 Natalia Malek
Natalia Malek Katarzyna Starowicz
Katarzyna Starowicz- Laboratory of Pain Pathophysiology, Department of Pain Pharmacology, Institute of Pharmacology, Polish Academy of Sciences, Krakow, Poland
Compared with acute pain that arises suddenly in response to a specific injury and is usually treatable, chronic pain persists over time, and is often resistant to medical treatment. Because of the heterogeneity of chronic pain origins, satisfactory therapies for its treatment are lacking, leading to an urgent need for the development of new treatments. The leading approach in drug design is selective compounds, though they are often less effective and require chronic dosing with many side effects. Herein, we review novel approaches to drug design for the treatment of chronic pain represented by dual-acting compounds, which operate at more than one biological target. A number of studies suggest the involvement of the cannabinoid and vanilloid receptors in pain. Interestingly cannabinoid system is in interrelation with other systems that comprise lipid mediators: prostaglandins, produced by COX enzyme. Therefore, in the present review, we summarize the role of dual-acting molecules (FAAH/TRPV1 and FAAH/COX-2 inhibitors) that interact with endocannabinoid and endovanillinoid systems and act as analgesics by elevating the endogenously produced endocannabinoids and dampening the production of pro-inflammatory prostaglandins. The plasticity of the endocannabinoid system (ECS) and the ability of a single chemical entity to exert an activity on two receptor systems has been developed and extensively investigated. Here, we review up-to-date pharmacological studies on compounds interacting with FAAH enzyme together with TRPV1 receptor or COX-2 enzyme respectively. Multi-target pharmacological intervention for treating pain may lead to the development of original and efficient treatments.
Introduction
Pain is defined by the International Association for the Study of Pain as an unpleasant sensory and/or emotional experience associated with actual or potential tissue damage (Bonica, 1979). Acute pain is important for the diagnosis and localization of the disease process and to avoid or minimize tissue damage. In a situation where the pain sensation remains despite mitigating the immediate cause, it loses its warning and defensive characteristics and becomes a disease itself, which is challenging to treat because of the heterogeneity of its origin. None of the currently available therapies provide sufficient relief for patients; at the same time, they all have risks of complications and a number of adverse effects as incidence increases with age (Ray et al., 2007; Gallagher and Rosenthal, 2008; Williams et al., 2013). Due to the aging population in developed countries and the lack of satisfactory therapies, chronic pain is a worldwide disease that requires the development of new treatments. One possible approach focuses on the modulation of endogenously produced compounds. The lipophilic molecule anandamide (AEA), that is an integral part of the endocannabinoid system (ECS), also binds to the transient receptor potential cation channel subfamily V member 1 (TRPV1); consequently, AEA is now frequently referred to as an “endovanilloid.” A promising approach to retain the analgesic effects of cannabinoid activation, while avoiding the undesirable results of its global action is to elevate endogenously produced endocannabinoids by inhibiting their hydrolytic degradation. Fatty acid amide hydrolase (FAAH) hydrolyzes AEA and other lipid signaling molecules, which makes it a good target for this new therapeutic method. Moreover, ECS is interrelated with other systems that comprise lipid mediators such as prostaglandins/leukotrienes, contributing to the chronic pain development (Huwiler and Pfeilschifter, 2009). Additionally, compounds with primary actions upon cyclooxygenase (COX), involved in pain processing, also interact with ECS, including having the ability to directly inhibit FAAH (Fowler, 2007; Guindon and Hohmann, 2008).
Recent studies suggest that inhibiting FAAH will not have as significant efficacy as expected in chronic pain patient groups (Huggins et al., 2012). Therefore, it could be more relevant for future research to take into account a new paradigm—a multi-therapeutic strategy combining the action on FAAH, TRPV1, and COX-2. Here, we review up-to-date studies on dual-acting compounds that interact with the endocannabionid/endovanilloid and COX systems, which may be beneficial for the treatment of chronic pain (the basis of molecular interactions between the systems is shown in Figure 1).
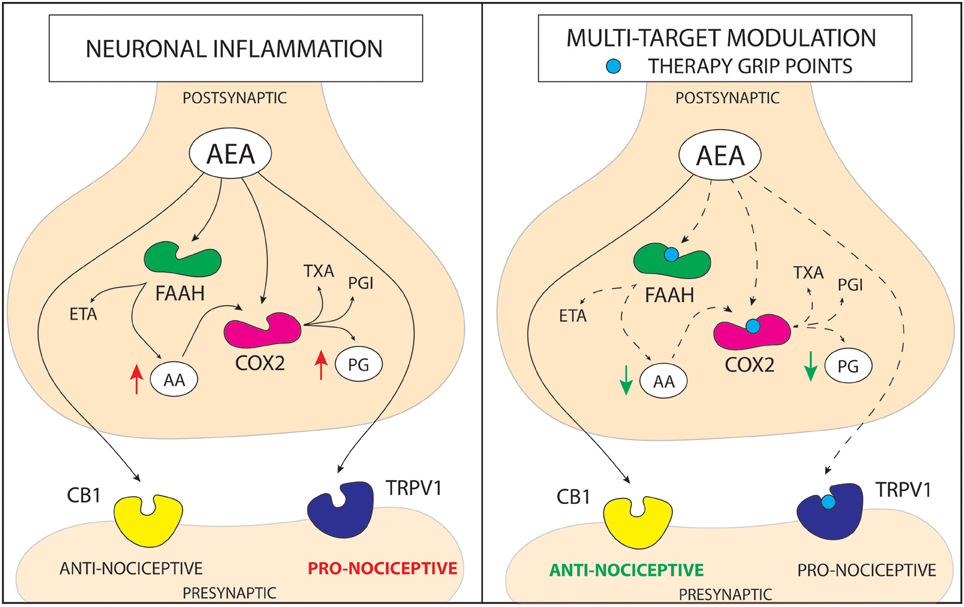
Figure 1. Schematic representation of a mechanism of action for cannabinoid based multi-target drugs. The novel strategy for the pain treatment is based on altering more than one enzymatic reaction and/or nociceptive pathway. Anandamide (AEA) is hydrolyzed into arachidonic acid (AA) and ethanolamine (ETA) principally by fatty acid amide hydrolase (FAAH). In addition to its hydrolysis by FAAH, AEA is metabolized by COX-2. On the other hand, COX metabolizes AA, leading to increase in pro-inflammatory prostaglandins (PG). AEA can act on transient receptor potential cation channel subfamily V member 1 (TRPV1) activating pro-nociceptive pathways or through cannabinoid receptor 1/2 (CB1/2) pathway leading to alleviation of pain. Although, in the molecular stress conditions elevation of AEA occurs, FAAH and COX quickly metabolize it. Therefore, there are three pathways that may increase anti-nociceptive properties of AEA; (1) inhibition of FAAH enzyme, that leads to an increase in AEA level and a decrease in AA level; (2) inhibition of COX enzyme, that leads to an increase in AEA level and a decrease in PG level; (3) antagonism of TRPV1 receptor, which prevents activation of pro-nociceptive pathway by AEA. Action on more than one molecular target lowers the redundancy of the system and may lead to obtaining more stable and robust response in pain alleviation. (PGI—prostacyclins; TXA—thromboxanes).
Single Target: Success or Defeat?
CB1 and CB2 Agonism
In the recent years, considerable progress has been made in understanding the role of the ECS in the modulation of pain processing. The system includes cannabinoid receptors 1 (CB1) and 2 (CB2), endogenous agonists called endocannabinoids: AEA and 2-arachidonyl glycerol (2-AG) and enzymes involved in their biosynthesis and degradation. Endocannabinoids are produced in injured tissues to suppress sensitization and inflammation by activation of CB1 and CB2 (Piomelli and Sasso, 2014). The key enzyme responsible for the catabolism of AEA is FAAH. However, AEA can also be metabolized by COX-2 and lipoxygenase 12/15 (LOX12/15). COX-2 metabolizes AEA to prostaglandins (PG), which are involved in inflammatory response (Kozak et al., 2002; Yang et al., 2005; Fowler, 2007). CB1 receptors expressed in the peripheral and central nervous system (CNS) are responsible for the conducting of pain signaling (Richardson et al., 1998). CB2 receptors expressed in immune cells (also microglia), regulate the neuro-immune interactions and interfere with the inflammatory hyperalgesia, thus also playing an important role in nociception (Malek et al., 2015b; Mecha et al., 2015). Endocannabinoids have been shown to behave as analgesics in models of both acute nociception and chronic pain such as inflammation (Pernía-Andrade et al., 2009) and painful neuropathy (Toth et al., 2010; La Porta et al., 2013). The main limitation of exogenous cannabinoid receptor agonists is the number of CNS side effects, including dysphoria, movement disorders, dizziness, memory loss, drug abuse potential, and dependence (Thaler et al., 2011). An alternative approach that may prevent the occurrence of these side effects is the stimulation of endogenous mechanisms that regulate ECS activity (Lichtman and Chapman, 2011; Lomazzo et al., 2015).
FAAH Inhibition
Endogenous cannabinoids are synthesized “on demand” in areas of cellular stress and therefore do not produce CB1 agonist side effects (Di Marzo and Petrosino, 2007). Unfortunately, locally released endocannabinoids have a very short half-life due to efficient enzymatic degradation (Deutsch and Chin, 1993; Ahn et al., 2008). Therefore, a subsequent grip point for pain therapies is FAAH, which is responsible for the degradation of AEA and other related fatty acid amides including N-palmitoylethanolamine (PEA), oleoylethanolamine (OEA), N-arachidonoyl glycine (NAGly; Bisogno et al., 2002; Khanna and Alexander, 2011). A series of in vivo experiments proved that genetic or pharmacological inactivation of FAAH results in elevation of endocannabinoids in the spinal cord and brain stem (Lichtman et al., 2004; De Lago et al., 2005). FAAH inhibition, rather than its deletion, may offer a distinctive strategy for the treatment of chronic pain, because no genotypic differences in pain behavior were evident in chronic pain models (Lichtman et al., 2004; Kinsey et al., 2009). FAAH inhibition was shown to cause anti-nociceptive, anti-inflammatory or anti-edemic effects in numerous acute (Holt et al., 2005), chronic (Jayamanne et al., 2006), and neuropathic pain animal models (Jhaveri et al., 2006; Kinsey et al., 2009; Guindon et al., 2013). Nevertheless, FAAH inhibitors (like URB957) are not optimal due to some limitations. Tissue-specific changes in the sensitivity to URB597 in neuropathic pain in rats, which may arise as a result of changes in FAAH activity, metabolic pathways, and tissue pH were reported (Chang et al., 2006; Paylor et al., 2006). Due to the differential effects of URB597 in carrageenan-induced inflammation and spinal nerve ligation models, efforts aimed at optimizing the clinical efficacy of FAAH inhibitors should be revised and redesigned (Di Marzo, 2012; Okine et al., 2012). Similarly, despite many promising preclinical results in various chronic pain models (Ahn et al., 2011), one of the most potent FAAH inhibitors, PF-04457845, has failed to show efficacy in humans in a randomized, placebo-controlled phase II clinical trial (Di Marzo, 2012; Huggins et al., 2012). Moreover, safety of FAAH inhibitors became questioned after “first-in-human” trial to test safety of BIA 10-2474 in healthy volunteers, which concluded with one person dead and five more hospitalized. The probable cause of the failure is unjustified dose, 80 times higher than that presumed to induce total FAAH inhibition, used in the study1.
TRPV1 Antagonism
TRPV1 has emerged as a promising target for the development of new analgesic and anti-inflammatory drugs. TRPV1 is a non-selective ion channel that is highly associated with pain nociception and linked to ECS through the common agonist AEA (Zygmunt et al., 1999; Van der Stelt et al., 2005; Lizanecz et al., 2006). This polymodal pain transducer is known to be expressed in peripheral sensory afferents (Singh Tahim et al., 2005; Ikeda-Miyagawa et al., 2015), spinal cord (Kanai et al., 2006; Horvath et al., 2008), and some brain stem nuclei involved in nociception, including periaqueductal gray matter (PAG) and cingulate cortex (Roberts et al., 2004; Cristino et al., 2006; Starowicz et al., 2007).
A growing body of evidence suggests that TRPV1 is essential in driving nociceptive response (Davis et al., 2000; Immke and Gavva, 2006; Horvath et al., 2008). TRPV1-knockout mice exhibited attenuated inflammation-induced hyperalgesia, while receptor blockade and desensitization caused analgesia (Karai et al., 2004; Bölcskei et al., 2005). Furthermore, enhanced TRPV1 expression and increased TRPV1 sensitivity to AEA during inflammation and neuropathic conditions has been described previously (De Petrocellis et al., 2001; Baamonde et al., 2005; Singh Tahim et al., 2005). Unfortunately, off-target effects of TRPV1 modulation on thermoregulation are widely described. TRPV1 activation results in hypothermia, whereas antagonizing TRPV1 causes hyperthermia, which accounted for the failure of AMG517 during a phase I clinical trial (Di Marzo et al., 2000; Swanson et al., 2005; Gavva et al., 2008). Nevertheless, TRPV1 antagonism has still emerged as an interesting strategy to alleviate pain, especially with simultaneous action on either FAAH and/or COX-2, which could allow lower doses to be used (Lee et al., 2015; Malek et al., 2015a, 2016).
COX-2 Antagonism
The basis for the use of non-steroidal anti-inflammatory drugs (NSAIDs) for the treatment of pain and inflammation is COX inhibition. Cyclooxygenases are enzymes that catalyze the conversion of membrane phospholipids to prostanoids, which include PG, prostacyclins (PGI) essential for intestine and kidney functioning, and thromboxanes (TXA), responsible for platelet aggregation. The two COX isozymes are COX-1 and COX-2. COX-1 is characterized by constitutive expression in tissues and COX-2 is an induced isoform with low baseline expression in human tissues that can be induced during a response to extracellular and intracellular stimuli. The increase in COX-2 expression elevates proinflammatory cytokine, mitogen and growth factor levels (Smith et al., 1996), as well it plays a key role in the transmission of pain to the brain and spinal cord (Ito et al., 2001). Moreover, blocking COXs can influence the ECS by stopping the conversion of endocannabinoids into prostanoid-like derivatives, which may be of benefit for the treatment of chronic pain (Jhaveri et al., 2007; Hu et al., 2008). The inflammatory process can dramatically reduce the level of endocannabinoids, while its inhibition may contribute to increased analgesic action. Although widely used in the treatment of pain, NSAIDs do not always provide adequate relief and have the risk for serious complications, including death by internal hemorrhaging (5–10% of chronic NSAIDs therapy) and severe kidney damage (Rahme and Bernatsky, 2010; Altman, 2011). However, it should be noted that some of the NSAIDs have the ability to inhibit FAAH (Fowler et al., 1999; Seidel et al., 2003; Guindon et al., 2006). Therefore, interaction of NSAIDs with ECS may be a novel approach for the treatment of chronic pain.
Two Targets: Improvement or Deterioration?
The aging population in developing countries, the lack of satisfactory therapies, and worldwide increase of chronic pain are burdens that require the development of new treatments for pain. A leading approach in the treatment of chronic pain is using single-target drugs, which are not always effective and can be associated with many side effects from chronic dosing. Multi-functional drugs may be advantageous for poorly controlled chronic pain and may improve safety and efficacy properties with fewer side effects compared to single-target molecules (Csermely et al., 2005; Stahl, 2009). Dual-acting compounds also have much more potential than single-target and highly specific agents due to better safety profiles and are less likely to exhibit pharmacokinetic and pharmacological interactions (compared to combined therapy with two single-target drugs). With respect to pain relief, there are some reports about multi-target therapeutics that will be discussed here, including piperazinyl carbamates that simultaneously interact with FAAH and TRPV1 or FAAH and COX inhibitors. We will determine the status of in vivo preclinical drug research and consider whether multi-target drugs are the current trend in search for better pain therapy (summarized information on the dual-acting compounds can be found in the Table 1).

Table 1. Dual-acting compounds: summary of in vivo studies.
Acting on FAAH and TRPV1
Endogenous levels of AEA can activate but not desensitize TRPV1 in non-pathological conditions, leading to stimulation of pain pathways. Elevation of endocannabinoid tone by FAAH inhibitors may lead to further activation of this channel, implying that the development of combined FAAH/TRPV1 blockers may have essential therapeutic applications in directing AEA's action toward the analgesic CB1/2 pathway. Lack of efficacy of FAAH inhibitors in patients (as described above) may be associated with activation of the alternative AEA biotransformation pathways after completely blocking the FAAH enzyme and the ability of AEA to induce pro-nociceptive signaling through TRPV1 receptors (Maione et al., 2006; Piscitelli and Di Marzo, 2012; Starowicz et al., 2013). The most promising aforementioned agent is the endogenous lipid signaling molecule N-arachidonoyl-5-hydroxytryptamine (AA-5-HT), which was originally described as an FAAH inhibitor and called an “unique dual FAAH/TRPV1 blocker” about a decade later (Maione et al., 2007). In the context of its inhibitory activity, similar to other FAAH blockers, AA-5-HT elevates AEA levels in the CNS and on the periphery (Capasso et al., 2005; Maione et al., 2007). Moreover, AA-5-HT is able to antagonize human and rat recombinant TRPV1 receptors with an affinity slightly stronger than capsaicin, though, it is not clear whether AA-5-HT is a competitive or non-competitive antagonist (Maione et al., 2007). AA-5-HT appears to be less effective at TRPV1 blockade when the pH of the cellular environment is reduced, meaning that it may be less effective under inflammatory conditions, although this hypothesis has not been tested in vivo yet (Fowler et al., 2003; Paylor et al., 2006). Nevertheless, studies involving a variety of animal models have been conducted to better understand the mechanism of action of AA-5-HT. A significant contribution was completed by Maione and colleagues, who indicated the significant analgesic impact exerted by the compound in acute and chronic pain models in rodents following systemic administration (Maione et al., 2007). A subsequent study showed that intra-PAG AA-5-HT also causes analgesia and that the effect is mimicked by co-injection of URB597 and the selective TRPV1 antagonist iodoresiniferatoxin (I-RTX). Furthermore, antagonizing the CB1 and TRPV1 receptors abolished AA-5-HT-exerted analgesia, suggesting that these co-localized receptors are AA-5-HT molecular targets in the PAG and cortex. Moreover, single AA-5-HT dosages exhibited anti-allodynic actions more effectively than URB597 or I-RTX (De Novellis et al., 2008, 2011). In the same series of experiments, de Novellis et al. determined the role of AA-5-HT in cognitive and emotional functions (i.e., decision making, goal-directed behavior and working memory) in neuropathic pain. The authors reported that AA-5-HT is able to restore the balance between excitatory and inhibitory responses in prefrontal cortex neurons, highlighting the potential of dual-acting drugs for mitigating the central sequelae associated with neuropathic pain (De Novellis et al., 2011). AA-5-HT was also proven to dose-dependently relieve pain in a model of acute inflammation in mice after systemic administration (Costa et al., 2010). Its anti-nociceptive action was associated with an increase in AEA levels in both inflamed paws and spinal cord, though CB1 and TRPV1 receptors were differentially involved; TRPV1 was responsible for the anti-inflammatory properties of AA-5-HT, while both CB1 and TRPV1 receptors mediated its anti-hyperalgesic activity (Costa et al., 2010). Our most recent research indicates that CB2 receptors are also involved in the anti-nociceptive actions of AA-5-HT, after intrathecal administration (Malek et al., 2016). Despite the fact that AA-5-HT was proven to be active it is unstable due to effective hydrolysis, therefore efforts to synthesize AA-5-HT analogs with improved modes of action have been undertaken (Ortar et al., 2007). The chemical synthesis led to discovery of piperazinyl carbamates and ureas that are characterized by higher stability than AA-5-HT, nevertheless none of them have shown higher efficacy than AA-5-HT (Ortar et al., 2007, 2013; Morera et al., 2009). Of the many synthesized compounds, OMDM-198 is best described and tested in vivo for its anti-nociceptive and analgesic properties. OMDM-198 exhibits anti-nociceptive, anti-hyperalgesic and anti-edemic actions in acute and inflammatory pain. It inhibits the second phase of formalin-induced nociceptive behavior in rodents and blocks carrageenan-induced thermal hyperalgesia and paw edema in mice (Maione et al., 2013). The anti-hyperalgesic effects of OMDM-198 were also evaluated in an animal model of osteoarthritic pain. It was shown that the effect of OMDM-198 depends on CB1 receptor activation and TRPV1 receptor blockade (Malek et al., 2015a). Moreover, OMDM-198 required lower doses to achieve a comparable analgesic effect than the two single-target compounds used in the study: URB597 and SB-366,791 (TRPV1 antagonist; Malek et al., 2015a). These data suggest that the use of dual-acting compounds may be beneficial in a matter of side effects characteristic for TRPV1 antagonists (hyperthermia) and FAAH inhibitors (lack of efficacy in clinics), because of significant decrease of the working doses. It was shown that hyperthermia was not observed in animals upon AA-5-HT treatment (Maione et al., 2007; Costa et al., 2010; Malek et al., 2016). The same observations were made for OMDM-198 (unpublished data). The field of FAAH/TRPV1 blockers is still developing. A recently synthesized series of arylboronic acids characterized by high efficacy on FAAH and TRPV1 have been reported, though these hybrid compounds have not yet been evaluated in animal studies for pain (Morera et al., 2016).
Acting on FAAH and COXs
Interactions between COX and FAAH enzymes that control endogenous lipid (including endocannabinoids) levels were reported (Guindon and Beaulieu, 2006; Jhaveri et al., 2008). AEA metabolism is of special interest because blocking FAAH enzyme activates AEA biotransformation by COX-2 pathway, leading to decreases in endocannabinoid tone. This may be another mechanism responsible for lack of analgesic action from selective FAAH inhibitors (Starowicz et al., 2013; Starowicz and Di Marzo, 2013). Indeed, co-administration of diclofenac (NSAID) and URB597 showed synergic analgesic effects in abdominal pain dependent on both CB1 activation and blocking prostaglandin production (Naidu et al., 2010). Another study demonstrated an additive anti-nociceptive activity of PF-3845 and diclofenac in the animal models of inflammatory and neuropathic pain (Grim et al., 2014). Moreover, ketorolac (NSAID) was shown to have an additive anti-nociceptive effect with WIN-55,212-2 (non-selective cannabinoid agonist; Ulugöl et al., 2006). In these studies, AEA levels were shown to be elevated with simultaneous inhibition of PG synthesis. Studies have shown that common NSAIDs (ibuprofen and flurbiprofen) act also as FAAH inhibitors (Fowler et al., 1999; Seidel et al., 2003; Guindon et al., 2006). The synergistic effect of ibuprofen and AEA was proven in experimental models for acute and neuropathic pain (Guindon and Beaulieu, 2006). Those data demonstrate the rationale underlying design of novel compounds that can interact with both COXs and FAAH enzymes. Indeed, numerous novel compounds with promising analgesic characteristics were developed recently, i.e., ARN2508, which was tested and effective in suppressing intestinal inflammation (Sasso et al., 2015). It also causes no gastric damage, which is often a case of chronic NSAID treatment and the protective effect was dependent on FAAH inhibition. This suggests that COX/FAAH inhibitors shouldn't exhibit NSAIDs side effects present during chronic treatment, although clinical studies would be needed to verify this hypothesis. A panel of novel ARN2508 derivatives was synthesized, of which (S)-(+)-ARN2508 demonstrated increased OEA plasma levels and decreased PGI and TXA levels after intravenous administration (Migliore et al., 2016). The in vivo results suggest that this compound may be useful for FAAH-COX related pathologies, such as pain.
Summary
In this review, we have described the complex network between TRPV1 and CB1/2 receptors and FAAH and COX-2 enzymes, which are all involved in anti-nociceptive action of AEA. It seems that the “single-target” approach is not as effective for the AEA elevation as this has been characterized by redundancy of metabolic pathways (by i.e., FAAH, COX). Inhibition of one enzyme can activate others, leading to absence of anticipated analgesic effects. The paradigm shift from selective drugs to multi-target compounds has led to promising results in the treatment of pain, which has been summarized herein. This approach may be beneficial for the treatment of pain, which poses a difficult challenge due to the heterogeneity of its origin. Compounds, acting on more than one molecular target may have higher efficacies and better safety profiles than currently used drugs (and preclinically studied compounds) that act on a single biological target (Fowler et al., 2009; Boran and Iyengar, 2010). Acting on more than one target brings a question of selectivity of dual-acting compounds, which is of concern during the design phase (Hwang et al., 2013; Aiello et al., 2016). Moreover, as these molecules are designed to be used for chronic pain treatment it is crucial to investigate their ability to develop tolerance and (possibly) dependence, as this was not investigated yet. For the reviewed compounds no side effects were reported in the preclinical models of chronic pain, probably due to low doses necessary to obtain satisfactory pharmacological effect (analgesic, anti-nociceptive or anti-allodynic). Nevertheless, disadvantages of the use of multitarget therapy cannot be omitted and its limitation need to be considered and studied, especially when safety and efficacy in clinical trials is a major concern (Brodie et al., 2015).
Author Contributions
NM: preparing of the figure and table, writing of the manuscript, KS: review conception, writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Center for Research and Development, Poland by grant LIDER/29/60/L-2/10/NCBiR/2011, the National Science Center, Poland by grant SONATA BIS NCN/2012/07/E/NZ7/01269, ETIUDA NCN/2015/16/T/NZ7/00052, and statutory funds. NM is a recipient of a scholarship from KNOW, which is sponsored by the Ministry of Science and Higher Education, Poland.
Footnotes
1. ^“Version anglaise: Minutes of the Temporary Specialist Scientific Committee (TSSC) meeting on “FAAH (Fatty Acid Amide Hydrolase) Inhibitors” of 15/02/2016 (08/03/2016).” Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM).
Abbreviations
2-AG, 2-arachidonoyl-glycerol; AA-5-HT, N-arachidonoyl-serotonin; AEA, N-arachidonylethanolamide, anandamide; AMG517, N-[4-[[6-[4-(trifluoromethyl)phenyl]-4-pyrimidinyl]oxy]-2-benzothiazolyl]-acetamide; ARN2508, 10r (±)-2-[3-fluoro-4-[3-(hexylcarbamoyloxy)phenyl]phenyl]propanoic acid; CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; CNS, central nervous system; COX, cyclooxygenase; ECS, endocannabinoid system; FAAH, fatty acid amide hydrolase; I-RTX, iodoresiniferatoxin; LOX, lipoxygenase; NAGly, N-arachidonoyl glycine; NSAID, non-steroidal anti-inflammatory drug; OEA, oleoylethanolamide; OMDM-198, 3-[((chloro)pyridin-2-yl)-3-phenyl]-carbamate; PAG, periaqueductal gray; PEA, palmitoylethanolamide; PF-3845, 4-(3-(5-(trifluoromethyl)pyridin-2-yloxy)benzyl)-N-(pyridin-3-yl)piperidine-1-carboxamide; PF04457845, N-pyridazin-3-yl-4-[(3-[5-(trifluoromethyl)pyridin-2-yl]oxyphenyl)methylidene]piperidine-1-carboxamide; PG, prostaglandins; PGI, prostacyclins; SB-366, 791, (E)-3-(4-Chlorophenyl)-N-(3-methoxyphenyl)prop-2-enamide; TRPV1, transient receptor potential cation channel subfamily V member 1; TXA, thromboxanes; URB597, cyclohexylcarbamic acid 3-carbamoyl-biphenyl-3-yl ester.
References
Ahn, K., McKinney, M. K., and Cravatt, B. F. (2008). Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 108, 1687–1707. doi: 10.1021/cr0782067
Ahn, K., Smith, S. E., Liimatta, M. B., Beidler, D., Sadagopan, N., Dudley, D. T., et al. (2011). Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 338, 114–124. doi: 10.1124/jpet.111.180257
Aiello, F., Carullo, G., Badolato, M., and Brizzi, A. (2016). TRPV1-FAAH-COX: the couples game in pain treatment. ChemMedChem. doi: 10.1002/cmdc.201600111. [Epub ahead of print].
Altman, R. D. (2011). Safety advantages of topical versus oral nonsteroidal antiinflammatory drugs. J. Rheumatol. 38, 572–573. doi: 10.3899/jrheum.100748
Baamonde, A., Lastra, A., Juarez, L., Hidalgo, A., and Menéndez, L. (2005). TRPV1 desensitisation and endogenous vanilloid involvement in the enhanced analgesia induced by capsaicin in inflamed tissues. Brain Res. Bull. 67, 476–481. doi: 10.1016/j.brainresbull.2005.07.001
Bisogno, T., De Petrocellis, L., and Di Marzo, V. (2002). Fatty acid amide hydrolase, an enzyme with many bioactive substrates. Possible therapeutic implications. Curr. Pharm. Des. 8, 533–547. doi: 10.2174/1381612023395655
Bölcskei, K., Helyes, Z., Szabó, A., Sándor, K., Elekes, K., Németh, J., et al. (2005). Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain 117, 368–376. doi: 10.1016/j.pain.2005.06.024
Boran, A. D. W., and Iyengar, R. (2010). Systems approaches to polypharmacology and drug discovery. Curr. Opin. Drug Discov. Devel. 13, 297–309.
Brodie, J. S., Di Marzo, V., and Guy, G. W. (2015). Polypharmacology shakes hands with complex aetiopathology. Trends Pharmacol. Sci. 36, 802–821. doi: 10.1016/j.tips.2015.08.010
Capasso, R., Matias, I., Lutz, B., Borrelli, F., Capasso, F., Marsicano, G., et al. (2005). Fatty acid amide hydrolase controls mouse intestinal motility in vivo. Gastroenterology 129, 941–951. doi: 10.1053/j.gastro.2005.06.018
Chang, L., Luo, L., Palmer, J. A., Sutton, S., Wilson, S. J., Barbier, A. J., et al. (2006). Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br. J. Pharmacol. 148, 102–113. doi: 10.1038/sj.bjp.0706699
Costa, B., Bettoni, I., Petrosino, S., Comelli, F., Giagnoni, G., and Di Marzo, V. (2010). The dual fatty acid amide hydrolase/TRPV1 blocker, N-arachidonoyl-serotonin, relieves carrageenan-induced inflammation and hyperalgesia in mice. Pharmacol. Res. 61, 537–546. doi: 10.1016/j.phrs.2010.02.001
Cristino, L., De Petrocellis, L., Pryce, G., Baker, D., Guglielmotti, V., and Di Marzo, V. (2006). Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 139, 1405–1415. doi: 10.1016/j.neuroscience.2006.02.074
Csermely, P., Agoston, V., and Pongor, S. (2005). The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol. Sci. 26, 178–182. doi: 10.1016/j.tips.2005.02.007
Davis, J. B., Gray, J., Gunthorpe, M. J., Hatcher, J. P., Davey, P. T., Overend, P., et al. (2000). Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183–187. doi: 10.1038/35012076
De Lago, E., Petrosino, S., Valenti, M., Morera, E., Ortega-Gutierrez, S., Fernandez-Ruiz, J., et al. (2005). Effect of repeated systemic administration of selective inhibitors of endocannabinoid inactivation on rat brain endocannabinoid levels. Biochem. Pharmacol. 70, 446–452. doi: 10.1016/j.bcp.2005.05.011
De Novellis, V., Palazzo, E., Rossi, F., De Petrocellis, L., Petrosino, S., Guida, F., et al. (2008). The analgesic effect of N-arachidonoyl-serotonin, a FAAH inhibitor and TRPV1 receptor antagonist, associated with changes in rostral ventromedial medulla and locus coeruleus cell activity in rats. Neuropharmacology 55, 1105–1113. doi: 10.1016/j.neuropharm.2008.06.023
De Novellis, V., Vita, D., Gatta, L., Luongo, L., Bellini, G., De Chiaro, M., et al. (2011). The blockade of the transient receptor potential vanilloid type 1 and fatty acid amide hydrolase decreases symptoms and central sequelae in the medial prefrontal cortex of neuropathic rats. Mol. Pain 7:7. doi: 10.1186/1744-8069-7-7
De Petrocellis, L., Bisogno, T., Maccarrone, M., Davis, J. B., Finazzi-Agro, A., and Di Marzo, V. (2001). The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J. Biol. Chem. 276, 12856–12863. doi: 10.1074/jbc.M008555200
Deutsch, D. G., and Chin, S. A. (1993). Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 46, 791–796. doi: 10.1016/0006-2952(93)90486-G
Di Marzo, V. (2012). Inhibitors of endocannabinoid breakdown for pain: not so FA(AH)cile, after all. Pain 153, 1785–1786. doi: 10.1016/j.pain.2012.06.016
Di Marzo, V., Breivogel, C., Bisogno, T., Melck, D., Patrick, G., Tao, Q., et al. (2000). Neurobehavioral activity in mice of N-vanillyl-arachidonyl-amide. Eur. J. Pharmacol. 406, 363–374. doi: 10.1016/S0014-2999(00)00687-7
Di Marzo, V., and Petrosino, S. (2007). Endocannabinoids and the regulation of their levels in health and disease. Curr. Opin. Lipidol. 18, 129–140. doi: 10.1097/MOL.0b013e32803dbdec
Fowler, C. J. (2007). The contribution of cyclooxygenase-2 to endocannabinoid metabolism and action. Br. J. Pharmacol. 152, 594–601. doi: 10.1038/sj.bjp.0707379
Fowler, C. J., Janson, U., Johnson, R. M., Wahlström, G., Stenström, A., Norström, K., et al. (1999). Inhibition of anandamide hydrolysis by the enantiomers of ibuprofen, ketorolac, and flurbiprofen. Arch. Biochem. Biophys. 362, 191–196. doi: 10.1006/abbi.1998.1025
Fowler, C. J., Naidu, P. S., Lichtman, A., and Onnis, V. (2009). The case for the development of novel analgesic agents targeting both fatty acid amide hydrolase and either cyclooxygenase or TRPV1. Br. J. Pharmacol. 156, 412–419. doi: 10.1111/j.1476-5381.2008.00029.x
Fowler, C. J., Tiger, G., López-Rodríguez, M. L., Viso, A., Ortega-Gutiérrez, S., and Ramos, J. A. (2003). Inhibition of fatty acid amidohydrolase, the enzyme responsible for the metabolism of the endocannabinoid anandamide, by analogues of arachidonoyl-serotonin. J. Enzyme Inhib. Med. Chem. 18, 225–231. doi: 10.1080/1475636031000080216
Gallagher, R. M., and Rosenthal, L. J. (2008). Chronic pain and opiates: balancing pain control and risks in long-term opioid treatment. Arch. Phys. Med. Rehabil. 89, S77–S82. doi: 10.1016/j.apmr.2007.12.003
Gavva, N. R., Treanor, J. J. S., Garami, A., Fang, L., Surapaneni, S., Akrami, A., et al. (2008). Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 136, 202–210. doi: 10.1016/j.pain.2008.01.024
Grim, T. W., Ghosh, S., Hsu, K.-L., Cravatt, B. F., Kinsey, S. G., and Lichtman, A. H. (2014). Combined inhibition of FAAH and COX produces enhanced anti-allodynic effects in mouse neuropathic and inflammatory pain models. Pharmacol. Biochem. Behav. 124, 405–411. doi: 10.1016/j.pbb.2014.07.008
Guindon, J., and Beaulieu, P. (2006). Antihyperalgesic effects of local injections of anandamide, ibuprofen, rofecoxib and their combinations in a model of neuropathic pain. Neuropharmacology 50, 814–823. doi: 10.1016/j.neuropharm.2005.12.002
Guindon, J., De Léan, A., and Beaulieu, P. (2006). Local interactions between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute and inflammatory pain. Pain 121, 85–93. doi: 10.1016/j.pain.2005.12.007
Guindon, J., and Hohmann, A. G. (2008). A physiological role for endocannabinoid-derived products of cyclooxygenase-2-mediated oxidative metabolism. Br. J. Pharmacol. 153, 1341–1343. doi: 10.1038/bjp.2008.41
Guindon, J., Lai, Y., Takacs, S. M., Bradshaw, H. B., and Hohmann, A. G. (2013). Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacol. Res. 67, 94–109. doi: 10.1016/j.phrs.2012.10.013
Holt, S., Comelli, F., Costa, B., and Fowler, C. J. (2005). Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br. J. Pharmacol. 146, 467–476. doi: 10.1038/sj.bjp.0706348
Horvath, G., Kekesi, G., Nagy, E., and Benedek, G. (2008). The role of TRPV1 receptors in the antinociceptive effect of anandamide at spinal level. Pain 134, 277–284. doi: 10.1016/j.pain.2007.04.032
Hu, S. S.-J., Bradshaw, H. B., Chen, J. S.-C., Tan, B., and Walker, J. M. (2008). Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFkappaB activity. Br. J. Pharmacol. 153, 1538–1549. doi: 10.1038/bjp.2008.33
Huggins, J. P., Smart, T. S., Langman, S., Taylor, L., and Young, T. (2012). An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 153, 1837–1846. doi: 10.1016/j.pain.2012.04.020
Huwiler, A., and Pfeilschifter, J. (2009). Lipids as targets for novel anti-inflammatory therapies. Pharmacol. Ther. 124, 96–112. doi: 10.1016/j.pharmthera.2009.06.008
Hwang, S. H., Wecksler, A. T., Wagner, K., and Hammock, B. D. (2013). Rationally Designed Multitarget Agents Against Inflammation and Pain. Curr. Med. Chem. 20, 1783–1799. doi: 10.2174/0929867311320130013
Ikeda-Miyagawa, Y., Kobayashi, K., Yamanaka, H., Okubo, M., Wang, S., Dai, Y., et al. (2015). Peripherally increased artemin is a key regulator of TRPA1/V1 expression in primary afferent neurons. Mol. Pain 8, 8–11. doi: 10.1186/s12990-015-0004-7
Immke, D. C., and Gavva, N. R. (2006). The TRPV1 receptor and nociception. Semin. Cell Dev. Biol. 17, 582–591. doi: 10.1016/j.semcdb.2006.09.004
Ito, S., Okuda-Ashitaka, E., and Minami, T. (2001). Central and peripheral roles of prostaglandins in pain and their interactions with novel neuropeptides nociceptin and nocistatin. Neurosci. Res. 41, 299–332. doi: 10.1016/S0168-0102(01)00289-9
Jayamanne, A., Greenwood, R., Mitchell, V. A., Aslan, S., Piomelli, D., and Vaughan, C. W. (2006). Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br. J. Pharmacol. 147, 281–288. doi: 10.1038/sj.bjp.0706510
Jhaveri, M. D., Richardson, D., and Chapman, V. (2007). Endocannabinoid metabolism and uptake: novel targets for neuropathic and inflammatory pain. Br. J. Pharmacol. 152, 624–632. doi: 10.1038/sj.bjp.0707433
Jhaveri, M. D., Richardson, D., Kendall, D. A., Barrett, D. A., and Chapman, V. (2006). Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. J. Neurosci. 26, 13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006
Jhaveri, M. D., Richardson, D., Robinson, I., Garle, M. J., Patel, A., Sun, Y., et al. (2008). Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacology 55, 85–93. doi: 10.1016/j.neuropharm.2008.04.018
Kanai, Y., Hara, T., and Imai, A. (2006). Participation of the spinal TRPV1 receptors in formalin-evoked pain transduction: a study using a selective TRPV1 antagonist, iodo-resiniferatoxin. J. Pharm. Pharmacol. 58, 489–493. doi: 10.1211/jpp.58.4.0008
Karai, L., Brown, D. C., Mannes, A. J., Connelly, S. T., Brown, J., Gandal, M., et al. (2004). Deletion of vanilloid receptor 1-expressing primary afferent neurons for pain control. J. Clin. Invest. 113, 1344–1352. doi: 10.1172/JCI20449
Khanna, I. K., and Alexander, C. W. (2011). Fatty acid amide hydrolase inhibitors-progress and potential. CNS Neurol. Disord. Drug Targets 10, 545–558. doi: 10.2174/187152711796234989
Kinsey, S. G., Long, J. Z., O'Neal, S. T., Abdullah, R. A., Poklis, J. L., Boger, D. L., et al. (2009). Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J. Pharmacol. Exp. Ther. 330, 902–910. doi: 10.1124/jpet.109.155465
Kozak, K. R., Crews, B. C., Morrow, J. D., Wang, L.-H., Ma, Y. H., Weinander, R., et al. (2002). Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J. Biol. Chem. 277, 44877–44885. doi: 10.1074/jbc.M206788200
La Porta, C., Bura, S. A., Aracil-Fernández, A., Manzanares, J., and Maldonado, R. (2013). Role of CB1 and CB2 cannabinoid receptors in the development of joint pain induced by monosodium iodoacetate. Pain 154, 160–174. doi: 10.1016/j.pain.2012.10.009
Lee, Y., Hong, S., Cui, M., Sharma, P. K., Lee, J., and Choi, S. (2015). Transient receptor potential vanilloid type 1 antagonists: a patent review (2011 - 2014). Expert Opin. Ther. Pat. 25, 291–318. doi: 10.1517/13543776.2015.1008449
Lichtman, A. H., and Chapman, V. (2011). A FAAH-fetched approach to treat osteoarthritis pain. Pain 152, 959–960. doi: 10.1016/j.pain.2011.02.034
Lichtman, A. H., Leung, D., Shelton, C. C., Saghatelian, A., Hardouin, C., Boger, D. L., et al. (2004). Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J. Pharmacol. Exp. Ther. 311, 441–448. doi: 10.1124/jpet.104.069401
Lizanecz, E., Bagi, Z., Pásztor, E. T., Papp, Z., Edes, I., Kedei, N., et al. (2006). Phosphorylation-dependent desensitization by anandamide of vanilloid receptor-1 (TRPV1) function in rat skeletal muscle arterioles and in Chinese hamster ovary cells expressing TRPV1. Mol. Pharmacol. 69, 1015–1023. doi: 10.1124/mol.105.015644
Lomazzo, E., Bindila, L., Remmers, F., Lerner, R., Schwitter, C., Hoheisel, U., et al. (2015). Therapeutic potential of inhibitors of endocannabinoid degradation for the treatment of stress-related hyperalgesia in an animal model of chronic pain. Neuropsychopharmacology 40, 488–501. doi: 10.1038/npp.2014.198
Maione, S., Bisogno, T., De Novellis, V., Palazzo, E., Cristino, L., Valenti, M., et al. (2006). Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J. Pharmacol. Exp. Ther. 316, 969–982. doi: 10.1124/jpet.105.093286
Maione, S., Costa, B., Piscitelli, F., Morera, E., De Chiaro, M., Comelli, F., et al. (2013). Piperazinyl carbamate fatty acid amide hydrolase inhibitors and transient receptor potential channel modulators as “dual-target” analgesics. Pharmacol. Res. 76, 98–105. doi: 10.1016/j.phrs.2013.07.003
Maione, S., De Petrocellis, L., De Novellis, V., Moriello, A. S., Petrosino, S., Palazzo, E., et al. (2007). Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br. J. Pharmacol. 150, 766–781. doi: 10.1038/sj.bjp.0707145
Malek, N., Kostrzewa, M., Makuch, W., Pajak, A., Kucharczyk, M., Piscitelli, F., et al. (2016). The multiplicity of spinal AA-5-HT anti-nociceptive action in a rat model of neuropathic pain. Pharmacol. Res. 111, 251–263. doi: 10.1016/j.phrs.2016.06.012
Malek, N., Mrugala, M., Makuch, W., Kolosowska, N., Przewlocka, B., Binkowski, M., et al. (2015a). A multi-target approach for pain treatment - dual inhibition of fatty acid amide hydrolase and TRPV1 in a rat model of osteoarthritis. Pain. 156, 890–903. doi: 10.1097/j.pain.0000000000000132
Malek, N., Popiolek-Barczyk, K., Mika, J., Przewlocka, B., and Starowicz, K. (2015b). Anandamide, Acting via CB2 Receptors, Alleviates LPS-Induced Neuroinflammation in Rat Primary Microglial Cultures. Neural Plast. 2015:130639. doi: 10.1155/2015/130639
Mecha, M., Feliú, A., Carrillo-Salinas, F. J., Rueda-Zubiaurre, A., Ortega-Gutiérrez, S., De Sola, R. G., et al. (2015). Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 49, 233–245. doi: 10.1016/j.bbi.2015.06.002
Migliore, M., Habrant, D., Sasso, O., Albani, C., Bertozzi, S. M., Armirotti, A., et al. (2016). Potent multitarget FAAH-COX inhibitors: design and structure-activity relationship studies. Eur. J. Med. Chem. 109, 216–237. doi: 10.1016/j.ejmech.2015.12.036
Morera, E., De Petrocellis, L., Morera, L., Moriello, A. S., Ligresti, A., Nalli, M., et al. (2009). Synthesis and biological evaluation of piperazinyl carbamates and ureas as fatty acid amide hydrolase (FAAH) and transient receptor potential (TRP) channel dual ligands. Bioorg. Med. Chem. Lett. 19, 6806–6809. doi: 10.1016/j.bmcl.2009.09.033
Morera, E., Di Marzo, V., Monti, L., Allarà, M., Schiano Moriello, A., Nalli, M., et al. (2016). Arylboronic acids as dual-action FAAH and TRPV1 ligands. Bioorg. Med. Chem. Lett. 26, 1401–1405. doi: 10.1016/j.bmcl.2016.01.071
Naidu, P. S., Kinsey, S. G., Guo, T. L., Cravatt, B. F., and Lichtman, A. H. (2010). Regulation of inflammatory pain by inhibition of fatty acid amide hydrolase. J. Pharmacol. Exp. Ther. 334, 182–190. doi: 10.1124/jpet.109.164806
Okine, B. N., Norris, L. M., Woodhams, S., Burston, J., Patel, A., Alexander, S. P., et al. (2012). Lack of effect of chronic pre-treatment with the FAAH inhibitor URB597 on inflammatory pain behaviour: evidence for plastic changes in the endocannabinoid system. Br. J. Pharmacol. 167, 627–640. doi: 10.1111/j.1476-5381.2012.02028.x
Ortar, G., Cascio, M. G., De Petrocellis, L., Morera, E., Rossi, F., Schiano-Moriello, A., et al. (2007). New N-arachidonoylserotonin analogues with potential “dual” mechanism of action against pain. J. Med. Chem. 50, 6554–6569. doi: 10.1021/jm070678q
Ortar, G., De Petrocellis, L., Moriello, A. S., Allarà, M., Morera, E., Nalli, M., et al. (2013). Tetrahydro-β-carboline derivatives targeting fatty acid amide hydrolase (FAAH) and transient receptor potential (TRP) channels. Bioorg. Med. Chem. Lett. 23, 138–142. doi: 10.1016/j.bmcl.2012.10.137
Paylor, B., Holt, S., and Fowler, C. J. (2006). The potency of the fatty acid amide hydrolase inhibitor URB597 is dependent upon the assay pH. Pharmacol. Res. 54, 481–485. doi: 10.1016/j.phrs.2006.07.006
Pernía-Andrade, A. J., Kato, A., Witschi, R., Nyilas, R., Katona, I., Freund, T. F., et al. (2009). Spinal endocannabinoids and CB1 receptors mediate C-fiber-induced heterosynaptic pain sensitization. Science 325, 760–764. doi: 10.1126/science.1171870
Piomelli, D., and Sasso, O. (2014). Peripheral gating of pain signals by endogenous lipid mediators. Nat. Neurosci. 17:1287. doi: 10.1038/nn0914-1287d
Piscitelli, F., and Di Marzo, V. (2012). “Redundancy” of endocannabinoid inactivation: new challenges and opportunities for pain control. ACS Chem. Neurosci. 3, 356–363. doi: 10.1021/cn300015x
Rahme, E., and Bernatsky, S. (2010). NSAIDs and risk of lower gastrointestinal bleeding. Lancet 376, 146–148. doi: 10.1016/S0140-6736(10)60839-2
Ray, W. A., Chung, C. P., Stein, C. M., Smalley, W. E., Hall, K., Arbogast, P. G., et al. (2007). Risk of peptic ulcer hospitalizations in users of NSAIDs with gastroprotective cotherapy versus coxibs. Gastroenterology 133, 790–798. doi: 10.1053/j.gastro.2007.06.058
Richardson, J. D., Kilo, S., and Hargreaves, K. M. (1998). Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain 75, 111–119. doi: 10.1016/S0304-3959(97)00213-3
Roberts, J. C., Davis, J. B., and Benham, C. D. (2004). [3H]Resiniferatoxin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution. Brain Res. 995, 176–183. doi: 10.1016/j.brainres.2003.10.001
Sasso, O., Migliore, M., Habrant, D., Armirotti, A., Albani, C., Summa, M., et al. (2015). Multitarget fatty acid amide hydrolase/cyclooxygenase blockade suppresses intestinal inflammation and protects against nonsteroidal anti-inflammatory drug-dependent gastrointestinal damage. FASEB J. 29, 2616–2627. doi: 10.1096/fj.15-270637
Seidel, K., Hamza, M., Ates, M., and Gühring, H. (2003). Flurbiprofen inhibits capsaicin induced calcitonin gene related peptide release from rat spinal cord via an endocannabinoid dependent mechanism. Neurosci. Lett. 338, 99–102. doi: 10.1016/S0304-3940(02)01366-6
Singh Tahim, A., Sántha, P., and Nagy, I. (2005). Inflammatory mediators convert anandamide into a potent activator of the vanilloid type 1 transient receptor potential receptor in nociceptive primary sensory neurons. Neuroscience 136, 539–548. doi: 10.1016/j.neuroscience.2005.08.005
Smith, W. L., Garavito, R. M., and DeWitt, D. L. (1996). Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 271, 33157–33160. doi: 10.1074/jbc.271.52.33157
Stahl, S. M. (2009). Multifunctional drugs: a novel concept for psychopharmacology. CNS Spectr. 14, 71–73. doi: 10.1017/S1092852900000213
Starowicz, K., and Di Marzo, V. (2013). Non-psychotropic analgesic drugs from the endocannabinoid system: “magic bullet” or “multiple-target” strategies? Eur. J. Pharmacol. 716, 41–53. doi: 10.1016/j.ejphar.2013.01.075
Starowicz, K., Maione, S., Cristino, L., Palazzo, E., Marabese, I., Rossi, F., et al. (2007). Tonic endovanilloid facilitation of glutamate release in brainstem descending antinociceptive pathways. J. Neurosci. 27, 13739–13749. doi: 10.1523/JNEUROSCI.3258-07.2007
Starowicz, K., Makuch, W., Korostynski, M., Malek, N., Slezak, M., Zychowska, M., et al. (2013). Full inhibition of spinal FAAH leads to TRPV1-mediated analgesic effects in neuropathic rats and possible lipoxygenase-mediated remodeling of anandamide metabolism. PLoS ONE 8:e60040. doi: 10.1371/journal.pone.0060040
Swanson, D. M., Dubin, A. E., Shah, C., Nasser, N., Chang, L., Dax, S. L., et al. (2005). Identification and biological evaluation of 4-(3-trifluoromethylpyridin-2-yl)piperazine-1-carboxylic acid (5-trifluoromethylpyridin-2-yl)amide, a high affinity TRPV1 (VR1) vanilloid receptor antagonist. J. Med. Chem. 48, 1857–1872. doi: 10.1021/jm0495071
Thaler, A., Gupta, A., and Cohen, S. P. (2011). Cannabinoids for pain management. Adv. Psychosom. Med. 30, 125–138. doi: 10.1159/000324070
Toth, C. C., Jedrzejewski, N. M., Ellis, C. L., and Frey, W. H. II. (2010). Cannabinoid-mediated modulation of neuropathic pain and microglial accumulation in a model of murine type I diabetic peripheral neuropathic pain. Mol. Pain 6:16. doi: 10.1186/1744-8069-6-16
Ulugöl, A., Ozyigit, F., Yesilyurt, O., and Dogrul, A. (2006). The additive antinociceptive interaction between WIN 55,212-2, a cannabinoid agonist, and ketorolac. Anesth. Analg. 102, 443–447. doi: 10.1213/01.ane.0000194587.94260.1d
Van der Stelt, M., Trevisani, M., Vellani, V., De Petrocellis, L., Schiano Moriello, A., Campi, B., et al. (2005). Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J. 24, 3026–3037. doi: 10.1038/sj.emboj.7600784
Williams, J. T., Ingram, S. L., Henderson, G., Chavkin, C., Von Zastrow, M., Schulz, S., et al. (2013). Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 65, 223–254. doi: 10.1124/pr.112.005942
Yang, W., Ni, J., Woodward, D. F., Tang-Liu, D. D.-S., and Ling, K.-H. J. (2005). Enzymatic formation of prostamide F2alpha from anandamide involves a newly identified intermediate metabolite, prostamide H2. J. Lipid Res. 46, 2745–2751. doi: 10.1194/jlr.M500374-JLR200
Keywords: FAAH, TRPV1, COXs, endocannabinoid system, dual acting compounds, pain
Citation: Malek N and Starowicz K (2016) Dual-Acting Compounds Targeting Endocannabinoid and Endovanilloid Systems—A Novel Treatment Option for Chronic Pain Management. Front. Pharmacol. 7:257. doi: 10.3389/fphar.2016.00257
Received: 16 May 2016; Accepted: 02 August 2016;
Published: 17 August 2016.
Edited by:
Allyn C. Howlett, Wake Forest School of Medicine, USAReviewed by:
Roberto Coccurello, National Research Council, ItalyGaurav Bedse, Vanderbilt University, USA
Copyright © 2016 Malek and Starowicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katarzyna Starowicz, starow@if-pan.krakow.pl