 Lesley D. Schurman
Lesley D. Schurman Aron H. Lichtman
Aron H. Lichtman- Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, VA, USA
The endogenous cannabinoid (endocannabinoid) system regulates a diverse array of physiological processes and unsurprisingly possesses considerable potential targets for the potential treatment of numerous disease states, including two receptors (i.e., CB1 and CB2 receptors) and enzymes regulating their endogenous ligands N-arachidonoylethanolamine (anandamide) and 2-arachidonyl glycerol (2-AG). Increases in brain levels of endocannabinoids to pathogenic events suggest this system plays a role in compensatory repair mechanisms. Traumatic brain injury (TBI) pathology remains mostly refractory to currently available drugs, perhaps due to its heterogeneous nature in etiology, clinical presentation, and severity. Here, we review pre-clinical studies assessing the therapeutic potential of cannabinoids and manipulations of the endocannabinoid system to ameliorate TBI pathology. Specifically, manipulations of endocannabinoid degradative enzymes (e.g., fatty acid amide hydrolase, monoacylglycerol lipase, and α/β-hydrolase domain-6), CB1 and CB2 receptors, and their endogenous ligands have shown promise in modulating cellular and molecular hallmarks of TBI pathology such as; cell death, excitotoxicity, neuroinflammation, cerebrovascular breakdown, and cell structure and remodeling. TBI-induced behavioral deficits, such as learning and memory, neurological motor impairments, post-traumatic convulsions or seizures, and anxiety also respond to manipulations of the endocannabinoid system. As such, the endocannabinoid system possesses potential drugable receptor and enzyme targets for the treatment of diverse TBI pathology. Yet, full characterization of TBI-induced changes in endocannabinoid ligands, enzymes, and receptor populations will be important to understand that role this system plays in TBI pathology. Promising classes of compounds, such as the plant-derived phytocannabinoids, synthetic cannabinoids, and endocannabinoids, as well as their non-cannabinoid receptor targets, such as TRPV1 receptors, represent important areas of basic research and potential therapeutic interest to treat TBI.
Introduction
Traumatic brain injury accounts for approximately 10 million deaths and/or hospitalizations annually in the world, and approximately 1.5 million annual emergency room visits and hospitalizations in the US (Langlois et al., 2006). Young men are consistently over-represented as being at greatest risk for TBI (Langlois et al., 2006). While half of all traumatic deaths in the USA are due to brain injury (Mayer and Badjatia, 2010), the majority of head injuries are considered mild and often never receive medical treatment (Corrigan et al., 2010). Survivors of TBI are at risk for lowered life expectancy, dying at a 3⋅2 times more rapid rate than the general population (Baguley et al., 2012). Survivors also face long term physical, cognitive, and psychological disorders that greatly diminish quality of life. Even so-called mild TBI without notable cell death may lead to enduring cognitive deficits (Niogi et al., 2008; Rubovitch et al., 2011). A 2007 study estimated that TBI results in $330,827 of average lifetime costs associated with disability and lost productivity, and greatly outweighs the $65,504 estimated costs for initial medical care and rehabilitation (Faul et al., 2007), demonstrating both the long term financial and human toll of TBI.
The development of management protocols in major trauma centers (Brain Trauma Foundation et al., 2007) has improved mortality and functional outcomes (Stein et al., 2010). Monitoring of intracranial pressure is now standard practice (Bratton et al., 2007), and advanced MRI technologies help define the extent of brain injury in some cases (Shah et al., 2012). Current treatment of major TBI is primarily managed through surgical intervention by decompressive craniotomy (Bullock et al., 2006) which involves the removal of skull segments to reduce intracranial pressure. Delayed decompressive craniotomy is also increasingly used for intractable intracranial hypertension (Sahuquillo and Arikan, 2006). The craniotomy procedure is associated with considerable complications, such as hematoma, subdural hygroma, and hydrocephalus (Stiver, 2009). At present, the pathology associated with TBI remains refractive to currently available pharmacotherapies (Meyer et al., 2010) and as such represents an area of great research interest and in need of new potential targets. Effective TBI drug therapies have yet to be proven, despite promising preclinical data (Lu et al., 2007; Mbye et al., 2009; Sen and Gulati, 2010) plagued by translational problems once reaching clinical trials (Temkin et al., 2007; Tapia-Perez et al., 2008; Mazzeo et al., 2009).
The many biochemical events that occur in the hours and months following TBI have yielded preclinical studies directed toward a single injury mechanism. However, an underlying premise of the present review is an important need to address the multiple targets associated with secondary injury cascades following TBI. A growing body of published scientific research indicates that the endogenous cannabinoid (endocannabinoid; eCB) system possesses several targets uniquely positioned to modulate several key secondary events associated with TBI. Here, we review the preclinical work examining the roles that the different components of the eCB system play in ameliorating pathologies associated with TBI.
The Endocannabinoid (eCB) System
Originally, “Cannabinoid” was the collective name assigned to the set of naturally occurring aromatic hydrocarbon compounds in the Cannabis sativa plant (Mechoulam and Goani, 1967). Cannabinoid now more generally refers to a much more broad set of chemicals of diverse structure whose pharmacological actions or structure closely mimic that of plant-derived cannabinoids. Three predominant categories are currently in use; plant-derived phytocannabinoids (reviewed in Gertsch et al., 2010), synthetically produced cannabinoids used as research (Wiley et al., 2014) or recreational drugs (Mills et al., 2015), and the endogenous cannabinoids, N-arachidonoylethanolamine (anandamide) (Devane et al., 1992) and 2-AG (Mechoulam et al., 1995; Sugiura et al., 1995).
These three broad categories of cannabinoids generally act through cannabinoid receptors, two types of which have so far been identified, CB1 (Devane et al., 1988) and CB2 (Munro et al., 1993). Both CB1 and CB2 receptors are coupled to signaling cascades predominantly through Gi/o-coupled proteins. CB1 receptors mediate most of the psychomimetic effects of cannabis, its chief psychoactive constituent THC, and many other CNS active cannabinoids. These receptors are predominantly expressed on pre-synaptic axon terminals (Alger and Kim, 2011), are activated by endogenous cannabinoids that function as retrograde messengers, which are released from post-synaptic cells, and their activation ultimately dampens pre-synaptic neurotransmitter release (Mackie, 2006). Acting as a neuromodulatory network, the outcome of cannabinoid receptor signaling depends on cell type and location. CB1 receptors are highly expressed on neurons in the central nervous system (CNS) in areas such as cerebral cortex, hippocampus, caudate-putamen (Herkenham et al., 1991). In contrast, CB2 receptors are predominantly expressed on immune cells, microglia in the CNS, and macrophages, monocytes, CD4+ and CD8+ T cells, and B cells in the periphery (Cabral et al., 2008). Additionally, CB2 receptors are expressed on neurons, but to a much less extent than CB1 receptors (Atwood and MacKie, 2010). The abundant, yet heterogeneous, distribution of CB1 and CB2 receptors throughout the brain and periphery likely accounts for their ability to impact a wide variety of physiological and psychological processes (e.g., memory, anxiety, and pain perception, reviewed in Di Marzo, 2008) many of which are impacted following TBI.
Another unique property of the eCB system is the functional selectivity produced by its endogenous ligands. Traditional neurotransmitter systems elicit differential activation of signaling pathways through activation of receptor subtypes by one neurotransmitter (Siegel, 1999). However, it is the endogenous ligands of eCB receptors which produce such signaling specificity. Although several endogenous cannabinoids have been described (Porter et al., 2002; Chu et al., 2003; Heimann et al., 2007) the two most studied are anandamide (Devane et al., 1992) and 2-AG (Mechoulam et al., 1995; Sugiura et al., 1995). 2-AG levels are three orders of magnitude higher than those of anandamide in brain (Béquet et al., 2007). Additionally, their receptor affinity (Pertwee and Ross, 2002; Reggio, 2002) and efficacy differ, with 2-AG acting as a high efficacy agonist at CB1 and CB2 receptors, while anandamide behaves as a partial agonist (Hillard, 2000a). In addition, anandamide binds and activates TRPV1 receptors (Melck et al., 1999; Zygmunt et al., 1999; Smart et al., 2000), whereas 2-AG also binds GABAA receptors (Sigel et al., 2011). As such, cannabinoid ligands differentially modulate similar physiological and pathological processes.
Distinct sets of enzymes, which regulate the biosynthesis and degradation of the eCBs and possess distinct anatomical distributions (see Figure 1), exert control over CB1 and CB2 receptor signaling. Inactivation of anandamide occurs predominantly through FAAH (Cravatt et al., 1996, 2001), localized to intracellular membranes of postsynaptic somata and dendrites (Gulyas et al., 2004), in areas such as the neocortex, cerebellar cortex, and hippocampus (Egertová et al., 1998). Inactivation of 2-AG proceeds primarily via MAGL (Dinh et al., 2002; Blankman et al., 2007), expressed on presynaptic axon terminals (Gulyas et al., 2004), and demonstrates highest expression in areas such as the thalamus, hippocampus, cortex, and cerebellum (Dinh et al., 2002). The availability of pharmacological inhibitors for eCB catabolic enzymes has allowed the selective amplification of anandamide and 2-AG levels following brain injury as a key strategy to enhance eCB signaling and to investigate their potential neuroprotective effects.
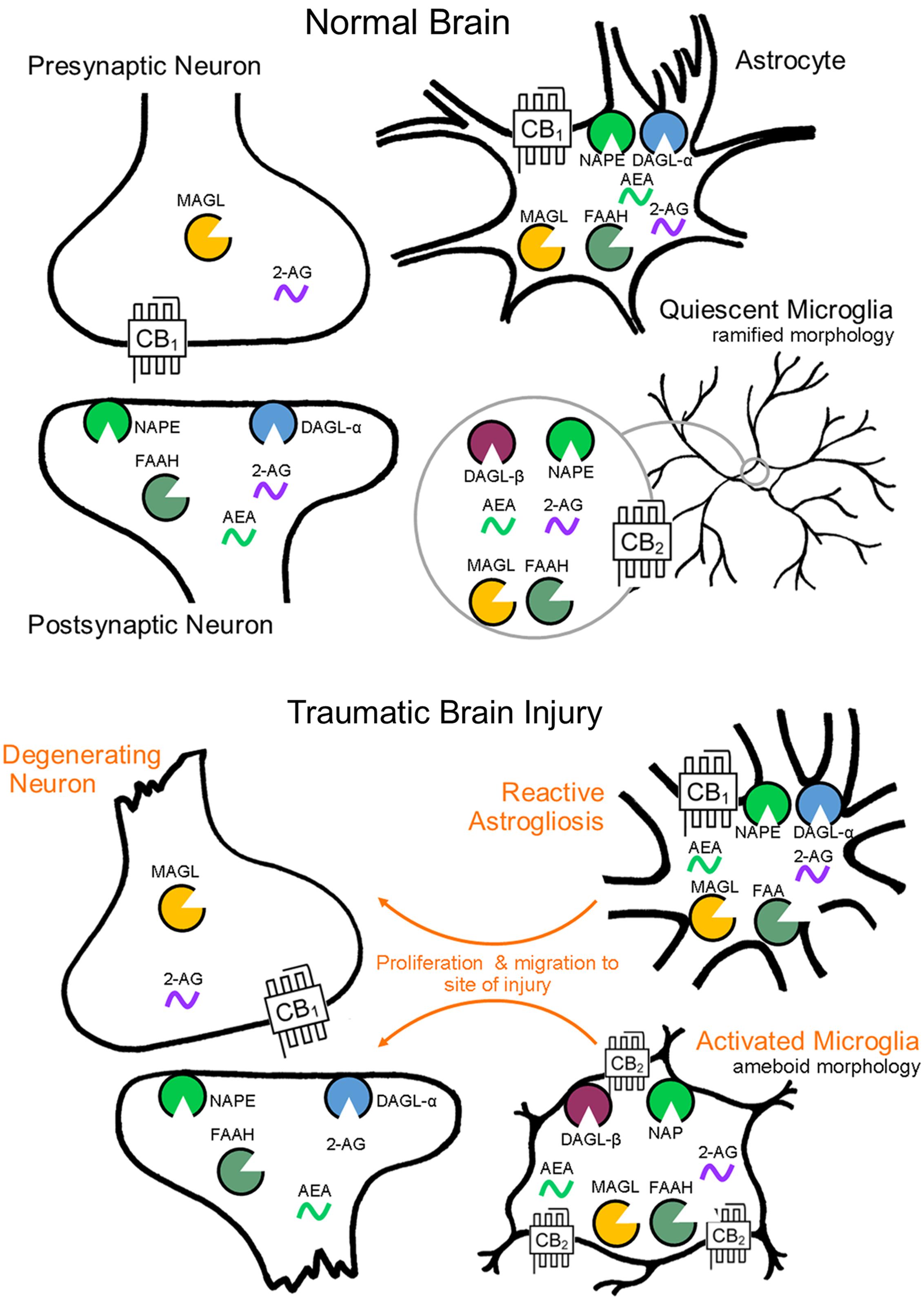
FIGURE 1. Endocannabinoid system cell localization by CNS cell type. Endocannabinoid functional specialization among CNS cell types is determined by the cellular compartmentalization of biosynthetic and catabolic enzymes (biosynthesis by NAPE and DAGL-α, -β, catabolism by FAAH and MAGL). Cellular level changes in eCB biosynthetic and catabolic enzymes as a result of brain injury have yet to be investigated, though morphological and molecular reactivity by cell type is well documented.
Finally, 2-AG functions not only as a major cannabinoid receptor signaling molecule, but also serves as a major precursor for AA, and therefore plays a role in inflammatory pathways (see Figure 2). Although AA is a degradative product of both 2-AG (Bell et al., 1979) and anandamide (Deutsch et al., 1997), MAGL represents a rate-limiting biosynthetic enzyme of highly bioactive lipid in brain, liver, and lung (Nomura et al., 2011). Historically, cPLA2 was considered to be the primary rate-limiting enzyme in AA production (reviewed in Buczynski et al., 2009). However, MAGL contributes ∼80% and cPLA2 ∼20% of LPS-stimulated eicosanoids in mouse brain. In contrast, cPLA2 is the dominant enzyme to control AA production in spleen (Nomura et al., 2011). Therefore, MAGL and cPLA2 appear to play differential roles in AA production, and concomitantly its eicosanoid metabolites in a tissue-specific manner (Nomura et al., 2011). As such, 2-AG functions not only as an endogenous CB1 and CB2 receptor ligand, but also an immunomodulator by virtue of its being a major precursor for AA, making it a versatile target for the treatment of TBI related pro-inflammatory pathologies. Understanding the biosynthesis mechanisms of eCBs may prove useful in modulating their entry into pro-inflammatory pathways. While 2-AG is known to be synthesized by DAGL-α and DAGL-β (Bisogno et al., 2003), the mechanisms mediating anandamide production are incompletely understood (Blankman and Cravatt, 2013).
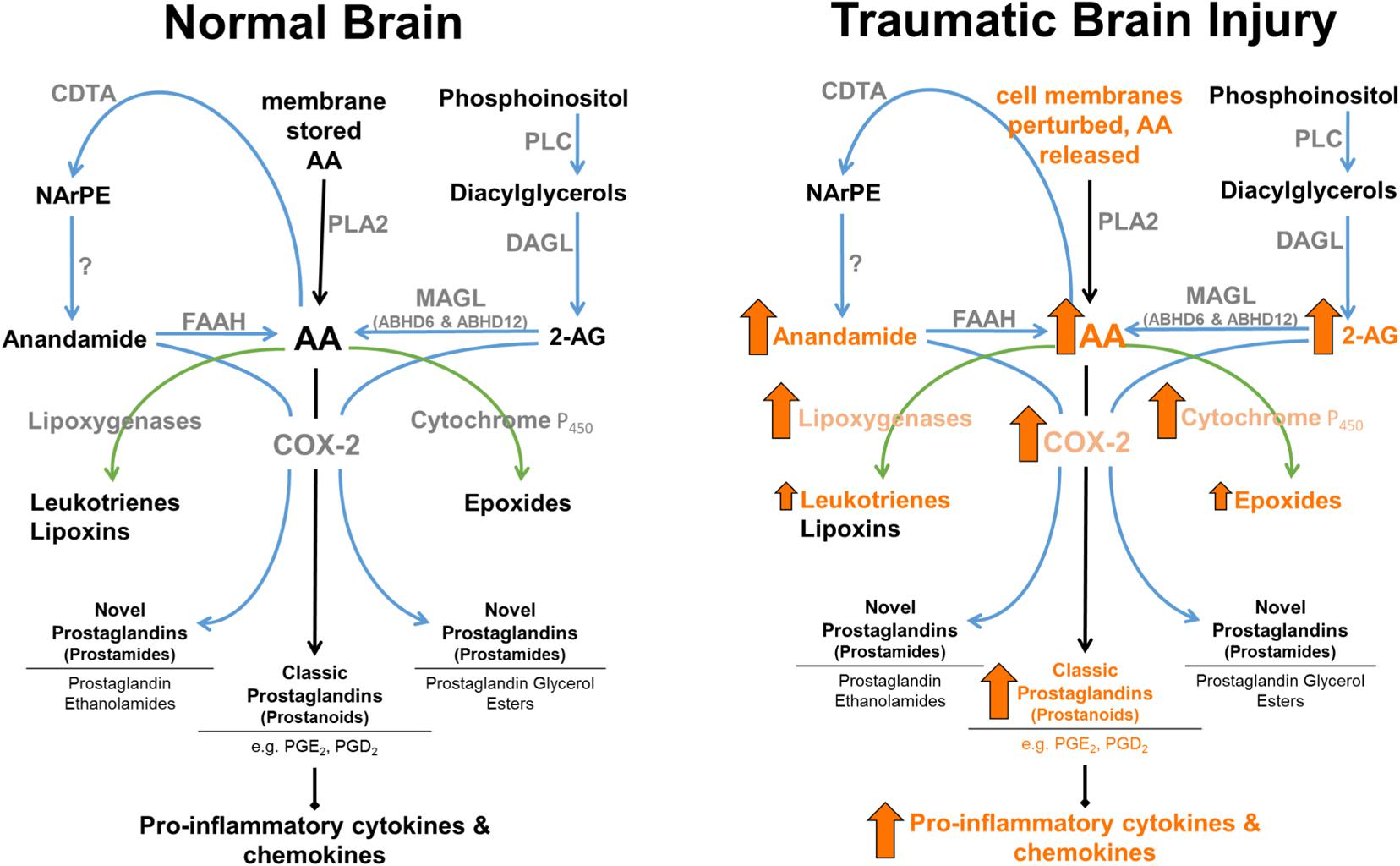
FIGURE 2. Enzymatic regulation of anandamide and 2-AG in normal brain, and following TBI. 2-AG levels are approximately 1,000-fold higher than anandamide levels in brain. MAGL plays a rate-limited role in the production of AA in brain, lung, and liver (Nomura et al., 2008). Arrows represent known TBI-induced changes in eCBs, catabolic and downstream enzymes, and their metabolic products (arrow size has no relation to magnitude of change).
Traumatic Brain Injury Pathology
Traumatic brain injuries are heterogeneous in their etiology, clinical presentation, severity, and pathology. The sequelae of molecular, biochemical, and physiological events that follow the application of an external mechanical force produce interacting acute and delayed pathologies, described as primary and secondary injuries. The initial insult produces an immediate mechanical disruption of brain tissue (Reilly, 2001). This primary injury consists of contusion, blood vessel disruption and brain oedema, localized necrotic cell death, as well as diffuse axonal injury producing degeneration of cerebral white matter (Adams et al., 1989; Gaetz, 2004).
Secondary injury mechanisms are initiated within minutes, in which necrotic and apoptotic cell death in contused areas and pericontusional penumbra continue over a period of days to months (Raghupathi, 2004). Neuronal disruption spills excitatory amino acids into the interstitial space, producing glutamate-mediated excitotoxicity (Bullock et al., 1998). Massive influx of Ca2+ into cells (Floyd et al., 2010) produce mitochondrial dysfunction and the release of ROS which lead to further apoptosis (Zhao et al., 2005). Injury-induced activation of CNS resident glial cells, microglia, as well as recruitment of circulating inflammatory cells, e.g., macrophages, then produce secretion of inflammatory mediators, cytokines and chemokines (reviewed in Woodcock and Morganti-Kossmann, 2013). Increased intracranial pressure leads to reductions in cerebral blood flow (Shiina et al., 1998), while injury-induced breakdown of the cerebrovascular endothelium contributes to dysfunction of the BBB (Chodobski et al., 2012). Extracranial pathologies are also evident following TBI with pulmonary complications being the most common (Pelosi et al., 2005). NPE often develops early after brain injury, producing hypoxemia and further aggravating secondary brain injury (Brambrink and Dick, 1997; Oddo et al., 2010). These varied and interacting disease processes highlight the necessity to address the multiple targets associated with secondary injury cascades following TBI.
While there are many types of CNS injury models [e.g., spinal cord injury, lesion studies, focal and global ischemic injury etc. (Arai and Lo, 2009; Titomanlio et al., 2015)], this review will focus primarily on the work investigating manipulations of the eCB system in preclinical models of TBI.
Pre-Clinical Evaluation of Cannabinoids to Treat TBI
While basal anandamide and 2-AG levels differ within various structures in the CNS, levels increase on demand in response to a given stimuli [e.g., the induction of nausea (Sticht et al., 2016) or pain states (Costa et al., 2008)]. eCBs are lipid messengers not stored in synaptic vesicles (likely due to their hydrophobicity) but rather synthesized in an activity-dependent manner from membrane phospholipid precursors (Alger and Kim, 2011). Consequently, eCB signaling is enhanced by a stimulus-response synthesis and release mechanism.
Endocannabinoid levels increase in selected CNS tissue following neuronal damage, which may reflect a self-neuroprotective response. NMDA excitotoxicity produces elevations of anandamide in ipsilateral cortex of rats by 4-fold at 4 h and 14-fold at 24 h, but with no changes in 2-AG levels (Hansen et al., 2002). Concussive head trauma in rats produces a similar pattern of findings in which modest increases of anandamide levels occur in ipsilateral cortex, and again with no change in 2-AG levels (Hansen et al., 2002). This pattern was replicated by Tchantchou et al. (2014), who found a 1.5-fold increase of anandamide levels at 3 days post-TBI in ipsilateral mouse brain, and with no change in 2-AG. In contrast, Panikashvili et al. (2001) reported that TBI in mice led to increases of 2-AG in ipsilateral brain from 1 to 24 h with elevations as high as 10-fold. Thus, further research is needed to discern whether species differences, the model used to elicit neurotrauma, and/or other procedural considerations contribute to the differential elevation of these eCBs (Mechoulam and Lichtman, 2003).
A lack of studies systematically investigating the consequences of TBI on changes in eCB levels in specific brain regions perhaps point to the difficulty in measuring changes in the volatile eCBs, prone to rapid degradation (Deutsch and Chin, 1993; Dinh et al., 2002). While pharmacological and genetic manipulations of the eCB system continue to be evaluated following TBI; full characterization of how eCB biosynthetic and degradative enzymes, receptors, and endogenous ligands, their precursors and catabolic products, change as a consequence of TBI remains to be fully illuminated.
Treatment of Cellular and Molecular Pathophysiology of TBI
In this section, we review preclinical studies of cannabinoids in the context of their potential to protect against cellular and molecular TBI pathology (see Table 1).
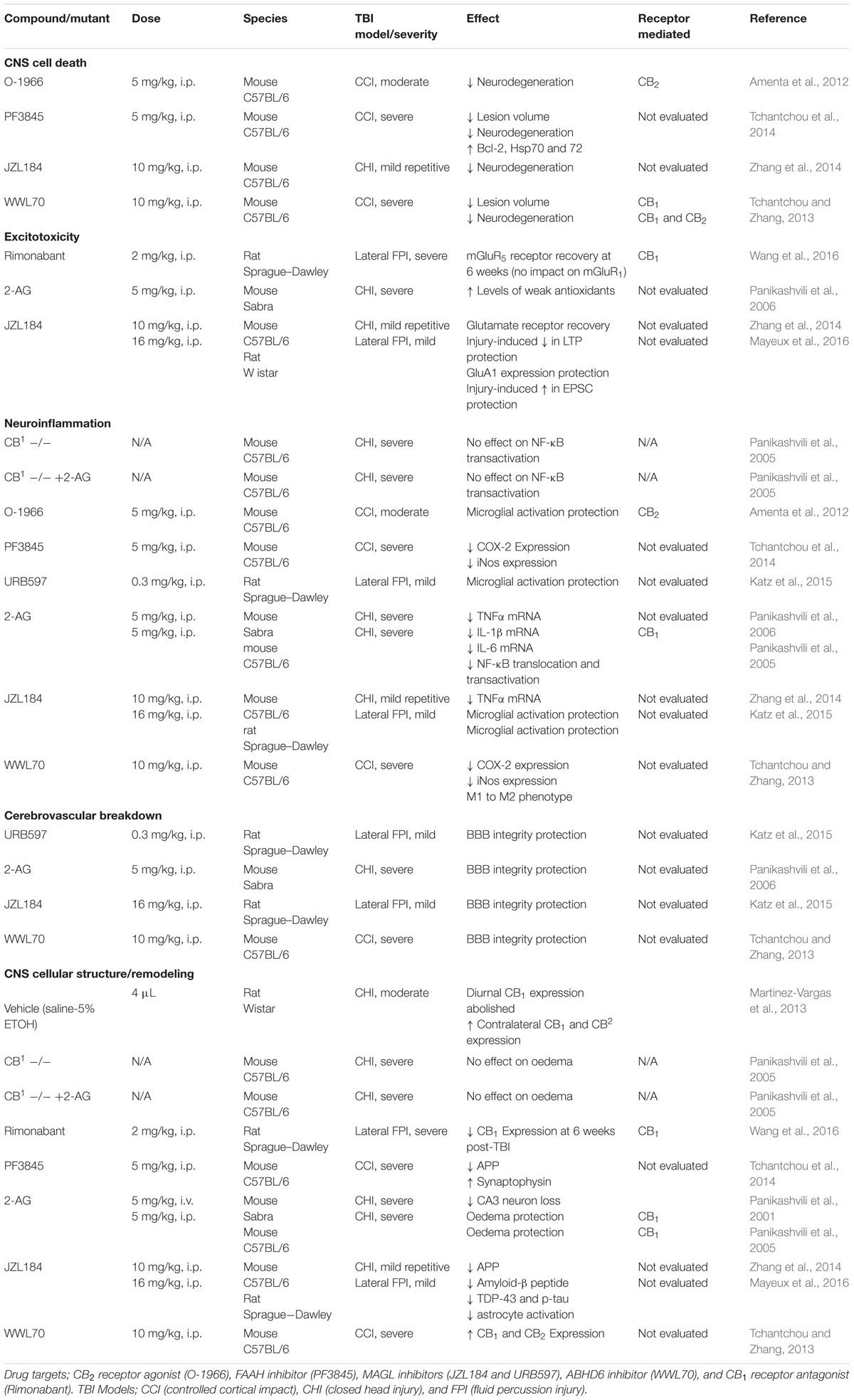
TABLE 1. Effect of cannabinoids on TBI-induced cellular and molecular pathophysiology.
CNS Cell Death
Traumatic brain injury-induced neuronal loss occurs almost immediately as necrotic cell death and continues for months following the initial insult via both necrotic and apoptotic cell death (Raghupathi, 2004). From a traumatic insult, the initial contused area forms a regional primary lesion or infarct surrounding which is the pericontusional penumbra, the area immediately adjacent to the primary lesion and at risk for further neurodegeneration. The evolution of the pericontusional penumbra occurs largely due to secondary injury mechanisms and has long been considered a candidate for interventions to protect against, or salvage from, further injury (Wang et al., 2014). The investigation of cannabinoids on traumatic CNS cell death have thus far demonstrated efficacy in two areas; attenuated neurodegeneration and reduced lesion volume.
Neurodegeneration, commonly measured by reductions in the neuronal marker fluoro-jade C, has been found to be readily attenuated in mice by CB2 receptor agonists (Amenta et al., 2012), as well as by inhibitors of FAAH (Tchantchou et al., 2014) and MAGL (Zhang et al., 2014). Additionally, FAAH inhibitors produce reductions in lesion volume, and increased production of the heat shock proteins Hsp70, known to be structurally protective, and Hsp72, a negative regulator of apoptosis (Tchantchou et al., 2014). Tchantchou et al. (2014) also showed that FAAH inhibition increased expression of the anti-apoptotic protein Bcl-2.
Several enzymes hydrolyze 2-AG including MAGL, which accounts for an estimated 85% of its total hydrolysis, as well as ABHD6 and ABHD12, which are responsible for much of the remaining 15% (Blankman et al., 2007). Tchantchou and Zhang (2013) found that inhibition of ABHD6 also reduced lesion volume and lowered neurodegeneration in a mouse CCI model. A CB1 receptor antagonist attenuated the protective effects on lesion volume, while CB1 and CB2 receptor antagonists prevented the protective effects on neurodegeneration (Tchantchou and Zhang, 2013).
Combined, this evidence suggests that inhibitors of eCB hydrolysis offer protection against TBI-induced cell death which involve CB1 and CB2 receptors, though the distinction between the eCBs remains to be clarified. Few studies have evaluated interactions between anandamide and 2-AG in laboratory models of TBI. One study using a model of cerebral focal ischemia found that exogenously administered anandamide and 2-AG in combination reduced infarct size in rats, but with no facilitatory effects beyond anandamide or 2-AG alone (Wang et al., 2009). Given the recent availability of dual FAAH/MAGL inhibitors (Long et al., 2009; Niphakis et al., 2012), simultaneous blockade of these enzymes following TBI may further reveal some insight into the relationship between anandamide and 2-AG on TBI-induced cell death.
Excitotoxicity
Previous efforts to attenuate the effects of excitotoxicity following brain injury focused on NMDA receptor antagonists, presumably with the understanding that the induction of depressed NMDA receptor function would counteract TBI-induced excitotoxicity. This class of drugs showed promise in laboratory animal models of TBI (Shohami et al., 1995), but failed to produce long-term beneficial outcomes in clinical trials, despite some acute benefits of improved intracranial pressure and cerebral perfusion pressure (Knoller et al., 2002; Maas et al., 2006). Research investigating manipulations of the eCB system on glutamatergic functioning following TBI have thus far focused primarily on 2-AG, and paradoxically, its effectiveness to protect the integrity of glutamate receptor function.
Several studies investigating the effects of cannabinoids in laboratory animal models of TBI have focused on expression changes of metabotropic (mGluRbbb, mGluR5), AMPA (GluA1, GluA2), and NMDA (GluN1, GluN2A, GluN2B) glutamatergic receptors. Specifically, post-injury administration of the MAGL inhibitor JZL184 reversed TBI-induced reductions of GluN2A, GluN2B, and GluA1 receptor expression, but with no impact on GluN1 or GluA2 receptors (Zhang et al., 2014). The CB1 receptor antagonist Rimonabant did not alter injury-induced lowered expression of mGluRbbb, but surprisingly reversed reduced mGluR5 receptor expression 6 weeks following TBI (Wang et al., 2016). Both findings were completed 30 days post injury (Zhang et al., 2014; Wang et al., 2016), suggesting long term changes in glutamatergic function following acute administration of cannabinoids post-injury. However, little overlap is found between receptor expression endpoints across papers. In an example of contradictory patterns of GluA1 expression after injury, GluA1 expression was reduced in a study that subjected mice to a daily mild CHI on three consecutive days (Zhang et al., 2014), and was increased in rats subjected to a single lateral fluid percussion brain injury (Mayeux et al., 2016). In these studies, MAGL inhibition ameliorated both the reduced (Zhang et al., 2014) and increased (Mayeux et al., 2016) GluA1 expression. As discussed above (see Pre-Clinical Evaluation of Cannabinoids to Treat TBI), systematic investigation of species (mice vs. rat), brain injury model, number of injuries, and other experimental variables are needed to understand the consequences of brain injury on glutamate receptor changes.
Endocannabinoids are known to depress glutamate release from pre-synaptic terminals, and in particular, 2-AG has been explored in its ability to influence the functioning of electrochemical neurotransmission. MAGL inhibition has been found to protect against injury-induced increases in frequency and amplitude of EPSC in pyramidal neurons at the site of injury (Mayeux et al., 2016), which may suggest changes in pre-synaptic transmitter release or post-synaptic strength (Zhang et al., 2005). MAGL inhibition has also protected against injury-induced LTP impairments at hippocampal CA3–CA1 synapses (Zhang et al., 2014), implicating the restoration of glutamate receptor function in protection against TBI-induced memory impairments.
Finally, the excitotoxicity resulting from TBI is part of the sequelae of events that lead to release of damaging ROS. Antioxidants are known to prevent oxidation of free radicals and thus protects against the cellular damage in response to sudden ROS elevation. eCBs have been linked to the neuroprotective production of antioxidants as the administration of exogenous 2-AG following injury has been found to increase levels of antioxidants (Panikashvili et al., 2006).
Combined, these data suggest that MAGL represents a promising target to reduce the damaging effects of injury-induced excitotoxicity through complimentary molecular pathways.
Neuroinflammation
Hydrolytic enzymes of anandamide and 2-AG produce a shared metabolic product in the formation of free AA, the major substrate of the biosynthetic enzymes of pro-inflammatory eicosanoids (Nomura et al., 2011). Therefore, eCB oxidation not only produces inactivation at cannabinoid receptors, but also leads to the production of bioactive lipids involved in inflammatory responses during the early stages of injury. Manipulations of the eCB system have proved effective in downregulating inflammation in many experimental models, such as inflammatory pain (Ahn et al., 2009), and multiple sclerosis (Mestre et al., 2005). The use of cannabinoids following TBI have thus far been linked to two predominant features of inflammation; decreased inflammatory cell activation, and decreases in pro-inflammatory cytokine production.
Pro-inflammatory activated microglia are known to exacerbate TBI-induced neuroinflammation (Kigerl et al., 2009). Thus, decreasing TBI-inductions of inflammatory cell activation is an attractive treatment strategy. MAGL inhibition protects against TBI-induced microglial activation (Zhang et al., 2014; Katz et al., 2015), while ABHD6 inhibition promotes microglia/macrophage shift from a pro-inflammatory M1 to an anti-inflammatory M2 phenotype (Tchantchou and Zhang, 2013). A parsimonious explanation for these findings is that prevention of 2-AG hydrolysis leads to reduced levels of AA and concomitant reductions of pro-inflammatory mediators. Given the contribution of 2-AG catabolism to eicosanoid production, it is unsurprising that several studies have reported eCBs as demonstrating pro-inflammatory roles, some examples of which include models of nephropathy (Mukhopadhyay et al., 2010a), cardiomyopathy (Mukhopadhyay et al., 2010b), and experimental dermatitis (Oka et al., 2006). Most of such pro-inflammatory effects are attributed to 2-AG and not anandamide, likely due to its considerable abundance over anandamide. However, FAAH inhibition, similarly has been found to protect against TBI-induced microglial activation (Katz et al., 2015), as too has activation of CB2 receptors (Amenta et al., 2012). Thus, a need exists to disentangle the potential contributions of 2-AG to pro-inflammatory processes from its role as a substrate for AA production, versus anti-inflammatory effects through cannabinoid receptors, following TBI.
Inhibition of eCB degradative enzymes has also produced decreases in TBI-induced pro-inflammatory mediators. Reductions in the expression of inducible enzymes that trigger eicosanoid production following brain injury, COX-2 enzyme (which converts free AA to prostaglandins) and iNos (which produces the free radical nitric oxide in response to cytokine signaling), are seen in response to ABHD6 inhibition (Tchantchou and Zhang, 2013) and FAAH inhibition (Tchantchou et al., 2014). Reductions in TBI-induced pro-inflammatory cytokine mRNA (Il-1β, TNFα, and IL-6) have also been found following treatment with exogenous 2-AG (Panikashvili et al., 2006). These findings seem counter-intuitive given the possibility of the rapid oxidation of 2-AG and its consequent contribution to eicosanoid production. However, exogenous 2-AG has also been shown to ameliorate TBI-induced transactivation of the nuclear factor NF-kB (linked to cytokine production) in wild type mice, but not in CB1 knockout mice, suggesting that CB1 receptors mediate the protective effects of exogenous 2-AG (Panikashvili et al., 2005).
Cerebrovascular Breakdown
The blood vessels which carry oxygen rich blood to the brain are lined by endothelial cells as well as astrocytes. These cells, combined with specific transport proteins and enzymes, strictly regulate movement between the general circulation and CNS extracellular fluid, and are collectively known as the BBB. TBI has been well documented in producing cerebral blood flow pathology (Kelly et al., 1997) as well as interfering with BBB integrity (Baskaya et al., 1997). Given that cannabinoids are known to exert vascular effects, producing vasodilation as well as hypotension (reviewed in Hillard, 2000b), their manipulation may hold promise as protectants against cerebrovascular damage. Below, we review studies examining the effects of cannabinoids on TBI-induced disruption of BBB integrity.
Exogenous administration of 2-AG (Panikashvili et al., 2006), as well as MAGL inhibition (Katz et al., 2015), and ABHD6 inhibition (Tchantchou and Zhang, 2013) administered post-injury protect against BBB breakdown. However, Panikashvili et al., 2006 found that the expression of proteolytic enzymes implicated in BBB breakdown were unaffected by exogenous 2-AG post-injury. These enzymes include matrix metallopeptidase-9 (MMP9) involved in extracellular matrix degradation, and tumor necrosis factor-α-converting enzyme (TACE), which cleaves membrane-bound proteins. The mechanism by which 2-AG acts as a protectant of BBB integrity following traumatic insult is yet to be resolved.
One study found that post-surgery administration of a FAAH inhibitor protected against BBB breakdown (Katz et al., 2015), suggesting that anandamide and/or other substrates of this enzyme play a protective role. While the mechanism underlying the structural protection of the BBB was not explored following TBI, anandamide has been found to decrease BBB permeability in a model of ischaemic stroke by transient receptor potential cation channel, subfamily V, member 1 (TRPV1) (Hind et al., 2015). Given that activation of TRPV1 receptors disrupts BBB integrity (Hu et al., 2005), it is possible that anandamide, as a partial agonist at TRPV1 channels (Pertwee and Ross, 2002), maybe be acting as a functional antagonist against a high efficacy endogenous agonist to produce its structurally protective effects of the cerebral microvascular endothelium. The exploration of how anandamide may be exerting its protective effects of BBB integrity may yet yield further novel targets for the treatment of TBI.
In cerebral circulation, CB1 receptor activation produces vasodilation. Indeed, the CB1 receptor antagonist rimonabant inhibited hypotension induced by endotoxin shock and hemorrhagic shock, as well as increasing survival (Varga et al., 1998). Though cannabinoids are yet to be explored in the context of TBI-induced changes in cerebral blood flow, CB1 receptor antagonism may prove to be a potential target for the treatment of TBI-induced hypotension.
Cell Structure/Remodeling
The key biological idea that structure dictates function also holds true for the neurophysiology of TBI. The shearing and tearing forces of TBI and subsequent secondary injury cascades produce changes in cell architecture, extracellular matrices, and the balance of fluid homeostasis, that impair neuronal function often both in a focal and/or diffuse manner throughout the brain (Gaetz, 2004). The use of cannabinoids has thus far been linked to protection against several of the CNS structural changes associated with TBI, with 2-AG being the most frequently studied eCB in this area.
While a traumatic insult can result in the rapid onset of cerebral oedema, exogenously administered 2-AG protects against TBI-induced oedema (Panikashvili et al., 2001, 2005). The observation that no such oedema protection was found following 2-AG administration in CB1 receptor-/- mice (Panikashvili et al., 2005) suggests that this protection requires CB1 receptor activation. Changes in protein physiology have also been found to occur following TBI. Specifically, the presence of protein aggregates such as amyloid-β plaques (Johnson et al., 2010), p-tau (Goldstein et al., 2012), and TDP-43 (Smith et al., 1999), have been found within hours following TBI. These proteins are thought to accumulate from damaged axons and as a result of a disturbed balance between genesis and catabolism (Johnson et al., 2010). MAGL inhibitors decrease amyloid-β protein and its precursor molecule APP, as well as p-tau and TDP-43 (Zhang et al., 2014). MAGL inhibition also decreases astrocyte activation (Mayeux et al., 2016), while exogenous 2-AG following TBI reduces hippocampal CA-3 neuron loss (Panikashvili et al., 2001). These consistent protective effects of 2-AG across varied TBI-related structural pathologies point to its important role in maintaining cell structure and promoting remodeling.
Protective roles played by anandamide in injury-induced structural changes are yet to be ascertained. Though FAAH inhibition decreases APP expression post-injury, as well as increases synaptophysin (Tchantchou et al., 2014), a synaptic vesicle protein whose elimination impairs object recognition and spatial learning in mice (Schmitt et al., 2009). Furthermore, eCBs may not be working alone to offer protection from TBI-induced structural impairments. For example, estradiol decreased the number of TBI-induced immunoreactive astrocytes, which was inhibited by CB1 and CB2 receptor antagonists, while also increasing cerebral cortex mRNA levels of CB2 receptors (Lopez Rodriguez et al., 2011). These findings suggest that the regulatory activity of the eCB receptors in response to TBI may be mediated by endocrine as well as paracrine signaling mechanisms.
Traumatic brain injury is well described to increase CB1 and CB2 receptor expression, which includes disruption of diurnal rhythms of CB1 receptor expression (Martinez-Vargas et al., 2013). Post-injury treatment with a CB1 receptor antagonist reduces CB1 receptor expression at 6 weeks following injury (Wang et al., 2016), whereas ABHD6 inhibition produces increased CB1 and CB2 receptor expression (Tchantchou and Zhang, 2013). As such, TBI-induced increases in cannabinoid receptor expression are perhaps facilitated by 2-AG.
Neurogenic Pulmonary Oedema
Pulmonary complications are reported in 20–25% of TBI patients (Holland et al., 2003), and its severity is related to brain injury magnitude (Alvarez et al., 2015). The exact CNS circuits involved in NPE have yet to be identified, though a sudden rise in intracranial pressure, rapid sympathetic surge, increased systemic vascular resistance and increase in hydrostatic pressure in the pulmonary vasculature, as well as release of pro-inflammatory mediators may all contribute to interstitial pulmonary oedema formation (Brambrink and Dick, 1997). NPE rapidly occurs within hours of TBI onset in clinical populations (Alvarez et al., 2015), and within minutes in animal models (Atkinson et al., 1998), producing CNS hypoxia (Oddo et al., 2010) which further contributes to secondary injury. NPE is a much needed area of interest in the study of TBI.
While at the present time there are no studies evaluating the contributions of, or protection by, the eCB system to NPE following TBI, this may prove an interesting area of future investigation. Specifically, the lung possesses a basal tone of 2-AG (Avraham et al., 2008; Nomura et al., 2008), and recently it has been shown that resident lung macrophages express major components of the eCB system, CB1 and CB2 receptors as well as anandamide and 2-AG (Staiano et al., 2015). Furthermore, MAGL inhibition has already been found to be protective against LPS-induced acute lung injury in mice, and attenuated with CB1 and CB2 receptor antagonists (Costola-de-Souza et al., 2013).
Treatment of Behavioral Deficits of TBI
The heterogeneous clinical presentation of TBI pathology in populations of survivors is reminiscent of its cellular and molecular pathophysiology described above. TBI patients report changes in mental health (depression, irritability, anxiety, and personality changes), sleep disturbance, post-traumatic headaches, persistent fatigue, epilepsy, learning and memory deficits (manifested also as impairments in attention and processing speed [Vakil, 2005]), and balance disorders (Stéfan et al., 2016). Most frequently investigated measures in the pre-clinical TBI literature include neurological motor, and learning and memory impairments, leaving a wide breadth of TBI clinical effects yet to be studied. Once again, components of the eCB system may become active to compensate for TBI symptomology given what is currently known of its regulatory effects within these areas, two examples being pain, and anxiety and depression (Corcoran et al., 2015).
In this section, we review what is currently known of cannabinoids in the context of their ability to alter post-traumatic animal behavior (see Table 2).
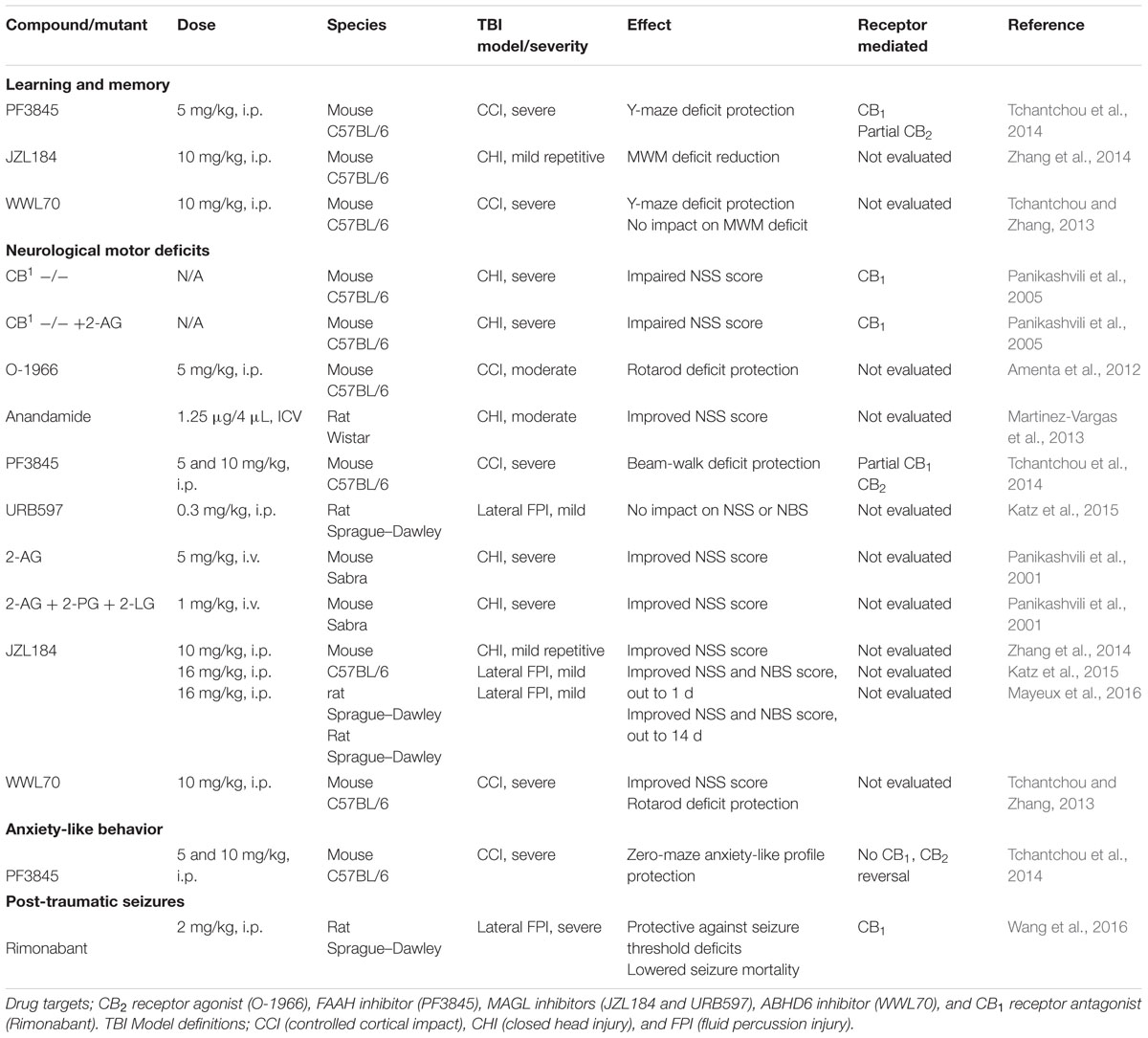
TABLE 2. Effect of cannabinoids on TBI-induced behavioral impairments.
Learning and Memory
Learning and memory impairments are among the most frequently reported symptoms following TBI, and are slow to recover with deficiencies reported 10 years later (Zec et al., 2001). The eCB system has been shown to play a well-documented role in memory regulation (reviewed in Mechoulam and Parker, 2011), and as such its manipulation holds considerable promise to address such a profound consequence of TBI.
Inhibition of the eCB hydrolytic enzymes FAAH (Tchantchou et al., 2014), MAGL (Zhang et al., 2014), and ABHD6 (Tchantchou and Zhang, 2013) have been shown to protect against TBI-induced memory impairments, suggesting that anandamide and 2-AG elevation post-TBI may offer protection from TBI-induced learning and memory deficits. The protective effects of 2-AG appear to be task specific, with ABHD6 inhibition showing learning and memory protection in a Y-maze task, but not a Morris water maze task. To date, only a Y-maze task has been used to evaluate the memory protective effects of FAAH inhibition, and this task-specific effect did not occur with a MAGL inhibitor. Mice are a well-used pre-clinical model organism to study the memory effects of TBI; however, they are known to perform behavioral tasks more readily, and with less error, when the task does not rely on aversive motivation (Stranahan, 2011). This attribute of mice may, in some part, contribute to the task-related differences seen between the Y-maze task (which uses exploratory behaviors associated with novelty) and the aversively motivated escape behavior necessary in the Morris water maze. Regardless, in clinical populations the most common memory process vulnerable to TBI involves difficulties applying active or effortful strategy’s in the learning or retrieval process (Vakil, 2005). Moving forward, the use of behavioral tasks able to selectively assess such frontal lobe-type memory impairments might improve the translational capacity of eCB TBI pre-clinical assessments (one such example being the Morris water maze Reversal Task, which evaluates cognitive flexibility).
Neurological Motor
Traumatic brain injury-induced neurological motor impairments currently represent the most frequently studied behavioral outcome measure in the TBI-cannabinoid literature. In clinical populations, neurological motor impairments seen as a result of TBI show spontaneous improvement over time, but one third of patients continue to experience neuromotor abnormalities 2 years after injury (Walker and Pickett, 2007). A variety of eCB system manipulations have thus far been found to be protective against the neurological motor deficits associated with murine models of TBI.
Both 2-AG and anandamide elevation provide protection against TBI-induced neurological motor deficits. MAGL inhibitors (Zhang et al., 2014; Katz et al., 2015; Mayeux et al., 2016), ABHD6 inhibitors (Tchantchou and Zhang, 2013), and exogenous 2-AG administration (Panikashvili et al., 2001), improve NSS in laboratory animal models of TBI. Moreover, ABHD6 inhibition also protects against TBI-induced rotarod deficits (Tchantchou and Zhang, 2013). Administration of exogenous 2-AG did not enhance NSS scores in CB1 receptor knockout mice subjected to TBI (Panikashvili et al., 2005), suggesting a CB1 receptor mechanism of action. FAAH inhibition has produced mixed findings in neurological motor tests, such as beam-walk deficit protection (Tchantchou et al., 2014) but no improvement on TBI-induced NSS deficits (Katz et al., 2015). In support of anandamide being protective against TBI-induced motor deficits, exogenous anandamide has also produced improved NSS performance (Martinez-Vargas et al., 2013). Full reversal, and partial reversal, of FAAH inhibitor mediated beam-walk deficit protection by respective CB2 and CB1 receptor antagonists (Tchantchou et al., 2014), suggest a role of both of these receptors in anandamide’s neuromotor deficit sparing effects. The involvement of the CB2 receptor is further supported by rotarod deficit protection from a CB2 receptor agonist (Amenta et al., 2012).
The role of entourage effects has also been evaluated in the area of TBI-induced neurological motor impairments. Co-release of endogenous fatty acid derivatives can potentiate 2-AG signaling, termed an entourage effect (Ben-Shabat et al., 1998; Lambert and Di Marzo, 1999; Lichtman et al., 2002). Administration of 2-AG with two related lipids that do not bind cannabinoid receptors, 2-LG and 2-PG, enhances recovery from TBI-induced NSS deficits (Panikashvili et al., 2001). Given FAAH is responsible for the degradation of various fatty acid amides in addition to anandamide (Boger et al., 2000), its various substrates may work in concert to ameliorate pathologies related to TBI. Thus any inferences drawn about anandamide through the use of FAAH inhibition needs to consider contributions of non-cannabinoid fatty acid amides.
Anxiety and Post-Traumatic Seizures
The signs of post-traumatic anxiety have been difficult to replicate in murine models of TBI (Tucker et al., 2016). Also, as there is a limited number of studies evaluating eCBs in this area, no definitive conclusions can be made. Thus far, only FAAH inhibition has been explored to address post-traumatic anxiety, and was found to protect against TBI-induced increases in anxiety-like behavior in mice (Tchantchou et al., 2014). This protection in the zero maze was unaffected by either CB1 or CB2 receptor antagonists, suggesting that these receptors are dispensable. Modeling post-traumatic epilepsy is time consuming and faces other challenges such as a low percentage of animals that develop epilepsy (Mazarati, 2006), however, recent models that produce consistent replication of spontaneous seizure activity following a TBI are available (Ping and Jin, 2016). Contrary to preclinical research demonstrating that the eCB system plays a protective roles against seizures (Wallace et al., 2001; Marsicano et al., 2003), a CB1 receptor antagonist has protected against injury-induced seizure threshold deficits as well as lowered seizure mortality (Wang et al., 2016), potentially through the disinhibition of GABAergic terminals.
This nascent body of data, suggests that eCB manipulations hold promise to treat injury-induced clinical symptoms outside of the more popular areas of learning and memory and neurological motor impairments.
Primary Phytocannabinoids and Traumatic Brain Injury
Although currently well over one hundred phytocannabinoids have been elucidated from the Cannabis sativa plant (Elsohly et al., 2017), the most extensively studied of these are THC and cannabidiol (CBD). The investigation of phytocannabinoids on TBI pathology not only holds topical relevance, but also but also holds promise as potential treatment for TBI and other disorders.
Without exception, all of the experimental work reviewed and listed in Tables 1 and 2 have used post-injury drug administration times ranging from 15 min to several days, clearly an attempt to simulate clinical intervention timing possibilities. However, clinical and pre-clinical findings provide evidence suggesting that the primary psychoactive constituent of Cannabis sativa, THC, is neuroprotective when administered prior to a traumatic insult. In a 3 year retrospective study of patients who had sustained a TBI, urine toxicology screen results showed decreased mortality in individuals with a positive THC screen (Nguyen et al., 2014). In two mouse models of CNS injury that yield cognitive deficits, pentylenetetrazole (an excitotoxic agent) and carbon monoxide induced hypoxic injury, prior administration of THC provided impairment protection (Assaf et al., 2011). Curiously, an extraordinarily a low dose of THC (i.e., 0.002 mg.kg-1) reduced injury-induced cognitive deficits in mice (Assaf et al., 2011). The authors explained this effect through the known biphasic effects of THC producing analgesia, acute hypothermia, and decreased locomotion at high doses (10 mg.kg-1), and producing hyperalgesia, hyperthermia, and increased locomotion at a low dose (0.002 mg.kg-1) (reviewed in Sarne et al., 2011). Such low dose effects of THC have been found to potentiate calcium entry into cells in vitro (Okada et al., 1992), increasing glutamate release, and thus may be mildly neurotoxic. Therefore, Assaf et al. (2011) hypothesized that low dose THC pre-treatment produced a pre-conditioning effect, where a mildly noxious stimulus becomes protective against a more severe subsequent insult, an effect known to occur in cardiology (Dirnagl et al., 2003) as well as cerebral ischaemia (Kitagawa et al., 1991). Moreover, the molecular signaling cascades behind cardiac and cerebral ischaemia preconditioning include activation of ERK and Akt (Dirnagl et al., 2003; Gidday, 2006), also shown to mediate the protective effects of ABHDB (Tchantchou and Zhang, 2013) and MAGL (Mayeux et al., 2016) inhibition following TBI.
Even though 80–90% of THC is excreted from individuals within 5 days of administration, the remaining slow release of lipophilic THC from lipid-storage compartments result in its long terminal half-life in plasma (Huestis, 2007). As such, individuals may experience very low plasma THC concentrations for prolonged periods after each application. Although the clinical study of TBI-induced mortality reported no data to quantify levels of THC in the THC positive individuals, the low dose THC in CNS injured mice may mimic the pharmacokinetics of THC in humans. This presumed prolonged exposure of THC due to its pharmacokinetics, as well as other potentially neuroprotective cannabinoids, such as CBD (Perez et al., 2013), may be responsible for the survival effects found in cannabis-exposed TBI patients. A finding of increased clinical relevance, is that post-conditioning (when the mildly noxious stimulus is applied after the insult) with low dose THC also produced cognitive sparing effects in mice (Assaf et al., 2011). These findings, however, remain controversial, and are yet to be replicated in animal models of TBI.
The phytocannabinoid CBD, currently being investigated in clinical trials for its seizure reduction potential in Tuberous Sclerosis Complex (Gw Research Ltd, 2016), has known anti-inflammatory properties. Although CBD does not bind CB1 and CB2 receptors, it activates the g-protein coupled receptor GPR55 (Ryberg et al., 2007), inhibits nucleoside transporter 1 (Carrier et al., 2006), inhibits sodium channels (Hill et al., 2014), and produces increased extracellular adenosine concentrations that consequently downregulate inflammatory cells through the adenosine A2A receptor (Ohta and Sitkovsky, 2001; Hasko and Pacher, 2008). While there are no studies at present which have investigated the anti-inflammatory effects of CBD following TBI, CBD has reduced FosB expression following cryogenic spinal cord injury (Kwiatkoski et al., 2012), and lowered iNos expression in a mouse model of tauopathy (Casarejos et al., 2013). As such CBD may be a promising future avenue of investigation in the study of neuroinflammation in response to brain injury.
Concluding Remarks and Future Directions
The eCB system, through release of its endogenous ligands or by changes in cannabinoid receptor constitutive activity, possesses promise in the treatment of diverse TBI pathology. An important step forward in understanding the role that the eCB system plays in TBI pathology includes not only the full characterization of ligands targeting cannabinoid receptors and eCB regulating enzymes, but also changes in cannabinoid receptors, eCB levels, and eCB regulating enzymes as a consequence of TBI. Another future area of therapeutic interest is non-CB1/CB2 receptor targets, such as TRPV1 receptors, and their potential contribution to the protective effects following TBI. Furthermore, alternative activation of CB1/CB2 receptors, such as potential entourage effects from other fatty acid derivatives, antagonism, or allosteric modulation, might impact functional selectivity and thus TBI-related outcomes also warrants further investigation. So too do the plant-derived phytocannabinoids represent an understudied yet promising group of compounds given the neuroprotective results obtained from other types of CNS injury. In particular, CBD as well as other phytocannabinoids which do not bind cannabinoid receptors, represent promising molecules to treat TBI.
To date, the only reported cannabinoid to be specifically evaluated for the treatment of TBI in patient populations is Dexanabinol, also known as HU211. While HU211 showed promise in animal models of TBI (Shohami et al., 1995), it failed to produce long term patient outcomes in one clinical trial despite some acute benefits (Knoller et al., 2002), and in a second study showed no short or long term benefits (Maas et al., 2006). Although HU211 has been described as a cannabinoid by virtue that it is an enantiomer of the potent synthetic cannabinoid agonist HU210, it does not bind or activate cannabinoid receptors. Instead, HU211acts as a non-competitive NMDA receptor antagonist (Feigenbaum et al., 1989). This therefore brings to light an important consideration of the classification of cannabinoids.
One consistently overlooked area across the study of TBI is the evaluation of the central penetration of systemically administered drugs. Pharmacological treatments will need to be assessed for their ability to cross the BBB. Also, it should be noted that TBI rapidly disrupts the BBB and lasts for three days post-injury (Baskaya et al., 1997). Furthermore, given the often biphasic nature of cannabinoid drugs, it is critical to move away from single dose pharmacology to full dose-response assessments, which may yield an increased understanding of the mechanism and potential of cannabinoids to treat TBI. Overall, the abundant and growing pre-clinical research suggests that the eCB system possesses many promising targets for new and existing drugs that may ameliorate diverse TBI pathology.
Author Contributions
LS performed the literature review and composed the article; AL contributed to the composition of the article.
Funding
This research was funded by a Ruth L. Kirshchstein Institutional National Research Service Award 1F31NS095628 (LS), and NIH award R01 DA032933 (AL), R01DA039942 (AL), P30DA033934 (AL).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
2-AG, 2-arachidonyl glycerol; 2-LG, 2-linoleoyl-glycerol; 2-PG, 2-palmitoyl-glycerol; AA, arachidonic acid; ABHD6, α/β-hydrolase domain-6; ABHD12, α/β-hydrolase domain-12; AEA, anandamide; APP, amyloid precursor protein; BBB, blood brain barrier; CCI, controlled cortical impact model of TBI; CDTA, calcium-dependent transacylase enzyme; CHI, closed head injury model of TBI; COX-2, cyclooxygenase-2; cPLA2, cytosolic phospholipase A2; DAGL-α, diacylglycerol lipase-α; DAGL-β, diacylglycerol lipase-β; eCB, endocannabinoid; EPSC, excitatory post-synaptic current; FAAH, fatty acid amide hydrolase; FPI, fluid percussion injury model of TBI; LTP, long term potentiation; MAGL, monoacylglycerol lipase; NArPE, N-arachidonoyl phosphatidylethanolamine; NBS, neurological behavioral score; NMDA, N-methyl-D-aspartate; NPE, neurogenic pulmonary oedema; NSS, neurological severity score; PLA2, phospholipase A2 enzyme; PLC, phospholipase C enzyme; p-tau, hyperphosphorylated tau; ROS, reactive oxygen species; TBI, traumatic brain injury; TDP-43, TAR DNA-binding protein; THC, Δ9-tetrahydrocannabinol.
References
Adams, J. H., Doyle, D., Ford, I., Gennarelli, T. A., Graham, D. I., and McLellan, D. R. (1989). Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 15, 49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x
Ahn, K., Johnson, D. S., Mileni, M., Beidler, D., Long, J. Z., McKinney, M. K., et al. (2009). Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 16, 411–420. doi: 10.1016/j.chembiol.2009.02.013
Alger, B. E., and Kim, J. (2011). Supply and demand for endocannabinoids. Trends Neurosci. 34, 304–315. doi: 10.1016/j.tins.2011.03.003
Alvarez, J., Quevedo, O., Furelos, L., Gonzalez, I., Llapur, E., Valeron, M., et al. (2015). Pulmonary complications in patients with severe brain injury. Pulm. Res. Respir. Med. 2, 69–74. doi: 10.17140/PRRMOJ-2-110
Amenta, P. S., Jallo, J. I., Tuma, R. F., and Elliott, M. B. (2012). A cannabinoid type 2 receptor agonist attenuates blood-brain barrier damage and neurodegeneration in a murine model of traumatic brain injury. J. Neurosci. Res. 90, 2293–2305. doi: 10.1002/jnr.23114
Arai, K., and Lo, E. H. (2009). Experimental models for analysis of oligodendrocyte pathophysiology in stroke. Exp. Transl. Stroke Med. 1:6. doi: 10.1186/2040-7378-1-6
Assaf, F., Fishbein, M., Gafni, M., Keren, O., and Sarne, Y. (2011). Pre- and post-conditioning treatment with an ultra-low dose of Δ9-tetrahydrocannabinol (THC) protects against pentylenetetrazole (PTZ)-induced cognitive damage. Behav. Brain Res. 220, 194–201. doi: 10.1016/j.bbr.2011.02.005
Atkinson, J. L. D., Anderson, R. E., and Murray, M. J. (1998). The early critical phase of severe head injury: importance of apnea and dysfunctional respiration. J. Trauma 45, 941–945. doi: 10.1097/00005373-199811000-00016
Atwood, B. K., and MacKie, K. (2010). CB 2: a cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 160, 467–479. doi: 10.1111/j.1476-5381.2010.00729.x
Avraham, Y., Magen, I., Zolotarev, O., Vorobiav, L., Nachmias, A., Pappo, O., et al. (2008). 2-Arachidonoylglycerol, an endogenous cannabinoid receptor agonist, in various rat tissues during the evolution of experimental cholestatic liver disease. Prostaglandins Leukot. Essent. Fatty Acids 79, 35–40. doi: 10.1016/j.plefa.2008.07.003
Baguley, I. J., Nott, M. T., Howle, A. A., Simpson, G. K., Browne, S., King, A. C., et al. (2012). Late mortality after severe traumatic brain injury in New South Wales: a multicentre study. Med. J. Aust. 196, 40–45. doi: 10.5694/mja11.10090
Baskaya, M. K., Rao, A. M., Doğan, A., Donaldson, D., and Dempsey, R. J. (1997). The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci. Lett. 226, 33–36. doi: 10.1016/S0304-3940(97)00239-5
Bell, R. L., Kennerly, D. A., Stanford, N., and Majerus, P. W. (1979). Diglyceride lipase: a pathway for arachidonate release from human platelets. Proc. Natl. Acad. Sci. U.S.A. 76, 3238–3241. doi: 10.1073/pnas.76.7.3238
Ben-Shabat, S., Fride, E., Sheskin, T., Tamiri, T., Rhee, M. H., Vogel, Z., et al. (1998). An entourage effect: inactive endogenous fatty acid glycerol esters enhance 2-arachidonoyl-glycerol cannabinoid activity. Eur. J. Pharmacol. 353, 23–31. doi: 10.1016/S0014-2999(98)00392-6
Béquet, F., Uzabiaga, F., Desbazeille, M., Ludwiczak, P., Maftouh, M., Picard, C., et al. (2007). Late mortality after severe traumatic brain injury in New South Wales: a multicentre studyrelease of endocannabinoids (as assessed by microdialysis coupled with LC/MS) in the rat hypothalamus. Eur. J. Neurosci. 26, 3458–3464. doi: 10.1111/j.1460-9568.2007.05900.x
Bisogno, T., Howell, F., Williams, G., Minassi, A., Cascio, M. G., Ligresti, A., et al. (2003). Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 163, 463–468. doi: 10.1083/jcb.200305129
Blankman, J. L., and Cravatt, B. F. (2013). Chemical probes of endocannabinoid metabolism. Pharmacol. Rev. 65, 849–871. doi: 10.1124/pr.112.006387
Blankman, J. L., Simon, G. M., and Cravatt, B. F. (2007). A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 14, 1347–1356. doi: 10.1016/j.chembiol.2007.11.006
Boger, D., Fecik, R., and Patterson, J. (2000). Fatty acid amide hydrolase substrate specificity. Bioorg. Med. Chem. Lett. 10, 2613–2616. doi: 10.1016/S0960-894X(00)00528-X
Brain Trauma Foundation, American Association of Neurological Surgeons, and Congress of Neurological Surgeons (2007). Guidelines for the management of severe traumatic brain injury. J Neurotrauma 24(Suppl.), S1–S106. doi: 10.1089/neu.2007.9990
Brambrink, A. M., and Dick, W. F. (1997). Neurogenic pulmonary edema. Pathogenesis, clinical picture and therapy. Anaesthesist 46, 953–963. doi: 10.1007/s001010050492
Bratton, S. L., Chestnut, R. M., Ghajar, J., McConnell Hammond, F. F., Harris, O. A., Hartl, R., et al. (2007). VIII. Intracranial pressure thresholds. J. Neurotrauma 24, S55–S58. doi: 10.1089/neu.2007.9988
Buczynski, M. W., Dumlao, D. S., and Dennis, E. A. (2009). Thematic review series: proteomics. an integrated omics analysis of eicosanoid biology. J. Lipid Res. 50, 1015–1038. doi: 10.1194/jlr.R900004-JLR200
Bullock, R., Chestnut, R., Ghajar, J., Gordon, D., Hartl, R., Newell, D. W., et al. (2006). Guidelines for the surgical management of traumatic brain injury. Neurosurgery 58, S2–S62.
Bullock, R., Zauner, A., Woodward, J. J., Myseros, J., Choi, S. C., Ward, J. D., et al. (1998). Factors affecting excitatory amino acid release following severe human head injury. J. Neurosurg. 89, 507–518. doi: 10.3171/jns.1998.89.4.0507
Cabral, G. A., Raborn, E. S., Griffin, L., Dennis, J., and Marciano-Cabral, F. (2008). CB2 receptors in the brain: role in central immune function. Br. J. Pharmacol. 153, 240–251. doi: 10.1038/sj.bjp.0707584
Carrier, E. J., Auchampach, J. A., and Hillard, C. J. (2006). Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc. Natl. Acad. Sci. U.S.A. 103, 7895–7900. doi: 10.1073/pnas.0511232103
Casarejos, M. J., Perucho, J., Gomez, A., Muñoz, M. P., Fernandez-Estevez, M., Sagredo, O., et al. (2013). Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. J. Alzheimers Dis. 35, 525–539. doi: 10.3233/JAD-130050
Chodobski, A., Zink, B. J., and Szmydynger-chodobska, J. (2012). Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2, 492–516. doi: 10.1007/s12975-011-0125-x
Chu, C. J., Huang, S. M., De Petrocellis, L., Bisogno, T., Ewing, S. A., Miller, J. D., et al. (2003). N-oleoyldopamine, a novel endogenous capsaicin-like lipid that produces hyperalgesia. J. Biol. Chem. 278, 13633–13639. doi: 10.1074/jbc.M211231200
Corcoran, L., Roche, M., and Finn, D. P. (2015). The role of the brain’s endocannabinoid system in pain and its modulation by stress. Int. Rev. Neurobiol. 125, 203–255. doi: 10.1016/bs.irn.2015.10.003
Corrigan, J. D., Selassie, A. W., and Orman, J. A. (2010). The epidemiology of traumatic brain injury. J. Head Trauma Rehabil. 25, 72–80. doi: 10.1097/HTR.0b013e3181ccc8b4
Costa, B., Comelli, F., Bettoni, I., Colleoni, M., and Giagnoni, G. (2008). The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: involvement of CB1, TRPV1 and PPARgamma receptors and neurotrophic factors. Pain 139, 541–550. doi: 10.1016/j.pain.2008.06.003
Costola-de-Souza, C., Ribeiro, A., Ferraz-de-Paula, V., Calefi, A. S., Aloia, T. P. A., Gimenes-Júnior, J. A., et al. (2013). Monoacylglycerol lipase (MAGL) inhibition attenuates acute lung injury in mice. PLoS ONE 8:e77706. doi: 10.1371/journal.pone.0077706
Cravatt, B. F., Demarest, K., Patricelli, M. P., Bracey, M. H., Giang, D. K., Martin, B. R., et al. (2001). Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 98, 9371–9376. doi: 10.1073/pnas.161191698
Cravatt, B. F., Giang, D. K., Mayfield, S. P., Boger, D. L., Lerner, R. A., and Gilula, N. B. (1996). Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384, 83–87. doi: 10.1038/384083a0
Deutsch, D., and Chin, S. (1993). Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 46, 791–796. doi: 10.1016/0006-2952(93)90486-G
Deutsch, D. G., Goligorsky, M. S., Schmid, P. C., Krebsbach, R. J., Schmid, H. H. O., Das, S. K., et al. (1997). Production and physiological actions of anandamide in the vasculature of the rat kidney. J. Clin. Invest. 100, 1538–1546. doi: 10.1172/JCI119677
Devane, W. A., Dysarz, F. A. III, Johnson, M. R., Melvin, L. S., and Howlett, A. C. (1988). Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 34, 605–613.
Devane, W. A., Hanus, L., Breuer, A., Pertwee, R. G., Stevenson, L. A., Griffin, G., et al. (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258, 1946–1949. doi: 10.1126/science.1470919
Di Marzo, V. (2008). Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 7, 438–455. doi: 10.1038/nrd2553
Dinh, T. P., Carpenter, D., Leslie, F. M., Freund, T. F., Katona, I., Sensi, S. L., et al. (2002). Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. U.S.A. 99, 10819–10824. doi: 10.1073/pnas.152334899
Dirnagl, U., Simon, R. P., and Hallenbeck, J. M. (2003). Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 26, 248–254. doi: 10.1016/S0166-2236(03)00071-7
Egertová, M., Giang, D. K., Cravatt, B. F., and Elphick, M. R. (1998). A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc. Biol. Sci. 265, 2081–2085. doi: 10.1098/rspb.1998.0543
Elsohly, M. A., Radwan, M. M., Gul, W., Chandra, S., and Galal, A. (2017). “Phytochemistry of Cannabis sativa L,” in Phytocannabinoids, eds A. D. Kinghorn, H. Falk, S. Gibbons, and J. Kobayashi (Cham: Springer International Publishing). doi: 10.1007/978-3-319-45541-9
Faul, M., Wald, M. M., Rutland-Brown, W., Sullivent, E. E., and Sattin, R. W. (2007). Using a cost-benefit analysis to estimate outcomes of a clinical treatment guideline: testing the Brain Trauma Foundation guidelines for the treatment of severe traumatic brain injury. J. Trauma 63, 1271–1278. doi: 10.1097/TA.0b013e3181493080
Feigenbaum, J. J., Bergmann, F., Richmond, S. A., Mechoulam, R., Nadler, V., Kloog, Y., et al. (1989). Nonpsychotropic cannabinoid acts as a functional N-methyl-D-aspartate receptor blocker. Proc. Natl. Acad. Sci. U.S.A. 86, 9584–9587. doi: 10.1073/pnas.86.23.9584
Floyd, C. L., Gorin, F. A., and Lyeth, B. G. (2010). Mechanical strain injury increases intracellular sodium and reverses Na+ /Ca2+ exchange in cortical astrocytes. Glia 51, 35–46. doi: 10.1002/glia.20183
Gaetz, M. (2004). The neurophysiology of brain injury. Clin. Neurophysiol. 115, 4–18. doi: 10.1016/S1388-2457(03)00258-X
Gertsch, J., Pertwee, R. G., and Di Marzo, V. (2010). Phytocannabinoids beyond the Cannabis plant – do they exist? Br. J. Pharmacol. 160, 523–529. doi: 10.1111/j.1476-5381.2010.00745.x
Gidday, J. M. (2006). Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 7, 437–448. doi: 10.1038/nrn1927
Goldstein, L. E., Fisher, A. M., Tagge, C. A., Zhang, X.-L., Velisek, L., Sullivan, J. A., et al. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4:134ra60. doi: 10.1126/scitranslmed.3003716
Gulyas, A. I., Cravatt, B. F., Bracey, M. H., Dinh, T. P., Piomelli, D., Boscia, F., et al. (2004). Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur. J. Neurosci. 20, 441–458. doi: 10.1111/j.1460-9568.2004.03428.x
Gw Research Ltd (2016). A Randomized Controlled Trial of Cannabidiol (GWP42003-P, CBD) for Seizures in Tuberous Sclerosis Complex (GWPCARE6). Available at: https://clinicaltrials.gov/show/NCT02544763
Hansen, H. H., Schmid, P. C., Bittigau, P., Lastres-Becker, I., Berrendero, F., Manzanares, J., et al. (2002). Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem. 78, 1415–1427. doi: 10.1046/j.1471-4159.2001.00542.x
Hasko, G., and Pacher, P. (2008). A2A receptors in inflammation and injury: lessons learned from transgenic animals. J. Leukoc. Biol. 83, 447–455. doi: 10.1189/jlb.0607359
Heimann, A. S., Gomes, I., Dale, C. S., Pagano, R. L., Gupta, A., de Souza, L. L., et al. (2007). Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 20588–20593. doi: 10.1073/pnas.0706980105
Herkenham, M., Lynn, A. B., Johnson, M. R., Melvin, L. S., de Costa, B. R., and Rice, K. C. (1991). Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci. 11, 563–583.
Hill, A. J., Jones, N. A., Smith, I., Hill, C. L., Williams, C. M., Stephens, G. J., et al. (2014). Voltage-gated sodium (NaV) channel blockade by plant cannabinoids does not confer anticonvulsant effects per se. Neurosci. Lett. 566, 269–274. doi: 10.1016/j.neulet.2014.03.013
Hillard, C. J. (2000a). Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 61, 3–18. doi: 10.1016/S0090-6980(00)00051-4
Hind, W. H., Tufarelli, C., Neophytou, M., Anderson, S. I., England, T. J., and O’Sullivan, S. E. (2015). Endocannabinoids modulate human blood-brain barrier permeability in vitro. Br. J. Pharmacol. 172, 3015–3027. doi: 10.1111/bph.13106
Holland, M. C., Mackersie, R. C., Morabito, D., Campbell, A. R., Kivett, V. A., Patel, R., et al. (2003). The development of acute lung injury is associated with worse neurologic outcome in patients with severe traumatic brain injury. J. Trauma 55, 106–111. doi: 10.1097/01.TA.0000071620.27375.BE
Hu, D.-E., Easton, A. S., and Fraser, P. A. (2005). TRPV1 activation results in disruption of the blood-brain barrier in the rat. Br. J. Pharmacol. 146, 576–584. doi: 10.1038/sj.bjp.0706350
Huestis, M. (2007). Human cannabinoid pharmacokinetics. Chem. Biodivers. 4, 1770–1804. doi: 10.1002/cbdv.200790152
Johnson, V. E., Stewart, W., and Smith, D. H. (2010). Traumatic brain injury and amyloid-β pathology: a link to Alzheimer’s disease? Nat. Rev. Neurosci. 11, 361–370. doi: 10.1038/nrn2808
Katz, P. S., Sulzer, J. K., Impastato, R. A., Teng, S. X., Rogers, E. K., and Molina, P. E. (2015). Endocannabinoid degradation inhibition improves neurobehavioral function, blood-brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J. Neurotrauma 32, 297–306. doi: 10.1089/neu.2014.3508
Kelly, D. F., Martin, N. A., Kordestani, R., Counelis, G., Hovda, D. A., Bergsneider, M., et al. (1997). Cerebral blood flow as a predictor of outcome following traumatic brain injury. J. Neurosurg. 86, 633–641. doi: 10.3171/jns.1997.86.4.0633
Kigerl, K. A., Gensel, J. C., Ankeny, D. P., Alexander, J. K., Donnelly, D. J., and Popovich, P. G. (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009
Kitagawa, K., Matsumoto, M., Kuwabara, K., Tagaya, M., Ohtsuki, T., Hata, R., et al. (1991). “Ischemic tolerance” phenomenon detected in various brain regions. Brain Res. 561, 203–211. doi: 10.1016/0006-8993(91)91596-S
Knoller, N., Levi, L., Shoshan, I., Reichenthal, E., Razon, N., Rappaport, Z. H., et al. (2002). Dexanabinol (HU-211) in the treatment of severe closed head injury: a randomized, placebo-controlled, phase II clinical trial. Crit. Care Med. 30, 548–554. doi: 10.1097/00003246-200203000-00009
Kwiatkoski, M., Guimaraes, F. S., and Del-Bel, E. (2012). Cannabidiol-treated rats exhibited higher motor score after cryogenic spinal cord injury. Neurotox. Res. 21, 271–280. doi: 10.1007/s12640-011-9273-8
Lambert, D. M., and Di Marzo, V. (1999). The palmitoylethanolamide and oleamide enigmas: Are these two fatty acid amides cannabimimetic? Curr. Med. Chem. 6, 757–773.
Langlois, J. A., Rutland-Brown, W., and Wald, M. M. (2006). The epidemiology and impact of traumatic brain injury: a brief overview. J. Head Trauma Rehabil. 21, 375–378. doi: 10.1097/00001199-200609000-00001
Lichtman, A. H., Hawkins, E. G., Griffin, G., and Cravatt, B. F. (2002). Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J. Pharmacol. Exp. Ther. 302, 73–79. doi: 10.1124/jpet.302.1.73
Long, J. Z., Nomura, D. K., Vann, R. E., Walentiny, D. M., Booker, L., Jin, X., et al. (2009). Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 20270–20275. doi: 10.1073/pnas.0909411106
Lopez Rodriguez, A., Belen, B., Romero-Zerbo, S. Y., Rodriguez-Rodriguez, N., Bellini, M. J., Rodriguez De Fonseca, F., et al. (2011). Estradiol decreases cortical reactive astrogliosis after brain injury by a mechanism involving cannabinoid receptors. Cereb. Cortex 21, 2046–2055. doi: 10.1093/cercor/bhq277
Lu, D., Qu, C., Goussev, A., Jiang, H., Lu, C., Schallert, T., et al. (2007). Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J. Neurotrauma 24, 1132–1146. doi: 10.1089/neu.2007.0288
Maas, A. I. R., Murray, G., Henney, H., Kassem, N., Legrand, V., Mangelus, M., et al. (2006). Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol. 5, 38–45. doi: 10.1016/S1474-4422(05)70253-2
Mackie, K. (2006). Mechanisms of CB1 receptor signaling: endocannabinoid modulation of synaptic strength. Int. J. Obes. 30(Suppl. 1), S19–S23. doi: 10.1038/sj.ijo.0803273
Marsicano, G., Goodenough, S., Monory, K., Hermann, H., Eder, M., Cannich, A., et al. (2003). CB1 cannabinoid receptors and on-demand defense. Science 84, 84–88. doi: 10.1126/science.1088208
Martinez-Vargas, M., Morales-Gomez, J., Gonzalez-Rivera, R., Hernandez-Enriquez, C., Perez-Arredondo, A., Estrada-Rojo, F., et al. (2013). Does the neuroprotective role of anandamide display diurnal variations? Int. J. Mol. Sci. 14, 23341–23355. doi: 10.3390/ijms141223341
Mayer, S. A., and Badjatia, N. (2010). Merritt’s Neurology, eds L. P. Rowland and T. A. Pedley. Philadelphia, PA: Wolters Kluwer Lippincott Williams & Wilkins.
Mayeux, J. P., Katz, P. S., Edwards, S., Middleton, J., and Molina, P. (2016). Inhibition of endocannabinoid degradation improves outcomes from mild traumatic brain injury: a mechanistic role for synaptic hyperexcitability. J. Neurotrauma 34, 436–443. doi: 10.1089/neu.2016.4452
Mazarati, A. (2006). Is posttraumatic epilepsy the best model of posttraumatic epilepsy? Epilepsy Curr. 6, 213–214. doi: 10.1111/j.1535-7511.2006.00149.x
Mazzeo, A. T., Brophy, G. M., Gilman, C. B., Alves, O. L., Robles, J. R., Hayes, R. L., et al. (2009). Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: results from a prospective randomized trial. J. Neurotrauma 26, 2195–2206. doi: 10.1089/neu.2009.1012
Mbye, L. H. A. N., Singh, I. N., Carrico, K. M., Saatman, K. E., and Hall, E. D. (2009). Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. J. Cereb. Blood Flow Metab. 29, 87–97. doi: 10.1038/jcbfm.2008.93
Mechoulam, R., Ben-Shabat, S., Hanus, L., Ligumsky, M., Kaminski, N. E., Schatz, A. R., et al. (1995). Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 50, 83–90. doi: 10.1016/0006-2952(95)00109-D
Mechoulam, R., and Goani, Y. (1967). Recent advances in the chemistry of hashish. Fortschr. Chem. Org. Naurst. 25, 175–213. doi: 10.1007/978-3-7091-8164-5_6
Mechoulam, R., and Lichtman, A. H. (2003). Stout guards of the central nervous system. Science 302, 65–67. doi: 10.1126/science.1091256
Mechoulam, R., and Parker, L. A. (2011). The endocannabinoid system and the brain. Annu. Rev. Psychol. 64, 21–47. doi: 10.1146/annurev-psych-113011-143739
Melck, D., Bisogno, T., De Petrocellis, L., Chuang, H., Julius, D., Bifulco, M., et al. (1999). Unsaturated long-chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB1 cannabinoid receptors. Biochem. Biophys. Res. Commun. 262, 275–284. doi: 10.1006/bbrc.1999.1105
Mestre, L., Correa, F., Arévalo-Martín, A., Molina-Holgado, E., Valenti, M., Ortar, G., et al. (2005). Pharmacological modulation of the endocannabinoid system in a viral model of multiple sclerosis. J. Neurochem. 92, 1327–1339. doi: 10.1111/j.1471-4159.2004.02979.x
Meyer, M. J., Megyesi, J., Meythaler, J., Murie-Fernandez, M., Aubut, J.-A., Foley, N., et al. (2010). Acute management of acquired brain injury part II: an evidence-based review of pharmacological interventions. Brain Inj. 24, 706–721. doi: 10.3109/02699051003692126
Mills, B., Yepes, A., and Nugent, K. (2015). Synthetic cannabinoids. Can. Med. Assoc. J. 350, 59–62. doi: 10.1503/cmaj.130510
Mukhopadhyay, P., Pan, H., Rajesh, M., Bátkai, S., Patel, V., Harvey-White, J., et al. (2010a). CB 1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br. J. Pharmacol. 160, 657–668. doi: 10.1111/j.1476-5381.2010.00769.x
Mukhopadhyay, P., Rajesh, M., Bátkai, S., Patel, V., Kashiwaya, Y., Liaudet, L., et al. (2010b). CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc. Res. 85, 773–784. doi: 10.1093/cvr/cvp369
Munro, S., Thomas, K. L., and Abu-Shaar, M. (1993). Molecular characterization of a peripheral receptor for cannabinoids. Nature 365, 61–65. doi: 10.1038/365061a0
Nguyen, B. M., Kim, D., Bricker, S., Bongard, F., Neville, A., Putnam, B., et al. (2014). Effect of marijuana use on outcomes in traumatic brain injury. Am. Surg. 80, 979–983.
Niogi, S. N., Mukherjee, P., Ghajar, J., Johnson, C., Kolster, R. A., Sarkar, R., et al. (2008). Extent of microstructural white matter injury in postconcussive syndrome correlates with impaired cognitive reaction time: a 3T diffusion tensor imaging study of mild traumatic brain injury. Am. J. Neuroradiol. 29, 967–973. doi: 10.3174/ajnr.A0970
Niphakis, M. J., Johnson, D. S., Ballard, T. E., Stiff, C., and Cravatt, B. F. (2012). O -Hydroxyacetamide carbamates as a highly potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 3, 418–426. doi: 10.1021/cn200089j
Nomura, D. K., Hudak, C. S. S., Ward, A. M., Burston, J. J., Issa, R. S., Fisher, K. J., et al. (2008). Monoacylglycerol lipase regulates 2-arachidonoylglycerol action and arachidonic acid levels. Bioorg. Med. Chem. Lett. 18, 5875–5878. doi: 10.1016/j.bmcl.2008.08.007
Nomura, D. K., Morrison, B. E., Blankman, J. L., Jonathan, Z., Kinsey, S. G., Marcondes, M. C. G., et al. (2011). Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334, 809–813. doi: 10.1126/science.1209200.Endocannabinoid
Oddo, M., Nduom, E., Frangos, S., MacKenzie, L., Chen, I., Maloney-Wilensky, E., et al. (2010). Acute lung injury is an independent risk factor for brain hypoxia after severe traumatic brain injury. Neurosurgery 67, 338–344. doi: 10.1227/01.NEU.0000371979.48809.D9
Ohta, A., and Sitkovsky, M. (2001). Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414, 916–920. doi: 10.1038/414916a
Oka, S., Wakui, J., Ikeda, S., Yanagimoto, S., Kishimoto, S., Gokoh, M., et al. (2006). Involvement of the cannabinoid CB2 receptor and its endogenous ligand 2-arachidonoylglycerol in oxazolone-induced contact dermatitis in mice. J. Immunol. 177, 8796–8805. doi: 10.4049/jimmunol.177.12.8796
Okada, M., Urae, A., Mine, K., Shoyama, Y., Iwasaki, K., and Fujiwara, M. (1992). The facilitating and suppressing effects of delta 9-tetrahydrocannabinol on the rise in intrasynaptosomal Ca2+ concentration in rats. Neurosci. Lett. 140, 55–58. doi: 10.1038/sj.jcbfm.9600047
Panikashvili, D., Mechoulam, R., Beni, S. M., Alexandrovich, A., and Shohami, E. (2005). CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J. Cereb. Blood Flow Metab. 25, 477–484. doi: 10.1038/sj.jcbfm.9600047
Panikashvili, D., Shein, N. A., Mechoulam, R., Trembovler, V., Kohen, R., Alexandrovich, A., et al. (2006). The endocannabinoid 2-AG protects the blood-brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol. Dis. 22, 257–264. doi: 10.1016/j.nbd.2005.11.004
Panikashvili, D., Simeonidou, C., Ben-Shabat, S., Hanus, L., Breuer, A., Mechoulam, R., et al. (2001). An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413, 527–531. doi: 10.1038/35097089
Pelosi, P., Severgnini, P., and Chiaranda, M. (2005). An integrated approach to prevent and treat respiratory failure in brain-injured patients. Curr. Opin. Crit. Care 11, 37–42. doi: 10.1097/00075198-200502000-00006
Perez, M., Benitez, S. U., Cartarozzi, L. P., del Bel, E., Guimarães, F. S., and Oliveira, A. L. R. (2013). Neuroprotection and reduction of glial reaction by cannabidiol treatment after sciatic nerve transection in neonatal rats. Eur. J. Neurosci. 38, 3424–3434. doi: 10.1111/ejn.12341
Pertwee, R. G., and Ross, R. A. (2002). Cannabinoid receptors and their ligands. Prostaglandins Leukot. Essent. Fatty Acids 66, 101–121. doi: 10.1054/plef.2001.0341
Ping, X., and Jin, X. (2016). Chronic posttraumatic epilepsy following neocortical undercut lesion in mice. PLoS ONE 11:e0158231. doi: 10.1371/journal.pone.0158231
Porter, A. C., Sauer, J. M., Knierman, M. D., Becker, G. W., Berna, M. J., Bao, J., et al. (2002). Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J. Pharmacol. Exp. Ther. 301, 1020–1024. doi: 10.1124/jpet.301.3.1020
Raghupathi, R. (2004). Cell death mechanisms following traumatic brain injury. Brain Pathol. 14, 215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x
Reggio, P. H. (2002). Endocannabinoid structure-activity relationships for interaction at the cannabinoid receptors. Prostaglandins Leukot. Essent. Fatty Acids 66, 143–160. doi: 10.1054/plef.2001.0343
Reilly, P. L. (2001). Brain injury: the pathophysiology of the first hours.’Talk and Die revisited’. J. Clin. Neurosci. 8, 398–403. doi: 10.1054/jocn.2001.0916
Rubovitch, V., Ten-Bosch, M., Zohar, O., Harrison, C. R., Tempel-Brami, C., Stein, E., et al. (2011). A mouse model of blast-induced mild traumatic brain injury. Exp. Neurol. 232, 280–289. doi: 10.1016/j.expneurol.2011.09.018
Ryberg, E., Larsson, N., Sjögren, S., Hjorth, S., Hermansson, N.-O., Leonova, J., et al. (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 152, 1092–1101. doi: 10.1038/sj.bjp.0707460
Sahuquillo, J., and Arikan, F. (2006). Decompressive craniectomy for the treatment of refractory high intracranial pressure in traumatic brain injury. Cochrane database Syst. Rev. 1:CD003983. doi: 10.1002/14651858.CD003983.pub2
Sarne, Y., Asaf, F., Fishbein, M., Gafni, M., and Keren, O. (2011). The dual neuroprotective-neurotoxic profile of cannabinoid drugs. Br. J. Pharmacol. 163, 1391–1401. doi: 10.1111/bph.2011.163
Schmitt, U., Tanimoto, N., Seeliger, M., Schaeffel, F., and Leube, R. E. (2009). Detection of behavioral alterations and learning deficits in mice lacking synaptophysin. Neuroscience 162, 234–243. doi: 10.1016/j.neuroscience.2009.04.046
Sen, A. P., and Gulati, A. (2010). Use of magnesium in traumatic brain injury. Neurotherapeutics 7, 91–99. doi: 10.1016/j.nurt.2009.10.014
Shah, S., Yallampalli, R., Merkley, T. L., McCauley, S. R., Bigler, E. D., Macleod, M., et al. (2012). Diffusion tensor imaging and volumetric analysis of the ventral striatum in adults with traumatic brain injury. Brain Inj. 26, 201–210. doi: 10.3109/02699052.2012.654591
Shiina, G., Onuma, T., Kameyama, M., Shimosegawa, Y., Ishii, K., Shirane, R., et al. (1998). Sequential assessment of cerebral blood flow in diffuse brain injury by 123I-iodoamphetamine single-photon emission CT. Am. J. Neuroradiol. 19, 297–302.
Shohami, E., Novikov, M., and Bass, R. (1995). Long-term effect of Hu-211, a novel noncompetitive nmda antagonist, on motor and memory functions after closed-head injury in the rat. Brain Res. 674, 55–62. doi: 10.1016/0006-8993(94)01433-I
Siegel, G. J. (1999). “Synaptic transmission and cellular signaling: an overview”, in Basic Neurochemistry, eds M. D. Agranoff, B. W. Albers, R. W. Fisher, and S. K. Uhler (Philadelphia, PA: Lippincott-Raven).
Sigel, E., Baur, R., Rácz, I., Marazzi, J., Smart, T. G., Zimmer, A., et al. (2011). The major central endocannabinoid directly acts at GABA(A) receptors. Proc. Natl. Acad. Sci. U.S.A. 108, 18150–18155. doi: 10.1073/pnas.1113444108
Smart, D., Gunthorpe, M. J., Jerman, J. C., Nasir, S., Gray, J., Muir, A. I., et al. (2000). The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br. J. Pharmacol. 129, 227–230. doi: 10.1038/sj.bjp.0703050
Smith, D. H., Chen, X., Nonaka, M., Trojanowski, J. Q., Lee, V. M., Saatman, K. E., et al. (1999). Accumulation of amyloid β and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J. Neuropathol. Exp. Neurol. 58, 982–992. doi: 10.1097/00005072-199909000-00008
Staiano, R. I., Loffredo, S., Borriello, F., Iannotti, F. A., Piscitelli, F., Orlando, P., et al. (2015). Human lung-resident macrophages express CB1 and CB2 receptors whose activation inhibits the release of angiogenic and lymphangiogenic factors. J. Leukoc. Biol. 99, 531–540. doi: 10.1189/jlb.3HI1214-584R
Stéfan, A., Mathé, J. F., Dhenain, M., Blanchard, P., Blondet, E., Mathé, J. F., et al. (2016). What are the disruptive symptoms of behavioral disorders after traumatic brain injury? A systematic review leading to recommendations for good practices. Ann. Phys. Rehabil. Med. 59, 5–17. doi: 10.1016/j.rehab.2015.11.002
Stein, S. C., Georgoff, P., Meghan, S., Mizra, K., and Sonnad, S. S. (2010). 150 years of treating severe traumatic brain injury: a systematic review of progress in mortality. J. Neurotrauma 27, 1343–1353. doi: 10.1089/neu.2009.1206
Sticht, M. A., Limebeer, C. L., Rafla, B. R., Abdullah, R. A., Poklis, J. L., Ho, W., et al. (2016). Endocannabinoid regulation of nausea is mediated by 2-arachidonoylglycerol (2-AG) in the rat visceral insular cortex. Neuropharmacology 102, 92–102. doi: 10.1016/j.neuropharm.2015.10.039
Stiver, S. I. (2009). Complications of decompressive craniectomy for traumatic brain injury. Neurosurg. Focus 26:E7. doi: 10.3171/2009.4.FOCUS0965
Stranahan, A. (2011). Similarities and differences in spatial learning and object recognition between young male C57Bl/6J mice and Sprague-Dawley rats. Behav. Neurosci. 125, 791–795. doi: 10.1037/a0025133
Sugiura, T., Kondo, S., Sukagawa, A., Nakane, S., Shinoda, A., Itoh, K., et al. (1995). 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 215, 89–97. doi: 10.1006/bbrc.1995.2437
Tapia-Perez, J. H., Sanchez-Aguilar, M., Torres-Corzo, J. G., Gordillo-Moscoso, A., Martinez-Perez, P., Madeville, P., et al. (2008). Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758). J. Neurotrauma 25, 1011–1017. doi: 10.1089/neu.2008.0554
Tchantchou, F., Tucker, L. B., Fu, A. H., Bluett, R. J., McCabe, J. T., Patel, S., et al. (2014). The fatty acid amide hydrolase inhibitor PF-3845 promotes neuronal survival, attenuates inflammation and improves functional recovery in mice with traumatic brain injury. Neuropharmacology 85, 427–439. doi: 10.1016/j.neuropharm.2014.06.006
Tchantchou, F., and Zhang, Y. (2013). Selective inhibition of alpha/beta-hydrolase domain 6 attenuates neurodegeneration, alleviates blood brain barrier breakdown, and improves functional recovery in a mouse model of traumatic brain injury. J. Neurotrauma 30, 565–579. doi: 10.1089/neu.2012.2647
Temkin, N. R., Anderson, G. D., Winn, H. R., Ellenbogen, R. G., Britz, G. W., Schuster, J., et al. (2007). Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 6, 29–38. doi: 10.1016/S1474-4422(06)70630-5
Titomanlio, L., Fernández-López, D., Manganozzi, L., Moretti, R., Vexler, Z. S., and Gressens, P. (2015). Pathophysiology and neuroprotection of global and focal perinatal brain injury: lessons from animal models. Pediatr. Neurol. 52, 566–584. doi: 10.1016/j.pediatrneurol.2015.01.016
Tucker, L. B., Burke, J. F., Fu, A. H., and McCabe, J. T. (2016). Neuropsychiatric symptom modeling in male and female C57BL/6J mice after experimental traumatic brain injury. J. Neurotrauma doi: 10.1089/neu.2016.4508 [Epub ahead of print]
Vakil, E. (2005). The effect of moderate to severe traumatic brain injury (TBI) on different aspects of memory: a selective review. J. Clin. Exp. Neuropsychol. 27, 977–1021. doi: 10.1080/13803390490919245
Varga, R., Wagner, J. A., Bridgen, D. T., and Kunos, G. (1998). Platelet- and macrophage-derived endogenous cannabinoids are involved in endotoxin-induced hypotension. FASEB 12, 1035–1044.
Walker, W. C., and Pickett, T. C. (2007). Motor impairment after severe traumatic brain injury: a longitudinal multicenter study. J. Rehabil. Res. Dev. 44, 975–982. doi: 10.1682/JRRD.2006.12.0158
Wallace, M. J., Wiley, J. L., Martin, B. R., and DeLorenzo, R. J. (2001). Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. Eur. J. Pharmacol. 428, 51–57. doi: 10.1016/S0014-2999(01)01243-2
Wang, K., Liu, B. Y., and Ma, J. (2014). Research progress in traumatic brain penumbra. Chin. Med. J. 127, 1964–1968. doi: 10.3760/cma.j.issn.0366-6999.20120638
Wang, Q., Peng, Y., Chen, S., Gou, X., Hu, B., Du, J., et al. (2009). Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke 40, 2157–2164. doi: 10.1161/STROKEAHA.108.541490
Wang, X., Wang, Y., Zhang, C., Liu, C., Zhao, B., Wei, N., et al. (2016). CB1 receptor antagonism prevents long-term hyperexcitability after head injury by regulation of dynorphin-KOR system and mGluR5 in rat hippocampus. Brain Res. 1646, 174–181. doi: 10.1016/j.brainres.2016.05.055
Wiley, J. L., Marusich, J. A., and Huffman, J. W. (2014). Moving around the molecule: relationship between chemical structure and in vivo activity of synthetic cannabinoids. Life Sci. 97, 55–63. doi: 10.1016/j.lfs.2013.09.011
Woodcock, T., and Morganti-Kossmann, M. C. (2013). The role of markers of inflammation in traumatic brain injury. Front. Neurol. 4:18. doi: 10.3389/fneur.2013.00018
Zec, R. F., Zellers, D., Belman, J., Miller, J., Matthews, J., Femeau-Belman, D., et al. (2001). Long-term consequences of severe closed head injury on episodic memory. J. Clin. Exp. Neuropsychol. 23, 671–691. doi: 10.1076/jcen.23.5.671.1247
Zhang, J., Teng, Z., Song, Y., Hu, M., and Chen, C. (2014). Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J. Cereb. Blood Flow Metab. 35, 443–453. doi: 10.1038/jcbfm.2014.216
Zhang, J., Yang, Y., Li, H., Cao, J., and Xu, L. (2005). Amplitude/frequency of spontaneous mEPSC correlates to the degree of long-term depression in the CA1 region of the hippocampal slice. Brain Res. 1050, 110–117. doi: 10.1016/j.brainres.2005.05.032
Zhao, Z. C., Li, F., and Maiese, K. (2005). Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog. Neurobiol. 75, 207–246. doi: 10.1016/j.pneurobio.2005.02.004
Keywords: traumatic brain injury, cannabinoid, endocannabinoid, neuroprotection, phytocannabinoid
Citation: Schurman LD and Lichtman AH (2017) Endocannabinoids: A Promising Impact for Traumatic Brain Injury. Front. Pharmacol. 8:69. doi: 10.3389/fphar.2017.00069
Received: 15 October 2016; Accepted: 02 February 2017;
Published: 17 February 2017.
Edited by:
Rukiyah Van Dross-Anderson, The Brody School of Medicine at East Carolina University, USACopyright © 2017 Schurman and Lichtman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aron H. Lichtman, aron.lichtman@vcuhealth.org