 Xiaojuan Sun
Xiaojuan Sun Wei-Dong Chen
Wei-Dong Chen Yan-Dong Wang
Yan-Dong Wang- 1Key Laboratory of Receptors-Mediated Gene Regulation and Drug Discovery, School of Medicine, Henan University, Kaifeng, China
- 2State Key Laboratory of Chemical Resource Engineering, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing, China
The amyloid β peptide (Aβ) is a critical initiator that triggers the progression of Alzheimer’s Disease (AD) via accumulation and aggregation, of which the process may be caused by Aβ overproduction or perturbation clearance. Aβ is generated from amyloid precursor protein through sequential cleavage of β- and γ-secretases while Aβ removal is dependent on the proteolysis and lysosome degradation system. Here, we overviewed the biogenesis and toxicity of Aβ as well as the regulation of Aβ production and clearance. Moreover, we also summarized the animal models correlated with Aβ that are essential in AD research. In addition, we discussed current immunotherapeutic approaches targeting Aβ to give some clues for exploring the more potentially efficient drugs for treatment of AD.
Introduction
Alzheimer’s disease (AD), also known as Senile Dementia, is a most common age-related neurodegenerative disorder. More than 11 million people per year are estimated to suffer from this disease by 2050, leading to higher cost as well as more burdens on public health and society (Alzheimer’s, 2014, 2015). Featured by progressive memory loss and cognitive dysfunction, AD induces the loss of motor functions and personality changes, and eventually leads to death. Histopathologically, AD is mainly characterized by extracellular senile plaques (SPs) and intracellular neurofibrillary tangles (NFTs), which results in the loss of neurons and synapses and finally causes gross atrophy of the brain. NFTs are formed by the regulation of the abnormally hyperphosphorylated and glycosylated microtubule-related tau protein, whereas SPs are associated with the aggregation and deposition of amyloid β peptides (Aβ) (Mattson, 2004).
Aβ accumulation is considered to be the distinct morphological hallmark of early onset of AD and it is also proposed to be an activator to induce the sequential lesion events induced by the aggregation of P-Tau. Therefore, Aβ is predicted to be the most potentially efficient target of the drug therapies (Karran et al., 2011). Here, this review will focus on this peptide with the aspects of its biogenesis, regulations, as well as degradation and clearance to elucidate the potential significance of these processes for the clinic treatment of AD.
The Aβ Biogenesis, Toxicity, Production, and Clearance
The Biogenesis of Aβ
The factors involved in the pathogenesis of AD have been intensely investigated, however, the mechanisms governing this disease are not fully understood and remain debated. One prevailing proposal is the amyloid cascade hypothesis positing Aβ as the initiator of subsequent events that leads to AD (Figure 1A) (Hardy and Selkoe, 2002): Aβ peptides spontaneously aggregate and deposit into soluble oligomers, fibrils and SPs, which then induces oxidative injury, microglial and astrocytic activity as well as alters kinase/phosphatase activity, eventually leading to the neuronal death. Howerver, whether Aβ acts on tau aggregation is still debated (Musiek and Holtzman, 2015).
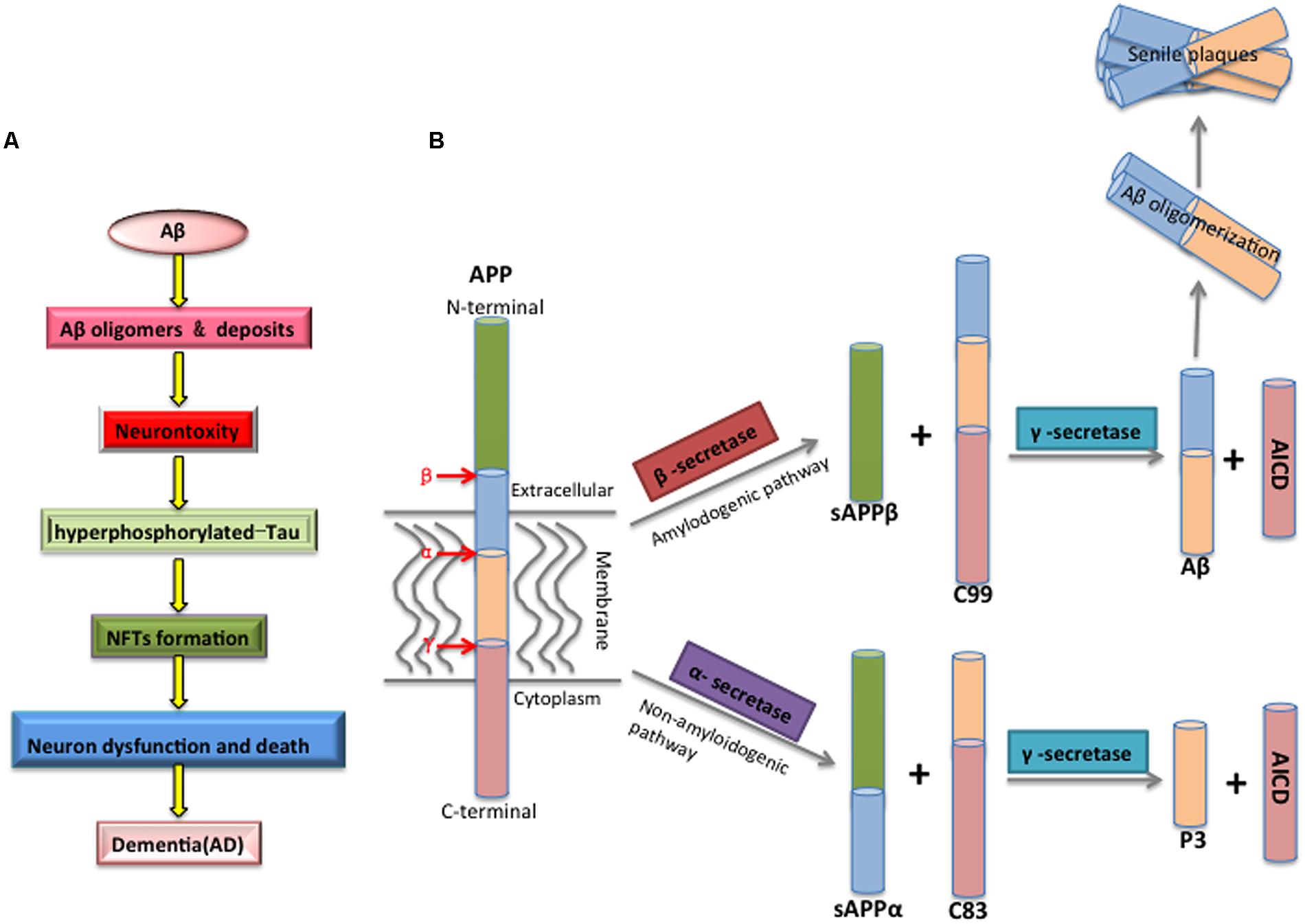
FIGURE 1. (A) The mechanism of Aβ toxicity. Accumulating Aβ will initially results in Aβ oligomerization, gradually deposits as the forms of fibrils and senile plaques. Furthermore, Aβ aggregation alters the kinase/phosphatase activity that leads to the Tau protein hyperphosphorylated, which causes the formation of neurofibrillary tangles (NFTs), and eventual synaptic and neuronal dysfunction and AD. (B) The proteolytic processing of the amyloid precursor protein (APP) and Aβ biogenesis. APP is a transmembrane glycoprotein with a large luminal domain and a short cytoplasmic domain, and it is processed through amyloidogenic or non-amyloidogenic pathway. The amyloidogenic pathway is the process of Aβ biogenesis: APP is firstly cleaved by β-secretase, producing soluble β-APP fragments (sAPPβ) and C-terminal β fragment (CTFβ, C99), and C99 is further cleaved by γ-secretase, generating APP intracellular domain (AICD) and Aβ. The non-amyloidogenic pathway is an innate way to prevent the generation of Aβ, as APP is firstly recognized by α-secretase within Aβ domain, generating soluble α-APP fragments (sAPPα) and C-terminal fragment α (CTFα, C83), C83 is then cleaved by γ-secretase, producing non-toxic P3 and AICD fragments.
Aβ is a small protein composed of 39–43 amino acids with a variety of biophysical states. There are two major isoforms of Aβ, soluble Aβ40, and insoluble Aβ42, and the latter peptide showing higher percentage concentration in AD patients is more prone to aggregate (Burdick et al., 1992; Gravina et al., 1995; Kim et al., 2007). In a physiological condition, more than 90% of Aβ is in the form of Aβ40 while less than 5% is generated as the longer form of Aβ42.
Aβ is derived by proteolysis of an evolutionary conserved large transmembrane amyloid precursor protein (APP) through cleavage of β-secretase followed by γ-secretase. Mutations in the gene encoding APP are the main causes of familial AD (FAD; Chartier-Harlin et al., 1991; Goate et al., 1991). APP can also be processed by α-secretase via non-amyloidogenic pathways to produce non-toxic fragments, which is thought to antagonize Aβ generation (Figure 1B; Gandy et al., 1994; Sahlin et al., 2007).
Most of intracellular Aβ normally distribute in the neuronal cytosol, but it is also colocalized with different organelles dependent on where APP, β- and γ-secretase reside. In particular, it has been reported to be produced in the secretory pathway related organelles including endoplasmic reticulum (ER), medial Golgi saccules as well as trans-Golgi network (Hartmann et al., 1997; Greenfield et al., 1999). It has also been found to be correlated with the endocytic endosomes/lysosomes and autophagic vacuoles (Koo and Squazzo, 1994; Yu et al., 2004). Besides, Aβ also resides in mitochondria (Muirhead et al., 2010).
Toxicity of Aβ
The physiological role of Aβ is still unknown, but it indeed exists throughout life in healthy individuals. One possible function is to inhibit the activity of γ-secretase in a negative feedback way (Kamenetz et al., 2003).
Aβ aggregation is considered to be the primary reason for the neurotoxicity in the classic view, and Aβ oligomers are the most neurotoxic form (Walsh et al., 2002). Three “developed-stimulators” may facilitate the aggregation process. The absolute levels of Aβ42 increased by production via APP processing, the ratio of Aβ42 to Aβ40 elevated due to the decreased level of Aβ40 or the soluble oligomeric Aβ (Glabe, 2005; Walsh and Selkoe, 2007). These potential stimulators further promote the accumulation and deposition of Aβ to develop into SPs, which eventually contributes to AD pathology. Moreover, Aβ-induced apoptosis through interaction with cell surface receptors and proteins is also thought to dedicate to the dysfunction of neuronal system (Small et al., 2001; Zhu et al., 2015).
The aggregation of Aβ might also promote free radicals such as reactive oxidative species (ROS) to react rapidly with several moieties of proteins and lipids, whose structures or functions are then altered to potential “toxic” oxidized proteins and peroxided lipids. Protein oxidation may cause harm to the membrane integrity or damage the sensitivity to oxidative modification of the enzymes such as glutamine synthetase (GS) and creatine kinase (CK), which are critical to neuronal function (Aksenov et al., 1995; Yatin et al., 1999). Lipids peroxidation usually causes the toxic product such as 4-hydroxy-2-nonenal (HNE) and 2-propenal (acrolein) that migrates to different parts of the neurons to cause multiple deleterious alterations of cellular function. It includes loss of Ca2+ homoeostasis, inhibition of ion-motive ATPases and glial cell Na+-dependent glutamate and disruption of signaling pathways, all of which are associated with neuronal death (Mark et al., 1995; Varadarajan et al., 2000; Ezeani and Omabe, 2015). Aβ-induced oxidative stress has also been reported to cause the DNA oxidation, leading to DNA damage (Varadarajan et al., 2000).
Continuous Aβ aggregation or sustained elevation of Aβ would cause a chronic response of the innate immune system by activating microglia through some immunological receptors such as Toll-like Receptors 2 (TLR2), TLR4, TLR6, their co-receptors CD14, CD36, and CD47, which can probably destroy functional neurons by direct phagocytosis (Weggen et al., 2001; Neniskyte et al., 2011; Liu et al., 2012). Besides, it also results in inflammatory response, concomitantly releasing a lot of inflammation related mediators including complement factors, eicosanoids, chemokines, and proinflammatory cytokines, which can impair microglial clearance of Aβ and the neuronal debris and increase microglia-mediated neuronal death and loss of neuronal synapses, contributing greatly to AD pathogenesis. Aβ deposition also induces tau pathology by promoting the intraneuronal formation of NFTs which consist of hyperphosphorylated tau proteins. It influences the late-stage of AD pathogenesis. The process is probably mediated by the microglia-driven neuroinflammatory response or by indirectly regulating kinase/phosphatase activity (Heneka et al., 2015).
In addition, Aβ precursor APP accumulation at mitochondria membrane can cause mitochondrial dysfunction by blocking the translocation of other mitochondrial inner molecules/proteins and disrupting the electron-transport chain (ETC; Anandatheerthavarada et al., 2003; Devi et al., 2006), which may in turn increase excessive Aβ generation to result in more toxicity. Excessive Aβ can also increase mitochondrial ROS production to induce mitochondrial fragmentation by activating mitochondrial fission proteins Drp1 and Fis1 (Barsoum et al., 2006). Aβ localized in mitochondria can bind to two pro-apoptotic factors including Aβ-binding alcohol dehydrogenase (ABAD) and cyclophin D (CypD), consequently increasing neurodegenerative cell death that may be toxic to neurons (Lustbader et al., 2004; de Moura et al., 2010). Hence, there may be a vicious feedback loop between increased Aβ production and mitochondria dysfunction.
Regulations of Aβ Production and Clearance
Because of the key role of Aβ in AD pathogenesis, it has been well accepted that reducing Aβ production or enhancing Aβ clearance may be a putative way to inhibit the cascade of Aβ-induced pathological events.
Aβ biogenesis is tightly correlated with APP metabolism, including processing and trafficking. There are three isoforms of APP, APP695, APP751, and APP770 (Goate et al., 1991). APP695 lacking the Kunitz-type protease inhibitor (KPI) domain is predominantly expressed in neurons while the other two isoforms are distributed in most tissues (Kang and Muller-Hill, 1990; Rohan de Silva et al., 1997). Some evidence show that APP751 and APP770 up-expression in brains are primarily associated to Aβ deposition (Menendez-Gonzalez et al., 2005; Bordji et al., 2010). APP processing is mainly regulated by α, β, and γ-secretases (Table 1A). Alpha-secretase plays an essential role in precluding the generation of intact Aβ on account of the cleavage site within the Aβ domain. As a membrane-bound endoprotease, α-secretase usually cleaves APP at plasma membrane (Sisodia, 1992). Several members of the a Disintegrin and metalloproteinase (ADAM) family listed in Table 1A have been reported to possess α-secretase activity, which is responsible for APP processing (Koike et al., 1999; Harold et al., 2007; Tanabe et al., 2007; Kim et al., 2009). β- and γ-secretases are devoted to Aβ production via amylodogenic pathway (Figure 1B). Beta-site APP cleaving enzyme 1 (BACE1) and BACE2 are the β-secretases while γ-secretase is complex and composed of presenilins (PSEN1 or PSEN2), Nicastrin, Presenilin enhancer 2 (PEN2), and anterior pharynx defective 1 (APH-1; De Strooper, 2003). Substantial evidence has shown that manipulation of these secretases could perturb the generation of Aβ. For example, with the α-cleavage abolished in ADAM17-deficient cells affectd Aβ generation (Buxbaum et al., 1998). Knock-out of BACE1 in mice completely depleted neuronal Aβ secretion (Cai et al., 2001). Mutations of PSEN1 and PSEN2 affected APP cleavage, thereby altering Aβ production (Wang et al., 2010). Moreover, regulators related with these secratase, such as the γ-secretase activating protein (GSAP) and CD147, are also likely to be involved in the generation of Aβ (Zhou et al., 2005; He et al., 2010).

TABLE 1A. Member proteins of three secretases.
Like other type I transmembrane proteins, APP is synthesized and translocated into ER followed by matured in the Golgi apparatus where APP is mainly concentrated in neurons at the steady state (Hartmann et al., 1997; Xu et al., 1997; Greenfield et al., 1999; Caster and Kahn, 2013). And then APP would traffic through the constitutive secretory pathway. Once reaching the cell surface, it is either cleaved by α-secretase to produce sAPPα (Sisodia, 1992) or rapidly re-internalized by recognition of a “YENPTY” motif and subsequently recycled back to the cell surface by the recycling compartments or delivered to the lysosome for degradation through the endosomal–lysosomal systems (Golde et al., 1992; Caster and Kahn, 2013). Generally, promoting APP delivery or inhibiting APP internalization from the cell surface favors the non-amyloidogenic processing, thereafter antagonizing the generation of Aβ. Elevating retention of APP in acidic compartments such as endosomes greatly adds the chances for amyloidogenic processing and consequent Aβ production. Mutations within the “YENPTY” internalization motif have been addressed to block APP internalization and consequently decrease Aβ generation (Perez et al., 1999). In contrast, mutation within extracellular KPI domain existing in APP751 and APP770 that assists APP sorting to plasma membrane causes APP retention in the ER, thereby elevating the Aβ production (Ben Khalifa et al., 2012). Synaptic transmission indicated to accelerate APP endocytosis has also been shown to result in increasing the level of secreted Aβ (Cirrito et al., 2005). Some general modulators that could regulate APP trafficking, such as dynamin I (Carey et al., 2005), the RAB GTPase family including RAB1B, RAB6, RAB8, and RAB11 (Huber et al., 1993; Dugan et al., 1995; McConlogue et al., 1996; Thyrock et al., 2013; Udayar et al., 2013), and the SNX family including SNX17 and SNX33 (Lee et al., 2008; Schobel et al., 2008), have also been found to be associated with Aβ generation. In addition, factors that function in the trafficking of the three secretases may also change the production of Aβ by affecting APP processing (Wahle et al., 2006; Wen et al., 2011; Bhalla et al., 2012). The G-protein-coupled receptor protein GPR3, which is responsible for the cell surface localization of matured γ-secretase, stimulates Aβ production when it is overexpression (Thathiah et al., 2009).
Proteolytic degradation is thought to take a large part of responsibility in preventing Aβ aggregation or deposition into plaques. The enzymes or proteases in proteolytic degradation play important roles by cleaving Aβ into shorter soluble fragments without neurotoxic effect. The proteases including cathepsin B (CatB), cathepsin D (CatD), Gelatinase A, serine protease factor Xia (FXIa), matrix metalloprotein-9 (MMP-9), neprilysin (NEP), presequence protease (Prep) and the α2M complex are involved in Aβ clearance (Saporito-Irwin and Van Nostrand, 1995; Yamada et al., 1995; Hamazaki, 1996; Carvalho et al., 1997; Iwata et al., 2001; Mueller-Steiner et al., 2006; King et al., 2014), while enzymes such as angiotensin-converting enzyme (ACE), endothelin-converting enzyme (ECE), insulin-degrading enzyme (IDE), and uPT and tPA have been found to be involved in the degradation of Aβ (Table 1B; Ledesma et al., 2000; Tucker et al., 2002; Eckman et al., 2003; Farris et al., 2003; Hemming and Selkoe, 2005; Baranello et al., 2015).
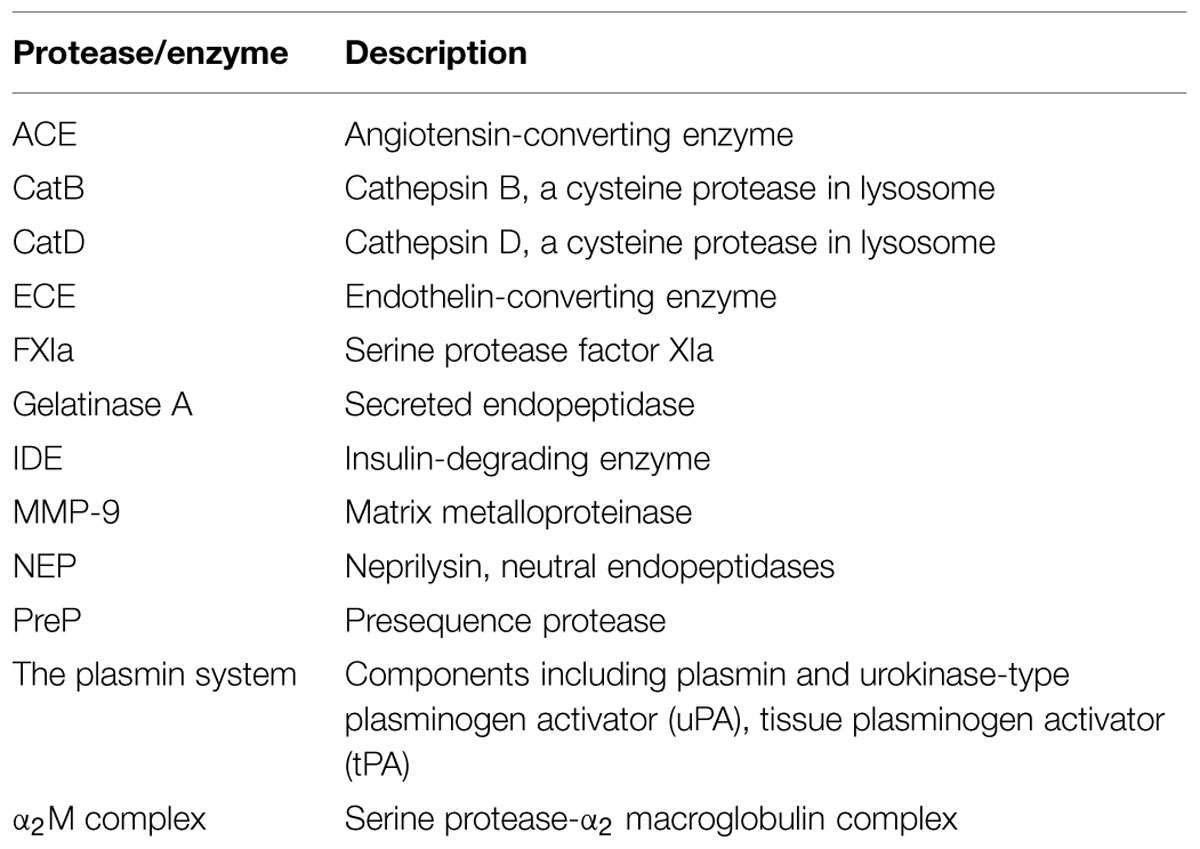
TABLE 1B. Proteases/enzymes involved in the cleavage of Aβ peptide.
Besides the proteolysis for Aβ degradation, receptor-mediated endocytosis of Aβ that delivers Aβ to lysosome for degradation also contributes to the clearance of toxic Aβ peptide and Aβ deposits. Low-density lipoprotein receptor-related protein 1(LRP1) is considered to be the vital modulator in this process by probably direct binding to Aβ for uptake (Li et al., 2000) or through Aβ receptor such as heparin sulphate proteoglycan (HSPG; Kanekiyo et al., 2011) and GPI-anchored cellular prion protein (PrPc; Taylor and Hooper, 2007) to facilitate Aβ trafficking. In addition, Aβ aggregates may also undergo maropinocytosis or phagocytosis for clearance, of which the critical step about actin polymerization is regulated by LRP1(Kanekiyo and Bu, 2014). Apolipoprotein E (ApoE), as a major ligand for LRP1 and an important partner of Aβ, plays dual roles in Aβ clearance (Li et al., 2012; Kanekiyo and Bu, 2014). Moreover, induction of another degrading pathway of autophagy serves to accelerate the clearance of both soluble Aβ and Aβ aggregates (Nixon, 2007).
Animal Models Related with Aβ for AD
Various types of animal models related to Aβ have been created to dissect the mechanisms for the development and progression of AD, the majority are overexpression transgenic lines (see the summary in Table 1C; Oakley et al., 2006; Elder et al., 2010; Do Carmo and Cuello, 2013; Lublin and Link, 2013; Lim et al., 2016).
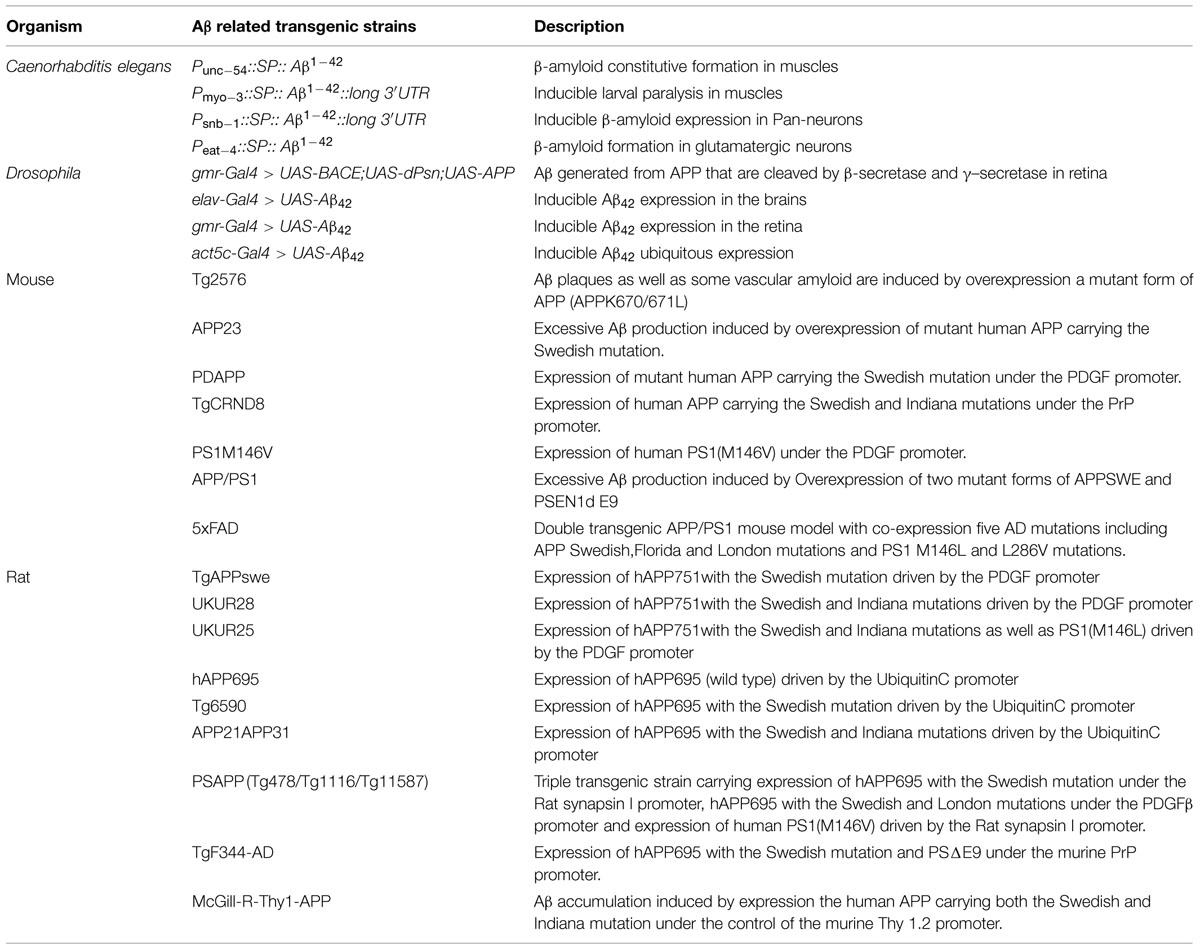
TABLE 1C. Summary for Aβ related transgenic animal models.
Despite the existing innate disadvantages. e.g., the transgenic flies that express both human APP and β-secretase BACE1 displayed Aβ accumulation, the animal models are useful to screen genes involved in APP processing (Ye and Fortini, 1999; Greeve et al., 2004), making a great contribution to the development of this field. The secreted-Aβ model in Drosophila is a direct approach to investigate the toxicity caused by Aβ (Finelli et al., 2004; Crowther et al., 2005; Iijima et al., 2008). The Caenorhabditis elegans Aβ-expressing models developed in different tissues are also helpful for examining genes involved in Aβ-induced toxicity (Link, 2006; Wu et al., 2006). Phenotypes were also analyzed in zebrafish through high-throughout screen by treatment with Alzheimer’s γ-secretase inhibitors to determine efficient compounds for blocking Aβ generation (Arslanova et al., 2010).
Aβ infusion models are that different species of Aβ peptides is directly injected in the rodent brains. They could mimic the most aspects of AD and deliver experimental results for analysis in a relatively short time (Nag et al., 1999; Harkany et al., 2001; Nakamura et al., 2001). However, these approaches usually induce much higher levels of Aβ in the brains than that exists in the patients, and the results usually vary due to differences in methodology and the concentration of Aβ and the duration treatment. Although most of the models do not show Tau pathology and other shedding fragments from APP processing may also influence neuron systems, transgenic rodent models with overexpression of wild type or mutated human APP can recapitulate some features of AD pathology and provide great convenience to discover more regulators involved in the onset of AD (Clarke et al., 2007; Agca et al., 2008; Leon et al., 2010; Rosen et al., 2012). Nevertheless, no model system is impeccable, further understanding of the molecular mechanisms for Aβ-initiated AD pathology would still be desirable.
Overviews of Current Therapeutics Targeting Aβ
According to the conventional approaches targeting Aβ, therapeutic strategies focus on reducing Aβ production via inhibition of β- and γ-secretases to prevent Aβ aggregation and facilitate Aβ clearance. However, the results are not so inspiring, as all the strategies have failed in clinical trials. Recently, immunotherapies by two monoclonal antibodies against Aβ have been tried. One is Bapineuzumab that could recognize both soluble and insoluble forms of Aβ; the other one is Solanezumab that targets Aβ central domain and recognizes only soluble Aβ. Yet both of them failed to improve the clinical outcomes in patients in phage III trials (Doody et al., 2014a,b; Salloway et al., 2014), which suggests that targeting Aβ alone might not be enough to impede AD progression and multiple steps of Aβ modulations should be taken into consideration according to the different clinical phenotypes in AD patients. e.g., the activity of Foxp3+ regulatory T cells (Tregs) has been reported to be related with Aβ plaque clearance, suggesting novel immunosuppression curing way (Baruch et al., 2015). Moreover, other approaches besides immunotherapy also need to be explored in order to understand multiple regulations of Aβ for the development of therapies for treating AD.
Conclusion and Perspectives
The vital role of Aβ as an initiator in the pathology of AD has been well accepted. Aβ production mainly depends on APP processing, whereas Aβ removal is largely associated with proteases and lysosomal enzymes. Subcellularlly, Aβ production together with Aβ precursor protein APP seems closely related with mitochondria, the major source of energy for the brain. Mitochondrial changes including increasing ROS production and reducing ATP generation are in an age-dependent manner. ROS-related oxidative stress induces more Aβ production, while Aβ and APP localized to mitochondrial membranes cause mitochondrial damage by elevating ROS production, blocking the transport of nuclear-encoded mitochondrial protein and disrupting ETC activities. However, the mechanisms of Aβ and APP transport into mitochondrial membranes are still unknown. Future work focus on this part might provide well understanding between mitochondria and APP as well as Aβ production, which might be helpful for exploring new compounds.
On the other hand, microglial cells play very important roles in the removal of accumulated Aβ not only by phagocytosis but also by releasing proteases such as IDE for degradation, and it is also associated with the innate immune system induced by the aggregated Aβ. Therefore, further researches are needed to find how to keep the clearance function of microglial cells without being impaired by the proinflammatory cytokines.
Although tremendous progress has been made in the development therapeutic strategies targeting Aβ, more work are still needed to find efficient drugs for curing AD. Network regulations of Aβ should be taken into consideration for the therapy approaches, and it would be instrumental to create good animal models and find more specific biomarkers for the Aβ-mediated pathogenesis of AD.
Funding
This work is supported by the National Natural Science Foundation of China (Grant No. 81370537) and the Fundamental Research Funds for the Central Universities (Grant No. YS1407 and 2050205) to YW, the National Natural Science Foundation of China (Grant No. 81270522 and Grant No. 81472232), Program for Science & Technology Innovation Talents in Universities of Henan Province (HASTIT, Grant No. 13HASTIT024) and Plan for Scientific Innovation Talent of Henan Province to WC, and the Scientific Research foundation of Henan University (Grant No. 2013YBZR036) to XS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agca, C., Fritz, J. J., Walker, L. C., Levey, A. I., Chan, A. W., Lah, J. J., et al. (2008). Development of transgenic rats producing human beta-amyloid precursor protein as a model for Alzheimer’s disease: transgene and endogenous APP genes are regulated tissue-specifically. BMC Neurosci. 9:28. doi: 10.1186/1471-2202-9-28
Aksenov, M., Aksenova, M. V., Harris, M. E., Hensley, K., Butterfield, D. A., and Carney, J. M. (1995). Enhancement of beta-amyloid peptide A beta(1-40)-mediated neurotoxicity by glutamine synthetase. J. Neurochem. 65, 1899–1902. doi: 10.1046/j.1471-4159.1995.65041899.x
Alzheimer’s, A. (2014). 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 10, e47–e92. doi: 10.1016/j.jalz.2014.02.001
Alzheimer’s, A. (2015). 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 11, 332–384. doi: 10.1016/j.jalz.2015.02.003
Anandatheerthavarada, H. K., Biswas, G., Robin, M. A., and Avadhani, N. G. (2003). Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 161, 41–54. doi: 10.1083/jcb.200207030
Arslanova, D., Yang, T., Xu, X., Wong, S. T., Augelli-Szafran, C. E., and Xia, W. (2010). Phenotypic analysis of images of zebrafish treated with Alzheimer’s gamma-secretase inhibitors. BMC Biotechnol. 10:24. doi: 10.1186/1472-6750-10-24
Baranello, R. J., Bharani, K. L., Padmaraju, V., Chopra, N., Lahiri, D. K., Greig, N. H., et al. (2015). Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr. Alzheimer Res. 12, 32–46. doi: 10.2174/1567205012666141218140953
Barsoum, M. J., Yuan, H., Gerencser, A. A., Liot, G., Kushnareva, Y., Graber, S., et al. (2006). Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25, 3900–3911. doi: 10.1038/sj.emboj.7601253
Baruch, K., Rosenzweig, N., Kertser, A., Deczkowska, A., Sharif, A. M., Spinrad, A., et al. (2015). Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 6, 7967. doi: 10.1038/ncomms8967
Ben Khalifa, N., Tyteca, D., Marinangeli, C., Depuydt, M., Collet, J. F., Courtoy, P. J., et al. (2012). Structural features of the KPI domain control APP dimerization, trafficking, and processing. FASEB J. 26, 855–867. doi: 10.1096/fj.11-190207
Bhalla, A., Vetanovetz, C. P., Morel, E., Chamoun, Z., Di Paolo, G., and Small, S. A. (2012). The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol. Dis. 47, 126–134. doi: 10.1016/j.nbd.2012.03.030
Bordji, K., Becerril-Ortega, J., Nicole, O., and Buisson, A. (2010). Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ss production. J. Neurosci. 30, 15927–15942.
Burdick, D., Soreghan, B., Kwon, M., Kosmoski, J., Knauer, M., Henschen, A., et al. (1992). Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J. Biol. Chem. 267, 546–554.
Buxbaum, J. D., Liu, K. N., Luo, Y., Slack, J. L., Stocking, K. L., Peschon, J. J., et al. (1998). Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273, 27765–27767. doi: 10.1074/jbc.273.43.27765
Cai, H., Wang, Y., McCarthy, D., Wen, H., Borchelt, D. R., Price, D. L., et al. (2001). BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 4, 233–234. doi: 10.1038/85064
Carey, R. M., Balcz, B. A., Lopez-Coviella, I., and Slack, B. E. (2005). Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid beta protein. BMC Cell Biol. 6:30. doi: 10.1186/1471-2121-6-30
Carvalho, K. M., Franca, M. S., Camarao, G. C., and Ruchon, A. F. (1997). A new brain metalloendopeptidase which degrades the Alzheimer beta-amyloid 1-40 peptide producing soluble fragments without neurotoxic effects. Braz. J. Med. Biol. Res. 30, 1153–1156.
Caster, A. H., and Kahn, R. A. (2013). Recruitment of the Mint3 adaptor is necessary for export of the amyloid precursor protein (APP) from the Golgi complex. J. Biol. Chem. 288, 28567–28580. doi: 10.1074/jbc.M113.481101
Chartier-Harlin, M. C., Crawford, F., Houlden, H., Warren, A., Hughes, D., Fidani, L., et al. (1991). Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353, 844–846. doi: 10.1038/353844a0
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Clarke, J., Thornell, A., Corbett, D., Soininen, H., Hiltunen, M., and Jolkkonen, J. (2007). Overexpression of APP provides neuroprotection in the absence of functional benefit following middle cerebral artery occlusion in rats. Eur. J. Neurosci. 26, 1845–1852. doi: 10.1111/j.1460-9568.2007.05807.x
Crowther, D. C., Kinghorn, K. J., Miranda, E., Page, R., Curry, J. A., Duthie, F. A., et al. (2005). Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 132, 123–135. doi: 10.1016/j.neuroscience.2004.12.025
de Moura, M. B., dos Santos, L. S., and Van Houten, B. (2010). Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ. Mol. Mutagen. 51, 391–405. doi: 10.1002/em.20575
De Strooper, B. (2003). Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 38, 9–12.
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006
Do Carmo, S., and Cuello, A. C. (2013). Modeling Alzheimer’s disease in transgenic rats. Mol. Neurodegener. 8, 37. doi: 10.1186/1750-1326-8-37
Doody, R. S., Farlow, M., Aisen, P. S., Alzheimer’s Disease Cooperative Study Data, A., and Publication, C. (2014a). Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N. Engl. J. Med. 370, 1460. doi: 10.1056/NEJMoa1312889
Doody, R. S., Thomas, R. G., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2014b). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321. doi: 10.1056/NEJMoa1312889
Dugan, J. M., deWit, C., McConlogue, L., and Maltese, W. A. (1995). The Ras-related GTP-binding protein, Rab1B, regulates early steps in exocytic transport and processing of beta-amyloid precursor protein. J. Biol. Chem. 270, 10982–10989. doi: 10.1074/jbc.270.18.10982
Eckman, E. A., Watson, M., Marlow, L., Sambamurti, K., and Eckman, C. B. (2003). Alzheimer’s disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J. Biol. Chem. 278, 2081–2084. doi: 10.1074/jbc.C200642200
Elder, G. A., Gama Sosa, M. A., and De Gasperi, R. (2010). Transgenic mouse models of Alzheimer’s disease. Mt. Sinai J. Med. N. Y. 77, 69–81. doi: 10.1002/msj.20159
Ezeani, M., and Omabe, M. (2015). A new perspective of lysosomal cation channel-dependent homeostasis in Alzheimer’s disease. Mol. Neurobiol. doi: 10.1007/s12035-015-9108-3 [Epub ahead of print].
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., et al. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U.S.A. 100, 4162–4167. doi: 10.1073/pnas.0230450100
Finelli, A., Kelkar, A., Song, H. J., Yang, H., and Konsolaki, M. (2004). A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol. Cell. Neurosci. 26, 365–375. doi: 10.1016/j.mcn.2004.03.001
Gandy, S., Caporaso, G., Buxbaum, J., Frangione, B., and Greengard, P. (1994). APP processing, A beta-amyloidogenesis, and the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 15, 253–256.
Glabe, C. C. (2005). Amyloid accumulation and pathogensis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell. Biochem. 38, 167–177. doi: 10.1007/0-387-23226-5_8
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. doi: 10.1038/349704a0
Golde, T. E., Estus, S., Younkin, L. H., Selkoe, D. J., and Younkin, S. G. (1992). Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science 255, 728–730. doi: 10.1126/science.1738847
Gravina, S. A., Ho, L., Eckman, C. B., Long, K. E., Otvos, L. Jr., Younkin, L. H., et al. (1995). Amyloid beta protein. (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J. Biol. Chem. 270, 7013–7016.
Greenfield, J. P., Tsai, J., Gouras, G. K., Hai, B., Thinakaran, G., Checler, F., et al. (1999). Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. U.S.A. 96, 742–747. doi: 10.1073/pnas.96.2.742
Greeve, I., Kretzschmar, D., Tschape, J. A., Beyn, A., Brellinger, C., Schweizer, M., et al. (2004). Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J. Neurosci. 24, 3899–3906. doi: 10.1523/JNEUROSCI.0283-04.2004
Hamazaki, H. (1996). Cathepsin D is involved in the clearance of Alzheimer’s beta-amyloid protein. FEBS Lett. 396, 139–142. doi: 10.1016/0014-5793(96)01087-3
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Harkany, T., O’Mahony, S., Keijser, J., Kelly, J. P., Konya, C., Borostyankoi, Z. A., et al. (2001). Beta-amyloid(1-42)-induced cholinergic lesions in rat nucleus basalis bidirectionally modulate serotonergic innervation of the basal forebrain and cerebral cortex. Neurobiol. Dis. 8, 667–678. doi: 10.1006/nbdi.2001.0398
Harold, D., Jehu, L., Turic, D., Hollingworth, P., Moore, P., Summerhayes, P., et al. (2007). Interaction between the ADAM12 and SH3MD1 genes may confer susceptibility to late-onset Alzheimer’s disease. Am. J. Med. Genet. 144B, 448–452. doi: 10.1002/ajmg.b.30456
Hartmann, T., Bieger, S. C., Bruhl, B., Tienari, P. J., Ida, N., Allsop, D., et al. (1997). Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat. Med. 3, 1016–1020. doi: 10.1038/nm0997-1016
He, G., Luo, W., Li, P., Remmers, C., Netzer, W. J., Hendrick, J., et al. (2010). Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467, 95–98. doi: 10.1038/nature09325
Hemming, M. L., and Selkoe, D. J. (2005). Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J. Biol. Chem. 280, 37644–37650. doi: 10.1074/jbc.M508460200
Heneka, M. T., Golenbock, D. T., and Latz, E. (2015). Innate immunity in Alzheimer’s disease. Nat. Immunol. 16, 229–236. doi: 10.1038/ni.3102
Huber, L. A., Pimplikar, S., Parton, R. G., Virta, H., Zerial, M., and Simons, K. (1993). Rab8, a small GTPase involved in vesicular traffic between the TGN and the basolateral plasma membrane. J. Cell Biol. 123, 35–45. doi: 10.1083/jcb.123.1.35
Iijima, K., Chiang, H. C., Hearn, S. A., Hakker, I., Gatt, A., Shenton, C., et al. (2008). Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS ONE 3:e1703. doi: 10.1371/journal.pone.0001703
Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N. P., et al. (2001). Metabolic regulation of brain Abeta by neprilysin. Science 292, 1550–1552. doi: 10.1126/science.1059946
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/S0896-6273(03)00124-7
Kanekiyo, T., and Bu, G. (2014). The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in Alzheimer’s disease. Front. Aging Neurosci. 6:93. doi: 10.3389/fnagi.2014.00093
Kanekiyo, T., Zhang, J., Liu, Q., Liu, C. C., Zhang, L., and Bu, G. (2011). Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J. Neurosci. 31, 1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011
Kang, J., and Muller-Hill, B. (1990). Differential splicing of Alzheimer’s disease amyloid A4 precursor RNA in rat tissues: preA4(695) mRNA is predominantly produced in rat and human brain. Biochem. Biophys. Res. Commun. 166, 1192–1200. doi: 10.1016/0006-291X(90)90992-V
Karran, E., Mercken, M., and De Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. 10, 698–712. doi: 10.1038/nrd3505
Kim, J., Onstead, L., Randle, S., Price, R., Smithson, L., Zwizinski, C., et al. (2007). Abeta40 inhibits amyloid deposition in vivo. J. Neurosci. 27, 627–633. doi: 10.1523/JNEUROSCI.4849-06.2007
Kim, M., Suh, J., Romano, D., Truong, M. H., Mullin, K., Hooli, B., et al. (2009). Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum. Mol. Genet. 18, 3987–3996. doi: 10.1093/hmg/ddp323
King, J. V., Liang, W. G., Scherpelz, K. P., Schilling, A. B., Meredith, S. C., and Tang, W. J. (2014). Molecular basis of substrate recognition and degradation by human presequence protease. Structure 22, 996–1007. doi: 10.1016/j.str.2014.05.003
Koike, H., Tomioka, S., Sorimachi, H., Saido, T. C., Maruyama, K., Okuyama, A., et al. (1999). Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem. J. 343(Pt 2), 371–375. doi: 10.1042/0264-6021:3430371
Koo, E. H., and Squazzo, S. L. (1994). Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389.
Ledesma, M. D., Da Silva, J. S., Crassaerts, K., Delacourte, A., De Strooper, B., and Dotti, C. G. (2000). Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer’s disease brains. EMBO Rep. 1, 530–535. doi: 10.1093/embo-reports/kvd107
Lee, J., Retamal, C., Cuitino, L., Caruano-Yzermans, A., Shin, J. E., van Kerkhof, P., et al. (2008). Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J. Biol. Chem. 283, 11501–11508. doi: 10.1074/jbc.M800642200
Leon, W. C., Canneva, F., Partridge, V., Allard, S., Ferretti, M. T., DeWilde, A., et al. (2010). A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J. Alzheimers Dis. 0, 113–126. doi: 10.3233/JAD-2010-1349
Li, J., Kanekiyo, T., Shinohara, M., Zhang, Y., LaDu, M. J., Xu, H., et al. (2012). Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem. 287, 44593–44601. doi: 10.1074/jbc.M112.420224
Li, Y., Marzolo, M. P., van Kerkhof, P., Strous, G. J., and Bu, G. (2000). The YXXL motif, but not the two NPXY motifs, serves as the dominant endocytosis signal for low density lipoprotein receptor-related protein. J. Biol. Chem. 275, 17187–17194. doi: 10.1074/jbc.M000490200
Lim, J. Y., Ott, S., and Crowther, D. C. (2016). Drosophila melanogaster as a model for studies on the early stages of Alzheimer’s disease. Methods Mol. Biol. 1303, 227–239. doi: 10.1007/978-1-4939-2627-5_13
Link, C. D. (2006). C. elegans models of age-associated neurodegenerative diseases: lessons from transgenic worm models of Alzheimer’s disease. Exp. Gerontol. 41, 1007–1013. doi: 10.1016/j.exger.2006.06.059
Liu, S., Liu, Y., Hao, W., Wolf, L., Kiliaan, A. J., Penke, B., et al. (2012). TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J. Immunol. 188, 1098–1107. doi: 10.4049/jimmunol.1101121
Lublin, A. L., and Link, C. D. (2013). Alzheimer’s disease drug discovery: in vivo screening using Caenorhabditis elegans as a model for beta-amyloid peptide-induced toxicity. Drug Discov. Today Technol. 10, e115–e119. doi: 10.1016/j.ddtec.2012.02.002
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Mark, R. J., Hensley, K., Butterfield, D. A., and Mattson, M. P. (1995). Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J. Neurosci. 15, 6239–6249.
Mattson, M. P. (2004). Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639. doi: 10.1038/nature02621
McConlogue, L., Castellano, F., deWit, C., Schenk, D., and Maltese, W. A. (1996). Differential effects of a Rab6 mutant on secretory versus amyloidogenic processing of Alzheimer’s beta-amyloid precursor protein. J. Biol. Chem. 271, 1343–1348. doi: 10.1074/jbc.271.3.1343
Menendez-Gonzalez, M., Perez-Pinera, P., Martinez-Rivera, M., Calatayud, M. T., and Blazquez Menes, B. (2005). APP processing and the APP-KPI domain involvement in the amyloid cascade. Neurodegener. Dis. 2, 277–283. doi: 10.1159/000092315
Mueller-Steiner, S., Zhou, Y., Arai, H., Roberson, E. D., Sun, B., Chen, J., et al. (2006). Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron 51, 703–714. doi: 10.1016/j.neuron.2006.07.027
Muirhead, K. E., Borger, E., Aitken, L., Conway, S. J., and Gunn-Moore, F. J. (2010). The consequences of mitochondrial amyloid beta-peptide in Alzheimer’s disease. Biochem. J. 426, 255–270. doi: 10.1042/BJ20091941
Musiek, E. S., and Holtzman, D. M. (2015). Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat. Neurosci. 18, 800–806. doi: 10.1038/nn.4018
Nag, S., Yee, B. K., and Tang, F. (1999). Chronic intracerebroventricular infusion of beta-amyloid (1-40) results in a selective loss of neuropeptides in addition to a reduction in choline acetyltransferase activity in the cortical mantle and hippocampus in the rat. Ann. N. Y. Acad. Sci. 897, 420–422. doi: 10.1111/j.1749-6632.1999.tb07911.x
Nakamura, S., Murayama, N., Noshita, T., Annoura, H., and Ohno, T. (2001). Progressive brain dysfunction following intracerebroventricular infusion of beta(1-42)-amyloid peptide. Brain Res. 912, 128–136. doi: 10.1016/S0006-8993(01)02704-4
Neniskyte, U., Neher, J. J., and Brown, G. C. (2011). Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J. Biol. Chem. 286, 39904–39913. doi: 10.1074/jbc.M111.267583
Nixon, R. A. (2007). Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 120, 4081–4091. doi: 10.1242/jcs.019265
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Perez, R. G., Soriano, S., Hayes, J. D., Ostaszewski, B., Xia, W., Selkoe, D. J., et al. (1999). Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J. Biol. Chem. 274, 18851–18856. doi: 10.1074/jbc.274.27.18851
Rohan de Silva, H. A., Jen, A., Wickenden, C., Jen, L. S., Wilkinson, S. L., and Patel, A. J. (1997). Cell-specific expression of beta-amyloid precursor protein isoform mRNAs and proteins in neurons and astrocytes. Brain Res. Mol. Brain Res. 47, 147–156. doi: 10.1016/S0169-328X(97)00045-4
Rosen, R. F., Fritz, J. J., Dooyema, J., Cintron, A. F., Hamaguchi, T., Lah, J. J., et al. (2012). Exogenous seeding of cerebral beta-amyloid deposition in betaAPP-transgenic rats. J. Neurochem. 120, 660–666. doi: 10.1111/j.1471-4159.2011.07551.x
Sahlin, C., Pettersson, F. E., Nilsson, L. N., Lannfelt, L., and Johansson, A. S. (2007). Docosahexaenoic acid stimulates non-amyloidogenic APP processing resulting in reduced Abeta levels in cellular models of Alzheimer’s disease. Eur. J. Neurosci. 26, 882–889. doi: 10.1111/j.1460-9568.2007.05719.x
Salloway, S., Sperling, R., Fox, N. C., Blennow, K., Klunk, W., Raskind, M., et al. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333. doi: 10.1056/NEJMoa1304839
Saporito-Irwin, S. M., and Van Nostrand, W. E. (1995). Coagulation factor XIa cleaves the RHDS sequence and abolishes the cell adhesive properties of the amyloid beta-protein. J. Biol. Chem. 270, 26265–26269. doi: 10.1074/jbc.270.44.26265
Schobel, S., Neumann, S., Hertweck, M., Dislich, B., Kuhn, P. H., Kremmer, E., et al. (2008). A novel sorting nexin modulates endocytic trafficking and alpha-secretase cleavage of the amyloid precursor protein. J. Biol. Chem. 283, 14257–14268. doi: 10.1074/jbc.M801531200
Sisodia, S. S. (1992). Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. U.S.A. 89, 6075–6079. doi: 10.1073/pnas.89.13.6075
Small, D. H., Mok, S. S., and Bornstein, J. C. (2001). Alzheimer’s disease and Abeta toxicity: from top to bottom. Nat. Rev. Neurosci. 2, 595–598. doi: 10.1038/35086072
Tanabe, C., Hotoda, N., Sasagawa, N., Sehara-Fujisawa, A., Maruyama, K., and Ishiura, S. (2007). ADAM19 is tightly associated with constitutive Alzheimer’s disease APP alpha-secretase in A172 cells. Biochem. Biophys. Res. Commun. 352, 111–117. doi: 10.1016/j.bbrc.2006.10.181
Taylor, D. R., and Hooper, N. M. (2007). The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem. J. 402, 17–23. doi: 10.1042/BJ20061736
Thathiah, A., Spittaels, K., Hoffmann, M., Staes, M., Cohen, A., Horre, K., et al. (2009). The orphan G protein-coupled receptor 3 modulates amyloid-beta peptide generation in neurons. Science 323, 946–951. doi: 10.1126/science.1160649
Thyrock, A., Ossendorf, E., Stehling, M., Kail, M., Kurtz, T., Pohlentz, G., et al. (2013). A new Mint1 isoform, but not the conventional Mint1, interacts with the small GTPase Rab6. PLoS ONE 8:e64149. doi: 10.1371/journal.pone.0064149
Tucker, H. M., Kihiko-Ehmann, M., and Estus, S. (2002). Urokinase-type plasminogen activator inhibits amyloid-beta neurotoxicity and fibrillogenesis via plasminogen. J. Neurosci. Res. 70, 249–255. doi: 10.1002/jnr.10417
Udayar, V., Buggia-Prevot, V., Guerreiro, R. L., Siegel, G., Rambabu, N., Soohoo, A. L., et al. (2013). A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of beta-amyloid production. Cell Rep. 5, 1536–1551. doi: 10.1016/j.celrep.2013.12.005
Varadarajan, S., Yatin, S., Aksenova, M., and Butterfield, D. A. (2000). Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol. 130, 184–208. doi: 10.1006/jsbi.2000.4274
Wahle, T., Thal, D. R., Sastre, M., Rentmeister, A., Bogdanovic, N., Famulok, M., et al. (2006). GGA1 is expressed in the human brain and affects the generation of amyloid beta-peptide. J. Neurosci. 26, 12838–12846. doi: 10.1523/JNEUROSCI.1982-06.2006
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Walsh, D. M., and Selkoe, D. J. (2007). A beta oligomers - a decade of discovery. J. Neurochem. 101, 1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x
Wang, B., Yang, W., Wen, W., Sun, J., Su, B., Liu, B., et al. (2010). Gamma-secretase gene mutations in familial acne inversa. Science 330, 1065. doi: 10.1126/science.1196284
Weggen, S., Eriksen, J. L., Das, P., Sagi, S. A., Wang, R., Pietrzik, C. U., et al. (2001). A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414, 212–216. doi: 10.1038/35102591
Wen, L., Tang, F. L., Hong, Y., Luo, S. W., Wang, C. L., He, W., et al. (2011). VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J. Cell Biol. 195, 765–779. doi: 10.1083/jcb.201105109
Wu, Y., Wu, Z., Butko, P., Christen, Y., Lambert, M. P., Klein, W. L., et al. (2006). Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J. Neurosci. 26, 13102–13113. doi: 10.1523/JNEUROSCI.3448-06.2006
Xu, H., Sweeney, D., Wang, R., Thinakaran, G., Lo, A. C., Sisodia, S. S., et al. (1997). Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc. Natl. Acad. Sci. U.S.A. 94, 3748–3752. doi: 10.1073/pnas.94.8.3748
Yamada, T., Miyazaki, K., Koshikawa, N., Takahashi, M., Akatsu, H., and Yamamoto, T. (1995). Selective localization of gelatinase A, an enzyme degrading beta-amyloid protein, in white matter microglia and in Schwann cells. Acta Neuropathol. 89, 199–203. doi: 10.1007/BF00309334
Yatin, S. M., Aksenov, M., and Butterfield, D. A. (1999). The antioxidant vitamin E modulates amyloid beta-peptide-induced creatine kinase activity inhibition and increased protein oxidation: implications for the free radical hypothesis of Alzheimer’s disease. Neurochem. Res. 24, 427–435. doi: 10.1023/A:1020997903147
Ye, Y., and Fortini, M. E. (1999). Apoptotic activities of wild-type and Alzheimer’s disease-related mutant presenilins in Drosophila melanogaster. J. Cell Biol. 146, 1351–1364. doi: 10.1083/jcb.146.6.1351
Yu, W. H., Kumar, A., Peterhoff, C., Shapiro Kulnane, L., Uchiyama, Y., Lamb, B. T., et al. (2004). Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 36, 2531–2540. doi: 10.1016/j.biocel.2004.05.010
Zhou, S., Zhou, H., Walian, P. J., and Jap, B. K. (2005). CD147 is a regulatory subunit of the gamma-secretase complex in Alzheimer’s disease amyloid beta-peptide production. Proc. Natl. Acad. Sci. U.S.A. 102, 7499–7504. doi: 10.1073/pnas.0502768102
Keywords: amyloid β peptide, Alzheimer’s disease, amyloid precursor protein, biogenesis, animal models
Citation: Sun X, Chen W-D and Wang Y-D (2015) β-Amyloid: the key peptide in the pathogenesis of Alzheimer’s disease Front. Pharmacol. 6:221. doi: 10.3389/fphar.2015.00221
Received: 02 August 2015; Accepted: 17 September 2015;
Published: 30 September 2015.
Edited by:
Chiranjib Chakraborty, Galgotias University, IndiaReviewed by:
Jiang Liu, University of Southern California, USAGhanshyam Upadhyay, City College of New York – CUNY, USA
Copyright © 2015 Sun, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Dong Chen, Key Laboratory of Receptors-Mediated Gene Regulation and Drug Discovery, School of Medicine, Henan University, Jinming Road, Kaifeng 475004, Henan, China, wdchen666@163.com; Yan-Dong Wang, State Key Laboratory of Chemical Resource Engineering, College of Life Science and Technology, Beijing University of Chemical Technology, No. 15 Beisanhuan East Road, Beijing 100029, China, ydwangbuct2009@163.com