 Cedric J. Laedermann
Cedric J. Laedermann Hugues Abriel
Hugues Abriel Isabelle Decosterd
Isabelle Decosterd- 1F.M. Kirby Neurobiology Research Center, Boston Children’s Hospital, Harvard Medical School, Boston, MA, USA
- 2Department of Clinical Research, University of Bern, Bern, Switzerland
- 3Pain Center, Department of Anesthesiology, Lausanne University Hospital (CHUV) and University of Lausanne, Lausanne, Switzerland
- 4Department of Fundamental Neurosciences, University of Lausanne, Lausanne, Switzerland
In the peripheral sensory nervous system the neuronal expression of voltage-gated sodium channels (Navs) is very important for the transmission of nociceptive information since they give rise to the upstroke of the action potential (AP). Navs are composed of nine different isoforms with distinct biophysical properties. Studying the mutations associated with the increase or absence of pain sensitivity in humans, as well as other expression studies, have highlighted Nav1.7, Nav1.8, and Nav1.9 as being the most important contributors to the control of nociceptive neuronal electrogenesis. Modulating their expression and/or function can impact the shape of the AP and consequently modify nociceptive transmission, a process that is observed in persistent pain conditions. Post-translational modification (PTM) of Navs is a well-known process that modifies their expression and function. In chronic pain syndromes, the release of inflammatory molecules into the direct environment of dorsal root ganglia (DRG) sensory neurons leads to an abnormal activation of enzymes that induce Navs PTM. The addition of small molecules, i.e., peptides, phosphoryl groups, ubiquitin moieties and/or carbohydrates, can modify the function of Navs in two different ways: via direct physical interference with Nav gating, or via the control of Nav trafficking. Both mechanisms have a profound impact on neuronal excitability. In this review we will discuss the role of Protein Kinase A, B, and C, Mitogen Activated Protein Kinases and Ca++/Calmodulin-dependent Kinase II in peripheral chronic pain syndromes. We will also discuss more recent findings that the ubiquitination of Nav1.7 by Nedd4-2 and the effect of methylglyoxal on Nav1.8 are also implicated in the development of experimental neuropathic pain. We will address the potential roles of other PTMs in chronic pain and highlight the need for further investigation of PTMs of Navs in order to develop new pharmacological tools to alleviate pain.
Pain, NOCICEPTION, INFLAMMATORY AND NEUROPATHIC PAIN
The ability to recognize and remember danger is one of the major evolutionary steps necessary for survival in a hostile environment. The primary line of defense for organisms relies on the primary nociceptive neurons, which are activated when stimuli intensities reach the noxious range. The peripheral endings of nociceptive neurons are found in almost all tissue of the body, i.e., skin, muscle and internal organs (Kruger et al., 2003). Their cell bodies are located in the dorsal root ganglia (DRG) or the trigeminal ganglia (TG) for the innervations of the face. Central terminals of primary nociceptive neurons project to the dorsal horn of the spinal cord where they make their first synapse with secondary order sensory neurons and/or interneurons (Millan, 1999). At this level, the transmission of the signal by the secondary order projection neurons to the supra spinal centers is modulated by a complex network of the dorsal horn. A barrage of activity or neurotransmitter release from the periphery can highly modulate the excitability of the secondary order neurons (Latremoliere and Woolf, 2009). Spinal neurons are subjected to change by microglial, astrocytic and oligodendrocitic cells (Fotia et al., 2002; Lou et al., 2005). Furthermore, descending projections from supra-spinal centers can affect the central terminal of the primary sensory neuron, the projection neuron and the inhibitory and excitatory interneurons. The signal is eventually processed and reaches supra-spinal centers, including brain areas involved in sensory and emotional pain perception. At this point, complex circuitries process and integrate the pain signal and allow for appropriate behavioral and motor responses. These circuitries also modulate pain signaling via descending pathways that ultimately reach the dorsal horn neurons.
For the most part, primary nociceptive neurons are polymodal (Dubin and Patapoutian, 2010), in that they are able to detect a wide range of stimuli, such as heat and mechanical or chemical stimuli. To integrate these wide ranges of stimuli, nociceptive neurons express a multitude of receptors and ion channels in their free endings (Basbaum et al., 2009). There are many review articles that extensively discuss these so called transducers, i.e., TRP channel family members or acid-sensing ion channels (ASICs; Basbaum et al., 2009; Stucky et al., 2009; Deval et al., 2010). Once transducers are activated, these non-selective ion channels open and contribute to cell depolarization, thus eliciting an action potential (AP) and allowing for the transmission of a noxious signal along pain pathways.
Nociception enables the organism to react quickly, and by virtue of the emotionally offensive component of pain, helps the organism avoid similar situations in the future. When the noxious stimulus is acute, pain is transient and the nociceptive neurons should subsequently return to a resting state. This process is referred to as nociception. On the other hand, tissue injury leads to longer lasting inflammatory pain, characterized by peripheral sensitization. Inflammatory pain is due to the modification of the chemical environment surrounding nociceptive neurons and the accumulation of several factors secreted by recruited non-neural cells, such as mast cells, macrophages, neutrophils, inflammatory cells, fibroblasts and keratinocytes, as well as by the nociceptive neurons themselves. These factors are of diverse origins but include protons (H+), nerve growth factors (NGFs), cytokines (such as IL-1β, IL-6), tumor necrosis factor alpha (TNF-α), prostaglandins (PGE2), several neurotransmitters (serotonin, ATP) and peptides (bradykinin, substance P, CGRP). This mixture is commonly referred to as “the inflammatory soup” (Basbaum et al., 2009), and will increase spontaneous neuronal firing, usually decrease the threshold of nociceptive neurons and increase firing in response to suprathreshold stimuli. The mechanisms by which inflammatory pain increases pain transmission include the activation of kinases that phosphorylate membrane channels and receptors, which subsequently alter their function, and the genetic regulation of primary sensory neurons (Woolf and Costigan, 1999). Inflammatory pain, by the virtue of central sensitization, is also accompanied by the local loss of inhibition (Julius and Basbaum, 2001) and enhanced postsynaptic transmission (Galan et al., 2004).
Inflammatory pain is linked to the persistence of inflammation, but should fade away when the tissue is healed. In some cases, however, abnormal activity from the peripheral neurons can also occur in the absence of tissue inflammation. This abnormal activity occurs when nociceptive neurons are damaged and elicit long-term molecular modifications that eventually lead to neuropathic pain. Neuropathic pain is defined as the “pain caused by a lesion or disease of the somatosensory system” (Heikamp et al., 2014). This definition reflects that not only nerve injury, but also degenerative, infectious or metabolic conditions can lead to neuropathic pain, accounting for the distinct etiologies of peripheral neuropathic pain (e.g., compressive disk herniation, diabetic neuropathy, chemotherapeutic-induced neuropathy, post-herpetic neuralgia, etc.) (Woolf and Mannion, 1999). The prevalence of neuropathic pain varies from 7 (Bouhassira et al., 2008) to 18% of the population (Toth et al., 2009). Most of these patients are often resistant to treatment (Brower, 2000). Neuropathic pain cardinal positive symptoms are spontaneous pain, allodynia and hyperalgesia, but can be also associated with negative symptoms such as hyposensitivity in a related nerve territory. Spontaneous pain is thought to arise from ectopic activity, which can be driven by C- (Djouhri et al., 2006) and A-fibers (Liu et al., 2000); where as allodynia and hyperalgesia are related to reduced activation thresholds or an increased response of primary afferent neurons (von Hehn et al., 2012). The peripheral mechanisms underlying neuropathic pain associated hyperexcitability have been extensively reviewed in other articles (Julius and Basbaum, 2001; Woolf, 2004; Campbell and Meyer, 2006; Hucho and Levine, 2007). These mechanisms include altered gene expression, dysregulation of membrane channel expression, migration of inflammatory cells and activation of satellite cells in DRG neurons. The increased peripheral input leads to activity-dependent mechanisms of central sensitization in the spinal cord and supra-spinal levels (Woolf and Salter, 2000; Latremoliere and Woolf, 2009; Woolf, 2011). Although they share similar pain symptoms and some common mechanisms, inflammatory and neuropathic pain differ fundamentally by their respective pharmacology and resolution. Altogether, peripheral and central mechanisms will lead to enhanced and long-lasting pain perception, which will ultimately result in debilitating chronic conditions, sleep disturbances, depression, anxiety and social withdrawal (Bair et al., 2003; Turk et al., 2010). On the other side of the spectrum, in diseases where patients can no longer experience pain appropriately, such as in congenital insensitivity to pain, the pain signal is permanently shut off. This results in an inappropriate behavioral response and a reduced life expectancy in a subset of individuals (Nagasako et al., 2003).
Navs: Structure And Function
After activation via transducers, primary nociceptive neurons transmit the signal along the axon to the spinal cord. Voltage-gated sodium channels (Navs) play a critical role in this process. Navs are activated upon depolarization of the transmembrane voltage, generating a fast, transient and massive inward sodium current, which accounts for the rising phase of the AP. Navs give rise to the upstroke of the AP and contribute to setting the resting membrane potential of nociceptive neurons (Herzog et al., 2001; Rush et al., 2007).
Since the first biochemical characterization of Navs (Beneski and Catterall, 1980), tremendous effort has been made to unravel the structure and function of the sodium channel. The α-subunit is encoded by a single gene, which is structurally divided into four homologous domains (I-IV) connected by an intra and/or extracellular loop, thus referred to as heterotetramere. Each domain is composed of six α-helical transmembrane segments. S5 and S6 compose the pore of the channel, whereas S1 – S4 are the voltage sensors. The crystallography structure of NavAb, a bacterial sodium channel from Arcobacter butzleri, was identified in 2011 (Payandeh et al., 2011). Although NavAb is a homotetramere and the mammalian Navs is heterotetrameric, they share similar pharmacological profiles (Ren et al., 2001). Most of the inferences made based on previous biochemical and electrophysiological experiments have been confirmed (Payandeh et al., 2011).
Nine discrete genes (SCNxA) encode for the α-subunits (Nav1.1 to Nav1.9 isoforms) (Catterall et al., 2005) and another atypical tenth isoform, NaX (Akopian et al., 1997; Noda and Hiyama, 2014). Each isoform has its own biophysical properties and particular expression pattern across the nervous system. A single amino acid substitution in the S5–S6 linker renders Nav1.5, Nav1.8, and Nav1.9 resistance to TTX.
α-subunits are accompanied by associated β-subunits with an assumed stoichiometry for an α-β association of 1:1 (Catterall, 1992). There are four different identified genes coding for the different β-subunits: SCN1B codes for β1 (Isom et al., 1992) and its associated splice variant β1A (Kazen-Gillespie et al., 2000); SCN2B codes for β2 (Isom et al., 1995a); SCN3B codes for β3 (Morgan et al., 2000) and SCN4B codes for β4 (Yu et al., 2003). The pore-forming α-subunit enables for Na+ conductance, but the β–subunits can modulate the biophysical properties and plasma membrane stabilization of Navs (Isom et al., 1995b).
Nav1.7, Nav1.8, and Nav1.9 Are Expressed In Nociceptive Neurons
Navs are broadly expressed in excitable cells throughout the body, with some isoforms ubiquitously expressed and others expressed in specific tissues. In nociceptive neurons, many of the different Navs isoforms are present and collaborate with one another for electrogenesis. With the exception of Nav1.2 and Nav1.4, all the Nav isoforms are expressed in DRG nociceptive neurons (Black et al., 1996; Rush et al., 2007; Berta et al., 2008; Fukuoka et al., 2008; Fukuoka and Noguchi, 2011; Ho and O’Leary, 2011). The role of Nav1.5 in adult small DRG neurons has not been fully unraveled (Renganathan et al., 2002). As compared to Nav1.1 and Nav1.6, the Nav1.7 isoform is the most expressed TTX-sensitive isoform among the DRG neurons (Black et al., 1996; Toledo-Aral et al., 1997; Berta et al., 2008; Ho and O’Leary, 2011; Dib-Hajj et al., 2013). The two TTX-resistant isoforms, Nav1.8 and Nav1.9, are also highly expressed in nociceptive neurons (Akopian et al., 1996; Dib-Hajj et al., 1998). During the last decade, mutations in Nav1.7, Nav1.8, and Nav1.9 have been linked with human pain disorders (see Inherited Pain Syndromes). The possible variable combinations of each of these “pain specialized” isoforms, as well as their relative expression levels, differentially shape the AP and firing properties, accounting for the heterogeneity among DRG neurons (Rush et al., 2007; Theriault and Chahine, 2014). In this review, we focus on the roles of Nav1.7, Nav1.8, and Nav1.9 in chronic pain states.
Navs’ Implication In Pain Syndromes
Reviewing all the studies that have investigated the Nav1.7, Nav1.8, and Nav1.9 mutations associated with painful channelopathies and those studies that have investigated the expression of Navs in human and animal models of pathological pain is beyond the scope of this review and can already be found in other recent reviews (Brouwer et al., 2014; Waxman et al., 2014; Waxman and Zamponi, 2014; Hoeijmakers et al., 2015). In the next two chapters we will summarize some of the key findings demonstrating the role of Navs in channelopathies and in animal models of inflammatory pain, and we will discuss conflicting results observed in neuropathic pain in both human and animal studies.
Inherited Pain Syndromes
The contribution of Nav1.7, Nav1.8, and Nav1.9 in chronic pain syndromes is exemplified through human mutations (familial and de novo mutations) of these channels and their associated pathologies, being either pain hypersensitivity or congenital insensitivity to pain (Dib-Hajj et al., 2009; Liu and Wood, 2011; Waxman et al., 2014). Seminal studies have linked Nav1.7 to altered pain sensitivity. A gain of function for this gene leads to inherited painful channelopathies, such as erythromelalgia (Cummins et al., 2004; Yang et al., 2004) and paroxysmal extreme pain disorder (Fertleman et al., 2006). Conversely, a loss of function for Nav1.7 was reported to be associated with congenital insensitivity to pain (Cox et al., 2006). Many other studies have identified Nav1.7 mutations as being implicated in numerous altered pain sensation pathologies (Dib-Hajj et al., 2005, 2013; Waxman and Dib-Hajj, 2005; Fertleman et al., 2006; Novella et al., 2007; Cheng et al., 2008). A gain of function mutation of Nav1.9 was linked to an episodic pain disorder (Zhang et al., 2013); while another gain of function mutation of Nav1.9 was reported to cause a loss of pain perception (Leipold et al., 2013). In the latter the authors showed that the excessive activity of Nav1.9 at resting voltages caused sustained depolarization of nociceptive neurons, leading to the inactivation of other Navs and subsequently to the impairment of the AP generation. Other studies also highlighted a role of Nav1.7 in idiopathic small-fiber neuropathy (I-SFN); nearly 30% of patients suffering from this pathology had a gain of function mutation in Nav1.7 (Faber et al., 2012a). Small fiber peripheral neuropathy is a type of peripheral neuropathy that occurs from damage to C-fibers and A-δ fibers, which can often lead to exaggerated pain sensitivity (Hoeijmakers et al., 2012). Since these first studies, similar gain of function mutations in Nav1.8 and Nav1.9 have been reported in I-SFN (Faber et al., 2012b; Han et al., 2014; Huang et al., 2014). None of the previous gain of function mutations of Nav1.7, Nav1.8, and Nav1.9 have been studied in animal models of chronic pain.
Acquired Pain Syndromes
Apart from intrinsic modifications of Nav channel function, a modification of expression levels will also impact neuronal excitability. Most of the studies that are discussed here investigated Nav expression while assuming that an increased sodium channel expression and conductance would cause neuronal hyperexcitability, something already demonstrated by computer simulations (Matzner and Devor, 1992). However, the link between increased sodium channel expression and hyperexcitability is likely more complex. For instance, computational studies revealed that increasing the sodium conductance might actually decrease the firing rate of neurons (Kispersky et al., 2012). In addition, a gain of function mutation of Nav1.9 was recently shown to be associated with congenital insensitivity to pain (Leipold et al., 2013).
Modifications in Nav1.7, Nav1.8, and Nav1.9 expression have been observed in several chronic pain syndromes. Both human studies and experimental pain model studies have helped unravel the role of these isoforms in chronic pain syndromes, including both inflammatory and neuropathic pain.
There is substantial evidence linking Nav1.7 to inflammatory pain in animal studies. Studies have reported an increase of Nav1.7 expression after injection of pro-inflammatory mediators (Gould et al., 2000; Black et al., 2004). Knocking-down Nav1.7 in a model of inflammatory pain with a viral vector in primary afferents (Yeomans et al., 2005) led to the attenuated development of hyperalgesia, a result confirmed by another study using Nav1.7 knockout mice (Nassar et al., 2004). Similarly to Nav1.7, knocking down (Khasar et al., 1998) or knocking out Nav1.8 (Khasar et al., 1998; Akopian et al., 1999) prevented full development of pain hypersensitivity in inflammatory pain models. Nav1.8 was also shown to be increased in inflammatory pain models (Coggeshall et al., 2004; Strickland et al., 2008), and its role was confirmed in knockdown studies (Yu et al., 2011) and in studies using specific blockers (Jarvis et al., 2007; Moon et al., 2012). Other studies also support a role for Nav1.9 in inflammatory pain (Dib-Hajj et al., 2010). Nav1.9 knockout mice or knockdown rats have a weaker response to inflammatory mediator application (Hirade et al., 1999; Priest et al., 2005; Amaya et al., 2006; Lolignier et al., 2011; Hockley et al., 2014).
Dysregulated Nav expression, by altering the intrinsic electrical properties of neuronal plasma membranes, is largely accountable for neuropathic pain-associated hyperexcitability (Matzner and Devor, 1994; Zhang et al., 1997). Navs’ contribution to neuropathic pain is also demonstrated by the application of local anesthetics known to block sodium channels, which suppresses ectopic discharges and attenuates allodynia and hyperalgesia (Mao and Chen, 2000; Suter et al., 2003; Scholz et al., 2005).
The development of nerve injury-induced neuropathic pain animal models has significantly contributed to the discovery of mechanisms that contribute to neuropathic pain syndromes, but also gave rise to conflicting results. For instance, the role of Nav1.7 in neuropathic pain is still being debated. Nav1.7 mRNA is reduced after peripheral nerve injury-induced neuropathic pain in rats (Berta et al., 2008; Laedermann et al., 2014b; Casals-Diaz et al., 2015), an observation confirmed by reduced levels of the Nav1.7 protein. Furthermore, Nav1.7 knockout mice still develop neuropathic pain-mediated mechanical allodynia (Nassar et al., 2004). More recent studies suggest that Nav1.7 is actually implicated in neuropathic pain by virtue of its concomitant expression in both sympathetic ganglion neurons and nociceptive neurons, rather than solely in nociceptive neurons (Minett et al., 2012). In contrast, other studies have reported an increased expression of Nav1.7 mRNA in DRG neurons (Liu et al., 2012), as well as an increased protein expression in the sciatic nerve of animal experimental neuropathic pain models (Laedermann et al., 2013a). In humans, many studies reported an increased expression of this isoform, such as in intervertebral disk injury (Sadamasu et al., 2014), human dental pulp neuromas (Luo et al., 2008b) and other neuromas (Coward et al., 2001a; Kretschmer et al., 2002; Bird et al., 2007; Persson et al., 2011).
Nav1.8 regulation is also controversial in experimental nerve-injury induced neuropathic pain. Many studies have reported a downregulation of Nav1.8 mRNA (Waxman, 1999; Berta et al., 2008), protein (Decosterd et al., 2002) and currents (Cummins and Waxman, 1997; Berta et al., 2008) in rodent models. Another group, however, reported an increase of the Nav1.8-mediated current (Abdulla and Smith, 2002). To reconcile these contradictory findings, it was suggested that a decrease in the expression of Nav1.8 mRNA and protein in the cell soma of nociceptive neurons could be due to a redistribution of this isoform in the sciatic nerve (Gold et al., 2003; Thakor et al., 2009). Controlling Nav1.8 expression in mice also leads to controversial results. Gene knockout studies did not find a role for Nav1.8 in neuropathic pain development (Akopian et al., 1999; Kerr et al., 2001; Abrahamsen et al., 2008), whereas gene knockdown speaks in favor of such a role (Lai et al., 2002; Dong et al., 2007; Leo et al., 2010). It is likely that Nav1.8 involvement depends on the type of lesion and the model of chronic pain (Joshi et al., 2006). A few studies carried out in humans showed that Nav1.8 expression was increased in neuromas (Kretschmer et al., 2002; Black et al., 2008; Bird et al., 2013).
Nav1.9 implication in neuropathic pain has been scarcely investigated. A few studies reported a downregulation of Nav1.9 mRNA and protein in animal models of neuropathic pain (Dib-Hajj et al., 1998; Decosterd et al., 2002; Berta et al., 2008; Laedermann et al., 2014b; Casals-Diaz et al., 2015; Yin et al., 2015). A human study reported no modification of Nav1.9 protein in patients with lingual nerve neuromas (Bird et al., 2013).
The conflicting observations of sodium channel regulation in animal models of neuropathic pain underscores that further research is necessary to clarify the mechanisms leading to Navs dysregulation and to those that generate hyperexcitability. Some of the aforementioned discrepant results can be attributed to the use of different models of pathological pain, the different species used, the relocalization of mRNA or protein, and/or various compensation mechanisms. Some of these studies analyzed the total pool of cellular Navs, but only Navs anchored at the membrane regulate the electrogenesis of nociceptive neurons. There is a large pool of intracellular Navs in the trafficking pathway (Schmidt et al., 1985), and it is possible that a modification of the membrane fraction can be overshadowed if one looks at the overall cellular pool of sodium channels. Furthermore, it remains possible, that an apparent decrease of total Nav expression in a cell is concomitant with an increase membrane expression of sodium channels. Studying mechanisms that regulate the trafficking of a channel, or mechanisms that directly alter the biophysical properties of a channel, might reconcile these discrepant results.
Post Translational Modifications
Both acquired and inherited pain syndromes are manifestations of altered function and expression of Navs that result in electrical instabilities in the nociceptive pathway, ultimately leading to pathological pain. In channelopathies, the pain syndrome is due to a DNA mutation that can be either sporadic or inherited. In acquired pain syndromes, the altered expression and function can be due to a plethora of signaling pathway activations. Among them, post-translational modifications (PTMs) are important contributors to the development of chronic pain syndromes.
Post-translational modifications are protein modifications that occur either soon after the ribosome-mediated translation of the mRNA into a polypeptide chain or later in the secretory pathway. These are critical steps for protein maturation and function. In these processes, many different enzymes attach biochemical groups (acetylation, phosphorylation), polypeptides (ubiquitylation, SUMOylation) and complex molecules (glycosylation, isoprenylation), or cleave (proteolysis) a protein’s specific amino acid. The overall effect of PTMs leads to a modulation of the structure, function or localization of the given protein. PTMs were first identified in the study of kinases (Hunter, 2009) and protein degradation (Ciechanover, 2005; Kresge et al., 2006) many decades ago. Since then the number of different PTMs has risen to over 200 (Mann and Jensen, 2003). PTMs are involved in almost every cellular event, from precise gene expression regulation to broad signal integration (Deribe et al., 2010).
Post-translational modifications have a large spectrum of action on proteins, ranging from very stable modifications to very transient and reversible changes. For instance, glycosylation and disulfide bridge formation are directly implicated in the synthesis, maturation and folding of the protein. The covalent binding of molecules, such as the addition of a ubiquitin moiety, leads to quicker and stable protein modification. On the other side of the spectrum, some PTMs are versatile and are important for transient cellular signaling, as exemplified by the phosphorylation process.
There is precise coupling between the interaction sites of PTMs and a given amino acid sequence on the target protein, rendering the system very specific. This also allows for good spatial (many different amino acids can be targeted by the same PTM) and temporal (a given amino acid can be modified by different PTMs) control and allows neurons to fine tune the properties of a protein depending on the changes occurring in the direct environment.
Ptms Alter Nav Function And/Or Expression
Computational studies have shown that, depending on the site of phosphorylation, the addition of a phosphoryl group that carries two negative charges at physiological pH (Narayanan and Jacobson, 2009) can modify the structure and the function of the protein through an alteration of the free energy landscape. Navs possesses charged residues in the voltage sensor domain that can sense membrane potential oscillation. When the transmembrane voltage changes, these charged domains reorient in the electric field resulting in conformational changes, a process referred to as gating. The addition of charged groups on their intra-cellular, extra-cellular or transmembrane domains modifies protein intrinsic properties and functions. Apart from the direct electrostatic effect on gating of the channels, phosphorylation can also create or disrupt binding sites for interaction with other regulatory proteins that modulate Nav function.
Navs need to be at the interface between a high extracellular and low intracellular sodium concentration to open and drive sodium influx. For this reason they are only functional when anchored at the plasma membrane. There is also, however, a large intracellular pool of Navs in the secretory pathway of the cell (Schmidt et al., 1985; Ritchie et al., 1990). A tight balance between the membrane and the intracellular pool, a process referred to as trafficking, is crucial for the fine-tuning of cellular excitability in nociceptive neurons. Maintaining this equilibrium is largely mediated by PTMs that regulate the trafficking of Navs (Cusdin et al., 2008). Some enzymes will generate PTMs that are responsible for internalization and/or degradation, whereas others will promote externalization or stabilization of Navs at the membrane.
Peripheral Sensitization Triggers Ptms
We previously highlighted that peripheral sensitization is triggered by the inflammatory soup (Basbaum et al., 2009). There is a large body of evidence that there is a recruitment of macrophages (Perry et al., 1987) and neutrophils (Daemen et al., 1998), as well as degranulation of mast cells (Olsson, 1967; Zochodne et al., 1994) in inflammatory processes, such as the ones observed after nerve injury. Once recruited, these cells secrete peptides, such as prostaglandins (PGE2), bradykinin, NGF and serotonin (Cesare et al., 1999; Petho and Reeh, 2012). They also secret cytokines, such as TNF-α or IL-1β and IL-6 (Wells et al., 1992). Some of these mediators are not only secreted by immune or inflammatory cells, but also by the nociceptive neurons themselves (Daemen et al., 1998). When inflammation resolves, these molecules are slowly cleared from the extracellular liquid around nociceptive neurons. In some cases, however, pro-nociceptive molecules, such as NGF and cytokines, (Leung and Cahill, 2010; Dogrul et al., 2011; Gaudet et al., 2011) are found to persist at the site of injury. This phenomenon may partially explain any long-term changes.
When binding to receptors, which include neurotrophic tyrosine kinase receptors (NTKR), such as TrkA, and/or G-protein coupled receptors (GPCRs), such as bradykinin and PGE2 receptors, these aforementioned ligands (peptides and cytokines) will activate multiple intracellular pathways, including Protein Kinase A (PKA), Protein Kinase B (PKB), Protein Kinase C (PKC), Mitogen-activated protein Kinase (MAPK) and the Ca++/Calmodulin-dependent Kinase I and II (CamKI/II), among others (Figure 1). The concrete, but not sole, effect of these cascades is phosphorylation or other PTMs of Navs (Dib-Hajj et al., 2010), which lead to long-term increased neuronal excitability. It should, however, be noted that PTMs also control other ion channels, such as potassium channels, and even modulate the activation of some transcription factors. They might also regulate nociceptive neuronal excitability through these alternative mechanisms.
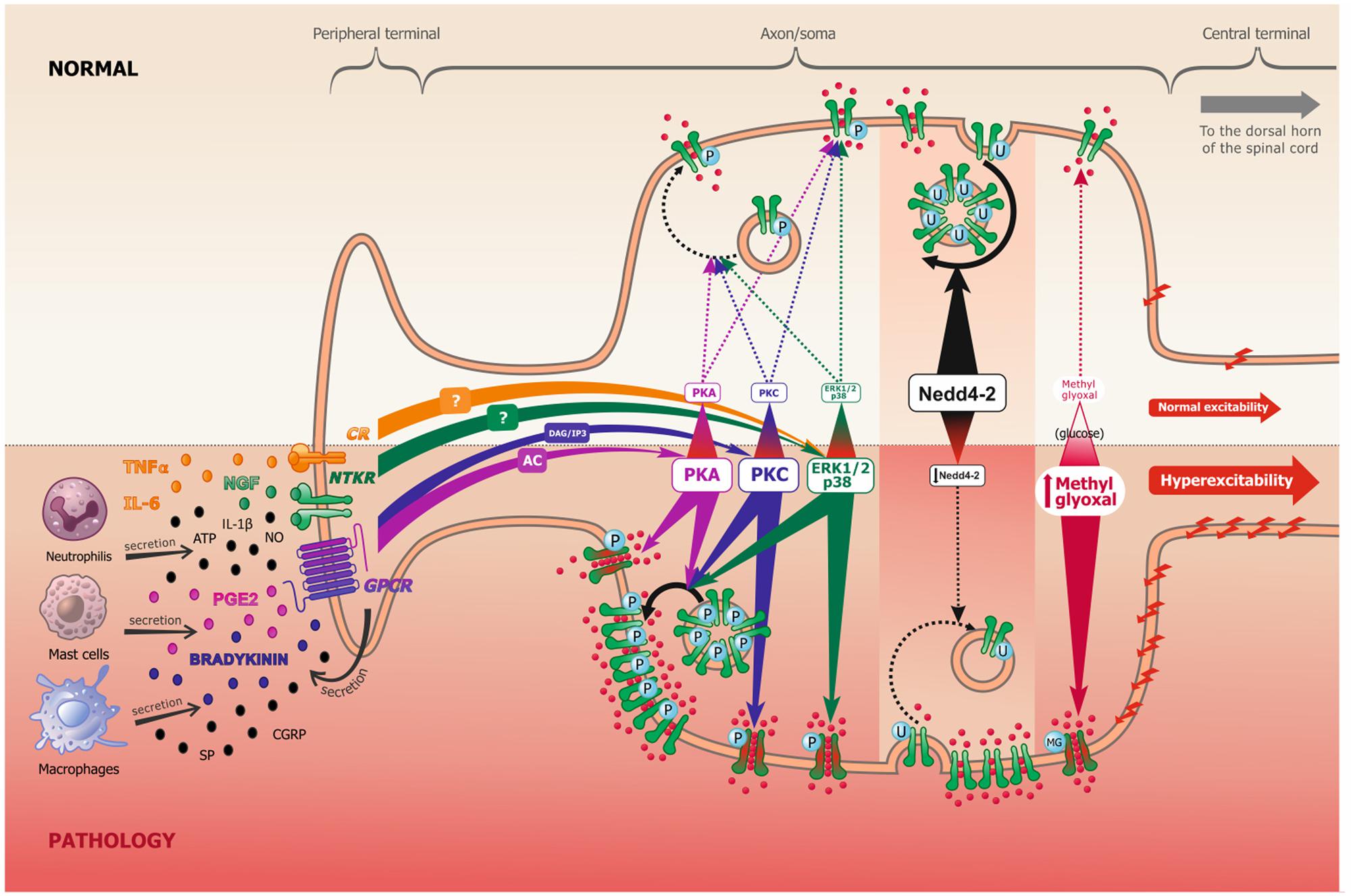
FIGURE 1. Scheme representing the peripheral terminal, axon and cell soma of a pseudo-unipolar dorsal root ganglia nociceptive neuron, in normal and pathological pain conditions. Following tissue damage and inflammation, recruited inflammatory cells secrete numerous pro-inflammatory molecules (referred to as the inflammatory soup). These chemical mediators activate many membrane proteins, including G-protein coupled receptors (GPCR), neurotrophic tyrosine kinase receptors (NTKR) and cytokines receptors (CR). GPCR binding by PGE2 and bradykinin mostly leads to protein kinase A (PKA) and protein kinase C (PKC) activation, through the adenylate cyclase (AC) and inositol 3-phosphate (IP3) secondary messengers, respectively. NTKR and CR binding by TNFα, IL-6 and NGF will activate ERK1/2 and p38 kinases via different potential secondary messengers. These signal transduction pathways can undergo cross communication one with another. In experimental neuropathic pain, nerve injury induces Nedd4-2 downregulation. In models of diabetic neuropathy, as well as in patients, methylglyoxal accumulation is related to an increased glucose concentration. The increased activation of kinases, the downregulation of Nedd4-2 and the accumulation of methylglyoxal all lead to an increase of Nav1.7/Nav1.8 function (shift of biophysical properties, making the channel more available, as represented in green and red in the figure) and/or to an increase of Nav1.7/Nav1.8 expression at the membrane (altered trafficking), which leads to increased sodium influx and consequently to nociceptive neuronal hyperexcitability.
Protein Kinases
Phosphorylation is an important PTM that affects up to 30% of proteins in vivo (Kreegipuu et al., 1999). The phosphate group is usually added to serine, threonine, histidine, and tyrosine residues in eukaryotic proteins. The effect of protein kinases (PKs) on Navs in peripheral chronic pain syndromes has been extensively reported in the literature. The reports are varied and complex, largely due to the important number of phosphorylation sites and to the large number of different PKs present in nociceptive neurons. PKs can modulate Nav function in a very short-term range upon electrostatic interferences. Phosphate groups carry a -2 charge at physiological pH that might interfere with the Navs voltage-sensing domain, or other domains implicated in the gating of the channel. PKs can also have long-term effects when regulating Nav trafficking, which is acheived by masking or highlighting targeting sequences, such as the endoplasmic reticulum (RE) retention signal of Navs (Zhou et al., 2002).
Protein Kinase A
The immediate increase in nociceptive neuronal excitability observed after tissue injury or inflammation results from G-protein-coupled receptor activation and the resulting intracellular signaling pathway (Woolf and Costigan, 1999). G-protein binding leads to an increase in cAMP production by adenylate cyclase (AC), which ultimately activates PKA. Triggering of this pathway by direct cAMP application or activation of AC is sufficient to elicit hyperalgesia in animals (Taiwo et al., 1989; Kress et al., 1996; Hucho and Levine, 2007). PKA activation not only plays a role in the initiation of pain hypersensitivity, but is also important for the maintenance of inflammatory pain (Aley and Levine, 1999). PGE2 is one of the best-known ligands that activates this pathway. PGE2 binds to the prostaglandin E2 receptor (E2) and increases cAMP in sensory neurons (Pierre et al., 2009). Many studies have reported that PGE2-induced hyperalgesia is due to PKA activation (Pitchford and Levine, 1991; Khasar et al., 1995; England et al., 1996).
Protein kinase A phosphorylates Nav1.2 in brain neurons (Costa and Catterall, 1984a; Rossie and Catterall, 1987), with the main effect being a 50% reduction in the peak current (Gershon et al., 1992), along with modifications of some biophysical properties (Li et al., 1992; Smith and Goldin, 1996). This suggests that both the function and expression of Nav1.2 are modified by PKA. Since these early studies, other groups have reported similar decreases in Nav1.1, Nav1.6, and Nav1.7 in different cell expression systems and intact cells (Gershon et al., 1992; Cantrell et al., 1997; Smith and Goldin, 1998; Zhou et al., 2000; Vijayaragavan et al., 2004a; Chen et al., 2008; Liu and Zheng, 2013). The phosphorylation sites of Nav1.2 by PKA were mostly investigated using traditional biochemical approaches (Murphy et al., 1993; Smith and Goldin, 1996; Cantrell et al., 1997). A lot of new predictive bioinformatic tools have helped identify phosphorylation sites on other isoforms.
Protein kinase A is increased after inflammation and in pathological pain states. Vijayaragavan et al. (2004a) showed that PKA decreases Nav1.7. One could hypothesize, therefore, that inflammation may decrease nociceptive neuronal excitability through Nav downregulation. However, Nav1.7, as well as other Nav isoforms, can undergo alternative splicing, with each splice variant possessing distinct biochemical and pharmacological properties (Schaller et al., 1992; Plummer et al., 1997; Dietrich et al., 1998; Schirmeyer et al., 2014). It was previously shown that a particular Nav1.7 splice variant, Nav1.7 11S, was upregulated and responsible for pain hypersensitivity in animal models of neuropathic pain (Raymond et al., 2004). Contrary to the lack of effect of PKA on three spice variants, the Nav1.7 11S splice variant activation curves were shifted to hyperpolarized potentials upon PKA activation, thus lowering the threshold for opening of the channel and presumably increasing neuronal excitability (Chatelier et al., 2008). The current hypothesis is that in chronic pain syndromes the increase of this splice variant, together with the modification of its biophysical properties by PKA, will lead to an increase in sodium conductance which results in nociceptive neuronal hyperexcitability. One must keep in mind that the effect of PKA on Nav1.7 was found in Xenopus oocytes and other mammalian cell lines, which have a different cellular background than nociceptive neurons, and that Nav regulation varies greatly depending on cell type (Cummins et al., 2001). This is highlighted by one Nav1.7 mutation that renders DRG sensory neurons hyperexcitable, but decreases sympathetic DRG neuron excitability (Rush et al., 2006). Studying an endogenous Nav1.7 current in adrenal chromaffin cells revealed that cAMP upregulates Nav1.7 (Yuhi et al., 1996). Thus, a careful characterization of PKA’s effect on the Nav1.7 current in nociceptive neurons needs to be performed.
The effect of PKA on TTX-resistant isoforms is the opposite to that aforementioned. PKA increases the TTX-resistant current (Gold et al., 1996, 1998) in DRG neurons, an effect at least partially due to PGE2 (England et al., 1996). A similar increase in the Nav1.8 current isolated from a recombinant protein expressed in Xenopus oocytes (Vijayaragavan et al., 2004a) and mammalian cells (Fitzgerald et al., 1999) was also reported, providing evidence that PKA activation in pathological pain states can generate nociceptive hyperexcitability through enhanced Nav1.8 expression and function. How PKA increases the Nav1.8 current is partially due to increased membrane trafficking, since blocking protein transports in DRG neurons prevented a PKA-induced increase of the Nav1.8 mediated current (Liu et al., 2010). Furthermore, using a site-directed mutagenesis approach, the authors proposed that the forward trafficking effects of Nav1.8 could be due to the phosphorylation of an identified ER retention signal.
Nav1.9 is also increased by inflammatory mediators (Maingret et al., 2008), such as PGE2. This mechanism is dependent on G-protein activation (Rush and Waxman, 2004) and GTPγS (Vanoye et al., 2013).
Protein Kinase B
The PKB (also referred to as Akt) is well-known for its role in neuronal plasticity in the brain (Sanna et al., 2002), but little is known about its role in the pain field. There is evidence, however, that PKB is activated in sensory neurons in animal models of neuropathic pain (Xu et al., 2007a; Shi et al., 2009) and inflammatory pain (Zhuang et al., 2004; Sun et al., 2006). Furthermore, intrathecal injection of a PKB inhibitor attenuated formalin and carrageenan-induced hypersensitivity (Xu et al., 2011). The authors demonstrated that this effect was due to mTOR signaling in the spinal cord, but the intrathecal injection mode of delivery cannot rule out an additional effect in the peripheral nervous system. A recent study showed that PKB activation in peripheral sensory neurons was necessary for the inflammatory-induced increased expression of both Nav1.7 and Nav1.8 (Liang et al., 2013) since blocking this kinase reversed the upregulation of both isoforms. The previous study did not demonstrate that PKB could directly phosphorylate Navs, nor has any other study to our knowledge. However, ASICS, another important ion channel implicated in pain processing, can be directly phosphorylated by PKB, leading to increased trafficking and enhanced expression of ASICs at the membrane (Duan et al., 2012). Further investigations are required to unravel the role of PKB in the phosphorylation of Nav channels in pathological pain.
Protein Kinase C
The implication of PKC activation in nociceptive neurons has been extensively studied. Activation of PKC by phorbol esters demonstrated an in vivo implication of this pathway in peripheral sensitization (Rang and Ritchie, 1988; Schepelmann et al., 1993; Souza et al., 2002). PKC inhibition decreased hyperalgesia in a model of diabetic neuropathy. Together with PGE2, bradykinins are also able to activate PKC pathways (Cesare and McNaughton, 1996; Ferreira et al., 2005, 2008). PKC is a serine/threonine kinase, which has at least 12 different isoforms (Way et al., 2000) that can be classified into different groups (Battaini, 2001). PKC𝜀 is one of the isoforms shown to be necessary for the development of hypersensitivity in animal models of peripheral chronic pain (Khasar et al., 1999; Aley et al., 2000; Dina et al., 2000, 2001). Inflammatory molecules enhance the translocation of PKC𝜀 to the membrane, where it contributes to peripheral sensitization (Khasar et al., 1999; Hucho and Levine, 2007; Zhang et al., 2007). Other PKC isoforms have also been implicated in chronic pain, but focus has been on their expression in the spinal cord.
As for PKA, the phosphorylation of Nav1.2 by PKC (Costa and Catterall, 1984b) could be responsible for a reduction of up to 80% of the current when expressed in Xenopus oocytes, with a concomitant slowing of its inactivation (Numann et al., 1991). This reduction is due, at least in part, to a positive shift in the voltage-dependence of activation (Dascal and Lotan, 1991). Similar findings on the total sodium current have been observed in rat brain neurons (Numann et al., 1991; Cantrell et al., 1996). Other studies in Xenopus oocytes have shown that PKC downregulates the skeletal muscle sodium channel Nav1.4 (Bendahhou et al., 1995), the cardiac channel Nav1.5 (Murray et al., 1997), as well as the two pain specialized isoforms Nav1.7 and Nav1.8 (Vijayaragavan et al., 2004a). It was initially hypothesized that PKC robustly downregulates Navs across species since a reduction in the Nav1.5 current was also observed in myocytes and CHO cells (Qu et al., 1994), and a reduction in the total sodium current was observed in neuroblastoma cells (Renganathan et al., 1995), hippocampal neurons (Cantrell et al., 1996), and cortical neurons (Mittmann and Alzheimer, 1998).
As with PKA activation, the decrease of the Nav1.7 current mediated by PKC activation (Vijayaragavan et al., 2004a) is discrepant with the increased excitability observed in pathological pain. Again, the cellular background studied could explain these discrepancies. Furthermore, a study reported that blocking PKC phosphorylation was concomitant with a decrease in Nav1.7 protein expression upon continuous opioid administration in a diabetic painful neuropathic model (Chattopadhyay et al., 2008). In addition, a recent study also showed that PMA activation of PKC pathways led to an increase of Nav1.7 resurgent currents in HEK cells (Tan et al., 2014), currents which have been implicated in pathological pain (Jarecki et al., 2010).
Gold et al. (1996, 1998) observed that the activation of PKC increased the TTX-resistant current in nociceptive neurons. This was later confirmed by another group (Ikeda et al., 2005). A later study determined which PKC isoform and which of the two TTX-resistant isoforms were responsible for the increased current, implicating PKC𝜀 and Nav1.8 (Cang et al., 2009). The PKC𝜀 mediated upregulation of Nav1.8 was confirmed in another study, demonstrating the causative link to hyperalgesia (Wu et al., 2012). Since PGE2 can activate the PKC pathway and increase the Nav1.9 current in nociceptive neurons (Rush and Waxman, 2004), it is likely that both Nav1.8 and Nav1.9 are implicated. Nav1.8 and Nav1.9 are well-identified sodium channels whose expression and function are increased upon pathological pain-driven PKC activation. The role of Nav1.7, however, remains to be fully elucidated. Since PKA and PKC pathways converge and co-regulate Nav1.2 function (Chahine et al., 2005), similar mechanisms might also account for the effects on Nav1.7, Nav1.8 (Gold et al., 1998) and Nav1.9.
MAPK Pathway
Mitogen-activated protein kinases are another family of kinases that play an important role in mammalian cell signaling. There are three major members in the MAPK family: ERK, p38 and c-JUN. Each member activates a specific intracellular pathway (Widmann et al., 1999). They regulate various cellular activities and have been implicated in numerous human diseases, including tissue injury (Kim and Choi, 2010). Since they are activated by proinflammatory cytokines (Ji et al., 2009), they have been shown to be important in pathological pain (Obata and Noguchi, 2004).
In transected axons of experimental neuromas, Nav1.7 accumulates with ERK1/2 at the site of injury (Persson et al., 2011). Since ERK1/2 was shown to phosphorylate Nav1.7, altering its biophysical properties and rendering it easier to open in response to stimuli (Stamboulian et al., 2010), it is hypothesized that a Nav1.7 and ERK1/2 co-accumulation would increase nociceptive neuronal firing. Another study proposed that IL-6 application enhances the excitability of trigeminal ganglion neurons via ERK-mediated phosphorylation of Nav1.7, a mechanism involved in the development of migraine-related pain behavior (Yan et al., 2012).
Another kinase of the MAPK, p38, was reported to be increased in animal models of neuropathic pain (Obata and Noguchi, 2004; Xu et al., 2007b), leading to the upregulation of TTX-resistant sodium channels in sensory neurons (Jin and Gereau, 2006). A study recently unraveled the role of TNF-α in activating p38, resulting in the modification of the slow inactivation and voltage dependence of activation of Nav1.8/Nav1.9. This, in turn, increases the TTX-resistant inward current and enhances nociceptive hyperexcitability (Gudes et al., 2015). Another study showed that p38 phosphorylates Nav1.8, increasing the trafficking of this channel at the membrane of DRG neurons (Hudmon et al., 2008).
Similar to the previous studies investigating MAPK activation in animal models of chronic pain and those focusing on the regulation of Nav1.7 and Nav1.8 by kinases in vitro, ERK1/2 and p38 are increased along with the Nav1.7 and Nav1.8 isoforms in painful human neuromas (Black et al., 2008), where they likely also contribute to neuronal hyperexcitability.
CamKII
Calmodulin (CaM) is a small calcium-binding protein that senses Ca2+ changes and drives cellular responses to rapid changes in intracellular calcium concentration. It is also known to be involved in regulating Ca2+-dependent neuronal plasticity (Solà et al., 2001).
The C-terminus of Navs contains a CaM-binding domain, known as the IQ motif (Mori et al., 2000), which is a recognizable site for these calcium-sensing proteins. Even though the functional meaning of such binding remains unknown, there are a few studies reporting the regulation of Nav currents by CaM. Despite the fact that all Navs possess a conserved IQ motif, the regulatory effect of CaM is isoform specific, as exemplified by the more potent regulation of Nav1.4 as compared to Nav1.5 by this protein (Deschênes et al., 2002; Young and Caldwell, 2005; Biswas et al., 2008; Ben-Johny et al., 2014). CaM also regulates Nav1.6, which is expressed in nociceptive neurons (Herzog et al., 2003). Interestingly, Nav1.7 is also bound by CaM (Herzog et al., 2003), but with a lower affinity than Nav1.6.
CaM Ca2+-dependent activation leads to the activation of many signaling molecules, one being the Ca2+/CaM-dependent serine/threonine kinase (CamK) (Nelson and Chazin, 1998). CamKII is expressed in nociceptive neurons and is involved in pain transmission (Hiruma et al., 1999; Brüggemann et al., 2000). It was proposed that CamKII is responsible for the transition from acute to chronic pain, a process involving PKC𝜀 activation (Ferrari et al., 2013) and links these two kinase pathways. Further evidence supporting a role for CamKII in neuropathic pain was demonstrated by using a CamKII inhibitor that reversed mechanical allodynia in animal models of both neuropathic pain (Chen et al., 2009) and inflammatory pain (Luo et al., 2008a).
Since the CaM binding effect on Navs regulation is thought to be due to the recruitment and subsequent phosphorylation of Navs by CamKII (Deschênes et al., 2002; Maltsev et al., 2008), it is likely that the activation of this kinase in pathological pain states is partially responsible for hyperexcitability through the regulation of Nav. This hypothesis remains to be confirmed.
Glycosylation
Glycosylation is another important PTM that affects sodium channel function and expression. Glycosylation is a crucial step for protein biosynthesis and folding, but it is also involved in cell signaling, cell-cell adhesion, protection against proteolysis, and cellular development and immunity (Moremen et al., 2012). Nav α-subunits undergo important glycosylation steps in the endoplasmic reticulum and Golgi apparatus (Waechter et al., 1983; Schmidt and Catterall, 1987), a process involving the sequential addition of N-acetylglucosamines capped by sialic acid residues, and the further addition of diverse oligosaccharide chains. Glycosylation can account for 5% (Cohen and Barchi, 1993) to 30% of the α-subunit’s molecular weight (Messner and Catterall, 1985), depending on the isoform, with an estimated stoichiometry of around 100 sialic acid molecules per channel (James and Agnew, 1987). Glycosylation is known to influence Nav gating properties (Recio-Pinto et al., 1990; Bennett et al., 1997; Zhang et al., 1999; Tyrrell et al., 2001) by interfering with the electric field near the gating sensors (Bennett et al., 1997; Cronin et al., 2004). It was proposed that extracellular sialic acid residues, which are negatively charged at physiological pH, influence the sensitivity of the voltage sensor domains to the transmembrane electrical potential difference (Ednie and Bennett, 2011).
Only a few studies have investigated glycosylation of Navs in the peripheral sensory nervous system. One reported that Nav1.9 is subject to important developmentally regulated glycosylation. This isoform is found in two different heavily glycosylated forms in neonatal rats, which have different gating properties as compared to the less glycosylated form of Nav1.9 in adult tissue (Tyrrell et al., 2001). Another study reported that Nav1.7 is found in at least two different glycosylated forms in HEK293 cells: a heavily functional glycosylated form and a core-glycosylated immature form (Laedermann et al., 2013a). The same authors later reported that a third intermediate glycosylated form is also present in HEK293 cells (Laedermann et al., 2013b). Inhibition of glycosylation in Xenopus oocytes by tunicamycin also altered Nav1.3 gating properties (Xu et al., 2008). Whether these different patterns of α-subunit glycosylation lead to modification of Nav function has yet to be investigated in peripheral chronic pain syndromes.
Ubiquitylation
Ubiquitylation is another well-known PTM that negatively regulates the cell surface expression of many different plasma membrane proteins (Staub and Rotin, 2006). Ubiquitylated proteins that are internalized through this pathway are either degraded or recycled (Shih et al., 2000; Abriel and Staub, 2005; Ciechanover, 2005). Ubiquitin is a small and highly conserved polypeptide of 76 amino acids that is covalently attached to the lysine residues of the targeted protein. Three enzymatic successive steps are required to ubiquitylate a protein (Pickart, 2001): (1) ubiquitin is first activated by a ubiquitin-activating enzyme (E1) in an ATP-dependent manner, (2) ubiquitin is then transferred to a ubiquitin-conjugating enzyme (E2) via a thioester bond, (3) this complex further interacts with an ubiquitin-protein ligase (E3) that eventually ubiquitylates the substrate protein.
The first and probably best-described protein that undergoes ubiquitylation is ENaC (Abriel et al., 1999; Rossier et al., 2002). The ENaC subunit possesses a PY motif. Mutating this motif is sufficient to generate a hypertensive phenotype, known as Liddle’s Syndrome (Schild et al., 1996), which involves increased ENaC function (Firsov et al., 1996). Nedd4 and Nedd4-2 proteins where shown to bind to ENaC’s PY motif on large members of the E3 ubiquitin ligase family (Joazeiro and Weissman, 2000; Metzger et al., 2012), leading to its internalization (Abriel et al., 1999; Henry et al., 2003). This process is impaired in Liddle’s syndrome.
Most Navs possess a PY motif at their α-subunit C-terminal, making them potential substrates for Nedd4-2 dependent ubiquitylation (Abriel et al., 2000; Laedermann et al., 2014a). The first sodium channel isoform described to be regulated by Nedd4-2 was Nav1.5. Regulation was demonstrated in both cell expression systems and cardiac tissue (van Bemmelen et al., 2004). Nav1.7 and Nav1.8 also possess a PY motif and were shown to be negatively regulated by Nedd4-2 in both Xenopus oocytes (Fotia et al., 2004) and HEK293 cells (Laedermann et al., 2013a). The functional significance of Nav regulation by Nedd4-2 was previously demonstrated in chronic pain syndromes. In an animal model of neuropathic pain, ubiquitin ligase expression was robustly reduced in both mice (Laedermann et al., 2013a) and rats (Cachemaille et al., 2012). Nav1.7 and Nav1.8 expression were increased as a consequence of Nedd4-2 downregulation. The causal link between Nedd4-2 downregulation and Nav1.7/Nav1.8 upregulation was demonstrated using both tissue specific knockout and viral overexpression of Nedd4-2, leading to hyper- and hypo- pain sensing phenotypes, respectively (Laedermann et al., 2013a). The reduction of Nedd4-2 might also increase membrane expression of other Navs, with the exception of Nav1.9 which lacks the PY motif, (Abriel et al., 2000), and other ion channels expressed in sensory neurons that are also substrates for this ubiquitin ligase (Bongiorno et al., 2011). Nav1.6 was also regulated by Nedd4-2 in mouse hippocampal neurons, a process dependent on the concomitant p38-mediated phosphorylation of this sodium channel isoform (Gasser et al., 2010). Whether Nedd4-2 downregulation in neuropathic pain is due to the inflammatory soup or to another mechanism remains to be investigated.
Methylglyoxal
A recent study unraveled a new mechanism accounting for painful peripheral neuropathy in diabetes. The authors showed that the concentration of methylglyoxal, an endogenous degradation product of excessive glycolysis (Thornalley, 2005), is increased in patients suffering from diabetes. Since peripheral nerves have low levels of enzymes that metabolize methylglyoxal (Bierhaus and Nawroth, 2009), its accumulation in the sensory system was proposed to account for pain hypersensitivity. The authors reported that the methylglyoxal effect on excitability was through its binding to Nav1.8 within the DIII-DIV linker on an arginine residue, which reduced channel inactivation (Bierhaus et al., 2012), leaving the channel in an excitable state. In sciatic nerve biopsies isolated from patients with diabetes and from those who had amputations due to peripheral artery disease, the authors observed an increase of Nav1.8 modification by methylglyoxal when compared to controls. It was proposed that this PTM was specific to Nav1.8 in regards to sodium channel regulation, but the authors demonstrated that methylglyoxal can also have an effect on pain pathways by depolarizing nociceptive neurons, and increasing GCRP release and COX-2 expression.
Post-Translational Modification Of Nav Protein Partners
Many protein partners are known to interact with typical pain isoforms and undergo PTM. Evidence is lacking which demonstrates a direct effect between protein partners’ PTMs and concomitant modification of Nav expression or function. In the following chapter, we will discuss the potential partners that undergo PTMs that are known to regulate Nav1.7, Nav1.8, and Nav1.9.
β–Subunits
β–subunits are important regulators of Navs. They are implicated in neuropathic pain and are subject to PTMs. β-subunits regulate α-subunit gating properties by direct steric interactions that interfere with the voltage-sensor (Zimmer and Benndorf, 2002). Even though the effects of the different β-subunits on biophysical properties give rise to conflicting results (Nuss et al., 1995; Sangameswaran et al., 1997; Smith and Goldin, 1998; Morgan et al., 2000; Fahmi et al., 2001; Vijayaragavan et al., 2001, 2004b; Zimmer and Benndorf, 2002), partially due to the different cell types used, the literature clearly demonstrates that β-subunits regulate Nav gating. β-subunits can also affect the Nav current in an ex vivo nociceptive neuron primary culture, as highlighted by the decreased INa current recorded from SCN1B and SCN2B knockout animals (Lopez-Santiago et al., 2006, 2011). SCNB knockout animals also demonstrated abnormal pain sensing, confirming that modulating Nav function ultimately modulates pain signaling (Pertin et al., 2005; Lopez-Santiago et al., 2006, 2011). Moreover, β1-, β2-, and β3-subunit expression levels are increased in different animal models of pathological pain (Shah et al., 2000; Coward et al., 2001b; Pertin et al., 2005), highlighting their potential implication in modulating cellular excitability. Auxiliary β-subunits are themselves substrates for glycosylation (Isom et al., 1992), which ultimately modulates α-subunit function (Johnson et al., 2004; Johnson and Bennett, 2006). β-subunits can also be phosphorylated, i.e., the phosphorylation of Tyr181 is necessary for the interaction with ankyrin, another Nav protein partner (Malhotra et al., 2002). Whether altered glycosylation or phosphorylation of the β-subunits could be implicated in pathological pain has, at least to our knowledge, never been investigated.
Nedd4-2
Nedd4-2, a potent regulator of Navs in the sensory nervous system, also undergoes several PTMs, particularly by kinases which alter its function. Most of these pathways have been investigated in the Nedd4-2 regulation of ENaC (Snyder, 2009). Whether a similar action also occurs on the sodium channels in the sensory system remains to be determined. Pathways that lead to Nedd4-2 PTMs are the same pathways as those activated in chronic pain syndromes. For instance, phosphorylation of Nedd4-2 by PKA was demonstrated to decrease Nedd4-2 ubiquitylating efficiency (Snyder et al., 2004). In addition to its direct effect on Navs, it is probable that PKA activation also indirectly leads to an increase of Nav channels at the membrane by impeding the downregulatory role of Nedd4-2. It has been well documented that cytokine signaling leads to NF-κB activation in inflammatory processes. These signals first activate the IκB kinase [an inhibitor of nuclear factor κB (NF-κB)] (IKK) via phosphorylation, which then phosphorylates and inactivates IκB inhibitory proteins. IKKβ, a subunit of IKK that is expressed in unmyelinated fibers, then binds and phosphorylates Nedd4-2 (Edinger et al., 2009). Finally, NGF, another inflammatory mediator (Leung and Cahill, 2010) also known to positively regulate Navs mRNA expression (Toledo-Aral et al., 1997; Fjell et al., 1999), triggers a cascade after binding to the TrkA receptor, which ultimately leads to Nedd4-2 phosphorylation (Arévalo et al., 2006). Pro-inflammatory molecules likely negatively regulate Nedd4-2 activity in nociceptive neurons following inflammation, leading to an increase in Nav expression at the membrane.
Collapsin Response Mediator Protein 2 (CRMP2)
Collapsin response mediator protein 2 (CRMP2) is a protein initially identified to be important for axonal outgrowth (Inagaki et al., 2001). It is now known to be important for modulating ion channel trafficking (Bretin et al., 2006; Chi et al., 2009). CRMP2 is the secondary target of the anti-epileptic drug lacosamide (Errington et al., 2006, 2008) and can directly bind to Navs (Wang et al., 2010), modulating the channel’s slow inactivation. In a recent study carried out in both HEK293 cells and sensory neurons, it was demonstrated that CRMP2 can be SUMOylated, which affects Nav1.7 trafficking (Dustrude et al., 2013). Whether SUMOylation of CRMP2 plays a role in pathological pain via the alteration of Nav1.7 trafficking remains to be investigated.
There are many other well described proteins, i.e., contactin (Ranscht, 1988; Kazarinova-Noyes et al., 2001), ankyrin (Malhotra et al., 2000), spectrin (Bennett and Baines, 2001), dystrophin, and syntrophin (Gee et al., 1998; Abriel and Kass, 2005) among others (Shao et al., 2009), that interact with Navs and modulate their function and cell surface expression. Kinases are themselves substrates for PTMs that can modify their function in important ways. All of these proteins are subject to PTMs, which may impact the control of Nav function and expression in peripheral chronic pain syndromes.
Ptms Modulate Nav Transcription
In studies carried out in adrenal chromaffin cells, which express relatively high levels of Nav1.7 (Klugbauer et al., 1995; Toledo-Aral et al., 1995, 1997; Goldin, 2001; Wada et al., 2004, 2008), a role for PTMs in controlling Nav transcription was proposed. ERK kinases were shown to positively regulate Nav1.7 expression at the membrane by modulating their mRNA stability (Yanagita et al., 2003). Conversely, PKC𝜀 kinase activation destabilized Nav1.7 mRNA, contributing to Nav1.7 negative regulation of the steady-state levels at the plasma membrane (Yanagita et al., 1996). Whether similar mechanisms occur in sensory neurons has not been reported.
Discussion
Navs expression and function are dysregulated in peripheral inflammatory pain, nerve injury induced neuropathic pain, metabolic, infectious, toxic or inherited painful neuropathies. Peripheral mechanisms for initiation and maintenance of pain is partially due to the release of inflammatory molecules that trigger different signaling cascades and leads to the activation of enzymes with diverse functions. In turn, these enzymes will post-translationally modify Nav function and expression, ultimately impacting nociceptive neuronal hyperexcitability and pain. Most knowledge of such mechanisms comes from the kinase field, but since Navs are large proteins composed of over 2000 amino acids, they possess a plethora of domains that could be subject to PTMs. It is likely that PTM’s contribution to chronic pain syndromes are still under-estimated and should be further investigated. In this review, we not only discussed proven Nav PTMs implications in pathological pain but also other potential relevant PTMs, which will eventually open new avenues for a deeper understanding of chronic pain syndromes. Together with the development of more efficient and specific pharmacological agonists and antagonists targeting PTM effectors, new genome engineering tools will facilitate the generation of knock-in mice with mutations on PTM sites or on the PTM effectors themselves. This will hopefully help confirm and identify new pathways that regulate Nav function and expression.
The pharmaceutical industry is attempting to develop highly selective drugs that block Nav1.7, Nav1.8, or Nav1.9, but with unfortunately little progress. This is partially due to the fact that Navs are well-conserved proteins, and non-specific blockers are bound to have many dramatic side effects. Rather than targeting and blocking the sodium channels themselves, regulating the function or expression of sodium channels, which may have more subtle effects on excitability, might prove to be an interesting alternative to treat chronic pain. From this perspective, more effort should be focused on targeting PTM pathways, as exemplified by the anti-TNF-α drug, which is promising and still in expansion (Leung and Cahill, 2010). Pharmacological targeting of the MAPK pathway has already been shown to have an analgesic effect (Tong et al., 2012), and many other kinases are currently in pre-clinical or clinical studies (Ji et al., 2007). Other promising avenues include the use of scavengers of methylglyoxal (Bierhaus et al., 2012) or the use of gene therapy to restore Nedd4-2 ubiquitylating function, which have proven to be efficient in relieving pain in animal models of chronic pain. Altogether, it is important to further characterize the known PTM effects on Navs and to identify new PTMs in order to gain insight into the development of pathological pain.
Funding
The authors are supported by grants from the Swiss National Science Foundation (Early-postdoc.mobility P2LAP3_151557 to C.J.L. 310030_159615/1 to ID and 310030B- 135693 to HA).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. E. Buttermore and Dr. A. Felley for their comments on the manuscript of this chapter. We thank Desi Ivanova for her help with Figure 1. Images from Motifolio drawing toolkit (www.motifolio.com) were used in Figure 1 preparation.
References
Abdulla, F. A., and Smith, P. A. (2002). Changes in Na(+) channel currents of rat dorsal root ganglion neurons following axotomy and axotomy-induced autotomy. J. Neurophysiol. 88, 2518–2529. doi: 10.1152/jn.00913.2001
Abrahamsen, B., Zhao, J., Asante, C. O., Cendan, C. M., Marsh, S., Martinez-Barbera, J. P., et al. (2008). The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321, 702–705.
Abriel, H., Kamynina, E., Horisberger, J. D., and Staub, O. (2000). Regulation of the cardiac voltage-gated Na+ channel (H1) by the ubiquitin-protein ligase NEDD4. FEBS Lett. 466, 377–380.
Abriel, H., and Kass, R. S. (2005). Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc. Med. 15, 35–40. doi: 10.1016/j.tcm.2005.01.001
Abriel, H., Loffing, J., Rebhun, J. F., Pratt, J. H., Schild, L., Horisberger, J. D., et al. (1999). Defective regulation of the epithelial Na+ channel by NEDD4 in Liddle’s syndrome. J. Clin. Invest. 103, 667–673. doi: 10.1172/JCI5713
Abriel, H., and Staub, O. (2005). Ubiquitylation of ion channels. Physiology (Bethesda) 20, 398–407. doi: 10.1152/physiol.00033.2005
Akopian, A., Sivilotti, L., and Wood, J. (1996). A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 379, 257–262.
Akopian, A. N., Souslova, V., England, S., Okuse, K., Ogata, N., Ure, J., et al. (1999). The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 2, 541–548.
Akopian, A. N., Souslova, V., Sivilotti, L., and Wood, J. N. (1997). Structure and distribution of a broadly expressed atypical sodium channel. FEBS Lett. 400, 183–187. doi: 10.1016/s0014-5793(96)01389-0
Aley, K. O., and Levine, J. D. (1999). Role of protein kinase A in the maintenance of inflammatory pain. J. Neurosci. 19, 2181–2186.
Aley, K. O., Messing, R. O., Mochly-Rosen, D., and Levine, J. D. (2000). Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the 𝜀 isozyme of protein kinase C. J. Neurosci. 20, 4680–4685.
Amaya, F., Wang, H., Costigan, M., Allchorne, A., Hatcher, J., Egerton, J., et al. (2006). The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 26, 12852–12860.
Arévalo, J. C., Waite, J., Rajagopal, R., Beyna, M., Chen, Z. Y., Lee, F. S., et al. (2006). Cell survival through trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron 50, 549–559. doi: 10.1016/j.neuron.2006.03.044
Bair, M. J., Robinson, R. L., Katon, W., and Kroenke, K. (2003). Depression and pain comorbidity: a literature review. Arch. Intern. Med. 163, 2433–2445. doi: 10.1001/archinte.163.20.2433
Basbaum, A., Bautista, D., Scherrer, G., and Julius, D. (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284.
Battaini, F. (2001). Protein kinase C isoforms as therapeutic targets in nervous system disease states. Pharmacol. Res. 44, 353–361.
Bendahhou, S., Cummins, T. R., Potts, J. F., Tong, J., and Agnew, W. S. (1995). Serine-1321-independent regulation of the mu 1 adult skeletal muscle Na+ channel by protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 92, 12003–12007.
Beneski, D. A., and Catterall, W. A. (1980). Covalent labeling of protein components of the sodium channel with a photoactivable derivative of scorpion toxin. Proc. Natl. Acad. Sci. U.S.A. 77, 639–643.
Ben-Johny, M., Yang, P. S., Niu, J., Yang, W., Joshi-Mukherjee, R., and Yue, D. T. (2014). Conservation of Ca2+/Calmodulin regulation across Na and Ca2+ channels. Cell 157, 1657–1670.
Bennett, E., Urcan, M. S., Tinkle, S. S., Koszowski, A. G., and Levinson, S. R. (1997). Contribution of sialic acid to the voltage dependence of sodium channel gating. J. Gen. Physiol. 109, 327–343. doi: 10.1085/jgp.109.3.327
Bennett, V., and Baines, A. J. (2001). Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol. Rev. 81, 1353–1392.
Berta, T., Poirot, O., Pertin, M., Ji, R., Kellenberger, S., and Decosterd, I. (2008). Transcriptional and functional profiles of voltage-gated Na(+) channels in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Mol. Cell. Neurosci. 37, 196–208.
Bierhaus, A., Fleming, T., Stoyanov, S., Leffler, A., Babes, A., Neacsu, C., et al. (2012). Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 18, 926–933. doi: 10.1038/nm.2750
Bierhaus, A., and Nawroth, P. P. (2009). Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 52, 2251–2263. doi: 10.1007/s00125-009-1458-9
Bird, E., Christmas, C., Loescher, A., Smith, K., Robinson, P., Black, J., et al. (2013). Correlation of Nav1.8 and Nav1.9 sodium channel expression with neuropathic pain in human subjects with lingual nerve neuromas. Mol. Pain 9:52.
Bird, E. V., Robinson, P. P., and Boissonade, F. M. (2007). Nav1.7 sodium channel expression in human lingual nerve neuromas. Arch. Oral Biol. 52, 494–502. doi: 10.1016/j.archoralbio.2006.11.011
Biswas, S., Deschênes, I., Disilvestre, D., Tian, Y., Halperin, V. L., and Tomaselli, G. F. (2008). Calmodulin regulation of NaV1.4 current: role of binding to the carboxyl terminus. J. Gen. Physiol. 131, 197–209. doi: 10.1085/jgp.200709863
Black, J. A., Dib-Hajj, S., Mcnabola, K., Jeste, S., Rizzo, M., Kocsis, J., et al. (1996). Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res. Mol. Brain Res. 43, 117–131.
Black, J. A., Liu, S., Tanaka, M., Cummins, T. R., and Waxman, S. G. (2004). Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108, 237–247. doi: 10.1016/j.pain.2003.12.035
Black, J. A., Nikolajsen, L., Kroner, K., Jensen, T., and Waxman, S. (2008). Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann. Neurol. 64, 644–653.
Bongiorno, D., Schuetz, F., Poronnik, P., and Adams, D. J. (2011). Regulation of voltage-gated ion channels in excitable cells by the ubiquitin ligases Nedd4 and NEDD4-2. Channels (Austin) 5, 79–88.
Bouhassira, D., Lanteri-Minet, M., Attal, N., Laurent, B., and Touboul, C. (2008). Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 136, 380–387. doi: 10.1016/j.pain.2007.08.013
Bretin, S., Rogemond, V., Marin, P., Maus, M., Torrens, Y., Honnorat, J., et al. (2006). Calpain product of WT-CRMP2 reduces the amount of surface NR2B NMDA receptor subunit. J. Neurochem. 98, 1252–1265. doi: 10.1111/j.1471-4159.2006.03969.x
Brouwer, B. A., Merkies, I. S. J., Gerrits, M. M., Waxman, S. G., Hoeijmakers, J. G. J., and Faber, C. G. (2014). Painful neuropathies: the emerging role of sodium channelopathies. J. Peripher. Nerv. Syst. 19, 53–65. doi: 10.1111/jns5.12071
Brüggemann, I., Schulz, S., Wiborny, D., and Höllt, V. (2000). Colocalization of the μ-opioid receptor and calcium/calmodulin-dependent kinase II in distinct pain-processing brain regions1. Mol. Brain Res. 85, 239–250. doi: 10.1016/S0169-328X(00)00265-5
Cachemaille, M., Laedermann, C. J., Pertin, M., Abriel, H., Gosselin, R. D., and Decosterd, I. (2012). Neuronal expression of the ubiquitin ligase NEDD4-2 in rat dorsal root ganglia: modulation in the spared nerve injury model of neuropathic pain. Neuroscience 227, 370–380. doi: 10.1016/j.neuroscience.2012.09.044
Cang, C.-L., Zhang, H., Zhang, Y.-Q., and Zhao, Z.-Q. (2009). PKCepsilon-dependent potentiation of TTX-resistant Nav1.8 current by neurokinin-1 receptor activation in rat dorsal root ganglion neurons. Mol. Pain 5:33.
Cantrell, A. R., Ma, J. Y., Scheuer, T., and Catterall, W. A. (1996). Muscarinic modulation of sodium current by activation of protein kinase C in rat hippocampal neurons. Neuron 16, 1019–1026. doi: 10.1016/S0896-6273(00)80125-7
Cantrell, A. R., Smith, R. D., Goldin, A. L., Scheuer, T., and Catterall, W. A. (1997). Dopaminergic modulation of sodium current in hippocampal neurons via camp-dependent phosphorylation of specific sites in the sodium channel α subunit. J. Neurosci. 17, 7330–7338.
Casals-Diaz, L., Casas, C., and Navarro, X. (2015). Changes of voltage-gated sodium channels in sensory nerve regeneration and neuropathic pain models. Restor. Neurol. Neurosci. 33, 321–324. doi: 10.3233/RNN-140444
Catterall, W., Goldin, A., and Waxman, S. (2005). International union of pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409.
Catterall, W. A. (1992). Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 72, S15–S48.
Cesare, P., Dekker, L. V., Sardini, A., Parker, P. J., and Mcnaughton, P. A. (1999). Specific Involvement of PKC-𝜀 in sensitization of the neuronal response to painful Heat. Neuron. 23, 617–624. doi: 10.1016/S0896-6273(00)80813-2
Cesare, P., and McNaughton, P. (1996). A novel heat-activated current in nociceptive neurons and its sensitization by bradykini. Proc. Natl. Acad. Sci. U.S.A. 93, 15435–15439.
Chahine, M., Ziane, R., Vijayaragavan, K., and Okamura, Y. (2005). Regulation of Na v channels in sensory neurons. Trends Pharmacol. Sci. 26, 496–502. doi: 10.1016/j.tips.2005.08.002
Chatelier, A., Dahllund, L., Eriksson, A., Krupp, J., and Chahine, M. (2008). Biophysical properties of human Nav1.7 splice variants and their regulation by protein kinase A. J. Neurophysiol. 99, 2241–2250.
Chattopadhyay, M., Mata, M., and Fink, D. J. (2008). Continuous δ-opioid receptor activation reduces neuronal voltage-gated sodium channel (NaV1.7) levels through activation of protein kinase C in painful diabetic neuropathy. J. Neurosci. 28, 6652–6658. doi: 10.1523/jneurosci.5530-07.2008
Chen, Y., Luo, F., Yang, C., Kirkmire, C. M., and Wang, Z. J. (2009). Acute inhibition of Ca2+/Calmodulin-dependent protein kinase II reverses experimental neuropathic pain in mice. J. Pharmacol. Exp. Ther. 330, 650–659. doi: 10.1124/jpet.109.152165
Chen, Y., Yu, F. H., Sharp, E. M., Beacham, D., Scheuer, T., and Catterall, W. A. (2008). Functional properties and differential neuromodulation of Na(v)1.6 Channels. Mol. Cell. Neurosci. 38, 607–615. doi: 10.1016/j.mcn.2008.05.009
Cheng, X., Dib-Hajj, S., Tyrrell, L., and Waxman, S. (2008). Mutation I136V alters electrophysiological properties of the NaV1.7 channel in a family with onset of erythromelalgia in the second decade. Mol. Pain 4:1.
Chi, X. X., Schmutzler, B. S., Brittain, J. M., Wang, Y., Hingtgen, C. M., Nicol, G. D., et al. (2009). Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J. Cell Sci. 122, 4351–4362. doi: 10.1242/jcs.053280
Ciechanover, A. (2005). Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 6, 79–87. doi: 10.1038/nrm1552
Coggeshall, R. E., Tate, S., and Carlton, S. M. (2004). Differential expression of tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9 in normal and inflamed rats. Neurosci. Lett. 355, 45–48. doi: 10.1016/j.neulet.2003.10.023
Cohen, S. A., and Barchi, R. L. (1993). Voltage-dependent sodium channels. Int. Rev. Cytol. 137C, 55–103.
Costa, M. R. C., and Catterall, W. A. (1984a). Cyclic AMP-dependent phosphorylation of the α subunit of the sodium channel in synaptic nerve ending particles. J. Biol. Chem. 259, 8210–8218.
Costa, M. R. C., and Catterall, W. A. (1984b). Phosphorylation of the α subunit of the sodium channel by protein kinase C. Cell Mol. Neurobiol. 4, 291–297. doi: 10.1007/BF00733592
Coward, K., Aitken, A., Powell, A., Plumpton, C., Birch, R., Tate, S., et al. (2001a). Plasticity of TTX-sensitive sodium channels PN1 and brain III in injured human nerves. NeuroReport 12, 495–500.
Coward, K., Jowett, A., Plumpton, C., Powell, A., Birch, R., Tate, S., et al. (2001b). Sodium channel beta 1 and beta 2 subunits parallel SNS/PN3 alpha-subunit changes in injured human sensory neurons. NeuroReport 12, 483–488. doi: 10.1097/00001756-200103050-00012
Cox, J. J., Reimann, F., Nicholas, A. K., Thornton, G., Roberts, E., Springell, K., et al. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898. doi: 10.1038/nature05413
Cronin, N. B., O’Reilly, A., Duclohier, H., and Wallace, B. A. (2004). Effects of Deglycosylation of sodium channels on their structure and function. Biochemistry 44, 441–449. doi: 10.1021/bi048741q
Cummins, T. R., Aglieco, F., Renganathan, M., Herzog, R. I., Dib-Hajj, S. D., and Waxman, S. G. (2001). Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 21, 5952–5961.
Cummins, T., Dib-Hajj, S., and Waxman, S. (2004). Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J. Neurosci. 24, 8232–8236.
Cummins, T. R., and Waxman, S. G. (1997). Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J. Neurosci. 17, 3503–3514.
Cusdin, F. S., Clare, J. J., and Jackson, A. P. (2008). Trafficking and cellular distribution of voltage-gated sodium channels. Traffic 9, 17–26. doi: 10.1111/j.1600-0854.2007.00673.x
Daemen, M., Kurvers, H., Kitslaar, P., Slaaf, D., Bullens, P., and Wildenberg, F. (1998). Neurogenic inflammation in an animal model of neuropathic pain. Neurol. Res. 20, 41–45.
Dascal, N., and Lotan, I. (1991). Activation of protein kinase C alters voltage dependence of a Na+ channel. Neuron 6, 165–175. doi: 10.1016/0896-6273(91)90131-I
Decosterd, I., Ji, R., Abdi, S., Tate, S., and Woolf, C. (2002). The pattern of expression of the voltage-gated sodium channels Na(v)1.8 and Na(v)1.9 does not change in uninjured primary sensory neurons in experimental neuropathic pain models. Pain 96, 269–277.
Deribe, Y. L., Pawson, T., and Dikic, I. (2010). Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672.
Deschênes, I., Neyroud, N., Disilvestre, D., Marbán, E., Yue, D. T., and Tomaselli, G. F. (2002). Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ. Res. 90, e49–e57. doi: 10.1161/01.res.0000012502.92751.e6
Deval, E., Gasull, X., Noël, J., Salinas, M., Baron, A., Diochot, S., et al. (2010). Acid-Sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol. Ther. 128, 549–558. doi: 10.1016/j.pharmthera.2010.08.006
Dib-Hajj, S. D., Binshtok, A. M., Cummins, T. R., Jarvis, M. F., Samad, T., and Zimmermann, K. (2009). Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment. Brain Res. Rev. 60, 65–83. doi: 10.1016/j.brainresrev.2008.12.005
Dib-Hajj, S. D., Cummins, T., Black, J., and Waxman, S. (2010). Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347.
Dib-Hajj, S. D., Rush, A. M., Cummins, T. R., Hisama, F. M., Novella, S., Tyrrell, L., et al. (2005). Gain-of-function mutation in NaV1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 128, 1847–1854.
Dib-Hajj, S. D., Tyrrell, L., Black, J., and Waxman, S. (1998). NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc. Natl. Acad. Sci. U.S.A. 95, 8963–8968.
Dib-Hajj, S. D., Yang, Y., Black, J., and Waxman, S. (2013). The Na(V)1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62.
Dietrich, P. S., Mcgivern, J. G., Delgado, S. G., Koch, B. D., Eglen, R. M., Hunter, J. C., et al. (1998). Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglia. J. Neurochem. 70, 2262–2272. doi: 10.1046/j.1471-4159.1998.70062262.x
Dina, O. A., Barletta, J., Chen, X., Mutero, A., Martin, A., Messing, R. O., et al. (2000). Key role for the epsilon isoform of protein kinase C in painful alcoholic neuropathy in the rat. J. Neurosci. 20, 8614–8619.
Dina, O. A., Chen, X., Reichling, D., and Levine, J. D. (2001). Role of protein kinase C𝜀 and protein kinase A in a model of paclitaxel-induced painful peripheral neuropathy in the rat. Neuroscience 108, 507–515. doi: 10.1016/S0306-4522(01)00425-0
Djouhri, L., Koutsikou, S., Fang, X., Mcmullan, S., and Lawson, S. N. (2006). Spontaneous pain, both neuropathic and inflammatory, is related to frequency of spontaneous firing in intact C-fiber nociceptors. J. Neurosci. 26, 1281–1292. doi: 10.1523/JNEUROSCI.3388-05.2006
Dogrul, A., Gul, H., Yesilyurt, O., Ulas, U., and Yildiz, O. (2011). Systemic and spinal administration of etanercept, a tumor necrosis factor alpha inhibitor, blocks tactile allodynia in diabetic mice. Acta Diabetol. 48, 135–142. doi: 10.1007/s00592-010-0237-x
Dong, X. W., Goregoaker, S., Engler, H., Zhou, X., Mark, L., Crona, J., et al. (2007). Small interfering RNA-mediated selective knockdown of NaV1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience 146, 812–821. doi: 10.1016/j.neuroscience.2007.01.054
Duan, B., Liu, D.-S., Huang, Y., Zeng, W.-Z., Wang, X., Yu, H., et al. (2012). PI3-kinase/Akt pathway-regulated membrane insertion of acid-sensing ion channel 1a underlies BDNF-induced pain hypersensitivity. J. Neurosci. 32, 6351–6363. doi: 10.1523/jneurosci.4479-11.2012
Dubin, A. E., and Patapoutian, A. (2010). Nociceptors: the sensors of the pain pathway. J. Clin. Invest. 120, 3760–3772. doi: 10.1172/jci42843
Dustrude, E. T., Wilson, S. M., Ju, W., Xiao, Y., and Khanna, R. (2013). CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J. Biol. Chem. 288, 24316–24331. doi: 10.1074/jbc.M113.474924
Edinger, R. S., Lebowitz, J., Li, H., Alzamora, R., Wang, H., Johnson, J. P., et al. (2009). Functional regulation of the epithelial Na+ channel by IκB kinase-β occurs via phosphorylation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 284, 150–157. doi: 10.1074/jbc.M807358200
Ednie, A. R., and Bennett, E. S. (2011). “Modulation of voltage-gated ion channels by sialylation,” in Comprehensive Physiology, ed. D. M. Pollock (New York, NY: John Wiley & Sons, Inc.).
England, S., Bevan, S., and Docherty, R. J. (1996). PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J. Physiol. 495, 429–440. doi: 10.1113/jphysiol.1996.sp021604
Errington, A. C., Coyne, L., Stöhr, T., Selve, N., and Lees, G. (2006). Seeking a mechanism of action for the novel anticonvulsant lacosamide. Neuropharmacology 50, 1016–1029. doi: 10.1016/j.neuropharm.2006.02.002
Errington, A. C., Stöhr, T., Heers, C., and Lees, G. (2008). The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 73, 157–169. doi: 10.1124/mol.107.039867
Faber, C. G., Hoeijmakers, J. G., Ahn, H. S., Cheng, X., Han, C., Choi, J. S., et al. (2012a). Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 71, 26–39. doi: 10.1002/ana.22485
Faber, C. G., Lauria, G., Merkies, I. S., Cheng, X., Han, C., Ahn, H. S., et al. (2012b). Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. U.S.A. 109, 19444–19449. doi: 10.1073/pnas.1216080109
Fahmi, A. I., Patel, M., Stevens, E. B., Fowden, A. L., John, J. E., Lee, K., et al. (2001). The sodium channel -subunit SCN3b modulates the kinetics of SCN5a and is expressed heterogeneously in sheep heart. J. Physiol. 537, 693–700. doi: 10.1113/jphysiol.2001.012691
Ferrari, L. F., Bogen, O., and Levine, J. D. (2013). Role of nociceptor αCaMKII in transition from acute to chronic pain (Hyperalgesic Priming) in male and female rats. J. Neurosci. 33, 11002–11011. doi: 10.1523/jneurosci.1785-13.2013
Ferreira, J., Trichês, K. M., Medeiros, R., Cabrini, D. A., Mori, M. A. S., Pesquero, J. B., et al. (2008). The role of kinin B1 receptors in the nociception produced by peripheral protein kinase C activation in mice. Neuropharmacology 54, 597–604. doi: 10.1016/j.neuropharm.2007.11.008
Ferreira, J., Trichês, K. M., Medeiros, R., and Calixto, J. B. (2005). Mechanisms involved in the nociception produced by peripheral protein kinase C activation in mice. Pain 117, 171–181. doi: 10.1016/j.pain.2005.06.001
Fertleman, C. R., Baker, M. D., Parker, K. A., Moffatt, S., Elmslie, F. V., Abrahamsen, B., et al. (2006). SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774. doi: 10.1016/j.neuron.2006.10.006
Firsov, D., Schild, L., Gautschi, I., MéRillat, A. M., Schneeberger, E., and Rossier, B. C. (1996). Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc. Natl. Acad. Sci. U.S.A. 93, 15370–15375.
Fitzgerald, E. M., Okuse, K., Wood, J. N., Dolphin, A. C., and Moss, S. J. (1999). cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS. J. Physiol. 516, 433–446. doi: 10.1111/j.1469-7793.1999.0433v.x
Fjell, J., Cummins, T. R., Dib-Hajj, S. D., Fried, K., Black, J. A., and Waxman, S. G. (1999). Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Mol. Brain Res. 67, 267–282. doi: 10.1016/S0169-328X(99)00070-4
Fotia, A. B., Dinudom, A., Shearwin, K. E., Koch, J.-P., Korbmacher, C., Cook, D. I., et al. (2002). The role of individual NEDD4-2 (KIAA0439) WW domains in binding and regulating epithelial sodium channels. FASEB J. 17, 70–72. doi: 10.1096/fj.02-0497fje
Fotia, A. B., Ekberg, J., Adams, D. J., Cook, D. I., Poronnik, P., and Kumar, S. (2004). Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and NEDD4-2. J. Biol. Chem. 279, 28930–28935. doi: 10.1074/jbc.M402820200
Fukuoka, T., Kobayashi, K., Yamanaka, H., Obata, K., Dai, Y., and Noguchi, K. (2008). Comparative study of the distribution of the alpha-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J. Comp. Neurol. 510, 188–206. doi: 10.1002/cne.21786
Fukuoka, T., and Noguchi, K. (2011). Comparative study of voltage-gated sodium channel alpha-subunits in non-overlapping four neuronal populations in the rat dorsal root ganglion. Neurosci. Res. 70, 164–171. doi: 10.1016/j.neures.2011.01.020
Galan, A., Laird, J. M. A., and Cervero, F. (2004). In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain 112, 315–323. doi: 10.1016/j.pain.2004.09.011
Gasser, A., Cheng, X., Gilmore, E. S., Tyrrell, L., Waxman, S. G., and Dib-Hajj, S. D. (2010). Two Nedd4-binding motifs underlie modulation of sodium channel Nav1.6 by p38 MAPK. J. Biol. Chem. 285, 26149–26161. doi: 10.1074/jbc.M109.098681
Gaudet, A., Popovich, P., and Ramer, M. (2011). Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflamm. 8:110.
Gee, S., Madhavan, R., Levinson, S., Caldwell, J., Sealock, R., and Froehner, S. (1998). Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J. Neurosci. 18, 128–137.
Gershon, E., Weigl, L., Lotan, I., Schreibmayer, W., and Dascal, N. (1992). Protein kinase A reduces voltage-dependent Na+ current in Xenopus oocytes. J. Neurosci. 12, 3743–3752.
Gold, M., Levine, J., and Correa, A. (1998). Modulation of TTX-R I Na by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J. Neurosci. 18, 10345–10355.
Gold, M. S., Reichling, D. B., Shuster, M. J., and Levine, J. D. (1996). Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc. Natl. Acad. Sci. U.S.A. 93, 1108–1112.
Gold, M. S., Weinreich, D., Kim, C. S., Wang, R., Treanor, J., Porreca, F., et al. (2003). Redistribution of NaV1.8 in uninjured axons enables neuropathic pain. J. Neurosci. 23, 158–166.
Goldin, A. L. (2001). Resurgence of sodium channel research. Annu. Rev. Physiol. 63, 871–894. doi: 10.1146/annurev.physiol.63.1.871
Gould, H. J., Gould, T. N., England, J. D., Paul, D., Liu, Z. P., and Levinson, S. R. (2000). A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain. Brain Res. 854, 19–29. doi: 10.1016/s0006-8993(99)02216-7
Gudes, S., Barkai, O., Caspi, Y., Katz, B., Lev, S., and Binshtok, A. M. (2015). The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor-α-mediated increase in nociceptors excitability. J. Neurophysiol. 113, 601–619. doi: 10.1152/jn.00652.2014
Han, C., Vasylyev, D., Macala, L. J., Gerrits, M. M., Hoeijmakers, J. G. J., Bekelaar, K. J., et al. (2014). The G1662S NaV1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. J. Neurol. Neurosurg. Psychiatry 85, 499–505. doi: 10.1136/jnnp-2013-306095
Heikamp, E. B., Patel, C. H., Collins, S., Waickman, A., Oh, M.-H., Sun, I.-H., et al. (2014). The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat. Immunol. 15, 457–464. doi: 10.1038/ni.2867
Henry, P. C., Kanelis, V., O’Brien, M. C., Kim, B., Gautschi, I., Forman-Kay, J., et al. (2003). Affinity and specificity of interactions between Nedd4 isoforms and the epithelial Na+ channel. J. Biol. Chem. 278, 20019–20028. doi: 10.1074/jbc.M211153200
Herzog, R. I., Cummins, T. R., and Waxman, S. G. (2001). Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J. Neurophysiol. 86, 1351–1364.
Herzog, R. I., Liu, C., Waxman, S. G., and Cummins, T. R. (2003). Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J. Neurosci. 23, 8261–8270.
Hirade, M., Yasuda, H., Omatsu-Kanbe, M., Kikkawa, R., and Kitasato, H. (1999). Tetrodotoxin-resistant sodium channels of dorsal root ganglion neurons are readily activated in diabetic rats. Neuroscience 90, 933–939.
Hiruma, H., Maruyama, H., Katakura, T., Simada, Z. I. B., Nishida, S., Hoka, S., et al. (1999). Axonal transport is inhibited by a protein kinase C inhibitor in cultured isolated mouse dorsal root ganglion cells. Brain Res. 826, 135–138. doi: 10.1016/S0006-8993(99)01249-4
Ho, C., and O’Leary, M. E. (2011). Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol. Cell. Neurosci. 46, 159–166. doi: 10.1016/j.mcn.2010.08.017
Hockley, J. R. F., Boundouki, G., Cibert-Goton, V., Mcguire, C., Yip, P. K., Chan, C., et al. (2014). Multiple roles for Na(V)1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease–derived stimuli. Pain 155, 1962–1975. doi: 10.1016/j.pain.2014.06.015
Hoeijmakers, J. G. J., Faber, C. G., Merkies, I. S. J., and Waxman, S. G. (2015). Painful peripheral neuropathy and sodium channel mutations. Neurosci. Lett. 596, 51–59. doi: 10.1016/j.neulet.2014.12.056
Hoeijmakers, J. G. J., Merkies, I. S. J., Gerrits, M. M., Waxman, S. G., and Faber, C. G. (2012). Genetic aspects of sodium channelopathy in small fiber neuropathy. Clin. Gen. 82, 351–358. doi: 10.1111/j.1399-0004.2012.01937.x
Huang, J., Han, C., Estacion, M., Vasylyev, D., Hoeijmakers, J. G. J., Gerrits, M. M., et al. (2014). Gain-of-function mutations in sodium channel NaV1.9 in painful neuropathy. Brain. 137(Pt 6), 1627–1642. doi: 10.1093/brain/awu079
Hucho, T., and Levine, J. D. (2007). Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron 55, 365–376. doi: 10.1016/j.neuron.2007.07.008
Hudmon, A., Choi, J.-S., Tyrrell, L., Black, J. A., Rush, A. M., Waxman, S. G., et al. (2008). Phosphorylation of sodium channel Nav1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root Ganglion Neurons. J. Neurosci. 28, 3190–3201. doi: 10.1523/jneurosci.4403-07.2008
Hunter, T. (2009). Tyrosine phosphorylation: thirty years and counting. Curr. Opin. Cell Biol. 21, 140–146. doi: 10.1016/j.ceb.2009.01.028
Ikeda, M., Yoshida, S., Kadoi, J., Nakano, Y., and Mastumoto, S. (2005). The effect of PKC activity on the TTX-R sodium currents from rat nodose ganglion neurons. Life Sci. 78, 47–53. doi: 10.1016/j.lfs.2005.04.053
Inagaki, N., Chihara, K., Arimura, N., Menager, C., Kawano, Y., Matsuo, N., et al. (2001). CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 4, 781–782.
Isom, L., De Jongh, K., Patton, D., Reber, B., Offord, J., Charbonneau, H., et al. (1992). Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256, 839–842. doi: 10.1126/science.256.5058.839
Isom, L. L., Ragsdale, D. S., De Jongh, K. S., Westenbroek, R. E., Reber, B. F. X., Scheuer, T., et al. (1995a). Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83, 433–442. doi: 10.1016/0092-8674(95)90121-3
Isom, L. L., Scheuer, T., Brownstein, A. B., Ragsdale, D. S., Murphy, B. J., and Catterall, W. A. (1995b). Functional co-expression of the beta 1 and type IIA alpha subunits of sodium channels in a mammalian cell line. J. Biol. Chem. 270, 3306–3312.
James, W. M., and Agnew, W. S. (1987). Multiple oligosaccharide chains in the voltage-sensitive Na channel from Electrophorus electricus: evidence for α-2,8-linked polysialic acid. Biochem. Biophys. Res. Commun. 148, 817–826. doi: 10.1016/0006-291x(87)90949-1
Jarecki, B. W., Piekarz, A. D., Jackson, J. O. II, and Cummins, T. R. (2010). Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J. Clin. Invest. 120, 369–378. doi: 10.1172/JCI40801
Jarvis, M. F., Honore, P., Shieh, C.-C., Chapman, M., Joshi, S., Zhang, X.-F., et al. (2007). A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. 104, 8520–8525. doi: 10.1073/pnas.0611364104
Ji, R. R., Gereau Iv, R. W., Malcangio, M., and Strichartz, G. R. (2009). MAP kinase and pain. Brain Res. Rev. 60, 135–148. doi: 10.1016/j.brainresrev.2008.12.011
Ji, R. R., Kawasaki, Y., Zhuang, Z. Y., Wen, Y. R., and Zhang, Y. Q. (2007). “Protein kinases as potential targets for the treatment of pathological pain,” in Analgesia, ed. C. Stein (Berlin: Springer), 359–389.
Jin, X., and Gereau, R. (2006). Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J. Neurosci. 26, 246–255.
Joazeiro, C. A., and Weissman, A. M. (2000). RING finger proteins: mediators of ubiquitin ligase activity. Cell 102, 549–552. doi: 10.1016/S0092-8674(00)00077-5
Johnson, D., and Bennett, E. S. (2006). Isoform-specific effects of the β2 Subunit on voltage-gated sodium channel gating. J. Biol. Chem. 281, 25875–25881. doi: 10.1074/jbc.M605060200
Johnson, D., Montpetit, M. L., Stocker, P. J., and Bennett, E. S. (2004). The sialic acid component of the β1 subunit modulates voltage-gated sodium channel function. J. Biol. Chem. 279, 44303–44310. doi: 10.1074/jbc.M408900200
Joshi, S. K., Mikusa, J. P., Hernandez, G., Baker, S., Shieh, C. C., Neelands, T., et al. (2006). Involvement of the TTX-resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain 123, 75–82.
Kazarinova-Noyes, K., Malhotra, J. D., Mcewen, D. P., Mattei, L. N., Berglund, E. O., Ranscht, B., et al. (2001). Contactin associates with Na+ channels and increases their functional expression. J. Neurosci. 21, 7517–7525.
Kazen-Gillespie, K. A., Ragsdale, D. S., D’Andrea, M. R., Mattei, L. N., Rogers, K. E., and Isom, L. L. (2000). Cloning, localization, and functional expression of sodium channel beta1A subunits. J. Biol. Chem. 275, 1079–1088. doi: 10.1074/jbc.275.2.1079
Kerr, B. J., Souslova, V., Mcmahon, S. B., and Wood, J. N. (2001). A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. NeuroReport 12, 3077–3080.
Khasar, S. G., Gold, M. S., and Levine, J. D. (1998). A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci. Lett. 256, 17–20. doi: 10.1016/S0304-3940(98)00738-1
Khasar, S. G., Lin, Y., Martin, A., Dadgar, J., Mcmahon, T., Wang, D., et al. (1999). A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron 24, 253–260.
Khasar, S. G., Ouseph, A. K., Chou, B., Ho, T., Green, P. G., and Levine, J. D. (1995). Is there more than one prostaglandin E receptor subtype mediating hyperalgesia in the rat hindpaw? Neuroscience 64, 1161–1165. doi: 10.1016/0306-4522(94)00423-3
Kim, E. K., and Choi, E.-J. (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405. doi: 10.1016/j.bbadis.2009.12.009
Kispersky, T. J., Caplan, J. S., and Marder, E. (2012). Increase in sodium conductance decreases firing rate and gain in model neurons. J. Neurosci. 32, 10995–11004. doi: 10.1523/jneurosci.2045-12.2012
Klugbauer, N., Lacinova, L., Flockerzi, V., and Hofmann, F. (1995). Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium-channel family from human neuroendocrine cells. EMBO J. 14, 1084–1090.
Kreegipuu, A., Blom, N., and Brunak, S. (1999). PhosphoBase, a database of phosphorylation sites: release 2.0. Nucleic Acids Res. 27, 237–239. doi: 10.1093/nar/27.1.237
Kresge, N., Simoni, R. D., and Hill, R. L. (2006). The discovery of ubiquitin-mediated proteolysis by Aaron Ciechanover, Avram Hershko, and Irwin Rose. J. Biol. Chem. 281:e32.
Kress, M., Rödl, J., and Reeh, P. W. (1996). Stable analogues of cyclic AMP but not cyclic GMP sensitize unmyelinated primary afferents in rat skin to heat stimulation but not to inflammatory mediators, in vitro. Neuroscience 74, 609–617. doi: 10.1016/0306-4522(96)00181-9
Kretschmer, T., Happel, L. T., England, J. D., Nguyen, D. H., Tiel, R. L., Beuerman, R. W., et al. (2002). Clinical article accumulation of PN1 and PN3 sodium channels in painful human neuroma-evidence from immunocytochemistry. Acta Neurochir. 144, 803–810. doi: 10.1007/s00701-002-0970-1
Kruger, L., Light, A., and Schweizer, F. (2003). Axonal terminals of sensory neurons and their morphological diversity. J. Neurocytol. 32, 205–216. doi: 10.1023/B:NEUR.0000010080.62031.f0
Laedermann, C. J., Cachemaille, M., Kirschmann, G., Pertin, M., Gosselin, R. D., Chang, I., et al. (2013a). Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J. Clin. Invest. 123, 3002–3013. doi: 10.1172/JCI68996
Laedermann, C. J., Syam, N., Pertin, M., Decosterd, I., and Abriel, H. (2013b). beta1- and beta3- voltage-gated sodium channel subunits modulate cell surface expression and glycosylation of Nav1.7 in HEK293 cells. Front. Cell Neurosci. 7:137. doi: 10.3389/fncel.2013.00137
Laedermann, C. J., Decosterd, I., and Abriel, H. (2014a). “Ubiquitylation of voltage-gated sodium channels,” in Voltage Gated Sodium Channels, ed. P. C. Ruben (Berlin: Springer), 231–250.
Laedermann, C. J., Pertin, M., Suter, M., and Decosterd, I. (2014b). Voltage-gated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol. Pain 10:19.
Lai, J., Gold, M. S., Kim, C. S., Bian, D., Ossipov, M. H., Hunter, J. C., et al. (2002). Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain 95, 143–152. doi: 10.1016/s0304-3959(01)00391-8
Latremoliere, A., and Woolf, C. J. (2009). Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J. Pain 10, 895–926. doi: 10.1016/j.jpain.2009.06.012
Leipold, E., Liebmann, L., Korenke, G. C., Heinrich, T., Gieszelmann, S., Baets, J., et al. (2013). A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 45, 1399–1404. doi: 10.1038/ng.2767
Leo, S., D’Hooge, R., and Meert, T. (2010). Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav. Brain Res. 208, 149–157. doi: 10.1016/j.bbr.2009.11.023
Li, M., West, J. W., Lai, Y., Scheuer, T., and Catterall, W. A. (1992). Functional modulation of brain sodium channels by cAMP-dependent phosphorylation. Neuron 8, 1151–1159. doi: 10.1016/0896-6273(92)90135-Z
Liang, L., Fan, L., Tao, B., Yaster, M., and Tao, Y.-X. (2013). Protein kinase B/Akt is required for complete freund’s adjuvant-induced upregulation of Nav1.7 and Nav1.8 in primary sensory neurons. J. Pain 14, 638–647. doi: 10.1016/j.jpain.2013.01.778
Liu, C., Cao, J., Ren, X., and Zang, W. (2012). Na(v)1.7 protein and mRNA expression in the dorsal root ganglia of rats with chronic neuropathic pain. Neural Regen. Res. 7, 1540–1544. doi: 10.3969/j.issn.1673-5374.2012.20.003
Liu, C., Li, Q., Su, Y., and Bao, L. (2010). Prostaglandin E2 promotes Nav1.8 trafficking via its intracellular RRR Motif through the protein kinase A pathway. Traffic 11, 405–417. doi: 10.1111/j.1600-0854.2009.01027.x
Liu, C.-N., Wall, P. D., Ben-Dor, E., Michaelis, M., Amir, R., and Devor, M. (2000). Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain 85, 503–521. doi: 10.1016/S0304-3959(00)00251-7
Liu, M., and Wood, J. N. (2011). The roles of sodium channels in nociception: implications for mechanisms of neuropathic pain. Pain. Med. 12(Suppl. 3), S93–S99. doi: 10.1111/j.1526-4637.2011.01158.x
Liu, S., and Zheng, P. (2013). Altered PKA modulation in the Na(v)1.1 epilepsy variant I1656M. J. Neurophysiol. 110, 2090–2098.
Lolignier, S., Amsalem, M., Maingret, F., Padilla, F., Gabriac, M., Chapuy, E., et al. (2011). Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS ONE 6:e23083. doi: 10.1371/journal.pone.0023083
Lopez-Santiago, L. F., Brackenbury, W. J., Chen, C., and Isom, L. L. (2011). Na+ channel scn1b gene regulates dorsal root ganglion nociceptor excitability in vivo. J. Biol. Chem. 286, 22913–22923. doi: 10.1074/jbc.M111.242370
Lopez-Santiago, L. F., Pertin, M., Morisod, X., Chen, C., Hong, S., Wiley, J., et al. (2006). Sodium channel beta2 subunits regulate tetrodotoxin-sensitive sodium channels in small dorsal root ganglion neurons and modulate the response to pain. J. Neurosci. 26, 7984–7994. doi: 10.1523/JNEUROSCI.2211-06.2006
Lou, J.-Y., Laezza, F., Gerber, B. R., Xiao, M., Yamada, K. A., Hartmann, H., et al. (2005). Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J. Physiol. 569, 179–193. doi: 10.1113/jphysiol.2005.097220
Luo, F., Yang, C., Chen, Y., Shukla, P., Tang, L., Wang, L. X., et al. (2008a). Reversal of chronic inflammatory pain by acute inhibition of Ca2+/Calmodulin-dependent protein kinase II. J. Pharmacol. Exp. Ther. 325, 267–275. doi: 10.1124/jpet.107.132167
Luo, S., Perry, G., Levinson, S. R., and Henry, M. (2008b). Nav1.7 expression is increased in painful human dental pulp. Molecular Pain 4:16.
Maingret, F., Coste, B., Padilla, F., Clerc, N., Crest, M., Korogod, S., et al. (2008). Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J. Gen. Physiol. 131, 211–225.
Malhotra, J. D., Kazen-Gillespie, K., Hortsch, M., and Isom, L. L. (2000). Sodium channel +Ý subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J. Biol. Chem. 275, 11383–11388.
Malhotra, J. D., Koopmann, M. C., Kazen-Gillespie, K. A., Fettman, N., Hortsch, M., and Isom, L. L. (2002). Structural requirements for interaction of sodium channel β1 subunits with ankyrin. J. Biol. Chem. 277, 26681–26688. doi: 10.1074/jbc.M202354200
Maltsev, V. A., Reznikov, V., Undrovinas, N. A., Sabbah, H. N., and Undrovinas, A. (2008). Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am. J. Physiol. Heart Circ. Physiol. 294, H1597–H1608.
Mann, M., and Jensen, O. N. (2003). Proteomic analysis of post-translational modifications. Nat. Biotech. 21, 255–261.
Mao, J., and Chen, L. L. (2000). Systemic lidocaine for neuropathic pain relief. Pain 87, 7–17. doi: 10.1016/s0304-3959(00)00229-3
Matzner, O., and Devor, M. (1992). Na+ conductance and the threshold for repetitive neuronal firing. Brain Res. 597, 92–98. doi: 10.1016/0006-8993(92)91509-D
Matzner, O., and Devor, M. (1994). Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J. Neurophysiol. 72, 349–359.
Messner, D. J., and Catterall, W. A. (1985). The sodium channel from rat brain. Separation and characterization of subunits. J. Biol. Chem. 260, 10597–10604.
Metzger, M. B., Hristova, V. A., and Weissman, A. M. (2012). HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 125, 531–537. doi: 10.1242/jcs.091777
Millan, M. J. (1999). The induction of pain: an integrative review. Prog. Neurobiol. 57, 1–164. doi: 10.1016/s0301-0082(98)00048-3
Minett, M., Nassar, M., Clark, A., Passmore, G., Dickenson, A., Wang, F., et al. (2012). Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 3:791.
Mittmann, T., and Alzheimer, C. (1998). Muscarinic inhibition of persistent Na+ current in rat neocortical pyramidal neurons. J. Physiol. 535(Pt 2), 383–396.
Moon, J.-Y., Song, S., Yoon, S.-Y., Roh, D.-H., Kang, S.-Y., Park, J.-H., et al. (2012). The differential effect of intrathecal Nav1.8 blockers on the induction and maintenance of capsaicin- and peripheral ischemia-induced mechanical allodynia and thermal hyperalgesia. Anesth. Analg. 114, 215–223. doi: 10.1213/ANE.0b013e318238002e
Moremen, K. W., Tiemeyer, M., and Nairn, A. V. (2012). Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462.
Morgan, K., Stevens, E. B., Shah, B., Cox, P. J., Dixon, A. K., Lee, K., et al. (2000). β3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc. Natl. Acad. Sci. U.S.A. 97, 2308–2313. doi: 10.1073/pnas.030362197
Mori, M., Konno, T., Ozawa, T., Murata, M., Imoto, K., and Nagayama, K. (2000). Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+-Sensitivity. Biochemistry 39, 1316–1323. doi: 10.1021/bi9912600
Murphy, B. J., Rossie, S., De Jongh, K. S., and Catterall, W. A. (1993). Identification of the sites of selective phosphorylation and dephosphorylation of the rat brain Na+ channel α subunit by cAMP-dependent protein kinase and phosphoprotein phosphatases. J. Biol. Chem. 268, 27355–27362.
Murray, K. T., Hu, N. N., Daw, J. R., Shin, H. G., Watson, M. T., Mashburn, A. B., et al. (1997). Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circ. Res. 80, 370–376.
Nagasako, E. M., Oaklander, A. L., and Dworkin, R. H. (2003). Congenital insensitivity to pain: an update. Pain 101, 213–219. doi: 10.1016/s0304-3959(02)00482-7
Narayanan, A., and Jacobson, M. P. (2009). Computational studies of protein regulation by post-translational phosphorylation. Curr. Opin. Struct. Biol. 19, 156–163. doi: 10.1016/j.sbi.2009.02.007
Nassar, M., Stirling, L., Forlani, G., Baker, M., Matthews, E., Dickenson, A., et al. (2004). Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. U.S.A. 101, 12706–12711.
Nelson, M. R., and Chazin, W. J. (1998). An interaction-based analysis of calcium-induced conformational changes in Ca2+ sensor proteins. Protein Sci. 7, 270–282. doi: 10.1002/pro.5560070206
Noda, M., and Hiyama, T. Y. (2014). The nax channel: what it is and what it does. Neuroscientist 21, 399–412. doi: 10.1177/1073858414541009
Novella, S. P., Hisama, F. M., Dib-Hajj, S. D., and Waxman, S. G. (2007). A case of inherited erythromelalgia. Nat. Clin. Pract. Neurol. 3, 229–234.
Numann, R., Catterall, W. A., and Scheuer, T. (1991). Functional modulation of brain sodium channels by protein kinase C phosphorylation. Science 254, 115–118.
Nuss, H. B., Chiamvimonvat, N., Perez-Garcia, M. T., Tomaselli, G. F., and Marban, E. (1995). Functional association of the beta 1 subunit with human cardiac (hH1) and rat skeletal muscle (mu 1) sodium channel alpha subunits expressed in Xenopus oocytes. J. Gen. Physiol. 106, 1171–1191. doi: 10.1085/jgp.106.6.1171
Obata, K., and Noguchi, K. (2004). MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 74, 2643–2653. doi: 10.1016/j.lfs.2004.01.007
Olsson, Y. (1967). Degranulation of mast cells in peripheral nerve injuries. Acta Neurol. Scand. 43, 365–374.
Payandeh, J., Scheuer, T., Zheng, N., and Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. doi: 10.1038/nature10238
Perry, V. H., Brown, M. C., and Gordon, S. (1987). The macrophage response to central and peripheral nerve injury. A possible role for macrophages in regeneration. J. Exp. Med. 165, 1218–1223. doi: 10.1084/jem.165.4.1218
Persson, A., Gasser, A., Black, J., and Waxman, S. (2011). Nav1.7 accumulates and co-localizes with phosphorylated ERK1/2 within transected axons in early experimental neuromas. Exp. Neurol. 230, 273–279.
Pertin, M., Ji, R., Berta, T., Powell, A., Karchewski, L., Tate, S., et al. (2005). Upregulation of the voltage-gated sodium channel beta2 subunit in neuropathic pain models: characterization of expression in injured and non-injured primary sensory neurons. J. Neurosci. 25, 10970–10980.
Petho, G., and Reeh, P. W. (2012). Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol. Rev. 92, 1699–1775. doi: 10.1152/physrev.00048.2010
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Pierre, S., Eschenhagen, T., Geisslinger, G., and Scholich, K. (2009). Capturing adenylyl cyclases as potential drug targets. Nat. Rev. Drug Discov. 8, 321–335.
Pitchford, S., and Levine, J. D. (1991). Prostaglandins sensitize nociceptors in cell culture. Neurosci. Lett. 132, 105–108.
Plummer, N. W., Mcburney, M. W., and Meisler, M. H. (1997). Alternative splicing of the sodium channel SCN8A predicts a truncated two-domain protein in fetal brain and non-neuronal cells. J. Biol. Chem. 272, 24008–24015. doi: 10.1074/jbc.272.38.24008
Priest, B. T., Murphy, B. A., Lindia, J. A., Diaz, C., Abbadie, C., Ritter, A. M., et al. (2005). Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc. Natl. Acad. Sci. U.S.A. 102, 9382–9387. doi: 10.1073/pnas.0501549102
Qu, Y., Rogers, J., Tanada, T., Scheuer, T., and Catterall, W. A. (1994). Modulation of cardiac Na+ channels expressed in a mammalian cell line and in ventricular myocytes by protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 91, 3289–3293.
Rang, H., and Ritchie, J. (1988). Depolarization of nonmyelinated fibers of the rat vagus nerve produced by activation of protein kinase C. J. Neurosci. 8, 2606–2617.
Ranscht, B. (1988). Sequence of contactin, a 130-kD glycoprotein concentrated in areas of interneuronal contact, defines a new member of the immunoglobulin supergene family in the nervous system. J. Cell Biol. 107, 1561–1573. doi: 10.1083/jcb.107.4.1561
Raymond, C. K., Castle, J., Garrett-Engele, P., Armour, C. D., Kan, Z., Tsinoremas, N., et al. (2004). Expression of alternatively spliced sodium channel α-subunit genes: unique splicing patterns are observed in dorsal root ganglia. J. Biol. Chem. 279, 46234–46241. doi: 10.1074/jbc.M406387200
Recio-Pinto, E., Thornhill, W. B., Duch, D. S., Levinson, S. R., and Urban, B. W. (1990). Neuraminidase treatment modifies the function of electroplax sodium channels in planar lipid bilayers. Neuron 5, 675–684. doi: 10.1016/0896-6273(90)90221-z
Ren, D., Navarro, B., Xu, H., Yue, L., Shi, Q., and Clapham, D. E. (2001). A prokaryotic voltage-gated sodium channel. Science 294, 2372–2375. doi: 10.1126/science.1065635
Renganathan, M., Dib-Hajj, S., and Waxman, S. G. (2002). Nav1.5 underlies the ‘third TTX-R sodium current’ in rat small DRG neurons. Mol. Brain Res. 106, 70–82. doi: 10.1016/s0169-328x(02)00411-4
Renganathan, M., Godoy, C. M., and Cukierman, S. (1995). Direct modulation of Na+ currents by protein kinase C activators in mouse neuroblastoma cells. J. Membr. Biol. 144, 59–69.
Ritchie, J. M., Black, J. A., Waxman, S. G., and Angelides, K. J. (1990). Sodium channels in the cytoplasm of Schwann cells. Proc. Natl. Acad. Sci. U.S.A. 87, 9290–9294.
Rossie, S., and Catterall, W. A. (1987). Cyclic-AMP-dependent phosphorylation of voltage-sensitive sodium channels in primary cultures of rat brain neurons. J. Biol. Chem. 262, 12735–12744.
Rossier, B. C., Pradervand, S., Schild, L., and Hummler, E. (2002). Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu. Rev. Physiol. 64, 877–897. doi: 10.1146/annurev.physiol.64.082101.143243
Rush, A. M., Cummins, T., and Waxman, S. (2007). Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 579, 1–14.
Rush, A. M., Dib-Hajj, S. D., Liu, S., Cummins, T. R., Black, J. A., and Waxman, S. G. (2006). A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 8245–8250.
Rush, A. M., and Waxman, S. G. (2004). PGE2 increases the tetrodotoxin-resistant Nav1.9 sodium current in mouse DRG neurons via G-proteins. Brain Res. 1023, 264–271. doi: 10.1016/j.brainres.2004.07.042
Sadamasu, A., Sakuma, Y., Suzuki, M., Orita, S., Yamauchi, K., Inoue, G., et al. (2014). Upregulation of NaV1.7 in dorsal root ganglia after intervertebral disc injury in rats. Spine 39, E421–E426. doi: 10.1097/brs.0000000000000229
Sangameswaran, L., Fish, L. M., Koch, B. D., Rabert, D. K., Delgado, S. G., Ilnicka, M., et al. (1997). A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J. Biol. Chem. 272, 14805–14809.
Sanna, P. P., Cammalleri, M., Berton, F., Simpson, C., Lutjens, R., Bloom, F. E., et al. (2002). Phosphatidylinositol 3-Kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J. Neurosci. 22, 3359–3365.
Schaller, K. L., Krzemien, D. M., Mckenna, N. M., and Caldwell, J. H. (1992). Alternatively spliced sodium channel transcripts in brain and muscle. J. Neurosci. 12, 1370–1381.
Schepelmann, K., Messlinger, K., and Schmidt, R. F. (1993). The effects of phorbol ester on slowly conducting afferents of the cat’s knee joint. Exp. Brain Res. 92, 391–398.
Schild, L., Lu, Y., Gautschi, I., Schneeberger, E., Lifton, R. P., and Rossier, B. C. (1996). Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO J. 15, 2381–2387.
Schirmeyer, J., Szafranski, K., Leipold, E., Mawrin, C., Platzer, M., and Heinemann, S. H. (2014). Exon 11 skipping of SCN10A coding for voltage-gated sodium channels in dorsal root ganglia. Channels 8, 210–215. doi: 10.4161/chan.28146
Schmidt, J., Rossie, S., and Catterall, W. A. (1985). A large intracellular pool of inactive Na channel alpha subunits in developing rat brain. Proc. Natl. Acad. Sci. U.S.A. 82, 4847–4851.
Schmidt, J. W., and Catterall, W. A. (1987). Palmitylation, sulfation, and glycosylation of the alpha subunit of the sodium channel. Role of post-translational modifications in channel assembly. J. Biol. Chem. 262, 13713–13723.
Scholz, J., Broom, D. C., Youn, D. H., Mills, C. D., Kohno, T., Suter, M. R., et al. (2005). Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina ii of the dorsal horn after peripheral nerve injury. J. Neurosci. 25, 7317–7323.
Shah, B. S., Stevens, E. B., Gonzalez, M. I., Bramwell, S., Pinnock, R. D., Lee, K., et al. (2000). beta3, a novel auxiliary subunit for the voltage-gated sodium channel, is expressed preferentially in sensory neurons and is upregulated in the chronic constriction injury model of neuropathic pain. Euro. J. Neurosci. 12, 3985–3990. doi: 10.1046/j.1460-9568.2000.00294.x
Shao, D., Okuse, K., and Djamgoz, M. B. A. (2009). Protein–protein interactions involving voltage-gated sodium channels: post-translational regulation, intracellular trafficking and functional expression. Int. J. Biochem. Cell Biol. 41, 1471–1481. doi: 10.1016/j.biocel.2009.01.016
Shi, T. J. S., Huang, P., Mulder, J., Ceccatelli, S., and Hökfelt, T. (2009). Expression of p-Akt in sensory neurons and spinal cord after peripheral nerve injury. Neurosignals 17, 203–212.
Shih, S. C., Sloper-Mould, K. E., and Hicke, L. (2000). Monoubiquitin carries a novel internalization signal that is appended to activated receptors. EMBO J. 19, 187–198. doi: 10.1093/emboj/19.2.187
Smith, R. D., and Goldin, A. L. (1996). Phosphorylation of brain sodium channels in the I-II linker modulates channel function in Xenopus oocytes. J. Neurosci. 16, 1965–1974.
Smith, R. D., and Goldin, A. L. (1998). Functional analysis of the rat I sodium channel in Xenopus oocytes. J. Neurosci. 18, 811–820.
Snyder, P. M. (2009). Down-regulating destruction: phosphorylation regulates the E3 ubiquitin ligase NEDD4-2. Sci. Signal. 2:pe41. doi: 10.1126/scisignal.279pe41
Snyder, P. M., Olson, D. R., Kabra, R., Zhou, R., and Steines, J. C. (2004). cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of NEDD4-2. J. Biol. Chem. 279, 45753–45758. doi: 10.1074/jbc.M407858200
Solà, C., Barrón, S., Tusell, J. M., and Serratosa, J. (2001). The Ca2+/calmodulin system in neuronal hyperexcitability. Int. J. Biochem. Cell Biol. 33, 439–455. doi: 10.1016/S1357-2725(01)00030-9
Souza, A. L. S., Moreira, F. A., Almeida, K. R., Bertollo, C. M., Costa, K. A., and Coelho, M. M. (2002). In vivo evidence for a role of protein kinase C in peripheral nociceptive processing. Br. J. Pharmacol. 135, 239–247. doi: 10.1038/sj.bjp.0704434
Stamboulian, S., Choi, J., Ahn, H., Chang, Y., Tyrrell, L., Black, J., et al. (2010). ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. J. Neurosci. 30, 1637–1647.
Staub, O., and Rotin, D. (2006). Role of ubiquitylation in cellular membrane transport. Physiol. Rev. 86, 669–707. doi: 10.1152/physrev.00020.2005
Strickland, I. T., Martindale, J. C., Woodhams, P. L., Reeve, A. J., Chessell, I. P., and Mcqueen, D. S. (2008). Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Euro. J. Pain 12, 564–572. doi: 10.1016/j.ejpain.2007.09.001
Stucky, C. L., Dubin, A. E., Jeske, N. A., Malin, S. A., Mckemy, D. D., and Story, G. M. (2009). Roles of transient receptor potential channels in pain. Brain Res. Rev. 60, 2–23. doi: 10.1016/j.brainresrev.2008.12.018
Sun, R.-Q., Tu, Y.-J., Yan, J.-Y., and Willis, W. D. (2006). Activation of protein kinase B/Akt signaling pathway contributes to mechanical hypersensitivity induced by capsaicin. Pain 120, 86–96. doi: 10.1016/j.pain.2005.10.017
Suter, M. R., Papaloïzos, M., Berde, C. B., Woolf, C. J., Gilliard, N., Spahn, D. R., et al. (2003). Development of neuropathic pain in the rat spared nerve injury model is not prevented by a peripheral nerve block. Anesthesiology 99, 1402–1408.
Taiwo, Y. O., Bjerknes, L. K., Goetzl, E. J., and Levine, J. D. (1989). Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience 32, 577–580.
Tan, Z.-Y., Priest, B. T., Krajewski, J. L., Knopp, K. L., Nisenbaum, E. S., and Cummins, T. R. (2014). Protein kinase C enhances human sodium channel hNav1.7 resurgent currents via a serine residue in the domain III–IV linker. FEBS Lett. 588, 3964–3969. doi: 10.1016/j.febslet.2014.09.011
Thakor, D., Lin, A., Matsuka, Y., Meyer, E., Ruangsri, S., Nishimura, I., et al. (2009). Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol. Pain 5:14.
Theriault, O., and Chahine, M. (2014). Correlation of the electrophysiological profiles and sodium channel transcripts of individual rat dorsal root ganglia neurons. Front. Cell Neurosci. 8:285. doi: 10.3389/fncel.2014.00285
Thornalley, P. J. (2005). Dicarbonyl intermediates in the maillard reaction. Ann. N. Y. Acad. Sci. 1043, 111–117. doi: 10.1196/annals.1333.014
Toledo-Aral, J. J., Brehm, P., Halegoua, S., and Mandel, G. (1995). A single pulse of nerve growth factor triggers long-term neuronal excitability through sodium channel gene induction. Neuron 14, 607–611. doi: 10.1016/0896-6273(95)90317-8
Toledo-Aral, J. J., Moss, B. L., He, Z. J., Koszowski, A. G., Whisenand, T., Levinson, S. R., et al. (1997). Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 1527–1532.
Tong, S. E., Daniels, S. E., Black, P., Chang, S., Protter, A., and Desjardins, P. J. (2012). Novel p38α mitogen-activated protein kinase inhibitor shows analgesic efficacy in acute postsurgical dental pain. J. Clin. Pharmacol. 52, 717–728. doi: 10.1177/0091270011405496
Toth, C., Lander, J., and Wiebe, S. (2009). The prevalence and impact of chronic pain with neuropathic pain symptoms in the general population. Pain Med. 10, 918–929. doi: 10.1111/j.1526-4637.2009.00655.x
Turk, D. C., Audette, J., Levy, R. M., Mackey, S. C., and Stanos, S. (2010). Assessment and treatment of psychosocial comorbidities in patients with neuropathic pain. Mayo Clin. Proc. 85, S42–S50. doi: 10.4065/mcp.2009.0648
Tyrrell, L., Renganathan, M., Dib-Hajj, S. D., and Waxman, S. G. (2001). Glycosylation alters steady-state inactivation of sodium channel Nav1.9/nan in dorsal root ganglion neurons and is developmentally regulated. J. Neurosci. 21, 9629–9637.
van Bemmelen, M. X., Rougier, J. S., Gavillet, B., Apotheloz, F., Daidie, D., Tateyama, M., et al. (2004). Cardiac voltage-gated sodium channel Nav1.5 is regulated by NEDD4-2 mediated ubiquitination. Circ. Res. 95, 284–291.
Vanoye, C. G., Kunic, J. D., Ehring, G. R., and George, A. L. (2013). Mechanism of sodium channel NaV1.9 potentiation by G-protein signaling. J. Gen. Physiol. 141, 193–202. doi: 10.1085/jgp.201210919
Vijayaragavan, K., Boutjdir, M., and Chahine, M. (2004a). Modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by protein kinase A and protein kinase C. J. Neurophysiol. 91, 1556–1569. doi: 10.1152/jn.00676.2003
Vijayaragavan, K., Powell, A. J., Kinghorn, I. J., and Chahine, M. (2004b). Role of auxiliary beta1-, beta2-, and beta3-subunits and their interaction with Na(v)1.8 voltage-gated sodium channel. Biochem. Biophys. Res. Commun. 319, 531–540. doi: 10.1016/j.bbrc.2004.05.026
Vijayaragavan, K., O’Leary, M. E., and Chahine, M. (2001). Gating properties of Nav1.7 and Nav1.8 peripheral nerve sodium channels. J. Neurosci. 21, 7909–7918.
von Hehn, C. A., Baron, R., and Woolf, C. J. (2012). Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 73, 638–652. doi: 10.1016/j.neuron.2012.02.008
Wada, A., Wanke, E., Gullo, F., and Schiavon, E. (2008). Voltage-dependent Nav1.7 sodium channels: multiple roles in adrenal chromaffin cells and peripheral nervous system. Acta Physiol. 192, 221–231. doi: 10.1111/j.1748-1716.2007.01810.x
Wada, A., Yanagita, T., Yokoo, H., and Kobayashi, H. (2004). Regulation of cell surface expression of voltage-dependent Nav1.7 sodium channels: mRNA stability and posttranscriptional control in adrenal chromaffin cells. Front. Biosci. 9:1954–1966. doi: 10.1371/journal.pone.0023083
Waechter, C. J., Schmidt, J. W., and Catterall, W. A. (1983). Glycosylation is required for maintenance of functional sodium channels in neuroblastoma cells. J. Biol. Chem. 258, 5117–5123.
Wang, Y., Brittain, J. M., Jarecki, B. W., Park, K. D., Wilson, S. M., Wang, B., et al. (2010). In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator Protein-2 identifies a pocket important in modulating sodium channel slow inactivation. J. Biol. Chem. 285, 25296–25307. doi: 10.1074/jbc.M110.128801
Waxman, S. G. (1999). The molecular pathophysiology of pain: abnormal expression of sodium channel genes and its contributions to hyperexcitability of primary sensory neurons. Pain 82, S133–S140. doi: 10.1016/s0304-3959(99)00147-5
Waxman, S. G., and Dib-Hajj, S. (2005). Erythermalgia: molecular basis for an inherited pain syndrome. Trends Mol. Med. 11, 555–562.
Waxman, S. G., Merkies, I. S. J., Gerrits, M. M., Dib-Hajj, S. D., Lauria, G., Cox, J. J., et al. (2014). Sodium channel genes in pain-related disorders: phenotype–genotype associations and recommendations for clinical use. Lancet Neurol. 13, 1152–1160. doi: 10.1016/S1474-4422(14)70150-4
Waxman, S. G., and Zamponi, G. W. (2014). Regulating excitability of peripheral afferents: emerging ion channel targets. Nat. Neurosci. 17, 153–163. doi: 10.1038/nn.3602
Way, K. J., Chou, E., and King, G. L. (2000). Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol. Sci. 21, 181–187. doi: 10.1016/S0165-6147(00)01468-1
Wells, M., Racis, S., and Vaidya, U. (1992). Changes in plasma cytokines associated with peripheral nerve injury. J. Neuroimmunol. 39, 261–268.
Widmann, C., Gibson, S., Jarpe, M. B., and Johnson, G. L. (1999). Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 79, 143–180.
Woolf, C. J. (2004). Dissecting out mechanisms responsible for peripheral neuropathic pain: implications for diagnosis and therapy. Life Sci. 74, 2605–2610. doi: 10.1016/j.lfs.2004.01.003
Woolf, C. J. (2011). Central sensitization: implications for the diagnosis and treatment of pain. Pain 152, S2–S15. doi: 10.1016/j.pain.2010.09.030
Woolf, C. J., and Costigan, M. (1999). Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc. Natl. Acad. Sci. U.S.A. 96, 7723–7730. doi: 10.1073/pnas.96.14.7723
Woolf, C. J., and Mannion, R. J. (1999). Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353, 1959–1964.
Woolf, C. J., and Salter, M. W. (2000). Neuronal plasticity: increasing the gain in pain. Science 288, 1765–1768. doi: 10.1126/science.288.5472.1765
Wu, D.-F., Chandra, D., Mcmahon, T., Wang, D., Dadgar, J., Kharazia, V. N., et al. (2012). PKC𝜀 phosphorylation of the sodium channel NaV1.8 increases channel function and produces mechanical hyperalgesia in mice. J. Clin. Investig. 122, 1306–1315. doi: 10.1172/jci61934
Xu, J.-T., Tu, H.-Y., Xin, W.-J., Liu, X.-G., Zhang, G.-H., and Zhai, C.-H. (2007a). Activation of phosphatidylinositol 3-kinase and protein kinase B/Akt in dorsal root ganglia and spinal cord contributes to the neuropathic pain induced by spinal nerve ligation in rats. Exp. Neurol. 206, 269–279. doi: 10.1016/j.expneurol.2007.05.029
Xu, J.-T., Xin, W.-J., Wei, X.-H., Wu, C.-Y., Ge, Y.-X., Liu, Y.-L., et al. (2007b). p38 activation in uninjured primary afferent neurons and in spinal microglia contributes to the development of neuropathic pain induced by selective motor fiber injury. Exp. Neurol. 204, 355–365. doi: 10.1016/j.expneurol.2006.11.016
Xu, Q., Cheng, H.-W., He, H.-Q., Liu, Z.-R., He, M., Yang, H.-T., et al. (2008). Deglycosylation altered the gating properties of rNav1.3: glycosylation/deglycosylation homeostasis probably complicates the functional regulation of voltage-gated sodium channel. Neurosci. Bull. 24, 283–287. doi: 10.1007/s12264-008-0524-5
Xu, Q., Fitzsimmons, B., Steinauer, J., Neill, A. O., Newton, A. C., Hua, X.-Y., et al. (2011). Spinal phosphinositide 3-Kinase–Akt–Mammalian target of rapamycin signaling cascades in inflammation-induced hyperalgesia. J. Neurosci. 31, 2113–2124. doi: 10.1523/jneurosci.2139-10.2011
Yan, J., Melemedjian, O., Price, T., and Dussor, G. (2012). Sensitization of dural afferents underlies migraine-related behavior following meningeal application of interleukin-6 (IL-6). Mol. Pain 8:6.
Yanagita, T., Kobayashi, H., Uezono, Y., Yokoo, H., Sugano, T., Saitoh, T., et al. (2003). Destabilization of Nav1.7 sodium channel α-subunit mRNA by constitutive phosphorylation of extracellular signal-regulated kinase: negative regulation of steady-state level of cell surface functional sodium channels in adrenal chromaffin cells. Mol. Pharmacol. 63, 1125–1136. doi: 10.1124/mol.63.5.1125
Yanagita, T., Wada, A., Yamamoto, R., Kobayashi, H., Yuhi, T., Urabe, M., et al. (1996). Protein kinase C-Mediated Down-Regulation of voltage-dependent sodium channels in adrenal chromaffin cells. J. Neurochem. 66, 1249–1253. doi: 10.1046/j.1471-4159.1996.66031249.x
Yang, Y., Wang, Y., Li, S., Xu, Z., Li, H., Ma, L., et al. (2004). Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 41, 171–174.
Yeomans, D. C., Levinson, S. R., Peters, M. C., Koszowski, A. G., Tzabazis, A. Z., Gilly, W. F., et al. (2005). Decrease in inflammatory hyperalgesia by herpes vector-mediated knockdown of Nav1.7 sodium channels in primary afferents. Hum. Gene Ther. 16, 271–277. doi: 10.1089/hum.2005.16.271
Yin, R., Liu, D., Chhoa, M., Li, C. M., Luo, Y., Zhang, M., et al. (2015). Voltage-gated sodium channel function and expression in injured and uninjured rat dorsal root ganglia neurons. Int. J. Neurosci. doi: 10.3109/00207454.2015.1004172 [Epub ahead of print].
Young, K. A., and Caldwell, J. H. (2005). Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J. Physiol. 565, 349–370. doi: 10.1113/jphysiol.2004.081422
Yu, F. H., Westenbroek, R. E., Silos-Santiago, I., Mccormick, K. A., Lawson, D., Ge, P., et al. (2003). Sodium channel β4, a new disulfide-linked auxiliary subunit with similarity to β2. J. Neurosci. 23, 7577–7585.
Yu, Y.-Q., Zhao, F., Guan, S.-M., and Chen, J. (2011). Antisense-mediated knockdown of Nav1.8, but Not Nav1.9, generates inhibitory effects on complete freund’s adjuvant-induced inflammatory pain in rat. PLoS ONE 6:e19865. doi: 10.1371/journal.pone.0019865
Yuhi, T., Wada, A., Yamamoto, R., Yanagita, T., Niina, H., and Niina, H. (1996). Up-regulation of functional voltage-dependent sodium channels by cyclic AMP-dependent protein kinase in adrenal medulla. Brain Res. 709, 37–43. doi: 10.1016/0006-8993(95)01252-4
Zhang, H., Cang, C., Kawasaki, Y., Liang, L., Zhang, Y., Ji, R., et al. (2007). Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: a novel pathway for heat hyperalgesia. J. Neurosci. 27, 12067–12077.
Zhang, J. M., Donnelly, D. F., Song, X. J., and Lamotte, R. H. (1997). Axotomy increases the excitability of dorsal root ganglion cells with unmyelinated axons. J. Neurophysiol. 78, 2790–2794.
Zhang, X. y., Wen, J., Yang, W., Wang, C., Gao, L., Zheng, L. H., et al. (2013). Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet. 93, 957–966. doi: 10.1016/j.ajhg.2013.09.016
Zhang, Y., Hartmann, H. A., and Satin, J. (1999). Glycosylation influences voltage-dependent gating of cardiac and skeletal muscle sodium channels. J. Membr. Biol. 171, 195–207.
Zhou, J., Shin, H.-G., Yi, J., Shen, W., Williams, C. P., and Murray, K. T. (2002). Phosphorylation and putative ER retention signals are required for protein kinase A-mediated potentiation of cardiac sodium current. Circ. Res. 91, 540–546. doi: 10.1161/01.res.0000033598.00903.27
Zhou, J., Yi, J., Hu, N., George, A. L., and Murray, K. T. (2000). Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus Oocytes. Circ. Res. 87, 33–38. doi: 10.1161/01.res.87.1.33
Zhuang, Z.-Y., Xu, H., Clapham, D. E., and Ji, R.-R. (2004). Phosphatidylinositol 3-Kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J. Neurosci. 24, 8300–8309. doi: 10.1523/jneurosci.2893-04.2004
Zimmer, T., and Benndorf, K. (2002). The human heart and rat brain IIA Na+ channels interact with different molecular regions of the beta1 subunit. J. Gen. Physiol. 120, 887–895. doi: 10.1085/jgp.20028703
Keywords: voltage-gated sodium channels, post-translational modification, chronic pain, hyperexcitability, nociceptive neurons
Citation: Laedermann CJ, Abriel H and Decosterd I (2015) Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes. Front. Pharmacol. 6:263. doi: 10.3389/fphar.2015.00263
Received: 15 June 2015; Accepted: 23 October 2015;
Published: 05 November 2015.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Michael E. O’Leary, Cooper Medical School of Rowan University, USASulayman Dib-Hajj, Yale University, USA
Copyright © 2015 Laedermann, Abriel and Decosterd. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cedric J. Laedermann, cedric.laedermann@gmail.com