et al., 2004; McMullen et al., 2004; Gao et al., 2006; Soesanto et al., 2009; Zhang et al., 2010; Shende et al., 2011).
In the first study, Shen et al. (2008) generated mice with cardiomyocyte-specific overexpression of a dominant negative mTOR (mTORkd) by overexpressing a mutant mTOR (a point mutation at aspartic acid residue 2338 leds to preventing mTOR auto-phosphorylation and kinase activity) under the control of the α-MHC promoter. The phosphorylation of S6 was decreased in these mice hearts, suggesting the inhibition of mTORC1 activity. However, the phosphorylation of Akt (S473) was unchanged. At baseline, mTORkd mice had normal cardiac gross morphology and histology. The weight and the size of the heart were normal. However, cardiac function as assessed by fractional shortening (FS%) and ejection fraction (EF%) was gradually decreased. Additionally, the heart rate was decreased; P-duration, P-R interval, and RR interval were increased in mTORkd mice. Isoproterenol infusion in mice caused a similar degree of increases in HW/BW ratio and cardiomyocyte area, suggesting disruption of mTOR kinase activity does not change β-adrenergic signaling induced cardiac hypertrophy.
In the same study, mice with cardiomyocyte-specific overexpression of a constitutively active mTOR were generated (mTORca – mTOR mutant lacking amino acid residues 2430–2450 led to increased kinase activity). mTORca mice displayed normal cardiac morphology and cardiac function at the baseline (Shen et al., 2008). When treated with doxorubicin, mTORca overexpression alleviated doxorubicin-induced cardiac dysfunction and the reduction of cardiac mass, but did not decrease doxorubicin-induced cardiomyocyte apoptosis (Zhu et al., 2009).
In a second mouse model, mice with cardiomyocyte-specific conditional deletion of mTOR (αMHC-MCM/MTORfl/fl) were generated. The αMHC-MCM/MTORfl/fl mice gradually developed dilated cardiomyopathy 4 weeks after the induction of mTOR gene deletion (Zhang et al., 2010). This was associated with increased fibrosis, apoptosis, autophagy, and mitochondrial dysfunction in the heart. The activation of downstream targets of mTORC1 S6K1 and S6 was decreased; while the amount of 4E-BP1 (eukaryotic initiation factor 4E-dependent protein 1), a protein that inhibits cap-dependent initiation of protein translation (Beretta et al., 1996), was increased. The αMHC-MCM/MTORfl/fl mice rapidly developed dilated cardiomyopathy when they were subjected to TAC 1 week after the initiation of mTOR gene deletion, which was associated with increased amount of 4E-BP1. Cross-breeding of αMHC-MCM/MTORfl/fl with 4E-BP1 knockout mice improved survival and cardiac function in αMHC-MCM/MTORfl/fl mice. These results suggest that cardiac hypertrophy induced by TAC, which is an adaptive response of the heart, was abolished by mTOR gene deletion. Instead, the heart progressed rapidly to dilation and failure in αMHC-MCM/MTORfl/fl mice.
To study the cardiac effects specifically related to mTORC1 deletion, Shen et al. (2008) generated mice with cardiomyocyte-specific conditional knockout of raptor (αMHC-MCM/raptorfl/fl). Under the baseline condition, αMHC-MCM/raptorfl/fl mice gradually developed dilated cardiomyopathy 5–6 weeks after the induction of raptor gene deletion. Decreased palmitate oxidation and increased glucose oxidation were observed before cardiac dysfunction was evident. TAC induced a rapid development of dilated cardiomyopathy in αMHC-MCM/raptorfl/fl mice, which was associated with absence of hypertrophic response, increased fetal gene expressions, increased apoptosis, autophagy, and mitochondrial abnormalities.
The cardiac effects of inhibiting mTOR signaling were also studied using rapamycin. Rapamycin pre-treatment significantly reduced ascending aortic stenosis (AS) induced cardiac hypertrophic remodeling, as assessed by the HW/BW ratio and cardiac myocyte area, while cardiac function was not affected (Shioi et al., 2003). In mice with established LV hypertrophy induced by AS or TAC, or in spontaneously hypertensive rats, short term rapamycin injections (2 mg/kg, i.p. daily) partially reversed LV hypertrophy (McMullen et al., 2004; Gao et al., 2006; Soesanto et al., 2009). Rapamycin injections also reduced myocardial infarction (MI) induced cardiac remodeling, fibrosis, and cardiac dysfunction in mice (Di et al., 2012) (Table 2).
These findings in transgenic mice suggest that mTOR and mTORC1 are necessary for adaptive growth response for the heart under mechanical stress. mTOR is pivotal for protecting the cardiomyocyte from apoptosis and maintaining cardiac mitochondrial function. However, studies using rapamycin injections suggested a different role of mTOR complexes in cardiac hypertrophy and failure.
Conclusion
The HER2-PI3K pathway plays a pivotal role in cancer development as well as in maintaining the normal physiological function of the heart. Newly developed cancer therapies targeting this pathway will inevitably have a significant impact on the heart.
In addition to cancer control, the ultimate goal of cancer therapy is to prolong survival of cancer patients. Cancer treatment is no doubt a matter of urgency; however, toxic side-effects caused by cancer therapy, such as those affecting the cardiovascular system may be more life-threatening. In addition, the incidence of cardiac diseases caused by cancer therapy may significantly rise in cancer survivors. Currently, several strategies are in practice in order to avoid or minimize the cardiac toxicity which include screening patients for cardiac dysfunction before starting the treatment, closely monitoring cardiac function during the treatment, and stopping the treatment when cardiac dysfunction is detected. These strategies certainly will reduce the cardiac incidence of drugs in clinical trials. However, a significant number of patients will be excluded. The adaptation of these management strategies, at the same time, clearly suggests that cardiac side-effects have already become a major obstacle for effective cancer treatment. Reducing the cardiac toxicity caused by cancer therapy, therefore, is a major challenge in medical practice.
Since cancer cells frequently adopt survival signals that are equally important for the well being of normal tissues, it is hard to selectively target cancer cells while sparing normal tissues. Studies of the same molecules by oncologists and by cardiologists often result in opposing theories when it comes to clinical practice. For example, oncologists will inhibit growth signals (such as VEGF and Akt) in cancer, while cardiologists will increase these signals in a failing heart. It seems that these two fields cannot compromise with each other.
Experimental evidence, however, has provided us clues for reconsidering this view. First, results from transgenic mice may not represent the outcomes of pharmacological intervention. For example, transgenic mice with cardiomyocyte-specific overexpression of a dominant negative mTOR, or conditional deletion of mTOR or raptor, all developed cardiac dysfunction, especially in the presence of cardiac stress. However, rapamycin, an mTOR inhibitor, can reduce aortic stenosis or myocardial infarction-induced adverse cardiac remodeling and dysfunction. Second, studies have shown that feed-back loops, cross-talks, and redundant signals exist in the HER2-PI3K pathway. Inhibition of certain kinases of the pathway may reactivate others, or vice versa. For example, inhibition of S6K feed-back reactivates insulin signals in the cell (Um et al., 2004). Activation of Akt by β-ARs, in turn, can phosphorylate threonine residues on the insulin receptor β-subunit leading to desensitization of the insulin receptor and insulin resistance (Morisco et al., 2005; Tian, 2005). Third, inhibition of certain kinases within the HER2-PI3K, such as PI3Kp110γ, may be beneficial, as suggested by studies. Fourth, the dose and the duration of the pharmacological treatment may need to be adjusted. Treatment using a repeated low dose of inhibitors (metronomic chemotherapy) may significantly increase the therapeutic window (Klement et al., 2000; Francia et al., 2012).
In summary, to address this challenge, oncologists and cardiologists need to work closely together to further understand the biology of both diseases and develop new strategies to achieve effective cancer treatment and minimize cardiac toxicity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by the NIGMS GM093050 (Giannoula L. Klement), NASA NNX11AD22G (David Goukassian), NCI U54CA149233 (Lynn Hlatky), American Heart Association Grant-In-Aid 10GRNT4710003 (Xinhua Yan), and NHLBI HL106098 (Xinhua Yan). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute, NASA, AHF or the National Institutes of Health.
Footnotes
References
Aoyagi, T., and Matsui, T. (2011). Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 17, 1818–1824.
Beretta, L., Gingras, A. C., Svitkin, Y. V., Hall, M. N., and Sonenberg, N. (1996). Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 15, 658–664.
Boluyt, M. O., Li, Z. B., Loyd, A. M., Scalia, A. F., Cirrincione, G. M., and Jackson, R. R. (2004). The mTOR/p70S6K signal transduction pathway plays a role in cardiac hypertrophy and influences expression of myosin heavy chain genes in vivo. Cardiovasc. Drugs Ther. 18, 257–267.
Brazil, D. P., Yang, Z. Z., and Hemmings, B. A. (2004). Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem. Sci. 29, 233–242.
Bruning, J. C., Michael, M. D., Winnay, J. N., Hayashi, T., Horsch, D., Accili, D., Goodyear, L. J., and Kahn, C. R. (1998). A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2, 559–569.
Ceci, M., Ross, J. Jr., and Condorelli, G. (2004). Molecular determinants of the physiological adaptation to stress in the cardiomyocyte: a focus on AKT. J. Mol. Cell. Cardiol. 37, 905–912.
Chaanine, A. H., and Hajjar, R. J. (2011). AKT signalling in the failing heart. Eur. J. Heart Fail. 13, 825–829.
Chang, Z., Zhang, Q., Feng, Q., Xu, J., Teng, T., Luan, Q., Shan, C., Hu, Y., Hemmings, B. A., Gao, X., and Yang, Z. (2010). Deletion of Akt1 causes heart defects and abnormal cardiomyocyte proliferation. Dev. Biol. 347, 384–391.
Cho, H., Thorvaldsen, J. L., Chu, Q., Feng, F., and Birnbaum, M. J. (2001). Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 276, 38349–38352.
Citri, A., Skaria, K. B., and Yarden, Y. (2003). The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp. Cell Res. 284, 54–65.
Citri, A., and Yarden, Y. (2006). EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol. Cell Biol. 7, 505–516.
Condorelli, G., Drusco, A., Stassi, G., Bellacosa, A., Roncarati, R., Iaccarino, G., Russo, M. A., Gu, Y., Dalton, N., Chung, C., Latronico, M. V., Napoli, C., Sadoshima, J., Croce, C. M., and Ross, J. Jr. (2002). Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 12333–12338.
Crackower, M. A., Oudit, G. Y., Kozieradzki, I., Sarao, R., Sun, H., Sasaki, T., Hirsch, E., Suzuki, A., Shioi, T., Irie-Sasaki, J., Sah, R., Cheng, H. Y., Rybin, V. O., Lembo, G., Fratta, L., Oliveira-Dos-Santos, A. J., Benovic, J. L., Kahn, C. R., Izumo, S., Steinberg, S. F., Wymann, M. P., Backx, P. H., and Penninger, J. M. (2002). Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 110, 737–749.
Crone, S. A., Zhao, Y. Y., Fan, L., Gu, Y., Minamisawa, S., Liu, Y., Peterson, K. L., Chen, J., Kahn, R., Condorelli, G., Ross, J. Jr., Chien, K. R., and Lee, K. F. (2002). ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 8, 459–465.
DeBosch, B., Sambandam, N., Weinheimer, C., Courtois, M., and Muslin, A. J. (2006a). Akt2 regulates cardiac metabolism and cardiomyocyte survival. J. Biol. Chem. 281, 32841–32851.
DeBosch, B., Treskov, I., Lupu, T. S., Weinheimer, C., Kovacs, A., Courtois, M., and Muslin, A. J. (2006b). Akt1 is required for physiological cardiac growth. Circulation 113, 2097–2104.
Di, R., Wu, X., Chang, Z., Zhao, X., Feng, Q., Lu, S., Luan, Q., Hemmings, B. A., Li, X., and Yang, Z. (2012). S6K inhibition renders cardiac protection against myocardial infarction through PDK1 phosphorylation of Akt. Biochem. J. 441, 199–207.
Di, R. M., Feng, Q. T., Chang, Z., Luan, Q., Zhang, Y. Y., Huang, J., Li, X. L., and Yang, Z. Z. (2010). PDK1 plays a critical role in regulating cardiac function in mice and human. Chin. Med. J. 123, 2358–2363.
Dickerson, E. B., Blackburn, W. H., Smith, M. H., Kapa, L. B., Lyon, L. A., and Mcdonald, J. F. (2010). Chemosensitization of cancer cells by siRNA using targeted nanogel delivery. BMC Cancer 10, 10. doi:10.1186/1471-2407-10-10
Dorn, G. W. II, and Force, T. (2005). Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Invest. 115, 527–537.
Dummler, B., and Hemmings, B. A. (2007). Physiological roles of PKB/Akt isoforms in development and disease. Biochem. Soc. Trans. 35, 231–235.
Etzion, S., Etzion, Y., Debosch, B., Crawford, P. A., and Muslin, A. J. (2010). Akt2 deficiency promotes cardiac induction of Rab4a and myocardial beta-adrenergic hypersensitivity. J. Mol. Cell. Cardiol. 49, 931–940.
Francia, G., Shaked, Y., Hashimoto, K., Sun, J., Yin, M., Cesta, C., Xu, P., Man, S., Hackl, C., Stewart, J., Uhlik, M., Dantzig, A. H., Foster, S., and Kerbel, R. S. (2012). Low-dose metronomic oral dosing of a prodrug of Gemcitabine (LY2334737) causes anti-tumor effects in the absence of inhibition of systemic vasculogenesis. Mol. Cancer Ther. 11, 680–689.
Fruman, D. A., Meyers, R. E., and Cantley, L. C. (1998). Phosphoinositide kinases. Annu. Rev. Biochem. 67, 481–507.
Gaidarov, I., and Keen, J. H. (1999). Phosphoinositide-AP-2 interactions required for targeting to plasma membrane clathrin-coated pits. J. Cell Biol. 146, 755–764.
Gao, X. M., Wong, G., Wang, B., Kiriazis, H., Moore, X. L., Su, Y. D., Dart, A., and Du, X. J. (2006). Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J. Hypertens. 24, 1663–1670.
Garcia-Echeverria, C., and Sellers, W. R. (2008). Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 27, 5511–5526.
Ghigo, A., Morello, F., Perino, A., Damilano, F., and Hirsch, E. (2011). Specific PI3K isoform modulation in heart failure: lessons from transgenic mice. Curr. Heart Fail Rep. 8, 168–175.
Guy, P. M., Platko, J. V., Cantley, L. C., Cerione, R. A., and Carraway, K. L. III. (1994). Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc. Natl. Acad. Sci. U.S.A. 91, 8132–8136.
Hait, W. N., and Hambley, T. W. (2009). Targeted cancer therapeutics. Cancer Res. 69, 1263–1267; discussion 1267.
Haubner, B. J., Neely, G. G., Voelkl, J. G., Damilano, F., Kuba, K., Imai, Y., Komnenovic, V., Mayr, A., Pachinger, O., Hirsch, E., Penninger, J. M., and Metzler, B. (2010). PI3Kgamma protects from myocardial ischemia and reperfusion injury through a kinase-independent pathway. PLoS ONE 5, e9350. doi:10.1371/journal.pone.0009350
Hers, I., Vincent, E. E., and Tavare, J. M. (2011). Akt signalling in health and disease. Cell. Signal. 23, 1515–1527.
Heineke, J., and Molkentin, J. D. (2006). Regulation of cardiac hypertrophy by intracellular signaling pathways. Nat. Rev. Mol. Cell Biol. 7, 589–600.
Hershman, D. L., and Shao, T. (2009). Anthracycline cardiotoxicity after breast cancer treatment. Oncology 23, 227–234.
Hirsch, E., Katanaev, V. L., Garlanda, C., Azzolino, O., Pirola, L., Silengo, L., Sozzani, S., Mantovani, A., Altruda, F., and Wymann, M. P. (2000). Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287, 1049–1053.
Ito, K., Akazawa, H., Tamagawa, M., Furukawa, K., Ogawa, W., Yasuda, N., Kudo, Y., Liao, C. H., Yamamoto, R., Sato, T., Molkentin, J. D., Kasuga, M., Noda, T., Nakaya, H., and Komuro, I. (2009). PDK1 coordinates survival pathways and beta-adrenergic response in the heart. Proc. Natl. Acad. Sci. U.S.A. 106, 8689–8694.
Klapper, L. N., Glathe, S., Vaisman, N., Hynes, N. E., Andrews, G. C., Sela, M., and Yarden, Y. (1999). The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. U.S.A. 96, 4995–5000.
Klein, P. M., and Dybdal, N. (2003). Trastuzumab and cardiac dysfunction: update on preclinical studies. Semin. Oncol. 305 (5 Suppl. 16), 49–53.
Klement, G., Baruchel, S., Rak, J., Man, S., Clark, K., Hicklin, D. J., Bohlen, P., and Kerbel, R. S. (2000). Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J. Clin. Invest. 105, R15–R24.
Lawlor, M. A., and Alessi, D. R. (2001). PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell. Sci. 114, 2903–2910.
Lee, K. F., Simon, H., Chen, H., Bates, B., Hung, M. C., and Hauser, C. (1995). Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 378, 394–398.
Li, X., Mikhalkova, D., Gao, E., Zhang, J., Myers, V., Zincarelli, C., Lei, Y., Song, J., Koch, W. J., Peppel, K., Cheung, J. Y., Feldman, A. M., and Chan, T. O. (2011). Myocardial injury after ischemia-reperfusion in mice deficient in Akt2 is associated with increased cardiac macrophage density. Am. J. Physiol. Heart Circ. Physiol. 301, H1932–H1940.
Li, Z., Jiang, H., Xie, W., Zhang, Z., Smrcka, A. V., and Wu, D. (2000). Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science 287, 1046–1049.
Lin, R. C., Weeks, K. L., Gao, X. M., Williams, R. B., Bernardo, B. C., Kiriazis, H., Matthews, V. B., Woodcock, E. A., Bouwman, R. D., Mollica, J. P., Speirs, H. J., Dawes, I. W., Daly, R. J., Shioi, T., Izumo, S., Febbraio, M. A., Du, X. J., and Mcmullen, J. R. (2010). PI3K(p110 alpha) protects against myocardial infarction-induced heart failure: identification of PI3K-regulated miRNA and mRNA. Arterioscler. Thromb. Vasc. Biol. 30, 724–732.
Liu, P., Cheng, H., Roberts, T. M., and Zhao, J. J. (2009). Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8, 627–644.
Luo, J., Manning, B. D., and Cantley, L. C. (2003). Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4, 257–262.
Luo, J., Mcmullen, J. R., Sobkiw, C. L., Zhang, L., Dorfman, A. L., Sherwood, M. C., Logsdon, M. N., Horner, J. W., Depinho, R. A., Izumo, S., and Cantley, L. C. (2005). Class IA phosphoinositide 3-kinase regulates heart size and physiological cardiac hypertrophy. Mol. Cell. Biol. 25, 9491–9502.
Matsui, T., Li, L., Wu, J. C., Cook, S. A., Nagoshi, T., Picard, M. H., Liao, R., and Rosenzweig, A. (2002). Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 277, 22896–22901.
Matsui, T., and Rosenzweig, A. (2005). Convergent signal transduction pathways controlling cardiomyocyte survival and function: the role of PI 3-kinase and Akt. J. Mol. Cell. Cardiol. 38, 63–71.
McMullen, J. R., Amirahmadi, F., Woodcock, E. A., Schinke-Braun, M., Bouwman, R. D., Hewitt, K. A., Mollica, J. P., Zhang, L., Zhang, Y., Shioi, T., Buerger, A., Izumo, S., Jay, P. Y., and Jennings, G. L. (2007). Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 104, 612–617.
McMullen, J. R., Sherwood, M. C., Tarnavski, O., Zhang, L., Dorfman, A. L., Shioi, T., and Izumo, S. (2004). Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 109, 3050–3055.
McMullen, J. R., Shioi, T., Zhang, L., Tarnavski, O., Sherwood, M. C., Kang, P. M., and Izumo, S. (2003). Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 100, 12355–12360.
Mora, A., Davies, A. M., Bertrand, L., Sharif, I., Budas, G. R., Jovanovic, S., Mouton, V., Kahn, C. R., Lucocq, J. M., Gray, G. A., Jovanovic, A., and Alessi, D. R. (2003). Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J. 22, 4666–4676.
Morisco, C., Condorelli, G., Trimarco, V., Bellis, A., Marrone, C., Sadoshima, J., and Trimarco, B. (2005). Akt mediates the cross-talk between beta-adrenergic and insulin receptors in neonatal cardiomyocytes. Circ. Res. 96, 180–188.
Morris, J. K., Lin, W., Hauser, C., Marchuk, Y., Getman, D., and Lee, K. F. (1999). Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron 23, 273–283.
Muslin, A. J. (2011). Akt2: a critical regulator of cardiomyocyte survival and metabolism. Pediatr. Cardiol. 32, 317–322.
Naga Prasad, S. V., Laporte, S. A., Chamberlain, D., Caron, M. G., Barak, L., and Rockman, H. A. (2002). Phosphoinositide 3-kinase regulates beta2-adrenergic receptor endocytosis by AP-2 recruitment to the receptor/beta-arrestin complex. J. Cell Biol. 158, 563–575.
Nienaber, J. J., Tachibana, H., Naga Prasad, S. V., Esposito, G., Wu, D., Mao, L., and Rockman, H. A. (2003). Inhibition of receptor-localized PI3K preserves cardiac beta-adrenergic receptor function and ameliorates pressure overload heart failure. J. Clin. Invest. 112, 1067–1079.
Oudit, G. Y., Crackower, M. A., Eriksson, U., Sarao, R., Kozieradzki, I., Sasaki, T., Irie-Sasaki, J., Gidrewicz, D., Rybin, V. O., Wada, T., Steinberg, S. F., Backx, P. H., and Penninger, J. M. (2003). Phosphoinositide 3-kinase gamma-deficient mice are protected from isoproterenol-induced heart failure. Circulation 108, 2147–2152.
Oudit, G. Y., and Penninger, J. M. (2009). Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 82, 250–260.
Ozcelik, C., Erdmann, B., Pilz, B., Wettschureck, N., Britsch, S., Hubner, N., Chien, K. R., Birchmeier, C., and Garratt, A. N. (2002). Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 99, 8880–8885.
Patrucco, E., Notte, A., Barberis, L., Selvetella, G., Maffei, A., Brancaccio, M., Marengo, S., Russo, G., Azzolino, O., Rybalkin, S. D., Silengo, L., Altruda, F., Wetzker, R., Wymann, M. P., Lembo, G., and Hirsch, E. (2004). PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 118, 375–387.
Pretorius, L., Du, X. J., Woodcock, E. A., Kiriazis, H., Lin, R. C., Marasco, S., Medcalf, R. L., Ming, Z., Head, G. A., Tan, J. W., Cemerlang, N., Sadoshima, J., Shioi, T., Izumo, S., Lukoshkova, E. V., Dart, A. M., Jennings, G. L., and Mcmullen, J. R. (2009). Reduced phosphoinositide 3-kinase (p110alpha) activation increases the susceptibility to atrial fibrillation. Am. J. Pathol. 175, 998–1009.
Rota, M., Boni, A., Urbanek, K., Padin-Iruegas, M. E., Kajstura, T. J., Fiore, G., Kubo, H., Sonnenblick, E. H., Musso, E., Houser, S. R., Leri, A., Sussman, M. A., and Anversa, P. (2005). Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ. Res. 97, 1332–1341.
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101.
Shaw, R. J., and Cantley, L. C. (2006). Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441, 424–430.
Shen, W. H., Chen, Z., Shi, S., Chen, H., Zhu, W., Penner, A., Bu, G., Li, W., Boyle, D. W., Rubart, M., Field, L. J., Abraham, R., Liechty, E. A., and Shou, W. (2008). Cardiac restricted overexpression of kinase-dead mammalian target of rapamycin (mTOR) mutant impairs the mTOR-mediated signaling and cardiac function. J. Biol. Chem. 283, 13842–13849.
Shende, P., Plaisance, I., Morandi, C., Pellieux, C., Berthonneche, C., Zorzato, F., Krishnan, J., Lerch, R., Hall, M. N., Ruegg, M. A., Pedrazzini, T., and Brink, M. (2011). Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation 123, 1073–1082.
Shioi, T., Kang, P. M., Douglas, P. S., Hampe, J., Yballe, C. M., Lawitts, J., Cantley, L. C., and Izumo, S. (2000). The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 19, 2537–2548.
Shioi, T., Mcmullen, J. R., Kang, P. M., Douglas, P. S., Obata, T., Franke, T. F., Cantley, L. C., and Izumo, S. (2002). Akt/protein kinase B promotes organ growth in transgenic mice. Mol. Cell. Biol. 22, 2799–2809.
Shioi, T., Mcmullen, J. R., Tarnavski, O., Converso, K., Sherwood, M. C., Manning, W. J., and Izumo, S. (2003). Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation 107, 1664–1670.
Shiraishi, I., Melendez, J., Ahn, Y., Skavdahl, M., Murphy, E., Welch, S., Schaefer, E., Walsh, K., Rosenzweig, A., Torella, D., Nurzynska, D., Kajstura, J., Leri, A., Anversa, P., and Sussman, M. A. (2004). Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ. Res. 94, 884–891.
Slamon, D. J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., Fleming, T., Eiermann, W., Wolter, J., Pegram, M., Baselga, J., and Norton, L. (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792.
Soesanto, W., Lin, H. Y., Hu, E., Lefler, S., Litwin, S. E., Sena, S., Abel, E. D., Symons, J. D., and Jalili, T. (2009). Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension 54, 1321–1327.
Stephens, L. R., Eguinoa, A., Erdjument-Bromage, H., Lui, M., Cooke, F., Coadwell, J., Smrcka, A. S., Thelen, M., Cadwallader, K., Tempst, P., and Hawkins, P. T. (1997). The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 89, 105–114.
Stoyanov, B., Volinia, S., Hanck, T., Rubio, I., Loubtchenkov, M., Malek, D., Stoyanova, S., Vanhaesebroeck, B., Dhand, R., Nurnberg, B., Gierschik, P., Seedorf, K., Hsuan, J. J., Waterfield, M. D., and Wetzker, R. (1995). Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269, 690–693.
Tian, R. (2005). Another role for the celebrity: Akt and insulin resistance. Circ. Res. 96, 139–140.
Ueno, N. T., Bartholomeusz, C., Herrmann, J. L., Estrov, Z., Shao, R., Andreeff, M., Price, J., Paul, R. W., Anklesaria, P., Yu, D., and Hung, M. C. (2000). E1A-mediated paclitaxel sensitization in HER-2/neu-overexpressing ovarian cancer SKOV3.ip1 through apoptosis involving the caspase-3 pathway. Clin. Cancer Res. 6, 250–259.
Um, S. H., Frigerio, F., Watanabe, M., Picard, F., Joaquin, M., Sticker, M., Fumagalli, S., Allegrini, P. R., Kozma, S. C., Auwerx, J., and Thomas, G. (2004). Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205.
Yang, Z. Z., Tschopp, O., Hemmings-Mieszczak, M., Feng, J., Brodbeck, D., Perentes, E., and Hemmings, B. A. (2003). Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J. Biol. Chem. 278, 32124–32131.
Yap, T. A., Garrett, M. D., Walton, M. I., Raynaud, F., De Bono, J. S., and Workman, P. (2008). Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr. Opin. Pharmacol. 8, 393–412.
Yarden, Y., and Sliwkowski, M. X. (2001). Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137.
Yuan, T. L., and Cantley, L. C. (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510.
Zhang, D., Contu, R., Latronico, M. V., Zhang, J., Rizzi, R., Catalucci, D., Miyamoto, S., Huang, K., Ceci, M., Gu, Y., Dalton, N. D., Peterson, K. L., Guan, K. L., Brown, J. H., Chen, J., Sonenberg, N., and Condorelli, G. (2010). MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Invest. 120, 2805–2816.
Zhao, L., and Vogt, P. K. (2008). Class I PI3K in oncogenic cellular transformation. Oncogene 27, 5486–5496.
Zhu, W., Soonpaa, M. H., Chen, H., Shen, W., Payne, R. M., Liechty, E. A., Caldwell, R. L., Shou, W., and Field, L. J. (2009). Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119, 99–106.
Keywords: cardiotoxicity, cancer, HER2, PI3K, Akt, mTOR
Citation: Klement GL, Goukassian D, Hlatky L, Carrozza J, Morgan JP and Yan X (2012) Cancer therapy targeting the HER2-PI3K pathway: potential impact on the heart. Front. Pharmacol. 3:113. doi: 10.3389/fphar.2012.00113
Received: 30 April 2012; Accepted: 24 May 2012;
Published online: 27 June 2012.
Edited by:
Jufeng Wang, Waylandgreen, USAReviewed by:
Domenico Criscuolo, Genovax, ItalySreedharan Nair Sabarinath, Food and Drug Administration, USA
Copyright: © 2012 Klement, Goukassian, Hlatky, Carrozza, Morgan and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Xinhua Yan, Cardiovascular Medicine, Steward St. Elizabeth Medical Center, Tufts University School of Medicine, 736 Cambridge Street CBR 345, Boston, MA 02135, USA. e-mail: xinhua.yan@tufts.edu